 Melissa A. Colden
Melissa A. Colden Sushant Kumar1,2
Sushant Kumar1,2 Bolormaa Munkhbileg
Bolormaa Munkhbileg Daria V. Babushok
Daria V. Babushok- 1Division of Hematology-Oncology, Department of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 2Comprehensive Bone Marrow Failure Center, Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, PA, United States
Paroxysmal Nocturnal Hemoglobinuria (PNH) is a disease as simple as it is complex. PNH patients develop somatic loss-of-function mutations in phosphatidylinositol N-acetylglucosaminyltransferase subunit A gene (PIGA), required for the biosynthesis of glycosylphosphatidylinositol (GPI) anchors. Ubiquitous in eukaryotes, GPI anchors are a group of conserved glycolipid molecules responsible for attaching nearly 150 distinct proteins to the surface of cell membranes. The loss of two GPI-anchored surface proteins, CD55 and CD59, from red blood cells causes unregulated complement activation and hemolysis in classical PNH disease. In PNH patients, PIGA-mutant, GPI (-) hematopoietic cells clonally expand to make up a large portion of patients’ blood production, yet mechanisms leading to clonal expansion of GPI (-) cells remain enigmatic. Historical models of PNH in mice and the more recent PNH model in rhesus macaques showed that GPI (-) cells reconstitute near-normal hematopoiesis but have no intrinsic growth advantage and do not clonally expand over time. Landmark studies identified several potential mechanisms which can promote PNH clonal expansion. However, to what extent these contribute to PNH cell selection in patients continues to be a matter of active debate. Recent advancements in disease models and immunologic technologies, together with the growing understanding of autoimmune marrow failure, offer new opportunities to evaluate the mechanisms of clonal expansion in PNH. Here, we critically review published data on PNH cell biology and clonal expansion and highlight limitations and opportunities to further our understanding of the emergence of PNH clones.
Introduction
Paroxysmal Nocturnal Hemoglobinuria (PNH) is a rare, life-threatening blood disease characterized by hemolysis, propensity for blood clotting, and bone marrow failure (1). Although PNH was recognized in the clinic as early as 1882 by Dr. Paul Strübing, it was over a century later when the cause of PNH was identified as an absence of a group of membrane proteins attached to the cell surface by glycosylphosphatidylinositol (GPI) anchors (1–3). In most PNH patients, GPI-anchor deficiency is caused by somatic mutations in phosphatidylinositol glycan anchor biosynthesis class A (PIGA), an X-linked gene required for GPI anchor biosynthesis (4–6) (Figure 1). PIGA mutations occur in early hematopoiesis, and subsequent clonal expansion of PIGA-mutant cells lacking GPI-anchored proteins (GPI-APs) leads to clinical PNH disease. In this review, we will refer to PIGA-mutant cells as GPI (-) or PNH cells. Loss of GPI-APs is particularly detrimental for human erythrocytes, which in the absence of complement regulatory proteins CD55 and CD59 become susceptible to uncontrolled complement activation, leading to hemolytic anemia (Figure 2).
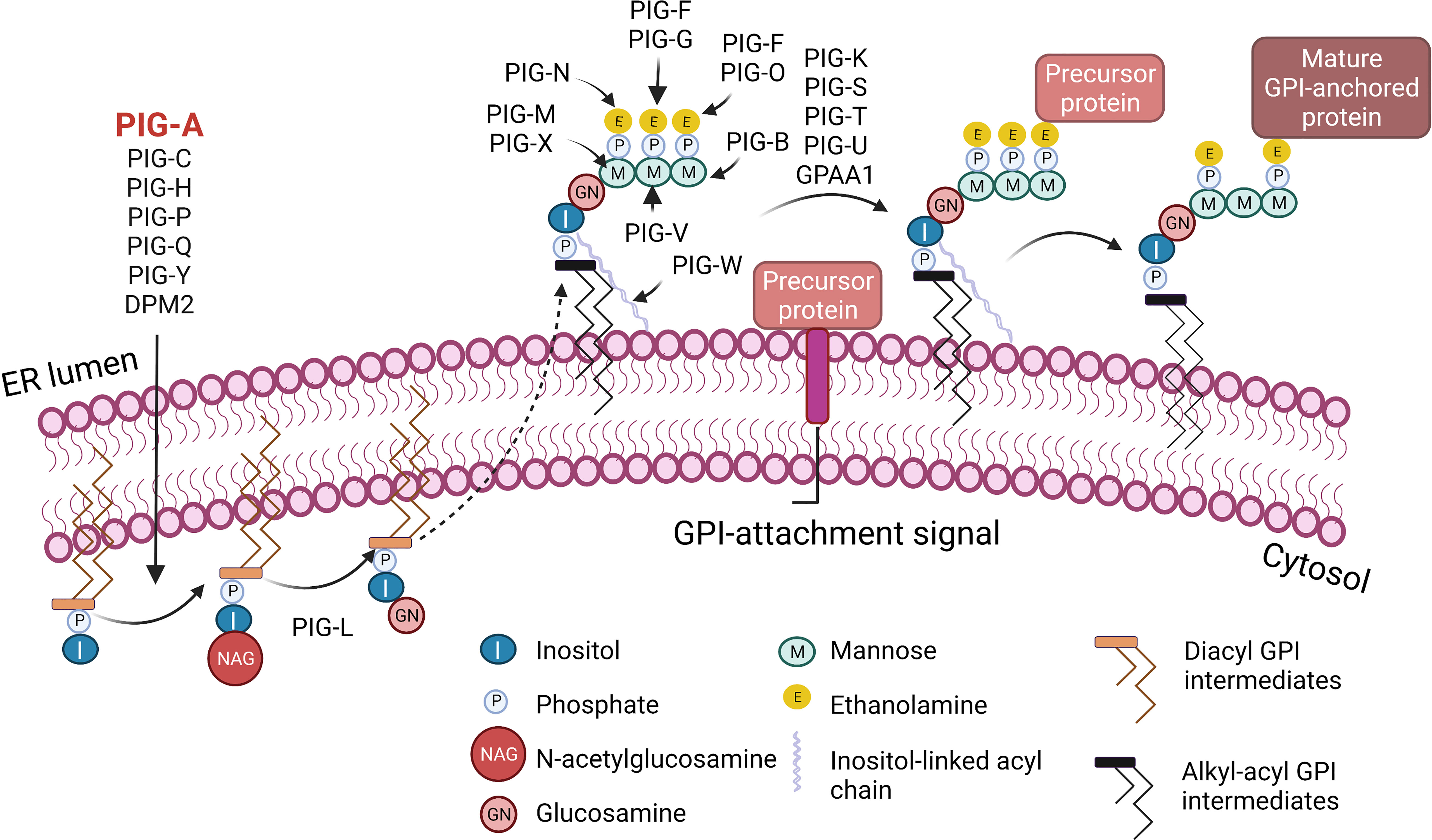
Figure 1 Biosynthesis of glycosylphosphatidylinositol (GPI)-anchored proteins. GPI-anchored proteins are surface proteins linked to the membrane through a GPI glycolipid attachment. GPI is synthesized through a series of steps on the membrane of the endoplasmic reticulum (ER). The initial step involves the transfer of N-acetylglucosamine to inositol on the cytoplasmic side of the ER membrane by a multi-subunit enzyme comprised of seven proteins that include PIGA. A series of subsequent steps, most of which occur on the luminal side of the ER membrane, produce the mature GPI moiety, which is then transferred to the C-terminus of the precursor protein that contains a GPI-anchor attachment signal. Following attachment of GPI, the GPI can be further modified and remodeled during its transport through the Golgi network on the way to the cell surface. I, Inositol; M, Mannose; P, Phosphate; E, Ethanolamine; NAG, N-acetylglucosamine; GN, Glucosamine. Figure created with BioRender.com.
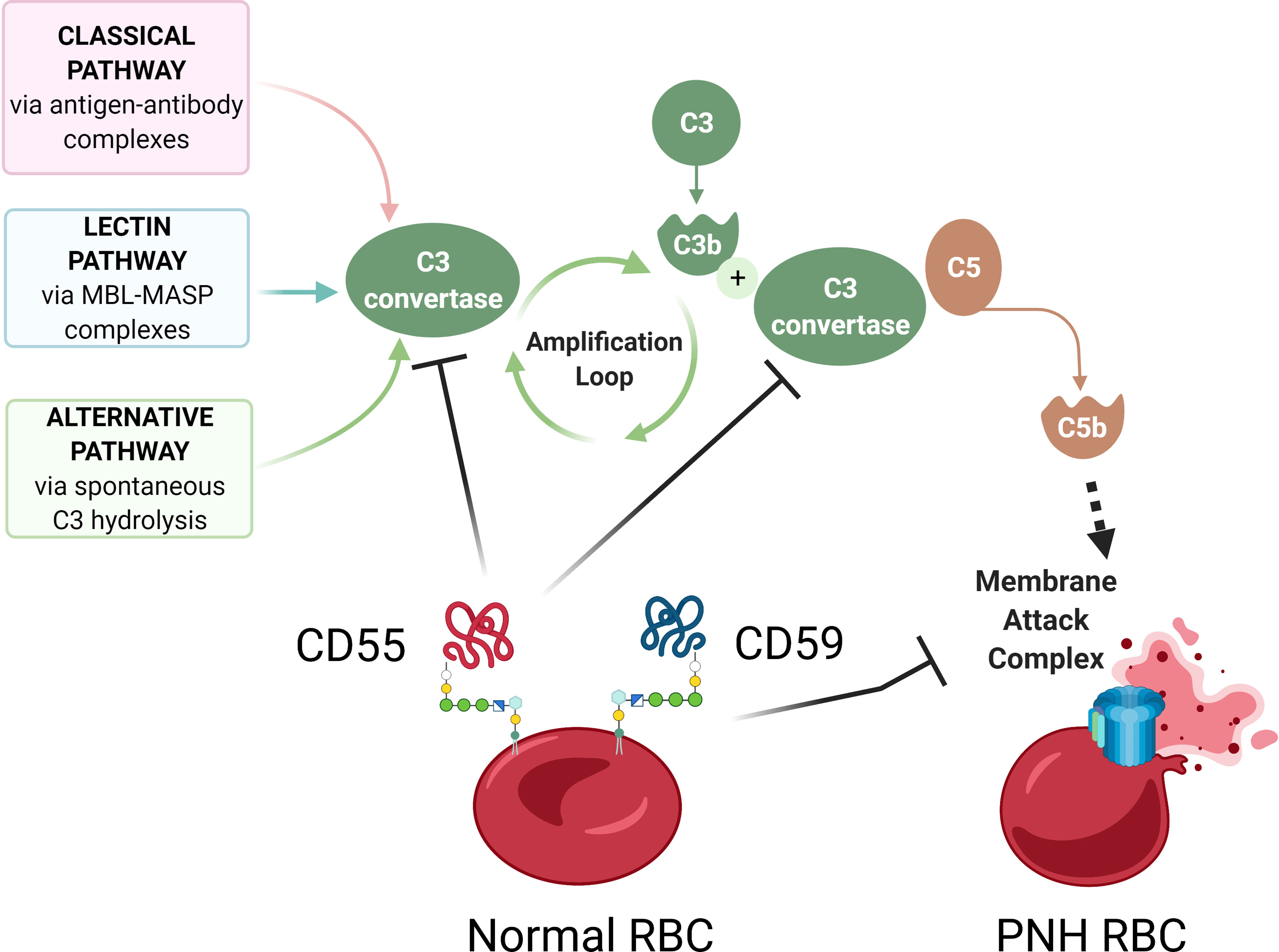
Figure 2 Complement-mediated hemolysis in PNH. A schematic diagram showing aberrant complement activation on PNH erythrocytes due to the deficiency of GPI-anchored proteins CD55 and CD59. The Classical, Lectin, and Alternative pathways of complement activation lead to the formation of C3 convertase, which cleaves C3 into C3a and C3b, leading to formation of C5 convertase, which cleaves C5 to C5b, to activate terminal complement components and form the membrane attack complex (MAC). In normal red blood cells (RBCs), GPI-anchored surface proteins CD55 and CD59 inhibit complement activation by blocking C3 convertase and membrane attack complex, respectively. In the absence of CD55 and CD59, GPI-deficient PNH RBCs have uncontrolled complement activation and lysis. Diagram made with BioRender.com; complement pathways adapted from “Roles of the Complement Cascade in Innate Immunity” by BioRender.com (2021).
While large clonal expansions of GPI (-) cells cause hemolytic PNH disease, tiny stable populations (~0.001% to 0.005%, 1 to 5 per 10 million cells) can be found in most healthy individuals (7, 8). In contrast, more expanded PNH clones develop in nearly 50% of patients with immune-mediated acquired aplastic anemia (AA), an autoimmune bone marrow aplasia caused by autoreactive T lymphocyte attack on hematopoietic stem and progenitor cells (HSPCs) (9–11). The close relationship between PNH and AA was first noted over 60 years ago by Dr. William Dameshek in his famous editorial (12), but the nature of the link between the two conditions continues to be a subject of intense speculation (1, 13–17). The frequent co-occurrence of PNH with immune-mediated AA and lack of several immunologically relevant GPI-anchored surface molecules on PNH HSPCs have led to the prevailing theory that PNH HSPCs may have a relative survival advantage during autoimmune attacks in AA patients, leading to clonal expansion of PNH HSPCs upon recovery in a subset of patients treated with immunosuppression (17, 18) (Figure 3).
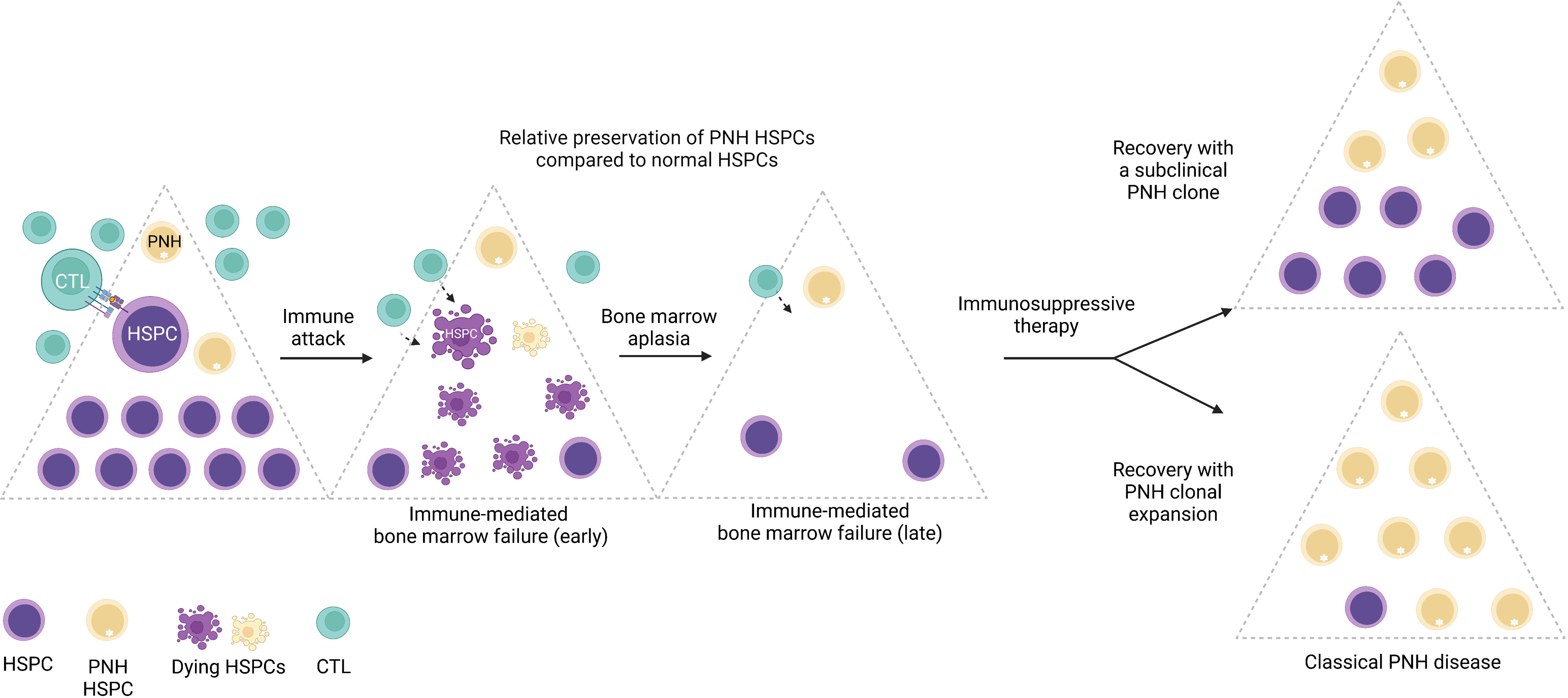
Figure 3 The emergence of PNH clones is closely related to immune-mediated bone marrow failure. A schematic diagram illustrating the relationship between immune-mediated bone marrow failure (acquired aplastic anemia) and clonal expansion of HSPCs with somatic mutations in the PIGA gene (PNH HSPCs). In acquired aplastic anemia, cytotoxic T lymphocytes (CTL) recognize an unknown autoantigen presented by the hematopoietic stem and progenitor cells (HSPCs). Aberrant recognition of the HSPCs leads to their autoimmune destruction (shown as dying HSPCs) and manifests clinically as bone marrow failure. PNH HSPCs are hypothesized to have relative resistance to autoimmune destruction in AA. In AA patients treated with immunosuppressive therapy, bone marrow may recover with a smaller subclinical PNH clone; however, in a subset of patients, PNH HSPCs undergo progressive clonal expansion, leading to classical PNH disease, characterized by hemolytic anemia and related symptoms, and an increased risk of thrombosis. Figure created with BioRender.com.
In this review, we critically examine existing animal models of PNH, comparing experimental observations with human PNH disease and discussing the challenges of cross-species modeling of PNH. We will highlight seminal results arising from animal models and studies of patient cells and discuss remaining controversies and opportunities to expand our understanding of the emergence of PNH disease and its relationship to AA.
Development of Mouse Models of PIGA Loss and GPI-Anchored Protein Deficiency
In 1993, somatic mutations in PIGA gene were identified as the cause of PNH (4–6, 19–21). Although over 20 distinct enzymes are required to produce mature GPI anchors (Figure 1), PIGA is the only gene in this pathway located on the X chromosome. A single inactivating PIGA mutation is sufficient to disrupt GPI biosynthesis and eliminate all GPI-APs from PIGA-mutant cells, both in males and females (due to X chromosome inactivation). Currently, there are 139 human proteins known to be GPI-anchored, and several more are predicted to be GPI-anchored based on computational and proteomic analyses (22–24). GPI-APs participate in various cellular processes, including immune response regulation, signal transduction, and cell-cell adhesion (25, 26). Multiple rare congenital syndromes of GPI-anchored protein deficiencies have been described, one of which is multiple congenital anomalies-hypotonia-seizures syndrome 2 (MCAHS2) (27). MCAHS2 is a rare X-linked disorder caused by germline hypomorphic mutations in PIGA, leading to early childhood lethality from severe anomalies that include structural malformations of the central nervous system (27–30). In contrast to the inherited PIGA mutations in MCAHS2, PIGA mutations in PNH are acquired somatically and are restricted to hematopoietic cells, which remain viable and can reconstitute trilineage hematopoiesis (31).
The evolutionary conservation of PIGA and the synteny of the mammalian X-chromosome led to the development of Piga-mutant mice to model PNH (Table 1). However, in cross-species modeling of PIGA deficiency, differences in GPI-anchoring status and functional divergence of various GPI-APs present a unique challenge. In contrast to mouse models of other single-gene defects, a knockout of Piga results in loss of not just Piga but all GPI-anchored proteins from the surface of mutant cells. A recent comparative analysis of GPI-AP conservation between humans and mice showed that the majority of GPI-APs are conserved, with 77% having one-to-one mouse GPI-AP orthologs and >90% of human GPI-AP genes having at least one GPI-anchored mouse ortholog (24). However, there were also several differences, with 13 human genes having no GPI-anchored orthologs in mice and, conversely, 16 mouse GPI-AP genes not having corresponding GPI-anchored orthologs in humans (Figure 4) (24). While divergence of GPI-AP genes with no expression in hematopoietic cells is less consequential to PNH modeling, an evolutionary divergence of those expressed in hematopoietic lineages (Figure 5) can lead to unanticipated phenotypic differences between PNH mice and PNH patients. Of practical relevance, the absence of GPI-APs has complicated the analysis of the HSPC compartment in mice because of their lack of classical GPI-anchored surface markers Sca1 and CD48 used for immunophenotypic analyses of murine HSPCs.

Table 1 Summary of animal models of paroxysmal nocturnal hemoglobinuria.
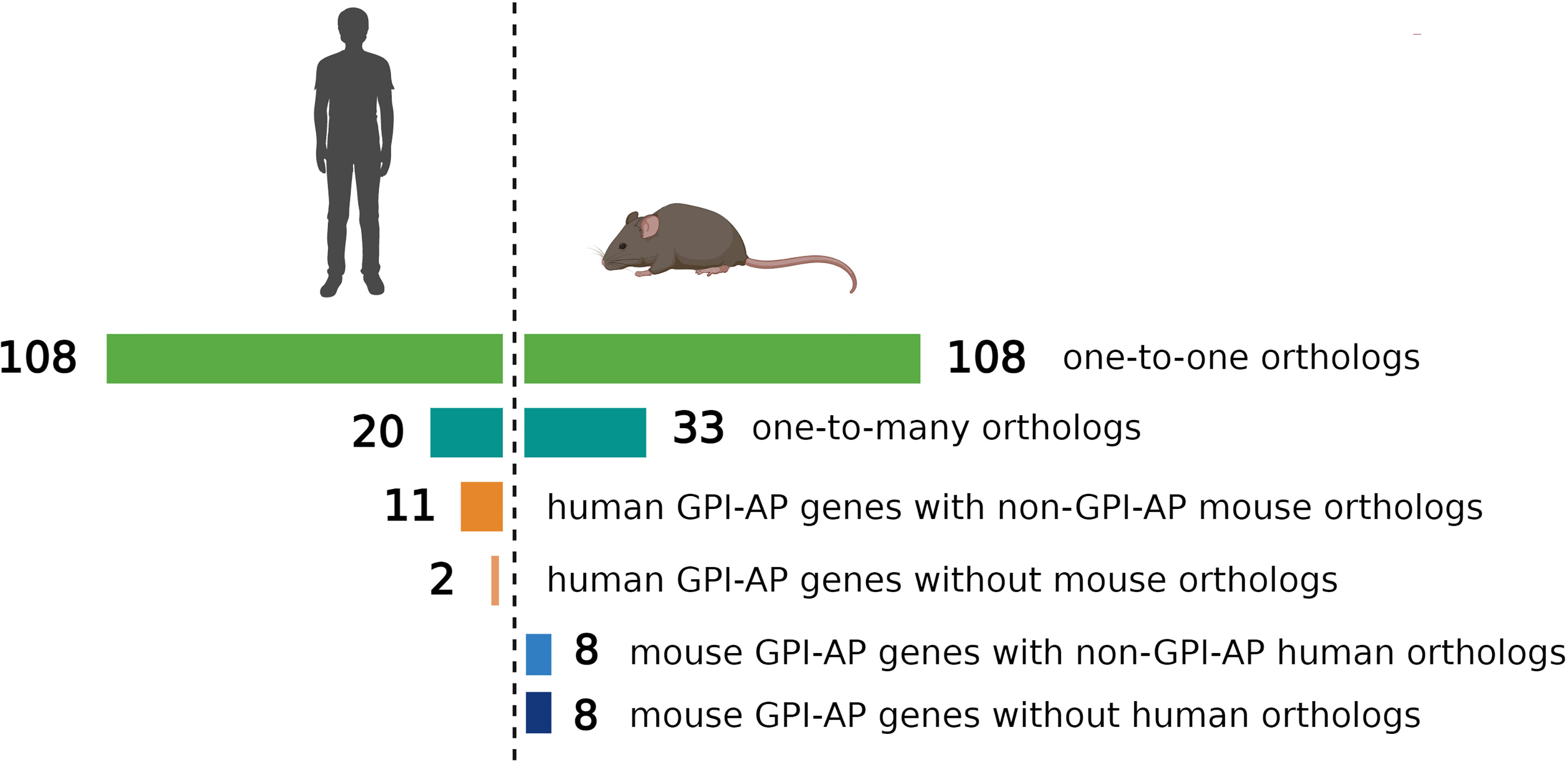
Figure 4 Comparison of evolutionary conservation of GPI-anchored proteins between humans and mice. 108 human GPI-AP genes had one-to-one orthologs in mouse, 20 human GPI-AP genes had more than one mouse ortholog (n=33), 11 human GPI-AP genes were orthologous to mouse genes whose products were not GPI-anchored, and 2 human GPI-AP genes lacked mouse orthologs. 8 mouse GPI-AP genes were orthologous to human genes whose products were not GPI-anchored, and 8 mouse GPI-AP genes lacked human orthologs. Figure adapted from Kumar and Babushok (24).
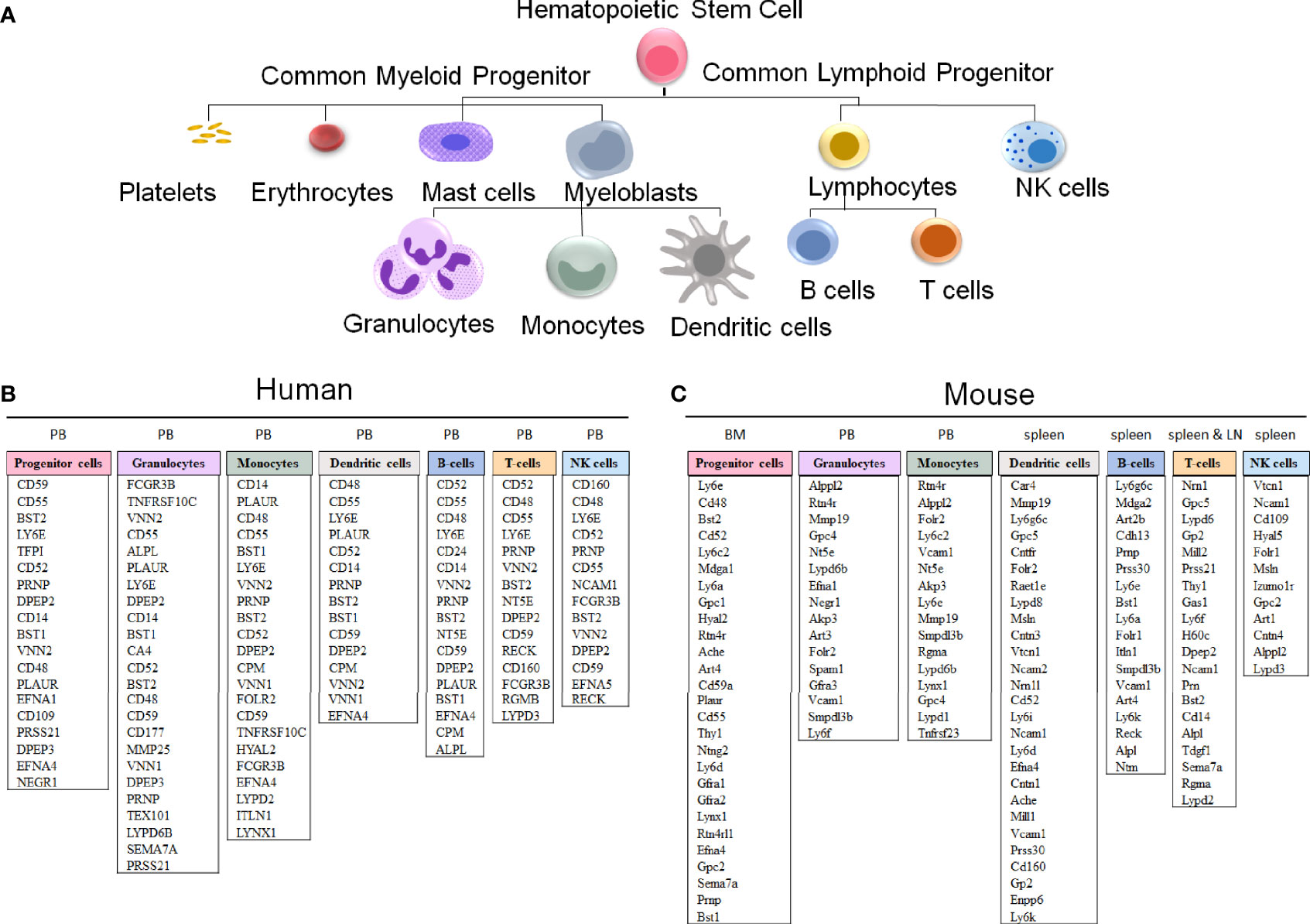
Figure 5 GPI-anchored protein genes in human and mouse hematopoietic cells. (A) A schematic representation of hematopoietic cell differentiation, with the corresponding lists of GPI-AP genes with the highest expression in sorted hematopoietic cell populations in humans (B) and mice (C). The subsets of the more highly expressed GPI-AP genes, defined as having ≥ 10 transcripts per million (tpm) in published RNA-sequencing datasets, are listed in the descending order of expression within each of the cell lineages. (B) The human GPI-AP gene expression are listed based on RNA-Seq from specified peripheral blood (PB) subsets by Monaco et al., 2019 (48), available at the Human Blood Atlas. (C) The mouse GPI-AP genes expressed at ≥ 10 tpm are shown based on RNA sequencing expression analysis of mouse (C57BL/6) hematopoietic cell subsets isolated from Bone Marrow (BM, progenitor cells; Bolden et al, 2018 (49)), PB (granulocytes and monocytes), spleen (dendritic cells and T cells published by Marsman et al, 2018 (50); B-cells published by Shi et al, 2015 (51)) and lymph nodes (LN) (T cells published by Marsman et al, 2018 (50))), as extracted from the Haemosphere data portal (52, 53).
Constitutional Piga Deletion in Mice Causes Embryonic Lethality Due to Multiple Extrahematopoietic Abnormalities
The early development of Piga-mutant mice was complicated by embryonic lethality. When a vector targeting Piga gene was transfected into murine embryonic stem cells (ESCs) followed by their injection into blastocysts to create chimeric mice (31, 32), litter sizes were smaller than expected. Surviving mice had very low (1-5%) chimerism with no germline transmission of Piga-null allele, suggesting that GPI-APs are essential for embryonic development (31). Piga-mutant ESCs also failed to produce normal embryoid body (EB) structures in vitro, with resultant EBs structurally disorganized and smaller than expected (31, 54).
Embryonic lethality was subsequently confirmed in the EIIa-Cre/Piga-LoxP model which deleted Piga in early embryogenesis under the control of the adenovirus EIIa promoter (34). EIIa-Cre is active in oocytes and preimplantation embryos, therefore female embryos with various degrees of mosaicism for Piga-mutant cells were produced. Heterozygous female mice with high levels of Cre recombination did not survive after birth due to multiple abnormalities, including orofacial malformations that caused feeding difficulties. X-inactivation analysis of mice with high Cre recombination rates revealed significantly skewed silencing of the X-chromosome carrying the null Piga allele in kidney, heart, lung, brain, and liver, suggesting that GPI (-) cells in these tissues die or are at a growth disadvantage. Partially recombined mice who survived to adulthood had complete Cre recombination within oocytes but produced no offspring bearing a Piga-null allele, consistent with embryonic lethality.
Subsequent studies using conditional Piga inactivation with keratin 5-Cre (K5Cre), expressed in basal cells of the skin, revealed lethality within a few days of birth due to abnormal skin development (55), reinforcing the importance of GPI-APs in nonhematopoietic tissues. The extrahematopoietic manifestations of Piga deletion explain the early childhood lethality seen with hypomorphic germline PIGA mutations in patients with MCAHS2 syndrome. However, hematopoietic tissues of EIIa-Cre/Piga-LoxP mice had nearly equal proportions of recombined and wild-type cells, consistent with no detrimental effect of Piga loss for hematopoietic development (34).
Piga-mutant Hematopoietic Cells Reconstitute Trilineage Hematopoiesis but Have No Cell-Intrinsic Growth Advantage
To model Piga loss in hematopoietic cells, several mouse models of hematopoietic cell-specific Piga deficiency have been developed (31, 33, 34, 36, 37, 40, 54, 56) (Table 1); of which two landmark studies were most informative in evaluating the role of GPI-AP deficiency in hematopoiesis (34, 40).
Murakami et al. created a transplant-based model of Piga deletion. In this model, Piga-LoxP/Y males were first mated with females bearing a broadly expressed human cytomegalovirus-Cre (hCMV-Cre) to produce heterozygous embryos mosaic for GPI-anchor deficiency due to X-inactivation (40, 57). Fetal liver cells from mosaic day 14 embryos were then transplanted into irradiated wild-type mice. The recipient mice were evaluated for reconstitution by donor GPI (+) and GPI (-) cells, and the relative contribution of GPI (-) cells to various peripheral blood cell lineages was tracked for evidence of clonal expansion over time (40).
In a separate model, Keller et al. produced a hematopoietic-specific knockout of Piga using a Cre/LoxP system under the control of the c-fes promoter region, which turns on in embryonic development and is active in definitive hematopoiesis (35). In this model, Piga excision was incomplete and occurred gradually, requiring up to eight months to achieve complete recombination in male fes-Cre homozygous, Piga-LoxP mice. Aged mice in this model had neuromuscular symptoms, attributed to the “leakiness” of the Cre expression, with recombination detected in some neural cells.
Both models demonstrated stable proportions of GPI (-) blood cells over the animals’ lifetime without clonal expansion in up to 42 weeks of follow-up. GPI (-) cells reconstituted near-normal hematopoiesis, with no or only minimal differences in blood counts between mutants and wild-type mice (35, 40). Transplant studies showed no apparent defects in repopulating capacity in primary and secondary transplantation assays (35, 40).
More recently, advances in gene editing allowed the creation of the first non-human primate model of PNH (47). Shin et al. used CRISPR/Cas9 gene editing to disrupt PIGA in CD34+ HSPCs in rhesus macaques. PIGA-mutant CD34+ HSPCs were then autologously transplanted into two animals, with the resultant GPI (-) cell proportions tracked by flow cytometry over time. Similar to the stability of PNH cell populations in mice, the GPI (-) cell populations persisted at a low, stable frequency of 0.2% to 0.6% for more than two years of follow-up without clonal expansion, confirming the absence of cell-intrinsic growth advantage of PNH cells.
Altered Lymphopoiesis in Mice Mosaic for Piga Deficiency
While the early mouse models of PNH were mired by difficulties in achieving complete Piga loss in hematopoietic cells, the ability to track GPI (-) cell populations by flow cytometry allowed studies of competitive advantage in animals mosaic for Piga loss. One crucial advantage of analyzing mosaic animals is the ability to track the relative contributions of GPI (-) and GPI (+) cells within various cell lineages, whereby GPI (+) cells serve as the “built-in” wild type control akin to a competitive situation in human patients with somatic PIGA mutations.
Immunophenotypic analysis of GPI (-) cell proportions in Piga mosaic mice unexpectedly revealed significant alterations in GPI (-) lymphopoiesis. T cell populations in peripheral blood of Piga mosaic animals in all PNH mouse models were comprised predominantly of GPI (-) cells, with the reverse skewing to GPI (+) cells in the B cell compartment (33, 34, 37, 40). Evaluation of GPI (-) and (+) cell proportions in immature thymocytes showed that the skewing to GPI (-) T cells occurred early in thymocyte development (40). The reasons for the disproportionate contribution of GPI (-) cells to T lymphopoiesis have not been experimentally defined but have been hypothesized to involve a possible inhibitory role of GPI-APs in thymic homing or T lineage commitment. Notably, a skewing to GPI (-) cells is not observed in the T cell lineage in PNH patients (Table 2), pointing to possible differences in GPI-AP involvement in human versus mouse lymphopoiesis. Alternative explanations for the smaller fractions of GPI (-) T lymphocytes in patients could involve factors such as the longevity of lymphocytes, acquisition of PIGA mutations later in life, and varying selection pressures on GPI (-) cells of lymphoid and myeloid lineages.
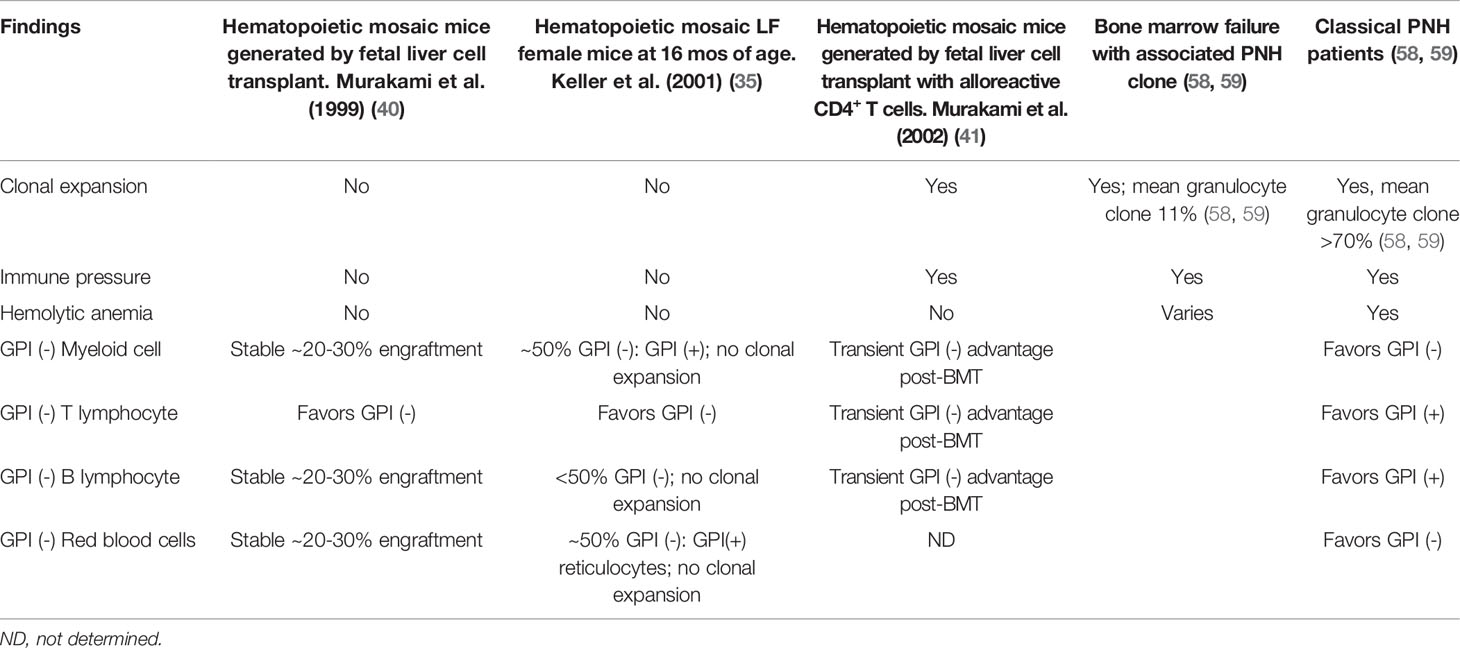
Table 2 Comparison of hematologic findings in PNH patients and mouse models of PNH.
Interestingly, non-mosaic mice with complete Piga deletion in T lymphocytes in the Lck-Cre/Piga-LoxP model showed no gross aberrations in T cell subsets (38), suggesting that the competitive environment in Piga mosaic mice likely exposes the differences between GPI (-) and GPI (+) lymphocytes. Functionally, GPI (-) T cells are competent in response to protein antigens, but have increased proliferation in response to lectin-based stimulant Concanavalin A (Con A) (38). Visconte et al. also reported abnormalities in T regulatory cell development, with smaller proportions of GPI (-) CD4+CD25+FoxP3+ T regulatory cells in mosaic mice (37).
GPI (-) Mouse Erythrocytes Have Mild Sensitivity to Complement Without Overt Hemolytic ANEMIA
While PNH patients suffer from severe hemolytic anemia, Piga-mutant mice have normal or near-normal red blood cell counts (33, 35, 40). Careful examination of hematologic parameters and complement sensitivity identified a shorter half-life and a heightened sensitivity to complement-mediated lysis of GPI (-) erythrocytes with compensatory reticulocytosis. However, these abnormalities were much milder than in human patients and did not result in hemolytic anemia (Table 2). An even milder phenotype was found in an erythroid-specific knockout of Piga driven by a GATA1-Cre. GATA1-Cre/Piga-LoxP knockout mice had only a partial GPI-AP deficiency, similar to type II erythrocytes in humans. The intermediate levels of GPI-APs were explained by GATA1-Cre turning on later in erythroid development, leading to the persistence of residual GPI-APs in mature erythrocytes (36).
The lack of intravascular hemolysis in Piga-mutant mice was the most notable cross-species difference in modeling PNH. Unlike humans, mice have a unique transmembrane complement regulatory molecule Crry, which inhibits C3 convertase at the cell surface, functioning similar to the human GPI-AP decay-accelerating factor (DAF, CD55) (60–62). Additionally, unlike the single GPI-anchored isoform for human DAF, mice also have a non-GPI-anchored DAF isoform. These cross-species differences in complement regulation explain why GPI (-) erythrocytes in mice are only mildly sensitive to complement. As a comparison, a combined knockout of Crry and CD59 led to 10 to 15 times increased erythrocyte complement sensitivity compared to control mice, and ~3-5 times higher complement sensitivity than Piga-mutant mice (40, 63). Notably, while these differences limit the utility of Piga-mutant mice for studies of complement-mediated hemolysis, the lack of hemolytic anemia and normal viability of PNH mice has been advantageous for long-term studies of PNH clonal expansion and HSPC biology.
The Search for External Factors Responsible for Clonal Expansion of PNH Cells
Piga-mutant cells’ lack of intrinsic growth advantage in animal models provides strong evidence that PIGA mutations alone are insufficient to cause PNH disease and that additional external factors are necessary for GPI (-) cell clonal expansion. Several possible hypotheses have been proposed; the most rigorously investigated were the reduced sensitivity to apoptosis and inflammatory cytokines by PNH cells and PNH cell resistance to various immune-mediated attacks.
GPI (-) and GPI (+) Cells in PNH Patients Have Different Responses to Apoptotic Stimuli
Resistance to apoptotic stimuli has been one of the most hotly debated mechanisms of PNH clonal expansion (64–76) (Table 3). Brodsky et al. reported improved preferential survival of GPI (-) cells (granulocytes, CD34+ progenitors, and lymphoblastoid B-cell lines) compared to GPI (+) cells under conditions of serum starvation. GPI (-) lymphoblastoid cells were also more resistant to radiation injury than cells corrected for PIGA (68). Follow-up studies using an inducible TF-1 erythroleukemia cell line model of PIGA loss revealed lower apoptosis rates of GPI (-) cells in multiple cell killing assays (using allogeneic mononuclear cells or NK92 natural killer cell as cytotoxic effectors, or inducing apoptosis by high dose TNF-α or γ-irradiation) (76). Other groups reported similar results, with reduced apoptosis rates and lower Fas antigen expression in GPI (-) cells (e.g., granulocytes or progenitors) compared to GPI (+) cells in PNH patients (67, 72, 75). However, subsequent observations challenged the conclusion that GPI (-) cells are intrinsically resistant to apoptosis.
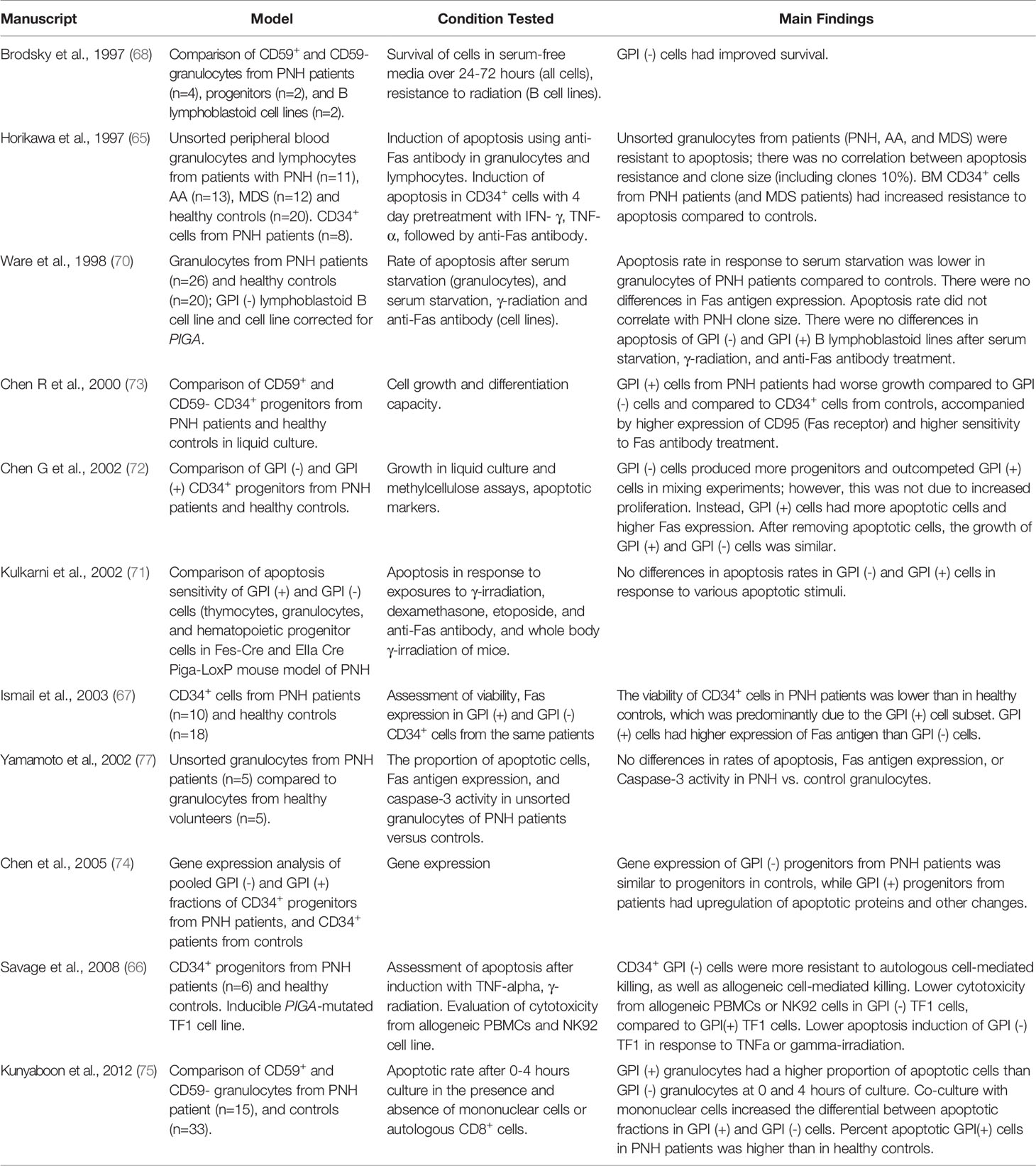
Table 3 Summary of studies evaluating response to apoptosis by PNH cells.
The first clue came from studies showing that while unfractionated granulocytes of PNH patients were more resistant to apoptosis, the apoptotic rates did not correlate with the patients’ PNH clone sizes (65, 70). Subsequent comparisons of apoptotic rates in cells from PNH patients and healthy controls showed that, unexpectedly, the differences in apoptosis between GPI (-) and GPI (+) cells were caused by higher cell death in “normal” GPI (+) cells of PNH patients (72–74). The dying GPI (+) cells were already present at the time of initial blood collection, and their removal eliminated all subsequent differences in apoptotic susceptibility and Fas antigen expression; thus, suggesting that the in vivo environment in patients led to GPI (+) cell death (72). Mouse models of PNH confirmed no intrinsic differences in apoptotic rates in Piga-mutant cells. Kulkarni et al. found no differences in cell death between GPI (-) and GPI (+) hematopoietic cells (thymocytes, granulocytes, and bone marrow hematopoietic progenitors from Fes-Cre/Piga-LoxP mice) exposed to γ-irradiation, etoposide, dexamethasone, anti-Fas antibody, or when mice were treated with whole-body γ-irradiation (71).
Together, these data suggest that GPI (+) and GPI (-) cells may have distinct responses to certain physiologic stimuli in vivo, leading to higher GPI (+) cell death and increased relative survival of GPI (-) cells in PNH patients. The inciting triggers have not been defined but could involve altered responsiveness to inflammatory cytokines, hematopoietic growth factors, or HSPC-directed autoimmune cytotoxicity, among other possibilities. Other factors not captured in animal or cell culture models of PNH are the cumulative differences between GPI (+) and GPI (-) cells, which may be present in patients due to varied clonal evolution, replicative history, telomere attrition, and cell senescence. Additionally, comparisons of apoptotic rates in GPI (+) and GPI (-) cells could be confounded by the known effects of GPI-APs on the externalization of phosphatidylserine unrelated to apoptosis, which could cause a relative increase in Annexin V staining in non-dying GPI (+) cells compared to GPI (-) cells (78).
Inflammatory Cytokine Responses by GPI (-) and GPI (+) Cells
The data on the effects of inflammatory cytokines on clonal expansion in PNH are conflicting but suggest that GPI (-) cells may respond differently to inflammatory cytokines under certain conditions in vitro (65, 66, 79, 80). For example, the addition of IFN-γ and TNF-α to methylcellulose media selectively reduced the fraction of GPI (+), favoring the growth of GPI (-) colonies in colony-forming assays using bone marrow mononuclear cells from PNH patients (79). Similarly, GPI (-) erythroleukemia TF1 cells had higher viability than GPI (+) TF1 cells when cultured with high doses of TNF-α (66).
In contrast to these in vitro studies of patient cells or TF1 cell lines, extensive studies of cytokine sensitivity in PNH mice showed no intrinsic differences in cytokine responses between GPI (-) and GPI (+) murine HSPCs. When treated with IFN-γ, TNF-α, MIP-1α, or TGF-β1, GPI (-) HSPCs from Fes-Cre/Piga-LoxP mice had no advantage over GPI (+) control cells in hematopoietic progenitor assays and did not outcompete GPI (+) cells in bone marrow transplants (80). Similarly, in vivo treatment with polyinosinic-polycytidylic acid (Poly I:C), which induces many proinflammatory cytokines, did not increase the proportion of GPI (-) cells in animals mosaic for Piga mutation (80).
Taken together, the available data suggest that there could be conditions under which patients’ PNH cells may respond differently to inflammatory cytokines. However, the molecular mechanisms underlying these differences and whether altered cytokine responsiveness in cultured cell assays is determined solely by PIGA loss or whether it depends partly on additional, unaccounted factors in GPI (+) versus GPI (-) patient cells are unknown. The degree to which cytokine-mediated selection contributes to PNH pathogenesis under physiologic conditions in vivo also remains unclear.
Mechanisms of Immune Escape by PNH Cells
Early observations of the unique association between PNH and immune-mediated AA have led to wide speculation that clonal expansion of GPI (-) cells occurs because GPI (-) cells escape HSPC-directed immune attack in AA (12, 17, 18). However, the search for immune escape mechanisms has been hindered by an incomplete understanding of the immune pathogenesis of AA. Candidate approaches led to several proof-of-concept demonstrations of immunologic scenarios where GPI (-) cells may have an upper edge, showing immunoselection of GPI (-) cells by 1) natural killer (NK) cell-mediated cytotoxicity (81–83); 2) NKT cell-mediated attack targeting GPI bound to CD1d molecules (84, 85); and 3) CD4+ T cell-mediated alloimmunity (41) (Figure 6). How these contribute to GPI (-) cell selection in AA and PNH patients is still a matter of active debate.
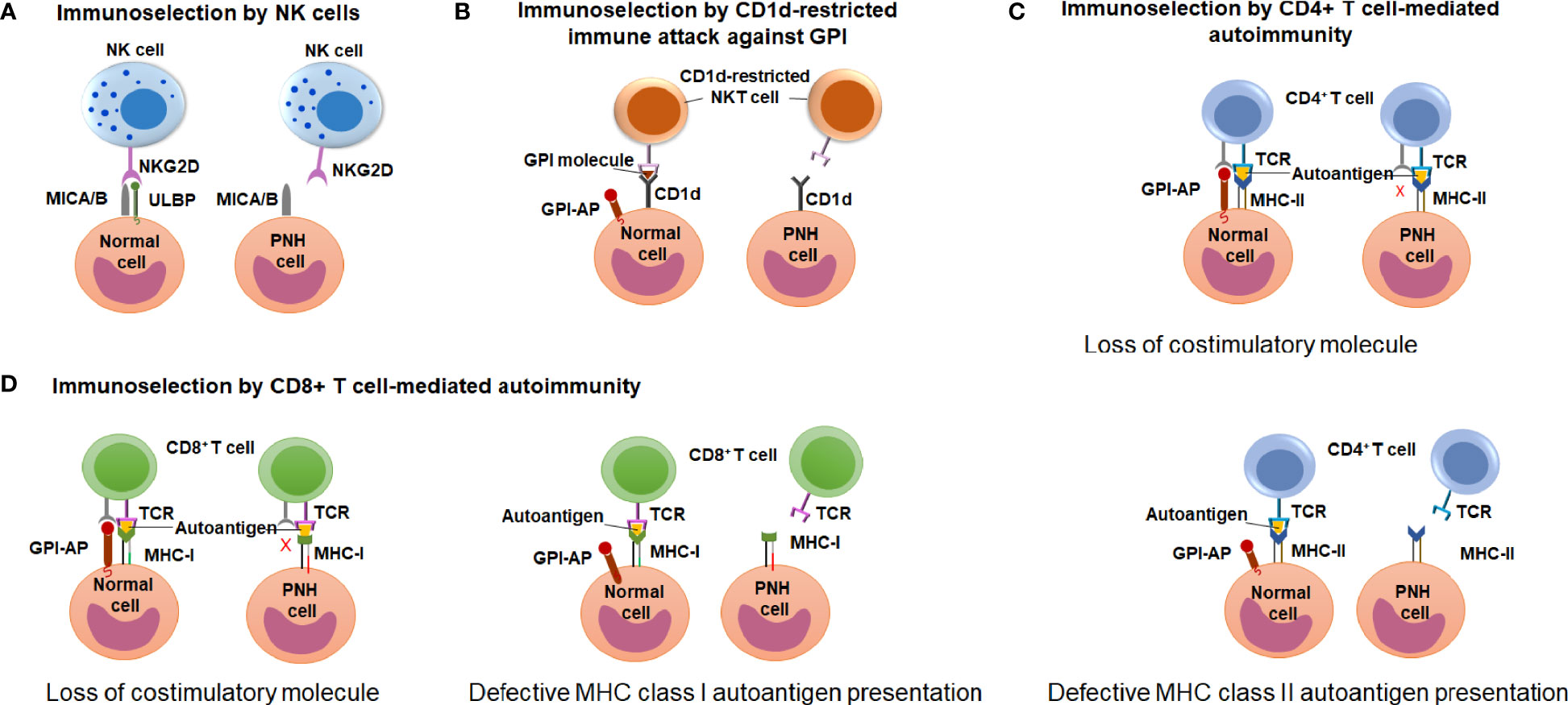
Figure 6 Potential mechanisms of immune-escape by the PNH cells. (A) A schematic diagram illustrating the hypothesis of immunoselection of GPI (-) cells by NK cells. Normal GPI (+) cells express NKG2D ligands ULBPs and MICA/B, and activate NK cells (left), whereas the lack of ULBPs on GPI (-) PNH cells leads to an impaired NK activation (right). (B) A schematic diagram illustrating the immunoselection of GPI (-) cells by CD1d-restricted immune attack against GPI. CD1d-restricted, GPI-specific NKT cells target GPI molecule presented by CD1d and attack GPI (+) normal HSPCs (left) but not the GPI (-) PNH HSPC (right). (C) A schematic diagram illustrating the hypothesis of immune selection of GPI (-) cells by CD4+ lymphocyte-mediated attack. GPI (-) cell may have the reduced ability to activate autoreactive CD4+ T lymphocytes due to missing GPI-anchored co-stimulatory molecules (top diagram). Alternatively, GPI (-) PNH cells may have reduced MHC class II presentation of hypothetical autoantigens compared to GPI (+) normal cells, and survive CD4+ T cell mediated immune attack due to their reduced recognition (bottom diagram). (D) A schematic diagram illustrating the hypothesis of immune selection of GPI (-) cells by CD8+ lymphocyte-mediated attack. GPI (-) cell may have the reduced ability to activate autoreactive CD4+ T lymphocytes due to missing GPI-anchored co-stimulatory molecules (left diagram). Alternatively, GPI (-) PNH cells may have reduced MHC class I presentation of hypothetical autoantigens compared to GPI (+) normal cells, and survive CD8+ T cell mediated immune attack due to their reduced recognition (right diagram).
Immunoselection of GPI (-) Cells by NK Cells
Nagakura et al. used PIGA-mutant and PIGA-corrected leukemia cell lines to test the sensitivity of GPI (-) cells to NK-mediated immune attack (81). When co-cultured with NK cells from healthy volunteers, PIGA-mutant cell lines were killed at lower rates than control cells. NK cytotoxicity does not involve Fas ligand but instead is dependent on perforin; accordingly, blocking perforin in these assays abrogated NK-mediated cytotoxicity. However, when purified perforin was used instead of NK cells, PIGA-mutant and control cell lines were killed at similar rates, indicating that differences in NK-mediated killing likely involved NK cell activation (81). The reasons for improved survival of PIGA-mutant lines were subsequently narrowed down to the lack of GPI-anchored stress-inducible surface molecules, ULBP1, ULBP2, and ULBP3 (83). ULBP proteins serve as ligands for the natural-killer group 2, member D (NKG2D) receptors on NK, natural killer T (NKT), and some conventional CD8+ T cells. Blocking ULBPs with monoclonal antibodies reduced NKG2D activation and NK-mediated cell killing in co-culture assays (83).
It has been more difficult to establish the role of ULBPs in autoimmune HSPC destruction and PNH clonal expansion in AA and PNH patients. Nearly 50% of PNH patients and 60% of AA patients have detectable ULBP 1, 2, or 3 on GPI (+) granulocytes, and all evaluated AA and PNH patients had detectable ULBPs on CD34+ hematopoietic progenitors (82, 83). However, ULBPs were almost always co-expressed with non-GPI-anchored stress-inducible NKG2D ligands MICA and MICB (82, 83). Cell killing assays using autologous NK cells and NKG2D-expressing T cells demonstrated killing of granulocytes from PNH patients expressing NKG2D ligands (83), and blocking NKG2D by antibodies improved hematopoietic progenitor colony formation by bone marrow mononuclear cells from AA and PNH patients (82). However, ULBP ligand dependence of NKG2D-mediated cytotoxicity could not be confirmed because patients cells also expressed MICA/B ligands. Moreover, in contrast to the preferential killing of GPI (+) cells in the K562 and two lymphoid leukemia cell line models of PNH, the same differential sensitivity of patients’ GPI (+) cells compared to GPI (-) cells was not demonstrated (82, 83). Discrepant results between NK-mediated killing of cell lines compared to patients’ cells could be at least partially explained by HLA class I expression differences. K562 and MOLT-4 cell lines, used in NK cytotoxicity studies, lack HLA class I expression and are thus more susceptible to NK-mediated killing (86–88). In contrast, increased inflammatory cytokines (e.g., interferon-γ) in AA patients upregulate HLA class I expression, inhibiting NK cell activation in vivo. Furthermore, detection of NKG2D ligands on mature granulocytes of AA patients (which are not targeted by autoimmune attack in AA) and the presence of ULBP3 on granulocytes in healthy controls demonstrate that NKG2D ligand expression alone is insufficient to induce NK-mediated cell killing in vivo. Indeed, from an evolutionary standpoint, unregulated NK cell activation by binding to NKG2D ligands on stressed hematopoietic cells would be highly detrimental.
In considering the role of NKG2D-mediated cytotoxicity in AA and PNH, additional questions remain. While NKG2D ligands aside from ULBP3 were not detected in healthy controls and disappeared in AA patients in remission, over a third of patients with myelodysplastic syndrome (MDS) had detectable NKG2D ligands. Although NKG2D ligand expression in patients experiencing inflammatory stress or patients with inherited marrow failure syndromes was not explored, the detection of NKG2D ligands in MDS raises a question of a more general NKG2D ligand induction in stress hematopoiesis. Broad activation of NKG2D ligands in stress hematopoiesis would not account for the unique association of PNH with AA, as PNH clonal expansion is not seen in other marrow failure diseases (89, 90). Additionally, NKG2D-mediated killing in AA and PNH does not account for the clinical efficacy of T cell-mediated therapies and the recently demonstrated MHC class I-restriction of autoimmune attack in AA (91–94).
Immunoselection of GPI (-) Cells by CD1d-Restricted Immune Attack Against GPI
Gargiulo et al. proposed that the target of autoimmunity in PNH and AA is the glycolipid molecule GPI itself, and that the absence of GPI in Piga-mutant cells allows them to escape GPI-directed autoimmune attack (85, 91, 92). Unlike peptide antigens presented by HLA molecules and recognized by conventional T cells, lipids are presented by monomorphic HLA class I-like molecules CD1d and recognized by a specialized family of lymphoid cells, the natural killer T cells (NKT cells) (93). GPI was previously identified as a natural ligand for CD1d (94), although its specific function as a CD1d ligand is still debated (94–97).
Gargiulo et al. found a small population of PNH patients’ CD8+ lymphocytes that could bind to CD1d dimers loaded with GPI molecules and demonstrated that this lymphocyte population was several-fold larger in PNH patients compared to controls (85). CD8+ T lymphocytes from PNH patients but not healthy controls were activated by co-culturing them with GPI-loaded CD1d-expressing leukemic cell lines (which lacked HLA class I expression). Lymphocyte activation was highest when both GPI and CD1d were present, suggesting that lymphocytes likely recognized GPI bound to CD1d (85). A parallel study of AA patients identified a similarly expanded CD1d-restricted T cell population in AA (84). However, not all AA patients had CD1d-restricted T cells, raising doubts about their role in AA pathogenesis (84). Moreover, the presence and relative amounts of CD1d-restricted T cells did not correlate with the presence and size of PNH clones, as would have been expected if CD1d-mediated autoimmunity was responsible for PNH immune selection (84).
Although Gargiulo et al.’s results demonstrated that PNH and AA patients have expanded populations of CD8+ lymphocytes capable of being activated by CD1d-GPI complexes in vitro, questions as to the identity of GPI/CD1d-reactive lymphocytes and their role in the pathogenesis of AA and PNH remain. While published studies did not distinguish NKT cells from conventional CD8+ T lymphocytes, the CD1d-restriction of the identified cell populations suggests that these cells are likely CD8+ NKT cells. Because the identified cells lacked the invariant Vα24-Jα18 T cell receptor (TCR) rearrangement that characterizes type I NKT cells in humans, the identified cells were likely type II NKT cells, which have a more varied TCR repertoire (93).
NKT cells arise during thymic development through a specialized differentiation process and are functionally distinct from conventional T cells. When a double-positive (DP) thymocyte produces a T cell receptor (TCR) rearrangement that can bind to CD1d on neighboring DP thymocytes, the CD1d-responsive thymocyte is selected to differentiate down the NKT cell lineage (98). Unlike HLA, CD1d lacks a binding site for CD4 or CD8 co-receptors, and most mature NKT cells are CD4- CD8- double-negative (DN) or CD4+; however, rare CD8+ NKT cells can occur (98). Because Gargiulo et al. focused their analysis on CD8+ cells, it is not known whether the identified CD1d-restricted CD8+ cells represent a fraction of the larger population of GPI/CD1d-reactive NKTs in AA and PNH patients or if CD8+ NKT cells are uniquely expanded. Notably, in an independent study, Mannick et al. had previously evaluated the frequency of CD3+ CD1d-tetramer+ NKT cells in two PNH patients (97). In agreement with Gargiulo’s findings, Mannick et al. also found an expanded population of NKT cells (0.28% and 0.095% of peripheral blood mononuclear cells) compared to 0.029% in 22 controls (97).
Importantly, the available data are insufficient to establish the role of CD1d-restricted GPI-reactive cells in HSPC-directed cytotoxicity in AA and the significance of expanded GPI-reactive NKT populations in AA and PNH pathogenesis remains uncertain. The proposed model of GPI-directed immune attack needs to account for the tissue specificity of autoimmunity in AA, despite the ubiquitous presence of GPI and the broad expression of CD1d on non-hematopoietic tissues (99). CD1d-restriction of NKT cell-mediated attack also contradicts the recent genetic evidence of MHC class I-restriction of autoimmune attack in AA (100, 101). An alternative explanation for increased CD1d-restricted NKT cells in AA and PNH is that they may have an immunomodulatory function. Type II NKT cells can modulate the activation of conventional T cells, type I NKT cells, B cells, and granulocytes and even affect the integrity of epithelial cells, causing suppression or activation of immune responses depending on context (93). In several models of autoimmunity, e.g., a model of experimental autoimmune encephalomyelitis (102), type II NKT cells have an immunomodulatory function and alleviate autoimmunity. Thus, additional studies are needed to clarify the function of the expanded GPI-reactive cells in AA and PNH patients.
Immune Selection of GPI (-) Cells by CD4+ Lymphocyte-Mediated Attack in a Murine Model of Graft-Versus-Host Disease
Murakami et al. used a mouse model of PNH to evaluate the ability of GPI (-) cells to outcompete GPI (+) cells in the setting of allogeneic graft-versus-host (GVH) response (41). The investigators specifically focused on CD4+ T cell mediated immunity because of previous reports linking HLA class II-restricted autoimmunity to AA—e.g., putative autoreactive CD4+ T cell clones were obtained by culturing AA patients’ T cells with autologous hematopoietic progenitors (103, 104), and multiple studies reported an association of HLA DR15 with AA (105, 106). Thus, Murakami et al. used an established model of lethal pancytopenia due to allogeneic incompatibility in MHC class II I-Ab gene (bm12) to test the sensitivity of GPI (-) cells to CD4+ T cell-mediated immune attack. A mixture of GPI (+) and GPI (-) murine fetal liver cells were transplanted alone or together with bm12-mismatched CD4+ T lymphocytes into lethally irradiated B6 control mice. In the presence of co-transplanted bm12 CD4+ T cells, the percentages of fetal liver-derived cells at 5 and 8 weeks post-transplant comprised only ~20%, consistent with T cell mediated killing of donor cells. However, GPI (-) cells made up most of the surviving donor cells, indicating that GPI (+) HSPCs were selectively killed, while GPI (-) cells were selectively spared by T cell mediated attack. As CD4+ bm12 T cells were gradually lost post-transplant, the fraction of GPI (+) cells increased. At 21 weeks post-transplant, the GPI (-) cell proportions in animals co-transplanted with CD4+ bm12 cells were the same as mice that did not receive bm12 CD4+ cells.
Murakami et al.’s results reinforced that PNH clonal expansion is not cell-intrinsic and occurs due to external selective pressure in the marrow environment (in this case, CD4+ T cell-mediated allogeneic immune attack). Reversion to the original proportions of GPI (-) cells following the disappearance of alloreactive T cells implies that the CD4+ T cells likely attacked multipotent progenitors but not the long-term hematopoietic stem cells (HSCs). The immune attack likely spared the long-term HSCs because of their lack of MHC-II expression. Once allogeneic T cells were gone, GPI (+) HSC were able to reconstitute hematopoiesis. While underlying reasons for the competitive advantage of PNH cells in the bm12 allogeneic T cell model were not explicitly studied, one likely contributing factor was the differential presence of CD48 on GPI (-) and GPI (+) cells. CD48 is a GPI-anchored costimulatory molecule on antigen-presenting cells, which contributes to the organization of the immune synapse by binding to CD2 on T cells (107). Pretreatment of mice with monoclonal antibodies against CD2 and CD48 significantly reduced the severity of CD4+ (but not CD8+) T lymphocyte-induced graft versus host disease (GVHD) and marrow aplasia, supporting a key role of CD48 costimulation in the induction of bone marrow failure by the bm12 allogeneic T cells (108). However, the extent to which the same mechanism contributes to PNH clonal expansion in humans is less certain. Unlike CD48 in mice, the high-affinity ligand for human CD2 is CD58 (LFA-3), which has some homology to CD48 but exists in both GPI-anchored and transmembrane forms (107, 109). Thus, CD48 is unlikely to play the same role in the immune selection of GPI (+) and GPI (-) cells in AA and PNH patients.
Discussion
The Greek philosopher Aristotle famously remarked on discovering the unknown: “The more you know, the more you realize you don’t know”. This certainly holds true for PNH, which is a fascinating case study of the complex interplay between cell-intrinsic changes due to PIGA loss and the external factors that together produce PNH clonal expansion in patients. Landmark studies identified several potential mechanisms that could promote PNH clonal expansion—whether due to the absence of GPI-anchored NKG2D ligands, a missing GPI antigen, loss of costimulatory molecules, or altered cytokine sensitivity. However, the key obstacle to identifying mechanisms relevant to PNH clonal expansion in patients has been an incomplete understanding of the nature of autoimmunity in AA and the connection between PNH and AA.
With recent advances in complement therapeutics (110–114) (115), have we reached the point where mechanistic understanding of PNH origins is no longer irrelevant? Certainly, many PNH patients can readily receive effective therapy to prevent complement-mediated intravascular hemolysis in as few as six infusions per year with current C5 inhibitor therapy (110, 111). Additionally, advances in proximal complement inhibition (115), including oral inhibitors of Factors B and D in late-stage clinical trials (115–119), hold great promise for more effective and convenient treatment of both intravascular and extravascular hemolysis. Yet, despite tremendous advancements, PNH continues to be a significant burden, requiring life-long, very costly immunosuppressive medications, which are not readily available to PNH patients in resource-poor countries. Furthermore, the close connection between PNH and AA suggests that uncovering the mechanisms of PNH emergence will improve our understanding of autoimmunity in AA. An improved understanding of AA and PNH holds promise for the future development of potentially curative therapies and means of prevention for these life-long relapsing and remitting blood diseases.
The recent discovery of MHC class I restriction of immune attack in AA (100, 101) presents new opportunities to revisit clonal expansion of PNH. As such, the ability of PNH cells to escape autoreactive CD8+ T lymphocytes should be investigated. PNH cells may evade MHC class I-restricted immune attack due to a deficiency of GPI-anchored costimulatory molecules, similar to the role of CD48 deficiency in promoting GPI (-) cell survival in bm12 CD4+ T cell-mediated mouse model of marrow aplasia (41). Alternatively, PNH cells may have a reduced expression of AA autoantigen (s) or other alterations in immune recognition. Indeed, given the absence of numerous GPI-APs with diverse functions in PNH cells and the more insidious alterations in intracellular protein trafficking and membrane structure, it is likely that several non-mutually-exclusive mechanisms contribute to immune escape and context-dependent enhancements of GPI (-) cell survival.
Although GPI (-) cell expansions in classical PNH and in marrow failure are believed to arise for similar reasons, mechanisms that lead to large expansions of PNH cells may differ from those that result in the more common smaller PNH clones in AA. Differences in clone size may be partly explained by the varying self-renewal potential of cells that incur the initial PIGA mutations. Random genetic drift, particularly when occurring in a hypocellular marrow with oligoclonal hematopoiesis, can also lead to stochastic expansions of PIGA-mutant cells (14, 41). However, the lack of PNH association with inherited bone marrow failure diseases or with aging, both of which are associated with oligoclonal hematopoiesis and the development of somatic mutations (9, 90), strongly argues that PNH clones, regardless of size, are etiologically related to AA. Proliferative mutations in several oncogenes (e.g., HMGA2, JAK2, and BCR-ABL) have been reported to cause rare cases of PNH disease (120–123). However, these are exceptional cases, while most PNH patients do not carry oncogenic mutations and most non-PIGA, non-HLA mutations in AA and PNH patients mirror age-related clonal hematopoiesis (e.g., DNMT3A, TET2, ASXL1, BCOR/BCORL1) (120, 124–127).
In sum, PNH is a unique example of a clonal blood disease whose development is fostered by AA’s autoimmune bone marrow environment. Recent advancements in disease models and immunologic technologies and the growing understanding of immunologic and marrow failure processes in AA offer new opportunities to crack the Dameshek Riddle (12) and unravel the mechanisms of clonal expansion of PNH cells and autoimmunity in AA.
Author Contributions
MC drafted the first version of the manuscript. MC, SK, BM, and DB performed critical literature review, wrote and revised the manuscript, and created figures and tables. All authors approved the final manuscript.
Funding
This work was supported by NIH K08 HL132101 and the Institute for Translational Medicine and Therapeutics of the Perelman School of Medicine at the University of Pennsylvania grant funded by NCATS UL1TR001878 to DB.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank patients and families for participating in studies of AA and PNH.
References
1. Brodsky RA. Paroxysmal Nocturnal Hemoglobinuria. Blood (2014) 124:2804–11. doi: 10.1182/blood-2014-02-522128
2. Socie G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, et al. Paroxysmal Nocturnal Haemoglobinuria: Long-Term Follow-Up and Prognostic Factors. French Soc Haematology. Lancet (1996) 348:573–7. doi: 10.1016/S0140-6736(95)12360-1
3. Nishimura JI, Kanakura Y, Ware RE, Shichishima T, Nakakuma H, Ninomiya H, et al. Clinical Course and Flow Cytometric Analysis of Paroxysmal Nocturnal Hemoglobinuria in the United States and Japan. Med (Baltimore) (2004) 83:193–207. doi: 10.1097/01.md.0000126763.68170.46
4. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI Anchor Caused by a Somatic Mutation of the PIG-a Gene in Paroxysmal Nocturnal Hemoglobinuria. Cell (1993) 73:703–11. doi: 10.1016/0092-8674(93)90250-T
5. Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda J, et al. Paroxysmal Nocturnal Haemoglobinuria (PNH) Is Caused by Somatic Mutations in the PIG-a Gene. EMBO J (1994) 13:110–7. doi: 10.1002/j.1460-2075.1994.tb06240.x
6. Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The Cloning of PIG-a, a Component in the Early Step of GPI-Anchor Biosynthesis. Science (1993) 259:1318–20. doi: 10.1126/science.7680492
7. Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA. PIG-A Mutations in Normal Hematopoiesis. Blood (2005) 105:3848–54. doi: 10.1182/blood-2004-04-1472
8. Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal Populations of Hematopoietic Cells With Paroxysmal Nocturnal Hemoglobinuria Genotype and Phenotype Are Present in Normal Individuals. Proc Natl Acad Sci USA (1999) 96:5209–14. doi: 10.1073/pnas.96.9.5209
9. Shah YB, Priore SF, Li Y, Tang CN, Nicholas P, Kurre P, et al. The Predictive Value of PNH Clones, 6p CN-LOH, and Clonal TCR Gene Rearrangement for Aplastic Anemia Diagnosis. Blood Adv (2021) 5(16):3216–26. doi: 10.1182/bloodadvances.2021004201
10. Fattizzo B, Ireland R, Dunlop A, Yallop D, Kassam S, Large J, et al. Clinical and Prognostic Significance of Small Paroxysmal Nocturnal Hemoglobinuria Clones in Myelodysplastic Syndrome and Aplastic Anemia. Leukemia (2021) 35(11):3223–31. doi: 10.1038/s41375-021-01190-9
11. Sugimori C, Mochizuki K, Qi Z, Sugimori N, Ishiyama K, Kondo Y, et al. Origin and Fate of Blood Cells Deficient in Glycosylphosphatidylinositol-Anchored Protein Among Patients With Bone Marrow Failure. Br J Haematol (2009) 147:102–12. doi: 10.1111/j.1365-2141.2009.07822.x
12. Dameshek W. Riddle: What do Aplastic Anemia, Paroxysmal Nocturnal Hemoglobinuria (PNH) and “Hypoplastic” Leukemia Have in Common? Blood (1967) 30:251–4. doi: 10.1182/blood.V30.2.251.251
13. Luzzatto L. Recent Advances in the Pathogenesis and Treatment of Paroxysmal Nocturnal Hemoglobinuria. F1000Res (2016) 5:F1000 Faculty Rev-209. doi: 10.12688/f1000research.7288.1
14. Dingli D, Luzzatto L, Pacheco JM. Neutral Evolution in Paroxysmal Nocturnal Hemoglobinuria. Proc Natl Acad Sci USA (2008) 105:18496–500. doi: 10.1073/pnas.0802749105
15. Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal Nocturnal Haemoglobinuria. Nat Rev Dis Primers (2017) 3:17028. doi: 10.1038/nrdp.2017.28
16. Savage WJ, Brodsky RA. New Insights Into Paroxysmal Nocturnal Hemoglobinuria. Hematology (2007) 12:371–6. doi: 10.1080/10245330701562634
17. Luzzatto L, Bessler M, Rotoli B. Somatic Mutations in Paroxysmal Nocturnal Hemoglobinuria: A Blessing in Disguise? Cell (1997) 88:1–4. doi: 10.1016/S0092-8674(00)81850-4
18. Luzzatto L, Notaro R. The “Escape” Model: A Versatile Mechanism for Clonal Expansion. Br J Haematol (2019) 184:465–6. doi: 10.1111/bjh.15111
19. Kawagoe K, Takeda J, Endo Y, Kinoshita T. Molecular Cloning of Murine Pig-a, a Gene for GPI-Anchor Biosynthesis, and Demonstration of Interspecies Conservation of Its Structure, Function, and Genetic Locus. Genomics (1994) 23:566–74. doi: 10.1006/geno.1994.1544
20. Hillmen P, Bessler M, Mason PJ, Watkins WM, Luzzatto L. Specific Defect in N-Acetylglucosamine Incorporation in the Biosynthesis of the Glycosylphosphatidylinositol Anchor in Cloned Cell Lines From Patients With Paroxysmal Nocturnal Hemoglobinuria. Proc Natl Acad Sci USA (1993) 90:5272–6. doi: 10.1073/pnas.90.11.5272
21. Iida Y, Takeda J, Miyata T, Inoue N, Nishimura J, Kitani T, et al. Characterization of Genomic PIG-a Gene: A Gene for Glycosylphosphatidylinositol-Anchor Biosynthesis and Paroxysmal Nocturnal Hemoglobinuria. Blood (1994) 83:3126–31. doi: 10.1182/blood.V83.11.3126.bloodjournal83113126
22. Masuishi Y, Kimura Y, Arakawa N, Hirano H. Identification of Glycosylphosphatidylinositol-Anchored Proteins and Omega-Sites Using Tio2-Based Affinity Purification Followed by Hydrogen Fluoride Treatment. J Proteomics (2016) 139:77–83. doi: 10.1016/j.jprot.2016.03.008
23. UniProt C. Uniprot: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res (2021) 49:D480–9. doi: 10.1093/nar/gkaa1100
24. Kumar S, Babushok DV. Analysis of Evolutionary Conservation of GPI-Anchored Proteins Between Humans and Mice. Blood Cells Mol Dis (2021) 92:102622. doi: 10.1016/j.bcmd.2021.102622
25. Paulick MG, Bertozzi CR. The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry (2008) 47:6991–7000. doi: 10.1021/bi8006324
26. Um JW, Ko J. Neural Glycosylphosphatidylinositol-Anchored Proteins in Synaptic Specification. Trends Cell Biol (2017) 27:931–45. doi: 10.1016/j.tcb.2017.06.007
27. Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF, et al. The Phenotype of a Germline Mutation in PIGA: The Gene Somatically Mutated in Paroxysmal Nocturnal Hemoglobinuria. Am J Hum Genet (2012) 90:295–300. doi: 10.1016/j.ajhg.2011.11.031
28. Belet S, Fieremans N, Yuan X, Van Esch H, Verbeeck J, Ye Z, et al. Early Frameshift Mutation in PIGA Identified in a Large XLID Family Without Neonatal Lethality. Hum Mutat (2014) 35:350–5. doi: 10.1002/humu.22498
29. Kato M, Saitsu H, Murakami Y, Kikuchi K, Watanabe S, Iai M, et al. PIGA Mutations Cause Early-Onset Epileptic Encephalopathies and Distinctive Features. Neurology (2014) 82:1587–96. doi: 10.1212/WNL.0000000000000389
30. Fauth C, Steindl K, Toutain A, Farrell S, Witsch-Baumgartner M, Karall D, et al. A Recurrent Germline Mutation in the PIGA Gene Causes Simpson-Golabi-Behmel Syndrome Type 2. Am J Med Genet A (2016) 170A:392–402. doi: 10.1002/ajmg.a.37452
31. Rosti V, Tremml G, Soares V, Pandolfi PP, Luzzatto L, Bessler M. Murine Embryonic Stem Cells Without Pig-a Gene Activity Are Competent for Hematopoiesis With the PNH Phenotype But Not for Clonal Expansion. J Clin Invest (1997) 100:1028–36. doi: 10.1172/JCI119613
32. Kawagoe K, Kitamura D, Okabe M, Taniuchi I, Ikawa M, Watanabe T, et al. Glycosylphosphatidylinositol-Anchor-Deficient Mice: Implications for Clonal Dominance of Mutant Cells in Paroxysmal Nocturnal Hemoglobinuria. Blood (1996) 87:3600–6. doi: 10.1182/blood.V87.9.3600.bloodjournal8793600
33. Tremml G, Dominguez C, Rosti V, Zhang Z, Pandolfi PP, Keller P, et al. Increased Sensitivity to Complement and a Decreased Red Blood Cell Life Span in Mice Mosaic for a Nonfunctional Piga Gene. Blood (1999) 94:2945–54. doi: 10.1182/blood.V94.9.2945.421k35_2945_2954
34. Keller P, Tremml G, Rosti V, Bessler M. X Inactivation and Somatic Cell Selection Rescue Female Mice Carrying a Piga-Null Mutation. Proc Natl Acad Sci USA (1999) 96:7479–83. doi: 10.1073/pnas.96.13.7479
35. Keller P, Payne JL, Tremml G, Greer PA, Gaboli M, Pandolfi PP, et al. FES-Cre Targets Phosphatidylinositol Glycan Class a (PIGA) Inactivation to Hematopoietic Stem Cells in the Bone Marrow. J Exp Med (2001) 194:581–9. doi: 10.1084/jem.194.5.581
36. Jasinski M, Keller P, Fujiwara Y, Orkin SH, Bessler M. GATA1-Cre Mediates Piga Gene Inactivation in the Erythroid/Megakaryocytic Lineage and Leads to Circulating Red Cells With a Partial Deficiency in Glycosyl Phosphatidylinositol-Linked Proteins (Paroxysmal Nocturnal Hemoglobinuria Type II Cells). Blood (2001) 98:2248–55. doi: 10.1182/blood.V98.7.2248
37. Visconte V, Raghavachari N, Liu D, Keyvanfar K, Desierto MJ, Chen J, et al. Phenotypic and Functional Characterization of a Mouse Model of Targeted Pig-a Deletion in Hematopoietic Cells. Haematologica (2010) 95:214–23. doi: 10.3324/haematol.2009.011650
38. Hazenbos WL, Murakami Y, Nishimura J, Takeda J, Kinoshita T. Enhanced Responses of Glycosylphosphatidylinositol Anchor-Deficient T Lymphocytes. J Immunol (2004) 173:3810–5. doi: 10.4049/jimmunol.173.6.3810
39. Hazenbos WL, Wu P, Eastham-Anderson J, Kinoshita T, Brown EJ. Impaired Fcepsilonri Stability, Signaling, and Effector Functions in Murine Mast Cells Lacking Glycosylphosphatidylinositol-Anchored Proteins. Blood (2011) 118:4377–83. doi: 10.1182/blood-2011-02-338053
40. Murakami Y, Kinoshita T, Maeda Y, Nakano T, Kosaka H, Takeda J. Different Roles of Glycosylphosphatidylinositol in Various Hematopoietic Cells as Revealed by a Mouse Model of Paroxysmal Nocturnal Hemoglobinuria. Blood (1999) 94:2963–70. doi: 10.1182/blood.V94.9.2963
41. Murakami Y, Kosaka H, Maeda Y, Nishimura J, Inoue N, Ohishi K, et al. Inefficient Response of T Lymphocytes to Glycosylphosphatidylinositol Anchor-Negative Cells: Implications for Paroxysmal Nocturnal Hemoglobinuria. Blood (2002) 100:4116–22. doi: 10.1182/blood-2002-06-1669
42. Sun X, Funk CD, Deng C, Sahu A, Lambris JD, Song WC. Role of Decay-Accelerating Factor in Regulating Complement Activation on the Erythrocyte Surface as Revealed by Gene Targeting. Proc Natl Acad Sci USA (1999) 96:628–33. doi: 10.1073/pnas.96.2.628
43. Lin F, Fukuoka Y, Spicer A, Ohta R, Okada N, Harris CL, et al. Tissue Distribution of Products of the Mouse Decay-Accelerating Factor (DAF) Genes. Exploitation of a Daf1 Knock-Out Mouse and Site-Specific Monoclonal Antibodies. Immunology (2001) 104:215–25. doi: 10.1046/j.1365-2567.2001.01287.x
44. Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeted Deletion of the CD59 Gene Causes Spontaneous Intravascular Hemolysis and Hemoglobinuria. Blood (2001) 98:442–9. doi: 10.1182/blood.V98.2.442
45. Miwa T, Zhou L, Hilliard B, Molina H, Song WC. Crry, But Not CD59 and DAF, Is Indispensable for Murine Erythrocyte Protection In Vivo From Spontaneous Complement Attack. Blood (2002) 99:3707–16. doi: 10.1182/blood.V99.10.3707
46. Qin X, Krumrei N, Grubissich L, Dobarro M, Aktas H, Perez G, et al. Deficiency of the Mouse Complement Regulatory Protein Mcd59b Results in Spontaneous Hemolytic Anemia With Platelet Activation and Progressive Male Infertility. Immunity (2003) 18:217–27. doi: 10.1016/S1074-7613(03)00022-0
47. Shin TH, Baek EJ, Corat MAF, Chen S, Metais JY, AlJanahi AA, et al. CRISPR/Cas9 PIG -A Gene Editing in Nonhuman Primate Model Demonstrates No Intrinsic Clonal Expansion of PNH Hspcs. Blood (2019) 133:2542–5. doi: 10.1182/blood.2019000800
48. Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep (2019) 26:1627–40.e7. doi: 10.1016/j.celrep.2019.01.041
49. Bolden JE, Lucas EC, Zhou G, O’Sullivan JA, de Graaf CA, McKenzie MD, et al. Identification of a Siglec-F+ Granulocyte-Macrophage Progenitor. J Leukoc Biol (2018) 104:123–33. doi: 10.1002/JLB.1MA1217-475R
50. Marsman C, Lafouresse F, Liao Y, Baldwin TM, Mielke LA, Hu Y, et al. Plasmacytoid Dendritic Cell Heterogeneity Is Defined by CXCL10 Expression Following TLR7 Stimulation. Immunol Cell Biol (2018) 96:1083–94. doi: 10.1111/imcb.12173
51. Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional Profiling of Mouse B Cell Terminal Differentiation Defines a Signature for Antibody-Secreting Plasma Cells. Nat Immunol (2015) 16:663–73. doi: 10.1038/ni.3154
52. de Graaf CA, Choi J, Baldwin TM, Bolden JE, Fairfax KA, Robinson AJ, et al. Haemopedia: An Expression Atlas of Murine Hematopoietic Cells. Stem Cell Rep (2016) 7:571–82. doi: 10.1016/j.stemcr.2016.07.007
53. Choi J, Baldwin TM, Wong M, Bolden JE, Fairfax KA, Lucas EC, et al. Haemopedia RNA-Seq: A Database of Gene Expression During Haematopoiesis in Mice and Humans. Nucleic Acids Res (2019) 47:D780–5. doi: 10.1093/nar/gky1020
54. Dunn DE, Yu J, Nagarajan S, Devetten M, Weichold FF, Medof ME, et al. A Knock-Out Model of Paroxysmal Nocturnal Hemoglobinuria: Pig-a(-) Hematopoiesis Is Reconstituted Following Intercellular Transfer of GPI-Anchored Proteins. Proc Natl Acad Sci USA (1996) 93:7938–43. doi: 10.1073/pnas.93.15.7938
55. Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K, et al. Tissue-Specific Knockout of the Mouse Pig-a Gene Reveals Important Roles for GPI-Anchored Proteins in Skin Development. Proc Natl Acad Sci USA (1997) 94:7400–5. doi: 10.1073/pnas.94.14.7400
56. Chen Y, Rong F. Advances in the Creation of Animal Models of Paroxysmal Nocturnal Hemoglobinuria. Hematology (2021) 26:491–6. doi: 10.1080/16078454.2021.1945244
57. Nozaki M, Ohishi K, Yamada N, Kinoshita T, Nagy A, Takeda J. Developmental Abnormalities of Glycosylphosphatidylinositol-Anchor-Deficient Embryos Revealed by Cre/Loxp System. Lab Invest (1999) 79:293–9.
58. Richards SJ, Whitby L, Cullen MJ, Dickinson AJ, Granger V, Reilly JT, et al. Development and Evaluation of a Stabilized Whole-Blood Preparation as a Process Control Material for Screening of Paroxysmal Nocturnal Hemoglobinuria by Flow Cytometry. Cytometry B Clin Cytom (2009) 76:47–55. doi: 10.1002/cyto.b.20438
59. Cannizzo E, Raia M, De Propris MS, Triolo A, Scarpati B, Marfia A, et al. Features, Reason for Testing, and Changes With Time of 583 Paroxysmal Nocturnal Hemoglobinuria Clones From 529 Patients: A Multicenter Italian Study. Ann Hematol (2019) 98:1083–93. doi: 10.1007/s00277-019-03644-8
60. Molina H, Wong W, Kinoshita T, Brenner C, Foley S, Holers VM. Distinct Receptor and Regulatory Properties of Recombinant Mouse Complement Receptor 1 (CR1) and Crry, the Two Genetic Homologues of Human CR1. J Exp Med (1992) 175:121–9. doi: 10.1084/jem.175.1.121
61. Spicer AP, Seldin MF, Gendler SJ. Molecular Cloning and Chromosomal Localization of the Mouse Decay-Accelerating Factor Genes. Duplicated Genes Encode Glycosylphosphatidylinositol-Anchored and Transmembrane Forms. J Immunol (1995) 155:3079–91.
62. Ueda Y, Gullipalli D, Song WC. Modeling Complement-Driven Diseases in Transgenic Mice: Values and Limitations. Immunobiology (2016) 221:1080–90. doi: 10.1016/j.imbio.2016.06.007
63. Rosse WF, Adams JP, Thorpe AM. The Population of Cells in Paroxysmal Nocturnal Haemoglobinuria of Intermediate Sensitivity to Complement Lysis: Significance and Mechanism of Increased Immune Lysis. Br J Haematol (1974) 28:181–90. doi: 10.1111/j.1365-2141.1974.tb06652.x
64. Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to Apoptosis Caused by PIG-a Gene Mutations in Paroxysmal Nocturnal Hemoglobinuria. Proc Natl Acad Sci USA (1997) 94:8756–60. doi: 10.1073/pnas.94.16.8756
65. Horikawa K, Nakakuma H, Kawaguchi T, Iwamoto N, Nagakura S, Kagimoto T, et al. Apoptosis Resistance of Blood Cells From Patients With Paroxysmal Nocturnal Hemoglobinuria, Aplastic Anemia, and Myelodysplastic Syndrome. Blood (1997) 90:2716–22. doi: 10.1182/blood.V90.7.2716
66. Savage WJ, Barber JP, Mukhina GL, Hu R, Chen G, Matsui W, et al. Glycosylphosphatidylinositol-Anchored Protein Deficiency Confers Resistance to Apoptosis in PNH. Exp Hematol (2009) 37:42–51. doi: 10.1016/j.exphem.2008.09.002
67. Ismail MM, Tooze JA, Flynn JA, Gordon-Smith EC, Gibson FM, Rutherford TR, et al. Differential Apoptosis and Fas Expression on GPI-Negative and GPI-Positive Stem Cells: A Mechanism for the Evolution of Paroxysmal Nocturnal Haemoglobinuria. Br J Haematol (2003) 123:545–51. doi: 10.1046/j.1365-2141.2003.04643.x
68. Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to Apoptosis Caused by PIG-a Gene Mutations in Paroxysmal Nocturnal Hemoglobinuria. Proc Natl Acad Sci USA (1997) 94:8756–60. doi: 10.1073/pnas.94.16.8756
69. Shichishima T. [Investigation on Apoptosis in Paroxysmal Nocturnal Hemoglobinuria (PNH) Granulocytes]. Rinsho Byori (2001) 49:986–91.
70. Ware RE, Nishimura J, Moody MA, Smith C, Rosse WF, Howard TA. The PIG-a Mutation and Absence of Glycosylphosphatidylinositol-Linked Proteins do Not Confer Resistance to Apoptosis in Paroxysmal Nocturnal Hemoglobinuria. Blood (1998) 92:2541–50. doi: 10.1182/blood.V92.7.2541.2541_2541_2550
71. Kulkarni S, Bessler M. The Effect of GPI-Anchor Deficiency on Apoptosis in Mice Carrying a Piga Gene Mutation in Hematopoietic Cells. J Leukoc Biol (2002) 72:1228–33.
72. Chen G, Kirby M, Zeng W, Young NS, Maciejewski JP. Superior Growth of Glycophosphatidy Linositol-Anchored Protein-Deficient Progenitor Cells In Vitro Is Due to the Higher Apoptotic Rate of Progenitors With Normal Phenotype In Vivo. Exp Hematol (2002) 30:774–82. doi: 10.1016/S0301-472X(02)00811-1
73. Chen R, Nagarajan S, Prince GM, Maheshwari U, Terstappen LW, Kaplan DR, et al. Impaired Growth and Elevated Fas Receptor Expression in PIGA(+) Stem Cells in Primary Paroxysmal Nocturnal Hemoglobinuria. J Clin Invest (2000) 106:689–96. doi: 10.1172/JCI8328
74. Chen G, Zeng W, Maciejewski JP, Kcyvanfar K, Billings EM, Young NS. Differential Gene Expression in Hematopoietic Progenitors From Paroxysmal Nocturnal Hemoglobinuria Patients Reveals an Apoptosis/Immune Response in ‘Normal’ Phenotype Cells. Leukemia (2005) 19:862–8. doi: 10.1038/sj.leu.2403678
75. Kunyaboon R, Wanachiwanawin W, Thedsawad UPYA, Taka O. Mechanism of Paroxysmal Nocturnal Hemoglobinuria Clonal Dominance: Possible Roles of Different Apoptosis and CD8+ Lymphocytes in the Selection of Paroxysmal Nocturnal Hemoglobinuria Clones. Hematol Oncol Stem Cell Ther (2012) 5:138–45. doi: 10.5144/1658-3876.2012.138
76. Savage WJ, Barber JP, Mukhina GL, Hu R, Chen G, Matsui W, et al. Glycosylphosphatidylinositol-Anchored Protein Deficiency Confers Resistance to Apoptosis in PNH. Exp Hematol (2009) 37:42–51. doi: 10.1016/j.exphem.2008.09.002
77. Yamamoto T, Shichishima T, Shikama Y, Saitoh Y, Ogawa K, Maruyama Y. Granulocytes From Patients With Paroxysmal Nocturnal Hemoglobinuria and Normal Individuals Have the Same Sensitivity to Spontaneous Apoptosis. Exp Hematol (2002) 30:187–94. doi: 10.1016/S0301-472X(01)00783-4
78. Smrz D, Draberova L, Draber P. Non-Apoptotic Phosphatidylserine Externalization Induced by Engagement of Glycosylphosphatidylinositol-Anchored Proteins. J Biol Chem (2007) 282:10487–97. doi: 10.1074/jbc.M611090200
79. Barcellini W, Fermo E, Guia Imperiali F, Zaninoni A, Bianchi P, Boschetti C, et al. Increased Resistance of PIG-a- Bone Marrow Progenitors to Tumor Necrosis Factor a and Interferon Gamma: Possible Implications for the In Vivo Dominance of Paroxysmal Nocturnal Hemoglobinuria Clones. Haematologica (2004) 89:651–6.
80. Kulkarni S, Bessler M. Effect of Proinflammatory Cytokines on PIGA- Hematopoiesis. Exp Hematol (2003) 31:770–8. doi: 10.1016/S0301-472X(03)00189-9
81. Nagakura S, Ishihara S, Dunn DE, Nishimura J, Kawaguchi T, Horikawa K, et al. Decreased Susceptibility of Leukemic Cells With PIG-a Mutation to Natural Killer Cells In Vitro. Blood (2002) 100:1031–7. doi: 10.1182/blood.V100.3.1031
82. Hanaoka N, Nakakuma H, Horikawa K, Nagakura S, Tsuzuki Y, Shimanuki M, et al. NKG2D-Mediated Immunity Underlying Paroxysmal Nocturnal Haemoglobinuria and Related Bone Marrow Failure Syndromes. Br J Haematol (2009) 146:538–45. doi: 10.1111/j.1365-2141.2009.07795.x
83. Hanaoka N, Kawaguchi T, Horikawa K, Nagakura S, Mitsuya H, Nakakuma H. Immunoselection by Natural Killer Cells of PIGA Mutant Cells Missing Stress-Inducible ULBP. Blood (2006) 107:1184–91. doi: 10.1182/blood-2005-03-1337
84. Gargiulo L, Zaimoku Y, Scappini B, Maruyama H, Ohumi R, Luzzatto L, et al. Glycosylphosphatidylinositol-Specific T Cells,Ifngamma-Producing T Cells,and Pathogenesis of Idiopathic Aplastic Anemia. Blood (2016) 129(3):388–92. doi: 10.1182/blood-2016-09-740845
85. Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, et al. Glycosylphosphatidylinositol-Specific, CD1d-Restricted T Cells in Paroxysmal Nocturnal Hemoglobinuria. Blood (2013) 121:2753–61. doi: 10.1182/blood-2012-11-469353
86. Boegel S, Lower M, Bukur T, Sahin U, Castle JC. A Catalog of HLA Type, HLA Expression, and Neo-Epitope Candidates in Human Cancer Cell Lines. Oncoimmunology (2014) 3:e954893. doi: 10.4161/21624011.2014.954893
87. Britten CM, Meyer RG, Kreer T, Drexler I, Wolfel T, Herr W. The Use of HLA-a*0201-Transfected K562 as Standard Antigen-Presenting Cells for CD8(+) T Lymphocytes in IFN-Gamma ELISPOT Assays. J Immunol Methods (2002) 259:95–110. doi: 10.1016/S0022-1759(01)00499-9
88. Erusalimsky JD, Kefford RF, Gilmore DJ, Milstein C. Phorbol Esters Potentiate the Induction of Class I HLA Expression by Interferon Alpha. Proc Natl Acad Sci USA (1989) 86:1973–6. doi: 10.1073/pnas.86.6.1973
89. Shah YB, Priore SF, Li Y, Tang CN, Nicholas P, Kurre P, et al. The Predictive Value of PNH Clones, 6p CN-LOH, and Clonal TCR Gene Rearrangement for Aplastic Anemia Diagnosis. Blood Adv (2021) 5:3216–26. doi: 10.1182/bloodadvances.2021004201
90. DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA. Detection of Paroxysmal Nocturnal Hemoglobinuria Clones to Exclude Inherited Bone Marrow Failure Syndromes. Eur J Haematol (2014) 92:467–70. doi: 10.1111/ejh.12299
91. Gargiulo L, Lastraioli S, Cerruti G, Serra M, Loiacono F, Zupo S, et al. Highly Homologous T-Cell Receptor Beta Sequences Support a Common Target for Autoreactive T Cells in Most Patients With Paroxysmal Nocturnal Hemoglobinuria. Blood (2007) 109:5036–42. doi: 10.1182/blood-2006-10-052381
92. Gargiulo L, Zaimoku Y, Scappini B, Maruyama H, Ohumi R, Luzzatto L, et al. Glycosylphosphatidylinositol-Specific T Cells, IFN-Gamma-Producing T Cells, and Pathogenesis of Idiopathic Aplastic Anemia. Blood (2017) 129:388–92. doi: 10.1182/blood-2016-09-740845
93. Macho-Fernandez E, Brigl M. The Extended Family of CD1d-Restricted NKT Cells: Sifting Through a Mixed Bag of Tcrs, Antigens, and Functions. Front Immunol (2015) 6:362. doi: 10.3389/fimmu.2015.00362
94. Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva AD, Boesteanu A, et al. Natural Ligand of Mouse CD1d1: Cellular Glycosylphosphatidylinositol. Science (1998) 279:1541–4. doi: 10.1126/science.279.5356.1541
95. Schofield L, McConville MJ, Hansen D, Campbell AS, Fraser-Reid B, Grusby MJ, et al. CD1d-Restricted Immunoglobulin G Formation to GPI-Anchored Antigens Mediated by NKT Cells. Science (1999) 283:225–9. doi: 10.1126/science.283.5399.225
96. Procopio DO, Almeida IC, Torrecilhas AC, Cardoso JE, Teyton L, Travassos LR, et al. Glycosylphosphatidylinositol-Anchored Mucin-Like Glycoproteins From Trypanosoma Cruzi Bind to CD1d But do Not Elicit Dominant Innate or Adaptive Immune Responses via the CD1d/NKT Cell Pathway. J Immunol (2002) 169:3926–33. doi: 10.4049/jimmunol.169.7.3926
97. Mannik LA, Chin-Yee I, Sharif S, Van Kaer L, Delovitch TL, Haeryfar SM. Engagement of Glycosylphosphatidylinositol-Anchored Proteins Results in Enhanced Mouse and Human Invariant Natural Killer T Cell Responses. Immunology (2011) 132:361–75. doi: 10.1111/j.1365-2567.2010.03369.x
98. Godfrey DI, Pellicci DG, Patel O, Kjer-Nielsen L, McCluskey J, Rossjohn J. Antigen Recognition by CD1d-Restricted NKT T Cell Receptors. Semin Immunol (2010) 22:61–7. doi: 10.1016/j.smim.2009.10.004
99. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-Based Map of the Human Proteome. Science (2015) 347:1260419.
100. Zaimoku Y, Takamatsu H, Hosomichi K, Ozawa T, Nakagawa N, Imi T, et al. Identification of an HLA Class I Allele Closely Involved in the Autoantigen Presentation in Acquired Aplastic Anemia. Blood (2017) 129:2908–16. doi: 10.1182/blood-2016-11-752378
101. Babushok DV, Duke JL, Xie HM, Stanley N, Atienza J, Perdigones N, et al. Somatic HLA Mutations Expose the Role of Class I-Mediated Autoimmunity in Aplastic Anemia and Its Clonal Complications. Blood Adv (2017) 1:1900–10. doi: 10.1182/bloodadvances.2017010918
102. Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of Autoimmunity by Targeting a Distinct, Noninvariant CD1d-Reactive T Cell Population Reactive to Sulfatide. J Exp Med (2004) 199:947–57. doi: 10.1084/jem.20031389
103. Nakao S, Takamatsu H, Yachie A, Itoh T, Yamaguchi M, Ueda M, et al. Establishment of a CD4+ T Cell Clone Recognizing Autologous Hematopoietic Progenitor Cells From a Patient With Immune-Mediated Aplastic Anemia. Exp Hematol (1995) 23:433–8.
104. Nakao S, Takami A, Takamatsu H, Zeng W, Sugimori N, Yamazaki H, et al. Isolation of a T-Cell Clone Showing HLA-DRB1*0405-Restricted Cytotoxicity for Hematopoietic Cells in a Patient With Aplastic Anemia. Blood (1997) 89:3691–9. doi: 10.1182/blood.V89.10.3691
105. Maciejewski JP, Follmann D, Nakamura R, Saunthararajah Y, Rivera CE, Simonis T, et al. Increased Frequency of HLA-DR2 in Patients With Paroxysmal Nocturnal Hemoglobinuria and the PNH/Aplastic Anemia Syndrome. Blood (2001) 98:3513–9. doi: 10.1182/blood.V98.13.3513
106. Saunthararajah Y, Nakamura R, Nam JM, Robyn J, Loberiza F, Maciejewski JP, et al. HLA-DR15 (DR2) Is Overrepresented in Myelodysplastic Syndrome and Aplastic Anemia and Predicts a Response to Immunosuppression in Myelodysplastic Syndrome. Blood (2002) 100:1570–4. doi: 10.1182/blood.V100.5.1570.h81702001570_1570_1574
107. McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in Regulating Immunity and Tolerance. Clin Immunol (2016) 164:10–20. doi: 10.1016/j.clim.2016.01.008
108. Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Yagita H, Bromberg JS, Vallera DA. A Critical Role for CD48 Antigen in Regulating Alloengraftment and Lymphohematopoietic Recovery After Bone Marrow Transplantation. Blood (1998) 92:4453–63. doi: 10.1182/blood.V92.11.4453
109. Zhang Y, Liu Q, Yang S, Liao Q. CD58 Immunobiology at a Glance. Front Immunol (2021) 12:705260. doi: 10.3389/fimmu.2021.705260
110. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Pessoa V, Gualandro S, Fureder W, et al. Ravulizumab (ALXN1210) vs Eculizumab in Adult Patients With PNH Naive to Complement Inhibitors: The 301 Study. Blood (2019) 133:530–9. doi: 10.1182/blood-2018-09-876136
111. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, et al. Ravulizumab (ALXN1210) vs Eculizumab in C5-Inhibitor-Experienced Adult Patients With PNH: The 302 Study. Blood (2019) 133:540–9. doi: 10.1182/blood-2018-09-876805
112. Hillmen P, Szer J, Weitz I, Roth A, Hochsmann B, Panse J, et al. Pegcetacoplan Versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med (2021) 384:1028–37. doi: 10.1056/NEJMoa2029073
113. Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The Complement Inhibitor Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med (2006) 355:1233–43. doi: 10.1056/NEJMoa061648
114. Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter Phase 3 Study of the Complement Inhibitor Eculizumab for the Treatment of Patients With Paroxysmal Nocturnal Hemoglobinuria. Blood (2008) 111:1840–7. doi: 10.1182/blood-2007-06-094136
115. Gavriilaki E, Peffault de Latour R, Risitano AM. Advancing Therapeutic Complement Inhibition in Hematologic Diseases: PNH and Beyond. Blood (2021). doi: 10.1182/blood.2021012860
116. Kulasekararaj AG, Risitano AM, Maciejewski JP, Notaro R, Browett P, Lee JW, et al. Phase 2 Study of Danicopan in Patients With Paroxysmal Nocturnal Hemoglobinuria With an Inadequate Response to Eculizumab. Blood (2021) 138:1928–38. doi: 10.1182/blood.2021011388
117. Risitano AM, Kulasekararaj AG, Lee JW, Maciejewski JP, Notaro R, Brodsky R, et al. Danicopan: An Oral Complement Factor D Inhibitor for Paroxysmal Nocturnal Hemoglobinuria. Haematologica (2020) 106(12):3188–97. doi: 10.3324/haematol.2020.261826
118. Jang JH, Wong L, Ko B-H, Yoon S-S, Li KJ, Rozenberg I, et al. First-Line Treatment of PNH Patients With Iptacopan Leads to Rapid and Durable Hemoglobin Increase by Controlling Both Intra- and Extravascular Hemolysis, EHA 2021, Hemasphere, Virtual Conference. Abstract Book (2021). p. 42. Abstract Book. doi: 10.1097/HS9.0000000000000566
119. Risitano AM, Kulasekararaj A, De Castro C, Nakao S, Scheinberg P, Levitch R, et al. PH3 Study of Efficacy and Safety of Iptacopan (LNP023), an Oral Complement Factor B Inhibitor, in PTS With Paroxysmal Nocturnal Hemoglobinuria and Residual Anemia Despite Anti-C5 Antibody Treatment, EHA, Hemasphere, Virtual Conference. (2021). p. 704. Abstract Book. doi: 10.1097/HS9.0000000000000566
120. Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, et al. Deep Sequencing Reveals Stepwise Mutation Acquisition in Paroxysmal Nocturnal Hemoglobinuria. J Clin Invest (2014) 124:4529–38. doi: 10.1172/JCI74747
121. Tominaga R, Katagiri T, Kataoka K, Kataoka K, Wee RK, Maeda A, et al. Paroxysmal Nocturnal Hemoglobinuria Induced by the Occurrence of BCR-ABL in a PIGA Mutant Hematopoietic Progenitor Cell. Leukemia (2016) 30:1208–10. doi: 10.1038/leu.2015.268
122. Murakami Y, Inoue N, Shichishima T, Ohta R, Noji H, Maeda Y, et al. Deregulated Expression of HMGA2 Is Implicated in Clonal Expansion of PIGA Deficient Cells in Paroxysmal Nocturnal Haemoglobinuria. Br J Haematol (2012) 156:383–7. doi: 10.1111/j.1365-2141.2011.08914.x
123. Hanaoka N, Murakami Y, Nagata M, Horikawa K, Nagakura S, Yonemura Y, et al. Occupancy of Whole Blood Cells by a Single PIGA-Mutant Clone With HMGA2 Amplification in a Paroxysmal Nocturnal Haemoglobinuria Patient Having Blood Cells With NKG2D Ligands. Br J Haematol (2013) 160:114–6. doi: 10.1111/bjh.12093
124. Babushok DV, Perdigones N, Perin JC, Olson TS, Ye W, Roth JJ, et al. Emergence of Clonal Hematopoiesis in the Majority of Patients With Acquired Aplastic Anemia. Cancer Genet (2015) 208:115–28. doi: 10.1016/j.cancergen.2015.01.007
125. Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic Mutations Identify a Subgroup of Aplastic Anemia Patients Who Progress to Myelodysplastic Syndrome. Blood (2014) 124:2698–704. doi: 10.1182/blood-2014-05-574889
126. Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med (2015) 373:35–47. doi: 10.1056/NEJMoa1414799
Keywords: paroxysmal nocturnal hemoglobinuria, PIGA, GPI-anchored, aplastic anemia (AA), bone marrow failure (BMF), autoimmunity, complement, clonal hematopoeisis
Citation: Colden MA, Kumar S, Munkhbileg B and Babushok DV (2022) Insights Into the Emergence of Paroxysmal Nocturnal Hemoglobinuria. Front. Immunol. 12:830172. doi: 10.3389/fimmu.2021.830172
Received: 06 December 2021; Accepted: 30 December 2021;
Published: 28 January 2022.
Edited by:
Amy DeZern, Johns Hopkins Medicine, United StatesReviewed by:
Jeffrey J. Pu, University of Arizona, United StatesBhumika Patel, Cleveland Clinic, United States
Copyright © 2022 Colden, Kumar, Munkhbileg and Babushok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daria V. Babushok, ZGFyaWEuYmFidXNob2tAcGVubm1lZGljaW5lLnVwZW5uLmVkdQ==