 Justyna Sakowska1†
Justyna Sakowska1† Łukasz Arcimowicz2†
Łukasz Arcimowicz2† Martyna Jankowiak1
Martyna Jankowiak1 Ines Papak2
Ines Papak2 Aleksandra Markiewicz3
Aleksandra Markiewicz3 Katarzyna Dziubek2
Katarzyna Dziubek2 Małgorzata Kurkowiak2
Małgorzata Kurkowiak2 Sachin Kote2
Sachin Kote2 Karolina Kaźmierczak-Siedlecka4
Karolina Kaźmierczak-Siedlecka4 Karol Połom4
Karol Połom4 Natalia Marek-Trzonkowska2,5*
Natalia Marek-Trzonkowska2,5* Piotr Trzonkowski1*
Piotr Trzonkowski1*- 1Department of Medical Immunology, Medical University of Gdańsk, Gdańsk, Poland
- 2International Centre for Cancer Vaccine Science, University of Gdańsk, Gdańsk, Poland
- 3Laboratory of Translational Oncology, Intercollegiate Faculty of Biotechnology, University of Gdańsk and Medical University of Gdańsk, Gdańsk, Poland
- 4Department of Surgical Oncology, Medical University of Gdańsk, Gdańsk, Poland
- 5Laboratory of Immunoregulation and Cellular Therapies, Department of Family Medicine, Medical University of Gdańsk, Gdańsk, Poland
Autoimmune disease results from the immune response against self-antigens, while cancer develops when the immune system does not respond to malignant cells. Thus, for years, autoimmunity and cancer have been considered as two separate fields of research that do not have a lot in common. However, the discovery of immune checkpoints and the development of anti-cancer drugs targeting PD-1 (programmed cell death receptor 1) and CTLA-4 (cytotoxic T lymphocyte antigen 4) pathways proved that studying autoimmune diseases can be extremely helpful in the development of novel anti-cancer drugs. Therefore, autoimmunity and cancer seem to be just two sides of the same coin. In the current review, we broadly discuss how various regulatory cell populations, effector molecules, genetic predisposition, and environmental factors contribute to the loss of self-tolerance in autoimmunity or tolerance induction to cancer. With the current paper, we also aim to convince the readers that the pathways involved in cancer and autoimmune disease development consist of similar molecular players working in opposite directions. Therefore, a deep understanding of the two sides of immune tolerance is crucial for the proper designing of novel and selective immunotherapies.
Introduction
Immune tolerance is a state of unresponsiveness of the immune system to self-tissues with a concomitant ability to identify and respond against non-self and dangerous antigens. Multiple mechanisms shape and control this state, including the elimination of autoreactive receptors from the system in bone marrow and the thymus (central tolerance). However, not all autoreactive cells are deleted in the primary lymphoid organs. For example, the naive T-cell repertoire that leaves the thymus contains up to 40% of low-avidity self-reactive T cells. These cells can potentially trigger an autoimmune response; therefore, several mechanisms of peripheral tolerance evolved to prevent their activation (1). Specialized cell subsets, such as regulatory T (Tregs) and B cells (Bregs), tolerogenic dendritic cells (tolDCs), and M2 macrophages, participate in keeping the balance between tolerance and activation. However, genetic predispositions and epigenetic modifications combined with exposure to environmental factors can disrupt this status, resulting in the development of autoimmunity. Therefore, an increasing number of approaches that boost the immune tolerance have been evaluated and were already implemented for the treatment of autoimmune diseases in humans. On the other hand, the same mechanisms can be exploited by cancer to set up cancer tolerance (2). In fact, the attraction of tolerogenic cell subsets and evading immune response is considered as one of the hallmarks of cancer. The malignant cells used to express immune checkpoint proteins show impaired antigen presentation, undergo epithelial-to-mesenchymal transition (EMT), or present alterations in RNA editing. In consequence, the presence of a tumor-specific antigen (TSA) or tumor-associated antigen (TAA) does not elicit immune responses to malignant cells (3). Therefore, multiple approaches have been already made to break cancer tolerance and awaken the immune system for the fight against cancer. These strategies were based on monoclonal antibodies, adoptive cell therapies, or therapeutic anti-cancer vaccines. Nevertheless, there is still a lack of full understanding of the complex network of mechanisms leading to tolerance induction or its breakdown. Therefore, with the current review, we aim to discuss the mechanisms involved in the development of autoimmunity and cancer, shedding a light simultaneously on two sides of the same coin. We hope that our paper will sort out the current knowledge in the field and inspire future studies on immune tolerance.
Microbiome and Immune Response
Gut microbiota imbalance is associated with the development and progression of multiple diseases, such as gastrointestinal cancers or inflammatory bowel disease. The link between gut dysbiosis and tumor development has been already reported with Helicobacter pylori being the best studied pathogen in this context (4, 5). However, this is definitely not the only component of the digestive tract microbiome involved in carcinogenesis. However, not only the composition of microbiota but also its activity have an impact on cancer development. Microbial metabolites, such as short-chain fatty acids (SCAFs) or N-nitroso compounds (NOCs) showed anti- and procarcinogenic effects, respectively (6, 7). The microbiome, as well as its metabolites, also affects the function of the immune system and, in this way, may contribute to cancer tolerance or the stimulation of anti-cancer responses. For instance, the fungal genus Candida, which is detected in 74% of oral cancer patients, was reported to increase the proliferation of myeloid-derived suppressor cells (MDSCs) known to dampen the anti-cancer response (8). Therefore, not surprisingly, gut microbiota may affect the efficacy of anti-cancer management as it was reported for immunotherapy with immune checkpoint inhibitors. For instance, the abundance of Bifidobacterium species or Akkermansia muciniphila (next-generation probiotic bacteria) was associated with slow tumor growth and beneficial responses to anti-PD-1 (programmed death receptor 1) therapy (9–11). Therefore, the modulation of gut microbiota may positively affect treatment efficiency and thus patient survival.
On the other hand, the interactions between immunological, microbial, and environmental factors in genetically susceptible individuals are involved in the etiopathogenesis of Crohn’s disease (12, 13). Dysbiotic microbial alterations, such as low gut microbiota diversity, as well as a decreased amount of bacteria belonging to the Firmicutes phylum, are observed in patients with Crohn’s disease (14). The link between mutations in TLR4 (Toll-like receptor 4) (rs4986790) and the IL-10 receptor with Mycobacterium avium subspecies paratuberculosis in these patients was also noted (15).
The nucleotide-binding oligomerization domain-containing protein 2/caspase recruitment domain-containing protein 15 (NOD2/CARD15) gene located on chromosome 16q12 was the first described gene connected with Crohn’s disease pathogenesis (16, 17). It encodes the NOD2 protein, which is mainly expressed not only by dendritic cells (DCs) and monocytes but also enterocytes and Paneth cells. The molecule is known to play a significant role in the intestinal innate immune response against the bacterial cell wall. More than 30 variants of the NOD2/CARD15 gene have been identified, while an increased risk of Crohn’s disease development was connected to R702W, G908R, and L1007fs variants, as well as P268S and IVS8+158 polymorphisms (17).
The role of microorganisms in autoimmunity development was also extensively studied for type 1 diabetes (T1D). Molecular mimicry is described as the structural similarity between self- and foreign (microbial) antigens and has been connected with the break of tolerance to pancreatic beta cells in T1D (18). Researchers described a number of homologies between the antigens of beta cells and microorganisms such as Coxsackievirus (19) or Rotaviruses (20). These data demonstrate a big dynamism of the immune status and suggest that tuning the microbial repertoire may skew the immune response to the desirable profile to fight the cancer or restore immune tolerance to self-antigens.
Escape From Central Tolerance Mechanisms and Cancer Immune Evasion
There is a considerable body of literature presenting the different genetic factors that are associated with specific disease phenotypes as well as with the risk of the disease occurrence (21). Various alleles of human leukocyte antigen (HLA) class I and class II molecules were reported to be associated with a particular autoimmune disease occurrence, including T1D, multiple sclerosis (MS), rheumatoid arthritis (RA), or celiac disease (22, 23). The exact mechanism of how HLA polymorphisms predispose to autoimmunity remains poorly understood. However, it is suggested that differences in the binding affinity of HLA molecules to autoantigens might be involved (24). Nevertheless, the association between autoimmune disorders and the polymorphisms of other genes involved in immune cell antigen recognition and activation like protein tyrosine phosphatase non-receptor type 22 (PTPN22), cytokines, chemokine receptors, costimulatory molecules, and inhibitory checkpoints were also identified (25).
The hallmark of autoimmunity is the presence of autoreactive T and B cells that were not deleted by the mechanisms of central tolerance (26). One of the most studied defects of T-cell-negative selection is mutations in the transcriptional autoimmune regulator gene (AIRE). AIRE is mainly expressed by the thymic medullary epithelial cells (mTECs) and is responsible for the expression of tissue-restricted antigens within the thymus. The T cells responding to these antigens are considered self-reactive and eliminated through negative selection. Thus, when AIRE is defective, the T cells specific to self-antigens leave the thymus and enter circulation. This results in a variety of autoimmune disorders (27, 28). The mouse models of Aire knockout showed that the AIRE expression prevents multiorgan lymphocyte infiltration, various organ-specific autoantibodies, and infertility (29). In humans, AIRE mutations lead to a severe condition called autoimmune polyendocrinopathy syndrome type 1 (APS1) (30, 31). In addition, it was observed that AIRE expression is regulated by sex hormones, leading to sexual dimorphism in autoimmune diseases (32, 33). For example, the castration of male animals led to a lower thymic expression of AIRE, while estrogen treatment resulted in the downregulation of AIRE in cultured human thymic epithelial cells (TECs). In addition, AIRE levels in the human thymus grafted into immunodeficient mice differed according to the sex of the recipient (32, 33). Therefore, AIRE has also been extensively studied in the context of reproductive system cancers. Kalra et al. reported that the AIRE expression in prostate cancer is responsible for resistance to anti-cancer therapy and increased invasiveness. AIRE+ prostate cancer cells were shown to secrete increased levels of IL-6 and prostaglandin 2 (PGE2), which polarized the tumor-associated macrophage toward the M2 phenotype with an increased expression of CD206 and CD163 antigens. In addition, prostate cancer growth and lymphadenopathy after subcutaneous tumor engraftment were only observed in the AIRE+/+ animal model. On the contrary, AIRE-/- mice showed small benign tumors (33).
The defects of the central tolerance mechanism of B cells, observed in a number of autoimmune diseases, result in the accumulation of autoreactive B cells in the periphery. The mutations of PTPN22, Bruton’s tyrosine kinase (BTK), adenosine deaminase (ADA), impaired BCR light-chain rearrangements, and Toll-like receptor (TLR) alterations were observed to contribute to the increase in autoreactive B cells (34). Recently, PTPN22 also emerged as a potential target for cancer immunotherapy. It is not surprising as PTPN22 plays an inhibitory role in the antigen-specific responses of both T and B cells; dectin-1 signaling in DCs; the development and function of Tregs; the macrophage functions mediated via TLRs, NOD2, and NLRP3; and neutrophil adherence and mast cell activation in an IgE-dependent manner (35). Several single-nucleotide polymorphisms in the PTPN22 gene were identified. The most extensively studied is a missense mutation at position 1858 (C3T), resulting in the substitution of an Arg (R) at position 620 to Trp (W). The generation of the Lyp620W variant (also identified as rs2476601) of the protein was found to impair the negative selection of autoreactive T and B cells during their development in the thymus and bone marrow, respectively, and the generation of self-reactive antibodies (36, 37). In consequence, the Lyp620W variant of PTPN22 was identified in multiple autoimmune diseases, including T1D, RA, systemic lupus erythematosus (SLE), Graves’ disease, and myasthenia gravis (38–41). On the other hand, the same variant of PTPN22 was reported to augment antitumor responses and be associated with lower cancer incidence (35, 42). For example, the carriers of the PTPN22(C1858T) variant have a lower risk of non-melanoma skin cancer, while the homozygotes for the PTPN22(C1858T) have improved survival when treated with atezolizumab (anti-PDL1 antibody). These data underline again that immune tolerance is indispensable for preventing autoimmunity, but lowering the threshold of T-cell activation can improve tumor control and the efficacy of anti-cancer treatment.
Cancer immune evasion and autoimmunity prevalence can also be affected by sex hormones. Differences in the male and female endocrine systems lead to discrepancies in the quality and quantity of their immune responses. It was reported that while the female immune system provides better antimicrobial and anticancer responses, it is also more prone to autoimmune diseases (43). Estrogen levels are higher during pregnancy and are correlated with an increased proportion of Tregs in peripheral blood (44). Accordingly, the incidence of relapses of MS in pregnant women decreases significantly (45). Both innate and adaptive immune cells express estrogen receptors α and β (higher expression was observed in B cells than T cells, NK cells, and monocytes) that activate protolerogenic effects (46). Estrogens drive the polarization of T cells into Th2 and Treg cells; increase the production of IL-4, IL-10, and transforming growth factor-β (TGF-β); induce the expression of GATA-3, FoxP3, PD-1, and CTLA-4 (cytotoxic T-lymphocyte antigen 4) on T cells; and reduce the Tfh (T follicular helper cell) response (47, 48). On the other hand, SLE patients experience more flares during pregnancy (49). Interestingly, B-cell tolerance is regulated by estrogens at the maturation stage by engaging estrogen receptor α. Estradiol was shown to be responsible for decreased B-cell lymphopoiesis while expanding the population of splenic marginal-zone B cells through the increase of BAFF concentration (50). Estrogens were also shown to influence immune cells in the tumor microenvironment (TME). Certain mutations in the estrogen receptor result in an increase of tumor-infiltrating Tregs and T helper cells (51). It was also reported that estrogens influence tumor-associated macrophages, directing their polarization into the M2 phenotype and thus promoting their immunosuppressive activity (52, 53).
Immune Checkpoints
Immune checkpoints are inhibitory receptors that convey negative signals to immune cells, preventing autoimmunity (54). The importance of immune checkpoints in supporting tolerance and preventing autoimmunity development is best observed in knockout mice models. For instance, the lack of CTLA-4, PD-1, BTLA (B- and T-lymphocyte attenuator), TIGIT (T-cell immunoreceptor with immunoglobulin and ITIM domain), and VISTA (V-domain Ig suppressor of T-cell activation) was shown to cause massive lymphoproliferation, an onset of autoimmune diseases, or fatal multiorgan tissue destruction (notably CTLA-4 deficiency) (55–61). In humans, several polymorphisms of immune checkpoint genes were identified and reported to be associated with susceptibility to autoimmune diseases (62–70).
CTLA-4 is a critical regulator of T-cell responses expressed by Tregs and activated conventional T cells. The main role of the receptor is to inhibit antigen presentation and the following activation of naive T cells by competitive binding to costimulatory receptors CD80 and CD86 on antigen-presenting cells (APCs) (71, 72). It was reported that CTLA-4 not only binds its ligands but also captures and removes them from APCs by a process of trans-endocytosis. In consequence, these costimulatory molecules are degraded inside CTLA-4-expressing cells resulting in a temporary lack of CD80/CD86 on APCs and thus impaired costimulation via CD28 (73). CTLA-4 is indispensable for preventing autoreactivity (74, 75). Its deficiency in humans is a common hallmark of primary immune deficiencies associated with immune dysregulation and prominent autoimmunity with highly variable features. The clinical symptoms probably result from the aberrant activation of polyclonal T cells. In addition, the deficiency of CTLA-4 results in increased CD28 co-stimulation that triggers self-reactive T cells against a variety of tissues. Treg dysfunction plays a vast role in the immune activation associated with CTLA-4 loss-of-function mutations (75). On the contrary, CTLA-4 expression on tumor cells was recognized as a prognostic factor of poor outcome in breast, pancreatic, and nasopharyngeal cancers (76–78). The application of therapeutic antibodies targeting CTLA-4 such as ipilimumab became a breakthrough in cancer therapy. Anti-CTLA-4 antibodies were shown to unlock the immune response to cancer, as well as lead to the depletion of tumor-infiltrating Tregs via antibody-dependent cell-mediated cytotoxicity. This way, anti-CTLA-4 demonstrated durable clinical activity in a subset of patients with solid malignancies including advanced melanoma (79–81).
Programmed cell death receptor 1 (PD-1) is another immune checkpoint significant for self-tolerance and the cessation of the immune response that became a target of cancer immunotherapy. Upon engagement by its ligand (PD-L1, Programmed cell death ligand 1), PD-1 acts as a brake to the immune system that induces the apoptosis of activated T cells (82). PD-L1 expression can be detected in pancreatic islets, vascular endothelial cells, and placenta where it is responsible for tissue protection from autoimmune responses (83). For example, in T1D, PD-L1 was observed to be upregulated in insulin-producing beta cells under an autoimmune attack and correlated with the intensity of CD8+ T-cell infiltration in the pancreas (84, 85). In addition, PD-1/PD-L1 interaction was reported to be involved in the generation of inducible Tregs (iTregs). Francisco et al. showed that PD-L1-negative APCs had an impaired ability to generate Tregs, either in vitro or in vivo (86). The failure of APCs isolated from SLE patients to upregulate PD-L1 expression validates these findings in humans (87). The blockade of PD-1 or PD-L1 in experimental models of autoimmunity led to disease onset and exacerbation (88, 89), indicating the essential role of these immune checkpoints in tolerance and, specifically, in Treg maintenance. Recent reports on autoimmune-related adverse events in oncologic patients treated with PD-1/PD-L1 axis blockers support these findings (90, 91).
In cancer, effector T cells, which are persistently exposed to antigen stimulation in TME, express PD-1 at high levels, in the long term, causing T-cell functional exhaustion. It results in the inability of T cells to eliminate tumor cells and facilitates cancer progression (34, 92). Additionally, cancer cells actively exploit PD-L1 to evade the immune system and hijack the immunosurveillance mechanisms with PD-L1 expression (93). Moreover, the results presented by Chen et al. (2018) revealed that apart from cell surface expression, PD-L1 was present in extracellular vesicles (exosomes) produced by melanoma cells, suggesting its systematic immunosuppressive impact (94). As a result, it leads to the transcriptomic changes and the exhaustion of CD4+ (95) and CD8+ (96) T cells that are unable to eliminate cancer cells effectively. In a vast number of cancers, lymphocyte infiltration is in positive correlation with PD-L1 expression, which is simply an adaptive mechanism of the tumor to escape an immune response. Even though tumor PD-L1 expression usually suggests poor prognosis, then higher levels of tumor PD-L1 expression correlate with a better efficiency of immunotherapy (97).
Another molecule involved in central and peripheral tolerance is Fas. Fas/FasL ligation on TCR-stimulated lymphocytes restricts the overactivation of immune cells after an antigenic challenge, called activation-induced cell death (AICD). It is one of the main mechanisms in restoring immune homeostasis (98). The Fas/FasL-induced apoptosis of B cells was shown to be important in germinal center reactions (98). FasL can be expressed on non-immune cells in immune-privileged sites such as the eye, brain, and placenta, restricting the access of activated immune cells to these tissues (99). Alterations in Fas-mediated apoptosis were implicated in the pathogenesis of autoimmune diseases. Mutations in Fas/FasL axis-related genes lead to a striking lymphoproliferation with autoimmune cytopenias in humans termed autoimmune lymphoproliferative syndrome (ALPS) (100, 101). An interesting feature of ALPS is an accumulation of double-negative T cells that are terminally differentiated, with the markers of immune exhaustion (102). On the other hand, increased expression of FasL was observed in T1D (103), autoimmune thyroid diseases (104), and in MS (105, 106). An interesting feature of Fas/FasL signaling is the opposite outcome of ligation with membrane-bound versus soluble forms of these molecules where the soluble Fas and FasL do not induce apoptosis (107, 108). This discovery prompted studies investigating the levels of serum Fas/FasL molecules in autoimmune diseases, revealing elevated levels in SLE patients (107, 109) and Sjögren’s syndrome (SS) (110). Excessive Fas signaling in the tumor microenvironment, majorly caused by high levels of the Fas ligand released by myeloid-derived suppressor cells (MDSCs), leads to the apoptosis of tumor-infiltrating lymphocytes (TILs) and was described as one of the core reasons for the failure of cancer immunotherapy (111). In addition, FasL was reported to be expressed in numerous cancer types with a potential to induce the apoptosis of immune cells in the TME and was associated with poor prognosis. On the other hand, there is still controversy when it comes to the role of Fas/FasL axis in cancer cells. Several in vitro studies suggest that the ultimate effect may depend on the level of FasL expression by tumor cells. As elevated levels of FasL cause neutrophil-mediated inflammation that leads to tumor rejection, surprisingly low levels of FasL seemed to facilitate tumor growth. The Fas/FasL role in cancer is still not fully understood and brings a lot of controversies but surely requires further investigation as targeting Fas may significantly improve the efficiency of immunotherapy and tumor rejection (112, 113).
Other known immune checkpoints include BTLA, T-cell immunoglobulin and mucin domain-3 (TIM-3), and TIGIT (114, 115). In general, all were shown to inhibit the responses of activated T cells, while BTLA also demonstrated an impact on B cells (116). It was observed that patients with SLE and MS present a low expression of BTLA on B and T cells (117–119). Its decreased expression on naïve B cells was associated with increased IFN-γ and autoantibody levels in SLE patients that could suggest alterations in B-cell activation during the course of the disease (118). In conditions where Th17/Treg balance is shifted, the involvement of immune checkpoint signaling pathways was also implicated. A study by Wu et al. described a lower frequency of TIM-3 positive T cells together with increased IL-17 levels in patients suffering from autoimmune hepatitis, and experiments on mice confirmed that the blockade of TIM-3 signaling aggravated liver injury (120). TIGIT has been recently associated with Treg biology through the transcriptional profiling of these cells. It was suggested to be a marker of natural thymus-derived Tregs (tTregs) with strong suppressive activity and lineage stability (121). It competes with the CD226 molecule for binding a costimulatory poliovirus receptor (PVR) CD155 and inhibitory CD112 (Nectin-2) expressed on DCs (121). TIGIT-CD226 signaling in T cells was shown to be implicated in the pathogenesis of experimental autoimmune encephalomyelitis (EAE). CD226 knockout EAE mice showed favorable Th17/Treg proportion and increased TIGIT and CTLA-4 expression on Tregs (122). On the other hand, the lack of TIGIT resulted in increased levels of proinflammatory cytokines and hindered IL-10 production by T cells (61). Recently, a novel ligand for TIGIT was discovered on cancer cells. Nectin4 was reported to bind exclusively to the TIGIT molecule (123). TIGIT-Nectin4 interaction inhibits natural killer (NK) cell activity, which is a crucial element of the anti-cancer immune response. In addition, antibodies blocking Nectin4 induced enhancement of tumor killing in vitro and in vivo (123).
Regulatory T Cells
Central tolerance is crucial for the development of a small subset of intermediate-affinity, self-reactive T-cell clones that are rescued from deletion and become (tTregs) (124, 125). Apart from tTregs, Tregs can be induced on the periphery from naïve or effector T cells, becoming peripheral Tregs (pTregs). In addition, specific Treg subpopulations can be distinguished based on secreted cytokines, such as type 1 regulatory T cells (Tr1), T-helper type 3 cells (Th3), and IL-35-producing regulatory T cells (iTr35). They secrete IL-10, TGF-β, and IL-35, respectively (126–128). Functionally, follicular Tregs (Tfr) can also be distinguished within the FoxP3+ population (129). Tfr cells have a TCR repertoire resembling tTregs and were shown to be able to control germinal center reactions and antibody production (130, 131).
Tregs exert their immune-suppressive effects using diverse mechanisms. The most important are (1) a high expression of immune checkpoint inhibitors; (2) infectious tolerance, where Tregs exert and transfer suppressive activity toward other immune cells when activated by autoantigens (132); (3) the secretion of anti-inflammatory cytokines (133), (4) IL-2 deprivation, and (5) adenosine accumulation via CD39 and CD73 activities (134). Apart from cytokines, extracellular vesicles are recently gaining attention as a way of efficient intercellular communication with a significant role in the regulation of the immune system (135, 136).
Tregs are crucial for preventing autoimmune reactions (Figure 1). They play an important role in immune tolerance maintenance, as their deficiency causes immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome, leading to multiorgan autoimmune damage when not treated (137, 138). Numerous studies described quantitative Treg changes in autoimmune diseases. A decrease in the Treg population was shown in juvenile idiopathic arthritis (139) and RA (140). However, in some diseases, such as systemic sclerosis (SSc), Tregs were shown to be increased (141). The results from SLE patients regarding Treg frequencies are conflicting, which may arise from differences in the analyzed phenotypes of Tregs (142). Numerous studies suggested the decreased immunosuppressive potential of Tregs in autoimmune diseases (143–147). The main limitation of studying Tregs in human organ-specific diseases is usually the lack of insight into the damaged tissue, as systemic and local immune responses may differ dramatically. Nevertheless, several studies pursued this problem. For instance, Marazuela et al. reported lower numbers of Tr1 and higher proportions of tTregs in the thyroid glands of patients with autoimmune thyroid disease (AITD) as compared with peripheral blood (145, 146). In patients with relapsing–remitting MS (RR-MS), higher frequencies of Tregs were present in cerebrospinal fluid (CSF) rather than in the peripheral blood. The same group of patients had decreased peripheral blood Treg levels compared to the patients with secondary-progressing MS and other neurological diseases, suggesting the migration of Tregs to the site of autoimmune inflammation (148). In addition, the primary role of tTregs, as opposed to pTregs, was demonstrated to control T1D development. However, the deficiency in pTregs increased the incidence of insulitis (149). In the synovial fluid of arthritis patients, high frequencies of iTregs and tTregs were present; however, tTregs presented an unstable FoxP3 expression. Moreover, FoxP3- Tregs were converted to IL-17-producing cells under the environment of the inflamed joint (150, 151). The Th17 cytokine profile (IL-17, IL-12, IFN-γ) influences the organ tissue environment, causing chronic inflammation and, ultimately, organ failure (152). Considering the close transcriptional programs of Th17 and Tregs, both depending on TGF-β, Tregs in the presence of IL-6 were shown to be converted into Th17 cells, or IL-17+ ex-regulatory T cells (exTregs). This plasticity of Tregs results in the blunting of suppressive capacity and the secretion of proinflammatory IL-17 and IFN-γ (153–155). On the other hand, cytokines IL-10 and TGF-β enable the differentiation of immune cells into anti-inflammatory Tregs, Bregs, tolDCs, and M2 macrophages (155).
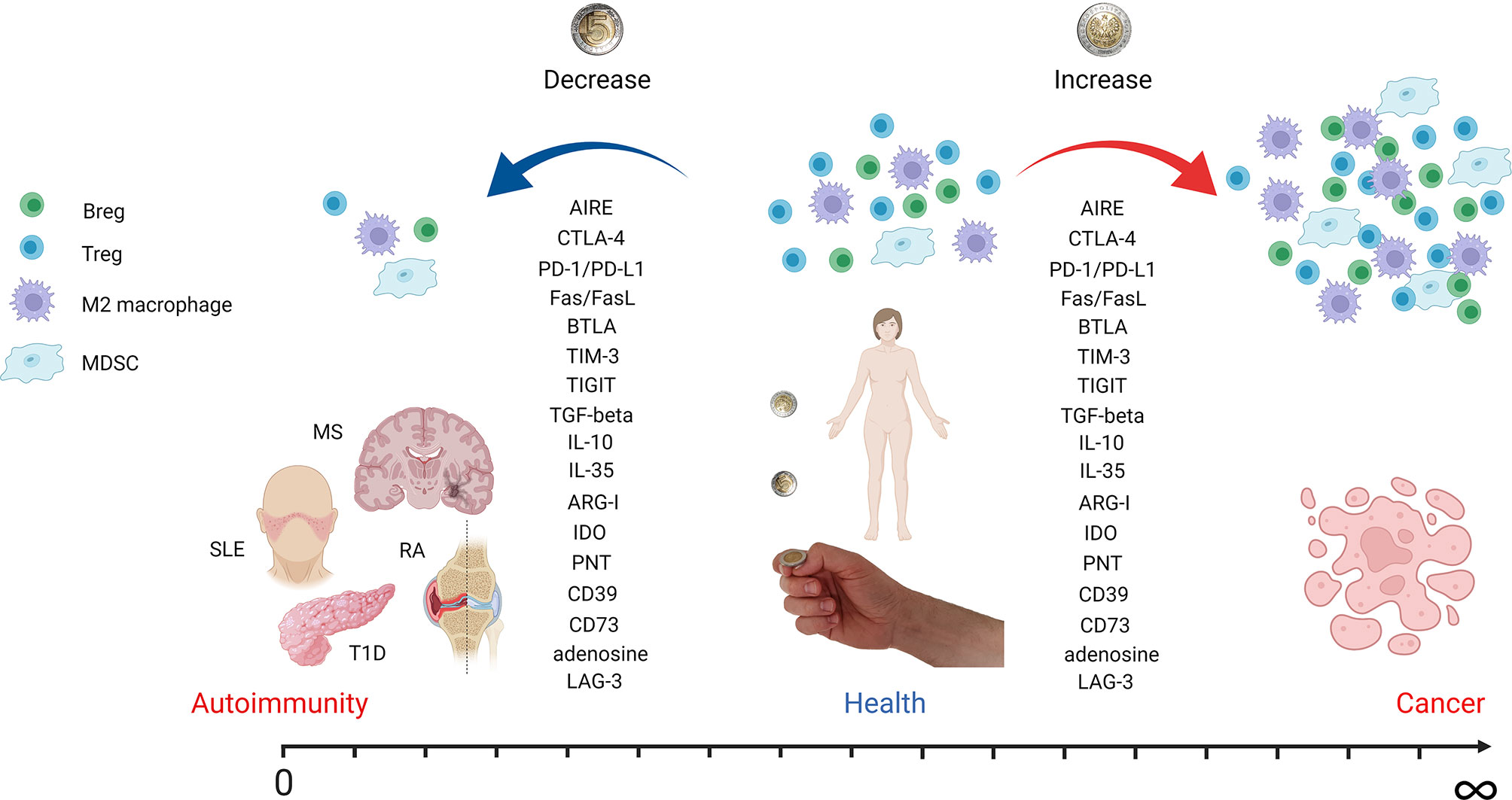
Figure 1 Autoimmunity and cancer as two sides of the same coin. The figure depicts how tuning of immune system regulatory mechanisms can contribute to autoimmunity, health, or cancer development. A decrease in regulatory cell populations like Tregs, Bregs, M2 macrophages, and MDSCs leads to autoimmune disease onset. However, an increase in the same cell subsets is associated with cancer development and progression. Effector molecules involved in immune tolerance induction are downregulated in autoimmunity but overexpressed in cancer. The most important molecules mentioned in the text are listed. AIRE, autoimmune regulator; CTLA-4, cytotoxic T-lymphocyte antigen 4; PD-1, programmed cell death receptor 1; PD-L1, programmed cell death ligand 1; BTLA, B- and T-lymphocyte attenuator; TIM-3, T-cell immunoreceptor with immunoglobulin and ITIM domain; TIGIT, T-cell immunoglobulin and ITIM domain; TGF-beta, transforming growth factor beta; IL, interleukin; ARG-I, arginase I; IDO, indoleamine-pyrrole 2,3-dioxygenase; PNT, peroxynitrite; LAG-3, lymphocyte-activation gene 3 (figure created with BioRender.com).
Indisputably, within the TME, Tregs are present in high frequencies. Treg presence is accommodated by the immunosuppressive cytokine milieu at the site as well as the chemotactic factors produced in TME. High numbers of FoxP3+-expressing Tregs infiltrating TME in lung, breast, and pancreatic cancers were associated with poor prognosis (156). Tregs express various chemokine receptors, like CCR4 and CCR5, that allow migration to TME more efficiently (157, 158). TME is rich in TGF-β and promotes the differentiation of conventional CD4+ T cells into pTregs (159). Resting Tregs are not immunosuppressive unless they become activated through TCR engagement and signaling molecules. The Tregs found in TME are, however, highly activated and immunosuppressive, characterized by upregulated levels of the master regulatory transcription factor FoxP3 (160). This subsequently leads to the suppression of CD8+ T cells, NK cells, NKT cells, and M1 macrophages and the maturation of DCs through IL-10, TGF-β, and indoleamine-pyrrole 2,3-dioxygenase (IDO) (161–163). In addition, Tregs not only bind IL-2 competitively to conventional T cells but also release soluble CD25 (IL-2R subunit) that eliminates IL-2 and alters cytotoxic T-cell functions. Tregs in TME may also release IL-35 that increases the expression of inhibitory receptors like PD1, TIM-3, and lymphocyte-activation gene 3 (LAG-3). This leads to the exhaustion of TILs (164–166). Interestingly, Treg elimination that was followed by cancer antigen vaccination generated effective anti-tumor CD4+ and CD8+ T-cell responses in cancer patients with advanced malignancies (167). However, as mentioned before, systemic Treg depletion would lead to severe autoimmune disorders, emphasizing the need for more selective methods that would specifically target intratumoral Tregs.
Regulatory B Cells
B-cell maturation mechanisms require consecutive checkpoints to develop tolerance: clonal deletion, receptor editing, and anergy. Immature B cells transmitting an overly strong signal through the B-cell receptor (BCR) in response to self-antigen undergo clonal deletion. A tolerance mechanism unique to B cells is the possibility of repeated immunoglobulin light-chain gene recombination. Such rearrangements lead to alterations in BCR specificity to ideally avoid the formation of self-reactive B-cell clones (168, 169). The subsets of B cells expressing PD-1 (170), TIM-3 (171), and BTLA (117) were described as Bregs, an important element for the maintenance of peripheral tolerance (Figure 1). However, a consensus regarding the definition and detailed phenotype of Bregs has not yet been reached. The distinct methods for identification in various disease models and different tissues complicate the general classification. IL-10, TGF-β, and IL-35 have been identified as the main suppressive cytokines produced by Bregs; thus, some authors used to classify the cells into IL-10+, TGF-β+, and IL-35+ Bregs (172). Among IL-10+ human Bregs, the following phenotypes of Bregs were reported: CD1dhi CD5+ (173), CD5+ (174), CD24hiCD27+ (175), CD24hiCD38hi (176–178), CD25+CD71+CD73−, and CD25+CD71+CD73lowPD-L1+ (179), CD154+ (180), CD5hiCD38lowPD-1hi (181), CD27intCD38+ (182). Up to now, 2 subsets of TGF-β+ Bregs have been identified in humans: CD25hiCD27hiCD86hi CD1dhi (183) and CD24hiCD38hi (178). Despite the fact that IL35+ B cells have been identified in humans, up to now, specific surface markers have not been reported for these cells in men (172, 184). The manipulation of the Breg compartment through the adoptive transfer of isolated or ex vivo-induced cells was explored in the murine models of autoimmune diseases. For example, IL-10+ Bregs were shown to suppress inflammation in the mice models of RA, EAE, and SLE. The most prominent therapeutic effects were observed when Bregs were administered early in the disease course (183, 185–188). The mechanisms used by Bregs have not been studied extensively. Nevertheless, in vitro studies performed by Kessel et al. resulted in several interesting observations. Human Bregs defined as CD25high CD27high CD86high CD1dhigh IL-10high TGF-βhigh cells were shown to significantly decrease the proliferation of autologous conventional CD4+ T cells in a dose-dependent manner. In addition, Bregs were found to upregulate FoxP3 and CTLA-4 expression in Tregs in cell-to-cell-dependent contact. The effect was even stronger when Bregs were pretreated with a TLR-9 agonist (oligodeoxynucleotide) and CD40L (183). The other groups also reported the suppressive effects of Bregs on DC and macrophage cytokine production and antigen presentation (175, 189). Increased frequencies of IL-10+ B cells and their progenitors were found in patients with various autoimmune diseases, such as SLE, RA, SS, autoimmune vesiculobullous skin disease, and MS. However, the significance of Bregs in the pathogenesis of human autoimmune diseases is yet to be determined (175).
The impact of B cells in cancer is still unclear and ambiguous as they were shown to play a role in both cancer promotion and anti-cancer responses (190). Significant B-cell infiltration was found in breast cancer, non-small cell lung cancer (NSCLC), ovarian cancer, melanoma, and renal cell carcinoma. Bregs have been also identified in a number of cancers including lung (191), gastric (192), and breast cancers (193). Increased infiltration with Bregs results in the inhibition of effector T-cell responses and their impaired proliferation. It was suggested that the tumor and TME can direct tumor-infiltrating B cells into tumor-induced Bregs (tBregs) (194) by the direct tumor cell: B-cell contact (195). Lindner et al. reported that tumor-infiltrating Bregs use Granzyme B for the degradation of the CD3 ζ-chain in CD4+ T cells. The phenomenon results in a limited proliferation of the target CD4+ T cells (196). Interestingly, tBregs were also shown to play a substantial role in the education of MDSCs, enhancing cancer-induced immune suppression (197). In addition, Breg-derived IL-10 leads to the conversion of conventional B cells into Bregs and contributes to Treg expansion (183). tBregs were also found to direct conventional CD4+ T cells into Tregs in breast and gastric cancers (177, 198). Another study utilizing a mouse model showed that tumor-educated Bregs suppress not only the proliferation of helper and cytotoxic T cells but also the secretion of Th-1 cytokines and the expansion of NK cells in a TGF-β- or PD-L1-dependent manner (195). A similar immunosuppressive activity was reported for IL-35+ Bregs. Breg-derived IL-35 was shown to stimulate cancer (199), as well as convert both T and B cells into Tregs and Bregs, respectively. Several surface molecules have been identified to be involved in direct cell-to-cell interactions between Bregs and the target immune cells, like Bregs CD40/CD40L, CTLA-4/CD80 and CD86, PD-L1/PD-1, or Fas/FasL (200–203).
Myeloid-Derived Suppressor Cells
A significant population of cells identified within the tumor was described as activated immature myeloid cells with immunosuppressive function, termed myeloid-derived suppressor cells (MDSCs). These cells, in general, can be divided into 2 populations: mononuclear (M-MDSCs; CD11b+Ly6G−Ly6Chi) and polymorphonuclear/granulocytic MDSCs (PMN-MDSCs; CD11b+Ly6G+/hiLy6Clow/int) (204). The granulocyte monocyte-colony stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), stem cell factor (SCF), prostaglandins, TNF-α, IFN-γ, and IL-18 were shown to promote the differentiation of functional MDSCs that contributed to the establishment of immunosuppressive niche and tumor progression (205–209). MDSCs were shown to be engaged in the suppression of TIL activity, EMT, and angiogenesis and participate in establishing a pre-metastatic niche (210, 211). In addition, the increased production of nitric oxide (NO) by MDSCs resulting from inducible nitric oxide synthase (iNOS) overexpression was reported to be responsible for T-cell apoptosis and proliferation suppression, as well as the inhibition of antigen presentation by DCs (212, 213). Moreover, MDSCs isolated from tumor-bearing animals showed significantly higher levels of reactive oxygen species (ROS) than the cells isolated from healthy controls. Further studies demonstrated that ROS are crucial for the MDSC suppression of T-cell proliferation, survival, and TCR signaling (214–216). It was also reported that MDSCs express elevated levels of arginase I (ARG-I; Figure 1). This way, they can deplete TME from indispensable amino acids, such as L-arginine or cysteine affecting T-cell activation and proliferation (217, 218). One of the mechanisms that stands behind this T-cell suppression is the downregulation of the CD3 ζ-chain of the TCR complex (219). Tumor-derived MDSCs are also a potent source of IDO, an L-tryptophan-degrading enzyme that induces the suppression of T-cell proliferation and survival, as well as promotes Treg induction (220–222). Another important effector molecule used by MDSCs is peroxynitrite (PNT). The production of PNT in TME was shown to nitrate the TCR complex, leading to the unresponsiveness of tumor-infiltrating cytotoxic T lymphocytes to the specific antigens presented by MDSCs (223–225).
It is also recognized that MDSCs participate in the generation of immunosuppressive adenosine (226, 227). MDSCs express the ectoenzymes triphosphate diphosphohydrolase 1 (NTPDase 1/CD39) and ecto-5’-nucleotidase (5’-NT/CD73). The first ectoenzyme is responsible for the hydrolysis of extracellular ATP or ADP into AMP, which is then degraded by CD73 into adenosine. Adenosine is known to inhibit the activation and effector function of T cells, mainly by A2A and A3 adenosine receptors (228). However, these receptors can also be found at the surface of MDSCs. The blockade of the A2B receptor was shown to reduce the secretion of IL-10 and monocyte chemoattractant protein 1 (MCP-1) by MDSCs in mice with melanoma (229). Aside from IL-10, TGF-β is another cytokine important for MDSC function. MDSC-derived IL-10 and TGF-β promote the differentiation of T cells into Tregs and suppress T- and NK-cell activation as well as DC function (230, 231). TGF-β was reported to induce EMT in cancer cells (211, 232), generate pro-tumorigenic M2 macrophages (233), and drive pro-tumorigenic neutrophil polarization (234). In NSCLC, higher levels of TGF-β were associated with an increased expression of inhibitory molecules such as CTLA-4 and TIM3 on cancer cells (235). It was suggested that MDSCs are responsible for the induction and recruitment of the Treg population in the TME. While the process of Treg induction is not fully elucidated and was suggested to depend on cytokine milieu and cell-to-cell contact, the Treg recruitment was shown to be largely dependent on the production of CCL2 and CCL5 chemokines (236, 237). On the other hand, MDSCs may also limit the T-cell infiltration of the tumor by metalloproteinase 17 (ADAM17), which cleaves L-selectin (CD62L) present on the surface of naïve T cells. In consequence, T cells are not able to infiltrate tumor or enter peripheral lymph nodes (238).
The growing body of research on MDSCs and their suppressive capacity in TME sparked interest for the exploration of their role and potential therapeutic use in autoimmune diseases. In the aim to diminish the heterogeneity of studied MDSCs, they used to be divided into M-MDSC (CD11b+Ly6G−Ly6Chi) and PMN-MDSC (CD11b+Ly6G+/hiLy6Clow/int) subsets as in cancer studies (239, 240). Multiple studies on the animal model of MS have pointed to the beneficial role of MDSCs in autoimmunity. Moliné-Velázquez et al. identified ARG-I positive MDSCs in the spinal cord during the course of EAE. The cells showed tropism to demyelinated areas in CNS. The density of ARG-I+ MDSC infiltrate, as well as the local proportion of the apoptotic T cells, correlated with the disease course and clinical state. They peaked in parallel with the clinical score, which were decreased significantly during remission, and was not detectable in the chronic phase (240). These data correspond with the previous studies that reported the presence of ARG-I+ cells exclusively when the switch from proinflammatory to anti-inflammatory conditions occurred and the active phase was about to end (241–243). These data indicate that MDSCs are involved in limiting inflammatory damage in MS and contribute to relative recovery in the remitting phase of the disease.
In humans, as in previously described animal studies, the numbers of MDSCs were found to be an indicator of the disease phase. For example, RR-MS was characterized by significantly higher levels of the PMN-MDSC subset in the peripheral blood at relapse than in the remission period or in healthy individuals. Experiments in vitro revealed that PMN-MDSCs from patients with RR-MS suppress autologous T-cell proliferation, suggesting their beneficial role for remission induction (244).
However, higher proportions of M-MDSCs were observed to be positively correlated with proinflammatory Th17 and Th1 cells, as well as with a worsened metabolic profile in the patients with T1D and their relatives at elevated risk for the disease (245). Similar patterns were described in RA (246) and SLE (247). These data indicate that a detailed characterization of MDSC subsets and their further stratification is inevitable if MDSCs are planned to be harnessed to stop autoimmune diseases. Nonetheless, the idea of utilizing the suppressive activity of MDSCs in therapy prompted the experiments of adoptive transfer of MDSCs to diabetes-prone mice that successfully prevented the onset of autoimmune diabetes and established tolerance to self-antigens via Treg induction (248).
Macrophages
Macrophages can be divided into two main groups, classically activated, proinflammatory macrophages (M1) and alternatively activated macrophages (M2) with anti-inflammatory and regenerative properties. M1 and M2 cells can be distinguished by secreted cytokines, for example, INF-γ, IL-1, IL-6, IL-12 and IL-10, and TGF-β, respectively. However, macrophages exhibit exceptional plasticity depending on the microenvironment (249). It has been reported that tumor-associated macrophages (TAMs) are recruited to TME by chemokines, such as CCL2 in different tumors, including glioblastoma and breast and lung cancers (250–252). Moreover, TAMs start to produce CCL2 and thus recruit more macrophages and stimulate their polarization toward a pro-tumoral M2 phenotype (253–256). Targeting TAMs in pancreatic ductal adenocarcinoma by inhibiting CCR2 has shown a therapeutic benefit by restoring anti-tumor immunity in preclinical models (257). Although TAMs can produce IL-8, a chemotactic factor for T cells, high levels of IL-8 in plasma, peripheral mononuclear cells, and TAMs were negatively correlated with clinical prognosis regardless of high CD8+ T-cell infiltration in the tumor (258). TAM-derived cytokines include IL-6, IL-10, and TGF-β. IL-6 combined with IL-6R can activate anti-apoptotic pathways in tumor cells and prolong their survival (259). A meta-analysis revealed that the serum levels of IL-10 are positively correlated with tumor progression, showing the importance of TAMs in the promotion of tumor development (Figure 1) (260). Additionally, TAMs secrete inflammatory mediators, including prostaglandin E2 (PGE2) and matrix metalloproteinase-7 (MMP-7). These molecules interfered with TLR-mediated or IFN-γ-mediated DC and macrophage activation. In addition, a direct induction of genes that suppress APC function was observed. Thus, TAMs indirectly impair the T-cell recognition of tumor antigens (261).
Macrophages are constantly present in peripheral tissues, where they can rapidly act as APC, as shown in the T1D animal model (262). In autoimmune diseases, the overreaction of the immune system and the resulting highly proinflammatory environment lead to tissue damage. Therefore, the imbalance in M1/M2 macrophage subsets was observed in several autoimmune diseases, both organ specific (MS) (263) and systemic with in-tissue manifestations (RA, SLE, SSc; Figure 1) (264). Recent studies on human pancreata from T1D patients, using multiparametric analyses, revealed the presence of macrophages of mixed M1/M2 characteristics, confirming the high plasticity of these cells (265, 266). Studies on EAE showed that the polarization of macrophages follows the natural pattern of the disease with the increase of M2 macrophages during the remission phase (263). The adoptive transfer of M2 macrophages in the mouse model of SLE decreased the disease severity score (267) and prevented diabetes in NOD mice (268). Importantly, these transferred cells were homed to the site of ongoing insulitis (268). These results suggest an attractive therapeutic opportunity.
Fibroblasts
TME contains a special subpopulation of fibroblasts with a myofibroblastic phenotype. Cancer-associated fibroblasts (CAFs) are activated, but unlike in a physiological wound-healing process and tissue repair, they remain constantly activated, leading to pathological fibrosis. Active fibroblasts and myofibroblasts are the main effectors involved in the initiation of fibrosis due to excessive collagen deposition and the modulation of extracellular matrix (ECM) (269, 270). Multiple mechanisms can be involved in their activation, like the composition of the (ECM), DNA damage, physiological stress (mediated by ROS), inflammatory signals (e.g., IL-1 and IL-6), and growth factors, fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) (271–273). Once activated, they are sufficient not only to promote tumor growth but also to further model ECM; produce proinflammatory cytokines, proangiogenic VEGF, and the chemokine ligand CXCL12 that is responsible for attracting immunosuppressive cells into TME that indirectly assist in immune tolerance establishment” as this part of the sentence is continuation of the role of CXCL12 (274). It was reported that throughout the secretion of TGF-β, CAFs induce the occurrence of EMT and promote lung metastasis in breast cancer (275). Moreover, the cytokine is involved in the synthesis of collagen and matrix modification by macrophages and fibroblasts, leading to local tissue scarification, like pulmonary fibrosis (276). Tissue fibrosis and the contractile properties of myofibroblasts stiffen ECM subsequently, lowering blood circulation and leading to local tissue hypoxia (277). These effects also reduce the possibility of cytotoxic effectors to reach cancer cells, therefore reducing immune surveillance and therapy efficacy. While using the combinations of multiple biomarkers to help identify cell subsets in TME, it has been found that the presence of CAFs is negatively correlated with the prognosis in patients receiving PD-1 immunotherapy in metastatic melanoma (266). This shows that the combination of different biomarkers can not only help us target CAFs as a potential clinical marker for the success of therapy, but targeting CAFs can also improve the efficacy of immunotherapy. Inhibiting the growth and proliferation of CAFs and preventing or reversing their activation status are potential ways to target CAFs in cancer therapy.
The therapeutic application of fibroblasts in autoimmune diseases has not been extensively studied. Jalili et al. reported tolerance induction by fibroblasts in the animal model of T1D and pancreatic islet transplantation. However, the therapeutic fibroblasts were transduced with a lentiviral vector carrying IDO cDNA. Thus, the cells artificially overexpressed IDO and efficiently suppressed immune responses (278, 279) Nevertheless, the data of Khosravi-Maharlooei et al. suggest the potential therapeutic use of fibroblasts in autoimmune diseases. They showed that fibroblasts can condition DCs to express higher levels of co-inhibitory molecules and anti-inflammatory cytokines. In addition, fibroblasts arrested the ability of DCs to induce the proliferation of T cells in both direct and indirect pathways. Fibroblast-primed DCs were also reported to migrate to the regional lymph nodes and present fibroblast-derived antigens. This study sheds light on the role of fibroblasts in the maintenance of self-tolerance and regulation of immune responses (280). Finally, the data provide inspiration for the future therapeutic approaches.
Epithelial-to-Mesenchymal Transition in Cancer and Autoimmune Disorders
Another complex phenomenon modulating immunity is EMT, which induces morphological changes in epithelial cells, after which, they start to resemble mesenchymal cells—fibroblasts (281–284). As a result, cells undergoing EMT show increased motility and invasiveness due to the degradation of extracellular matrix, but it can also acquire other features, like stem cell properties or the ability to escape the immune system, which overall contributes to the aggressive phenotype of cancers (281, 285, 286). A direct connection between immunotolerance and EMT was shown in breast and lung cancer in vitro studies, where upon EMT induction, the expression of PD-L1 in cancer cells increased (287, 288). Moreover, cells with a mesenchymal phenotype showed higher levels of PD-L1 than cells of epithelial phenotype (288). Hypoxic hepatoma cells, which undergo EMT, induce IDO expression in monocyte-derived macrophages and further suppress the proliferation of T cells as well as promote the expansion of Tregs (289). Pancreatic tumors with EMT features co-express PD-L1, and melanoma cells with EMT features show increased NK immunosuppressive function in comparison to epithelial melanomas (290), which overall indicates that EMT in cancer cells leads to a decreased immune response. On the other hand, the EMT inducers present in the tumor microenvironment can modify the activity and composition of the immune cells in the tumor niche. TGF-β, a potent inducer of EMT in multiple cancers, including breast (291, 292), lung (235, 293, 294), and colon (295, 296) cancers, exerts immunosuppressive function (235). In lung adenocarcinoma, the EMT signature of the tumor was associated with increased infiltration by CD4+ FoxP3+ Tregs (297), a decreased infiltration of activated effector T cells (including Th17 cells), and higher levels of activated B cells and γδ T-cells (235). Similarly, in patients with pancreatic ductal adenocarcinoma, tumors with mesenchymal features have decreased the number of CD8+ T cells and increased the frequencies of Tregs (298).
EMT develops in response to chronic inflammation where it can lead to pathological fibrosis-the generation of myofibroblasts, which actively deposit ECM, leading to a decreased functionality of the affected organs (299–301). The triggers for EMT and fibrosis are overlapping; most importantly, both require TGF-β (302, 303). Chronic inflammation in autoimmune disorders such as RA, Crohn’s disease, SLE, or scleroderma have been associated with fibrotic tissue remodeling (300, 304, 305). The local proinflammatory environment is not neutral for tissue-resident mesenchymal cells/fibroblasts that become activated and, as ECM-producing cells, exacerbate fibrosis. Signaling through the proinflammatory IL-17A receptor was responsible for fibroblast activation and the fibrosis of lung tissue in RA-associated lung disease and idiopathic pulmonary fibrosis (306). It seems that during chronic inflammation, overridden tolerance mechanisms interfere in the natural process of healing and repair mediated by fibroblasts, which can additionally support inflammation.
RNA Editing
One of the mechanisms used by the innate immune response for self- vs. non-self-recognition is the RNA-editing process. There are two main types of RNA editing: (i) adenosine-to-inosine (A-to-I) conversion catalyzed by adenosine deaminases acting on RNA (ADAR) enzymes and (ii) cytidine to uridine (C-to-U) deamination by apolipoprotein B mRNA-editing catalytic polypeptide-like (APOBEC) family. A-to-I RNA editing allows cells to mark the host RNA as self. This way, the cell is able to recognize and tolerate edited self-RNAs with viral dsRNA sensors (such as PKR, MDA5, and RIG-I) and simultaneously discriminate non-edited dsRNAs present in the cells as viral genetic material (307). This launches an innate immune response, and results in death of cells where non-edited dsRNA was detected. Defects in RNA editing may contribute to autoimmune diseases and are observed in various cancers (308, 309).
The role of RNA editing and the enzymes involved in this process in cancer are currently being explored (308). Potentially, RNA editing may lead to presentation of edited and thus changed peptides by the MHC class I molecules of malignant cells. This phenomenon was recently shown in melanoma, where TILs were able to recognize the peptides derived from the ADAR1-edited form of cyclin I (CCNI) presented on the surface of cancer cells (310). These findings suggest that either the absence of or a higher expression of ADAR1 can result in novel ADAR1-dependent neoantigens that may be used as biomarkers in cancer or as potential targets for cancer immunotherapy. The study of Asaoka et al. supports this hypothesis. The increase of APOBEC3-mediated RNA editing in breast cancer was correlated with a higher T-cell infiltration of the tumor, improved survival, and better prognosis (311). The role of RNA editing in immune regulation is also proven by the fact that the expression of some RNA-editing enzymes is dependent on IFN (312). The knockdown of Adar1 in mouse B16 melanoma cells was shown to increase the susceptibility of the tumor cells to anti-PD1 therapy after engraftment to animal model (313). Interestingly, Adar1 knockout does not disturb growth of B16 cells in culture but mediates killing of B16 Adar1-/- cells by T lymphocytes in vivo. This effect is determined by abnormal activation of the intracellular dsRNA sensors (Mda5 and PKR) by unedited intracellular dsRNA mimicking virus infection (314). In contrary to B16 mouse melanoma cells, in many human cancer cell lines, loss of ADAR1 results in cell death, even in the absence of innate immune cells. These ADAR1-dependent tumors usually show high IFN induction, probably through the innate immune DNA sensor STING (315) and have a higher expression of both: IFN-stimulated genes (ISGs, including ADAR1) and innate immune sensors for dsRNA, than other types of tumor cells. In addition, they are sensitive to elevated levels of dsRNAs while ADAR1 knockdown is lethal for these cells through the Mda5/MAVS and PKR pathways (315).
RNA editing is also involved in autoimmune diseases connected to the dysregulation of IFN signaling. For instance, mutations in the ADAR1 gene were identified to be involved in the development of type I interferonopathies, including Aicardi–Goutieres syndrome (316), dyschromatosis symmetrica hereditiaria (317), bilateral striatal necrosis (318), and spastic paraplegia (319). ADAR1 expression was shown to be also involved in RA or SLE (320, 321). The enzyme was over-expressed in synovium of RA patients regardless of the disease duration. In addition, the ADAR1p150 isoform was found to be elevated in the blood of the patients with active RA. Interestingly, decreased baseline ADAR1p150 expression and the individual adenosine RNA editing rate of cathepsin S AluSx+ in RA were indicators of a good clinical response to the treatment (320).
Discussion
Immune response and tolerance are vital for proper reaction against pathogens and maintaining internal homeostasis. For years, immunologists have been studying the mechanisms’ underlying tolerance to fight autoimmune diseases. However, a deeper understanding of immune tolerance in TME as well as the mechanisms underlying autoimmunity may help to generate an antitumor response and break tolerance to cancer. Phenomena, such as the generation of tolerogenic immune cell populations or EMT, are revealing pathways that lead to immunological changes in the tumor milieu. Anti-cancer immunotherapies should attempt to break immune tolerance toward the tumor; otherwise, the efficacy of such treatments is greatly limited. On the other hand, the immunotherapies aiming to combat autoimmune diseases seek to induce immunological tolerance, therefore, to limit the pathological immune reaction against self-antigens. As potent tolerance to cancer and the lack of self-tolerance in autoimmune diseases stand on two sides of the same coin (Figure 1; Table 1), certain lessons can be learned from the understanding of these two fields of medicine. We believe that combining knowledge from research on autoimmune diseases and cancer therapies will lead to a considerable progress in both areas. The advantages of exchanging knowledge between these two research fields can already be observed in the therapeutic strategies that are being developed. For instance, while genetically engineered super-activated CAR T cells have been successfully applied for the therapy of non-solid malignancies (322), the depletion of autoreactive immune cells gives promising results in the treatment of autoimmune diseases (323, 324). Moreover, the therapeutic potential of CAR Tregs is being explored in the context of autoimmunity (325), as antigen-specific Tregs proved to have better control over autoreactive effector cells than polyclonal Tregs (326). The strategy has already proved its efficacy in the animal models of MS (327), colitis (328), and T1D (329). Another example of a similar therapeutic approach in cancer and autoimmune diseases are adoptive cellular therapies, such as those that use mature DCs in cancer and tolDCs in autoimmune diseases. In cancer research, DCs loaded with tumor antigens are used as a cancer vaccine (330). In the therapy of autoimmune diseases, tolDCs presenting synovial fluid-derived peptides have been recently tested in a phase I clinical trial in RA patients (331). Many of the immune regulatory axes can be targeted in both autoimmune diseases and cancer, usually in an opposite manner—targeting different cytokines (including IL-2, IL-6, IL-10, IL-15, IL-17, and TNF-α) to manipulate the tolerance and increasing or decreasing the regulatory populations of the cells. As presented in this review, cytokine imbalance is a vital component of TME or autoimmune disorders that creates an opportunity for therapeutic intervention. On the other hand, therapies depleting or promoting the expansion of effector subsets of immune cells are also valid therapeutical strategies, for example, the depletion of effector cells in autoimmune diseases and adoptive cell therapy in cancer patients (322).
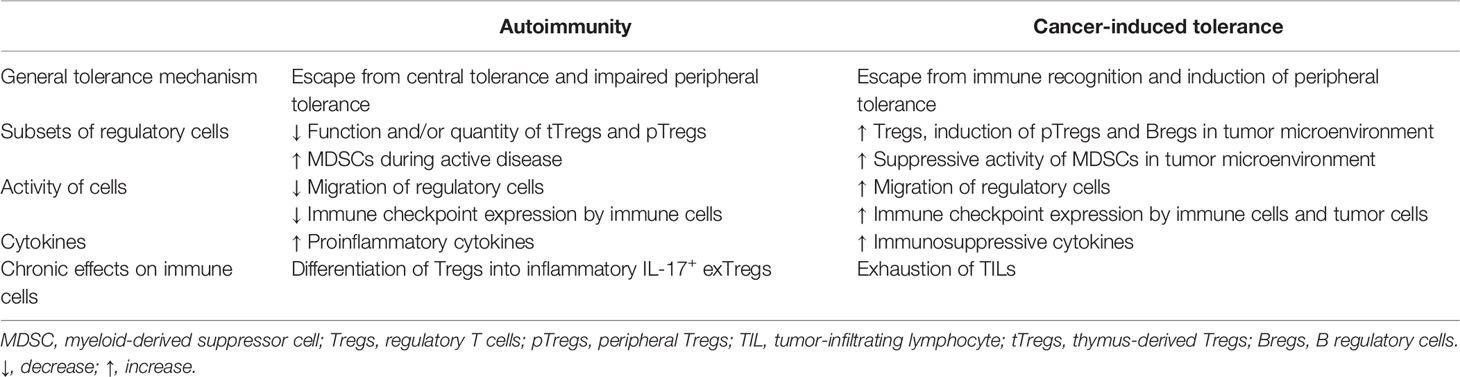
Table 1 Mechanisms involved in breaking tolerance to self-tissues and in induction of cancer tolerance.
Immune checkpoint inhibitors were found to be a milestone in cancer therapy. Ipilimumab, the first immune checkpoint-blocking antibody targeting CTLA-4, was approved by the FDA. It was used for the first time in 2002 and later approved in 2011 for treating unresectable melanoma (332). PD-1-inhibiting antibodies have been also successfully used for the treatment of multiple cancer types as they are at least partially able to reinvigorate exhausted T-cells that regain the cytotoxicity against the cancer (333). Mechanistically, PD-1 signaling acts as a brake to the immune system but it can be stopped by implementation of either PD-1 or PD-L1 blocking monoclonal antibodies that are able to directly inactivate the PD-L1 inhibitory signaling in TME, reverse T-cell exhaustion, and ultimately induce tumor regression (334, 335). Nivolumab, pembrolizumab, and cemiplimab are FDA-approved PD-1-blocking antibodies for the treatment of various cancers including melanoma, renal cell carcinoma, NSCLC, and squamous cell carcinoma. However, many other indications are waiting for the approval (336). When it comes to PD-L1 inhibitors, currently, FDA has approved the following three: atezolizumab, durvalumab, and avelumab (337). At the same time, immune checkpoint fusion proteins are arising as a tool in the treatment of autoimmune diseases. The first promising results of exploiting the inhibitory activity of CTLA-4 in animal models of autoimmune diseases were presented over 25 years ago (338, 339). Successful clinical trials in human patients with psoriasis vulgaris, RA, and juvenile idiopathic arthritis led to the FDA approval of abatacept in 2005 (340–342). CTLA-4Ig is also tested in MS (343) and T1D (344, 345); however, these organ-specific diseases were far less responsive to this therapeutic agent. Experimental studies revealed that CTLA-4Ig induced the suppression of tolDCs (346) and Treg differentiation (347), improved the Treg function (348), and decreased the numbers of Th2 cells (349). The fusion proteins of PD-1 also convey immunomodulatory properties (350). Consequently, other immune checkpoint fusion proteins or agonistic antibodies, such as TIGIT-Fc, TIGIT mAb, and VISTA mAb, are evaluated in pre-clinical and clinical trials (351–353).
A particularly attractive therapeutic approach is the generation of an antigen-specific response with antigen-based and cell-based anti-cancer vaccines (354). These type of vaccines also constitute an extensively investigated strategy to induce tolerance in autoimmune diseases (355). Noteworthy, the combined use of different therapeutic strategies proved to be a valid option for enhancing the response to therapy in both—cancer and autoimmune disease (356, 357). However, therapeutic strategies need to be focused on restoring balance in the immune system and be applied with caution, as the overstimulation of the immune system in cancer may lead to the development of autoimmune disorders (358, 359). On the other hand, over-suppression in the treatment of autoimmune diseases might create a window of opportunity for cancer growth and progression (360, 361).
We hope that with the current paper, we were able to give a glimpse into the mechanisms that regulate tolerance to self-tissues and cancer. A dynamic balance between the resting and activation states is crucial to keep the organism safe from external and internal threats like pathogenic microorganisms, cancer cells, or hypersensitivity. We believe that a better understanding of these mechanisms opens the opportunities for novel and selective immunotherapies.
Author Contributions
JS, ŁA, MJ, NM-T, and PT contributed to the concept of the review. All authors wrote the manuscript. NM-T revised the manuscript with the contribution of all authors. All authors read and approved the manuscript.
Funding
The study was funded by project ”International Centre for Cancer Vaccine Science” that is carried out within the International Research Agendas Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (NM-T), National Science Centre, Poland (funding decision no. DEC-2011/01/D/NZ3/00262, granted to NM-T), and by internal grant of Medical University of Gdańsk ST-49 (PT). This work was supported by the National Science Centre grant number 2016/21/D/NZ3/02629 (AM). The publication of the article was supported by the project POWR.03.05.00-00-z082/18 (JS) cofinanced by the European Union through the European Social Fund under the Operational Programme Knowledge Education Development 2014–2020.
Conflict of Interest
NM-T and PT are the co-authors of 2 patents related to the presented content and are shareholders of PolTREG S.A. company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. ElTanbouly MA, Noelle RJ. Rethinking Peripheral T Cell Tolerance: Checkpoints Across a T Cell’s Journey. Nat Rev Immunol (2021) 21:257–67. doi: 10.1038/s41577-020-00454-2
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
3. Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of Immune Evasion in Breast Cancer. BMC Cancer (2018) 18:556. doi: 10.1186/s12885-018-4441-3
4. Kaźmierczak-Siedlecka K, Daca A, Fic M, van de Wetering T, Folwarski M, Makarewicz W. Therapeutic Methods of Gut Microbiota Modification in Colorectal Cancer Management – Fecal Microbiota Transplantation, Prebiotics, Probiotics, and Synbiotics. Gut Microbes (2020) 11:1518–30. doi: 10.1080/19490976.2020.1764309
5. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
6. Kaźmierczak-Siedlecka K, Daca A, Roviello G, Catalano M, Połom K. Interdisciplinary Insights Into the Link Between Gut Microbiome and Gastric Carcinogenesis—What is Currently Known? Gastric Cancer (2022) 25:1–10. doi: 10.1007/s10120-021-01260-y
7. Bakhti SZ, Latifi-Navid S. Interplay and Cooperation of Helicobacter Pylori and Gut Microbiota in Gastric Carcinogenesis. BMC Microbiol (2021) 21:258. doi: 10.1186/s12866-021-02315-x
8. Mäkinen A, Nawaz A, Mäkitie A, Meurman JH. Role of Non-Albicans Candida and Candida Albicans in Oral Squamous Cell Cancer Patients. J Oral Maxillofac Surg (2018) 76:2564–71. doi: 10.1016/j.joms.2018.06.012
9. Matson V, Chervin CS, Gajewski TF. Cancer and the Microbiome—Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology (2021) 160:600–13. doi: 10.1053/j.gastro.2020.11.041
10. Li W, Deng Y, Chu Q, Zhang P. Gut Microbiome and Cancer Immunotherapy. Cancer Lett (2019) 447:41–7. doi: 10.1016/j.canlet.2019.01.015
11. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut Microbiome Influences Efficacy of PD-1–Based Immunotherapy Against Epithelial Tumors. Science (80- ) (2018) 359:91–7. doi: 10.1126/science.aan3706
12. Michail S, Bultron, Depaolo W. Genetic Variants Associated With Crohn’s Disease. Appl Clin Genet (2013) 6:25. doi: 10.2147/TACG.S33966
13. Caparrós E, Wiest R, Scharl M, Rogler G, Gutiérrez Casbas A, Yilmaz B, et al. Dysbiotic Microbiota Interactions in Crohn’s Disease. Gut Microbes (2021) 13:e1949096. doi: 10.1080/19490976.2021.1949096
14. Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in Diversity of the Colonic Mucosa Associated Bacterial Microflora in Patients With Active Inflammatory Bowel Disease. Gut (2004) 53:685–93. doi: 10.1136/gut.2003.025403
15. Wagner J, Skinner NA, Catto-Smith AG, Cameron DJS, Michalski WP, Visvanathan K, et al. TLR4, IL10RA, and NOD2 Mutation in Paediatric Crohn’s Disease Patients: An Association With Mycobacterium Avium Subspecies Paratuberculosis and TLR4 and IL10RA Expression. Med Microbiol Immunol (2013) 202:267–76. doi: 10.1007/s00430-013-0290-5
16. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 Leucine-Rich Repeat Variants With Susceptibility to Crohn’s Disease. Nature (2001) 411:599–603. doi: 10.1038/35079107
17. Abdelnaby H, Ndiaye N, D’Amico F, Fouad A, Hassan S, Elshafey A, et al. NOD2/CARD15 Polymorphisms (P268S, IVS8 +158 , G908R, L1007fs, R702W) Among Kuwaiti Patients With Crohn’s Disease: A Case-Control Study. Saudi J Gastroenterol (2021) 27:249. doi: 10.4103/sjg.sjg_613_20
18. Cusick MF, Libbey JE, Fujinami RS. Molecular Mimicry as a Mechanism of Autoimmune Disease. Clin Rev Allergy Immunol (2012) 42:102–11. doi: 10.1007/s12016-011-8294-7
19. Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to Two Forms of Glutamate Decarboxylase in Insulin-Dependent Diabetes Mellitus. J Clin Invest (1992) 89:283–92. doi: 10.1172/JCI115573
20. Coppieters KT, Wiberg A, von Herrath MG. Viral Infections and Molecular Mimicry in Type 1 Diabetes. APMIS (2012) 120:941–9. doi: 10.1111/apm.12011
21. Ceccarelli F, Agmon-Levin N, Perricone C. Genetic Factors of Autoimmune Diseases 2017. J Immunol Res (2017) 2017:1–2. doi: 10.1155/2017/2789242
22. Wucherpfennig KW. “HLA and Autoimmunity”. In: Immunogenetics of Autoimmune Disease. Boston, MA: Springer US (2006). p. 1–12. doi: 10.1007/978-0-387-39926-3_1
23. Noble JA, Valdes AM. Genetics of the HLA Region in the Prediction of Type 1 Diabetes. Curr Diabetes Rep (2011) 11:533–42. doi: 10.1007/s11892-011-0223-x
24. Gregersen PK. HLA Class II Polymorphism: Implications for Genetic Susceptibility to Autoimmune Disease. Lab Invest (1989) 61:5–19.
25. Mehra P, Wells AD. Variant to Gene Mapping to Discover New Targets for Immune Tolerance. Front Immunol (2021) 12:633219. doi: 10.3389/fimmu.2021.633219
26. Musette P, Bouaziz JD. B Cell Modulation Strategies in Autoimmune Diseases: New Concepts. Front Immunol (2018) 9:622. doi: 10.3389/fimmu.2018.00622
27. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The Cellular Mechanism of Aire Control of T Cell Tolerance. Immunity (2005) 23:227–39. doi: 10.1016/j.immuni.2005.07.005
28. Liston A, Gray DHD, Lesage S, Fletcher AL, Wilson J, Webster KE, et al. Gene Dosage–limiting Role of Aire in Thymic Expression, Clonal Deletion, and Organ-Specific Autoimmunity. J Exp Med (2004) 200:1015–26. doi: 10.1084/jem.20040581
29. Ramsey C. Aire Deficient Mice Develop Multiple Features of APECED Phenotype and Show Altered Immune Response. Hum Mol Genet (2002) 11:397–409. doi: 10.1093/hmg/11.4.397
30. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17:393–8. doi: 10.1038/ng1297-393
31. Peterson P, Peltonen L. Autoimmune Polyendocrinopathy Syndrome Type 1 (APS1) and AIRE Gene: New Views on Molecular Basis of Autoimmunity. J Autoimmun (2005) 25:49–55. doi: 10.1016/j.jaut.2005.09.022
32. Zhu M-L, Bakhru P, Conley B, Nelson JS, Free M, Martin A, et al. Sex Bias in CNS Autoimmune Disease Mediated by Androgen Control of Autoimmune Regulator. Nat Commun (2016) 7:11350. doi: 10.1038/ncomms11350
33. Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, Serraf A, et al. Estrogen-Mediated Downregulation of AIRE Influences Sexual Dimorphism in Autoimmune Diseases. J Clin Invest (2016) 126:1525–37. doi: 10.1172/JCI81894
34. Chemnitz JM, Riley JL, Frauwirth KA, Braunstein I, Kobayashi SV, Linsley PS, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Blood (2004) 104:2657–7. doi: 10.1182/blood.V104.11.2657.2657
35. Cubas R, Khan Z, Gong Q, Moskalenko M, Xiong H, Ou Q, et al. Autoimmunity Linked Protein Phosphatase PTPN22 as a Target for Cancer Immunotherapy. J Immunother Cancer (2020) 8:e001439. doi: 10.1136/jitc-2020-001439
36. Arechiga AF, Habib T, He Y, Zhang X, Zhang Z, Funk A, et al. Cutting Edge: The PTPN22 Allelic Variant Associated With Autoimmunity Impairs B Cell Signaling. J Immunol (2009) 182:3343–7. doi: 10.4049/jimmunol.0713370
37. Gregersen PK. Gaining Insight Into PTPN22 and Autoimmunity. Nat Genet (2005) 37:1300–2. doi: 10.1038/ng1205-1300
38. Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A Missense Single-Nucleotide Polymorphism in a Gene Encoding a Protein Tyrosine Phosphatase (PTPN22) is Associated With Rheumatoid Arthritis. Am J Hum Genet (2004) 75:330–7. doi: 10.1086/422827
39. Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, et al. The Codon 620 Tryptophan Allele of the Lymphoid Tyrosine Phosphatase (LYP) Gene is a Major Determinant of Graves’ Disease. J Clin Endocrinol Metab (2004) 89:5862–5. doi: 10.1210/jc.2004-1108
40. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JMM, et al. Replication of an Association Between the Lymphoid Tyrosine Phosphatase Locus (LYP/PTPN22) With Type 1 Diabetes, and Evidence for its Role as a General Autoimmunity Locus. Diabetes (2004) 53:3020–3. doi: 10.2337/diabetes.53.11.3020
41. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A Functional Variant of Lymphoid Tyrosine Phosphatase is Associated With Type I Diabetes. Nat Genet (2004) 36:337–8. doi: 10.1038/ng1323
42. Ho WJ, Croessmann S, Lin J, Phyo ZH, Charmsaz S, Danilova L, et al. Systemic Inhibition of PTPN22 Augments Anticancer Immunity. J Clin Invest (2021) 131:e146950. doi: 10.1172/JCI146950
43. Jaillon S, Berthenet K, Garlanda C. Sexual Dimorphism in Innate Immunity. Clin Rev Allergy Immunol (2019) 56:308–21. doi: 10.1007/s12016-017-8648-x
44. Xiong YH, Yuan Z, He L. Effects of Estrogen on CD4+ CD25+ Regulatory T Cell in Peripheral Blood During Pregnancy. Asian Pac J Trop Med (2013) 6:748–52. doi: 10.1016/S1995-7645(13)60131-5
45. Finkelsztejn A, Brooks JBB, Paschoal FM, Fragoso YD. What can We Really Tell Women With Multiple Sclerosis Regarding Pregnancy? A Systematic Review and Meta-Analysis of the Literature. BJOG Int J Obstet Gynaecol (2011) 118:790–7. doi: 10.1111/j.1471-0528.2011.02931.x
46. Kovats S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell Immunol (2015) 294:63–9. doi: 10.1016/j.cellimm.2015.01.018
47. Maglione A, Rolla S, De Mercanti SF, Cutrupi S, Clerico M. The Adaptive Immune System in Multiple Sclerosis: An Estrogen-Mediated Point of View. Cells (2019) 8:1280. doi: 10.3390/cells8101280
48. Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, et al. Induction of Regulatory T Cells by Physiological Level Estrogen. J Cell Physiol (2008) 214:456–64. doi: 10.1002/jcp.21221
49. Smyth A, Oliveira GHM, Lahr BD, Bailey KR, Norby SM, Garovic VD. A Systematic Review and Meta-Analysis of Pregnancy Outcomes in Patients With Systemic Lupus Erythematosus and Lupus Nephritis. Clin J Am Soc Nephrol (2010) 5:2060–8. doi: 10.2215/CJN.00240110
50. Hill L, Jeganathan V, Chinnasamy P, Grimaldi C, Diamond B. Differential Roles of Estrogen Receptors α and β in Control of B-Cell Maturation and Selection. Mol Med (2011) 17:211–20. doi: 10.2119/molmed.2010.00172
51. Williams MM, Spoelstra NS, Arnesen S, O’Neill KI, Christenson JL, Reese J, et al. Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer. Cancer Res (2021) 81:732–46. doi: 10.1158/0008-5472.CAN-20-1200
52. Dou C, Ding N, Zhao C, Hou T, Kang F, Cao Z, et al. Estrogen Deficiency–Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J Bone Miner Res (2018) 33:899–908. doi: 10.1002/jbmr.3364
53. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, et al. Estrogen Receptor-Alpha Promotes Alternative Macrophage Activation During Cutaneous Repair. J Invest Dermatol (2014) 134:2447–57. doi: 10.1038/jid.2014.175
54. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not All Immune Checkpoints Are Created Equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
55. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
56. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders With Early Lethality in Mice Deficient in Ctla-4. Science (80- ) (1995) 270:985–8. doi: 10.1126/science.270.5238.985
57. Nishimura H. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science (80- ) (2001) 291:319–22. doi: 10.1126/science.291.5502.319
58. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
59. Oya Y, Watanabe N, Kobayashi Y, Owada T, Oki M, Ikeda K, et al. Lack of B and T Lymphocyte Attenuator Exacerbates Autoimmune Disorders and Induces Fas-Independent Liver Injury in MRL-Lpr/Lpr Mice. Int Immunol (2011) 23:335–44. doi: 10.1093/intimm/dxr017
60. Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the Immune-Checkpoint VISTA Gene Imparts a Proinflammatory Phenotype With Predisposition to the Development of Autoimmunity. Proc Natl Acad Sci (2014) 111:14846–51. doi: 10.1073/pnas.1407447111
61. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting Edge: TIGIT Has T Cell-Intrinsic Inhibitory Functions. J Immunol (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
62. Zhu J-M, Li B-K, Chen G-M, Feng C-C, Cen H, Fan Y-G, et al. CTLA-4 -1722t/C Polymorphism and Systemic Lupus Erythematosus Susceptibility: A Meta-Analysis Involving Ten Separate Studies. Immunol Invest (2013) 42:91–105. doi: 10.3109/08820139.2012.724752
63. Yu L, Shao M, Zhou T, Xie H, Wang F, Kong J, et al. Association of CTLA-4 (+49 A/G) Polymorphism With Susceptibility to Autoimmune Diseases: A Meta-Analysis With Trial Sequential Analysis. Int Immunopharmacol (2021) 96:107617. doi: 10.1016/j.intimp.2021.107617
64. Chen S, Li Y, Deng C, Li J, Wen X, Wu Z, et al. The Associations Between PD-1, CTLA-4 Gene Polymorphisms and Susceptibility to Ankylosing Spondylitis: A Meta-Analysis and Systemic Review. Rheumatol Int (2016) 36:33–44. doi: 10.1007/s00296-015-3327-9
65. Liu R, Wang X, Chen X, Wang S, Zhang H. TIM-3 Rs1036199 Polymorphism Increases Susceptibility to Autoimmune Diseases: Evidence Based on 4200 Subjects. Biosci Rep (2018) 38:20181235. doi: 10.1042/BSR20181235
66. Gao J, Gai N, Wang L, Liu K, Liu X-H, Wei L-T, et al. Meta-Analysis of Programmed Cell Death 1 Polymorphisms With Systemic Lupus Erythematosus Risk. Oncotarget (2017) 8:36885–97. doi: 10.18632/oncotarget.16378
67. Lee YH, Bae S-C, Kim J-H, Song GG. Meta-Analysis of Genetic Polymorphisms in Programmed Cell Death 1. Z Rheumatol (2015) 74:230–9. doi: 10.1007/s00393-014-1415-y
68. Song G, Bae S-C, Choi S, Ji J, Lee Y. Association Between the CD226 Rs763361 Polymorphism and Susceptibility to Autoimmune Diseases: A Meta-Analysis. Lupus (2012) 21:1522–30. doi: 10.1177/0961203312458840
69. Pawlak-Adamska E, Nowak O, Karabon L, Pokryszko-Dragan A, Partyka A, Tomkiewicz A, et al. PD-1 Gene Polymorphic Variation is Linked With First Symptom of Disease and Severity of Relapsing-Remitting Form of MS. J Neuroimmunol (2017) 305:115–27. doi: 10.1016/j.jneuroim.2017.02.006
70. Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, Mäurer M, et al. A PD-1 Polymorphism is Associated With Disease Progression in Multiple Sclerosis. Ann Neurol (2005) 58:50–7. doi: 10.1002/ana.20514
71. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 Coreceptor Expression and Signal Transduction. Immunol Rev (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
72. Corse E, Allison JP. Cutting Edge: CTLA-4 on Effector T Cells Inhibits In Trans. J Immunol (2012) 189:1123–7. doi: 10.4049/jimmunol.1200695
73. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science (80- ) (2011) 332:600–3. doi: 10.1126/science.1202947
74. Jain N, Nguyen H, Chambers C, Kang J. Dual Function of CTLA-4 in Regulatory T Cells and Conventional T Cells to Prevent Multiorgan Autoimmunity. Proc Natl Acad Sci (2010) 107:1524–8. doi: 10.1073/pnas.0910341107
75. Verma N, Burns SO, Walker LSK, Sansom DM. Immune Deficiency and Autoimmunity in Patients With CTLA-4 (CD152) Mutations. Clin Exp Immunol (2017) 190:1–7. doi: 10.1111/cei.12997
76. Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol Biochem (2018) 47:721–34. doi: 10.1159/000490025
77. Yang M, Sun T, Zhou Y, Wang L, Liu L, Zhang X, et al. The Functional Cytotoxic T Lymphocyte-Associated Protein 4 49G-to-A Genetic Variant and Risk of Pancreatic Cancer. Cancer (2012) 118:4681–6. doi: 10.1002/cncr.27455
78. Roncella S, Laurent S, Fontana V, Ferro P, Franceschini MC, Salvi S, et al. CTLA-4 in Mesothelioma Patients: Tissue Expression, Body Fluid Levels and Possible Relevance as a Prognostic Factor. Cancer Immunol Immunother (2016) 65:909–17. doi: 10.1007/s00262-016-1844-3
79. Tong TML, van der Kooij MK, Speetjens FM, van Erkel AR, van der Meer RW, Lutjeboer J, et al. Combining Hepatic Percutaneous Perfusion With Ipilimumab Plus Nivolumab in Advanced Uveal Melanoma (CHOPIN): Study Protocol for a Phase Ib/Randomized Phase II Trial. Trials (2022) 23:137. doi: 10.1186/s13063-022-06036-y
80. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 Immunotherapy Does Not Deplete Foxp3 Þ Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res (2019) 25:1233–8. doi: 10.1158/1078-0432.CCR-18-0762
81. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-Dependent Cell-Mediated Cytotoxicity of Regulatory T Cells Ex Vivo by Nonclassical Monocytes in Melanoma Patients. Proc Natl Acad Sci USA (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
82. Francisco LM, Sage PT, Sharpe AH. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
83. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J Exp Med (2006) 203:883–95. doi: 10.1084/jem.20051776
84. Colli ML, Hill JLE, Marroquí L, Chaffey J, Dos Santos RS, Leete P, et al. PDL1 is Expressed in the Islets of People With Type 1 Diabetes and is Up-Regulated by Interferons-α and-γ via IRF1 Induction. EBioMedicine (2018) 36:367–75. doi: 10.1016/j.ebiom.2018.09.040
85. Osum KC, Burrack AL, Martinov T, Sahli NL, Mitchell JS, Tucker CG, et al. Interferon-Gamma Drives Programmed Death-Ligand 1 Expression on Islet β Cells to Limit T Cell Function During Autoimmune Diabetes. Sci Rep (2018) 8:8295. doi: 10.1038/s41598-018-26471-9
86. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/jem.20090847
87. Mozaffarian N, Wiedeman AE, Stevens AM. Active Systemic Lupus Erythematosus is Associated With Failure of Antigen-Presenting Cells to Express Programmed Death Ligand-1. Rheumatology (2008) 47:1335–41. doi: 10.1093/rheumatology/ken256
88. Pauken KE, Jenkins MK, Azuma M, Fife BT. PD-1, But Not PD-L1, Expressed by Islet-Reactive CD4+ T Cells Suppresses Infiltration of the Pancreas During Type 1 Diabetes. Diabetes (2013) 62:2859–69. doi: 10.2337/db12-1475
89. Ke Y, Sun D, Jiang G, Kaplan HJ, Shao H. PD-L1 Hi Retinal Pigment Epithelium (RPE) Cells Elicited by Inflammatory Cytokines Induce Regulatory Activity in Uveitogenic T Cells. J Leukoc Biol (2010) 88:1241–9. doi: 10.1189/jlb.0610332
90. Zhao Z, Wang X, Bao X, Ning J, Shang M, Zhang D. Autoimmune Polyendocrine Syndrome Induced by Immune Checkpoint Inhibitors: A Systematic Review. Cancer Immunol Immunother (2021) 70:1527–40. doi: 10.1007/s00262-020-02699-1
91. Schneider S, Potthast S, Komminoth P, Schwegler G, Böhm S. PD-1 Checkpoint Inhibitor Associated Autoimmune Encephalitis. Case Rep Oncol (2017) 10:473–8. doi: 10.1159/000477162
92. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
93. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 Interactions Inhibit Antitumor Immune Responses in a Murine Acute Myeloid Leukemia Model. Blood (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
94. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 Contributes to Immunosuppression and is Associated With Anti-PD-1 Response. Nature (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
95. Jancewicz I, Szarkowska J, Konopinski R, Stachowiak M, Swiatek M, Blachnio K, et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells. Cancers (2021) 13:4148. doi: 10.3390/CANCERS13164148
96. He Q-F, Xu Y, Li J, Huang Z-M, Li X-H, Wang X. CD8+ T-Cell Exhaustion in Cancer: Mechanisms and New Area for Cancer Immunotherapy. Brief Funct Genomics (2019) 18:99–106. doi: 10.1093/bfgp/ely006
97. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on Tumor Cells in the Escape From Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc Natl Acad Sci USA (2002) 99:12293–7. doi: 10.1073/pnas.192461099
98. Yamada A, Arakaki R, Saito M, Kudo Y, Ishimaru N. Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance. Front Immunol (2017) 8:403. doi: 10.3389/fimmu.2017.00403
99. Niederkorn JY. See No Evil, Hear No Evil, Do No Evil: The Lessons of Immune Privilege. Nat Immunol (2006) 7:354–9. doi: 10.1038/ni1328
100. Roths JB, Murphy ED, Eicher EM. A New Mutation, Gld, That Produces Lymphoproliferation and Autoimmunity in C3H/HeJ Mice. J Exp Med (1984) 159:1–20. doi: 10.1084/jem.159.1.1
101. Singer GG, Carrera AC, Marshak-Rothstein A, Martínez-A C, Abbas AK. Apoptosis, Fas and Systemic Autoimmunity: The MRL-Ipr/Ipr Model. Curr Opin Immunol (1994) 6:913–20. doi: 10.1016/0952-7915(94)90013-2
102. Rensing-Ehl A, Völkl S, Speckmann C, Lorenz MR, Ritter J, Janda A, et al. Abnormally Differentiated CD4+ or CD8+ T Cells With Phenotypic and Genetic Features of Double Negative T Cells in Human Fas Deficiency. Blood (2014) 124:851–60. doi: 10.1182/blood-2014-03-564286
103. Saxena A, Yagita H, Donner TW, Hamad ARA. Expansion of FasL-Expressing CD5+ B Cells in Type 1 Diabetes Patients. Front Immunol (2017) 8:402. doi: 10.3389/fimmu.2017.00402
104. Nakano A, Watanabe M, Iida T, Kuroda S, Matsuzuka F, Miyauchi A, et al. Apoptosis-Induced Decrease of Intrathyroidal CD4 + CD25 + Regulatory T Cells in Autoimmune Thyroid Diseases. Thyroid (2007) 17:25–31. doi: 10.1089/thy.2006.0231
105. de Oliveira GLV, Ferreira AF, Gasparotto EPL, Kashima S, Covas DT, Guerreiro CT, et al. Defective Expression of Apoptosis-Related Molecules in Multiple Sclerosis Patients is Normalized Early After Autologous Haematopoietic Stem Cell Transplantation. Clin Exp Immunol (2017) 187:383–98. doi: 10.1111/cei.12895
106. Huang W-X, Huang P, Gomes A, Hillert J. Apoptosis Mediators FasL and TRAIL are Upregulated in Peripheral Blood Mononuclear Cells in MS. Neurology (2000) 55:928–34. doi: 10.1212/WNL.55.7.928
107. Cheng J, Zhou T, Liu C, Shapiro J, Brauer M, Kiefer M, et al. Protection From Fas-Mediated Apoptosis by a Soluble Form of the Fas Molecule. Science (80- ) (1994) 263:1759–62. doi: 10.1126/science.7510905
108. Hohlbaum AM, Moe S, Marshak-Rothstein A. Opposing Effects of Transmembrane and Soluble FAS Ligand Expression on Inflammation and Tumor Cell Survival. J Exp Med (2000) 191:1209–20. doi: 10.1084/jem.191.7.1209
109. Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Harris J, Hoi AY, Mackay F, et al. Associations of Serum Soluble Fas and Fas Ligand (FasL) With Outcomes in Systemic Lupus Erythematosus. Lupus Sci Med (2020) 7:e000375. doi: 10.1136/lupus-2019-000375
110. Vincent FB, Bubicich M, Downie-Doyle S, Mackay F, Morand EF, Rischmueller M. Serum Soluble Fas and Fas Ligand (FasL) in Primary Sjögren’s Syndrome. Clin Exp Rheumatol (2019) 37 Suppl 1:254–6.
111. Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8 + T-Cell Apoptosis Is a Major Component of T-Cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol Res (2018) 6:14–24. doi: 10.1158/2326-6066.CIR-17-0249
112. Wada A, Tada Y, Kawamura K, Takiguchi Y, Tatsumi K, Kuriyama T, et al. The Effects of FasL on Inflammation and Tumor Survival are Dependent on its Expression Levels. Cancer Gene Ther (2007) 14:262–7. doi: 10.1038/sj.cgt.7701008
113. Zhu J, Powis de Tenbossche CG, Cané S, Colau D, van Baren N, Lurquin C, et al. Resistance to Cancer Immunotherapy Mediated by Apoptosis of Tumor-Infiltrating Lymphocytes. Nat Commun (2017) 8:1404. doi: 10.1038/s41467-017-00784-1
114. Seki M, Oomizu S, Sakata K, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 Suppresses the Generation of Th17, Promotes the Induction of Regulatory T Cells, and Regulates Experimental Autoimmune Arthritis. Clin Immunol (2008) 127:78–88. doi: 10.1016/j.clim.2008.01.006
115. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat Immunol (2005) 6:1245–52. doi: 10.1038/ni1271
116. Liu X, Alexiou M, Martin-Orozco N, Chung Y, Nurieva RI, Ma L, et al. Cutting Edge: A Critical Role of B and T Lymphocyte Attenuator in Peripheral T Cell Tolerance Induction. J Immunol (2009) 182:4516–20. doi: 10.4049/jimmunol.0803161
117. Piancone F, Saresella M, Marventano I, La Rosa F, Zoppis M, Agostini S, et al. B Lymphocytes in Multiple Sclerosis: Bregs and BTLA/CD272 Expressing-CD19+ Lymphocytes Modulate Disease Severity. Sci Rep (2016) 6:29699. doi: 10.1038/srep29699
118. Wiedemann A, Lettau M, Weißenberg SY, Stefanski A-L, Schrezenmeier E-V, Rincon-Arevalo H, et al. BTLA Expression and Function Are Impaired on SLE B Cells. Front Immunol (2021) 12:667991. doi: 10.3389/fimmu.2021.667991
119. Oster C, Wilde B, Specker C, Sun M, Kribben A, Witzke O, et al. BTLA Expression on Th1, Th2 and Th17 Effector T-Cells of Patients With Systemic Lupus Erythematosus Is Associated With Active Disease. Int J Mol Sci (2019) 20:4505. doi: 10.3390/ijms20184505
120. Wu H, Tang S, Zhou M, Xue J, Yu Z, Zhu J. Tim-3 Suppresses Autoimmune Hepatitis via the P38/MKP-1 Pathway in Th17 Cells. FEBS Open Bio (2021) 11:1406–16. doi: 10.1002/2211-5463.13148
121. Fuhrman CA, Yeh W-I, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
122. Wang N, Liang S, Jin J, Fang L, Ma Q, Wang X, et al. CD226 Attenuates Treg Suppressive Capacity via CTLA-4 and TIGIT During EAE. Immunol Res (2019) 67:486–96. doi: 10.1007/s12026-019-09112-9
123. Reches A, Ophir Y, Stein N, Kol I, Isaacson B, Charpak Amikam Y, et al. Nectin4 is a Novel TIGIT Ligand Which Combines Checkpoint Inhibition and Tumor Specificity. J Immunother Cancer (2020) 8:e000266. doi: 10.1136/jitc-2019-000266
124. Mouchess ML, Anderson M. “Central Tolerance Induction”. Berlin, Heidelberg: Springer Berlin Heidelberg (2013). p. 69–86. doi: 10.1007/82_2013_321
125. Caramalho Í, Nunes-Cabaço H, Foxall RB, Sousa AE. Regulatory T-Cell Development in the Human Thymus. Front Immunol (2015) 6:395. doi: 10.3389/fimmu.2015.00395
126. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-Mediated Induction of a Potent Regulatory T Cell Population. Nat Immunol (2010) 11:1093–101. doi: 10.1038/ni.1952
127. Zeng H, Zhang R, Jin B, Chen L. Type 1 Regulatory T Cells: A New Mechanism of Peripheral Immune Tolerance. Cell Mol Immunol (2015) 12:566–71. doi: 10.1038/cmi.2015.44
128. Weiner HL. Induction and Mechanism of Action of Transforming Growth Factor-β-Secreting Th3 Regulatory Cells. Immunol Rev (2001) 182:207–14. doi: 10.1034/j.1600-065X.2001.1820117.x
129. Ryba-Stanisławowska M, Sakowska J, Zieliński M, Ławrynowicz U, Trzonkowski P. Regulatory T Cells: The Future of Autoimmune Disease Treatment. Expert Rev Clin Immunol (2019) 15:777–89. doi: 10.1080/1744666X.2019.1620602
130. Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, et al. T Follicular Helper and T Follicular Regulatory Cells Have Different TCR Specificity. Nat Commun (2017) 8:15067. doi: 10.1038/ncomms15067
131. Vaeth M, Müller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular Regulatory T Cells Control Humoral Autoimmunity via NFAT2-Regulated CXCR5 Expression. J Exp Med (2014) 211:545–61. doi: 10.1084/jem.20130604
132. Fisson S, Darrasse-Jèze G, Litvinova E, Septier F, Klatzmann D, Liblau R, et al. Continuous Activation of Autoreactive CD4+ CD25+ Regulatory T Cells in the Steady State. J Exp Med (2003) 198:737–46. doi: 10.1084/jem.20030686
133. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil – CD4+CD25+ T Regulatory Cell Interactions: A Possible New Mechanism of Infectious Tolerance. Immunobiology (2013) 218:455–64. doi: 10.1016/j.imbio.2012.05.029
134. Piekarska K, Urban-Wójciuk Z, Kurkowiak M, Pelikant-Małecka I, Schumacher A, Sakowska J, et al. Mesenchymal Stem Cells Transfer Mitochondria to Allogeneic Tregs in an HLA-Dependent Manner Improving Their Immunosuppressive Activity. Nat Commun (2022) 13:1–20. doi: 10.1038/s41467-022-28338-0
135. Robbins PD, Morelli AE. Regulation of Immune Responses by Extracellular Vesicles. Nat Rev Immunol (2014) 14:195–208. doi: 10.1038/nri3622
136. Sullivan JA, Tomita Y, Jankowska-Gan E, Lema DA, Arvedson MP, Nair A, et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep (2020) 30:1039–51.e5. doi: 10.1016/j.celrep.2019.12.081
137. Barzaghi F, Passerini L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front Pediatr (2021) 9:612760. doi: 10.3389/fped.2021.612760
138. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova J-L, Buist N, et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome is the Human Equivalent of Mouse Scurfy. Nat Genet (2001) 27:18–20. doi: 10.1038/83707
139. de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4 + CD25 Bright Regulatory T Cells Actively Regulate Inflammation in the Joints of Patients With the Remitting Form of Juvenile Idiopathic Arthritis. J Immunol (2004) 172:6435–43. doi: 10.4049/jimmunol.172.10.6435
140. van Amelsfort JMR, Jacobs KMG, Bijlsma JWJ, Lafeber FPJG, Taams LS. CD4+CD25+ Regulatory T Cells in Rheumatoid Arthritis: Differences in the Presence, Phenotype, and Function Between Peripheral Blood and Synovial Fluid. Arthritis Rheum (2004) 50:2775–85. doi: 10.1002/art.20499
141. Slobodin G, Ahmad MS, Rosner I, Peri R, Rozenbaum M, Kessel A, et al. Regulatory T Cells (CD4+CD25brightFoxP3+) Expansion in Systemic Sclerosis Correlates With Disease Activity and Severity. Cell Immunol (2010) 261:77–80. doi: 10.1016/j.cellimm.2009.12.009
142. La Cava A. Tregs in SLE: An Update. Curr Rheumatol Rep (2018) 20:6. doi: 10.1007/s11926-018-0714-8
143. Bonelli M, Savitskaya A, von Dalwigk K, Steiner CW, Aletaha D, Smolen JS, et al. Quantitative and Qualitative Deficiencies of Regulatory T Cells in Patients With Systemic Lupus Erythematosus (SLE). Int Immunol (2008) 20:861–8. doi: 10.1093/intimm/dxn044
144. Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4+CD25+ Regulatory T-Cells in Autoimmune Liver Disease. J Hepatol (2004) 41:31–7. doi: 10.1016/j.jhep.2004.03.008
145. Vitales-Noyola M, Serrano-Somavilla A, Martínez-Hernández R, Sampedro-Nuñez M, Ramos-Levi AM, González-Amaro R, et al. Patients With Autoimmune Thyroiditis Show Diminished Levels and Defective Suppressive Function of Tr1 Regulatory Lymphocytes. J Clin Endocrinol Metab (2018) 103:3359–67. doi: 10.1210/jc.2018-00498
146. Marazuela M, García-López MA, Figueroa-Vega N, de la Fuente H, Alvarado-Sánchez B, Monsiváis-Urenda A, et al. Regulatory T Cells in Human Autoimmune Thyroid Disease. J Clin Endocrinol Metab (2006) 91:3639–46. doi: 10.1210/jc.2005-2337
147. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4+CD25+FoxP3+ T Lymphocytes Fail to Suppress Myelin Basic Protein-Induced Proliferation in Patients With Multiple Sclerosis. J Neuroimmunol (2006) 180:178–84. doi: 10.1016/j.jneuroim.2006.08.003
148. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens J-L, et al. Compromised CD4 + CD25 High Regulatory T-Cell Function in Patients With Relapsing-Remitting Multiple Sclerosis is Correlated With a Reduced Frequency of FOXP3-Positive Cells and Reduced FOXP3 Expression at the Single-Cell Level. Immunology (2008) 123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x
149. Holohan DR, Van Gool F, Bluestone JA. Thymically-Derived Foxp3+ Regulatory T Cells are the Primary Regulators of Type 1 Diabetes in the non-Obese Diabetic Mouse Model. PloS One (2019) 14:e0217728. doi: 10.1371/journal.pone.0217728
150. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial Regulatory T Cells Occupy a Discrete TCR Niche in Human Arthritis and Require Local Signals To Stabilize FOXP3 Protein Expression. J Immunol (2015) 195:5616–24. doi: 10.4049/jimmunol.1500391
151. Yang S, Zhang X, Chen J, Dang J, Liang R, Zeng D, et al. Induced, But Not Natural, Regulatory T Cells Retain Phenotype and Function Following Exposure to Inflamed Synovial Fibroblasts. Sci Adv (2020) 6:eabb0606. doi: 10.1126/sciadv.abb0606
152. Yasuda K, Takeuchi Y, Hirota K. The Pathogenicity of Th17 Cells in Autoimmune Diseases. Semin Immunopathol (2019) 41:283–97. doi: 10.1007/s00281-019-00733-8
153. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn LR. CD161 Defines the Subset of FoxP3+ T Cells Capable of Producing Proinflammatory Cytokines. Blood (2013) 121:2647–58. doi: 10.1182/blood-2012-08-443473
154. Qiu R, Zhou L, Ma Y, Zhou L, Liang T, Shi L, et al. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin Rev Allergy Immunol (2020) 58:52–70. doi: 10.1007/s12016-018-8721-0
155. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
156. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:1–9. doi: 10.1038/srep15179
157. De Oliveira CE, Gasparoto TH, Pinheiro CR, Amôr NG, Nogueira MRS, Kaneno R, et al. CCR5-Dependent Homing of T Regulatory Cells to the Tumor Microenvironment Contributes to Skin Squamous Cell Carcinoma Development. Mol Cancer Ther (2017) 16:2871–80. doi: 10.1158/1535-7163.MCT-17-0341
158. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 Chemokines Within Tumor Microenvironment are Related to Accumulation of Foxp3+ Regulatory T Cells in Gastric Cancer. Int J Cancer (2008) 122:2286–93. doi: 10.1002/ijc.23392
159. Lu L, Ma J, Li Z, Lan Q, Chen M, Liu Y, et al. All-Trans Retinoic Acid Promotes TGF-β-Induced Tregs via Histone Modification But Not DNA Demethylation on Foxp3 Gene Locus. PloS One (2011) 6:e24590. doi: 10.1371/journal.pone.0024590
160. Shitara K, Nishikawa H. Regulatory T Cells: A Potential Target in Cancer Immunotherapy. Ann N Y Acad Sci (2016) 1417:104–15. doi: 10.1111/nyas.13625
161. Chaudhary B, Elkord E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines (2016) 4:1–25. doi: 10.3390/vaccines4030028
162. Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of Tolerance by IL-10-Treated Dendritic Cells. J Immunol (1997) 159:4772–80.
163. Lazarova M, Steinle A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front Immunol (2019) 10:2689. doi: 10.3389/fimmu.2019.02689
164. Lindqvist CA, Christiansson LH, Simonsson B, Enblad G, Olsson-Strömberg U, Loskog ASI. T Regulatory Cells Control T-Cell Proliferation Partly by the Release of Soluble CD25 in Patients With B-Cell Malignancies. Immunology (2010) 131:371–6. doi: 10.1111/j.1365-2567.2010.03308.x
165. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive Plasticity of IL-10+ and IL-35+ Treg Cells Cooperatively Promotes Tumor T Cell Exhaustion. Nat Immunol (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
166. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 Limits Anti-Tumor Immunity. Immunity (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
167. Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Kim Lyerly H, et al. Depletion of Human Regulatory T Cells Specifically Enhances Antigen-Specific Immune Responses to Cancer Vaccines. Blood (2008) 112:610–8. doi: 10.1182/blood-2008-01-135319
168. Wang Y, Liu J, Burrows PD, Wang J-Y. “B Cell Development and Maturation”. Singapore: Springer Singapore (2020). p. 1–22. doi: 10.1007/978-981-15-3532-1_1
169. Nemazee D. Mechanisms of Central Tolerance for B Cells. Nat Rev Immunol (2017) 17:281–94. doi: 10.1038/nri.2017.19
170. Zhao Y, Shen M, Feng Y, He R, Xu X, Xie Y, et al. Regulatory B Cells Induced by Pancreatic Cancer Cell-Derived Interleukin-18 Promote Immune Tolerance via the PD-1/PD-L1 Pathway. Oncotarget (2018) 9:14803–14. doi: 10.18632/oncotarget.22976
171. Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, et al. Regulatory B Cells are Identified by Expression of TIM-1 and can be Induced Through TIM-1 Ligation to Promote Tolerance in Mice. J Clin Invest (2011) 121:3645–56. doi: 10.1172/JCI46274
172. Baba Y, Saito Y, Kotetsu Y. Heterogeneous Subsets of B-Lineage Regulatory Cells (Breg Cells). Int Immunol (2020) 32:155–62. doi: 10.1093/intimm/dxz068
173. Chen Y, Li C, Lu Y, Zhuang H, Gu W, Liu B, et al. IL-10-Producing CD1dhiCD5+ Regulatory B Cells May Play a Critical Role in Modulating Immune Homeostasis in Silicosis Patients. Front Immunol (2017) 8:110. doi: 10.3389/fimmu.2017.00110
174. Noh J, Lee JH, Noh G, Bang SY, Kim HS, Choi WS, et al. Characterisation of Allergen-Specific Responses of IL-10-Producing Regulatory B Cells (Br1) in Cow Milk Allergy. Cell Immunol (2010) 264:143–9. doi: 10.1016/j.cellimm.2010.05.013
175. Iwata Y, Matsushita T, Horikawa M, DiLillo DJ, Yanaba K, Venturi GM, et al. Characterization of a Rare IL-10–Competent B-Cell Subset in Humans That Parallels Mouse Regulatory B10 Cells. Blood (2011) 117:530–41. doi: 10.1182/blood-2010-07-294249
176. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals But Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity (2010) 32:129–40. doi: 10.1016/j.immuni.2009.11.009
177. Wang W, Yuan X, Chen H, Xie G, Ma Y, Zheng Y, et al. CD19+CD24hiCD38hiBregs Involved in Downregulate Helper T Cells and Upregulate Regulatory T Cells in Gastric Cancer. Oncotarget (2015) 6:33486–99. doi: 10.18632/oncotarget.5588
178. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B Cells Maintain Regulatory T Cells While Limiting TH1 and TH17 Differentiation. Sci Transl Med (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
179. Van De Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 Production is Confined to Human IL-10-Producing Regulatory B Cells That Suppress Antigen-Specific Immune Responses. J Allergy Clin Immunol (2013) 131:1204–12. doi: 10.1016/j.jaci.2013.01.014
180. Díaz-Alderete A, Crispin JC, Inés Vargas-Rojas M, Alcocer-Varela J. IL-10 Production in B Cells is Confined to CD154 + Cells in Patients With Systemic Lupus Erythematosus. J Autoimmun (2004) 23:379–83. doi: 10.1016/j.jaut.2004.10.001
181. Xiao X, Lao X-M, Chen M-M, Liu R-X, Wei Y, Ouyang F-Z, et al. PD-1hi Identifies a Novel Regulatory B-Cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov (2016) 6:546–59. doi: 10.1158/2159-8290.CD-15-1408
182. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity (2014) 41:1040–51. doi: 10.1016/j.immuni.2014.10.016
183. Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E, et al. Human CD19+CD25high B Regulatory Cells Suppress Proliferation of CD4+ T Cells and Enhance Foxp3 and CTLA-4 Expression in T-Regulatory Cells. Autoimmun Rev (2012) 11:670–7. doi: 10.1016/j.autrev.2011.11.018
184. Matsushita T. Regulatory and Effector B Cells: Friends or Foes? J Dermatol Sci (2019) 93:2–7. doi: 10.1016/j.jdermsci.2018.11.008
185. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B Cells Control T-Cell Autoimmunity Through IL-21-Dependent Cognate Interactions. Nature (2012) 491:264–8. doi: 10.1038/nature11501
186. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of Arthritis by Interleukin 10–Producing B Cells. J Exp Med (2003) 197:489–501. doi: 10.1084/jem.20021293
187. Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, et al. Regulatory B Cells (B10 Cells) Have a Suppressive Role in Murine Lupus: CD19 and B10 Cell Deficiency Exacerbates Systemic Autoimmunity. J Immunol (2010) 184:4801–9. doi: 10.4049/jimmunol.0902385
188. Matsushita T, Yanaba K, Bouaziz J-D, Fujimoto M, Tedder TF. Regulatory B Cells Inhibit EAE Initiation in Mice While Other B Cells Promote Disease Progression. J Clin Invest (2008) 118:3420–30. doi: 10.1172/JCI36030
189. Lo-Man R. Regulatory B Cells Control Dendritic Cell Functions. Immunotherapy (2011) 3:19–20. doi: 10.2217/imt.11.34
190. Yuen GJ, Demissie E, Pillai S. B Lymphocytes and Cancer: A Love–Hate Relationship. Trends Cancer (2016) 2:747–57. doi: 10.1016/j.trecan.2016.10.010
191. Zhou J, Min Z, Zhang D, Wang W, Marincola F, Wang X. Enhanced Frequency and Potential Mechanism of B Regulatory Cells in Patients With Lung Cancer. J Transl Med (2014) 12:1–11. doi: 10.1186/s12967-014-0304-0
192. Murakami Y, Saito H, Shimizu S, Kono Y, Shishido Y, Miyatani K, et al. Increased Regulatory B Cells are Involved in Immune Evasion in Patients With Gastric Cancer. Sci Rep (2019) 9:1–9. doi: 10.1038/s41598-019-49581-4
193. Ishigami E, Sakakibara M, Sakakibara J, Masuda T, Fujimoto H, Hayama S, et al. Coexistence of Regulatory B Cells and Regulatory T Cells in Tumor-Infiltrating Lymphocyte Aggregates is a Prognostic Factor in Patients With Breast Cancer. Breast Cancer (2019) 26:180–9. doi: 10.1007/s12282-018-0910-4
194. Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, et al. Multiparametric Profiling of Non–Small-Cell Lung Cancers Reveals Distinct Immunophenotypes. JCI Insight (2016) 1:1–18. doi: 10.1172/jci.insight.89014
195. Zhang Y, Morgan R, Chen C, Cai Y, Clark E, Khan WN, et al. Mammary-Tumor-Educated B Cells Acquire LAP/TGF-β and PD-L1 Expression and Suppress Anti-Tumor Immune Responses. Int Immunol (2016) 28:423–33. doi: 10.1093/intimm/dxw007
196. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TFE, et al. Interleukin 21–Induced Granzyme B–Expressing B Cells Infiltrate Tumors and Regulate T Cells. Cancer Res (2013) 73:2468–79. doi: 10.1158/0008-5472.CAN-12-3450
197. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, et al. Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely Upon Education From Tumor-Associated B Cells. Cancer Res (2015) 75:3456–65. doi: 10.1158/0008-5472.CAN-14-3077
198. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4 + T Cells to T-Regulatory Cells. Cancer Res (2011) 71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316
199. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discovery (2016) 6:247–55. doi: 10.1158/2159-8290.CD-15-0843
200. Guan H, Lan Y, Wan Y, Wang Q, Wang C, Xu L, et al. PD-L1 Mediated the Differentiation of Tumor-Infiltrating CD19 + B Lymphocytes and T Cells in Invasive Breast Cancer. Oncoimmunology (2016) 5:e1075112. doi: 10.1080/2162402X.2015.1075112
201. Klinker MW, Reed TJ, Fox DA, Lundy SK. Interleukin-5 Supports the Expansion of Fas Ligand-Expressing Killer B Cells That Induce Antigen-Specific Apoptosis of CD4+ T Cells and Secrete Interleukin-10. PloS One (2013) 8:e70131. doi: 10.1371/journal.pone.0070131
202. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory Effects of B Cells on Antitumor Immunity. Cancer Res (2006) 66:7741–7. doi: 10.1158/0008-5472.CAN-05-3766
203. Shao Y, Lo CM, Ling CC, Liu XB, Ng KT-P, Chu ACY, et al. Regulatory B Cells Accelerate Hepatocellular Carcinoma Progression via CD40/CD154 Signaling Pathway. Cancer Lett (2014) 355:264–72. doi: 10.1016/j.canlet.2014.09.026
204. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
205. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-Dose Granulocyte-Macrophage Colony-Stimulating Factor-Producing Vaccines Impair the Immune Response Through the Recruitment of Myeloid Suppressor Cells. Cancer Res (2004) 64:6337–43. doi: 10.1158/0008-5472.CAN-04-0757
206. Guo Q, Lv Z, Fu Q, Jiang C, Liu Y, Lai L, et al. IFN-γ Producing T Cells Contribute to the Increase of Myeloid Derived Suppressor Cells in Tumor-Bearing Mice After Cyclophosphamide Treatment. Int Immunopharmacol (2012) 12:425–32. doi: 10.1016/j.intimp.2011.12.016
207. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor Necrosis Factor-α Blocks Differentiation and Enhances Suppressive Activity of Immature Myeloid Cells During Chronic Inflammation. Immunity (2013) 38:541–54. doi: 10.1016/j.immuni.2013.02.007
208. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 Promotes Tumor Progression by Inducing Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
209. Nakamura K, Kassem S, Cleynen A, Avet-Loiseau H, Martinet L, Smyth Correspondence MJ. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell (2018) 33:634–48.e5. doi: 10.1016/j.ccell.2018.02.007
210. Li C, Liu T, Bazhin AV, Yang Y. The Sabotaging Role of Myeloid Cells in Anti-Angiogenic Therapy: Coordination of Angiogenesis and Immune Suppression by Hypoxia. J Cell Physiol (2017) 232:2312–22. doi: 10.1002/jcp.25726
211. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong W-C, et al. Mesenchymal Transition and Dissemination of Cancer Cells Is Driven by Myeloid-Derived Suppressor Cells Infiltrating the Primary Tumor. PloS Biol (2011) 9:e1001162. doi: 10.1371/journal.pbio.1001162
212. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of Myeloid-Derived Suppressor Cells Impair T Cell Responses Through Independent Nitric Oxide-Related Pathways. Int J Cancer (2014) 134:2853–64. doi: 10.1002/ijc.28622
213. Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, et al. Nitric Oxide Mediated Inhibition of Antigen Presentation From DCs to CD4+ T Cells in Cancer and Measurement of STAT1 Nitration. Sci Rep (2017) 7:15424. doi: 10.1038/s41598-017-14970-0
214. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-Specific Inhibition of CD8 + T Cell Response by Immature Myeloid Cells in Cancer Is Mediated by Reactive Oxygen Species. J Immunol (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
215. Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive Oxygen Species in the Immune System. Int Rev Immunol (2013) 32:249–70. doi: 10.3109/08830185.2012.755176
216. Chen X, Song M, Zhang B, Zhang Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev (2016) 2016:11–6. doi: 10.1155/2016/1580967
217. Rodríguez PC, Ochoa AC. Arginine Regulation by Myeloid Derived Suppressor Cells and Tolerance in Cancer: Mechanisms and Therapeutic Perspectives. Immunol Rev (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
218. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res (2010) 70:68–77. doi: 10.1158/0008-5472.CAN-09-2587
219. Baniyash M. TCR ζ-Chain Downregulation: Curtailing an Excessive Inflammatory Immune Response. Nat Rev Immunol (2004) 4:675–87. doi: 10.1038/nri1434
220. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses Through IDO Expression and Correlate With Lymph Node Metastasis in Patients With Breast Cancer. J Immunol (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
221. Platten M, Wick W, Van den Eynde BJ. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
222. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
223. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered Recognition of Antigen is a Mechanism of CD8+ T Cell Tolerance in Cancer. Nat Med (2007) 13:828–35. doi: 10.1038/nm1609
224. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting Antitumor Responses of T Lymphocytes Infiltrating Human Prostate Cancers. J Exp Med (2005) 201:1257–68. doi: 10.1084/jem.20042028
225. Lu T, Gabrilovich DI. Molecular Pathways: Tumor-Infiltrating Myeloid Cells and Reactive Oxygen Species in Regulation of Tumor Microenvironment: Figure 1. Clin Cancer Res (2012) 18:4877–82. doi: 10.1158/1078-0432.CCR-11-2939
226. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 Upregulation on Myeloid-Derived Suppressor Cells via TGF-β-mTOR-HIF-1 Signaling in Patients With non-Small Cell Lung Cancer. Oncoimmunology (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
227. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients With Ovarian Cancer. Cancer Res (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
228. Umansky V, Shevchenko I, Bazhin AV, Utikal J. Extracellular Adenosine Metabolism in Immune Cells in Melanoma. Cancer Immunol Immunother (2014) 63:1073–80. doi: 10.1007/s00262-014-1553-8
229. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b Adenosine Receptor Reduces Tumor Growth and Immune Suppression Mediated by Myeloid-Derived Suppressor Cells in a Mouse Model of Melanoma. Neoplasia (2013) 15:1400–IN10. doi: 10.1593/neo.131748
230. Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang G-J. Up-Regulated Myeloid-Derived Suppressor Cell Contributes to Hepatocellular Carcinoma Development by Impairing Dendritic Cell Function. Scand J Gastroenterol (2011) 46:156–64. doi: 10.3109/00365521.2010.516450
231. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells Through Membrane-Bound TGF-β1. J Immunol (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
232. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor Cells Convert Immature Myeloid Dendritic Cells Into TGF-β–Secreting Cells Inducing CD4+CD25+ Regulatory T Cell Proliferation. J Exp Med (2005) 202:919–29. doi: 10.1084/jem.20050463
233. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β Induces M2-Like Macrophage Polarization via SNAIL-Mediated Suppression of a Pro-Inflammatory Phenotype. Oncotarget (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
234. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” Versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
235. Chae YK, Chang S, Ko T, Anker J, Agte S, Iams W, et al. Epithelial-Mesenchymal Transition (EMT) Signature is Inversely Associated With T-Cell Infiltration in Non-Small Cell Lung Cancer (NSCLC). Sci Rep (2018) 8:2–9. doi: 10.1038/s41598-018-21061-1
236. Izhak L, Wildbaum G, Uri W, Shaked Y, Alami J, Dumont D, et al. Predominant Expression of CCL2 at the Tumor Site of Prostate Cancer Patients Directs a Selective Loss of Immunological Tolerance to CCL2 That Could Be Amplified in a Beneficial Manner. J Immunol (2010) 184:1092–101. doi: 10.4049/jimmunol.0902725
237. Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5 + Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res (2018) 78:157–67. doi: 10.1158/0008-5472.CAN-17-0348
238. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on CD4 + and CD8 + T Cells. J Immunol (2009) 183:937–44. doi: 10.4049/jimmunol.0804253
239. Melero-Jerez C, Ortega MC, Moliné-Velázquez V, Clemente D. Myeloid Derived Suppressor Cells in Inflammatory Conditions of the Central Nervous System. Biochim Biophys Acta - Mol Basis Dis (2016) 1862:368–80. doi: 10.1016/j.bbadis.2015.10.015
240. Moliné-Velázquez V, Cuervo H, Vila-Del Sol V, Ortega MC, Clemente D, De Castro F. Myeloid-Derived Suppressor Cells Limit the Inflammation by Promoting T Lymphocyte Apoptosis in the Spinal Cord of a Murine Model of Multiple Sclerosis. Brain Pathol (2011) 21:678–91. doi: 10.1111/j.1750-3639.2011.00495.x
241. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone Marrow Myeloid-Derived Suppressor Cells (MDSCs) Inhibit Graft-Versus-Host Disease (GVHD) via an Arginase-1-Dependent Mechanism That is Up-Regulated by Interleukin-13. Blood (2010) 116:5738–47. doi: 10.1182/blood-2010-06-287839
242. Angulo I, de las Heras FG, García-Bustos JF, Gargallo D, Muñoz-Fernández MA, Fresno M. Nitric Oxide-Producing CD11b+Ly-6g(Gr-1)+CD31(ER-MP12)+cells in the Spleen of Cyclophosphamide–Treated Mice: Implications for T-Cell Responses in Immunosuppressed Mice. Blood (2000) 95:212–20. doi: 10.1182/blood.v95.1.212
243. Stüve O, Youssef S, Slavin AJ, King CL, Patarroyo JC, Hirschberg DL, et al. The Role of the MHC Class II Transactivator in Class II Expression and Antigen Presentation by Astrocytes and in Susceptibility to Central Nervous System Autoimmune Disease. J Immunol (2002) 169:6720–32. doi: 10.4049/jimmunol.169.12.6720
244. Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, et al. Crucial Role of Granulocytic Myeloid-Derived Suppressor Cells in the Regulation of Central Nervous System Autoimmune Disease. J Immunol (2012) 188:1136–46. doi: 10.4049/jimmunol.1101816
245. Grohová A, Dáňová K, Adkins I, Šumník Z, Petruželková L, Obermannová B, et al. Myeloid - Derived Suppressor Cells in Type 1 Diabetes are an Expanded Population Exhibiting Diverse T-Cell Suppressor Mechanisms. PloS One (2020) 15:e0242092. doi: 10.1371/journal.pone.0242092
246. Jiao Z, Hua S, Wang W, Wang H, Gao J, Wang X. Increased Circulating Myeloid-Derived Suppressor Cells Correlated Negatively With Th17 Cells in Patients With Rheumatoid Arthritis. Scand J Rheumatol (2013) 42:85–90. doi: 10.3109/03009742.2012.716450
247. Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu Z-G, et al. Arginase-1–Dependent Promotion of T H 17 Differentiation and Disease Progression by MDSCs in Systemic Lupus Erythematosus. Sci Transl Med (2016) 8:331ra40–331ra40. doi: 10.1126/scitranslmed.aae0482
248. Yin B, Ma G, Yen C-Y, Zhou Z, Wang GX, Divino CM, et al. Myeloid-Derived Suppressor Cells Prevent Type 1 Diabetes in Murine Models. J Immunol (2010) 185:5828–34. doi: 10.4049/jimmunol.0903636
249. Cassetta L, Cassol E, Poli G. Macrophage Polarization in Health and Disease. Sci World J (2011) 11:2391–402. doi: 10.1100/2011/213962
250. Leung SY, Wong MP, Chung LP, Chan ASY, Yuen ST. Monocyte Chemoattractant Protein-1 Expression and Macrophage Infiltration in Gliomas. Acta Neuropathol (1997) 93:518–27. doi: 10.1007/s004010050647
251. Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, et al. Significance of Macrophage Chemoattractant Protein-1 in Macrophage Recruitment, Angiogenesis, and Survival in Human Breast Cancer. Clin Cancer Res (2000) 6:3282 LP – 89.
252. Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Strom SRB, Burdick MD, et al. Macrophage Infiltration in Human non-Small-Cell Lung Cancer: The Role of CC Chemokines. Cancer Immunol Immunother (2000) 49:63–70. doi: 10.1007/s002620050603
253. McClellan JL, Mark Davis J, Steiner JL, Enos RT, Jung SH, Carson JA, et al. Inking Tumor-Associated Macrophages, Inflammation, and Intestinal Tumorigenesis: Role of MCP-1. Am J Physiol - Gastrointest Liver Physiol (2012) 303:G1087–95. doi: 10.1152/ajpgi.00252.2012
254. Rhee I. Diverse Macrophages Polarization in Tumor Microenvironment. Arch Pharm Res (2016) 39:1588–96. doi: 10.1007/s12272-016-0820-y
255. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J Exp Med (2015) 212:1043–59. doi: 10.1084/jem.20141836
256. Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang LCS, et al. Monocyte Chemoattractant Protein-1 Blockade Inhibits Lung Cancer Tumor Growth by Altering Macrophage Phenotype and Activating CD8+ Cells. Am J Respir Cell Mol Biol (2011) 44:230–7. doi: 10.1165/rcmb.2010-0080OC
257. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Phase 1b Study Targeting Tumour Associated Macrophages With CCR2 Inhibition Plus FOLFIRINOX in Locally Advanced and Borderline Resectable Pancreatic Cancer HHS Public Access. Lancet Oncol (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4.Phase
258. Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High Systemic and Tumor-Associated IL-8 Correlates With Reduced Clinical Benefit of PD-L1 Blockade. Nat Med (2020) 26:693–8. doi: 10.1038/s41591-020-0860-1
259. Ara T, DeClerck YA. Interleukin-6 in Bone Metastasis and Cancer Progression. Eur J Cancer (2010) 46:1223–31. doi: 10.1016/j.ejca.2010.02.026
260. Zhao S, Wu D, Wu P, Wang Z, Huang J, Gao JX. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PloS One (2015) 10:1–15. doi: 10.1371/journal.pone.0139598
261. Mittal SK, Roche PA. Suppression of Antigen Presentation by IL-10. Curr Opin Immunol (2015) 34:22–7. doi: 10.1016/j.coi.2014.12.009
262. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident Macrophages of Pancreatic Islets Have a Seminal Role in the Initiation of Autoimmune Diabetes of NOD Mice. Proc Natl Acad Sci USA (2017) 114:E10418–27. doi: 10.1073/pnas.1713543114
263. Jiang Z, Jiang JX, Zhang G-X. Macrophages: A Double-Edged Sword in Experimental Autoimmune Encephalomyelitis. Immunol Lett (2014) 160:17–22. doi: 10.1016/j.imlet.2014.03.006
264. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of Macrophage Polarization in Autoimmunity. Immunology (2018) 154:186–95. doi: 10.1111/imm.12910
265. Weitz JR, Jacques-Silva C, Qadir MMF, Umland O, Pereira E, Qureshi F, et al. Secretory Functions of Macrophages in the Human Pancreatic Islet Are Regulated by Endogenous Purinergic Signaling. Diabetes (2020) 69:1206–18. doi: 10.2337/db19-0687
266. Wong PF, Wei W, Gupta S, Smithy JW, Zelterman D, Kluger HM, et al. Multiplex Quantitative Analysis of Cancer-Associated Fibroblasts and Immunotherapy Outcome in Metastatic Melanoma. J Immunother Cancer (2019) 7:1–10. doi: 10.1186/s40425-019-0675-0
267. Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage Polarization Modulates Development of Systemic Lupus Erythematosus. Cell Physiol Biochem (2015) 37:1279–88. doi: 10.1159/000430251
268. Parsa R, Andresen P, Gillett A, Mia S, Zhang X-M, Mayans S, et al. Adoptive Transfer of Immunomodulatory M2 Macrophages Prevents Type 1 Diabetes in NOD Mice. Diabetes (2012) 61:2881–92. doi: 10.2337/db11-1635
269. Vallée A, Lecarpentier Y. TGF-β in Fibrosis by Acting as a Conductor for Contractile Properties of Myofibroblasts. Cell Biosci (2019) 9:1–15. doi: 10.1186/s13578-019-0362-3
270. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-Associated Fibroblasts Drive the Progression of Metastasis Through Both Paracrine and Mechanical Pressure on Cancer Tissue. Mol Cancer Res (2012) 10:1403–18. doi: 10.1158/1541-7786.MCR-12-0307
271. Yoshida GJ. Regulation of Heterogeneous Cancer-Associated Fibroblasts: The Molecular Pathology of Activated Signaling Pathways. J Exp Clin Cancer Res (2020) 39:1–15. doi: 10.1186/s13046-020-01611-0
272. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic Switch Drives the Conversion of Fibroblasts Into Proinvasive Cancer-Associated Fibroblasts. Nat Commun (2015) 6:10204. doi: 10.1038/ncomms10204
273. Ping Q, Yan R, Cheng X, Wang W, Zhong Y, Hou Z, et al. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene Ther (2021) 28:984–99. doi: 10.1038/s41417-021-00318-4
274. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front Cell Dev Biol (2019) 7:60. doi: 10.3389/fcell.2019.00060
275. Hao Y, Baker D, Dijke PT. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci (2019) 20:2767. doi: 10.3390/ijms20112767
276. Wei P, Xie Y, Abel PW, Huang Y, Ma Q, Li L, et al. Transforming Growth Factor (TGF)-β1-Induced miR-133a Inhibits Myofibroblast Differentiation and Pulmonary Fibrosis. Cell Death Dis (2019) 10:670. doi: 10.1038/s41419-019-1873-x
277. Butcher DT, Alliston T, Weaver VM. A Tense Situation: Forcing Tumour Progression. Nat Rev Cancer (2009) 9:108–22. doi: 10.1038/nrc2544
278. Jalili RB, Forouzandeh F, Rezakhanlou AM, Hartwell R, Medina A, Warnock GL, et al. Local Expression of Indoleamine 2,3 Dioxygenase in Syngeneic Fibroblasts Significantly Prolongs Survival of an Engineered Three-Dimensional Islet Allograft. Diabetes (2010) 59:2219–27. doi: 10.2337/db09-1560
279. Jalili RB, Zhang Y, Hosseini-Tabatabaei A, Kilani RT, Khosravi Maharlooei M, Li Y, et al. Fibroblast Cell-Based Therapy for Experimental Autoimmune Diabetes. PloS One (2016) 11:1–16. doi: 10.1371/journal.pone.0146970
280. Khosravi-Maharlooei M, Pakyari M, Jalili RB, Salimi-Elizei S, Lai JCY, Poormasjedi-Meibod M, et al. Tolerogenic Effect of Mouse Fibroblasts on Dendritic Cells. Immunology (2016) 148:22–33. doi: 10.1111/imm.12584
281. Kalluri R, Weinberg RA. The Basics of Epithelial-Mesenchymal Transition. J Clin Invest (2009) 119:1420–8. doi: 10.1172/JCI39104
282. Polyak K, Weinberg RA. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat Rev Cancer (2009) 9:265–73. doi: 10.1038/nrc2620
283. Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-Mesenchymal Transition: A Hallmark in Metastasis Formation Linking Circulating Tumor Cells and Cancer Stem Cells. Pathobiology (2012) 79:195–208. doi: 10.1159/000337106
284. Weinberg RA. The Biology of Cancer. New York: W.W. Norton & Company (2013). doi: 10.1201/9780429258794
285. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells With Properties of Stem Cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
286. Ye X, Weinberg RA. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol (2015) 25:675–86. doi: 10.1016/j.tcb.2015.07.012
287. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y-H, Byers LA, et al. Metastasis is Regulated via microRNA-200/ZEB1 Axis Control of Tumour Cell PD-L1 Expression and Intratumoral Immunosuppression. Nat Commun (2014) 5:5241. doi: 10.1038/ncomms6241
288. Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T, et al. Bidirectional Crosstalk Between PD-L1 Expression and Epithelial to Mesenchymal Transition: Significance in Claudin-Low Breast Cancer Cells. Mol Cancer (2015) 14:149. doi: 10.1186/s12943-015-0421-2
289. Ye L-Y, Chen W, Bai X-L, Xu X-Y, Zhang Q, Xia X-F, et al. Hypoxia-Induced Epithelial-To-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res (2016) 76:818–30. doi: 10.1158/0008-5472.CAN-15-0977
290. Huergo-Zapico L, Parodi M, Cantoni C, Lavarello C, Fernández-Martínez JL, Petretto A, et al. NK-Cell Editing Mediates Epithelial-To-Mesenchymal Transition via Phenotypic and Proteomic Changes in Melanoma Cell Lines. Cancer Res (2018) 78:3913–25. doi: 10.1158/0008-5472.CAN-17-1891
291. Pang M-F, Georgoudaki A-M, Lambut L, Johansson J, Tabor V, Hagikura K, et al. TGF-β1-Induced EMT Promotes Targeted Migration of Breast Cancer Cells Through the Lymphatic System by the Activation of CCR7/CCL21-Mediated Chemotaxis. Oncogene (2016) 35:748–60. doi: 10.1038/onc.2015.133
292. Yu Y, Luo W, Yang Z-J, Chi J-R, Li Y-R, Ding Y, et al. miR-190 Suppresses Breast Cancer Metastasis by Regulation of TGF-β-Induced Epithelial–Mesenchymal Transition. Mol Cancer (2018) 17:70. doi: 10.1186/s12943-018-0818-9
293. Zu L, Xue Y, Wang J, Fu Y, Wang X, Xiao G, et al. The Feedback Loop Between miR-124 and TGF-β Pathway Plays a Significant Role in non-Small Cell Lung Cancer Metastasis. Carcinogenesis (2016) 37:333–43. doi: 10.1093/carcin/bgw011
294. Kim BN, Ahn DH, Kang N, Yeo CD, Kim YK, Lee KY, et al. TGF-β Induced EMT and Stemness Characteristics are Associated With Epigenetic Regulation in Lung Cancer. Sci Rep (2020) 10:10597. doi: 10.1038/s41598-020-67325-7
295. Zhu SM, Park YR, Seo SY, Kim IH, Lee ST, Kim SW. Parthenolide Inhibits Transforming Growth Factor β1-Induced Epithelial-Mesenchymal Transition in Colorectal Cancer Cells. Intest Res (2019) 17:527–36. doi: 10.5217/ir.2019.00031
296. Lu C, Yang Z, Yu D, Lin J, Cai W. RUNX1 Regulates TGF-β Induced Migration and EMT in Colorectal Cancer. Pathol Res Pract (2020) 216:153142. doi: 10.1016/j.prp.2020.153142
297. Lou Y, Diao L, Cuentas ERP, Denning WL, Chen L, Fan YH, et al. Epithelial–Mesenchymal Transition Is Associated With a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res (2016) 22:3630–42. doi: 10.1158/1078-0432.CCR-15-1434
298. Imai D, Yoshizumi T, Okano S, Itoh S, Ikegami T, Harada N, et al. IFN-γ Promotes Epithelial-Mesenchymal Transition and the Expression of PD-L1 in Pancreatic Cancer. J Surg Res (2019) 240:115–23. doi: 10.1016/j.jss.2019.02.038
299. Di Gregorio J, Robuffo I, Spalletta S, Giambuzzi G, De Iuliis V, Toniato E, et al. The Epithelial-To-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front Cell Dev Biol (2020) 8:607483. doi: 10.3389/fcell.2020.607483
300. Sisto M, Ribatti D, Lisi S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in Tgfβ-Dependent EMT. Biomolecules (2021) 11:1–26. doi: 10.3390/biom11020310
301. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence That Fibroblasts Derive From Epithelium During Tissue Fibrosis. J Clin Invest (2002) 110:341–50. doi: 10.1172/JCI200215518
302. López-Novoa JM, Nieto MA. Inflammation and EMT: An Alliance Towards Organ Fibrosis and Cancer Progression. EMBO Mol Med (2009) 1:303–14. doi: 10.1002/emmm.200900043
303. Moretti L, Stalfort J, Barker TH, Abebayehu D. The Interplay of Fibroblasts, the Extracellular Matrix, and Inflammation in Scar Formation. J Biol Chem (2022) 298:101530. doi: 10.1016/j.jbc.2021.101530
304. Wynn TA, Ramalingam TR. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
305. Connolly MK, Bedrosian AS, Mallen-St. Clair J, Mitchell AP, Ibrahim J, Stroud A, et al. In Liver Fibrosis, Dendritic Cells Govern Hepatic Inflammation in Mice via TNF-α. J Clin Invest (2009) 119:3213–25. doi: 10.1172/JCI37581
306. Zhang J, Wang D, Wang L, Wang S, Roden AC, Zhao H, et al. Profibrotic Effect of IL-17A and Elevated IL-17RA in Idiopathic Pulmonary Fibrosis and Rheumatoid Arthritis-Associated Lung Disease Support a Direct Role for IL-17a/IL-17RA in Human Fibrotic Interstitial Lung Disease. Am J Physiol Cell Mol Physiol (2019) 316:L487–97. doi: 10.1152/ajplung.00301.2018
307. Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, et al. The RNA-Editing Enzyme ADAR1 Controls Innate Immune Responses to RNA. Cell Rep (2014) 9:1482–94. doi: 10.1016/j.celrep.2014.10.041
308. Kurkowiak M, Arcimowicz Ł, Chruściel E, Urban-Wójciuk Z, Papak I, Keegan L, et al. The Effects of RNA Editing in Cancer Tissue at Different Stages in Carcinogenesis. RNA Biol (2021) 18:1524–39. doi: 10.1080/15476286.2021.1877024
309. Song B, Shiromoto Y, Minakuchi M, Nishikura K. The Role of RNA Editing Enzyme ADAR1 in Human Disease. Wiley Interdiscip Rev RNA (2022) 13:1–24. doi: 10.1002/wrna.1665
310. Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, et al. RNA Editing Derived Epitopes Function as Cancer Antigens to Elicit Immune Responses. Nat Commun (2018) 9:3919. doi: 10.1038/s41467-018-06405-9
311. Asaoka M, Ishikawa T, Takabe K, Patnaik SK. APOBEC3-Mediated RNA Editing in Breast Cancer is Associated With Heightened Immune Activity and Improved Survival. Int J Mol Sci (2019) 20:5621. doi: 10.3390/ijms20225621
312. George CX, Samuel CE. Human RNA-Specific Adenosine Deaminase ADAR1 Transcripts Possess Alternative Exon 1 Structures That Initiate From Different Promoters, One Constitutively Active and the Other Interferon Inducible. Proc Natl Acad Sci USA (1999) 96:4621–6. doi: 10.1073/pnas.96.8.4621
313. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in Tumours Overcomes Resistance to Immune Checkpoint Blockade. Nature (2019) 565:43–8. doi: 10.1038/s41586-018-0768-9
314. Bhate A, Sun T, Li JB. ADAR1: A New Target for Immuno-Oncology Therapy. Mol Cell (2019) 73:866–8. doi: 10.1016/j.molcel.2019.02.021
315. Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-Derived IFN Triggers Chronic Pathway Agonism and Sensitivity to ADAR Loss. Nat Med (2019) 25:95–102. doi: 10.1038/S41591-018-0302-5
316. Crow YJ, Manel N. Aicardi-Goutières Syndrome and the Type I Interferonopathies. Nat Rev Immunol (2015) 15:429–40. doi: 10.1038/nri3850
317. Kono M, Matsumoto F, Suzuki Y, Suganuma M, Saitsu H, Ito Y, et al. Dyschromatosis Symmetrica Hereditaria and Aicardi-Goutières Syndrome 6 Are Phenotypic Variants Caused by ADAR1 Mutations. J Invest Dermatol (2016) 136:875–8. doi: 10.1016/j.jid.2015.12.034
318. Livingston JH, Lin J-P, Dale RC, Gill D, Brogan P, Munnich A, et al. A Type I Interferon Signature Identifies Bilateral Striatal Necrosis Due to Mutations in ADAR1. J Med Genet (2014) 51:76–82. doi: 10.1136/jmedgenet-2013-102038
319. Crow Y, Zaki M, Abdel-Hamid M, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro N, et al. Mutations in ADAR1, IFIH1, and RNASEH2B Presenting As Spastic Paraplegia. Neuropediatrics (2014) 45:386–91. doi: 10.1055/s-0034-1389161
320. Vlachogiannis NI, Gatsiou A, Silvestris DA, Stamatelopoulos K, Tektonidou MG, Gallo A, et al. Increased Adenosine-to-Inosine RNA Editing in Rheumatoid Arthritis. J Autoimmun (2020) 106:102329. doi: 10.1016/j.jaut.2019.102329
321. Roth SH, Danan-Gotthold M, Ben-Izhak M, Rechavi G, Cohen CJ, Louzoun Y, et al. Increased RNA Editing May Provide a Source for Autoantigens in Systemic Lupus Erythematosus. Cell Rep (2018) 23:50–7. doi: 10.1016/j.celrep.2018.03.036
322. Chruściel E, Urban-Wójciuk Z, Arcimowicz Ł, Kurkowiak M, Kowalski J, Gliwiński M, et al. Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers (Basel) (2020) 12:683. doi: 10.3390/cancers12030683
323. Sadeqi Nezhad M, Seifalian A, Bagheri N, Yaghoubi S, Karimi MH, Adbollahpour-Alitappeh M. Chimeric Antigen Receptor Based Therapy as a Potential Approach in Autoimmune Diseases: How Close Are We to the Treatment? Front Immunol (2020) 11:603237. doi: 10.3389/fimmu.2020.603237
324. Florou D, Katsara M, Feehan J, Dardiotis E, Apostolopoulos V. Anti-Cd20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab. Brain Sci (2020) 10:1–13. doi: 10.3390/brainsci10100758
325. Zhang Q, Lu W, Liang C-L, Chen Y, Liu H, Qiu F, et al. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
326. Gliwiński M, Iwaszkiewicz-Grześ D, Wołoszyn-Durkiewicz A, Tarnowska M, Żalińska M, Hennig M, et al. Proinsulin-Specific T Regulatory Cells may Control Immune Responses in Type 1 Diabetes: Implications for Adoptive Therapy. BMJ Open Diabetes Res Care (2020) 8:e000873. doi: 10.1136/bmjdrc-2019-000873
327. De Paula Pohl A, Schmidt A, Zhang A-H, Maldonado T, Königs C, Scott DW. Engineered Regulatory T Cells Expressing Myelin-Specific Chimeric Antigen Receptors Suppress EAE Progression. Cell Immunol (2020) 358:104222. doi: 10.1016/j.cellimm.2020.104222
328. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of Colitis by Genetically Engineered Murine Regulatory T Cells Redirected by Antigen-Specific Chimeric Receptor. Gastroenterology (2009) 136:1721–31. doi: 10.1053/j.gastro.2009.01.049
329. Imam S, Dar P, Alfonso-Jaume MA, Jaume JC. 125-LB: Beta-Cell Antigen-Specific Chimeric Antigen Receptor Tregs for Type 1 Diabetes Prevention and Treatment. Diabetes (2020) 69:125–LB. doi: 10.2337/db20-125-LB
330. Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, Abolghasemi M, Poursaei E, Asadi M, et al. Dendritic Cell Therapy in Cancer Treatment; the State-of-the-Art. Life Sci (2020) 254:117580. doi: 10.1016/j.lfs.2020.117580
331. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous Tolerogenic Dendritic Cells for Rheumatoid and Inflammatory Arthritis. Ann Rheum Dis (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
332. Lipson EJ, Drake CG. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin Cancer Res (2011) 17:6958–62. doi: 10.1158/1078-0432.CCR-11-1595
333. Zarour HM. Reversing T-Cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res (2016) 22:1856 LP – 64. doi: 10.1158/1078-0432.CCR-15-1849
334. Deng C, Li Z, Guo S, Chen P, Chen X, Zhou Q, et al. Tumor PD-L1 Expression is Correlated With Increased TILs and Poor Prognosis in Penile Squamous Cell Carcinoma. Oncoimmunology (2017) 6:e1269047. doi: 10.1080/2162402X.2016.1269047
335. Xu Y, Wan B, Chen X, Zhan P, Zhao Y, Zhang T, et al. The Association of PD-L1 Expression With the Efficacy of Anti-PD-1/PD-L1 Immunotherapy and Survival of non-Small Cell Lung Cancer Patients: A Meta-Analysis of Randomized Controlled Trials. Transl Lung Cancer Res (2019) 8:413–28. doi: 10.21037/tlcr.2019.08.09
336. FDA Approves Opdivo as Single Agent for Melanoma. Oncol Times (2016) 38:41. doi: 10.1097/01.COT.0000479772.25205.6f
337. Twomey JD, Zhang B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J (2021) 23:1–11. doi: 10.1208/S12248-021-00574-0
338. Nishikawa K, Linsley PS, Collins AB, Stamenkovic I, McCluskey RT, Andres G. Effect of CTLA-4 Chimeric Protein on Rat Autoimmune Anti-Glomerular Basement Membrane Glomerulonephritis. Eur J Immunol (1994) 24:1249–54. doi: 10.1002/eji.1830240602
339. Dall’Era M, Davis J. CTLA4Ig: A Novel Inhibitor of Costimulation. Lupus (2004) 13:372–6. doi: 10.1191/0961203303lu1029oa
340. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. CTLA4Ig-Mediated Blockade of T-Cell Costimulation in Patients With Psoriasis Vulgaris. J Clin Invest (1999) 103:1243–52. doi: 10.1172/JCI5857
341. Moreland LW, Alten R, Van Den Bosch F, Appelboom T, Leon M, Emery P, et al. Costimulatory Blockade in Patients With Rheumatoid Arthritis: A Pilot, Dose-Finding, Double-Blind, Placebo-Controlled Clinical Trial Evaluating CTLA-4Ig and LEA29Y Eighty-Five Days After the First Infusion. Arthritis Rheum (2002) 46:1470–9. doi: 10.1002/art.10294
342. Ruperto N, Lovell DJ, Quartier P, Paz E, Rubio-Pérez N, Silva CA, et al. Abatacept in Children With Juvenile Idiopathic Arthritis: A Randomised, Double-Blind, Placebo-Controlled Withdrawal Trial. Lancet (2008) 372:383–91. doi: 10.1016/S0140-6736(08)60998-8
343. Viglietta V, Bourcier K, Buckle GJ, Healy B, Weiner HL, Hafler DA, et al. CTLA4Ig Treatment in Patients With Multiple Sclerosis: An Open-Label, Phase 1 Clinical Trial. Neurology (2008) 71:917–24. doi: 10.1212/01.wnl.0000325915.00112.61
344. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-Stimulation Modulation With Abatacept in Patients With Recent-Onset Type 1 Diabetes: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2011) 378:412–9. doi: 10.1016/S0140-6736(11)60886-6
345. Orban T, Beam CA, Xu P, Moore K, Jiang Q, Deng J, et al. Reduction in CD4 Central Memory T-Cell Subset in Costimulation Modulator Abatacept-Treated Patients With Recent-Onset Type 1 Diabetes Is Associated With Slower C-Peptide Decline. Diabetes (2014) 63:3449–57. doi: 10.2337/db14-0047
346. Fallarino F, Bianchi R, Orabona C, Vacca C, Belladonna ML, Fioretti MC, et al. CTLA-4–Ig Activates Forkhead Transcription Factors and Protects Dendritic Cells From Oxidative Stress in Nonobese Diabetic Mice. J Exp Med (2004) 200:1051–62. doi: 10.1084/jem.20040942
347. Razmara M, Hilliard B, Ziarani AK, Chen YH, Tykocinski ML. CTLA-4 X Ig Converts Naive CD4+CD25- T Cells Into CD4+CD25+ Regulatory T Cells. Int Immunol (2008) 20:471–83. doi: 10.1093/intimm/dxn007
348. Álvarez-Quiroga C, Abud-Mendoza C, Doníz-Padilla L, Juárez-Reyes A, Monsiváis-Urenda A, Baranda L, et al. CTLA-4-Ig Therapy Diminishes the Frequency But Enhances the Function of Treg Cells in Patients With Rheumatoid Arthritis. J Clin Immunol (2011) 31:588–95. doi: 10.1007/s10875-011-9527-5
349. Glatigny S, Höllbacher B, Motley SJ, Tan C, Hundhausen C, Buckner JH, et al. Abatacept Targets T Follicular Helper and Regulatory T Cells, Disrupting Molecular Pathways That Regulate Their Proliferation and Maintenance. J Immunol (2019) 202:1373–82. doi: 10.4049/jimmunol.1801425
350. Song M-Y, Hong C-P, Park SJ, Kim J-H, Yang B-G, Park Y, et al. Protective Effects of Fc-Fused PD-L1 on Two Different Animal Models of Colitis. Gut (2015) 64:260–71. doi: 10.1136/gutjnl-2014-307311
351. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J Immunol (2018) 200:3000–7. doi: 10.4049/jimmunol.1700407
352. Grebinoski S, Vignali DA. Inhibitory Receptor Agonists: The Future of Autoimmune Disease Therapeutics? Curr Opin Immunol (2020) 67:1–9. doi: 10.1016/j.coi.2020.06.001
353. ElTanbouly MA, Zhao Y, Schaafsma E, Burns CM, Mabaera R, Cheng C, et al. VISTA: A Target to Manage the Innate Cytokine Storm. Front Immunol (2021) 11:595950. doi: 10.3389/fimmu.2020.595950
354. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic Cancer Vaccines. Nat Rev Cancer (2021) 21:360–78. doi: 10.1038/s41568-021-00346-0
355. Moorman CD, Sohn SJ, Phee H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T Cell Therapy, and IL-2 Therapy. Front Immunol (2021) 0:657768. doi: 10.3389/FIMMU.2021.657768
356. Smilek DE, Ehlers MR, Nepom GT. Restoring the Balance: Immunotherapeutic Combinations for Autoimmune Disease. Dis Model Mech (2014) 7:503. doi: 10.1242/DMM.015099
357. Medler TR, Blair TC, Crittenden MR, Gough MJ. Defining Immunogenic and Radioimmunogenic Tumors. Front Oncol (2021) 0:667075. doi: 10.3389/FONC.2021.667075
358. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-Related Adverse Events With Immune Checkpoint Blockade: A Comprehensive Review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
359. Luu Q, Major G. Unleashing the Tiger – Iatrogenic Autoimmunity From Cancer Immunotherapy Drugs. JRSM Open (2018) 9:205427041774690. doi: 10.1177/2054270417746905
360. Opelz G, Döhler B. Lymphomas After Solid Organ Transplantation: A Collaborative Transplant Study Report. Am J Transplant (2004) 4:222–30. doi: 10.1046/j.1600-6143.2003.00325.x
Keywords: immune tolerance, autoimmune diseases, cancer immunology, tumor microenvironment, regulatory cells
Citation: Sakowska J, Arcimowicz Ł, Jankowiak M, Papak I, Markiewicz A, Dziubek K, Kurkowiak M, Kote S, Kaźmierczak-Siedlecka K, Połom K, Marek-Trzonkowska N and Trzonkowski P (2022) Autoimmunity and Cancer—Two Sides of the Same Coin. Front. Immunol. 13:793234. doi: 10.3389/fimmu.2022.793234
Received: 11 October 2021; Accepted: 12 April 2022;
Published: 13 May 2022.
Edited by:
Sophie Tourdot, Pfizer, United StatesReviewed by:
Michal Kuczma, Georgia State University, United StatesGeorgia Fousteri, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Sakowska, Arcimowicz, Jankowiak, Papak, Markiewicz, Dziubek, Kurkowiak, Kote, Kaźmierczak-Siedlecka, Połom, Marek-Trzonkowska and Trzonkowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia Marek-Trzonkowska, bmF0YWxpYS5tYXJlay10cnpvbmtvd3NrYUB1Zy5lZHUucGw=; Piotr Trzonkowski, cHRyem9uQGd1bWVkLmVkdS5wbA==
†These authors have contributed equally to this work and share first authorship