 Belinda S. Hall
Belinda S. Hall Louise Tzung-Harn Hsieh
Louise Tzung-Harn Hsieh Sandra Sacre
Sandra Sacre Rachel E. Simmonds
Rachel E. Simmonds- 1Department of Microbial Sciences, School of Bioscience and Medicine, Faculty of Health and Medical Sciences, University of Surrey, Guildford, United Kingdom
- 2Brighton and Sussex Medical School, University of Sussex, Brighton, United Kingdom
Buruli ulcer (BU), caused by Mycobacterium ulcerans, is a devastating necrotizing skin disease. Key to its pathogenesis is mycolactone, the exotoxin virulence factor that is both immunosuppressive and cytotoxic. The discovery that the essential Sec61 translocon is the major cellular target of mycolactone explains much of the disease pathology, including the immune blockade. Sec61 inhibition leads to a loss in production of nearly all cytokines from monocytes, macrophages, dendritic cells and T cells, as well as antigen presentation pathway proteins and costimulatory molecules. However, there has long been evidence that the immune system is not completely incapable of responding to M. ulcerans infection. In particular, IL-1β was recently shown to be present in BU lesions, and to be induced from M. ulcerans-exposed macrophages in a mycolactone-dependent manner. This has important implications for our understanding of BU, showing that mycolactone can act as the “second signal” for IL-1β production without inhibiting the pathways of unconventional secretion it uses for cellular release. In this Perspective article, we validate and discuss this recent advance, which is entirely in-line with our understanding of mycolactone’s inhibition of the Sec61 translocon. However, we also show that the IL-1 receptor, which uses the conventional secretory pathway, is sensitive to mycolactone blockade at Sec61. Hence, a more complete understanding of the mechanisms regulating IL-1β function in skin tissue, including the transient intra-macrophage stage of M. ulcerans infection, is urgently needed to uncover the double-edged sword of IL-1β in BU pathogenesis, treatment and wound healing.
Introduction
Buruli ulcer (BU) is a neglected tropical disease resulting from subcutaneous infection by Mycobacterium ulcerans. It typically presents as painless, ulcerative skin lesions or as pre-ulcerative nodules, plaques and in oedematous forms (1). This environmentally acquired infection is reported in 33 countries world-wide, but is most common in West Africa and Australia (2, 3). The disease has a high morbidity, causing both disfigurement and disability (1). Untreated, the infection can become chronic and extend up to 15% of body surface-area. When diagnosed early, treatment with antibiotics alone can be effective (4). More severe cases may also require surgery, including debridement, skin grafts or, in extreme cases, amputation. Physiotherapy and long-term rehabilitation are frequently required where the disease has affected joints and even in high resource settings wound healing can take over a year (5).
M. ulcerans and Mycolactone
In contrast to the granulomatous immune responses typical of mycobacterial infections such as TB and leprosy, BU lesions usually display clusters of extracellular bacilli within the necrotic tissue with a relative paucity of infiltrating leukocytes. This immunosuppression, affecting both innate and acquired immune responses, is due to mycolactone, the exotoxin virulence factor unique to M. ulcerans (6).
Mycolactone production is encoded by a large plasmid, acquired by M. ulcerans and the other mycolactone-producing mycobacteria during their evolutionary emergence from a common ancestor highly similar to M. marinum (7, 8). The chromosomal genome of M. ulcerans is ~98% identical to that of M. marinum (9), sharing more than 4,000 orthologous genes (10). However, extensive gene rearrangements, expansion of pseudogenes and frequent insertions of IS2404 and IS2606 have led to loss of several virulence factors. For example, M. ulcerans has lost two out of the five ESX secretion systems, including the ESX-1 and its substrate, ESAT-6, which facilitate mycobacterial phagosomal escape into the cytoplasm (9). It seems likely that these losses are compensated by the gain of mycolactone, since this diffusible, lipid-like molecule has wide ranging effects on host cells and tissues that replicate the pathological features of BU (6).
Mycolactone mimics the effects of M. ulcerans infection upon injection into tissue (6) inducing immunosuppression, analgaesia and endothelial cell dysfunction (11, 12). Mycolactone suppresses both innate and adaptive immune responses, ablating production of cytokines and chemokines by monocytes, macrophages, dendritic cells and T cells (13–19), preventing the induction of immune receptors and co-stimulatory proteins (13, 17), and restricting antigen presentation (13–16, 20). This wide-reaching immunosuppression is thought to be central to the lack of systemic inflammation seen in BU patients and explains the dearth of inflammatory cells close to the bacteria within lesions.
Furthermore, mycolactone is both cytopathic and cytotoxic, causing cytoskeletal rearrangements, rounding up and detachment of cells (6, 21), cell cycle arrest (6), disturbance of Ca2+ flux (17, 22), oxidative stress (23–25), activation of the integrated stress response (26, 27), autophagy (28, 29) and, eventually, apoptosis (26, 30). Importantly, in most cell types, apoptosis occurs at least several days after exposure in vitro (11, 26). In BU lesions there is strong evidence of mycolactone-induced apoptosis (31) and tissue necrosis, with debris from neutrophils and macrophages in close proximity to the bacteria (32–34). There is also cytotoxicity to neurons that may underpin the analgaesia (35, 36), although alternative explanations have also been proposed (37).
Sec61 Inhibition by Mycolactone
We discovered that mycolactone inhibits co-translational translocation of proteins into the endoplasmic reticulum (ER) by the Sec61 translocon (16) (Figure 1), and it is now clear that this mechanism underlies most of the biological actions of mycolactone laid out above. Crucially, this is the first mechanistic explanation for the biological effects of mycolactone that has been corroborated by multiple labs worldwide (16, 18, 38, 39), and is now accepted by the WHO as the mechanism that substantially explains the immunosuppression and cytotoxicity in BU (40).
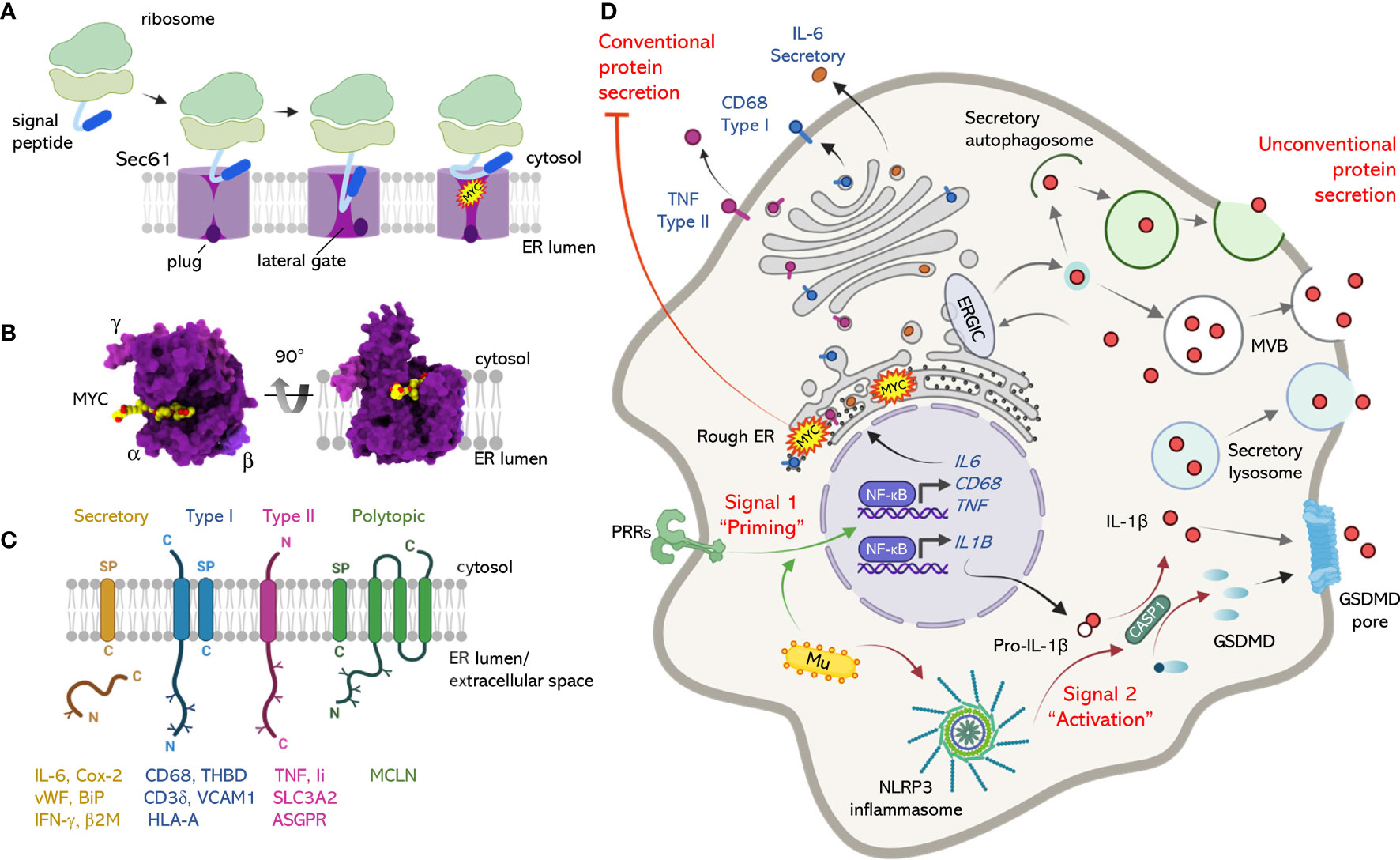
Figure 1 The mechanism of action of mycolactone at the Sec61 translocon and its influence over conventional and unconventional secretion. (A) Sec61-dependent co-translational translocation of proteins into the ER involves recognition of a signal peptide (SP) or signal anchor by the signal recognition particle and its receptor (not shown), which transfer it to Sec61. This results in reorganization of the translocon and movement the Sec61α plug domain, opening the central pore and allowing transit of the translating protein into the ER. Mycolactone binds Sec61α, preventing the SP from accessing its binding site at the lateral gate. Although the lateral gate is open, the plug remains closed and the translocon is locked in an inactive state. (B) The structure of inhibited Sec61, with mycolactone bound inside the lateral gate of Sec61α. Dark purple; Sec61α, light purple; Sec61β, pink; Sec61γ, yellow/red = mycolactone (from PDB:6Z3T). Two views are shown, looking down from the cytosol towards the ER and from the side, as in (A). (C) Mycolactone-dependent inhibition of Sec61 affects a wide range of proteins, including secretory proteins with SP, type I transmembrane proteins (N-terminal ‘out’, with SP), type II transmembrane proteins (N-terminal ‘in’, with signal anchor), and a subset of polytopic proteins (particularly those with an SP). (D) Conventional and unconventional secretion of cytokines and inflammatory mediators. Stimulation of macrophage pattern recognition receptors (PRRs) promotes activation of signalling pathways (Signal 1) e.g., NF-κB, leading to transcriptional induction of many genes, including conventionally secreted proteins (eg IL-6), type I transmembrane proteins (eg CD68), type II transmembrane proteins (eg TNF, cleaved from the cell surface by ADAM17, not shown), as well as unconventionally secreted proteins (eg IL-1β). Here, the pathways diverge, with conventionally exported inflammatory mediators entering the ER via Sec61 while pro-IL-1β translation occurs in the cytosol. Activation of the NLRP3 inflammasome (Signal 2) activates pro-caspase 1, which cleaves pro-IL-1β to its mature form. Mature IL-1β is secreted via unconventional secretion pathways involving gasdermin D (GSDMD) pore formation, and/or membrane-bound organelles such as multivesicular bodies (MVB), autophagosomes and secretory vesicles loaded via TMED10 (not shown) at the ER-Golgi intermediate compartment (ERGIC). M. ulcerans (Mu) was recently shown to provide Signal 1 by means of TLR2 activators on its cells surface, and Signal 2 via mycolactone (MYC), which also inhibits Sec61 at the ER membrane. Figures generated using BioRender.com and Chimera X (https://www.rbvi.ucsf.edu/chimerax).
It is useful to understand the details of Sec61 inhibition by mycolactone, as it applies across the nucleated cells of higher eukaryotic species that harbor M. ulcerans infections (41, 42). The Sec61 translocon consists of 3 subunits, Sec61α, Sec61β and Sec61γ, with Sec61α being the dynamic major pore forming subunit. It can open in two directions – via a central pore between the cytosol and ER, and sideways via a “lateral gate”. When the majority of secretory or membrane proteins are synthesized, they undergo a process of co-translational translocation. Here, their signal peptide or signal anchor engages with a specific site on Sec61α, which then opens both the lateral gate and the pore (Figure 1A). Mycolactone acts by binding to Sec61α (18, 43) and blocking the transit of proteins through the pore (38, 44). We recently resolved the structure of Sec61 bound to mycolactone using cryo-electron microscopy (43), which revealed that mycolactone occupies the entrance to the signal peptide binding site and, although the lateral gate is open, the channel remains closed (Figure 1B). Importantly, Sec61α is no longer able to move dynamically, and is instead trapped in this unproductive conformation.
This model fits well with the biochemical data, where mycolactone inhibits translocation of signal peptide-bearing proteins that use Sec61 to access the secretory pathway, i.e. co-translationally translocated secretory proteins, type I and type II transmembrane proteins, and a subset of polytopic proteins (18, 27, 44) (Hall, Hsieh, Simmonds et al., unpublished) (Figures 1C, D). Based on these features, and the high conservancy of Sec61-dependent translocation (45), the pattern of proteins affected by mycolactone in any given cell type is highly predictable. However, since constitutively expressed proteins are depleted at the turnover rate (11), which is highly variable, some proteins are lost more rapidly than others (27). On the other hand, induced (e.g. immune) responses are extremely sensitive to mycolactone’s effect on new protein production. Hence we and others (46) believe that many, if not all, of the cellular effects of mycolactone can be explained by Sec61 inhibition. Indeed, some observations previously ascribed to other mechanisms can retrospectively be ascribed to Sec61 inhibition, such as depletion of the type I transmembrane proteins e-Cadherin (21), L-selectin (20) and CD3 (17). Formal links have now been proven between Sec61 inhibition and many different aspects of mycolactone’s functions. These include loss of macrophage inflammatory mediators (16), T cell responses (18) and antigen presentation (47).
Translocation blockade is also intrinsically linked to mycolactone’s cytotoxicity, as forward genetic screening identified mutations in the gene for Sec61α, that reduce its ability to bind mycolactone (43). Cells carrying these resistance mutations survive and replicate in the presence of mycolactone (18, 26, 39, 43). Our working model is that the translocation blockade results in protein translation in the wrong cellular compartment (i.e. the cytoplasm), where they are degraded by the proteasome (16) or later removed by selective autophagy (28) (Figure 1D). Eventually these systems become overwhelmed, resulting in proteotoxic stress. Inhibition of Sec61 has been shown to be directly responsible for Ca2+ flux disturbance, induction of an integrated stress response and autophagy (22, 26, 28).
Evidence for an Immune Response to M. ulcerans
Despite the fundamental process that mycolactone inhibits, and the critical role that secreted proteins and membrane receptors play in the immune response, there is significant evidence for an immune response to infection with M. ulcerans. For instance, despite reports of T cell anergy in BU patients, T lymphocyte responses to M. ulcerans antigens can be found in the blood of most BU patients (48–51). Notably, these responses can also be detected in uninfected people living in endemic areas (52, 53), even if they bear no sign of clinical disease, suggesting previous subclinical exposure to M. ulcerans. Furthermore, granulomas may develop in BU patients with late-stage disease (54) and spontaneous healing without antibiotics has also been reported (55–57). Hence, it is possible that many BU cases could eventually self-heal without medical intervention.
Control of M. ulcerans infection is reported in both guinea pig (58) and FVB/N mouse strain models (59). Initial evidence suggested the latter involves innate but not acquired immune responses (59) but recently, Foulon and colleagues demonstrated a correlation with the emergence of plasma cells within the local skin tissue, and mycolactone inhibitory antibodies (60), as well as down-regulation of mycolactone synthesis (61), although the drivers of this are unclear.
While these observations might be explained by the spatial distribution and local concentrations of mycolactone in infected tissue, genetic studies provide further evidence that macrophage responses are important in M. ulcerans infection. Many of the polymorphisms that affect either the likelihood of developing BU or disease severity impact the innate response to intracellular infection (62–65). For example, SNPs in the gene for the macrophage activating cytokine IFN-γ (IFNG), and iNOS, the inducible nitric oxide synthase that generates bactericidal NO in macrophages, both increase susceptibility to infection. Interestingly, SNPs in three genes involved in the autophagy pathway also affect BU. Two polymorphisms have been identified in the E3 ligase PARK2 which increase susceptibility to BU while one in NOD2 increases the risk of severe disease (62). By contrast, a SNP in ATG16L1 is associated with reduced risk of ulceration (62, 65). This is particularly intriguing as we recently uncovered a protective role for autophagy in the cellular response to mycolactone (28).
The most recent evidence of an ongoing innate immune response to M. ulcerans bacteria is the discovery that macrophages produce IL-1β following exposure to mycolactone (66). Given that most other cytokines and chemokines made by macrophages are Sec61-dependent and therefore strongly suppressed by mycolactone, this discovery likely has especial importance for BU. We have therefore validated this important finding in our laboratory (Figure 2), as will be discussed in more detail below.
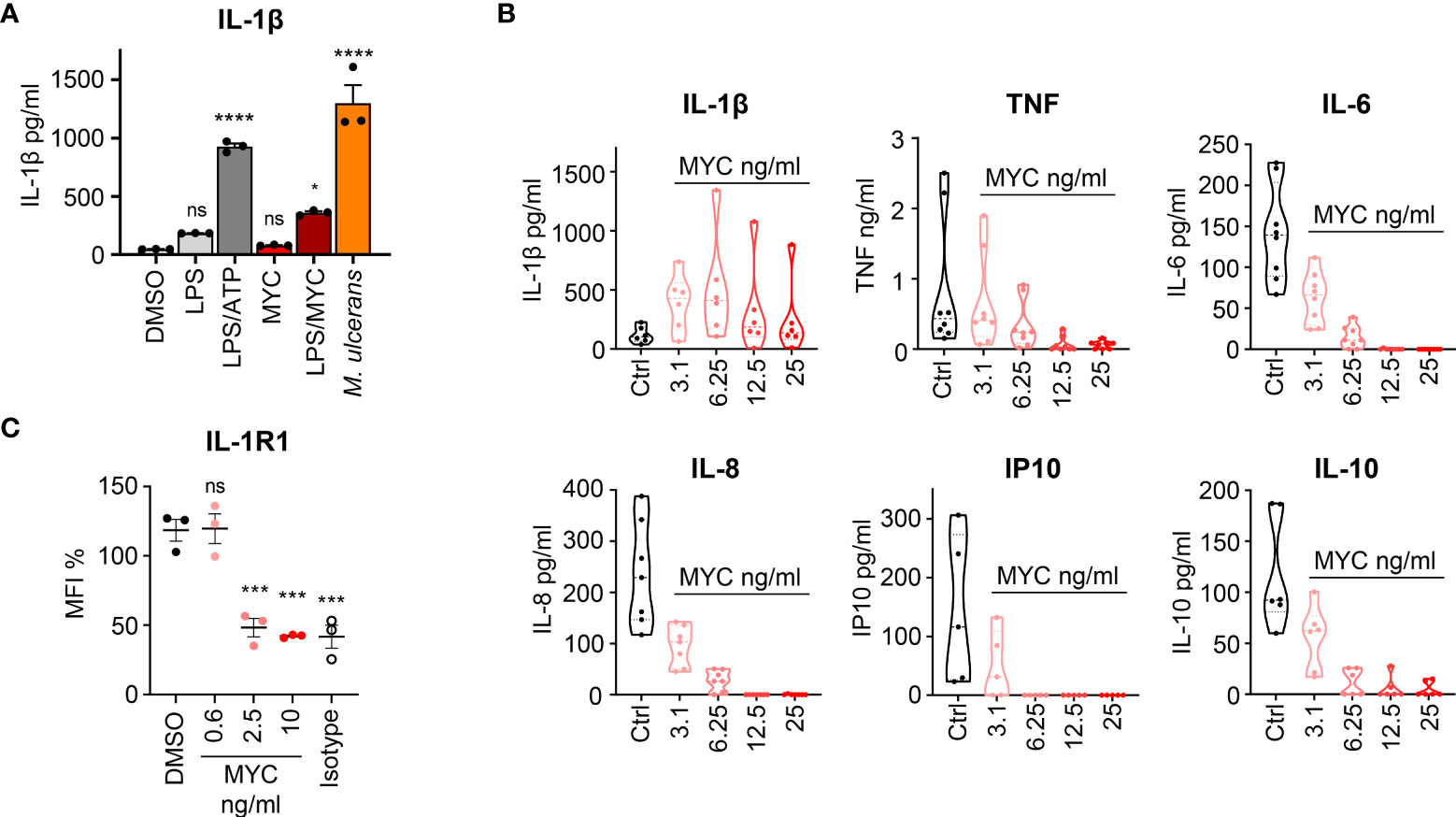
Figure 2 Induction of macrophage IL-1β activation and secretion by M. ulcerans. (A) IL-1β production by PMA-activated THP-1 cells incubated for 24 h with 0.05% DMSO, 10 ng/ml E.Coli-derived LPS (Enzo) with or without 1 mM ATP or 31.25 ng/ml synthetic mycolactone (MYC) or M. ulcerans Mu_1082. At this time point THP-1 cell viability was >95%. IL-1β was measured by ELISA (eBioscience) in triplicate (mean ± SD). Results are representative of duplicate experiments. (B) Rheumatoid arthritis (RA) synovial membrane cultures were isolated as in (67) and then cultured in medium alone or medium containing increasing concentrations of natural mycolactone (MYC) for 24 h. The concentration of IL-1β, TNF, IL-6, IL-8, IP-10 and IL-10 in the cell supernatants were measured by ELISA (68–70). This mixed cell population, including predominantly CD45- fibroblast-like cells, CD14+CD45+ macrophages and CD3+CD4+ T lymphocytes, are highly activated, and spontaneously produce cytokines without further stimulation. In addition to those shown, these cells also secrete other cytokines/chemokines/inflammatory mediators including CCL5, CCL2, GM-CSF, IL-23, IL-17, IL1RA, IL-11,TNFRSF1B (TNFR2), MMP1, MMP2, MMP3, MMP13, and TIMP1, but not IFNγ or lymphotoxin (67, 71–76). All RA patients gave written informed consent and the study was approved by the Riverside Research Ethics Committee, REC number: 07/H0706/81. Violin plots showing median and quartile for n = 5-8 patients; median control values for the measured cytokines were IL-1β (103 pg/ml), TNF (433 pg/ml), IL-6 (139 pg/ml), IL-8 (229 pg/ml), IP-10 (116 pg/ml) and IL-10 (92 pg/ml). (C) IL-1R1 surface expression in human dermal microvascular endothelial cells exposed to 0.02% DMSO, or increasing concentrations of synthetic mycolactone (MYC) for 24 h. Cells were dissociated, stained with anti-IL-1R1 antibody (PA546930, Invitrogen) or isotype goat IgG (AB-108-C, bio-techne), donkey anti-goat IgG FITC (A16000, Invitrogen), and subjected to flow cytometry analysis. Mean fluorescence intensity (MFI) is presented as a % of untreated control (mean ± SEM, n = 3). For all panels ns; not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001 using a one-way ANOVA and Dunnett’s multiple comparison test.
IL-1β
IL-1β, produced predominantly by monocytes and macrophages, is a potent pyrogen and one of the ‘alarm’ cytokines known to have widespread and systemic effects once induced (77). Foulon and colleagues detected IL-1β in lesions arising from M. ulcerans infection in both BU patients and in animal models. In vitro, IL-1β was induced in both murine and human primary macrophages, by a variety of different mycolactone-containing stimuli, including purified or synthetic mycolactone together with LPS, M. ulcerans bacteria, and mycolactone-containing extracellular vesicles (EVs) (66). This latter preparation, derived from the extracellular matrix of M. ulcerans, contains high concentrations of mycolactone, as well as a variety of proteins, carbohydrates and lipids (78).
The induction and biosynthesis of IL-1β differs from that of many other cytokines, chemokines and inflammatory mediators such as TNF, IL-8 and Cox-2 (Figure 1D). In common are the pathways that induce mRNA expression and therefore protein synthesis, such as NF-κB signaling (often referred to as “Priming” or “Signal 1”). Following M. ulcerans, or mycolactone-containing EV exposure, it has been demonstrated that priming of macrophages is probably via activation of TLR2, since genetic knockout of this receptor ablated the response (66). Although TLR production is presumably sensitive to mycolactone (47, 79), these priming signals occur very rapidly (minutes), before any depletion is likely (hours/days) (15, 16). Importantly, however, IL-1β lacks a signal peptide to direct the translating protein into the canonical secretory pathway utilizing Sec61 at the ER. Instead, IL-1β is translated as pro-IL-1β in the cytosol. A separate signaling pathway that culminates in the activation of a complex known as the inflammasome (referred to “Activation” or “Signal 2”) is responsible for cleaving pro-IL-1β into biologically active mature IL-1β. In macrophages, activation of the NLRP3 inflammasome (a complex formed of NLRP3, ASC and pro-caspase-1) cleaves and activates pro-caspase-1, which is then able to cleave pro-IL-1β, although alternative mechanisms also exist (80). Activators of the NLRP3 inflammasome are known to include microbial infection, particulate matter and ATP (81). While the precise mechanism involved in the mycolactone-dependent activation of the NLRP3 inflammasome is not known (66), it is highly likely that the toxin is providing “Signal 2”.
We now confirm that mycolactone induces IL-1β release by macrophages (Figure 2A). As before (66) mycolactone alone was not sufficient to induce IL-1β production, requiring LPS or intact M. ulcerans bacteria. However, mycolactone alone can super-induce IL-1β secretion in a well-defined ex vivo model of rheumatoid arthritis (RA) (67, 71–76), consisting of cultures of synovial membrane cells from RA patients who have undergone elective joint replacement surgery (Figure 2B). This mixed population of cells spontaneously releases a range of cytokines, chemokines and other mediators (See Figure 2 legend for details). Within these cultures, endogenous damage associated molecular patterns (DAMPs) activate TLRs providing the priming signal to induce pro-IL-1β [reviewed in (82)]. Consequently, low amounts of mycolactone are able to super-activate Signal 2 and boost the level of IL-1β released approximately 4-fold.
Once produced, several mechanisms for secretion of IL-1β have been proposed. All of these are independent of the canonical secretory pathway and Sec61-dependent translocation of proteins into the ER, and are known as unconventional secretory pathways (83) (Figure 1D). The precise mechanism taking place in a particular circumstance depends on a variety of factors. A major exit route relies on cleavage of gasdermin D (GSDMD) by caspase-1 following activation of the inflammasome. The N-terminal fragment oligomerizes, forming pores in the membrane that facilitate the release of IL-1β and that can also induce pyroptosis (84). Recent evidence has argued that GSDMD-mediated IL-1β release is a tightly regulated process independent of cell death (85), and while pyroptosis is a consequence of GSDMD pore formation, it is not a prerequisite for IL-1β release (83). An alternative pathway, involving different types of cellular vesicles and/or membrane-bound organelles, can also secrete IL-1β. TMED10 was recently identified as the translocon for IL-1β into the ER–Golgi intermediate compartment (ERGIC) (86). The autophagy pathway is also implicated in regulation of IL-1β production at multiple levels, controlling inflammasome activity, promoting processing of pro-IL-1β and mediating secretion (87).
Hence, the recent discovery of IL-1β in BU lesions, and its induction by mycolactone in macrophages is entirely in line with our current knowledge of the cell biology and biochemistry of Sec61 inhibition by mycolactone.
Discussion - Potential Implications of IL-1β Induction for Buruli Ulcer Pathogenesis
IL-1β is well known to induce inflammatory cytokines, chemokines and other mediators from a wide range of cell types (80, 88). The resultant local inflammation attracts an influx of immune cells to a site of infection, hence IL-1β release in BU could impact the ability of the host to respond to M. ulcerans infection, by enhancing the activation state of surrounding cells, including innate immune macrophages and neutrophils. In the mouse footpad model of M. ulcerans infection, treatment with the non-specific steroid anti-inflammatory dexamethasone reduced footpad swelling (66), suggesting that IL-1-driven proinflammatory responses may contribute to BU pathology. However, its potential role in controlling the infection has not yet been reported. Pre-existing IL-1β may also play a role in driving paradoxical responses that are observed at the outset of antibiotic treatment. As well as directing pathogenesis and antimicrobial immune responses, IL-1 also plays an important role in wound healing (89, 90), which likely becomes important during spontaneous healing. Moreover, other IL-1 family cytokines utilize the same strategies for protein expression and unconventional protein secretion, including IL-18, IL-33, IL-36, IL-37, and IL-38 (88, 91). Hence, these other cytokines may also bypass mycolactone’s blockade of co-translational translocation and may be present in M. ulcerans-infected tissues, further modulating immune responses. Indeed, IL-18 production by LPS-stimulated macrophages is increased, not blocked, by mycolactone (66).
A further downstream target of IL-1β that likely affects both pathological and protective responses to M. ulcerans infection is the vascular endothelium, which lines the blood vessels making up the skin microvasculature. Endothelial cells are extremely responsive to IL-1β, which modulates a range of functions, depleting junctional β-catenin and VE-cadherin (thus increasing vascular permeability) and down-regulating anticoagulant proteins such as thrombomodulin and the EPCR. IL-1β also induces procoagulant PAI-1, enhances tissue factor expression, and promotes angiogenesis via upregulation of adhesion molecule and vascular endothelial growth factor (VEGF) expression (91). Importantly, we have recently shown that mycolactone and IL-1β have an additive effect on both thrombomodulin depletion from endothelial cells and vascular permeability increase (12), suggesting that the local production of IL-1β may further worsen the endothelial cell dysfunction induced by mycolactone (11).
However, these downstream effects are all dependent on IL-1 receptor expression and are complicated by the fact that physiological responses to IL-1 are moderated by a complex system of receptors, co-receptors, ligands, and endogenous antagonists. IL-1β is recognised by its cognate receptor, IL-1R1, which then binds to a co-receptor IL-1R3/IL-1RAcP, triggering intracellular signalling. However, some cells, including neutrophils, express both IL-1R1 and the decoy receptor IL-1R2 (92) which increases the amount of IL-1β needed to activate them. On the other hand, highly sensitive cells, such as endothelial cells, express IL-1R1 but undetectable IL-1R2 (93, 94). A further problem in interpretation of the role of IL-1 family proteins in BU arises because the IL-1 receptors are all single-pass type I membrane proteins secreted through the conventional ER-Golgi pathway and, as such, are likely to be susceptible to mycolactone inhibition (Figures 1C, D) (27). Indeed, flow cytometry analysis of IL-1R1 expression by primary dermal microvascular endothelial cells shows that this receptor is susceptible to mycolactone-dependent loss (Figure 2C). The closer cells are to the infected tissue and the source of mycolactone production the more likely they are to be depleted of receptors and therefore insensitive to IL-1β.
In conclusion, IL-1β is likely to be a double-edged sword in BU. The question of whether it is driving the pathology, protecting against further expansion of the infection, or both, should be urgently addressed as it offers an attractive alternative approach to therapy. IL-1β targeting with canakinumab, or its cell surface receptors with Anakinra, are proposed treatments for a variety of conditions including vasculitis and diabetic foot ulcers (95–101). Similarly, diabetic mice receiving IL-1β neutralizing antibodies display reduced inflammation and enhanced re-epithelialization of skin wounds (99). However, in the context of the local inflammation, abnormal vascular phenotype and long-lasting wounds in Buruli ulcer, there are a number of issues that need to be considered. We should thoroughly examine whether the level of IL-1β and its receptor in affected tissues is sufficient to activate signaling, what happens during and after antibiotic treatment and whether its presence promotes or hinders wound healing. Understanding the role of IL-β in BU may allow us to develop new treatments that aid recovery from this devastating disease.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Riverside Research Ethics Committee REC number: 07/H0706/81. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
BH, LT-HH, and SS performed laboratory analysis. BH and LH prepared schematics with suggestions from SS and RS. RS prepared data figures and Chimera poses. BH, LT-HH, and RS wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
RS holds a Wellcome Trust Investigator Award in Science (202843/Z/16/Z). This work was also supported by a grant from the Kennedy Institute Trustees.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Prof Pamela Small (University of Knoxville, Tennessee) and Prof Yoshito Kishi (Harvard University) for providing natural and synthetic mycolactone, respectively. We are grateful to Prof Rajko Reljic and Dr Justice Boakye-Appiah (St George’s University London) for providing the M. ulcerans strain Mu_1082.
References
1. Yotsu RR, Suzuki K, Simmonds RE, Bedimo R, Ablordey A, Yeboah-Manu D, et al. Buruli Ulcer: A Review of the Current Knowledge. Curr Trop Med Rep (2018) 5(4):247–56. doi: 10.1007/s40475-018-0166-2
2. Tai AYC, Athan E, Friedman ND, Hughes A, Walton A, O'Brien DP. Increased Severity and Spread of Mycobacterium Ulcerans, Southeastern Australia. Emerging Infect Dis (2018) 24(1):58–64. doi: 10.3201/eid2401.171070
3. Simpson H, Deribe K, Tabah EN, Peters A, Maman I, Frimpong M, et al. Mapping the Global Distribution of Buruli Ulcer: A Systematic Review With Evidence Consensus. Lancet Glob Health (2019) 7(7):e912–22. doi: 10.1016/S2214-109X(19)30171-8
4. Phillips RO, Robert J, Abass KM, Thompson W, Sarfo FS, Wilson T, et al. Rifampicin and Clarithromycin (Extended Release) Versus Rifampicin and Streptomycin for Limited Buruli Ulcer Lesions: A Randomised, Open-Label, Non-Inferiority Phase 3 Trial. Lancet (2020) 395(10232):1259–67. doi: 10.1016/S0140-6736(20)30047-7
5. Johnson PDR. Buruli Ulcer: Cured by 8 Weeks of Oral Antibiotics? Lancet (2020) 395(10232):1231–2. doi: 10.1016/S0140-6736(20)30478-5
6. George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, et al. Mycolactone: A Polyketide Toxin From Mycobacterium Ulcerans Required for Virulence. Science (1999) 283(5403):854–7. doi: 10.1126/science.283.5403.854
7. Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R, et al. Giant Plasmid-Encoded Polyketide Synthases Produce the Macrolide Toxin of Mycobacterium Ulcerans. Proc Natl Acad Sci USA (2004) 101(5):1345–9. doi: 10.1073/pnas.0305877101
8. Porter JL, Tobias NJ, Pidot SJ, Falgner S, Tuck KL, Vettiger A, et al. The Cell Wall-Associated Mycolactone Polyketide Synthases are Necessary But Not Sufficient for Mycolactone Biosynthesis. PloS One (2013) 8(7):e70520. doi: 10.1371/journal.pone.0070520
9. Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, et al. Reductive Evolution and Niche Adaptation Inferred From the Genome of Mycobacterium Ulcerans, the Causative Agent of Buruli Ulcer. Genome Res (2007) 17(2):192–200. doi: 10.1101/gr.5942807
10. Demangel C, Stinear TP, Cole ST. Buruli Ulcer: Reductive Evolution Enhances Pathogenicity of Mycobacterium Ulcerans. Nat Rev Microbiol (2009) 7(1):50–60. doi: 10.1038/nrmicro2077
11. Ogbechi J, Ruf MT, Hall BS, Bodman-Smith K, Vogel M, Wu HL, et al. Mycolactone-Dependent Depletion of Endothelial Cell Thrombomodulin is Strongly Associated With Fibrin Deposition in Buruli Ulcer Lesions. PloS Pathog (2015) 11:e1005011. doi: 10.1371/journal.ppat.1005011
12. Hsieh LT-H, Dos Santos SJ, Ogbechi J, Loglo AD, Salguero FJ, Ruf M-T, et al. Aberrant Stromal Tissue Factor and Mycolactone-Driven Vascular Permeability, Exacerbated by IL-1β, Orchestrate Pathogenic Fibrin Formation in Buruli Ulcer Lesions. medRxiv (2021). 2021.08.04.21261598. doi: 10.1101/2021.08.04.21261598
13. Coutanceau E, Decalf J, Martino A, Babon A, Winter N, Cole ST, et al. Selective Suppression of Dendritic Cell Functions by Mycobacterium Ulcerans Toxin Mycolactone. J Exp Med (2007) 204(6):1395–403. doi: 10.1084/jem.20070234
14. Pahlevan AA, Wright DJ, Andrews C, George KM, Small PL, Foxwell BM. The Inhibitory Action of Mycobacterium Ulcerans Soluble Factor on Monocyte/T Cell Cytokine Production and NF-Kappa B Function. J Immunol (1999) 163(7):3928–35.
15. Simmonds RE, Lali FV, Smallie T, Small PL, Foxwell BM. Mycolactone Inhibits Monocyte Cytokine Production by a Posttranscriptional Mechanism. J Immunol (2009) 182(4):2194–202. doi: 10.4049/jimmunol.0802294
16. Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, et al. The Pathogenic Mechanism of the Mycobacterium Ulcerans Virulence Factor, Mycolactone, Depends on Blockade of Protein Translocation Into the ER. PloS Pathog (2014) 10(4):e1004061. doi: 10.1371/journal.ppat.1004061
17. Boulkroun S, Guenin-Mace L, Thoulouze MI, Monot M, Merckx A, Langsley G, et al. Mycolactone Suppresses T Cell Responsiveness by Altering Both Early Signaling and Posttranslational Events. J Immunol (2010) 184(3):1436–44. doi: 10.4049/jimmunol.0902854
18. Baron L, Paatero AO, Morel JD, Impens F, Guenin-Mace L, Saint-Auret S, et al. Mycolactone Subverts Immunity by Selectively Blocking the Sec61 Translocon. J Exp Med (2016) 213(13):2885–96. doi: 10.1084/jem.20160662
19. Torrado E, Adusumilli S, Fraga AG, Small PL, Castro AG, Pedrosa J. Mycolactone-Mediated Inhibition of Tumor Necrosis Factor Production by Macrophages Infected With Mycobacterium Ulcerans has Implications for the Control of Infection. Infect Immun (2007) 75(8):3979–88. doi: 10.1128/IAI.00290-07
20. Guenin-Mace L, Carrette F, Asperti-Boursin F, Le Bon A, Caleechurn L, Di Bartolo V, et al. Mycolactone Impairs T Cell Homing by Suppressing microRNA Control of L-Selectin Expression. Proc Natl Acad Sci USA (2011) 108(31):12833–8. doi: 10.1073/pnas.1016496108
21. Guenin-Mace L, Veyron-Churlet R, Thoulouze MI, Romet-Lemonne G, Hong H, Leadlay PF, et al. Mycolactone Activation of Wiskott-Aldrich Syndrome Proteins Underpins Buruli Ulcer Formation. J Clin Invest (2013) 123(4):1501–12. doi: 10.1172/JCI66576
22. Bhadra P, Dos Santos S, Gamayun I, Pick T, Neumann C, Ogbechi J, et al. Mycolactone Enhances the Ca2+ Leak From Endoplasmic Reticulum by Trapping Sec61 Translocons in a Ca2+ Permeable State. Biochem J (2021) 478(22):4005–24. doi: 10.1042/BCJ20210345
23. Förster B, Demangel C, Thye T. Mycolactone Induces Cell Death by SETD1B-Dependent Degradation of Glutathione. PloS Negl Trop Dis (2020) 14(10):e0008709. doi: 10.1371/journal.pntd.0008709
24. Kwaffo YA, Sarpong-Duah M, Owusu-Boateng K, Gbewonyo WS, Adjimani JP, Mosi L. Natural Antioxidants Attenuate Mycolactone Toxicity to RAW 264.7 Macrophages. Exp Biol Med (Maywood) (2021) 246(17):1884–94. doi: 10.1177/15353702211015628
25. Gronberg A, Zettergren L, Bergh K, Stahle M, Heilborn J, Angeby K, et al. Antioxidants Protect Keratinocytes Against M. Ulcerans Mycolactone Cytotoxicity. PloS One (2010) 5(11):e13839. doi: 10.1371/journal.pone.0013839
26. Ogbechi J, Hall BS, Sbarrato T, Taunton J, Willis AE, Wek RC, et al. Inhibition of Sec61-Dependent Translocation by Mycolactone Uncouples the Integrated Stress Response From ER Stress, Driving Cytotoxicity via Translational Activation of ATF4. Cell Death Dis (2018) 9(3):397. doi: 10.1038/s41419-018-0427-y
27. Morel JD, Paatero AO, Wei J, Yewdell JW, Guenin-Mace L, Van Haver D, et al. Proteomics Reveals Scope of Mycolactone-Mediated Sec61 Blockade and Distinctive Stress Signature. Mol Cell Proteomics (2018) 17(9):1750–65. doi: 10.1074/mcp.RA118.000824
28. Hall BS, Dos Santos SJ, Hsieh LT, Manifava M, Ruf MT, Pluschke G, et al. Inhibition of the SEC61 Translocon by Mycolactone Induces a Protective Autophagic Response Controlled by EIF2S1-Dependent Translation That Does Not Require ULK1 Activity. Autophagy (2021), 1–19. doi: 10.1080/15548627.2021.1961067
29. Gama JB, Ohlmeier S, Martins TG, Fraga AG, Sampaio-Marques B, Carvalho MA, et al. Proteomic Analysis of the Action of the Mycobacterium Ulcerans Toxin Mycolactone: Targeting Host Cells Cytoskeleton and Collagen. PloS Negl Trop Dis (2014) 8(8):e3066. doi: 10.1371/journal.pntd.0003066
30. Bieri R, Scherr N, Ruf MT, Dangy JP, Gersbach P, Gehringer M, et al. The Macrolide Toxin Mycolactone Promotes Bim-Dependent Apoptosis in Buruli Ulcer Through Inhibition of mTOR. ACS Chem Biol (2017) 12(5):1297–307. doi: 10.1021/acschembio.7b00053
31. George KM, Pascopella L, Welty DM, Small PL. A Mycobacterium Ulcerans Toxin, Mycolactone, Causes Apoptosis in Guinea Pig Ulcers and Tissue Culture Cells. Infection Immun (2000) 68(2):877–83. doi: 10.1128/IAI.68.2.877-883.2000
32. Adusumilli S, Mve-Obiang A, Sparer T, Meyers W, Hayman J, Small PL. Mycobacterium Ulcerans Toxic Macrolide, Mycolactone Modulates the Host Immune Response and Cellular Location of M. Ulcerans In Vitro and In Vivo. Cell Microbiol (2005) 7(9):1295–304. doi: 10.1111/j.1462-5822.2005.00557.x
33. Bolz M, Ruggli N, Borel N, Pluschke G, Ruf MT. Local Cellular Immune Responses and Pathogenesis of Buruli Ulcer Lesions in the Experimental Mycobacterium Ulcerans Pig Infection Model. PloS Negl Trop Dis (2016) 10(4):e0004678. doi: 10.1371/journal.pntd.0004678
34. Bolz M, Ruggli N, Ruf MT, Ricklin ME, Zimmer G, Pluschke G. Experimental Infection of the Pig With Mycobacterium Ulcerans: A Novel Model for Studying the Pathogenesis of Buruli Ulcer Disease. PloS Negl Trop Dis (2014) 8(7):e2968. doi: 10.1371/journal.pntd.0002968
35. Goto M, Nakanaga K, Aung T, Hamada T, Yamada N, Nomoto M, et al. Nerve Damage in Mycobacterium Ulcerans-Infected Mice: Probable Cause of Painlessness in Buruli Ulcer. Am J Pathol (2006) 168(3):805–11. doi: 10.2353/ajpath.2006.050375
36. En J, Goto M, Nakanaga K, Higashi M, Ishii N, Saito H, et al. Mycolactone is Responsible for the Painlessness of Mycobacterium Ulcerans Infection (Buruli Ulcer) in a Murine Study. Infect Immun (2008) 76(5):2002–7. doi: 10.1128/IAI.01588-07
37. Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, et al. Mycobacterial Toxin Induces Analgesia in Buruli Ulcer by Targeting the Angiotensin Pathways. Cell (2014) 157(7):1565–76. doi: 10.1016/j.cell.2014.04.040
38. McKenna M, Simmonds RE, High S. Mechanistic Insights Into the Inhibition of Sec61-Dependent Co- and Post-Translational Translocation by Mycolactone. J Cell Sci (2016) 129(7):1404–5. doi: 10.1242/jcs.182352
39. Zong G, Hu Z, O'Keefe S, Tranter D, Iannotti MJ, Baron L, et al. Ipomoeassin F Binds Sec61α to Inhibit Protein Translocation. J Am Chem Soc (2019) 141(21):8450–61. doi: 10.1021/jacs.8b13506
40. WorldHealthOrganisation. WHO Meeting on Buruli Ulcer and Other Skin NTDs. Final Report. Switzerland: WHO Headquarters Geneva (2019).
41. Muleta AJ, Lappan R, Stinear TP, Greening C. Understanding the Transmission of Mycobacterium Ulcerans: A Step Towards Controlling Buruli Ulcer. PloS Negl Trop Dis (2021) 15(8):e0009678. doi: 10.1371/journal.pntd.0009678
42. Receveur JP, Bauer A, Pechal JL, Picq S, Dogbe M, Jordan HR, et al. A Need for Null Models in Understanding Disease Transmission: The Example of Mycobacterium Ulcerans (Buruli Ulcer Disease). FEMS Microbiol Rev (2021). doi: 10.1093/femsre/fuab045
43. Gérard SF, Hall BS, Zaki AM, Corfield KA, Mayerhofer PU, Costa C, et al. Structure of the Inhibited State of the Sec Translocon. Mol Cell (2020) 79(3):406–15.e7. doi: 10.1016/j.molcel.2020.06.013
44. McKenna M, Simmonds RE, High S. Mycolactone Reveals the Substrate-Driven Complexity of Sec61-Dependent Transmembrane Protein Biogenesis. J Cell Sci (2017) 130(7):1307–20. doi: 10.1242/jcs.198655
45. Van Puyenbroeck V, Vermeire K. Inhibitors of Protein Translocation Across Membranes of the Secretory Pathway: Novel Antimicrobial and Anticancer Agents. Cell Mol Life Sci (2018) 75(9):1541–58. doi: 10.1007/s00018-017-2743-2
46. Demangel C, High S. Sec61 Blockade by Mycolactone: A Central Mechanism in Buruli Ulcer Disease. Biol Cell (2018) 110(11):237–48. doi: 10.1111/boc.201800030
47. Grotzke JE, Kozik P, Morel JD, Impens F, Pietrosemoli N, Cresswell P, et al. Sec61 Blockade by Mycolactone Inhibits Antigen Cross-Presentation Independently of Endosome-to-Cytosol Export. Proc Natl Acad Sci USA (2017) 114(29):E5910–9. doi: 10.1073/pnas.1705242114
48. Nausch N, Antwi-Berko D, Mubarik Y, Abass KM, Owusu W, Owusu-Dabo E, et al. Analysis of Mycobacterium Ulcerans-Specific T-Cell Cytokines for Diagnosis of Buruli Ulcer Disease and as Potential Indicator for Disease Progression. PloS Negl Trop Dis (2017) 11(2):e0005415. doi: 10.1371/journal.pntd.0005415
49. Gooding TM, Johnson PD, Campbell DE, Hayman JA, Hartland EL, Kemp AS, et al. Immune Response to Infection With Mycobacterium Ulcerans. Infection Immun (2001) 69(3):1704–7. doi: 10.1128/IAI.69.3.1704-1707.2001
50. Gooding TM, Kemp AS, Robins-Browne RM, Smith M, Johnson PD. Acquired T-Helper 1 Lymphocyte Anergy Following Infection With Mycobacterium Ulcerans. Clin Infect Dis an Off Publ Infect Dis Soc America (2003) 36(8):1076–7. doi: 10.1086/368315
51. Phillips R, Sarfo FS, Guenin-Mace L, Decalf J, Wansbrough-Jones M, Albert ML, et al. Immunosuppressive Signature of Cutaneous Mycobacterium Ulcerans Infection in the Peripheral Blood of Patients With Buruli Ulcer Disease. J Infect Dis (2009) 200(11):1675–84. doi: 10.1086/646615
52. Loglo AD, Frimpong M, Sarpong Duah M, Sarfo F, Sarpong FN, Agbavor B, et al. IFN-γ and IL-5 Whole Blood Response Directed Against Mycolactone Polyketide Synthase Domains in Patients With Mycobacterium Ulcerans Infection. PeerJ (2018) 6:e5294. doi: 10.7717/peerj.5294
53. Gooding TM, Johnson PD, Smith M, Kemp AS, Robins-Browne RM. Cytokine Profiles of Patients Infected With Mycobacterium Ulcerans and Unaffected Household Contacts. Infection Immun (2002) 70(10):5562–7. doi: 10.1128/IAI.70.10.5562-5567.2002
54. Guarner J, Bartlett J, Whitney EA, Raghunathan PL, Stienstra Y, Asamoa K, et al. Histopathologic Features of Mycobacterium Ulcerans Infection. Emerging Infect Dis (2003) 9(6):651–6. doi: 10.3201/eid0906.020485
55. Beissner M, Piten E, Maman I, Symank D, Jansson M, Nitschke J, et al. Spontaneous Clearance of a Secondary Buruli Ulcer Lesion Emerging Ten Months After Completion of Chemotherapy–a Case Report From Togo. PloS Negl Trop Dis (2012) 6(7):e1747. doi: 10.1371/journal.pntd.0001747
56. Gordon CL, Buntine JA, Hayman JA, Lavender CJ, Fyfe JA, Hosking P, et al. Spontaneous Clearance of Mycobacterium Ulcerans in a Case of Buruli Ulcer. PloS Negl Trop Dis (2011) 5(10):e1290. doi: 10.1371/journal.pntd.0001290
57. O'Brien DP, Murrie A, Meggyesy P, Priestley J, Rajcoomar A, Athan E. Spontaneous Healing of Mycobacterium Ulcerans Disease in Australian Patients. PloS Negl Trop Dis (2019) 13(2):e0007178. doi: 10.1371/journal.pntd.0007178
58. Silva-Gomes R, Marcq E, Trigo G, Gonçalves CM, Longatto-Filho A, Castro AG, et al. Spontaneous Healing of Mycobacterium Ulcerans Lesions in the Guinea Pig Model. PloS Negl Trop Dis (2015) 9(12):e0004265. doi: 10.1371/journal.pntd.0004265
59. Marion E, Jarry U, Cano C, Savary C, Beauvillain C, Robbe-Saule M, et al. FVB/N Mice Spontaneously Heal Ulcerative Lesions Induced by Mycobacterium Ulcerans and Switch M. Ulcerans Into a Low Mycolactone Producer. J Immunol (2016) 196(6):2690–8. doi: 10.4049/jimmunol.1502194
60. Foulon M, Pouchin A, Manry J, Khater F, Robbe-Saule M, Durand A, et al. Skin-Specific Antibodies Neutralizing Mycolactone Toxin During the Spontaneous Healing of Mycobacterium Ulcerans Infection. Sci Adv (2020) 6(9):eaax7781. doi: 10.1126/sciadv.aax7781
61. Robbe-Saule M, Foulon M, Poncin I, Esnault L, Varet H, Legendre R, et al. Transcriptional Adaptation of Mycobacterium Ulcerans in an Original Mouse Model: New Insights Into the Regulation of Mycolactone. Virulence (2021) 12(1):1438–51. doi: 10.1080/21505594.2021.1929749
62. Capela C, Dossou AD, Silva-Gomes R, Sopoh GE, Makoutode M, Menino JF, et al. Genetic Variation in Autophagy-Related Genes Influences the Risk and Phenotype of Buruli Ulcer. PloS Negl Trop Dis (2016) 10(4):e0004671. doi: 10.1371/journal.pntd.0004671
63. Stienstra Y, van der Werf TS, Oosterom E, Nolte IM, van der Graaf WT, Etuaful S, et al. Susceptibility to Buruli Ulcer is Associated With the SLC11A1 (NRAMP1) D543N Polymorphism. Genes Immun (2006) 7(3):185–9. doi: 10.1038/sj.gene.6364281
64. Bibert S, Bratschi MW, Aboagye SY, Collinet E, Scherr N, Yeboah-Manu D, et al. Susceptibility to Mycobacterium Ulcerans Disease (Buruli Ulcer) Is Associated With IFNG and iNOS Gene Polymorphisms. Front Microbiol (2017) 8:1903. doi: 10.3389/fmicb.2017.01903
65. Manry J, Vincent QB, Johnson C, Chrabieh M, Lorenzo L, Theodorou I, et al. Genome-Wide Association Study of Buruli Ulcer in Rural Benin Highlights Role of Two LncRNAs and the Autophagy Pathway. Commun Biol (2020) 3(1):177. doi: 10.1038/s42003-020-0920-6
66. Foulon M, Robbe-Saule M, Manry J, Esnault L, Boucaud Y, Alcaïs A, et al. Mycolactone Toxin Induces an Inflammatory Response by Targeting the IL-1β Pathway: Mechanistic Insight Into Buruli Ulcer Pathophysiology. PloS Pathog (2020) 16(12):e1009107. doi: 10.1371/journal.ppat.1009107
67. Brennan FM, Chantry D, Jackson AM, Maini RN, Feldmann M. Cytokine Production in Culture by Cells Isolated From the Synovial Membrane. J Autoimmun (1989) 2(Suppl):177–86. doi: 10.1016/0896-8411(89)90129-7
68. Sacre S, Lo A, Gregory B, Stephens M, Chamberlain G, Stott P, et al. Oligodeoxynucleotide Inhibition of Toll-Like Receptors 3, 7, 8, and 9 Suppresses Cytokine Production in a Human Rheumatoid Arthritis Model. Eur J Immunol (2016) 46(3):772–81. doi: 10.1002/eji.201546123
69. Sacre SM, Lundberg AM, Andreakos E, Taylor C, Feldmann M, Foxwell BM. Selective Use of TRAM in Lipopolysaccharide (LPS) and Lipoteichoic Acid (LTA) Induced NF-kappaB Activation and Cytokine Production in Primary Human Cells: TRAM is an Adaptor for LPS and LTA Signaling. J Immunol (2007) 178(4):2148–54. doi: 10.4049/jimmunol.178.4.2148
70. Thwaites RS, Unterberger S, Chamberlain G, Walker-Bone K, Davies KA, Sacre S. TLR1/2 and 5 Induce Elevated Cytokine Levels From Rheumatoid Arthritis Monocytes Independent of ACPA or RF Autoantibody Status. Rheumatol (Oxford) (2020) 59(11):3533–9. doi: 10.1093/rheumatology/keaa220
71. Feldmann M, Brennan FM, Maini RN. Role of Cytokines in Rheumatoid Arthritis. Annu Rev Immunol (1996) 14:397–440. doi: 10.1146/annurev.immunol.14.1.397
72. Hillyer P, Larché MJ, Bowman EP, McClanahan TK, de Waal Malefyt R, Schewitz LP, et al. Investigating the Role of the Interleukin-23/-17A Axis in Rheumatoid Arthritis. Rheumatol (Oxford) (2009) 48(12):1581–9. doi: 10.1093/rheumatology/kep293
73. Schmidt EM, Davies M, Mistry P, Green P, Giddins G, Feldmann M, et al. Selective Blockade of Tumor Necrosis Factor Receptor I Inhibits Proinflammatory Cytokine and Chemokine Production in Human Rheumatoid Arthritis Synovial Membrane Cell Cultures. Arthritis Rheum (2013) 65(9):2262–73. doi: 10.1002/art.38055
74. Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, et al. The Toll-Like Receptor Adaptor Proteins MyD88 and Mal/TIRAP Contribute to the Inflammatory and Destructive Processes in a Human Model of Rheumatoid Arthritis. Am J Pathol (2007) 170(2):518–25. doi: 10.2353/ajpath.2007.060657
75. Sacre SM, Lo A, Gregory B, Simmonds RE, Williams L, Feldmann M, et al. Inhibitors of TLR8 Reduce TNF Production From Human Rheumatoid Synovial Membrane Cultures. J Immunol (2008) 181(11):8002–9. doi: 10.4049/jimmunol.181.11.8002
76. Bondeson J, Foxwell B, Brennan F, Feldmann M. Defining Therapeutic Targets by Using Adenovirus: Blocking NF-kappaB Inhibits Both Inflammatory and Destructive Mechanisms in Rheumatoid Synovium But Spares Anti-Inflammatory Mediators. Proc Natl Acad Sci USA (1999) 96(10):5668–73. doi: 10.1073/pnas.96.10.5668
77. Fields JK, Günther S, Sundberg EJ. Structural Basis of IL-1 Family Cytokine Signaling. Front Immunol (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
78. Marsollier L, Brodin P, Jackson M, Kordulakova J, Tafelmeyer P, Carbonnelle E, et al. Impact of Mycobacterium Ulcerans Biofilm on Transmissibility to Ecological Niches and Buruli Ulcer Pathogenesis. PloS Pathog (2007) 3(5):e62. doi: 10.1371/journal.ppat.0030062
79. Isaac C, Mauborgne A, Grimaldi A, Ade K, Pohl M, Limatola C, et al. Mycolactone Displays Anti-Inflammatory Effects on the Nervous System. PloS Negl Trop Dis (2017) 11(11):e0006058. doi: 10.1371/journal.pntd.0006058
80. Netea MG, Simon A, van de Veerdonk F, Kullberg B-J, van der Meer JWM, Joosten LAB. IL-1β Processing in Host Defense: Beyond the Inflammasomes. PloS Pathog (2010) 6(2):e1000661. doi: 10.1371/journal.ppat.1000661
81. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of Inflammasome Signaling. Nat Immunol (2012) 13(4):333–2. doi: 10.1038/ni.2237
82. Unterberger S, Davies KA, Rambhatla SB, Sacre S. Contribution of Toll-Like Receptors and the NLRP3 Inflammasome in Rheumatoid Arthritis Pathophysiology. Immunotargets Ther (2021) 10:285–98. doi: 10.2147/ITT.S288547
83. Pallotta MT, Nickel W. FGF2 and IL-1β - Explorers of Unconventional Secretory Pathways at a Glance. J Cell Sci (2020) 133(21):jcs250449. doi: 10.1242/jcs.250449
84. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
85. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D Pore Structure Reveals Preferential Release of Mature Interleukin-1. Nature (2021) 593(7860):607–11. doi: 10.1038/s41586-021-03478-3
86. Zhang M, Liu L, Lin X, Wang Y, Li Y, Guo Q, et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell (2020) 181(3):637–52.e15. doi: 10.1016/j.cell.2020.03.031
87. Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and Autophagy Regulation - a Two-Way Street. Mol Med (2017) 23:188–95. doi: 10.2119/molmed.2017.00077
88. Dinarello CA. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
89. Artlett CM. Inflammasomes in Wound Healing and Fibrosis. J Pathol (2013) 229(2):157–67. doi: 10.1002/path.4116
90. Gushiken LFS, Beserra FP, Bastos JK, Jackson CJ, Pellizzon CH. Cutaneous Wound Healing: An Update From Physiopathology to Current Therapies. Life (Basel) (2021) 11(7):665. doi: 10.3390/life11070665
91. Fahey E, Doyle SL. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front Immunol (2019) 10:1426. doi: 10.3389/fimmu.2019.01426
92. Martin P, Palmer G, Vigne S, Lamacchia C, Rodriguez E, Talabot-Ayer D, et al. Mouse Neutrophils Express the Decoy Type 2 Interleukin-1 Receptor (IL-1R2) Constitutively and in Acute Inflammatory Conditions. J Leukoc Biol (2013) 94(4):791–802. doi: 10.1189/jlb.0113035
93. Barrett L, Chen J, Dai L, Plaisance-Bonstaff K, Del Valle L, Qin Z. Role of Interleukin-1 Family Members and Signaling Pathways in KSHV Pathogenesis. Front Cell infection Microbiol (2020) 10:587929. doi: 10.3389/fcimb.2020.587929
94. Song A, Zhu L, Gorantla G, Berdysz O, Amici SA, Guerau-de-Arellano M, et al. Salient Type 1 Interleukin 1 Receptor Expression in Peripheral non-Immune Cells. Sci Rep (2018) 8(1):723. doi: 10.1038/s41598-018-19248-7
95. Kay J, Calabrese L. The Role of Interleukin-1 in the Pathogenesis of Rheumatoid Arthritis. Rheumatol (Oxford) (2004) 43(Suppl 3):iii2–9. doi: 10.1093/rheumatology/keh201
96. Botsios C, Sfriso P, Punzi L, Todesco S. Non-Complementaemic Urticarial Vasculitis: Successful Treatment With the IL-1 Receptor Antagonist, Anakinra. Scand J Rheumatol (2007) 36(3):236–7. doi: 10.1080/03009740600938647
97. Krause K, Mahamed A, Weller K, Metz M, Zuberbier T, Maurer M. Efficacy and Safety of Canakinumab in Urticarial Vasculitis: An Open-Label Study. J Allergy Clin Immunol (2013) 132(3):751–4.e5. doi: 10.1016/j.jaci.2013.04.008
98. Dana MR, Zhu SN, Yamada J. Topical Modulation of Interleukin-1 Activity in Corneal Neovascularization. Cornea (1998) 17(4):403–9. doi: 10.1097/00003226-199807000-00011
99. Mirza RE, Fang MM, Ennis WJ, Koh TJ. Blocking Interleukin-1β Induces a Healing-Associated Wound Macrophage Phenotype and Improves Healing in Type 2 Diabetes. Diabetes (2013) 62(7):2579–87. doi: 10.2337/db12-1450
100. Grayson PC, Yazici Y, Merideth M, Sen HN, Davis M, Novakovich E, et al. Treatment of Mucocutaneous Manifestations in Behçet's Disease With Anakinra: A Pilot Open-Label Study. Arthritis Res Ther (2017) 19(1):69. doi: 10.1186/s13075-017-1222-3
Keywords: Buruli ulcer, Mycobacterium ulcerans, mycolactone, IL-1β, Sec61, protein translocation, macrophages
Citation: Hall BS, Hsieh LT-H, Sacre S and Simmonds RE (2022) The One That Got Away: How Macrophage-Derived IL-1β Escapes the Mycolactone-Dependent Sec61 Blockade in Buruli Ulcer. Front. Immunol. 12:788146. doi: 10.3389/fimmu.2021.788146
Received: 01 October 2021; Accepted: 27 December 2021;
Published: 26 January 2022.
Edited by:
Sangeeta Tiwari, The University of Texas at El Paso, United StatesReviewed by:
Tjip S. van der Werf, University of Groningen, NetherlandsYoshihiko Hoshino, National Institute of Infectious Diseases (NIID), Japan
Copyright © 2022 Hall, Hsieh, Sacre and Simmonds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel E. Simmonds, cmFjaGVsLnNpbW1vbmRzQHN1cnJleS5hYy51aw==