 Jiayue Ding1†
Jiayue Ding1† Baoying Song2,3Xiran Xie2,3Xaingyu Li4
Baoying Song2,3Xiran Xie2,3Xaingyu Li4 Zhiying Chen2,3
Zhiying Chen2,3 Zhongao Wang2,3
Zhongao Wang2,3 Liqun Pan2,3Duo Lan2
Liqun Pan2,3Duo Lan2 Ran Meng2,3*†
Ran Meng2,3*†- 1Department of Neurology, Tianjin Medical University General Hospital, Tianjin, China
- 2Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
- 3Advanced Center of Stroke, Beijing Institute for Brain Disorders, Beijing, China
- 4Department of Neurology, Tianjin Huanhu Hospital, Tianjin, China
Cerebral venous thrombosis (CVT) is a rare form of cerebrovascular disease that impairs people’s wellbeing and quality of life. Inflammation is considered to play an important role in CVT initiation and progression. Several studies have reported the important role of leukocytes, proinflammatory cytokines, and adherence molecules in the CVT-related inflammatory process. Moreover, inflammatory factors exacerbate CVT-induced brain tissue injury leading to poor prognosis. Based on clinical observations, emerging evidence shows that peripheral blood inflammatory biomarkers—especially neutrophil-to-lymphocyte ratio (NLR) and lymphocyte count—are correlated with CVT [mean difference (MD) (95%CI), 0.74 (0.11, 1.38), p = 0.02 and −0.29 (−0.51, −0.06), p = 0.01, respectively]. Moreover, increased NLR and systemic immune-inflammation index (SII) portend poor patient outcomes. Evidence accumulated since the outbreak of coronavirus disease-19 (COVID-19) indicates that COVID-19 infection and COVID-19 vaccine can induce CVT through inflammatory reactions. Given the poor understanding of the association between inflammation and CVT, many conundrums remain unsolved. Further investigations are needed to elucidate the exact relationship between inflammation and CVT in the future.
Introduction
Cerebral venous thrombosis (CVT) is a subtype of stroke, which accounts for 0.5%–1% of all cases of stroke; it mainly occurs in young and middle-aged individuals (1). Recent studies reported a CVT incidence of 1.3–1.6/100,000 in Western countries and even higher in Asia (2, 3). CVT usually involves either dural sinuses or cortical veins and results in variable pathogenic factors and complex clinical features (2). Similar to cerebral arterial ischemic stroke, inflammation is one of the well-known risk factors for CVT onset and development. Infection or autoimmune-mediated inflammatory reaction leads to localized hyper-coagulation and endothelial injury, which may cause CVT eventually; CVT-induced inflammation further aggravates brain tissue ischemic injury and leads to poor clinical outcomes (2, 3). Therefore, inflammation is a predisposing and exacerbating factor for CVT onset and development. The diagnostic and treatment strategies for CVT cannot be separated from those for inflammation in clinical settings. This review summarized the relationship between CVT and inflammation to serve as a reference for further studies.
Methods
We searched several literature databases, including PubMed, Embase, and Cochrane, for publications with specific keywords, “venous thrombosis/thrombus/thrombi” and “inflammation/inflammatory” or “immune/immunology/immunological,” that were published prior to December 2021. The references were subsequently thoroughly reviewed to retrieve articles for additional reports that we may have missed out on in our search. The adopted search strategy identified 125 articles in this study, including 62 studies focusing on the pathophysiology of inflammation associated with venous thromboembolism (VTE) and CVT, 11 clinical studies investigating the peripheral inflammatory biomarkers in the patients with CVT, and 52 studies on the inflammation in COVID-19 complicated with CVT (a study selection flowchart has been shown in Figure 1).
Figure 1 The selection flowchart for this review.
The clinical studies were further conducted with meta-analysis. Stata software (version 15.0 SE) was used for analysis in this study. The values are presented as mean ± SD. Estimating the sample mean and SD from the sample size, median, and interquartile range was based on the method provided by Luo et al. and Wan et al. (4, 5). Pooled analysis was conducted with the fixed-effects model using the Mantel–Haenszel method when the heterogeneity was expected to be available (I2 ≤ 50%). Otherwise, the random-effects model computed through the DerSimonian–Laird method was performed (I2 > 50%). p-Values < 0.05 were considered statistically significant.
Inflammation Promotes Cerebral Venous Thrombosis Onset
It is generally acknowledged that inflammation plays a central role in the occurrence of VTE, but the predisposing effect of inflammation on CVT remains poorly understood. However, known risk factors of CVT, such as infection, anticoagulant factor deficiency, and systematic autoimmune diseases, are linked with inflammatory reactions, indicating that inflammation may be involved in cerebral venous thrombogenesis (2, 3). Clinical studies that reported the conspicuous activation of serum and cerebrospinal fluid (CSF) inflammatory biomarkers in CVT further verified inflammation’s causative role in CVT onset (6–16). VTE and CVT are deemed to share similar inflammatory mechanisms clinically. Hence, the inflammatory pathogenesis in VTE seems to mimic the inflammatory reactions in CVT. The formation of venous thrombus of any type could be attributed mainly to three key elements—blood flow alteration, endothelial injury, and hypercoagulable state—collectively known as Virchow’s triad (17). Endothelial injury exposes collagen and tissue factors (TFs) that promote platelet aggravation and activate the immune system to participate in thrombus formation (18, 19). The prominent immunological factors for VTE mainly include leukocytes, cytokines, and adhesion molecules, all of which are likely to be implicated in the development of CVT (20). Given the paucity of reports on the effects of inflammation on the development of CVT, this study described the major inflammatory processes in VTE to mimic the possible inflammatory pathogenesis in CVT development.
Leukocytes
Currently, innate immune response in VTE has attracted tremendous attention worldwide. Neutrophils, which patrol the bloodstream and contribute to pathogen clearance by phagocytosis, serve as essential leukocytes in the innate immune response. Neutrophils are responsible for the inflammatory process in venous thrombus formation, regarded as a double-edged sword during thrombosis: early massive activation resulting in tissue damage promotes thrombus propagation, whereas late activation contributes to thrombus resolution (21, 22). Neutrophils extrude DNA in the form of neutrophil extracellular traps (NETs), which play an important role in the inflammatory pathophysiology of thrombus formation (23). NETs are comprised of myeloperoxidase and histones, which stimulate fibrin formation to trap and destroy invading microorganisms. In addition, NETs directly activate the Hageman factor (FXII), bind to von Willebrand factor (vWF), trigger platelet recruitment, and increase the concentration of enzymes such as neutrophil elastase and myeloperoxidase to promote intrinsic coagulation and thrombus formation (23–25). Besides, neutrophil elastase and myeloperoxidase can cleave and oxidize anticoagulants as well, leading to thrombus extension (25). Engelmann and Massberg coined the mechanism of NET-mediated microvascular thrombosis as “immunothrombosis” (25). Immunothrombosis inhibits the spread of infection through 1) capturing and ensnaring pathogens in the microvasculature, 2) restricting pathogens movement for bactericidal activity by the action of innate immune cells, and 3) recruiting other immune cells such as monocytes and macrophages to the invaded sites for further bactericidal activity (25). Thus, immunothrombosis may be an essential contributor to venous thrombus formation. Although it has not been proven in CVT pathogenesis, we hypothesized that immunothrombosis might be responsible for infection-mediated CVT, especially cortical venous thrombosis.
Besides neutrophils, monocytes play a role in immunothrombosis by producing microparticles with TFs (26). Accepted views consider thrombus formation in the venous vasculature to be driven by TF from the vessel wall when endothelial cells are destroyed (27, 28). However, Brühl et al. found that TF expressed by Ly6G+ monocytes showed a strong signal in VTE, indicating that both vessel wall TF and blood cell-derived TF critically contribute to venous thrombosis (29). Microparticles are encrypted in the monocyte genome and are in low content in normal physical conditions (26). When stimulated by pattern recognition receptors (PRRs), microparticles within monocytes deliver the activated TF to the sites of pathogen exposure by assisting with adherence of extruded NETs, which trigger the extrinsic pathway of coagulation finally (29, 30). The synergy between neutrophil-released NETs and monocyte-released microparticles participates in thrombus formation and elongation in veins, including cerebral veins.
Mast cells are another important component of the immune system that plays a significant role in venous thrombosis (31). Mast cells exert their prothrombotic actions by releasing the granular constituent, including histamine, cytokines, and proteases, which cause endothelial activation, platelet adhesion, vWF secretion, leukocyte recruitment, P-selection release, and intercellular adhesion molecule-1 (ICAM-1) expression (31, 32). During a hypersensitivity reaction, polyphosphate (polyP) serves as a potent proinflammatory signal released from mast cells assisting with coagulation by promoting factor V activation, reducing TF pathway inhibitor (TFPI) activity, and retarding thrombolysis by eliciting thrombin-activatable fibrinolysis inhibitor (TAFI) (33–35).
In summary, immune cells precipitate venous thrombosis through the following pathophysiological pathways: neutrophils extrude NETs and activate FXII to FXIIa to trigger intrinsic coagulation; monocytes release microparticles that deliver TF to trigger extrinsic coagulation; and mast cells promote platelet activation and adhesion to support immunothrombosis (Figure 2 and Table 1). In theory, the aforementioned mechanisms may apply to both arterial and venous thrombosis, including CVT.
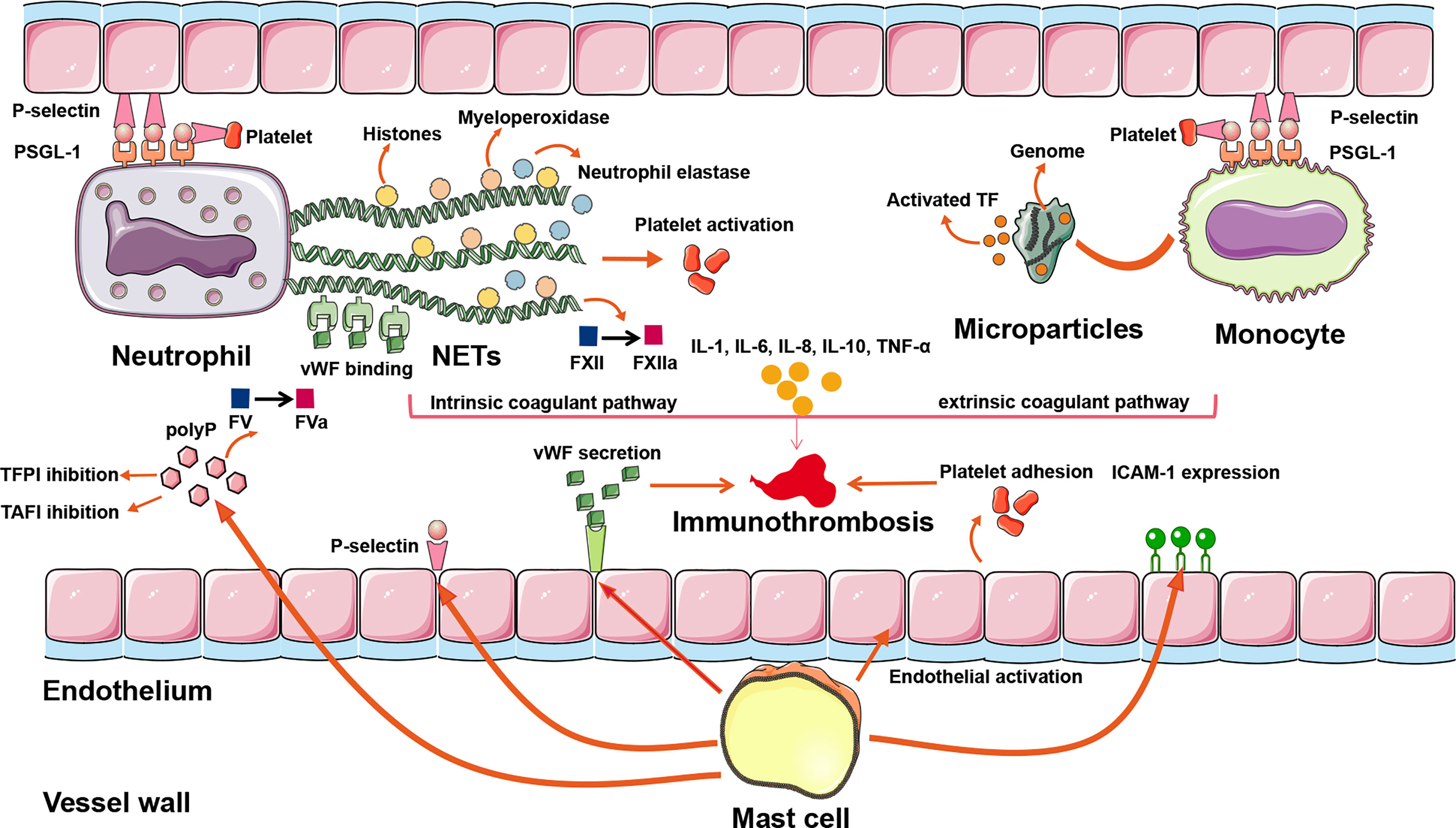
Figure 2 The inflammatory mechanisms of CVT formation. A plethora of inflammatory pathways are implicated in CVST formation: (1) NETs extruded by neutrophils can directly activate FXII, bind to vWF, trigger platelet recruitment, and concentrate enzymes such as neutrophil elastase and myeloperoxidase, so that intrinsic coagulant pathway is activated. (2) Activated TF is delivered by microparticles within monocytes to the sites of pathogen exposure. Assisting with adherence of extruded NETs, the extrinsic coagulant pathway is triggered. (3) Mast cells exert their prothrombotic actions through the release of a granular constituent, leading to endothelial activation, platelet adhesion, vWF secretion, leukocyte recruitment, P-selection release, and ICAM-1 expression. (4) Proinflammatory cytokines such as IL-1, IL-6, IL-8, IL-10, and TNF-α modulate the inflammatory reactions to affect the evolution or resolution of thrombosis. (5) Binding of P-selectin to PSGL-1 initiates the rolling leukocytes adhering on activated platelets and endothelial cells, renders neutrophils migrating to the sites of injury, promotes NETs formation in the setting of activated platelets, and promotes monocytes releasing TF-bearing microparticles. ICAM-1, intercellular adhesion molecule-1; NETs, neutrophil extracellular traps; polyP, polyphosphate; PSGL-1, P-selectin glycoprotein ligand 1; TAFI, thrombin-activatable fibrinolysis inhibitor; TF, tissue factor; TFPI, tissue factor pathway inhibitor; vWF, von Willebrand factor; CVT, cerebral venous thrombosis; CVST, cerebral venous sinus thrombosis.

Table 1 Proinflammatory factors implicated in venous thrombosis formation and development.
Proinflammatory Cytokines
Varieties of proinflammatory cytokines, such as interleukin-6 (IL-6), interleukin-1 (IL-1), interleukin-8 (IL-8), and tumor necrosis factor-alpha (TNF-α), are involved in venous thrombus formation. They modulate the inflammatory reactions to affect the development or resolution of thrombosis and play an essential role in thrombosis by promoting TF expression resulting in a procoagulant state (46, 71, 72). IL-6 is the most notable cytokine in boosting coagulation and has been used to evaluate the coagulant status and predict the clinical outcomes of CVT patients (7, 12, 73). A high level of IL-6 in VTE is strongly correlated with a high level of fibrinogen, high rates of complications and thrombosis recurrence, and poor clinical outcomes (7, 12, 74, 75). IL-6 increases the expression of TF, fibrinogen, factor VIII, and vWF, leading to endothelial cells activation, vessel wall damage, platelet aggregation, and recruitment and activation of leukocytes at the venous wall, all of which provoke the localized thrombus formation (39, 40). Additionally, IL-6 can reduce the concentration of the natural thrombosis inhibitors, such as protein S, antithrombin, and thrombomodulin, resulting in thrombus formation (40). Thus, IL-6 serves as an important promoter of thrombosis in VTE. Other risk factors for VTE mainly include IL-1, IL-8, IL-18, TNF-α, IFN-γ, and CCR2/CCL2, and their functions are shown in Table 1.
In contrast, IL-10 plays a beneficial role in patients with VTE by its downregulatory effect on the immune system (42). IL-10 is deemed an anti-inflammatory cytokine capable of inhibiting the activation of T cells, monocytes, and macrophages (43). In addition, P-selectin stored in platelets and endothelial cells is also significantly suppressed by IL-10 (44). Therefore, IL-10 restrains inflammatory events and inhibits thrombus formation. Although elevated proinflammatory cytokines’ beneficial or detrimental influences on CVT are uncertain, the indispensable role of cytokines in venous thrombus formation has been well-acknowledged (Figure 2 and Table 1).
Adhesion Molecules
Adhesion molecules are a critical component implicated in the inflammatory process and thrombus generation. P-selectin is one of the hot topic adhesion molecules for VTE pathogenesis and serves as an interaction mediator among platelets, leukocytes, and endothelial cells that modulate both hemostasis and inflammation (52). P-selectin is an extended protein with a membrane-distal C-type lection domain, always bonded to P-selectin glycoprotein ligand 1 (PSGL-1) mainly expressed in the neutrophils and monocytes (53). Endothelial cells and platelets activated by thrombin or other mediators rapidly mobilize P-selectin from secretory granules to the cell surface when infection or injury occurs (53, 54). The binding of P-selectin to PSGL-1 initiates the rolling leukocytes’ adhesion to activated platelets and endothelial cells and engenders platelets’ microparticles to form bridges among leukocytes (54). In addition, signaling by P-selectin through PSGL-1 aids neutrophils’ migration to the sites of injury and promotes NET formation in the setting of activated platelets (55, 56). Likewise, P-selectin can promote monocytes releasing TF-bearing microparticles, resulting in extrinsic coagulation initiation (29, 76). When blocking the binding of P-selectin to PSGL-1, leukocyte adhesion to platelets and endothelial cells is inhibited, and the expression of molecules that amplifies inflammation and thrombosis is also suppressed (76–79). Herein, P-selection is considered as an important mediator implicated in venous immune-related thrombosis and aggravation (Figure 2). Other adhesion molecules such as integrin, leukocyte integrin Mac-1, and CLEC-2 also play an important role in VTE initiation, shown in Table 1.
Summary
The factors mentioned above have been fully elucidated and are considered the major inflammatory reactions involved in VTE. Additionally, a growing body of proinflammatory factors such as complements, cellular markers, and secretory proteins is also implicated in venous thrombosis (Table 1) (23–25, 29, 30, 33–70). The processes are complex and varied, and many mechanisms remain poorly understood. Given that inflammation is essential in venous thrombus formation, inflammatory diseases that produce high immune system activation are definite risk factors for CVT. Usually, the incidence of VTE in patients with inflammatory diseases is threefold higher than in general populations (80). In particular, some acquired, multi-organ, inflammatory diseases such as systemic lupus erythematosus (SLE), Behçet’s disease (BD), and inflammatory bowel disease (IBD) are independent risk factors for CVT (81–83). Both central and peripheral inflammatory markers such as NETs, TF, P-selectin, IL, and C-reactive protein (CRP) are considered to be mediated by inflammatory diseases and accompany CVT onset and aggravation (84–87). In patients with inflammatory disease-related CVT, inflammatory reaction control cannot be overlooked. In these settings, immunosuppressives combined with anticoagulant therapies will be suggested. With the cooperation of leukocytes, cytokines, adhesion molecules, and other factors, the inflammation affects the onset and development of venous thrombosis and can be considered a paramount issue for CVT onset.
Inflammation in Cerebral Venous Thrombosis-Related Brain Damage
Similar to cerebral arterial infarction, an inflammatory reaction also occurs after a CVT attack (Figure 3). Blood–brain barrier (BBB) disruption inducing vasogenic edema is a pivotal pathological phenomenon after CVT, and leukocyte–endothelial cell adhesion contributes to this process (88). Nagai et al. reported that no BBB disruption or brain edema was detected 3 h post-CVT; however, increased leukocyte adhesion, elevated monocyte chemotactic protein 1 (MCP-1), and decreased IL-10 were observed, suggesting that inflammation might occur ahead of the edema (89). At hour 48 after CVT, the abnormal elevated IL-10 was restored, MCP-1 level remained high, and BBB disruption and edema were noted (89). On day 3 post-CVT, T-cell recruitment, microglia and macrophage activation, and astrocyte aggregation could be seen, and these phenomena were maintained for 1 to 2 weeks (88). Initially, neutrophils aggregate in the venous walls, followed by monocytes and macrophages. Then, cytokines, chemokines, and other inflammatory factors such as TNF-α elicit inflammatory reactions (90). Proinflammation and anti-inflammation are jointly mediated by the venous wall damage ultimately. P-selectin accelerates the inflammatory reaction in this process, and the deletion of P-selectin profoundly inhibits the venous wall inflammatory response (44, 91, 92). In addition, microparticles and TF further amplify the inflammatory reaction and promote thrombus growth and tissue damage (28).
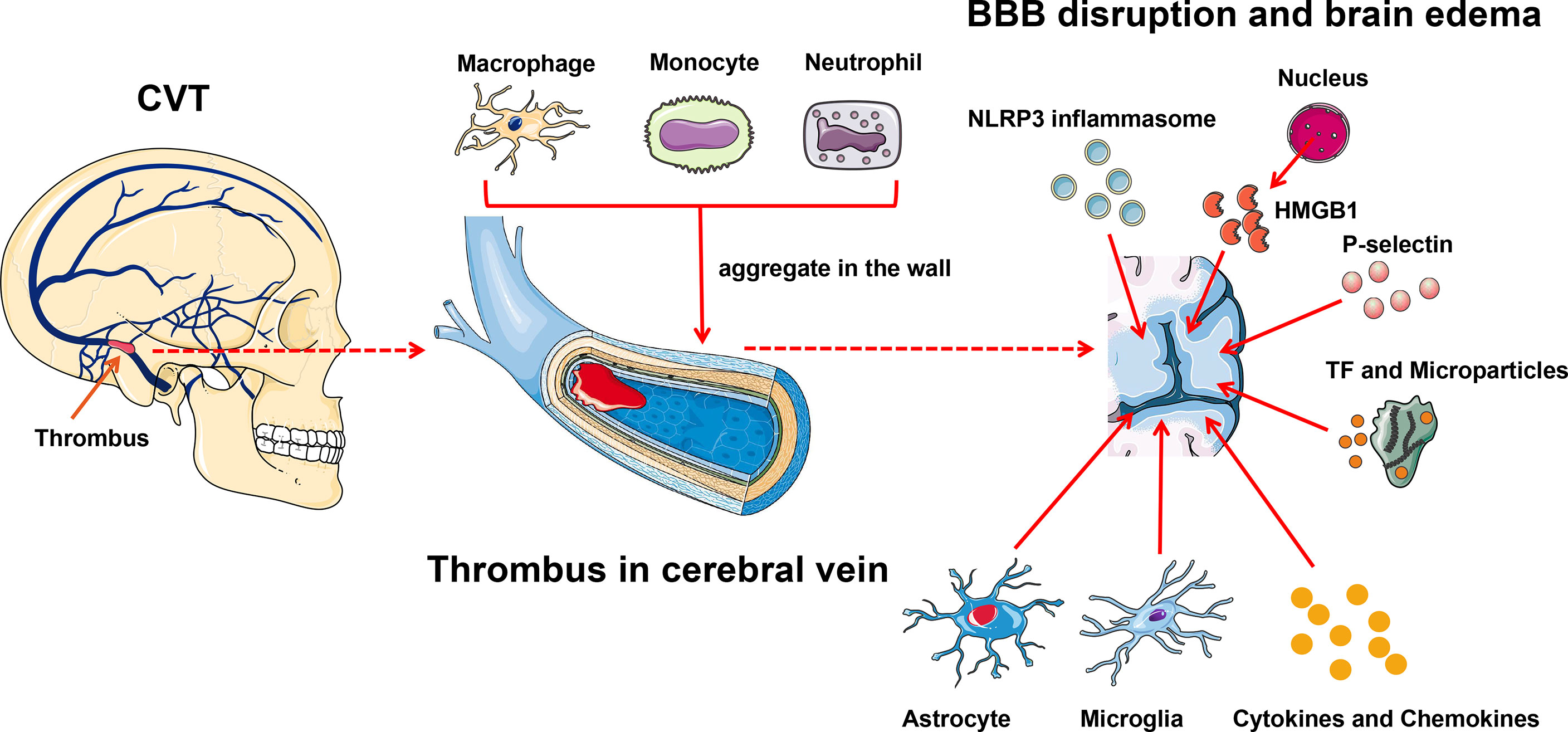
Figure 3 Inflammation exacerbates CVT-induced brain injury. Leukocyte–endothelial cell adhesion contributes to brain damage after CVT. Initially, neutrophils aggregate in the venous wall followed by monocytes and macrophages; meanwhile, microglia and astrocyte infiltrate into the damaged tissue. And then, cytokines, chemokines, and other inflammatory factors such as TNF-α are released to elicit an inflammatory reaction. P-selectin and activated TF within microparticles further amplify the brain tissue injury. NLRP3 is involved in the inflammatory reaction post-CVT through eliciting caspase-1 to cleave pro-IL-1β and pro-IL-18 to their active forms. HMGB1, which is released from the nucleus, can induce endocytosis and generate proinflammatory factors to further damage the brain tissue. BBB, blood–brain barrier; CVT, cerebral venous thrombosis; HMGB1, high-mobility group box 1; TF, tissue factor.
NLRP3, the most characterized inflammasome, is found abundantly in the brain (61, 88, 93). The role of NLRP3 in CVT is well documented. Unlike in arterial ischemia, where NLRP3 changes suddenly minutes to hours after stroke onset, NLRP3 in CVT activates in a subacute fashion, evident at day 3, and maintained until day 7 after CVT, which is in parallel with the immune cell activation and infiltration (88, 94). Thioredoxin-interacting protein (TXNIP) is a well-known binding partner to NLRP3 and is responsible for NLRP3 inflammasome activation under endoplasmic reticulum (ER) stress and oxidative stress (95, 96). ER stress and oxidative stress jointly boost the expression and activation of TXNIP (94). Meanwhile, peroxynitrite formation, which is elicited by oxidative stress, is also a pivotal trigger of NLRP3 inflammasome activation (94, 97). Therefore, TXNIP and peroxynitrite jointly facilitate the activation of NLRP3 inflammasome after CVT under ER stress and oxidative stress. NLRP3 can elicit caspase-1 to cleave pro-IL-1β and pro-IL-18 to their active forms, both of which are involved in the inflammatory reaction post-CVT (93, 98). Meanwhile, NLRP3 can lead to pyroptosis after CVT, validated by gasdermin D (GSDMD, an indicator of pyroptosis) (94).
High mobility group box 1 (HMGB1), a member of the damage-associated molecular pattern molecular family, is recently reported to be implicated in the inflammatory cascade-amplification reaction in the pathophysiology of CVT (67). Under normal physical conditions, HMGB1 exists in the nucleus and maintains the chromosomal structure and physiological activities of the DNA (99). On suffering inflammatory stimuli, HMGB1 is released into the extracellular space and immediately activates the innate immune response (99). Independently, HMBG1 can activate its downstream mediator, the receptor for advanced glycated end products (RAGE), to induce endocytosis and generate proinflammatory factors such as TNF-α, IL-1β, and IL-6 (67). The inhibition of HMGB1 and RAGE can reduce the expression of downstream inflammatory factors and attenuate CVT-mediated further damage (67).
Although the understanding is far from adequate, the pivotal role of inflammation on CVT-related brain tissue damage is undoubted. Currently, how to inhibit post-CVT inflammation progression presents a great interest in CVT correction. Since immunosuppressors have been gradually used in cerebral arterial ischemia, the inhibition of immunological reactions may be a novel promising strategy for CVT treatment (100).
Peripheral Inflammatory Biomarkers for Cerebral Venous Thrombosis
Several reports support the central role played by the inflammatory process in thrombosis development. Besides the focal inflammatory response described earlier, peripheral inflammatory markers also correlate with the pathogenesis of thrombosis. Peripheral neutrophils and lymphocytes are strongly associated with the severity and prognosis of cerebral arterial ischemia (101). Similarly, it is believed that the inflammatory parameters are also altered in CVT (7). Elevated inflammatory parameters can reflect the severity and predict the prognosis of CVT.
Peripheral Inflammatory Biomarkers Are Characterized in Cerebral Venous Thrombosis
A variety of peripheral inflammatory markers are associated with central venous thrombosis. The high neutrophil-to-lymphocyte ratio (NLR) is identified as the conspicuous index for cerebral arterial ischemia. However, the role of NLR in CVT is not clear. Four studies reported that CVT patients had a significantly elevated NLR value than controls (6, 7, 13, 16). However, a large retrospective study reported by Artoni et al., including 100 CVT patients and 299 controls, found no difference in NLR (15). We performed a pooled analysis in this review (shown in Figure 4A) and found that CVT patients had a higher level of NLR than controls [mean difference (MD) (95%CI), 0.74 (0.11, 1.38), p = 0.02]. Although our result conflicted with that of Artoni et al., we believe that NLR could be used as an assistant biomarker for CVT evaluation. Platelet-to-lymphocyte ratio (PLR) is also a predictive value for cerebral arterial ischemia (101). Two studies demonstrated a statistical correlation between higher PLR value and CVT in relation to cerebral arterial ischemia (6, 13). In contrast, Artoni et al. showed a lack of association between them (15). When pooling these results together (shown in Figure 4B), no significant difference in PLR values was obtained between CVT patients and controls [20.30 (−17.44, 58.04), p = 0.29]. Hence, PLR might not be suitable for CVT diagnosis. Neutrophil and lymphocyte counts were also analyzed in previous studies: Artoni et al. found higher neutrophil count in controls than in CVT patients (15). In contrast, three other studies did not find a statistical difference in neutrophil count between the two populations (6, 13, 16). Intriguingly, the 3 studies found significantly lower lymphocyte counts in CVT patients than in controls, but Artoni et al. did not (6, 13, 15, 16). The results regarding neutrophil and lymphocyte counts were also pooled together in this review. As shown in Figures 4C, D, lymphocyte count was significantly lower in CVT patients than controls [−0.29 (−0.51, −0.06), p = 0.01], but the neutrophil count [0.72 (−0.25, 1.69), p = 0.14] did not show a significant difference between the groups. In addition, pooled analysis for platelet count from the three studies did not find any difference between the 2 groups [20.30 (−17.44, 58.04), p = 0.29] (6, 13, 15). All results mentioned above uncovered the critical role of lymphocyte count as a laboratory feature for CVT. Although neutrophils and platelets were responsible for venous thrombus initiation and development, this review considered the possible characterization of NLR value and lymphocyte count with CVT.
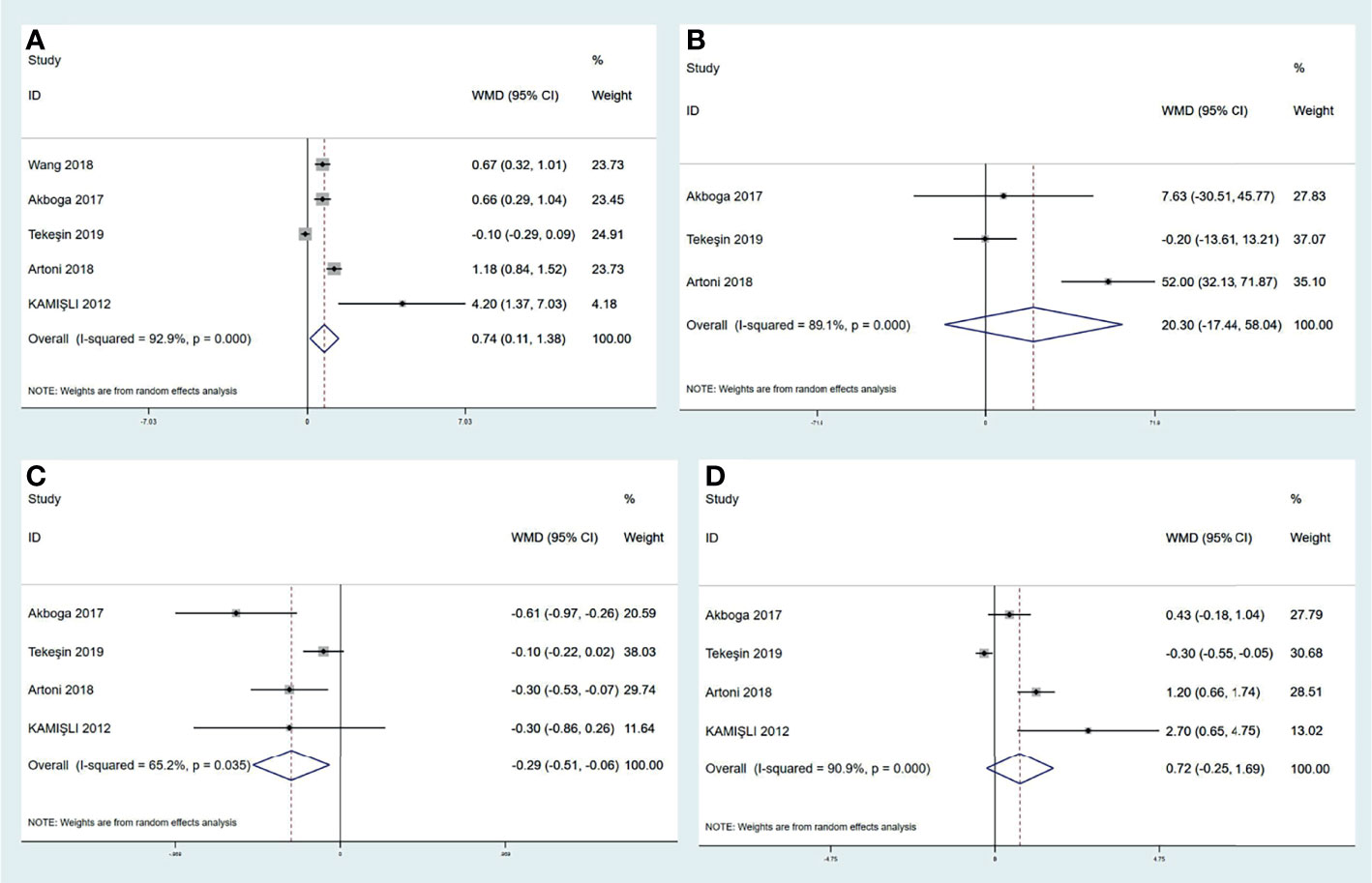
Figure 4 Meta-analysis of inflammatory biomarkers in CVT versus healthy controls, including NLR (A), PLR (B), lymphocyte count (C), and neutrophil count (D). CVT, cerebral venous thrombosis; NLR, neutrophil-to-lymphocyte ratio; PLR, platelet-to-lymphocyte ratio.
Peripheral Inflammatory Biomarkers Predict the Prognosis of Cerebral Venous Thrombosis
Inflammation aggravates CVT-related brain injury resulting from various etiologies, leading to poor prognosis. Except for leukocyte infiltrating into the focal lesion, peripheral leukocyte variations are more convenient to be detected in clinical settings. Since the NLR value is the most important inflammatory index characterized by CVT, it also seems to predict CVT’s prognosis. Wang et al. found that increased serum NLR value was associated with the severity of CVT patients at admission and reflected the short-term outcome at discharge (7). Sousa et al. indicated that increased serum NLR value had a significant association with the unfavorable functional outcome at 90 days (9). Apart from NLR, other inflammatory marker variations such as low serum lymphocyte-to-monocyte ratio (LMR), high serum IL-6 concentration, and high CSF IgM concentration correlate with poor outcomes of patients with CVT (7, 9, 11). To comprehensively reflect the balance of host immune and inflammatory status in CVT, Li et al. used systemic immune-inflammation index (SII) to predict the prognosis of patients with CVT. SII is defined as follows: platelet (/L) × neutrophil (/L)/lymphocyte (/L) (8). They demonstrated that higher SII presented lower survival rates and was a significant predictor of poor outcomes. Neutrophils are considered an important immune cell facilitating venous thrombus formation, and lymphocytes are also involved in the inflammatory process of developing blood clots (102). Given the interaction among neutrophils, lymphocytes, and platelets, SII may be more available to evaluate the inflammatory extent during CVT.
Peripheral Inflammation Biomarkers Fluctuate in Different Cerebral Venous Thrombosis Phases
Peripheral inflammation markers are different among acute, subacute, and chronic CVT phases. The acute and subacute phases of CVT have a higher neutrophil count, lower NLR value, and higher CRP levels than the chronic phase (7, 14). In addition, lower lymphocyte counts and higher NLR values are observed in patients with acute onset CVT than those with subacute and chronic symptoms (7, 14). Intriguingly, the monocyte count is higher in the subacute phase than in the chronic phase, but there is no significant difference between the acute and chronic phases (14). Antibodies in CSF such as IgM, IgA, and IgG are substantially lower in the chronic phase than in the acute and subacute phases (7). All results mentioned above indicate the inflammatory activity peaks at the acute phase, which is the most severe stage, decreases gradually at the subsequent phases, and returns close to normal at last.
Overall, this part indicates that peripheral inflammatory biomarkers are invariably associated with CVT progress and prognosis in clinical settings. The prominent markers such as NLR, lymphocyte count, and SII are widely applied to CVT diagnosis, monitoring, and evaluation of treatment. However, the variations in peripheral inflammatory biomarkers as either a contributor to or result of CVT may be less clear. Thus, the connections between central damage and peripheral inflammation are necessary to be investigated in the future.
COVID-19 Complicated With Cerebral Venous Thrombosis
COVID-19 Infection-Induced Cerebral Venous Thrombosis
The novel severe acute respiratory syndrome coronavirus-2 [SARS-CoV-2, also named coronavirus disease-19 (COVID-19)] pandemic currently challenges the global public health and healthcare sectors. The complications and prognosis of COVID-19 are manifold. Clinical observations show an increased risk of stroke and thrombotic complications in patients with COVID-19 infection (103). COVID-19 infection itself has a rare but demonstrated association with CVT. Inflammation is also implicated in this disease’s progression. A recent systematic review using data from 34,331 patients identified the frequency of CVT among patients hospitalized for COVID-19 infection to be 0.08%, which is potentially higher than the expected incidence of 5 to 20 per million per year in the general population (104). Besides, two retrospective studies also reported higher incidence rates of CVT in patients with COVID-19 infection than in the general population (2–39 patients per million versus 0.4 per million) (105, 106).
The underlying mechanisms of COVID-19 infection-induced CVT are complex. Inflammatory reactions contribute to this process substantially. Vascular endothelial dysfunction caused by COVID-19 infection is the most accepted pathophysiology to elucidate the observed high incidence of CVT (107). It has been documented that both the virion and the angiotensin-converting enzyme 2 receptors are found in endothelial cells (108, 109). Besides, recruitment of immune cells, either by the viral infection of the endothelium or immune-mediated, causes endothelial dysfunction (110). The dysfunctional endothelium releases vWF and exposes locally provided TF, consequently activating the extrinsic pathway (110). COVID-19 infection-induced endothelial dysfunction causes hypercoagulability and fibrinolysis shutdown, as evidenced by elevated D-dimer and complete failure of clot lysis on thromboelastography (111). In addition, excessive activity of cytokine, coined as “cytokine storm” with high concentrations of proinflammatory cytokines and chemokines (such as IL-1, IL-6, and TNF-α), can suppress anticoagulant systems, release vWF, and increase TF expression in the backdrop of COVID-19 infection (112, 113). Neutrophils with NETs, monocytes with microparticles, and complement systems are subsequently activated, predisposing the vessels to thrombus formation and propagation (114). As Virchow’s triad criteria are fulfilled in critically ill COVID-19 patients; other pathophysiologies such as altered flow dynamics, prothrombotic and fibrinolytic imbalance, and hypoxia-mediated vasoconstriction are also responsible for CVT initiation and development (114). Therefore, CVT in COVID-19 disease is a complex interplay between the endothelium and a range of inflammatory factors.
COVID-19 Vaccination-Induced Cerebral Venous Thrombosis
Apart from COVID-19 infection, the COVID-19 vaccine is also associated with CVT through immune thrombotic thrombocytopenia (115). One study from Germany estimated the incidence rate of CVT within 1 month after first dose administration of 1.52 per 100,000 person-months for ChAdOx1 vaccine (AstraZeneca) and 0.11 per 100,000 person-months for BNT162b2 (BioNTech/Pfizer) (116). Most of the patients had platelet-activating antibodies directed against platelet factor 4 (PF4), leading to prothrombotic status and thrombocytopenia. Similar to the pathophysiology of heparin‐induced thrombocytopenia (HIT), in the cases of vaccine-induced immune thrombotic thrombocytopenia (VITT), an unidentified polyanion in the adenoviral vaccines or expressed by the infected cells by the vaccine is likely binding to PF4 (115, 117). The complex crosslinks many FcγRIIa receptors on platelets, conducing thromboxane synthesis and platelet activation. More PF4 molecules and procoagulant microparticles are released from activated platelets, resulting in increased thrombin generation (118). In addition, increased expression of TF and NETs enhances coagulation by providing a scaffold for platelets, vWF, and red blood cells (118). The activated platelets and antibody-coated platelets are removed by the macrophages of the reticuloendothelial system, resulting in thrombocytopenia (117). In general, the COVID-19 vaccine can cause VITT, which initiates inflammatory reactions and contributes to the occurrence of CVT in patients with COVID-19 infection. Although both HIT and VITT present with antibodies to PF4, these thrombotic manifestations occur in VITT and not typically in HIT, a phenomenon yet to be explained (119).
Conclusions
For the first time, the pivotal role of inflammation in CVT was systematically recapitulated in this review. Mounting clinical and laboratory evidence has unraveled the complex interplay among inflammation, coagulant system, and venous thrombus formation. Assisting platelets and collagen, cells involved in innate immunity, such as ILs, leukocytes, adhesion molecules, and chemokines, activate intrinsic and extrinsic coagulation pathways, leading to CVT. Innate immunity also promotes thrombus propagation and accelerates the progression of CVT. Peripheral inflammatory biomarkers may serve as convenient and accurate indices to evaluate CVT severity and patient prognosis. Exploring the inflammatory background in time for the patients with CVT will be very useful for clinicians to make a proper individualized treatment strategy for the patients. In addition, COVID-19 infection and COVID-19 vaccine administration can also induce CVT through inflammatory reactions. Due to a poor understanding of inflammation with CVT, a plethora of conundrums are yet to be resolved. Further clinical and basic studies are needed to explore and establish the exact association between inflammation and CVT in the future.
Author Contributions
JD and RM formulated the conception of the review, drafted the manuscript, and prepared the figures. BS, XX, and XL completed the screening of the publications. ZW, LP, and DL were responsible for the statistical analysis. ZC made critical revisions to the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was sponsored by the National Key R&D Program of China (2017YFC1308401), the National Natural Science Foundation of China (82171297), and the Beijing Natural Science Foundation (7212047).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all patients and doctors who participated in this study for their cooperation.
References
1. Saposnik G, Barinagarrementeria F, Brown RD Jr, Bushnell CD, Cucchiara B, Cushman M, et al. Diagnosis and Management of Cerebral Venous Thrombosis: A Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke (2011) 42(4):1158–92. doi: 10.1161/STR.0b013e31820a8364
2. Silvis S, De Sousa DA, Ferro JM, Coutinho JM. Cerebral Venous Thrombosis. Nat Rev Neurol (2017) 13(9):555–65. doi: 10.1038/nrneurol.2017.104
3. Bousser MG, Ferro JM. Cerebral Venous Thrombosis: An Update. Lancet Neurol (2007) 6(2):162–70. doi: 10.1016/S1474-4422(07)70029-7
4. Luo D, Wan X, Liu J, Tong T. Optimally Estimating the Sample Mean From the Sample Size, Median, Mid-Range, and/or Mid-Quartile Range. Stat Methods Med Res (2018) 27(6):1785–805. doi: 10.1177/0962280216669183
5. Wan X, Wang W, Liu J, Tong T. Estimating the Sample Mean and Standard Deviation From the Sample Size, Median, Range and/or Interquartile Range. BMC Med Res Methodol (2014) 14:135. doi: 10.1186/1471-2288-14-135
6. Tekesin A, Tunç A. Inflammatory Markers Are Beneficial in the Early Stages of Cerebral Venous Thrombosis. Arq Neuropsiquiatr (2019) 77(2):101–5. doi: 10.1590/0004-282x20190001
7. Wang L, Duan J, Bian T, Meng R, Wu L, Zhang Z, et al. Inflammation Is Correlated With Severity and Outcome of Cerebral Venous Thrombosis. J Neuroinflamm (2018) 15(1):329. doi: 10.1186/s12974-018-1369-0
8. Li S, Liu K, Gao Y, Zhao L, Zhang R, Fang H, et al. Prognostic Value of Systemic Immune-Inflammation Index in Acute/Subacute Patients With Cerebral Venous Sinus Thrombosis. Stroke Vasc Neurol (2020) 5(4):368–73. doi: 10.1136/svn-2020-000362
9. Aguiar De Sousa D, Pereira-Santos MC, Serra-Caetano A, Lucas Neto L, Sousa AL, Gabriel D, et al. Blood Biomarkers Associated With Inflammation Predict Poor Prognosis in Cerebral Venous Thrombosis:: A Multicenter Prospective Observational Study. Eur J Neurol (2021) 28(1):202–8. doi: 10.1111/ene.14526
10. Duan J, Leng X, Han Z, Cai Y, Wang C, Rajah G, et al. Identifying Biomarkers Associated With Venous Infarction in Acute/Subacute Cerebral Venous Thrombosis. Aging Dis (2021) 12(1):93–101. doi: 10.14336/AD.2020.0405
11. Li S, Liu K, Zhang R, Gao Y, Fang H, Liu X, et al. Lower Lymphocyte to Monocyte Ratio Is A Potential Predictor of Poor Outcome in Patients With Cerebral Venous Sinus Thrombosis. Stroke Vasc Neurol (2019) 4(3):148–53. doi: 10.1136/svn-2018-000180
12. Akbari F, Ghorbani A, Fatehi F. The Assessment of Proinflammatory Cytokines in the Patients With the History of Cerebral Venous Sinus Thrombosis. Iran J Neurol (2016) 15(2):75–9.
13. Akboga Ye, Bektas H, Anlar O. Usefulness of Platelet to Lymphocyte and Neutrophil to Lymphocyte Ratios in Predicting the Presence of Cerebral Venous Sinus Thrombosis and In-Hospital Major Adverse Cerebral Events. J Neurol Sci (2017) 380:226–9. doi: 10.1016/j.jns.2017.07.036
14. Dias L, Pinto MJ, Castro P, Carvalho M. Inflammatory Biomarkers Correlate With Time Evolution in Cerebral Venous Thrombosis. J Stroke Cerebrovasc Dis (2021) 30(7):105844. doi: 10.1016/j.jstrokecerebrovasdis.2021.105844
15. Artoni A, Abbattista M, Bucciarelli P, Gianniello F, Scalambrino E, Pappalardo E, et al. Platelet to Lymphocyte Ratio and Neutrophil to Lymphocyte Ratio As Risk Factors for Venous Thrombosis. Clin Appl Thromb Hemost (2018) 24(5):808–14. doi: 10.1177/1076029617733039
16. Kamisli S, Kamisli O, Gonullu S, Kaplan Y, Ozcan C. The Prognostic Value of Increased Leukocyte and Neutrophil Counts in the Early Phase of Cerebral Venous Sinus Thrombosis. Turk Serebrovaskuler Hastaliklar Dergisi (2012) 18(2):35–8. doi: 10.5505/tbdhd.2012.43531
17. Kyrle Pa, Eichinger S. Deep Vein Thrombosis. Lancet (2005) 365(9465):1163–74. doi: 10.1016/S0140-6736(05)71880-8
18. Khakpour S, Wilhelmsen K, Hellman J. Vascular Endothelial Cell Toll-Like Receptor Pathways in Sepsis. Innate Immun (2015) 21(8):827–46. doi: 10.1177/1753425915606525
19. Samuels Pb, Webster Dr. The Role of Venous Endothelium in the Inception of Thrombosis. Ann Surg (1952) 136(3):422–38. doi: 10.1097/00000658-195209000-00010
20. Saghazadeh A, Hafizi S, Rezaei N. Inflammation in Venous Thromboembolism: Cause Or Consequence. Int Immunopharmacol (2015) 28(1):655–65. doi: 10.1016/j.intimp.2015.07.044
21. Granelli-Piperno A, Vassalli JD, Reich E. Secretion of Plasminogen Activator by Human Polymorphonuclear Leukocytes. Modulation by Glucocorticoids and Other Effectors. J Exp Med (1977) 146(6):1693–706. doi: 10.1084/jem.146.6.1693
22. Heiden M, Seitz R, Egbring R. The Role of Inflammatory Cells and Their Proteases in Extravascular Fibrinolysis. Semin Thromb Hemost (1996) 22(6):497–501. doi: 10.1055/s-2007-999050
23. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA Traps Promote Thrombosis. Proc Natl Acad Sci USA (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
24. Fuchs TA, Brill A, Wagner DD. Neutrophil Extracellular Trap (Net) Impact On Deep Vein Thrombosis. Arterioscler Thromb Vasc Biol (2012) 32(8):1777–83. doi: 10.1161/ATVBAHA.111.242859
25. Engelmann B, Massberg S. Thrombosis As An Intravascular Effector of Innate Immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
26. Kawata J, Aoki M, Ishimaru Y, Ono T, Sagara K, Narahara S, et al. Mechanism of Tissue Factor Production by Monocytes Stimulated With Neutrophil Elastase. Blood Cells Mol Dis (2015) 54(2):206–9. doi: 10.1016/j.bcmd.2014.10.005
27. Zhou J, May L, Liao P, Gross Pl, Weitz J. Inferior Vena Cava Ligation Rapidly Induces Tissue Factor Expression and Venous Thrombosis in Rats. Arterioscler Thromb Vasc Biol (2009) 29(6):863–9. doi: 10.1161/ATVBAHA.109.185678
28. Day Sm, Reeve Jl, Pedersen B, Farris DM, Myers DD, Im M, et al. Macrovascular Thrombosis Is Driven by Tissue Factor Derived Primarily From the Blood Vessel Wall. Blood (2005) 105(1):192–8. doi: 10.1182/blood-2004-06-2225
29. Von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice In Vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
30. Kambas K, Mitroulis I, Ritis K. The Emerging Role of Neutrophils in Thrombosis-The Journey of Tf Through Nets. Front Immunol (2012) 3:385. doi: 10.3389/fimmu.2012.00385
31. Ponomaryov T, Payne H, Fabritz L, Wagner DD, Brill A. Mast Cells Granular Contents Are Crucial for Deep Vein Thrombosis in Mice. Circ Res (2017) 121(8):941–50. doi: 10.1161/CIRCRESAHA.117.311185
32. Huang Al, Bosco JJ, Peter K. Mast Cell: An Unexpected Villain in Venous Thromboembolism. Circ Res (2017) 121(8):899–901. doi: 10.1161/CIRCRESAHA.117.311777
33. Choi SH, Smith SA, Morrissey JH. Polyphosphate Accelerates Factor V Activation by Factor Xia. Thromb Haemost (2015) 113(3):599–604. doi: 10.1160/TH14-06-0515
34. Docampo R. Polyphosphate: A Target for Thrombosis Attenuation. Blood (2014) 124(22):3177–8. doi: 10.1182/blood-2014-09-601641
35. Morrissey Jh. Polyphosphate: A Link Between Platelets, Coagulation and Inflammation. Int J Hematol (2012) 95(4):346–52. doi: 10.1007/s12185-012-1054-5
36. Orbe J, Chordá C, Montes R, Páramo J. Changes in the Fibrinolytic Components of Cultured Human Umbilical Vein Endothelial Cells Induced by Endotoxin, Tumor Necrosis Factor-Alpha and Interleukin-1alpha. Haematologica (1999) 84(4):306–11.
37. Esmon C. Possible Involvement of Cytokines in Diffuse Intravascular Coagulation and Thrombosis. Baillieres Clin Haematol (1994) 7(3):453–68. doi: 10.1016/S0950-3536(05)80093-9
38. Van Minkelen R, De Visser Mc, Houwing-Duistermaat Jj, Vos Hl, Bertina Rm, Rosendaal Fr. Haplotypes of Il1b, Il1rn, Il1r1, and Il1r2 and the Risk of Venous Thrombosis. Arterioscler Thromb Vasc Biol (2007) 27(6):1486–91. doi: 10.1161/ATVBAHA.107.140384
39. Jerjes-Sanchez C. Venous and Arterial Thrombosis: A Continuous Spectrum of the Same Disease. Eur Heart J (2005) 26(1):3–4. doi: 10.1093/eurheartj/ehi041
40. Vazquez-Garza E, Jerjes-Sanchez C, Navarrete A, Joya-Harrison J, Rodriguez D. Venous Thromboembolism: Thrombosis, Inflammation, and Immunothrombosis for Clinicians. J Thromb Thrombolysis (2017) 44(3):377–85. doi: 10.1007/s11239-017-1528-7
41. Henke PK, Wakefield TW, Kadell AM, Linn MJ, Varma MR, Sarkar M, et al. Interleukin-8 Administration Enhances Venous Thrombosis Resolution in a Rat Model. J Surg Res (2001) 99(1):84–91. doi: 10.1006/jsre.2001.6122
42. Myers DD Jr, Hawley AE, Farris DM, Chapman AM, Wrobleski SK, Henke PK, et al. Cellular Il-10 Is More Effective Than Viral Il-10 in Decreasing Venous Thrombosis. J Surg Res (2003) 112(2):168–74. doi: 10.1016/S0022-4804(03)00144-6
43. Ding Y, Qin L, Kotenko SV, Pestka S, Bromberg JS. A Single Amino Acid Determines the Immunostimulatory Activity of Interleukin 10. J Exp Med (2000) 191(2):213–24. doi: 10.1084/jem.191.2.213
44. Henke PK, Debrunye LA, Strieter RM, Bromberg JS, Prince M, Kadell AM, et al. Viral Il-10 Gene Transfer Decreases Inflammation and Cell Adhesion Molecule Expression in a Rat Model of Venous Thrombosis. J Immunol (2000) 164(4):2131–41. doi: 10.4049/jimmunol.164.4.2131
45. Li G, Zhou R, Zhao X, Liu R, Ye C. Correlation Between the Expression of Il−18 and Deep Venous Thrombosis. Int J Mol Med (2018) 42(2):2972. doi: 10.3892/ijmm.2018.3682
46. Wakefield TW, Greenfield LJ, Rolfe MW, DeLucia A 3rd, Strieter RM, Abrams GD, et al. Inflammatory and Procoagulant Mediator Interactions in an Experimental Baboon Model of Venous Thrombosis. Thromb Haemost (1993) 69(2):164–72. doi: 10.1055/s-0038-1651574
47. Luther N, Shahneh F, Brähler M, Krebs F, Jäckel S, Subramaniam S, et al. Innate Effector-Memory T-Cell Activation Regulates Post-Thrombotic Vein Wall Inflammation and Thrombus Resolution. Circ Res (2016) 119(12):1286–95. doi: 10.1161/CIRCRESAHA.116.309301
48. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Inui M, Mukaida N, et al. Absence of Ifn-Γ Accelerates Thrombus Resolution Through Enhanced Mmp-9 and Vegf Expression in Mice. J Clin Invest (2011) 121(7):2911–20. doi: 10.1172/JCI40782
49. Laurance S, Bertin FR, Ebrahimian T, Kassim Y, Rys RN, Lehoux S, et al. Gas6 Promotes Inflammatory (Ccr2(Hi)Cx3cr1(Lo)) Monocyte Recruitment in Venous Thrombosis. Arterioscler Thromb Vasc Biol (2017) 37(7):1315–22. doi: 10.1161/ATVBAHA.116.308925
50. Cohoon KP, Mazur M, Mcbane RD, Ammash N, Asirvatham SJ, Wysokinski We. Association of Soluble Cd40 Ligand With Duration of Atrial Fibrillation and With Intensity of Spontaneous Echocardiographic Contrast. JACC Clin Electrophysiol (2016) 2(5):623–32. doi: 10.1016/j.jacep.2016.03.006
51. Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, et al. Ectonucleotidase Tri(Di)Phosphohydrolase-1 (Entpd-1) Disrupts Inflammasome/Interleukin 1β-Driven Venous Thrombosis. J Clin Invest (2019) 129(7):2872–7. doi: 10.1172/JCI124804
52. Mcever RP. Adhesive Interactions of Leukocytes, Platelets, and the Vessel Wall During Hemostasis and Inflammation. Thromb Haemost (2001) 86(3):746–56. doi: 10.1055/s-0037-1616128
53. Mcever RP. Cummings RD. Role of Psgl-1 Binding to Selectins in Leukocyte Recruitment. J Clin Invest (1997) 100(11 Suppl):S97–103.
54. Mcever RD. P-Selectin and Psgl-1: Exploiting Connections Between Inflammation and Venous Thrombosis. Thromb Haemost (2002) 87(3):364–5. doi: 10.1055/s-0037-1613012
55. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-Selectin Promotes Neutrophil Extracellular Trap Formation in Mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
56. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils Scan for Activated Platelets to Initiate Inflammation. Science (2014) 346(6214):1234–8. doi: 10.1126/science.1256478
57. Wang LM, Duan QL, Yang F, Yi XH, Zeng Y, Tian HY, et al. Activation of Circulated Immune Cells and Inflammatory Immune Adherence Are Involved in the Whole Process of Acute Venous Thrombosis. Int J Clin Exp Med (2014) 7(3):566–72.
58. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, A Soloviev D, et al. Leukocyte Integrin Mac-1 Regulates Thrombosis Via Interaction With Platelet Gpibα. Nat Commun (2017) 8:15559. doi: 10.1038/ncomms15559
59. Payne H, Ponomaryov T, Watson SP, Brill A. Mice With A Deficiency in Clec-2 Are Protected Against Deep Vein Thrombosis. Blood (2017) 129(14):2013–20. doi: 10.1182/blood-2016-09-742999
60. Høiland Ii, Liang Ra, Hindberg K, Latysheva N, Brekke OL, Mollnes TE, et al. Associations Between Complement Pathways Activity, Mannose-Binding Lectin, and Odds of Unprovoked Venous Thromboembolism. Thromb Res (2018) 169:50–6. doi: 10.1016/j.thromres.2018.06.019
61. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of Nlrp3 Inflammasome Complex Potentiates Venous Thrombosis in Response to Hypoxia. Proc Natl Acad Sci USA (2017) 114(18):4763–8. doi: 10.1073/pnas.1620458114
62. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil Histone Modification by Peptidylarginine Deiminase 4 Is Critical for Deep Vein Thrombosis in Mice. Proc Natl Acad Sci USA (2013) 110(21):8674–9. doi: 10.1073/pnas.1301059110
63. Massberg S, Grahl L, Von Bruehl Ml, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal Coupling of Coagulation and Innate Immunity Via Neutrophil Serine Proteases. Nat Med (2010) 16(8):887–96. doi: 10.1038/nm.2184
64. Jasuja R, Furie B, Furie BC. Endothelium-Derived But Not Platelet-Derived Protein Disulfide Isomerase Is Required for Thrombus Formation In Vivo. Blood (2010) 116(22):4665–74. doi: 10.1182/blood-2010-04-278184
65. Henke PK. Plasmin and Matrix Metalloproteinase System in Deep Venous Thrombosis Resolution. Vascular (2007) 15(6):366–71. doi: 10.2310/6670.2007.00050
66. Hayashi H, Cherpokova D, Martinod K, Witsch T, Wong SL, Gallant M, et al. Sirt3 Deficiency Does Not Affect Venous Thrombosis Or Netosis Despite Mild Elevation of Intracellular Ros in Platelets and Neutrophils in Mice. PloS One (2017) 12(12):E0188341. doi: 10.1371/journal.pone.0188341
67. Mu SW, Dang Y, Fan YC, Zhang H, Zhang JH, Wang W, et al. Effect of Hmgb1 and Rage On Brain Injury and the Protective Mechanism of Glycyrrhizin in Intracranial−Sinus Occlusion Followed by Mechanical Thrombectomy Recanalization. Int J Mol Med (2019) 44(3):813–22.
68. Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, et al. High-Mobility Group Box 1 Protein Promotes Development of Microvascular Thrombosis in Rats. J Thromb Haemost (2007) 5(1):109–16. doi: 10.1111/j.1538-7836.2006.02255.x
69. Lippi G, Favaloro EJ, Montagnana M, Franchini M. C-Reactive Protein and Venous Thromboembolism: Causal Or Casual Association. Clin Chem Lab Med (2010) 48(12):1693–701. doi: 10.1515/CCLM.2010.335
70. Henke PK, Mitsuya M, Luke CE, Elfline MA, Baldwin JF, Deatrick KB, et al. Toll-Like Receptor 9 Signaling Is Critical for Early Experimental Deep Vein Thrombosis Resolution. Arterioscler Thromb Vasc Biol (2011) 31(1):43–9. doi: 10.1161/ATVBAHA.110.216317
72. Jezovnik MK, Poredos P. Factors Influencing the Recanalisation Rate of Deep Venous Thrombosis. Int Angiol (2012) 31(2):169–75.
73. Matos MF, Lourenço DM, Orikaza CM, Gouveia CP, Morelli VM. Abdominal Obesity and the Risk of Venous Thromboembolism Among Women: A Potential Role of Interleukin-6. Metab Syndr Relat Disord (2013) 11(1):29–34. doi: 10.1089/met.2012.0077
74. Shbaklo H, Holcroft CA, Kahn SR. Levels of Inflammatory Markers and the Development of the Post-Thrombotic Syndrome. Thromb Haemost (2009) 101(3):505–12. doi: 10.1160/TH08-08-0511
75. Maruna P, Kunstyr J, Plocova KM, Mlejnsky F, Hubacek J, Klein AA, et al. Predictors of Infection After Pulmonary Endarterectomy for Chronic Thrombo-Embolic Pulmonary Hypertension. Eur J Cardiothorac Surg (2011) 39(2):195–200. doi: 10.1016/j.ejcts.2010.05.018
76. André P, Hartwell D, Hrachovinová I, Saffaripour S, Wagner DD. Pro-Coagulant State Resulting From High Levels of Soluble P-Selectin in Blood. Proc Natl Acad Sci USA (2000) 97(25):13835–40. doi: 10.1073/pnas.250475997
77. Myers DD Jr, Schaub R, Wrobleski SK, Londy FJ 3rd, Fex BA, Chapman AM, et al. P-Selectin Antagonism Causes Dose-Dependent Venous Thrombosis Inhibition. Thromb Haemost (2001) 85(3):423–9.
78. Wakefield TW, Strieter RM, Downing LJ, Kadell AM, Wilke CA, Burdick MD, et al. P-Selectin and Tnf Inhibition Reduce Venous Thrombosis Inflammation. J Surg Res (1996) 64(1):26–31. doi: 10.1006/jsre.1996.0301
79. Eppihimer Mj, Schaub Rg. P-Selectin-Dependent Inhibition of Thrombosis During Venous Stasis. Arterioscler Thromb Vasc Biol (2000) 20(11):2483–8. doi: 10.1161/01.ATV.20.11.2483
80. Lee JJ, Pope JE. A Meta-Analysis of the Risk of Venous Thromboembolism in Inflammatory Rheumatic Diseases. Arthritis Res Ther (2014) 16(5):435. doi: 10.1186/s13075-014-0435-y
81. Gibson T, Myers AR. Nervous System Involvement in Systemic Lupus Erythematosus. Ann Rheum Dis (1975) 35(5):398–406. doi: 10.1136/ard.35.5.398
82. Masheter HC. Behcet’s Syndrome Complicated by Intracranial Thrombophlebitis. Proc R Soc Med (1959) 52:1039–40.
83. Talbot RW, Heppell J, Dozois RR, Beart RW Jr. Vascular Complications of Inflammatory Bowel Disease. Mayo Clin Proc (1986) 61(2):140–5. doi: 10.1016/S0025-6196(12)65200-8
84. Lood C, Eriksson S, Gullstrand B, Jönsen A, Sturfelt G, Truedsson L, et al. Increased C1q, C4 and C3 Deposition on Platelets in Patients With Systemic Lupus Erythematosus–A Possible Link to Venous Thrombosis. Lupus (2012) 21(13):1423–32. doi: 10.1177/0961203312457210
85. Ureten K, Ertenli I, Oztürk MA, Kiraz S, Onat AM, Tuncer M, et al. Neutrophil Cd64 Expression in Behçet’s Disease. J Rheumatol (2005) 32(5):849–52.
86. Sahin S, Akoğlu T, Direskeneli H, Sen LS, Lawrence R. Neutrophil Adhesion to Endothelial Cells and Factors Affecting Adhesion in Patients With Behçet’s Disease. Ann Rheum Dis (1996) 55(2):128–33. doi: 10.1136/ard.55.2.128
87. Danese S, Papa A, Saibeni S, Repici A, Malesci A, Vecchi M. Inflammation and Coagulation in Inflammatory Bowel Disease: the Clot Thickens. Am J Gastroenterol (2007) 102(1):174–86. doi: 10.1111/j.1572-0241.2006.00943.x
88. Rashad S, Niizuma K, Sato-Maeda M, Fujimura M, Mansour A, Endo H, et al. Early Bbb Breakdown and Subacute Inflammasome Activation and Pyroptosis as A Result of Cerebral Venous Thrombosis. Brain Res (2018) 1699:54–68. doi: 10.1016/j.brainres.2018.06.029
89. Nagai M, Terao S, Yilmaz G, Yilmaz CE, Esmon CT, Watanabe E, et al. Roles of Inflammation and the Activated Protein C Pathway in the Brain Edema Associated With Cerebral Venous Sinus Thrombosis. Stroke (2010) 41(1):147–52. doi: 10.1161/STROKEAHA.109.562983
90. Wakefield TW, Henke PK. The Role of Inflammation in Early and Late Venous Thrombosis: Are There Clinical Implications. Semin Vasc Surg (2005) 18(3):118–29. doi: 10.1053/j.semvascsurg.2005.05.003
91. Downing LJ, Wakefield TW, Strieter RM, Prince MR, Londy FJ, Fowlkes JB, et al. Anti-P-Selectin Antibody Decreases Inflammation and Thrombus Formation in Venous Thrombosis. J Vasc Surg (1997) 25(5):816–27; Discussion 828. doi: 10.1016/S0741-5214(97)70211-8
92. Myers D Jr, Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, et al. Selectins Influence Thrombosis in a Mouse Model of Experimental Deep Venous Thrombosis. J Surg Res (2002) 108(2):212–21. doi: 10.1006/jsre.2002.6552
93. Mamik MK, Power C. Inflammasomes in Neurological Diseases: Emerging Pathogenic and Therapeutic Concepts. Brain (2017) 140(9):2273–85. doi: 10.1093/brain/awx133
94. Ding R, Ou W, Chen C, Liu Y, Li H, Zhang X, et al. Endoplasmic Reticulum Stress and Oxidative Stress Contribute to Neuronal Pyroptosis Caused by Cerebral Venous Sinus Thrombosis in Rats: Involvement of Txnip/Peroxynitrite-Nlrp3 Inflammasome Activation. Neurochem Int (2020) 141:104856. doi: 10.1016/j.neuint.2020.104856
95. Chen D, Dixon BJ, Doycheva DM, Li B, Zhang Y, Hu Q, et al. Ire1α Inhibition Decreased Txnip/Nlrp3 Inflammasome Activation Through Mir-17-5p After Neonatal Hypoxic-Ischemic Brain Injury in Rats. J Neuroinflamm (2018) 15(1):32. doi: 10.1186/s12974-018-1077-9
96. Bharti V, Tan H, Zhou H, Wang Jf. Txnip Mediates Glucocorticoid-Activated Nlrp3 Inflammatory Signaling in Mouse Microglia. Neurochem Int (2019) 131:104564. doi: 10.1016/j.neuint.2019.104564
97. Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Neurovascular Protective Effect of Fetpps in N-Methyl-D-Aspartate Model: Similarities to Diabetes. Am J Pathol (2010) 177(3):1187–97. doi: 10.2353/ajpath.2010.091289
98. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
99. Andersson U, Yang H, Harris H. High-Mobility Group Box 1 Protein (Hmgb1) Operates as An Alarmin Outside As Well As Inside Cells. Semin Immunol (2018) 38:40–8. doi: 10.1016/j.smim.2018.02.011
100. Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of An Immune Modulator Fingolimod on Acute Ischemic Stroke. Proc Natl Acad Sci USA (2014) 111(51):18315–20. doi: 10.1073/pnas.1416166111
101. Song SY, Zhao XX, Rajah G, Hua C, Kang RJ, Han YP, et al. Clinical Significance of Baseline Neutrophil-To-Lymphocyte Ratio in Patients With Ischemic Stroke or Hemorrhagic Stroke: An Updated Meta-Analysis. Front Neurol (2019) 10:1032. doi: 10.3389/fneur.2019.01032
102. Esmon CT, Xu J, Lupu F. Innate Immunity and Coagulation. J Thromb Haemost (2011) 9 Suppl 1(Suppl 1):182–8. doi: 10.1111/j.1538-7836.2011.04323.x
103. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol (2020) 77(6):683–90. doi: 10.1001/jamaneurol.2020.1127
104. Baldini T, Asioli GM, Romoli M, Carvalho Dias M, Schulte EC, Hauer L, et al. Cerebral Venous Thrombosis and Severe Acute Respiratory Syndrome Coronavirus-2 Infection: A Systematic Review and Meta-Analysis. Eur J Neurol (2021) 28(10):3478–90. doi: 10.1111/ene.14727
105. Taquet M, Husain M, Geddes JR, Luciano S, Harrison PJ. Cerebral Venous Thrombosis and Portal Vein Thrombosis: A Retrospective Cohort Study of 537,913 Covid-19 Cases. Eclinicalmedicine (2021) 39:101061. doi: 10.1016/j.eclinm.2021.101061
106. Siegler JE, Cardona P, Arenillas JF, Talavera B, Guillen AN, Chavarría-Miranda A, et al. Cerebrovascular Events and Outcomes in Hospitalized Patients With Covid-19: the Svin Covid-19 Multinational Registry. Int J Stroke (2021) 16(4):437–47. doi: 10.1177/1747493020959216
107. Nagashima S, Mendes MC, Camargo Martins AP, Borges NH, Godoy TM, Miggiolaro A, et al. Endothelial Dysfunction and Thrombosis in Patients With Covid-19-Brief Report. Arterioscler Thromb Vasc Biol (2020) 40(10):2404–7. doi: 10.1161/ATVBAHA.120.314860
108. Ye J, Zhang B, Xu J, Chang Q, McNutt MA, Korteweg C, et al. Molecular Pathology in the Lungs of Severe Acute Respiratory Syndrome Patients. Am J Pathol (2007) 170(2):538–45. doi: 10.2353/ajpath.2007.060469
109. Hamming I, Timens W, Bulthuis Ml, Lely AT, Navis G, Van Goor H. Tissue Distribution of Ace2 Protein, the Functional Receptor for Sars Coronavirus. A First Step in Understanding Sars Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
110. Libby P, Lüscher T. Covid-19 Is, in the End, An Endothelial Disease. Eur Heart J (2020) 41(32):3038–44. doi: 10.1093/eurheartj/ehaa623
111. Wright Fl, Vogler To, Moore Ee, Moore HB, Wohlauer MV, Urban S, et al. Fibrinolysis Shutdown Correlation With Thromboembolic Events in Severe Covid-19 Infection. J Am Coll Surg (2020) 231(2):193–203.E1. doi: 10.1016/j.jamcollsurg.2020.05.007
112. Klok FA, Kruip M, van der Meer N, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the High Cumulative Incidence of Thrombotic Complications in Critically Ill Icu Patients With Covid-19: An Updated Analysis. Thromb Res (2020) 191:148–50. doi: 10.1016/j.thromres.2020.04.041
113. Mcgonagle D, Sharif K, Regan A, Bridgewood C. The Role of Cytokines Including Interleukin-6 in Covid-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
114. Speeckaert MM, Speeckaert R, Delanghe JR. Potential Underlying Mechanisms of Cerebral Venous Thrombosis Associated With Covid-19. J Neuroradiol (2020) 47(6):473–4. doi: 10.1016/j.neurad.2020.06.009
115. Rizk JG, Gupta A, Sardar P, Henry BM, Lewin JC, Lippi G, et al. Clinical Characteristics and Pharmacological Management of Covid-19 Vaccine-Induced Immune Thrombotic Thrombocytopenia With Cerebral Venous Sinus Thrombosis: A Review. JAMA Cardiol (2021) 6(12):1451–60. doi: 10.1001/jamacardio.2021.3444
116. Schulz JB, Berlit P, Diener Hc, Gerloff C, Greinacher A, Klein C, et al. Covid-19 Vaccine-Associated Cerebral Venous Thrombosis in Germany. Ann Neurol (2021) 90(4):627–39. doi: 10.1002/ana.26172
117. Lee GM, Arepally GM. Heparin-Induced Thrombocytopenia. Hematol Am Soc Hematol Educ Program (2013) 2013:668–74. doi: 10.1182/asheducation-2013.1.668
118. Thakur KT, Tamborska A, Wood GK, McNeill E, Roh D, Akpan IJ, et al. Clinical Review of Cerebral Venous Thrombosis in the Context of Covid-19 Vaccinations: Evaluation, Management, and Scientific Questions. J Neurol Sci (2021) 427:117532. doi: 10.1016/j.jns.2021.117532
Keywords: cerebral venous thrombosis, inflammation, brain injury, COVID-19, cytokines
Citation: Ding J, Song B, Xie X, Li X, Chen Z, Wang Z, Pan L, Lan D and Meng R (2022) Inflammation in Cerebral Venous Thrombosis. Front. Immunol. 13:833490. doi: 10.3389/fimmu.2022.833490
Received: 11 December 2021; Accepted: 28 February 2022;
Published: 04 April 2022.
Edited by:
Vijay Kumar, Duke University, United StatesReviewed by:
Mohammad Wasay, Aga Khan University, PakistanAleksandra Antovic, Karolinska Institutet, Sweden
Copyright © 2022 Ding, Song, Xie, Li, Chen, Wang, Pan, Lan and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ran Meng, dmljdG9yNjVAMTI2LmNvbQ==
†ORCID: Jiayue Ding, orcid.org/0000-0003-3562-2187
Ran Meng, orcid.org/0000-0003-1190-4710