 I-Hsin Huang1,2,3†
I-Hsin Huang1,2,3† Wen-Hung Chung1,2,4,5,6,7,8,9,10,11†
Wen-Hung Chung1,2,4,5,6,7,8,9,10,11† Po-Chien Wu1,2,3
Po-Chien Wu1,2,3 Chun-Bing Chen1,2,4,5,6,7,8,9,10,11,12,13*
Chun-Bing Chen1,2,4,5,6,7,8,9,10,11,12,13*- 1Drug Hypersensitivity Clinical and Research Center, Chang Gung Memorial Hospital, Linkou, Taipei, and Keelung, Taoyuan, Taiwan
- 2Department of Dermatology, Chang Gung Memorial Hospital, Linkou, Taipei and Keelung, Taiwan
- 3Research Center of Big Data and Meta-analysis, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan
- 4Cancer Vaccine and Immune Cell Therapy Core Laboratory, Chang Gung Memorial Hospital, Linkou, Taiwan
- 5Chang Gung Immunology Consortium, Chang Gung Memorial Hospital, Linkou, and Chang Gung University, Taoyuan, Taiwan
- 6Department of Dermatology, Xiamen Chang Gung Hospital, Xiamen, China
- 7Xiamen Chang Gung Allergology Consortium, Xiamen, Xiamen Chang Gung Hospital, Xiamen, China
- 8College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 9Whole-Genome Research Core Laboratory of Human Diseases, Chang Gung Memorial Hospital, Keelung, Taiwan
- 10Immune-Oncology Center of Excellence, Chang Gung Memorial Hospital, Linkou, Taiwan
- 11Genomic Medicine Core Laboratory, Chang Gung Memorial Hospital, Linkou, Taiwan
- 12Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 13School of Medicine, National Tsing Hua University, Hsinchu, Taiwan
Atopic dermatitis (AD) is a chronic, inflammatory, pruritic form of dermatosis with heterogeneous manifestations that can substantially affect patients' quality of life. AD has a complex pathogenesis, making treatment challenging for dermatologists. The Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway plays a central role in modulating multiple immune axes involved in the immunopathogenesis of AD. In particular, Th2 cytokines, including interleukin (IL)-4, IL-5, IL-13, IL-31, and thymic stromal lymphopoietin, which contribute to the symptoms of chronic inflammation and pruritus in AD, are mediated by JAK–STAT signal transduction. Furthermore, JAK–STAT is involved in the regulation of the epidermal barrier and the modulation of peripheral nerves related to the transduction of pruritus. Targeting the JAK–STAT pathway may attenuate these signals and show clinical efficacy through the suppression of various immune pathways associated with AD. Topical and oral JAK inhibitors with variable selectivity have emerged as promising therapeutic options for AD. Notably, topical ruxolitinib, oral upadacitinib, and oral abrocitinib were approved by the U.S. Food and Drug Administration for treating patients with AD. Accordingly, the present study reviewed the role of JAK–STAT pathways in the pathogenesis of AD and explored updated applications of JAK inhibitors in treating AD.
1. Introduction
Atopic dermatitis (AD), a chronic inflammatory dermatosis, clinically manifests as eczematous and itchy skin lesions with a predictable distribution on the basis of the patient’s age (1, 2). Chronic pruritus may result in sleep disturbance, decreased quality of life, low self-esteem, and a higher risk of anxiety or depression (3, 4). Long-term inflammation could result in lichenification and has been linked to frequent cutaneous infection, particularly Staphylococcus aureus colonization (5, 6). AD affects up to 20% of children, especially in families with atopic history, and it affects 10% of adults, although the rate varies by region (7–9). The interaction between genetic, environmental, and immunological factors was implicated in the pathogenesis of AD (10). AD comprises intrinsic and extrinsic variations and can be divided into subtypes on the basis of filaggrin mutation status, patient race, and age (11). Clinical manifestations and molecular profiles may vary from different endotypes. The immunopathogenesis of AD involves the interplay of innate and adaptive immune responses (12). The innate immune responses in AD are initiated from epithelial defects or defects in the expression levels of specific proteins or innate receptors, resulting in the stimulation of the adaptive immune system, and type 2 helper T (Th2) cells in particular (13, 14). The complex immune axes involved in AD complicate treatments with conventional medicines. Nevertheless, emerging biologics and small molecules have brought a new era in AD treatment (15, 16). Small molecules that target the Janus kinases (JAK) signal mediators, which regulate the proinflammatory cytokines, constitute a promising treatment for AD. Accordingly, this study reviews the role of the JAK–Signal Transducer and Activator of Transcription (STAT) pathway in the pathogenesis of AD and provides updated data supporting the applicability of JAK inhibitors in treating AD.
2. Role of the JAK–STAT pathway in the pathogenesis of AD
2.1. Pathogenesis of AD
The development of AD involves complex multifactorial pathophysiologies, comprising an impaired skin barrier, immunoglobulin E (IgE) sensitization, alterations of type 2 immune responses to cellular infiltration, and environmental and genetic factors (17). Dysfunction of the skin barrier is postulated to be a cause and a consequence of AD. Skin barrier abnormalities could be caused by a mutation of the filaggrin (FLG) gene, which encodes the key barrier protein FLG; irregularities of other structural proteins or lipids; or dysfunction of keratinocytes. Any of these abnormalities could lead to an impaired antimicrobial effect and a higher risk of antigen penetration (18). The disruption of the skin barrier triggers keratinocytes to release thymic stromal lymphopoietin (TSLP), which is a Th2-type immune-inducing factor (19). TSLP is also secreted by stromal cells, epithelial cells, fibroblasts, keratinocytes, and basophils, and is highly associated with other inflammatory diseases (20).
The immunopathogenesis of AD is suggestive of a biphasic T-cell-mediated inflammatory disease, wherein a Th2 immune response is responsible for the initial acute stage, and then the Th1 immune axis takes over as the disease progresses into its chronic stage (1) (Figure 1). In addition to Th cells, the presence of cytotoxic T cells, dendritic cells, eosinophils, and mast cells is markedly increased in AD skin lesions relative to healthy skin. During the acute phase, Th2-predominant immune responses trigger the release of Th2 cytokines such as interleukin (IL)-4, IL-5, IL-13, and IL-31 to promote local inflammation after stimulation by allergens, TSLP or IL-33. As the central cytokines in the pathogenesis of AD, IL-4 and IL-13 activate and promote Th2 cells, induce differentiation and activation of myeloid and atopic dendritic cells, activate B cells, stimulate IgE class-switching, and recruit eosinophils (21). Clinically, IL-4 and IL-13 contribute to skin barrier defects, cutaneous infections, inflammation, skin thickening, and itchiness (22). IL-4 and IL-13 are also associated with AD disease activity (23). As for IL-31, it is postulated to account for the symptom of pruritus in AD (24). Additionally, IL-31 delays the apoptosis of eosinophils through the expression of IL-31 receptor A IL 31RA on the surface of eosinophils (25). Another Th2 cytokine, IL-5, is a key mediator of eosinophil differentiation and proliferation in the bone marrow (26). Additionally, IL-5 can activate and prime eosinophils, increasing their sensitivity to allergic inflammatory reactions (27). In the chronic phase of AD, increased expression levels of IL-12, interferon-gamma (IFN-γ), and granulocyte–macrophage colony-stimulating factor (GM-CSF) are characteristic of a Th1 axis immune response (1). Additionally, Th17 and Th22 cytokines (IL-17, IL-19, and IL-22) contribute to the formation of chronic skin lesions in AD, especially in pediatric, Asian, and genetically predisposed patients (28). In contrast to the Th1 and Th2 immune responses, Th17 produces IL-17A, IL-17F, IL-22, and tumor necrosis factor-alpha (TNF-α). These cytokines promote cell proliferation and macrophage infiltration into damaged skin and stimulate eosinophils to secrete profibrotic cytokines (29). Although IL-17 is not as predominant in AD as it is in psoriasis, studies have revealed that IL-17 can stimulate the differentiation of B cells into IgE-producing plasma cells, promote the release of IL-8, TNF-α, and TSLP, activate keratinocytes to express adhesion molecules, and modulate the Th2 immune response, thus triggering the chronic phase of AD (29–31). Furthermore, T cells can promote keratinocyte hyperplasia and the production of antimicrobial peptides through IL-22 (32).
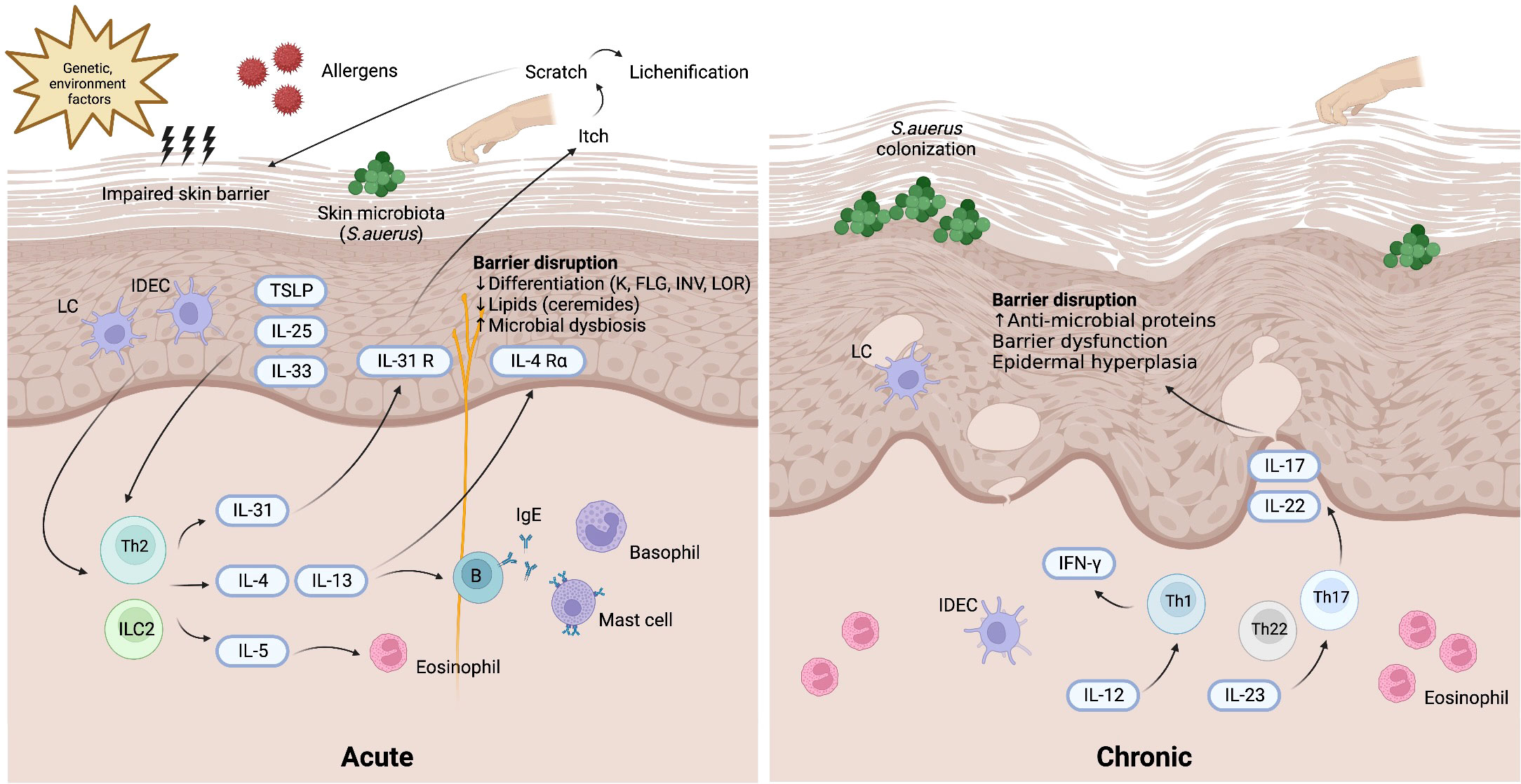
Figure 1 Pathogenesis of atopic dermatitis. Background genetic predisposition and environment factors may induce skin barrier impairment along with the invasion of allergens and skin microbiota (mostly Staphylococcus aureus). TSLP, IL-25, and IL-33 released from keratinocytes and activated skin LCs and IDECs lead to the activation of Th2 axis immune response through ILC2 and Th2 cells. In the acute phase of atopic dermatitis, the production of various Th2 cytokines including IL-4, IL-5, IL-13, IL-31 results in barrier dysfunction. In the chronic phase of atopic dermatitis, Th17 and Th22 cytokines cooperatively modulate local inflammation through upregulation of proinflammatory cytokines stimulating epidermal hyperplasia. Th1 immune response also plays a role in the chronic phase of atopic dermatitis. IDEC, inflammatory dendritic epidermal cell; IFN, interferon; IL, interleukin; ILC2, type 2 innate lymphoid cell; LC, Langerhans cell; R, receptor; S. aureus, Staphylococcus aureus; TSLP, thymic stromal lymphopoietin.
2.2. JAK-STAT pathway
The JAK–STAT signaling pathway is essential to the downstream regulation of various cytokines and growth factors that contribute to a diverse array of biological developmental and homeostatic processes, cell proliferation, and immune regulation. The JAK kinase family comprises four receptor-associated kinases: JAK1, JAK2, JAK3, and TYK2 (tyrosine kinase 2), and the STAT family comprises seven proteins: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (33). Generally, the JAK–STAT signal cascade is initiated by the ligation of extracellular cytokines with their cognate receptors. JAKs bind directly to the intracellular domains of type I and II cytokine receptor chains (Table 1) (34, 35), resulting in the apposition of the JAKs and the transphosphorylation and activation of the receptors. Accordingly, activated JAKs may phosphorylate tyrosine residues on the cytokine receptors and on other JAKs to create docking sites, thereby enabling the recruitment and phosphorylation of principal downstream signaling molecules such as STAT proteins (36). Once phosphorylated, the STATs dimerize and subsequently translocate into the nucleus, where they bind to DNA and regulate gene transcription (33). Different JAKs and STATs are recruited into this process according to the tissue specificity and the receptors involved in the signaling event (37).

Table 1 Families of interleukin transduction signals via the JAK–STAT pathway.
2.3. JAK-STAT signaling pathways in AD
2.3.1. Th2 axis immune response
In AD, epithelial cell–derived cytokines such as IL-25, IL-33, and TSLP promote the overexpression of Th2 cytokines, including IL-4, IL-5, IL-13, and IL-31, either directly through Th2 cytokine-secreting cells or indirectly through dendritic cell-mediated polarization (38). IL-4 performs its biological function by binding to an IL-4 receptor (IL-4R), which is a heterodimeric receptor complex on the cellular surface. IL-4R has two subtypes. Type I IL-4R comprises an IL-4 receptor alpha (IL-4Rα) chain and a common gamma (γ) chain, whereas type II IL-4R comprises an IL-4Rα and an IL-13 receptor alpha 1 (IL-13Rα1) chain (39, 40). IL-13Rα1, along with IL-13 receptor alpha 2 (IL-13Rα2), is a subtype of the IL-13 receptor alpha (IL-13Rα) chain. Unlike the ubiquitously presented IL-4R, IL-13Rα1 chains are expressed on limited cell surfaces including those of B cells, monocytes, macrophages, basophils, mast cells, and endothelial cells (39). Nonhematopoietic structural tissue cells such as keratinocytes also express relatively high amounts of IL-13Rα1, facilitating type II IL-4 receptor formation even with low levels of or absence of common γ chain (γc). Considering IL-4Rα as a functional component responsible for intercellular signal transduction, IL-13 may deliver its signal by binding type II IL-4R with the coexpression of the IL-4Rα and IL-13Rα1 chains (41). Different JAKs bind to different subunits of the receptors, accounting for the variability of JAK–STAT signaling pathways. For example, JAK1, JAK3, and TYK2 bind to IL-4Rα, γc chain, and IL-13Rα subunits of the IL-4R receptors, respectively.
In the IL-4 mediated immune response, IL-4 binds to type I IL-4R, stimulating the phosphorylation of JAK1 and JAK3, which in turn activate and phosphorylate IL-4Rα and STAT6 (Figure 2) (42). The phosphorylated STAT6 subsequently dimerizes and acts as a transcription factor by binding to specific DNA sequences of IL-4-responsive genes. Activated JAK1 can also phosphorylate Fes kinase, which then phosphorylates insulin receptor substrate-1/2 (IRS-1/2), a protein on the IL-4Rα chain linked to several downstream signaling pathways. Phosphorylated IRS-1/2 recruits p85 regulatory subunits, which activate the phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT) pathway. The PI3K–AKT pathway then promotes the proliferation, regeneration, and apoptosis of keratinocytes (43, 44). Conversely, IL-4 and IL-13 can bind to type II IL-4R, inducing the robust phosphorylation of JAK1 and TYK2, followed by the activation and phosphorylation of STAT3 and STAT6. This results in the downregulation of FLG expression and skin barrier dysfunction, and the increased production of TSLP, IL-25, and IL-33 in keratinocytes (45). The IL-4-induced JAK–STAT signal cascade can be inhibited by suppressors of cytokine signaling-1 (SOCS-1), which directly prevent JAK1/STAT6 phosphorylation and arrest IL-4-induced gene expression.
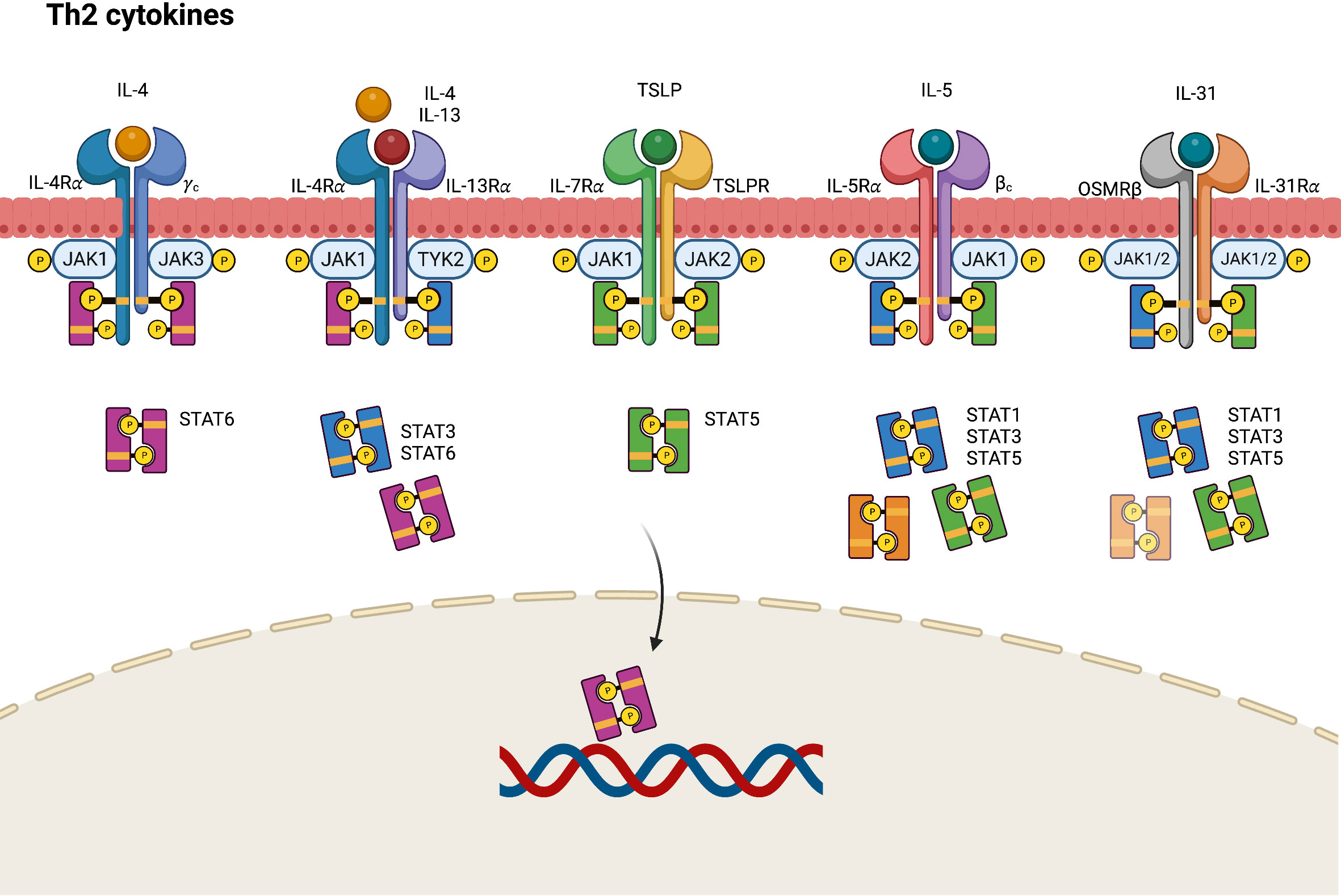
Figure 2 JAK-STAT signaling pathway in Th2 axis immune response of atopic dermatitis. IL, interleukin; JAK, Janus kinase; R, receptor; OSMR, oncostatin M receptor; STAT, signal transducer and activator of transcription; TSLP, thymic stromal lymphopoietin.
TSLP, a pro-Th2 cytokine, plays a key role in the activation and migration of dendritic cells, which drive the differentiation of Th2 cells to produce IL-4, IL-5, and IL-13 (46). TSLP is highly associated with IL-7, another stromal cell-derived cytokine. TSLP signals by binding to a heterodimeric receptor comprising IL-7 receptor alpha (IL-7Rα) and TSLPR, which interact with JAK1 and JAK2, respectively (20). Accordingly, the TSLP-mediated response activates STAT5 through the phosphorylation of JAK1 and JAK2 (20, 47).
The IL-5 receptor (IL-5R) comprises an alpha subunit (IL-5Rα), which specifically recognizes only IL-5 and a common beta (βc) heterodimeric chain, which can nonspecifically conjugate with not only IL-5 but also IL-3 and granulocyte-macrophage colony stimulating factor (GM-CSF) (27, 48, 49). In the JAK–STAT signaling pathway, the conjugation of IL-5 and the IL-5R triggers the phosphorylation of JAK1 and JAK2, resulting in the activation of STAT1, STAT3, and STAT5 (50).
IL-31 is a helical cytokine belonging to the glycoprotein 130 (gp130)/IL-6 family, a group of receptor proteins that share a common gp130 subunit in their receptor complex (51). IL-31 constitutively engages with the IL-31 receptor (IL-31R), a heterodimeric receptor comprising an IL-31 receptor alpha chain (IL-31Rα) coupled with an oncostatin M receptor beta chain (OSMRβ) (51). The intercellular signals of IL-31 are transmitted by the JAK–STAT pathway, mitogen-activated protein kinases (MAPKs), and the PI3K–AKT pathway. By engaging with the IL-31Rα–OSMRβ complex, IL-31 triggers the activation of JAK1 and JAK2, thereby stimulating STAT3, STAT5 and, to a lesser degree, STAT1 transcriptional activity (52).
2.3.2. Th17/Th22 axis immune response
During the chronic phase of AD, Th17 and Th22 cytokines cooperatively modulate local inflammation through upregulation of proinflammatory cytokines that stimulate epidermal hyperplasia (53). IL-17A and IL-17F are eponymous cytokines of the Th17 cell lineage, and IL-17A is believed to have a greater influence on driving autoimmunity than IL-17F because of its more potent signaling ability (54). The IL-17 receptor comprises IL-17RA and IL-17RC, which present different affinities to IL-17A and IL-17F, resulting in different ligand preferences depending on the ratios of IL-17RA and IL-17RC in the receptor complexes (54). In contrast with the classic Th1 and Th2 immune response through the JAK–STAT signaling pathway, Th17-derived IL-17 recruits ACT1, an IL-17 receptor complex adaptor protein, thus engaging with tumor necrosis factor receptor-associated factors (TRAF6) and activating nuclear factor (NF)-κB, CCAAT/enhancer binding protein (C/EBP), and MAPK pathways (Figure 3) (54). Generally, it seems that JAK–STAT pathway is not directly involved in the Th17 signaling and may only play a limited role. Nevertheless, STAT3 is found to orchestrate diverse aspects of Th17 differentiation and function (55). Via STAT3, a combination of Th17-promoting cytokines including IL-6, IL-21, and IL-23 would stimulate STAT3-dependent Th17 differentiation and enhance the production of IL-17 (56). STAT3 also mediate multiple epigenetic modification of Th17 gene transcription and is essential for Th17 proliferation and survival (57). As a result, selective JAK inhibitors may show the impact on the inhibition of Th17 immune response in AD via targeting upstream STAT3 activation.
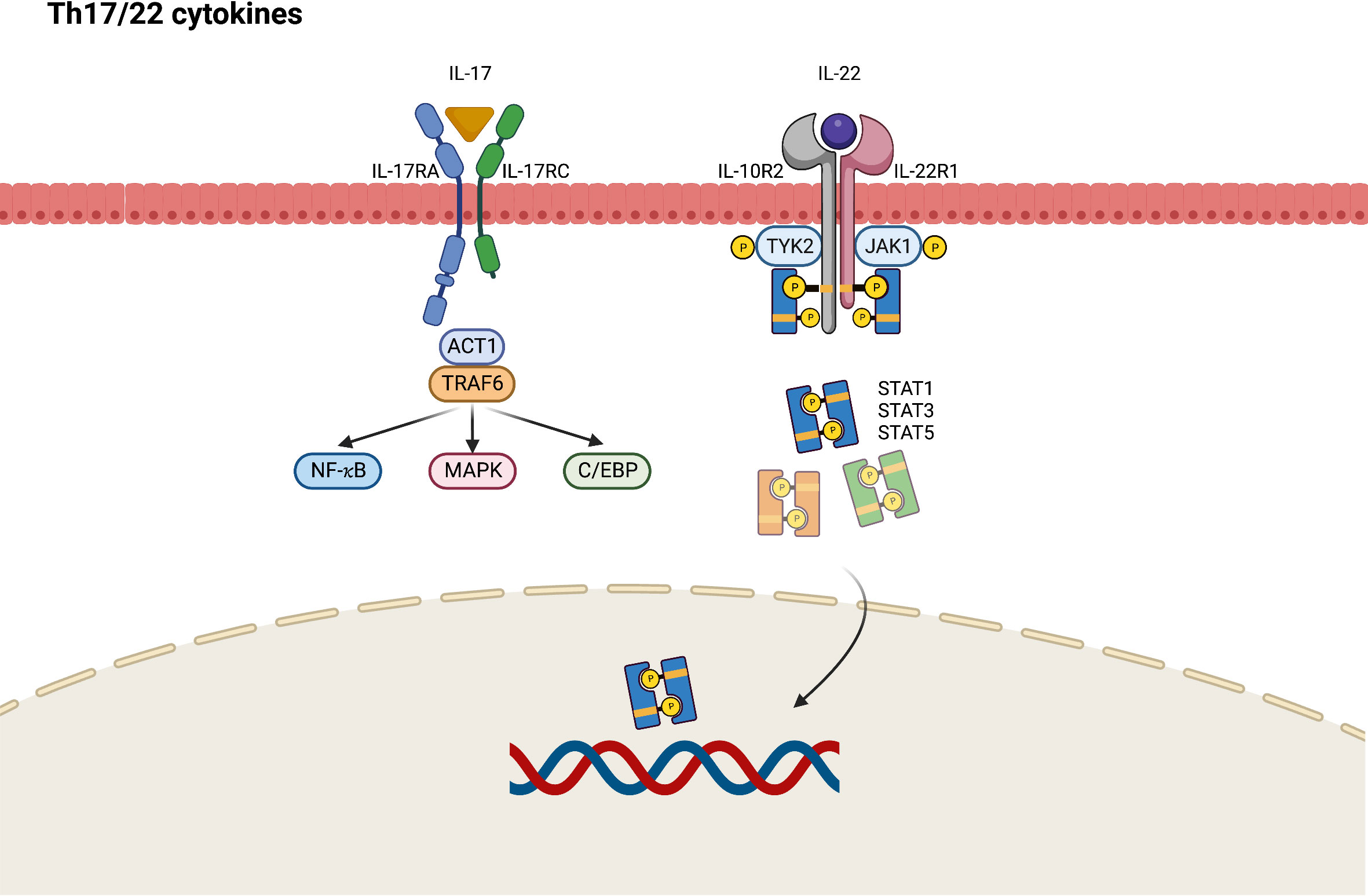
Figure 3 JAK-STAT signaling pathway in Th17/Th22 axis immune response of atopic dermatitis. C/EBP, CCAAT/enhancer binding protein; IL, interleukin; JAK, Janus kinase; MAPK, mitogen-activated protein kinases; NF, nuclear factor; R, receptor; STAT, signal transducer and activator of transcription; TRAF, tumor necrosis factor receptor-associated factors; TYK2, tyrosine kinase 2.
IL-22 was originally named IL-10-related T-cell-derived inducible factor (IL-TIF) and is identified as a key mediator of epidermal hyperplasia (58). Studies have primarily attributed IL-22 production to Th17- and Th22-cell subsets (59). IL-22 heterodimeric receptor complex comprises the IL-22 receptor (IL-22R1) and IL-10 receptor 2 (IL-10R2) subunits, which are expressed in small amounts by keratinocytes of the skin, gut, and lungs. IL-10R2 is also a subunit of the receptors for IL-10, IL-26, IL-28, and IL-29, whereas IL-22R1 is a component of IL-20 receptor 2 (IL-20R2) for IL-20 and IL-24 (58). After binding to IL-22R1, IL-22 induces the phosphorylation of JAK1 and TYK2. Although STAT3 is considered to be the primary mediator in this cascade, the activation of STAT1 and STAT5 has also been observed in IL-22 signaling (60). Apart from the JAK–STAT pathway, MAPK and p38 pathways are also engaged in IL-22 signaling (58).
2.3.3. Th1 axis immune response
Although Th1 cells are more predominant in the pathogenesis of psoriasis, Th1 skewed immune response also plays a role in the chronic phase of AD. Higher levels of Th1 cytokines including interferon-gamma (IFN- γ), IL-12, and granulocyte colony stimulating factor (G-CSF) are found in chronic AD lesions (61, 62). In the pathogenesis of AD, IL-12 secreted by dendritic cells can polarize Th1 cells from naïve T-helper cells and inhibit the activation of Th2 cells. The IL-12 signaling pathway is initiated by binding to a distinct heterodimeric receptor comprising interleukin 12 receptor beta (IL-12Rβ)1 and IL-12Rβ2 subunits (63). Subsequently, JAK2 and TYK2 are polarized, resulting in the mediation of signal transduction through the activation of STAT4, and to a lesser degree STAT1, STAT3, and STAT5 (Figure 4) (64).
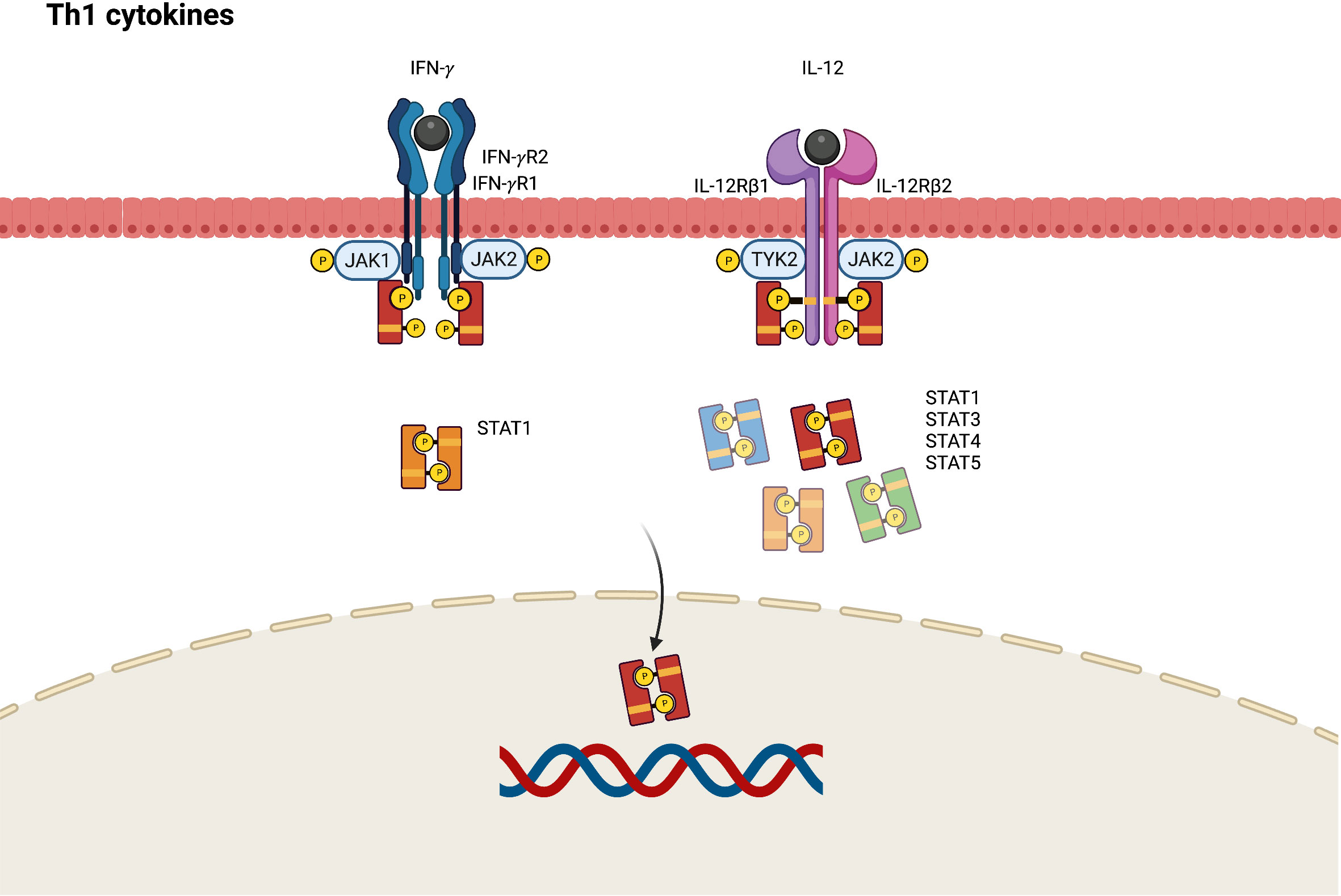
Figure 4 JAK-STAT signaling pathway in Th1 axis immune response of atopic dermatitis. IFN, interferon; IL, interleukin; JAK, Janus kinase; R, receptor; STAT, signal transducer and activator of transcription.
IFN-γ contributes to epidermal dysfunction by reducing the expression levels of ceramides and long-chain fatty acids (65). JAK1 and JAK2 are activated by the conjugation of IFN-γ and its receptor, which comprises IFN-gamma receptor (IFN-γR)1 and IFN-γR2. This phosphorylates STAT1 to transduce transcription signals into the nucleus and bind to the γ-IFN-activated sequence, affecting IFN-γ-induced gene expression (66).
2.3.4. Skin barrier dysfunction
JAK1 is involved in the pathogenesis of AD through the signaling pathways of IL-4, IL-5, IL-13, IL-22, TSLP, and IFN-γ. Hyperactivation of JAK1 results in the overexpression of cutaneous serine protease, causing skin barrier dysfunction (67). Furthermore, the STAT3 signal is a key transcriptional factor that modulates keratinocyte differentiation and maintains skin integrity. In a murine dry skin model, delgocitinib, a JAK inhibitor blocking JAK1, JAK2, and JAK3, inhibited the activation of STAT3 and improved skin barrier functionality by increasing epidermis-associated proteins such as FLG, loricrin, and natural moisturizing factors (68).
2.3.5. Pruritus and pain in AD
The etiology of pruritus in AD involves the interplay between hyperinnervation of the epidermis; increased pruritogens such as histamine, 5 hydroxytryptamine, nerve growth factor (NGF), and substance P; TSLP; and IL-31 (69, 70). The sensation of itchiness is transmitted by cutaneous unmyelinated C-fiber and myelinated Aδ fiber afferents originating from the dorsal root ganglion. The activation of STAT3 mediates astrogliosis in the spinal dorsal horn, acting as an amplifier of itchiness and leading to chronic pruritus (71). In this signaling pathway, lipocalin-2 (LCN2) was identified as the distinct upregulated factor that enhances pruritus signals and triggers the vicious itch-scratch cycle (72).
Notably, IL-31 was observed to stimulate nerve elongation and directly communicate with the primary cutaneous afferent neurons required by pruritogens to transduce itch signals (73, 74). In an AD-like murine model, topical delgocitinib was implicated to suppress IL-31-promoted nerve elongation in vivo and morphological changes of peripheral nerves from the dorsal root ganglion in vitro (75). The topical application of delgocitinib also resulted in substantially reduced scratching behavior in AD mouse models.
Patients with AD may also experience skin pain, demonstrating that the AD symptom burden extends beyond itchiness. The pain inflicted by AD can cause sleep disturbances and decrease quality of life. Evidence supports that the underlying mechanism for the pain response in AD is inflammation induced by proinflammatory cytokines and neuropathic pain of the peripheral nervous system (76). Cytokines involved in the JAK–STAT pathway also contribute to pain modulation. GM-CSF can induce hyperalgesia (77), whereas IL-17 may result in allodynia (78). TNF-α in the spinal cord is believed to modulate neuropathic pain sensations and sensitize pain-receptors for both mechanical and thermal hyperalgesia (79). Hence, patients receiving JAK inhibitors such as baricitinib reported reduced skin pain according to a numerical rating scale (NRS) (80).
3. JAK inhibitors for AD
Oral and topical JAK inhibitors are novel small molecules developed for treating various autoimmune and inflammatory diseases. Although few JAK inhibitors have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of dermatological diseases, emerging evidence demonstrated the therapeutic efficacy of JAK inhibitors in treating numerous skin diseases, including AD, alopecia areata, psoriasis, and vitiligo (81). The selective inhibition of various JAK inhibitors provides a diverse array of clinical applications through the modulation of various cytokine signaling pathways.
3.1. Topical JAK inhibitors for AD
3.1.1. Tofacitinib
Tofacitinib is a first-generation small molecules designed to selectively block JAK1 and JAK3, and to a lesser extent, JAK2 and TYK2. Tofacitinib has oral and topical formulations, but only the oral form of tofacitinib is commercially available as of writing this paper. Oral tofacitinib was approved by the U.S. FDA for the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, polyarticular juvenile idiopathic arthritis, and ulcerative colitis (82). Although the oral administration of tofacitinib has demonstrated clinical efficacy in the treatment of recalcitrant AD without significant AEs (83, 84), clinical application remains limited because of the paucity of randomized controlled trials (RCTs). Nevertheless, the relevant literature has reported that topical tofacitinib therapy for mild-to-moderate AD resulted in a significantly greater mean percentage change in Eczema Area and Severity Index (EASI) score, Physician’s Global Assessment (PGA) response, and body surface area than the vehicle treatment (Table 2) (85). Notably, topical tofacitinib demonstrated rapid efficacy during the first week of treatment and continuous improvement throughout a 4-week treatment course. The patients were reported to be well-tolerated to topical tofacitinib without any serious adverse events nor death (85).
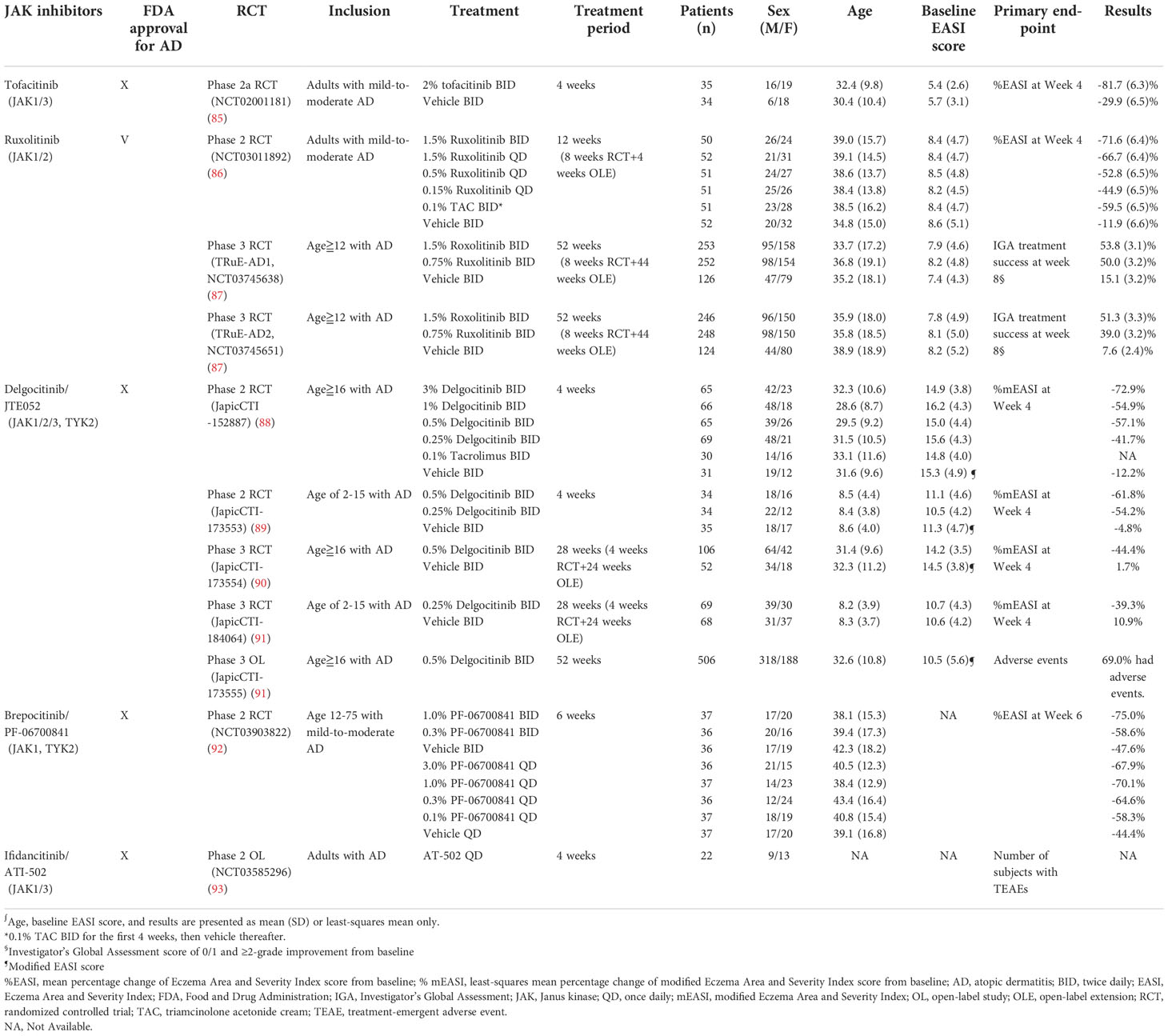
Table 2 Topical JAK inhibitors for atopic dermatitis.
3.1.2. Ruxolitinib
Ruxolitinib is a selective JAK1 and JAK2 inhibitor. Its oral formulation has been approved by the U.S. FDA for the treatment of myelofibrosis, polycythemia vera, and graft-versus-host disease (94). Topical 1.5% ruxolitinib cream has been approved by the U.S. FDA for the short-term and noncontinuous chronic treatment of mild-to-moderate AD in patients aged ≥12 years (95). Compared with the vehicle, ruxolitinib therapy resulted in significant improvement of EASI score and Investigator’s Global Assessment (IGA) scores among patients with AD (86). Notably, the rapid activation of ruxolitinib accounted for a significant reduction in itching within the first 12 hours after application, with sustainable efficacy throughout an 8-week treatment period (87). No differences in application site reactions were observed between ruxolitinib cream and the vehicle, indicating the favorable skin tolerability of ruxolitinib cream (87).
3.1.3. Delgocitinib (JTE-052)
As a pan-JAK inhibitor, topical delgocitinib (Corectim) has been approved by Japan Tobacco for the treatment of AD; however, this drug has yet to be approved by the U.S. FDA (96). Patients aged 2–15 and ≥16 years who were treated with 0.25% and 0.5% formulations of topical delgocitinib, respectively, had substantially higher modified EASI (mEASI) scores than the vehicle group. The majority of AEs were mild and well tolerated, such as nasopharyngitis, Kaposi varicelliform eruption, and acne (89–91). Application site reactions were rare and mostly occurred within the first 2 weeks of treatment, indicating high skin tolerability of topical delgocitinib (97). Furthermore, blood tests revealed no plasma concentration of delgocitinib in most patients, indicating minimal systemic absorption of delgocitinib (91).
3.1.4. Brepocitinib (PF-06700841)
Brepocitinib (PF-06700841) is a selective JAK1 and TYK2 inhibitor. Oral brepocitinib for the treatment of plaque psoriasis, alopecia areata, inflammatory bowel disease, and other inflammatory diseases remains under investigation (98–100). TYK2 is phosphorylated in the IL-4, IL-12, IL-13, and IL-22 cytokine signaling pathways; thus, brepocitinib is believed to be a potentially effective treatment for AD. The efficacy of topical brepocitinib in treating patients between the ages of 12–75 years with AD was evaluated by a dose-ranging, phase 2b RCT (92). In the RCT, brepocitinib met the primary endpoint of a mean change in EASI score at Week 6 and demonstrated superiority over the vehicle treatment. The rate of AEs in the brepocitinib group was generally lower than that in the vehicle group.
3.1.5. Ifidancitinib (ATI-502)
ATI-502, a topical JAK1 and JAK3 inhibitor, was investigated for efficacy and safety in the treatment of AD in a phase 2 open-label study (93). ATI-502 demonstrated high efficacy through improved EASI score, IGA score, and itch-NRS. ATI-502 was also demonstrated to be well tolerated, with primary safety endpoints met (93, 101).
3.2. Oral JAK inhibitors for AD
3.2.1. Upadacitinib (ABT-494)
Upadacitinib, an oral reversible JAK inhibitor with greater inhibitory potency for JAK1 than JAK2, JAK3, and TYK2, has been approved by the U.S. FDA for treating AD, rheumatoid arthritis, and psoriatic arthritis (102). According to the FDA recommendation, the strategy for upadacitinib treatment in patients with AD aged 12-64 years is initiation from 15 mg/day and titration to 30 mg/day if necessary (Table 3). The same guideline proposed a 15 mg/day dose for patients aged ≥65 years (102). In two consecutive phase 3 RCTs (Measure Up 1 and Measure Up 2), both the 30 and 15 mg doses of upadacitinib presented significantly higher EASI and IGA responses than the placebo (104). Furthermore, upadacitinib combined with TCS demonstrated high tolerability and sustainable efficacy over a 52-week treatment period (123).
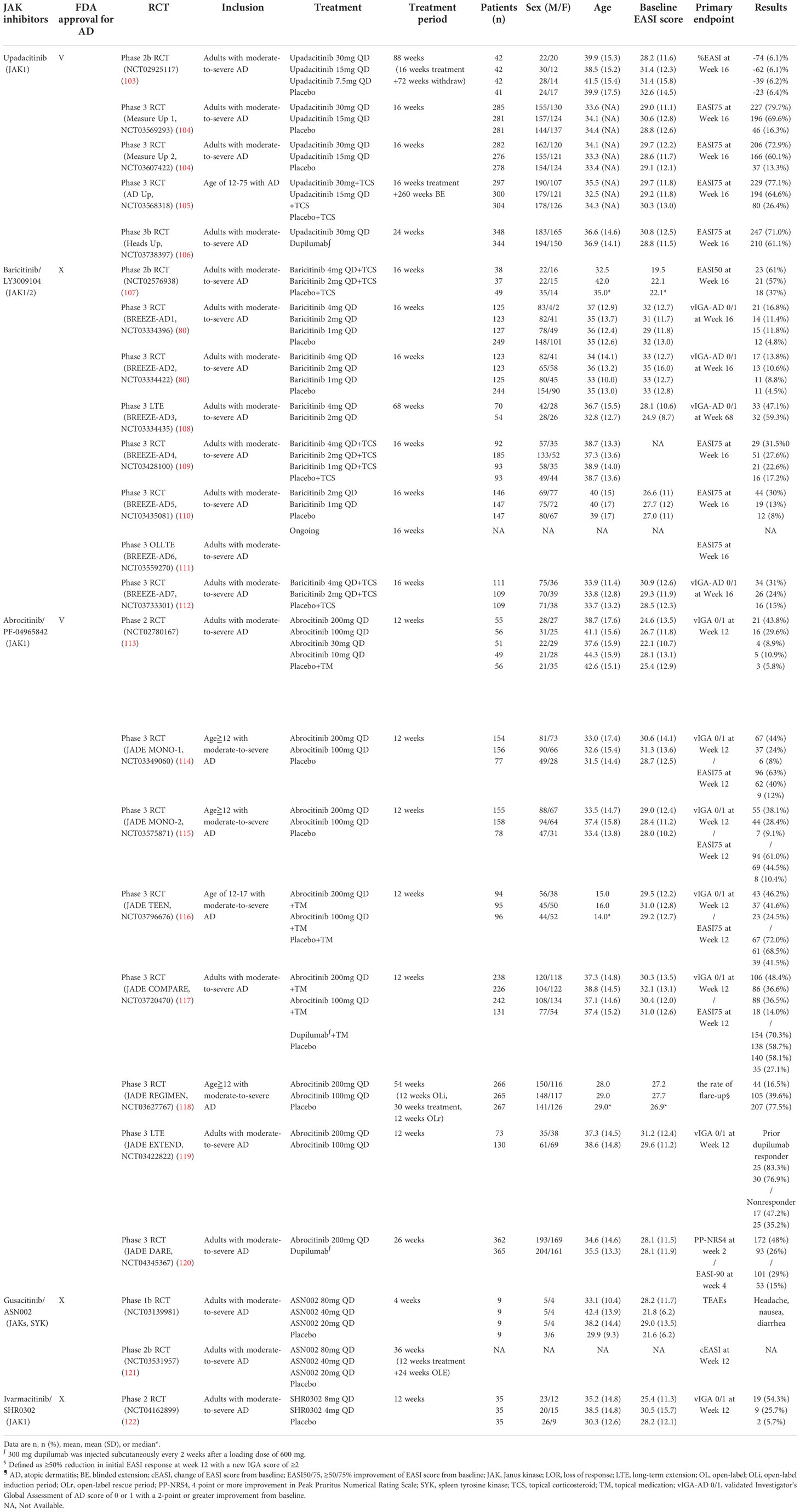
Table 3 Oral JAK inhibitors for atopic dermatitis.
Dupilumab was designed as a fully human monoclonal antibody targeting IL-4 and IL-13 receptors and was considered a promising biologics for AD. Nevertheless, less than 50% of patients achieved clear or almost clear skin during a 16-week course of dupilumab monotherapy (13), leading to the search for more effective therapeutic options. Compared with the 50% success rate for dupilumab, patients treated with upadacitinib presented substantially better results, with ≥75% of patients achieving an improvement in EASI score from baseline (EASI75) (106). Upadacitinib was also superior to dupilumab in terms of several secondary endpoints, including the percentage of patients achieving ≥90% and 100% improvement in EASI score (EASI90 and EASI100) and the percentage change of Worst Pruritus NRS at week 16 (106). In contrast, patients receiving upadacitinib presented higher rates of serious infection, eczema herpeticum, herpes zoster, and laboratory-related AEs than patients receiving dupilumab (106).
Overall, the rate of AEs associated with upadacitinib was relatively low, and the frequency of AEs was reported to be similar between the 15 and 30 mg doses. The 30 mg dose was associated with a slightly higher rate of treatment discontinuation because of AEs and serious infection than the 15 mg dose, which was considered to be attributed to the dosage effect (124). The most common treatment-emergent AEs (TEAEs) of upadacitinib were acne, upper respiratory tract infection, nasopharyngitis, and headache (104). Nevertheless, long-term use of upadacitinib demonstrated a favorable risk–benefit profile in treating patients with moderate-to-severe AD (124). Therefore, upadacitinib remains a promising orally systemic therapeutic alternative to conventional therapies and subcutaneous biologics for patients with moderate-to-severe AD.
3.2.2. Baricitinib (LY3009104)
Baricitinib was approved by the U.S. FDA as a selective JAK1 and JAK2 inhibitor for the indications of rheumatoid arthritis and Coronavirus Disease 2019 (COVID-19) but not yet for atopic dermatitis (125). Nevertheless, it has been approved in European union and Japan for adult patients with moderate to severe AD (126). Dosages of 4, 2, and 1 mg baricitinib were used in various RTCs to evaluate efficacy. In a phase 2 study, baricitinib 4 and 2 mg both effectively improved EASI scores when combined with TCS treatment (107). Among the patients with moderate-to-severe AD who did not respond to TCS therapy, monotherapy of baricitinib provided rapid itch reduction and demonstrated significant amelioration of AD severity relative to the placebo. Additionally, baricitinib improved the patients’ quality of life with fair tolerability (80). Furthermore, baricitinib demonstrated sustained long-term efficacy of up to 68 weeks and greater amelioration of itch-related symptoms throughout long-term administration (108). The frequency of serious infections and opportunistic infections was similar to that of the placebo (111). Notably, the 4-mg dose of baricitinib presented a higher risk of herpes simplex than the 2-mg dose, which was considered to be attributed to the dosage effect (111). Overall, baricitinib alone or in combination with TCS is an effective and well-tolerated systemic therapy for patients with moderate-to-severe AD.
3.2.3. Abrocitinib (PF-04965842)
Abrocitinib, a selective JAK1 inhibitor, is U.S. FDA approved for treating refractory, moderate-to-severe AD, with a recommended dosage of 100 mg/day (127). A dose-ranging phase 2b study reported that only abrocitinib 200 and 100 mg/day demonstrated significant efficacy and tolerability compared with the placebo, whereas the outcomes for the 30 and 10 mg doses of abrocitinib were statistically nonsignificant (113). Several phase 3 RCTs have demonstrated that the 200 and 100 mg doses of abrocitinib as monotherapy or concomitant with topical agents were highly beneficial treatments for patients aged ≥12 with moderate-to-severe AD (113–116). Compared with dupilumab, the 200 mg dose of abrocitinib showed a better IGA 0/1 or EASI75 response during the 16-week treatment period but did not reach statistical significance (117). The 200-mg dose of abrocitinib was superior to dupilumab with respect to itch response at week 2 (117). In a recent published phase 3 trial, abrocitinib 200 mg once per day provided higher rates of early itch reduction than dupilumab 300 mg (a 4 point or higher improvement in Peak Pruritus Numerical Rating Scale [PP-NRS4] response at week 2) which complements the above findings (120). Notably, a statistical superiority of abrocitinib versus dupilumab on the primary endpoints with faster onset of high-level improvement of disease signs (EASI-90 response at week 4), and the difference in EASI-90 response remained significant at week 16 (120). Furthermore, abrocitinib demonstrated sustainable long-term efficacy through 52 weeks (118). The results of these trials are supporting the potential important role of JAK1 inhibition in patients who need fast relief of pruritus and skin inflammation in atopic dermatitis (113–118, 120).
The most common TEAEs associated with abrocitinib were nausea, upper respiratory tract infection, nasopharyngitis, and headache, which were similar to the AEs reported for other JAK inhibitors (114). Nausea, the most frequent gastrointestinal AE, occurred only in patients with fasted status, indicating that the gastrointestinal symptoms may be caused by the local gastric concentration of abrocitinib (128). Accordingly, abrocitinib is an effective and safe small molecules for treating patients aged ≥12 years with moderate-to-severe AD.
3.2.4. Gusacitinib (ASN002)
ASN002 is a potent dual inhibitor, which simultaneously blocks both spleen tyrosine kinase (SYK) and JAKs, but is yet to receive U.S. FDA approval. SYK is a nonreceptor tyrosine kinase involved in the regulation of B cells, the differentiation of dendritic cells, the inhibition of keratinocyte differentiation, and Th17 signaling (129).
Patients with AD who were treated with ASN002 exhibited robust tissue improvement in cellular infiltration (AD lesions), inflammatory pathogenesis, and skin barrier dysfunction (129). Clinically, ASN002 is a safe and well-tolerated therapy for patients with moderate-to-severe AD. Mild AEs were observed, including headache, nausea, and diarrhea. However, only the highest dose (80 mg, as opposed to 40 or 20 mg) of ASN002 demonstrated significantly greater amelioration of pruritus than the placebo. A phase 2b RCT was completed but the data were unavailable at the time of publication.
3.2.5. Ivarmacitinib (SHR0302)
SHR0302 is a highly selective JAK1 inhibitor that also exercises a mild JAK2 inhibiting effect. In laboratory studies, SHR0302 was revealed to minimize the risk of AEs such as neutropenia and anemia (36). SHR0302 is easily absorbed through oral administration, and pharmacodynamic evidence supports a once-daily treatment strategy. A phase 2 study demonstrated that both 8 and 4 mg doses of SHR0302 were superior to a placebo in achieving IGA0/1, EASI75, and an improvement of ≥3 points on the NRS (NRS-3) (122). The oral therapy was also well tolerated, without clinically significant changes in hematology parameters. Further studies evaluating the efficacy and safety of SHR0302 are warranted.
3.3. Evidence of comparative effectiveness from network meta-analysis
Numerous novel small molecules and biologics have been developed. Choosing the most appropriate treatment for patients with AD recalcitrant to conventional treatment can be challenging. Among all steroid-sparing topical treatments for AD, twice-daily doses of tofacitinib 2%, delgocitinib 3%, and ruxolitinib 1.5% demonstrated superior efficacy when compared with other topical JAK and PDE4 inhibitors (130). Regarding the choice of systemic therapy, Drucker et al. conducted a network meta-analysis to compare the efficacy among all systemic immunomodulatory therapies for AD (131). In the reported analysis, the 200-mg dose of abrocitinib and 30-mg dose of upadacitinib demonstrated greater improvements in EASI scores than dupilumab, whereas the 15-mg dose of upadacitinib demonstrated similar efficacy to dupilumab.
3.4. Safety for JAK inhibitors in AD
In addition to the efficacy of these small molecules, drug safety is also a priority for clinicians to make the most appropriate decision in treating AD. The most commonly reported adverse events associated with JAK inhibitors are generally not serious, such as upper respiratory infections, urinary tract infections, and nasopharyngitis, hematological abnormalities, nausea, and headache (132, 133). Laboratory abnormalities include lymphopenia, neutropenia, anemia, dyslipidemia, and elevated liver enzymes; nonsignificant elevations of creatine phosphokinase and serum creatinine have also been reported (133). However, increased risks of serious infectious episodes, herpes zoster, tuberculosis, opportunistic infections were noted (133). Moreover, the U.S. FDA has added a black box warning on all approved JAK inhibitors indicated for the treatment of arthritis and other inflammatory conditions as well as those approved for AD, namely topical ruxolitinib, oral upadacitinib, and oral abrocitinib (95, 102, 127, 134). The black box warnings included serious infections, mortality, malignancies, major adverse cardiovascular events (MACEs), and thrombosis (95, 102, 127, 134).
Overall, apart from serious infections, other serious adverse events of JAK inhibitors were mostly reported in treating diseases other than AD, such as rheumatic arthritis (95, 102, 127, 134, 135). Among all immune-mediated inflammatory diseases, AD is generally considered as a subgroup with relatively less complicated comorbidity compared to other inflammatory conditions. The underlying inflammation of the disease may contribute to the further adverse events of the treatment (136). For example, regarding the risk of venous thromboembolism, several inflammatory diseases including psoriatic arthritis, rheumatic arthritis, and inflammatory bowel disease, but not AD are proved to be associated with higher risk of venous thromboembolism (137–139). Similarly, among all patients treated with JAK inhibitors, adverse events of venous thromboembolism were mostly reported by patients with rheumatic arthritis other than patients with AD (135). A recent systematic review and meta-analysis also provided the evidence that no increased risk of venous thromboembolism was detected in patients with AD treated with JAK inhibitors (140).
The black box warnings of JAK inhibitors including malignancies and MACEs were based on the ORAL Surveillance study of tofacitinib versus TNF-α inhibitors in rheumatoid arthritis (141). The patients with current or past smokers presented higher risk of malignancies and MACEs. In the population of AD, no significantly increased risk of malignancies or adjudicated MACEs were observed in patients treated with oral abrocitinib, oral upadacitinib, oral baricitinib and topical ruxolitinib in comparison to the control group (87, 106, 111, 115). Nevertheless, sporadic cases non-melanoma skin cancers have been reported in oral upadacitinib, oral baricitinib, oral abrocitinib and topical ruxolitinib (87, 106, 111, 115). Periodic skin examination is suggested for patients with increased risk of skin cancers. Further studies are warranted to evaluate the long-term safety of these JAK inhibitors for AD and their association to serious adverse events listed in black box warning.
Despite the less evidence proving the risk of malignancies and MACEs in patients with AD treated by JAK inhibitors, serious infection due to bacterial, viral, mycobacterial, and opportunistic infections should be always kept in mind in patients with AD treated with JAK inhibitors. Olivera et al. conducted a systematic review and meta-analysis revealed patients with various immune-mediated diseases treated by JAK inhibitors demonstrated higher risk of serious infection, especially herpes zoster (142). The most common serious infection of oral abrocitinib for AD were herpes simplex, herpes zoster, and pneumonia (127). As for oral upadacitinib for AD, the most frequent serious infections were reported as pneumonia and cellulitis (102). Serious lower respiratory tract infections have been reported in clinical studies evaluating the safety of topical ruxolitinib in treating AD (95).
Prior to treatment initiation of systemic JAK inhibitors, evaluations of tuberculosis infection, viral hepatitis, complete blood count, liver function tests, and lipid panel are recommended. Most patients do not require adjustment to therapy based on the laboratory findings (87, 106, 111, 115). Closely monitor patients for the signs of infection, and laboratory abnormalities and interrupt the treatment if necessary (87, 106, 111, 115). Additionally, JAK inhibitors are suggested not to be given in combination with biologic disease-modifying antirheumatic drugs or potent immunosuppressants such as cyclosporine, azathioprine and methotrexate (87, 106, 111, 115). Before the treatment, share decision making is suggested by discussing with the patients about the current evidence of efficacy and safety in order to tailor the optimal treatment strategy (134).
4. Conclusions
JAK–STAT signals play a pivotal role in the pathogenesis of AD. In addition to the involvement of various cytokine signaling pathways associated with mainly Th2, as well as Th1, Th17, and Th22 immune responses in AD, JAK–STAT is also engaged in the regulation of the skin barrier and the modulation of peripheral nerves related to the transduction of pruritus of AD. Oral and topically administered novel JAK inhibitors are now being used to treat patients with AD. Notably, topical ruxolitinib, oral upadacitinib, oral baricitinib and oral abrocitinib were approved for the treatment of AD. Considering the balance between efficacy and safety, the choice of the appropriate JAK inhibitors should be made in accordance with the individual patient’s condition. Further studies comparing the efficacy among conventional treatments, biologics, and all small molecules for AD are warranted, and the long-term safety of JAK inhibitors should also be investigated.
Author contributions
Conceptualization, I-HH, P-CW and C-BC; methodology, I-HH and C-BC; investigation, I-HH, P-CW and C-BC; resources, W-HC and C-BC; writing—original draft preparation, I-HH and C-BC; writing—review and editing, I-HH, P-CW and C-BC; visualization, I-HH; supervision, W-HC and C-BC; project administration, W-HC and C-BC. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This manuscript was edited by Wallace Academic Editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Zhang L WH, Tian XQ, Liu WL, Hao Y, Gao L. Identification of immune-related genes in atopic dermatitis, contact dermatitis, and psoriasis: A bioinformatics analysis. Dermatol Sin (2022) 40(3):162–67. doi: 10.4103/ds.ds_26_22
3. Thyssen JP, Hamann CR, Linneberg A, Dantoft TM, Skov L, Gislason GH, et al. Atopic dermatitis is associated with anxiety, depression, and suicidal ideation, but not with psychiatric hospitalization or suicide. Allergy (2018) 73(1):214–20. doi: 10.1111/all.13231
4. Wang C-H, Fu Y, Chi C-C. Association of atopic dermatitis with inflammatory bowel disease: A systematic review and meta-analysis. Dermatologica Sinica (2020) 38(3):159–65. doi: 10.4103/ds.ds_20_20
5. Breuer K, Kapp A, Werfel T. Bacterial infections and atopic dermatitis. Allergy (2001) 56(11):1034–41. doi: 10.1034/j.1398-9995.2001.00146.x
6. Sroka-Tomaszewska J, Trzeciak M. Molecular mechanisms of atopic dermatitis pathogenesis. Int J Mol Sci (2021) 22(8):4130. doi: 10.3390/ijms22084130
7. Wuthrich B, Schmid-Grendelmeier P. The atopic eczema/dermatitis syndrome. epidemiology, natural course, and immunology of the IgE-associated (“extrinsic”) and the nonallergic (“intrinsic”) AEDS. J Investig Allergol Clin Immunol (2003) 13(1):1–5.
8. Deckers IA, McLean S, Linssen S, Mommers M, van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: A systematic review of epidemiological studies. PloS One (2012) 7(7):e39803. doi: 10.1371/journal.pone.0039803
9. Ee S, Tay Y-K, Chu C-Y, Hon K-L, Leong K-F, Wananukul S. A study on the knowledge, attitudes, and practices of Asian dermatologists in the management of atopic dermatitis. Dermatologica Sinica (2020) 38(2):67–80. doi: 10.4103/ds.ds_31_19
10. Solomon I, Ilie MA, Draghici C, Voiculescu VM, Caruntu C, Boda D, et al. The impact of lifestyle factors on evolution of atopic dermatitis: An alternative approach. Exp Ther Med (2019) 17(2):1078–84. doi: 10.3892/etm.2018.6980
11. He H, Guttman-Yassky E. JAK inhibitors for atopic dermatitis: An update. Am J Clin Dermatol (2019) 20(2):181–92. doi: 10.1007/s40257-018-0413-2
12. De Benedetto A, Agnihothri R, McGirt LY, Bankova LG, Beck LA. Atopic dermatitis: A disease caused by innate immune defects? J Invest Dermatol (2009) 129(1):14–30. doi: 10.1038/jid.2008.259
13. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med (2016) 375(24):2335–48. doi: 10.1056/NEJMoa1610020
14. Gooderham MJ, Hong HC, Eshtiaghi P, Papp KA. Dupilumab: A review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol (2018) 78(3 Suppl 1):S28–36. doi: 10.1016/j.jaad.2017.12.022
15. Lee C-H, Huang Y-H, Chu C-Y. Taiwan Dermatological association recommendations for coronavirus disease of 2019 vaccination in patients treated with immunotherapeutics. Dermatologica Sinica (2021) 39(4):169–74. doi: 10.4103/ds.ds_50_21
16. Yu C-L, Lin Y-T, Chi C-C. Recommendations on use of systemic treatments for immune-mediated dermatologic disorders in patients with confirmed COVID-19 infection: A rapid review. Dermatologica Sinica (2022) 40(2):67–70. doi: 10.4103/1027-8117.349030
17. David Boothe W, Tarbox JA, Tarbox MB. Atopic dermatitis: Pathophysiology. Adv Exp Med Biol (2017) 1027:21–37. doi: 10.1007/978-3-319-64804-0_3
18. Peng W, Novak N. Pathogenesis of atopic dermatitis. Clin Exp Allergy (2015) 45(3):566–74. doi: 10.1111/cea.12495
19. Nakashima C, Yanagihara S, Otsuka A. Innovation in the treatment of atopic dermatitis: Emerging topical and oral janus kinase inhibitors. Allergol Int (2022) 71(1):40–6. doi: 10.1016/j.alit.2021.10.004
20. Rochman Y, Kashyap M, Robinson GW, Sakamoto K, Gomez-Rodriguez J, Wagner KU, et al. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc Natl Acad Sci USA (2010) 107(45):19455–60. doi: 10.1073/pnas.1008271107
21. Hamilton JD, Suarez-Farinas M, Dhingra N, Cardinale I, Li X, Kostic A, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol (2014) 134(6):1293–300. doi: 10.1016/j.jaci.2014.10.013
22. Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy (2020) 75(1):54–62. doi: 10.1111/all.13954
23. Leung DY, Guttman-Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol (2014) 134(4):769–79. doi: 10.1016/j.jaci.2014.08.008
24. Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol (2006) 117(2):411–7. doi: 10.1016/j.jaci.2005.10.033
25. Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT (2013) 2(3):e24137. doi: 10.4161/jkst.24137
26. Sanderson CJ. Interleukin-5, eosinophils, and disease. Blood (1992) 79(12):3101–9. doi: 10.1182/blood.V79.12.3101.bloodjournal79123101
27. Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med (1995) 182(4):1169–74. doi: 10.1084/jem.182.4.1169
28. Liu T, Li S, Ying S, Tang S, Ding Y, Li Y, et al. The IL-23/IL-17 pathway in inflammatory skin diseases: From bench to bedside. Front Immunol (2020) 11:594735. doi: 10.3389/fimmu.2020.594735
29. Sugaya M. The role of Th17-related cytokines in atopic dermatitis. Int J Mol Sci (2020) 21(4):1314. doi: 10.3390/ijms21041314
30. Brunner PM, Guttman-Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol (2017) 139(4S):S65–76. doi: 10.1016/j.jaci.2017.01.011
31. Klonowska J, Glen J, Nowicki RJ, Trzeciak M. New cytokines in the pathogenesis of atopic dermatitis-new therapeutic targets. Int J Mol Sci (2018) 19(10):3086. doi: 10.3390/ijms19103086
32. Simon D, Aeberhard C, Erdemoglu Y, Simon HU. Th17 cells and tissue remodeling in atopic and contact dermatitis. Allergy (2014) 69(1):125–31. doi: 10.1111/all.12351
33. Chapman S, Kwa M, Gold LS, Lim HW. Janus kinase inhibitors in dermatology: Part i. a comprehensive review. J Am Acad Dermatol (2022) 86(2):406–13. doi: 10.1016/j.jaad.2021.07.002
34. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery (2017) 17(1):78. doi: 10.1038/nrd.2017.201
35. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci (2018) 27(12):1984–2009. doi: 10.1002/pro.3519
36. Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation of janus kinase (JAK) activation. Biochem J (2014) 462(1):1–13. doi: 10.1042/BJ20140712
37. Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol (2008) 19(4):311–8. doi: 10.1016/j.semcdb.2008.08.010
38. Brandt EB, Sivaprasad U. Th2 cytokines and atopic dermatitis. J Clin Cell Immunol (2011) 2(3):110. doi: 10.4172/2155-9899.1000110
39. Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol (2000) 105(6 Pt 1):1063–70. doi: 10.1067/mai.2000.107604
40. Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, et al. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J Immunol (1998) 161(5):2317–24.
41. Zurawski SM, Chomarat P, Djossou O, Bidaud C, McKenzie AN, Miossec P, et al. The primary binding subunit of the human interleukin-4 receptor is also a component of the interleukin-13 receptor. J Biol Chem (1995) 270(23):13869–78. doi: 10.1074/jbc.270.23.13869
42. Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science (1994) 266(5187):1045–7. doi: 10.1126/science.7973659
43. Harb H, Chatila TA. Mechanisms of dupilumab. Clin Exp Allergy (2020) 50(1):5–14. doi: 10.1111/cea.13491
44. Teng Y, Fan Y, Ma J, Lu W, Liu N, Chen Y, et al. The PI3K/Akt pathway: Emerging roles in skin homeostasis and a group of non-malignant skin disorders. Cells (2021) 10(5):1219. doi: 10.3390/cells10051219
45. Furue M. Regulation of skin barrier function via competition between AHR axis versus IL-13/IL-4JAKSTAT6/STAT3 axis: Pathogenic and therapeutic implications in atopic dermatitis. J Clin Med (2020) 9(11):3741. doi: 10.3390/jcm9113741
46. Shi Z, Jiang W, Wang M, Wang X, Li X, Chen X, et al. Inhibition of JAK/STAT pathway restrains TSLP-activated dendritic cells mediated inflammatory T helper type 2 cell response in allergic rhinitis. Mol Cell Biochem (2017) 430(1-2):161–9. doi: 10.1007/s11010-017-2963-7
47. Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol (2000) 1(1):59–64. doi: 10.1038/76923
48. Pelaia C, Paoletti G, Puggioni F, Racca F, Pelaia G, Canonica GW, et al. Interleukin-5 in the pathophysiology of severe asthma. Front Physiol (2019) 10:1514. doi: 10.3389/fphys.2019.01514
49. Pelaia C, Vatrella A, Busceti MT, Gallelli L, Terracciano R, Savino R, et al. Severe eosinophilic asthma: from the pathogenic role of interleukin-5 to the therapeutic action of mepolizumab. Drug Des Devel Ther (2017) 11:3137–44. doi: 10.2147/DDDT.S150656
50. Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discovery (2016) 15(1):35–50. doi: 10.1038/nrd4624
51. Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev (2008) 19(5-6):347–56 doi: 10.1016/j.cytogfr.2008.08.003
52. Di Salvo E, Ventura-Spagnolo E, Casciaro M, Navarra M, Gangemi S. IL-33/IL-31 axis: A potential inflammatory pathway. Mediators Inflamm (2018) 2018:3858032. doi: 10.1155/2018/3858032
53. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol (2008) 128(11):2625–30. doi: 10.1038/jid.2008.111
54. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol (2009) 9(8):556–67. doi: 10.1038/nri2586
55. Howell MD, Fitzsimons C, Smith PA. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann Allergy Asthma Immunol (2018) 120(4):367–75. doi: 10.1016/j.anai.2018.02.012
56. Tripathi SK, Chen Z, Larjo A, Kanduri K, Nousiainen K, Aijo T, et al. Genome-wide analysis of STAT3-mediated transcription during early human Th17 cell differentiation. Cell Rep (2017) 19(9):1888–901. doi: 10.1016/j.celrep.2017.05.013
57. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity (2010) 32(5):605–15. doi: 10.1016/j.immuni.2010.05.003
58. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
59. Brunner PM, Pavel AB, Khattri S, Leonard A, Malik K, Rose S, et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol (2019) 143(1):142–54. doi: 10.1016/j.jaci.2018.07.028
60. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. pathways that are shared with and distinct from IL-10. J Biol Chem (2002) 277(37):33676–82. doi: 10.1074/jbc.M204204200
61. Grewe M, Gyufko K, Schopf E, Krutmann J. Lesional expression of interferon-gamma in atopic eczema. Lancet (1994) 343(8888):25–6. doi: 10.1016/S0140-6736(94)90879-6
62. Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest (1994) 94(2):870–6. doi: 10.1172/JCI117408
63. Bastian D, Wu Y, Betts BC, Yu XZ. The IL-12 cytokine and receptor family in graft-vs.-Host disease. Front Immunol (2019) 10:988. doi: 10.3389/fimmu.2019.00988
64. Gotthardt D, Trifinopoulos J, Sexl V, Putz EM. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front Immunol (2019) 10:2590. doi: 10.3389/fimmu.2019.02590
65. Tawada C, Kanoh H, Nakamura M, Mizutani Y, Fujisawa T, Banno Y, et al. Interferon-gamma decreases ceramides with long-chain fatty acids: possible involvement in atopic dermatitis and psoriasis. J Invest Dermatol (2014) 134(3):712–8. doi: 10.1038/jid.2013.364
66. Li H, Gade P, Xiao W, Kalvakolanu DV. The interferon signaling network and transcription factor C/EBP-beta. Cell Mol Immunol (2007) 4(6):407–18.
67. Yasuda T, Fukada T, Nishida K, Nakayama M, Matsuda M, Miura I, et al. Hyperactivation of JAK1 tyrosine kinase induces stepwise, progressive pruritic dermatitis. J Clin Invest (2016) 126(6):2064–76. doi: 10.1172/JCI82887
68. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol (2015) 136(3):667–77.e7. doi: 10.1016/j.jaci.2015.03.051
69. Mollanazar NK, Smith PK, Yosipovitch G. Mediators of chronic pruritus in atopic dermatitis: Getting the itch out? Clin Rev Allergy Immunol (2016) 51(3):263–92. doi: 10.1007/s12016-015-8488-5
70. Kido-Nakahara M, Furue M, Ulzii D, Nakahara T. Itch in atopic dermatitis. Immunol Allergy Clin North Am (2017) 37(1):113–22. doi: 10.1016/j.iac.2016.08.007
71. Shiratori-Hayashi M, Koga K, Tozaki-Saitoh H, Kohro Y, Toyonaga H, Yamaguchi C, et al. STAT3-dependent reactive astrogliosis in the spinal dorsal horn underlies chronic itch. Nat Med (2015) 21(8):927–31. doi: 10.1038/nm.3912
72. Rerknimitr P, Otsuka A, Nakashima C, Kabashima K. The etiopathogenesis of atopic dermatitis: barrier disruption, immunological derangement, and pruritus. Inflammation Regen (2017) 37:14. doi: 10.1186/s41232-017-0044-7
73. Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J Allergy Clin Immunol (2014) 133(2):448–60. doi: 10.1016/j.jaci.2013.10.048
74. Feld M, Garcia R, Buddenkotte J, Katayama S, Lewis K, Muirhead G, et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol (2016) 138(2):500–8 e24. doi: 10.1016/j.jaci.2016.02.020
75. Yamamoto Y, Otsuka A, Nakashima C, Ishida Y, Honda T, Egawa G, et al. Janus kinase inhibitor delgocitinib suppresses pruritus and nerve elongation in an atopic dermatitis murine model. J Dermatol Sci (2020) 97(2):161–4. doi: 10.1016/j.jdermsci.2019.12.008
76. Kwatra SG, Misery L, Clibborn C, Steinhoff M. Molecular and cellular mechanisms of itch and pain in atopic dermatitis and implications for novel therapeutics. Clin Transl Immunol (2022) 11(5):e1390. doi: 10.1002/cti2.1390
77. Nicol LSC, Thornton P, Hatcher JP, Glover CP, Webster CI, Burrell M, et al. Central inhibition of granulocyte-macrophage colony-stimulating factor is analgesic in experimental neuropathic pain. Pain (2018) 159(3):550–9. doi: 10.1097/j.pain.0000000000001130
78. Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain (2011) 12(3):370–83. doi: 10.1016/j.jpain.2010.08.003
79. Andrade P, Visser-Vandewalle V, Hoffmann C, Steinbusch HW, Daemen MA, Hoogland G. Role of TNF-alpha during central sensitization in preclinical studies. Neurol Sci (2011) 32(5):757–71. doi: 10.1007/s10072-011-0599-z
80. Simpson EL, Lacour JP, Spelman L, Galimberti R, Eichenfield LF, Bissonnette R, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol (2020) 183(2):242–55. doi: 10.1111/bjd.18898
81. Damsky W, King BA. JAK inhibitors in dermatology: The promise of a new drug class. J Am Acad Dermatol (2017) 76(4):736–44. doi: 10.1016/j.jaad.2016.12.005
82. U.S. Food and Drug Administration. XELJANZ. U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/203214s028,208246s013,213082s003lbl.pdf (Accessed Access date 08/06/2022).
83. Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol (2015) 73(3):395–9. doi: 10.1016/j.jaad.2015.06.045
84. Berbert Ferreira S, Berbert Ferreira R, Scheinberg MA. Atopic dermatitis: Tofacitinib, an option for refractory disease. Clin Case Rep (2020) 8(12):3244–7. doi: 10.1002/ccr3.3325
85. Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: A phase IIa randomized trial. Br J Dermatol (2016) 175(5):902–11. doi: 10.1111/bjd.14871
86. Kim BS, Howell MD, Sun K, Papp K, Nasir A, Kuligowski ME, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol (2020) 145(2):572–82. doi: 10.1016/j.jaci.2019.08.042
87. Papp K, Szepietowski JC, Kircik L, Toth D, Eichenfield LF, Leung DYM, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: Results from 2 phase 3, randomized, double-blind studies. J Am Acad Dermatol (2021) 85(4):863–72. doi: 10.1016/j.jaad.2021.04.085
88. Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: A phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol (2018) 178(2):424–32. doi: 10.1111/bjd.16014
89. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Oda M, Kabashima K, et al. Phase 2 clinical study of delgocitinib ointment in pediatric patients with atopic dermatitis. J Allergy Clin Immunol (2019) 144(6):1575–83. doi: 10.1016/j.jaci.2019.08.004
90. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J Am Acad Dermatol (2020) 82(4):823–31. doi: 10.1016/j.jaad.2019.12.015
91. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kabashima K, Oda M, et al. Delgocitinib ointment in pediatric patients with atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and a subsequent open-label, long-term study. J Am Acad Dermatol (2021) 85(4):854–62. doi: 10.1016/j.jaad.2021.06.014
92. ClinicalTrials.gov. Dose ranging study to assess efficacy, safety, tolerability and pharmacokinetics of PF-06700841 topical cream in participants with mild or moderate atopic dermatitis. Available at: https://clinicaltrials.gov/ct2/show/results/NCT03903822?term=PF-06700841&cond=atopic+dermatitis&draw=2&rank=1 (Accessed Access date 12/06/2022).
93. Stacy Smith NB, . Shanler SD, DeMoor R, Schnyder J. 16089 safety of ATI-502, a novel topical JAK1/3 inhibitor, in adults with moderate to severe atopic dermatitis: Results from a phase 2a open-label trial. J Am Acad Dermatol (2020) 83(6):AB170. doi: 10.1016/j.jaad.2020.06.770
94. U.S. Food and Drug Administration. JAKAFI (ruxolitinib). U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/202192s025lbl.pdf (Accessed Access date 09/06/2022).
95. U.S. Food and Drug Administration. OPZELURA (ruxolitinib). U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/215309Orig1s000ltr.pdf (Accessed Access date 09/06/2022).
96. Dhillon S. Delgocitinib: First approval. Drugs (2020) 80(6):609–15. doi: 10.1007/s40265-020-01291-2
97. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Murata R, Kaino H, et al. Long-term safety and efficacy of delgocitinib ointment, a topical janus kinase inhibitor, in adult patients with atopic dermatitis. J Dermatol (2020) 47(2):114–20. doi: 10.1111/1346-8138.15173
98. Forman SB, Pariser DM, Poulin Y, Vincent MS, Gilbert SA, Kieras EM, et al. TYK2/JAK1 inhibitor PF-06700841 in patients with plaque psoriasis: Phase IIa, randomized, double-blind, placebo-controlled trial. J Invest Dermatol (2020) 140(12):2359–70 e5. doi: 10.1016/j.jid.2020.03.962
99. King B, Guttman-Yassky E, Peeva E, Banerjee A, Sinclair R, Pavel AB, et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J Am Acad Dermatol (2021) 85(2):379–87. doi: 10.1016/j.jaad.2021.03.050
100. Danese S, Peyrin-Biroulet L. Selective tyrosine kinase 2 inhibition for treatment of inflammatory bowel disease: New hope on the rise. Inflammation Bowel Dis (2021) 27(12):2023–30. doi: 10.1093/ibd/izab135
101. ClinicalTrials.gov. A study of ATI-502 topical solution for the treatment of atopic dermatitis. Available at: https://clinicaltrials.gov/ct2/show/results/NCT03585296?term=ATI-502&cond=atopic+dermatitis&draw=2&rank=1 (Accessed Access date 10/06/2022).
102. U.S. Food and Drug Administration. RINVOQ (Upadacitinib). U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/211675s004lbl.pdf (Accessed Access date 10/06/2022).
103. Guttman-Yassky E, Thaci D, Pangan AL, Hong HC, Papp KA, Reich K, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol (2020) 145(3):877–84. doi: 10.1016/j.jaci.2019.11.025
104. Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure up 1 and measure up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet (2021) 397(10290):2151–68. doi: 10.1016/S0140-6736(21)00588-2
105. Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2021) 397(10290):2169–81. doi: 10.1016/S0140-6736(21)00589-4
106. Blauvelt A, Teixeira HD, Simpson EL, Costanzo A, De Bruin-Weller M, Barbarot S, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-Severe atopic dermatitis: A randomized clinical trial. JAMA Dermatol (2021) 157(9):1047–55. doi: 10.1001/jamadermatol.2021.3023
107. Guttman-Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol (2019) 80(4):913–21 e9. doi: 10.1016/j.jaad.2018.01.018
108. Silverberg JI, Simpson EL, Wollenberg A, Bissonnette R, Kabashima K, DeLozier AM, et al. Long-term efficacy of baricitinib in adults with moderate to severe atopic dermatitis who were treatment responders or partial responders: An extension study of 2 randomized clinical trials. JAMA Dermatol (2021) 157(6):691–9. doi: 10.1001/jamadermatol.2021.1273
109. Bieber T, Reich K, Paul C, Tsunemi Y, Augustin M, Lacour JP, et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in patients with moderate-to-Severe atopic dermatitis with inadequate response, intolerance, or contraindication to cyclosporine: Results from a randomized, placebo-controlled, phase III clinical trial (BREEZE-AD4). Br J Dermatol (2022) 187(3):338–352. doi: 10.1111/bjd.21630
110. Simpson EL, Forman S, Silverberg JI, Zirwas M, Maverakis E, Han G, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: Results from a randomized monotherapy phase 3 trial in the united states and Canada (BREEZE-AD5). J Am Acad Dermatol (2021) 85(1):62–70. doi: 10.1016/j.jaad.2021.02.028
111. Bieber T, Thyssen JP, Reich K, Simpson EL, Katoh N, Torrelo A, et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J Eur Acad Dermatol Venereol (2021) 35(2):476–85. doi: 10.1111/jdv.16948
112. Reich K, Kabashima K, Peris K, Silverberg JI, Eichenfield LF, Bieber T, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: A randomized clinical trial. JAMA Dermatol (2020) 156(12):1333–43. doi: 10.1001/jamadermatol.2020.3260
113. Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: A phase 2 randomized clinical trial. JAMA Dermatol (2019) 155(12):1371–9. doi: 10.1001/jamadermatol.2019.2855
114. Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet (2020) 396(10246):255–66. doi: 10.1016/S0140-6736(20)30732-7
115. Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-Severe atopic dermatitis: A randomized clinical trial. JAMA Dermatol (2020) 156(8):863–73. doi: 10.1001/jamadermatol.2020.1406
116. Eichenfield LF, Flohr C, Sidbury R, Siegfried E, Szalai Z, Galus R, et al. Efficacy and safety of abrocitinib in combination with topical therapy in adolescents with moderate-to-Severe atopic dermatitis: The JADE TEEN randomized clinical trial. JAMA Dermatol (2021) 157(10):1165–73. doi: 10.1001/jamadermatol.2021.2830
117. Bieber T, Simpson EL, Silverberg JI, Thaci D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med (2021) 384(12):1101–12. doi: 10.1056/NEJMoa2019380
118. Blauvelt A, Silverberg JI, Lynde CW, Bieber T, Eisman S, Zdybski J, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: Results from the JAK1 atopic dermatitis efficacy and safety (JADE) REGIMEN phase 3 trial. J Am Acad Dermatol (2022) 86(1):104–12. doi: 10.1016/j.jaad.2021.05.075
119. Shi VY, Bhutani T, Fonacier L, Deleuran M, Shumack S, Valdez H, et al. Phase 3 efficacy and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab (JADE EXTEND). J Am Acad Dermatol (2022) 87(2):351–8. doi: 10.1016/j.jaad.2022.04.009
120. Reich K, Thyssen JP, Blauvelt A, Eyerich K, Soong W, Rice ZP, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: A randomised, double-blind, multicentre phase 3 trial. Lancet (2022) 400(10348):273–82. doi: 10.1016/S0140-6736(22)01199-0
121. ClinicalTrials.gov. Phase 2B study to evaluate ASN002 in subjects with moderate to severe atopic dermatitis (RADIANT). Available at: https://clinicaltrials.gov/ct2/show/record/NCT03531957?view=record (Accessed Access date 11/06/2022).
122. Zhao Y, Zhang L, Ding Y, Tao X, Ji C, Dong X, et al. Efficacy and safety of SHR0302, a highly selective janus kinase 1 inhibitor, in patients with moderate to severe atopic dermatitis: A phase II randomized clinical trial. Am J Clin Dermatol (2021) 22(6):877–89. doi: 10.1007/s40257-021-00627-2
123. Silverberg JI, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, Costanzo A, et al. Upadacitinib plus topical corticosteroids in atopic dermatitis: Week 52 AD up study results. J Allergy Clin Immunol (2022) 149(3):977–87 e14. doi: 10.1016/j.jaci.2021.07.036
124. Simpson EL, Papp KA, Blauvelt A, Chu CY, Hong HC, Katoh N, et al. Efficacy and safety of upadacitinib in patients with moderate to severe atopic dermatitis: Analysis of follow-up data from the measure up 1 and measure up 2 randomized clinical trials. JAMA Dermatol (2022) 158(4):404–13. doi: 10.1001/jamadermatol.2022.0029
125. U.S. Food and Drug Administration. OLUMIANT (baricitinib). U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/207924s006lbl.pdf (Accessed Access date 10/06/2022).
126. Newswire CP. Updates on OLUMIANT® (baricitinib) phase 3 lupus program and FDA review for atopic dermatitis. Available at: https://www.prnewswire.com/news-releases/updates-on-olumiant-baricitinib-phase-3-lupus-program-and-fda-review-for-atopic-dermatitis-301470359.html.
127. U.S. Food and Drug Administration. CIBINQOTM (abrocitinib). U.S. food and drug administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf (Accessed Access date 11/06/2022).
128. Simpson EL, Silverberg JI, Nosbaum A, Winthrop KL, Guttman-Yassky E, Hoffmeister KM, et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-Severe atopic dermatitis from the phase II and phase III clinical trial program. Am J Clin Dermatol (2021) 22(5):693–707. doi: 10.1007/s40257-021-00618-3
129. Pavel AB, Song T, Kim HJ, Del Duca E, Krueger JG, Dubin C, et al. Oral janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J Allergy Clin Immunol (2019) 144(4):1011–24. doi: 10.1016/j.jaci.2019.07.013
130. Zhang L, Du D, Wang L, Guo L, Jiang X. Efficacy and safety of topical janus kinase and phosphodiesterase inhibitor-4 inhibitors for the treatment of atopic dermatitis: A network meta-analysis. J Dermatol (2021) 48(12):1877–83. doi: 10.1111/1346-8138.16126
131. Drucker AM, Morra DE, Prieto-Merino D, Ellis AG, Yiu ZZN, Rochwerg B, et al. Systemic immunomodulatory treatments for atopic dermatitis: Update of a living systematic review and network meta-analysis. JAMA Dermatol (2022) 158(5):523–32. doi: 10.1001/jamadermatol.2022.0455
132. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
133. Le M, Berman-Rosa M, Ghazawi FM, Bourcier M, Fiorillo L, Gooderham M, et al. Systematic review on the efficacy and safety of oral janus kinase inhibitors for the treatment of atopic dermatitis. Front Med (Lausanne) (2021) 8:682547. doi: 10.3389/fmed.2021.682547
134. Kragstrup TW, Glintborg B, Svensson AL, McMaster C, Robinson PC, Deleuran B, et al. Waiting for JAK inhibitor safety data. RMD Open (2022) 8(1):e002236. doi: 10.1136/rmdopen-2022-002236
135. Yates M, Mootoo A, Adas M, Bechman K, Rampes S, Patel V, et al. Venous thromboembolism risk with JAK inhibitors: A meta-analysis. Arthritis Rheumatol (2021) 73(5):779–88. doi: 10.1002/art.41580
136. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med (2019) 25(12):1822–32. doi: 10.1038/s41591-019-0675-0
137. Ogdie A, Kay McGill N, Shin DB, Takeshita J, Jon Love T, Noe MH, et al. Risk of venous thromboembolism in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: A general population-based cohort study. Eur Heart J (2018) 39(39):3608–14. doi: 10.1093/eurheartj/ehx145
138. Yuhara H, Steinmaus C, Corley D, Koike J, Igarashi M, Suzuki T, et al. Meta-analysis: the risk of venous thromboembolism in patients with inflammatory bowel disease. Aliment Pharmacol Ther (2013) 37(10):953–62. doi: 10.1111/apt.12294
139. Meyers KJ, Silverberg JI, Rueda MJ, Goodloe R, Pierce EJ, Deberdt W, et al. Risk of venous thromboembolism among patients with atopic dermatitis: A cohort study in a US administrative claims database. Dermatol Ther (Heidelb) (2021) 11(3):1041–52. doi: 10.1007/s13555-021-00538-4
140. Chen TL, Lee LL, Huang HK, Chen LY, Loh CH, Chi CC. Association of risk of incident venous thromboembolism with atopic dermatitis and treatment with janus kinase inhibitors: A systematic review and meta-analysis. JAMA Dermatol (2022) 158(11):1254–61. doi: 10.1001/jamadermatol.2022.3516
141. Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med (2022) 386(4):316–26. doi: 10.1056/NEJMoa2109927
Keywords: atopic dermatitis (AD), Janus kinase (JAK), Janus kinase inhibitor, signal transducer and activator of transcription (STAT), review
Citation: Huang I-H, Chung W-H, Wu P-C and Chen C-B (2022) JAK–STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front. Immunol. 13:1068260. doi: 10.3389/fimmu.2022.1068260
Received: 20 October 2022; Accepted: 22 November 2022;
Published: 08 December 2022.
Edited by:
Silvia Gregori, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), ItalyReviewed by:
Yasutomo Fujii, Astellas Pharma (Japan), JapanLaura Calabrese, Ludwig Maximilian University of Munich, Germany
Copyright © 2022 Huang, Chung, Wu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Bing Chen, Y2h1bmJpbmcuY2hlbkBnbWFpbC5jb20=
†These authors have contributed equally to this work