 Xiaochen Qi
Xiaochen Qi Qifei Wang
Qifei Wang Guangzhen Wu
Guangzhen Wu- First Affiliated Hospital, Dalian Medical University, Dalian, China
The development of cancer treatment methods is constantly changing. For common cancers, our treatment methods are still based on conventional treatment methods, such as chemotherapy, radiotherapy, and targeted drug therapy. Nevertheless, the emergence of tumor resistance has a negative impact on treatment. Regulated cell death is a gene-regulated mode of programmed cell death. After receiving specific signal transduction, cells change their physical and chemical properties and the extracellular microenvironment, resulting in structural destruction and decomposition. As research accumulates, we now know that by precisely inducing specific cell death patterns, we can treat cancer with less collateral damage than other treatments. Many newly discovered types of RCD are thought to be useful for cancer treatment. However, some experimental results suggest that some RCDs are not sensitive to cancer cell death, and some may even promote cancer progression. This review summarizes the discovered types of RCDs, reviews their clinical efficacy in cancer treatment, explores their anticancer mechanisms, and discusses the feasibility of some newly discovered RCDs for cancer treatment in combination with the immune and tumor microenvironment.
Introduction
With the increase in the incidence of various types of cancer such as breast, kidney, and lung cancers, cancer therapy has always been the focus of clinical development (1). With the improvement of the tumor gene spectrum, cancer treatment has been developed from early radiotherapy and chemotherapy to targeted therapy, immunotherapy, and other personalized therapeutic approaches (2). The earlier methods mainly focused on preventing the biosynthesis of cancer cells and reducing their ability to reproduce and metastasize. As research has progressed, many researchers have realized that promoting the death of cancer cells is also a feasible way to treat cancer (3). Cell death can be classified into accidental cell death (ACD) and regulated cell death (RCD) (4). ACD is generally unregulated and usually results from detrimental stimuli that exceed the cell’s ability to control. RCD is defined as programmed cell death (PCD) and is generally regulated by signaling pathways (5). Since apoptosis was discovered in 1972, more than 15 types of RCDs have been unraveled by researchers (5, 6). The Nomenclature Committee on Cell Death (NCCD), updated in 2018, has formulated the current classification, interpretations in addition to the morphological, biochemical, and functional definitions of cell death (7). RCD has been widely studied in the field of cancer treatment, including apoptosis, necroptosis, and other forms of RCD. Additionally, it has been proven to be feasible for guiding the new direction of cancer treatment (8). Because of their different molecular mechanisms, different types of RCDs can often be used as therapeutic alternatives to each other (9). Although the positive role of RCD in cancer treatment is well established, it is still a double-edged sword as some studies have shown that the RCD mechanism can also be utilized to promote tumor growth (10). Therefore, the selective manipulation of RCD to treat cancer is the focus of current research. In some cancers related to lipid accumulation, such as renal and breast cancers, lipid metabolism often regulates cancer development (11–13). Many studies have shown that lipid metabolism is closely associated with some RCDs, such as apoptosis and ferroptosis (14–16). Therefore, targeting lipid metabolism to induce cancer cell death in cancers that are sensitive to lipid metabolism can be an adequate therapeutic approach. In addition, some RCDs such as immunogenic cell death (ICD) have been considered to be associated with immunology (17). This review introduces different kinds of RCDs, examines their relationship with each other on one hand and their relationship with regards to cancer occurrence and development on the other hand. Additionally, this review discusses the possibility of application of various RCDs including lipid metabolism and cellular immunity in cancer treatment.
Classical RCD
Based on the functional differences, there are two main types of cell death: ACD and RCD (6). ACD is an uncontrolled mode of cell death triggered by external detrimental stimuli due to the inability of the affected cells to respond beyond their regulatory capacity (18). RCD is a cell death pathway regulated by genes or signaling molecules and involves a signaling cascade by effector molecules. RCD generally has unique biochemical and morphological characteristics as well as immunological consequences (19).
Since the discovery of apoptosis in 1972 (20, 21), research on RCD has shown continuous progress. By 2018, more than 10 different types of RCD were identified. These include necroptosis, pyroptosis, ferroptosis, parthanatos, immunogenic cell death (ICD), lysosome-dependent cell death (LCD), necrotic cell death (NCD), and autophagy-dependent cell death (5).
Apoptosis and Caspase Family
Apoptosis is an active programmed cell death caused by gene regulation that maintains homeostasis in the internal environment. The apoptosis activation pathway is diverse, including intrinsic and extrinsic pathways among others. These pathways will eventually promote the cysteinyl aspartate specific proteinase (caspase) cascade reaction; thus, apoptosis depends on the caspase family (22). The extrinsic pathway can be activated by binding of receptors, including type 1 TNF receptor (TNFR1) and related protein Fas (CD95) to their ligands, TNF and Fas ligand (FasL), respectively (23). When the intrinsic pathway is activated, the Bcl-2 pro-apoptotic protein family (Bax, Bak, BID, PUMA, etc.) is activated, releasing cytochrome C from the mitochondria. Cytochrome C then binds to apoptotic protease activating factor-1 (APAF-1), forming a polymer that activates and binds to caspase-9 forming apoptotic bodies (24). Activated caspase-9, in turn, activates caspase-3, which can shear poly (ADP-ribose) polymerase (PARP) that normally exhibits a negative regulation on the endonuclease activity. This results in increasing the endonuclease activity and DNA cleavage (25). The initiator caspase-3 and the effector caspase-9 play roles upstream and downstream of the apoptosis signaling pathway, respectively (22, 26). Fourteen different members of the caspase family were identified. Most caspases directly participate in the apoptosis process, while only a few caspases participate indirectly. The caspases involved in signal transduction not only affect apoptosis, but also affect a variety of RCDS, including pyroptosis and anoikis (27), in addition to affecting inflammation (27) (Figure 1). This review summarizes the members of the caspase family and their roles based on their relative importance (Table 1).
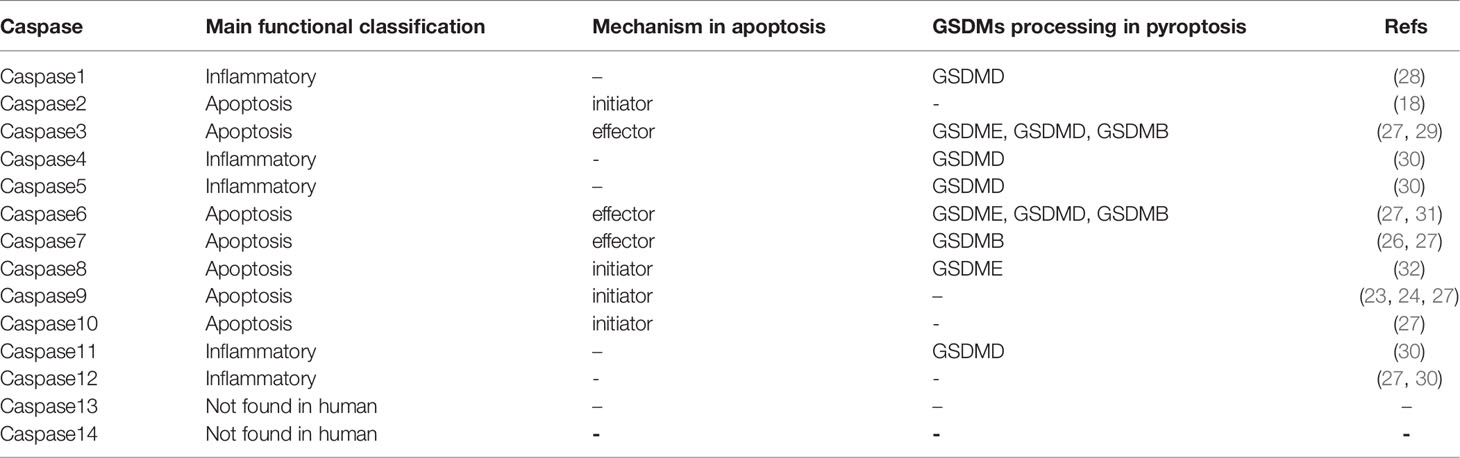
Table 1 The role and mechanism of Caspase family in Apoptosis and Pyroptosis.
Apoptosis in Cancer
In normal cells, detection of irreversible DNA damage can induce apoptosis. However, cells having an overexpression of the apoptosis inhibitor Bcl-2 (33) or p53 defects (34) do not undergo apoptosis and pass on these DNA mutations during cell division, leading to the accumulation of mutations and thus contributing to the occurrence of cancer. The current mainstream cancer therapy targeting apoptosis are the drugs targeting Bcl-2 family proteins, including Oblimersen sodium (Genasense Bcl-2 antisense oligonucleotide (35)) (36), inhibitors of Bcl-2 family (37), BH3 mimetics (38), and others. Silencing the anti-apoptotic Bcl family proteins/genes has also shown a promising therapeutic effect. Studies have shown that Bcl-2-specific siRNA can effectively inhibit the proliferation and promote apoptosis of pancreatic cancer cells (39). Moreover, silencing Bmi-1 expression in breast cancer cells can also promote apoptosis by down-regulating Bcl-2 expression (40). Studies have confirmed the feasibility of using the regulatory miR-15/16 of Bcl-2-associated X protein (BAX)/Bcl-2 homologous antagonist killer (BAK)-dependent pathway, the classical pathway of Bcl-2, to fight cancer (41). On the other hand, p53 plays a main role in ferroptosis (42) as discussed in this review. In general, the mechanisms by which tumor cells evade apoptosis can be roughly divided into 1. the balance of pro-apoptotic protein and anti-apoptotic protein is disrupted, 2. the function of caspase is reduced, 3. the death receptor signal is impaired (22).
Necroptosis
Apoptosis has conventionally been considered the only form of RCD, while necrosis has been considered a type of accidental death that is not regulated by molecular events. This misconception was later amended when necroptosis was discovered. It is a type of RCD resembling apoptosis in mechanism and necrosis in appearance (9). Necroptosis is a caspase-independent form of RCD that is driven by the activation of receptor-interacting protein kinases (RIPs) and mixed lineage kinase domain-like (MLKL) (43–45). The TNFα receptor superfamily, T cell receptors (TCRs), interferon receptors (IFNRs), and toll-like receptors (TLRs) bind to their ligands to initiate the necroptosis signaling process (8). For example, cylindromatosis (CYLD) is stimulated when the TNF-α binds to its receptor, the TNF-α receptor (46). RIP1 is then activated by CYLD to trigger the phosphorylation of RIP3 and MLKL. The complex formed by RIP1, RIP3, and MLKL, known as the necrosome or RIP1-RIP3-MLKL complex (47), regulates the transfer of the phosphorylated MLKL trimer to the plasma membrane, resulting in increased permeability of necrotic membranes (48). In addition to changing the osmotic pressure of the cell, necrotic bodies can also cause cell death by regulating ROS balance and intracellular ATP content (45). Moreover, a previous study reported that dimerization of RIP1 can lead to self-activation, followed by binding to the caspase-8–FADD complex to form complex IIa-RDA (RIP1-dependent apoptosis) (49). When caspase-8 is inactivated or inhibitors of apoptosis proteins (IAPs) are inhibited, RIP1 binds RIP3 and MLKL to form a necrosome, leading to necroptosis (3, 44, 50) (Figure 1). This shows that caspase-8 plays an important role in both apoptosis and necroptosis (51). A methylation study has shown that RIP3 expression is absent in cancer cells (52). Many key necroptosis factors are downregulated in cancer, including RIPs, MLKL, CYLD, and FADD. Stoll et al. (53) reported the downregulation of RIP3 in breast cancer leading to a poor prognosis. Other studies have showed that the upregulation of RIP1 in lung cancer can reduce the ROS, thus promoting oncogenesis (54). However, in head and neck squamous cell carcinoma, RIP1 was found to be downregulated and was correlated with enhanced tumorigenesis (55). Decreased CYLD expression found in melanoma and chronic lymphocytic cells can enhance tumor progression and reduce OS in patients (56, 57). Research has also shown that decreased MLKL expression decreases the overall survival and leads to poor prognosis in different types of cancer (58–60).
Apoptosis and necroptosis, the first two forms of RCD discovered, have been playing an active role in cancer. The development of various new small-molecule drugs is aimed at activating the apoptotic pathway of cancer cells, thereby promoting cell death. The current mainstream direction points to the gene-level regulation of cellular apoptosis and necroptosis, and inhibiting or overexpressing the expression levels of related genes promotes the occurrence of cellular apoptosis and necroptosis. Not only these two RCDs, but more newly discovered RCD types have also been confirmed to be applicable in cancer treatment.
Caspase-Dependent RCD in Target Therapy
Anoikis and Autophagy
Anoikis was first discovered in 1993 and explains the susceptibility of outlier cells to death (61, 62). Apoptosis occurs when tumor cells detach from the primary site and spread through the circulatory system owing to the biological relevance and function of anoikis (63). Anoikis includes endogenous and exogenous pathways. In the endogenous pathway, normal aggregation of cells can come in contact with the extracellular matrix (ECM), which in turn can activate the pro-apoptotic factor Bim/Bid, synthesize Bax/Bak oligomers, inhibit the anti-apoptotic protein Bcl-XL, and eventually induce apoptosis (64, 65). In the exogenous pathway, the Fas ligand and TNF receptor apoptosis-inducing ligand (TRAIL) and their corresponding receptors bind to polymerized receptors to recruit and activate the adaptor protein Fas-related death domain (FADD). Afterward, the death effect domain of FADD binds to the death receptor to recruit caspase-8, resulting in the formation of the death-inducing signal complex object (DISC). This in turn promotes the dimerization, activation, and cleavage of caspase-8, which is then released to the cytoplasm, activating the effector molecules caspase-3 and caspase-7. Effector caspases cleave and degrade different cellular proteins eventually leading to apoptosis. FADD-induced activation of caspase-8 can also activate and dissociate Bid, destroy the mitochondrial membrane, and lead to apoptosis (8, 66, 67). There is a less common pathway in which AES/TLE1 heterooligomers are translocated from the nucleus to the cytoplasm after cells lose contact with the ECM. Subsequent proteasomal downregulation of TLE1 leads to the activation of the death signaling pathway (63). Anoikis is thought to occur when cancer cells are detached and lose contact with the ECM. However, tumor cells may resist anoikis through mutation-induced secretion abnormalities after they lose the intercellular and ECM contact. Constructional activation of pro-survival signals such as PI3K, RAS-ERK, NF-κB, and Rho GTPase occurs frequently in cancer cells and antagonizes anoikis (68, 69). Autophagy has also been found to protect cancer cells from anoikis (70, 71). Therefore, treatment approaches targeting the inhibition of cancer cells’ resistance to anoikis is currently the mainstream research strategy.
Cancer cells acquire resistance against anoikis by regulating integrins and activating the EMT. Integrins play a key role in regulating cell contact with the ECM (72). Integrins are bidirectional signaling molecules with two different conformational states that determine their different affinities for the ECM. Closed integrins have a low affinity for ECM ligands, while open integrins have a high affinity for ECM and bind to ECM to induce downstream signal transduction (73). Previously, integrins and their cancer-regulating functions were thought to be limited to signal transduction in plasma membranes and focal adhesions. The loss of ECM tension during matrix degradation was suggested to allow the uptake of active integrin-binding ligand fragments. This explains why ECM ligands, such as active integrins, are easily detected in the endosomes of cancer cells (73, 74). Epithelial-mesenchymal transition (EMT) promotes epithelial cancer cells to acquire mesenchymal characteristics through inhibiting epithelial cell markers and upregulating mesenchymal markers. This facilitates the metastasis of cancer cells and is an important resistance mechanism of cancer cells to anoikis (75).
Autophagy, a type of cell death that is completely varies from apoptosis, has been recognized to have a dual role where it can either inhibit or promote cancer metastasis (8). The anti-metastatic effect of autophagy is mainly through four mechanisms. The first mechanism is through decreasing tumor necrosis induced by hypoxia and preventing the infiltration of inflammatory cells. The second mechanism is through regulating the release of HMGB1 from cancer cells, thus mediating the anticancer immune response. The third mechanism of autophagy involves the direct death of cancer cells. The last mechanism is through triggering apoptosis, thus resulting in cancer cell death (76). On the other hand, autophagy promotes cancer metastasis and has been linked to anoikis. Research has shown that autophagy provides a mechanism for stromal isolated premetastatic tumor cells to resist anoikis (71). In a hepatocellular carcinoma metastasis model, inhibition of autophagy did not affect cell invasion, migration, or EMT, but attenuated the anoikis resistance of hepatocellular carcinoma cells. This greatly enhanced the ability of hepatocellular carcinoma to metastasize (77). Similarly, other studies have shown that ECM detachment and β1 integrin inhibition can induce autophagy (78). Experiments have shown that there is a correlation between autophagy and anoikis.
Cancer treatment for anoikis are becoming available, and more in vitro and in vivo trials are supporting this treatment strategy. Studies have shown that carnitine palmityl transferase 1A (CPT1A)-mediated fatty acid oxidation (FAO) can help colon cancer cells resist anoikis and thus promote colon cancer metastasis. Researchers have identified CPT1A as a potential target for colon cancer treatment (79). Myosin heavy chain 9 (MYH9) has also been found to promote the transcription of catenin beta 1 (CTNNB1), thus rendering resistance to gastric cancer cells against anoikis both in vivo and in vitro (69). Anoikis has also been employed for the treatment of lung cancer. Researchers have found that the PLAG1-GDH1 axis improves the resistance of lung cancer cells against anoikis. Glutamate dehydrogenase 1 (GDH-1) can be upregulated by pleomorphic adenoma gene 1 (PLAG1), and its product, α-KG, activates calcium/calmodulin-dependent protein kinase kinase 2 (CamKK2) by enhancing the binding of CamKK2 to the substrate adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK). This binding contributes to energy production and thus to resistance against anoikis (80). Jin et al. showed that lactate dehydrogenase A (LDHA), an enzyme that catalyzes the conversion of pyruvate to lactate, is phosphorylated at tyrosine 10 by upstream kinases HER2 and Src, thus promoting anoikis resistance in breast cancer (81).
Pyroptosis
Pyroptosis, a type of RCD generally caused by the inflammasome, is mainly characterized by the expansion of the cells until the membrane is ruptured and the cellular contents overflow, thus inducing a potent inflammatory response (82). The occurrence of pyroptosis depends on the caspase family and the GSDM protein family. After the activated caspase cleaves the GSDM protein, the released GSDM n-terminal (GSDM-NT) (83) binds to and drills in the cell membrane, resulting in changes in the cellular osmotic pressure, cellular swelling, and eventually cell rupture (82, 84).
Currently, there are three main pathways of pyroptosis, namely the canonical inflammasome pathway, the non-canonical inflammasome pathway, and the extracellular fluid pathway (85). Caspases involved in these pathways include caspases-1, 4, 5, and 11 (85, 86). The canonical inflammasome pathway has a relatively clear mechanism. Inflammasomes in pyroptosis, including NLRP1, NLRP3, NLRC4, and AIM2, are usually activated by pathogens and their secretions such as thymodyl dipeptide, flagellin, and double-stranded DNA (dsDNA) (87–89) (Table 2). NLRP3, in particular, is activated by a wide range of stimuli, including ROS, extracellular RNA, uric acid, and cholesterol (Table 2) (90–92). The activated inflammasome combines with apoptosis-associated speck-like protein containing a CARD (ASC) and recruits procaspase-1, which activates caspase-1 (93, 94). Caspase-1 cleaves GSDMD and releases GSDMD-NT into the membrane, which leads to membrane drilling (95). Caspase 1 also cleaves proL-18 and proIL-1, which participate in the maturation of proIL-18 and proIL-1 β, which are in turn released into the extracellular space and trigger inflammatory responses (96, 97) (Figure 1). Pyroptosis is an extensive frontier of cancer. Current studies report that the role of pyroptosis in cancer cells is complicated; thus, the deployment of pyroptosis to combat cancer has always been a research focus area. In esophageal squamous cell carcinoma (ESCC), BI2536, an inhibitor of polo-like kinase 1(PLK1), was found to activate caspase-3 and BAX in combination with cisplatin, resulting in GSDME disruption and increased DNA damage. Meanwhile, GSDME was also found to be highly expressed in ESCC, suggesting that BI2536 can markedly increase the sensitivity of ESCC to chemotherapy (98). In gastric cancer (GC), the low expression of pyroptosis-affecting protein, GSDMD, is one of the reasons for promoting the proliferation of cancer cells. GSDMD was found to reduce the expression of cyclin A2 and cyclin-dependent kinase 2 (CDK2), which slowed down DNA synthesis and S-G2 phase progression in cyclin through ERK1/2, STAT3, and PI3K/AKT (99, 100), and ultimately slowed down GC cell proliferation (101, 102). In triple-negative breast cancer (TNBC), docosahexaenoic acid (DHA, an omega-3 fatty acid) induces caspase-1 activation, which leads to GSDMD division, secretion of IL-1β, and membrane pore formation, thus promoting cancer cell death (103). HMGB1, an important damage-associated molecular pattern (DAMP), is transferred from the nucleus to the cytoplasm after DHA-induced activation of caspase-1, and facilitates the progression of pyroptosis (104). A recent study on the role of NPD-L1 and TNFα in breast cancer showed that under hypoxia, PD-L1 can migrate to the nucleus and activate GSDMC, which is then cleaved by the TNFα activated-caspase-8, triggering pyroptosis and promoting the death of breast cancer cells (105). The current mainstream view of the non-canonical inflammasome pathway is that lipopolysaccharide (LPS) drives caspase-4, 5, and 11 to cleave GSDMD, thereby triggering pyroptosis (5). The main differences between the canonical and non-canonical inflammasome pathways are the activation of caspases and the types of caspases that cleave GSDMD. In addition to GSDMC, GSDMD, a new pyroptosis pathway was reported in recent studies. GSDMD cleaves GSDME by caspase-3 to form GSDME-NT, thus enabling it to perform its function (29). Unfortunately, the unique application of this mechanism in cancer has not been found, and it cannot be differentiated from other GSDMs.

Table 2 Different types of Inflammasome activate Necroptosis.
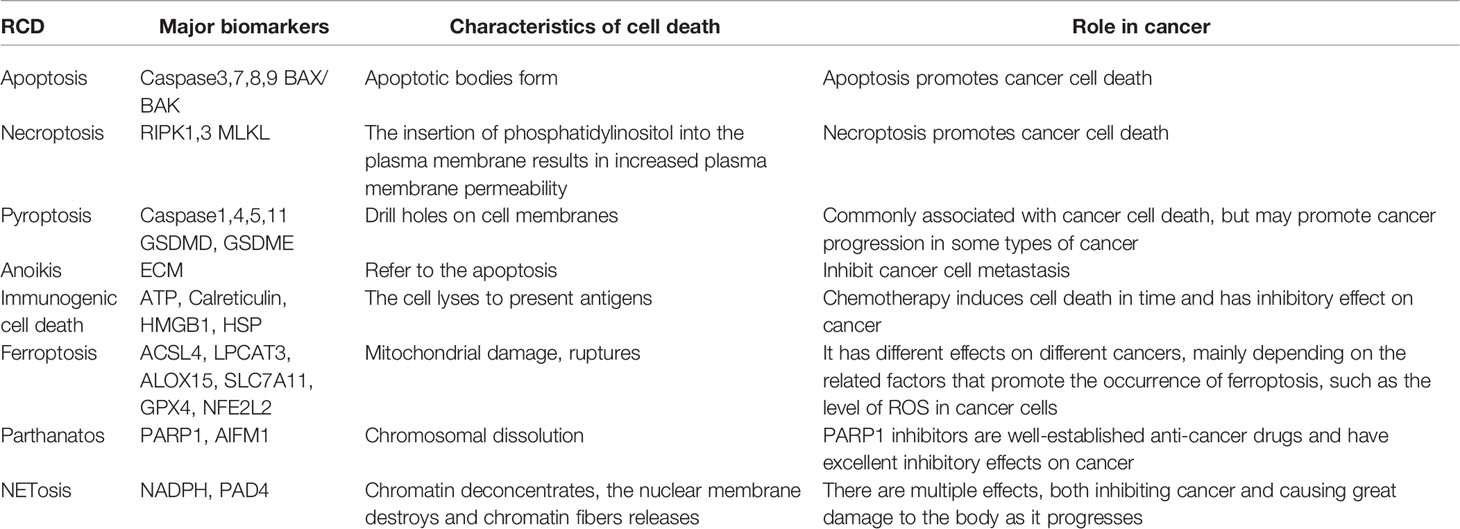
Table 3 Differences between different types of RCDS.
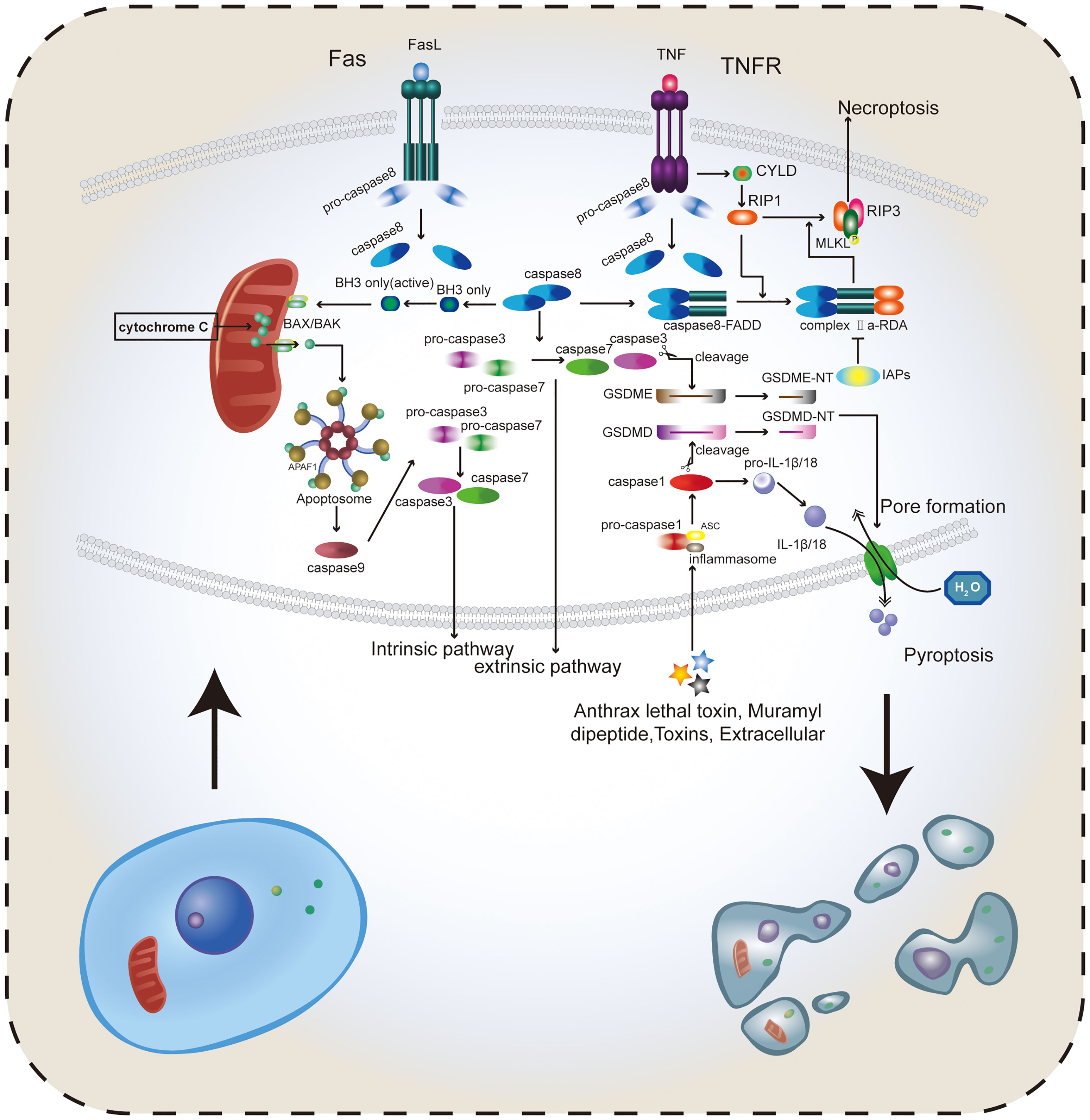
Figure 1 Several important types of RCD. Apoptosis includes both intrinsic and extrinsic pathways. The intrinsic pathway mainly depends on caspase-8 to activate BAX/BAK in the mitochondria, release cytochrome C, and promote the activation of pro-caspase-3 and pro-caspase-7 forming caspase-3 and caspase-7. In the extrinsic pathway, caspase-8 directly promotes the formation of caspase-3 and caspase-7, thus inducing apoptosis. Activated caspase-3 and caspase-7 can cleave GSDMD and GSDME to form GSDMD-NT and GSDME-NT, which can be adsorbed on the cell membrane and create holes, thus destroying the intracellular environmental homeostasis and inducing pyroptosis. TNF binding to receptors can activate CYLD and promote RIP1, RIP3, and MLKL to form trimers. Caspase-8 and the dimer formed by FADD in FAS bind to RIP1 to form complex IIa-RDA, which promotes this binding reaction and ultimately RIP1-RIP3-MLKL trimer binds to the cell membrane to induce necroptosis. The caspase family plays a major role in the pathogenesis of apoptosis, necroptosis, and pyroptosis. Apoptosis and necroptosis tend to change the cell membrane structure and intracellular physical and chemical properties, and pyroptosis can directly drill into the cell membrane through the GSDM protein, destroying the integrity of the cell membrane. Therefore, reasonably inducing the occurrence of these kinds of RCDs can effectively eradicate cancer cells. Caspase-8 plays an important role in caspase-dependent RCD.
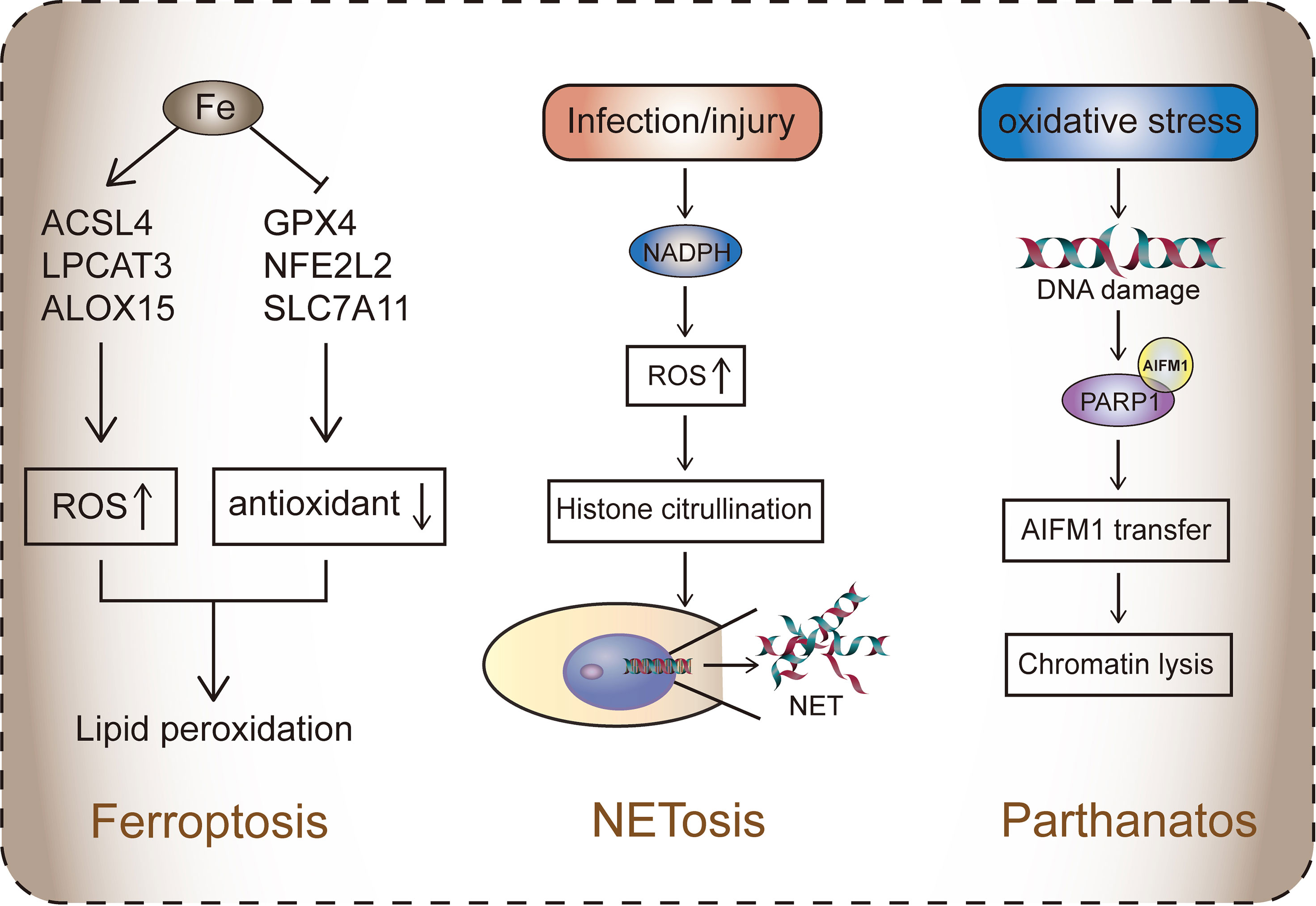
Figure 2 The three RCDs that do not rely on caspase. Ferroptosis is caused by the accumulation of iron, which is activated by the disruption of the balance between ROS and the antioxidant system. ACSL4, LPCAT3, ALOX15, and other genes mediate fatty acid oxidation, resulting in lipid toxicity in cells. Antioxidant systems such as Xc-, GPX4, and NFE2L2 protect cells from ROS. Ferroptosis is mainly dependent on the formation of PUFA-OOH. In system Xc-, GPX4 participates in the reduction of lipid peroxides (such as PUFA-OOH) and inhibits ferroptosis. In the lipid metabolism pathway, lipid droplets decompose to PUFA and AA/AdA. The latter is processed by ACSL4-LPCAT3-ALOX15 to form PUFA-OOH.NETosis: ROS mediated by NADPH activates histone citrullination, leading to the release of NET (chromatin in the nucleus), which blocks invading substances such as pathogens.Parthanatos: Oxidative stress leads to DNA damage. Activated PARP1 binds to AIFM1, causing the latter to migrate to the nucleus, leading to the dissolution of part of the chromosome.
To date, the role of pyroptosis in cancer is still not fully understood. Research studies on the signaling pathways related to the pyroptosis pathway are relatively limited; therefore, more studies and analyses are needed to confirm its effect.
From a molecular mechanism perspective, the main difference between apoptosis, necroptosis, and pyroptosis depends on the caspase family members they activate. This also implies an inter-relationship between the three caspase-dependent RCDs. Therefore, the combined effects of those pathways must be considered in studies involving cancer treatment approaches.
Caspase-Independent RCD in Targeted Therapy
The discovery of caspase and GSDM proteins promoted RCD-related research. However, many other RCD types can also exercise their functions without these two proteins. The imbalance of various cytokines, proteins, and cholesterol contained in the cancer cell microenvironment may cause cell death. Therefore, we reviewed other non-caspase/GSDM-dependent RCDs.
Ferroptosis
Ferroptosis depends on the balance between ROS, resulting from lipid peroxidation due to iron accumulation, and the antioxidant system. Lipid peroxidation results in mitochondrial diminution, mitochondrial crest reduction, increased membrane density, and membrane rupture (106). The pathways affecting iron-mediated death include GSH/GPX4 pathway, iron metabolism pathway, and lipid metabolism pathway (107).
The Xc- system is an amino acid anti-transporter located in cell membranes and is part of an important antioxidant system in cells. It is a heterodimer composed of two subunits, SLC7A11 and SLC3A2 (108). Xc- regulates the exchange of cysteine and glutamate. The ingested cystine is reduced in the cells to cysteine, which is involved in glutathione (GSH) synthesis. GSH reduces ROS and reactive nitrogen in the presence of glutathione peroxidase (GPX) (109). Therefore, when systemic Xc- inhibition occurs, the antioxidant capacity of cells decreases, and ROS accumulation eventually leads to ferroptosis (110). P53 can also inhibit the absorption of cystine by downregulating the expression of SLC7A11 through system Xc-, thus affecting GPX4 activity (42) (Figure 2).
VDACs are transmembrane transport channels of ions and metabolites that play an important regulatory role in ferroptosis (111). Erastin was found to act on VDACs, causing mitochondrial dysfunction and the release of large amounts of ROS, ultimately leading to iron-mediated ferroptosis (112). TF receptor 1 (TFR1) promotes iron absorption and increases the intracellular concentration of iron, which promotes iron-mediated ferroptosis (113). A study reported that overexpression of heat shock protein beta-1 (HSPB1) can significantly inhibit ferroptosis, mainly due to the inhibition of TFR1 membrane protein by HSPB1 (114).
Almost all the regulatory pathways of ferroptosis is dependent on ROS produced by lipid peroxidation (106). Polyunsaturated fatty acids (PUFAs) are sensitive to lipid peroxidation and are key substances in the mechanism of ferroptosis. The esterification and oxidation products of PUFAs, arachidonic acid (AA) and phosphatidylethanolamine (PE), can transmit ferroptosis signals and promote ferroptosis (115). Acyl-CoA synthase long-chain family member 4 (ACSL4) and lysophosphatidyl cholinyl transferase 3 (LPCAT3) participate in PE biosynthesis and remodeling, activate PUFAs, and affect the transmembrane properties of PUFAs. PUFA-PE is further oxidized by lipoxygenase (LOX) and eventually induces ferroptosis (116). Therefore, lowering the expression of ACSL4 and LPCAT3 can reduce the accumulation of lipid peroxide substrates in cells, thereby inhibiting ferroptosis.
Since ferroptosis has a special focus in cancer therapy, our team identified a large number of ferroptosis pathway genes that are highly expressed in various cancer patients through bioinformatics analysis (117). The gene expression of some of the proteins (such as ACSL4, SLC7A11, and ALOX15) is altered in different cancers. It is worth noting that our study found that ferroptosis often plays a dual role in tumor progression. This phenomenon is thought to be influenced by the balance between the release of damage-associated molecular patterns and the immune response induced by ferroptosis (118).
The P62-KEAP1-NrF2 pathway plays a key role in ferroptosis in hepatocellular carcinoma cells. P62 can destroy the structure of Keap1 and attenuate its degradation of NRF2, resulting in NRF2 accumulation in cells (119). NRF2 inhibits ferroptosis by upregulating quinone oxidoreductase 1(NQO1), heme oxygenase-1 (HO-1), and ferritin heavy chain 1(FTH1), thus promoting iron and ROS metabolism (120). Inhibition of NRF2 expression either by genetic tools or drugs significantly enhanced the antitumor effects of erastin and sorafenib in HCC, while activation of NRF2 expression resulted in hepatocellular carcinoma resistance to ferroptosis (121). Serramazine and lapatinib significantly increased iron-dependent ROS levels in breast cancer cells (122). However, cysteine dioxygenase type 1 (CDO1) overexpression can reduce GSH expression and ROS accumulation in breast cancer cells (123). Therefore, studies have shown that these two drugs have a better therapeutic effect in CDO1 overexpressing breast cancer cells (124).
This was also observed in clear cell renal carcinoma (ccRCC). Bioinformatics analysis showed that CDO1 promoter methylation was significantly correlated with poor prognosis of ccRCC patients, suggesting that CDO1 promoter methylation may be a new prognostic molecular marker of ccRCC (125). Studies have shown that p53 can prevent the aggregation of dipeptidyl peptidase-4 (DDP4) on the plasma membrane, weaken lipid peroxidation, and ultimately lead to ferroptosis (126). In colorectal cancer, p53 inhibits DDP4 activity, resulting in cancer cell resistance to ferroptosis. Our bioinformatics analysis showed that, as the core of ferroptosis, mutations in ROS-induced oxidative stress pathway-related regulatory genes are widely present in more than thirty types of cancer, and their high expression in ccRCC leads to a good prognosis in patients (127).
According to the current research, the development of anticancer drugs based on ferroptosis mainly focuses on two aspects; system Xc and GPX4. The survival and growth of cancer cells strongly depend on the transport activity of system Xc-, making system Xc- a potential target for anticancer drug development.
Systemic Xc- inhibitors can inhibit cystine uptake and interfere with cellular mechanisms that control protein folding, induce cellular stress, and thus lead to ferroptosis (128). Erastins is a prototype ferroptosis inducer that can directly inhibit system Xc-. Erastin has been shown to activate ferroptosis in tumor cells by upregulating the RAF/MEK/ERK signaling pathway (112, 129). It has also been shown to enhance the effectiveness of some traditional anticancer drugs in certain cases, such as doxorubicin (130) and cisplatin (131). Other studies have shown that erastin resistance can be induced by the knockout of its target gene, the voltage-dependent anion channel 2/3(VDAC) (132). Imidazole ketone Erastin, an Erastin derivative, has been successfully applied in heterogeneous animal models for the treatment of diffuse large B-cell lymphoma (DLBCL) owing to its excellent administration efficiency and anticancer performance (133).
Sorafenib is a clinically approved multikinase inhibitor for the treatment of advanced cancers, such as renal and liver cancers (134, 135). Research shows that sorafenib may inhibit systemic Xc by two potential mechanisms, which are either through inactivating kinases necessary for systemic Xc activity or interactions with non-kinase targets and their binding sites, which are similar to those of sorafenib-sensitive kinases (136). Several tumors are currently resistant to Sorafenib. Metallothionein-1g (MT-1G), a transcription target of the redox regulator NRF2, has been observed in drug-resistant cancer cells. Metallothioneins protect cells from the oxidative damage caused by heavy metals through binding to them. Therefore, inhibition of the MT-1G pathway during sorafenib treatment can reduce the risk of chemotherapy resistance and improve its therapeutic effect (137).
It is worth noting that some cancer cells can bypass system Xc- and synthesize cysteine through the transsulfation pathway. This suggests that inhibition of system Xc- therapy is not suitable for all cancers. GPX4 is a central regulator of iron function, and its inactivation leads to iron function loss, even at normal cysteine and GSH levels (138).
RSL3 targets enzymes with nucleophilic sites (e.g., cysteine serine selenocysteine) and inactivates GPX4 directly by alkylation of selenocysteine (139). One of the four diastereomers of RSL3, namely (1S, 3R)-RSL3, has been found to be more selective and lethal to cancer cells; thus, it is considered to be the optimal scheme for the application of RSL3 in cancer therapy (140). Ghoochani’s studies confirmed that prostate cancer cells were sensitive to (1S, 3R)-RSL3 and suggested that RSL3 could induce ferroptosis in tumor cells (141).
FIN56 is a ferroptosis inducer derived from CIL56. Compared with CIL56, FIN56 has a higher efficiency and specificity in inducing ferroptosis. FIN56 promotes GPX4 degradation, which requires the enzymatic activity of acetyl-CoA carboxylase (ACC) and simultaneously binds and activates squalene synthetase (SQS), leading to the depletion of the endogenous antioxidant CoQ10 and enhanced cell sensitivity to fin56-induced ferroptosis (142). Sun et al. showed that FIN56 can induce ferroptosis in bladder cancer cells and can be enhanced by combination with the mTOR inhibitor, Torin 2 (143). Zhang et al. reported that FIN56 can increase lysosomal membrane permeability through a tFeb-dependent pathway and thus promote glioblastoma cell death (144).
Since its discovery in 2012, ferroptosis has been widely studied as a therapeutic approach for the treatment of cancer; thus, its anticancer mechanism is worth further exploration.
Parthanatos and PARP Inhibitors
Parthanatos is a PARP1-dependent mode of cell death that is typically caused by poly ADP-ribose Polymerase-1 (PARP1) overactivation. PARP is a multifunctional post-translational modification enzyme that is present in most eukaryotic cells. It is activated by the recognition of structurally damaged DNA fragments and then performs DNA repair (145). PARP primarily transfers ADP-ribose units from nicotinamide adenine dinucleotides to receptor proteins, including histones, RNA polymerase, DNA polymerase, and DNA ligase (146). Under pathophysiological conditions, overactivation of PARP1 is usually caused by DNA damage, leading to the accumulation of poly ADP-Ribose (PAR) and the nuclear translocation of apoptosis-inducing factor (AIF). This in turn leads to the dissolution of part of the chromosome, ultimately triggering parthanatos (147). DNA damage that triggers PARP1 activation is usually induced by ultraviolet light, reactive oxygen species (ROS), or alkylation agents. In addition, activation of the calcium (Ca2+) signaling pathway or DNA modification (including phosphorylation, acetylation, etc.) can also induce PARP1 activation (148, 149) (Figure 2). Parthanatos is involved in several important pathological processes, including ischemia-reperfusion injury after cerebral ischemia or myocardial infarction (150, 151), and neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease (152).
Parthanatos is closely related to cancer, mainly because PARP1 plays an important role in the occurrence, development, and treatment of tumors. First, PARP1 has a dual nature, acting as both a promoter of DNA repair/replication and a stimulator of DNA fragments (153). The role of PARP1 in cancer is complicated. The loss of PARP1 often leads to impairment in the DNA repair machinery, which can contribute to cancer development (154, 155). However, inhibiting the function of PARP can also help in the treatment of cancer. The primary role of PARP inhibitors was to inhibit DNA repair and enhance the efficacy of chemotherapy and radiation. PARP inhibitors bind to the PARP1 or PARP2 catalytic site, preventing the PARP protein from shedding from the DNA damage site. This in turn leads to failure of DNA replication and activation of homologous recombination repair (HRR) as a compensatory mechanism (156). This involves a newly proposed principle of synthetic death.
Synthetic death implies that a cell dies when two genes or proteins are altered at the same time, but the cell is able to survive if only one of those genes or proteins is altered (157). BRCA1 and BRCA2 are the key HRR proteins; consequently, when this protein fails to function properly, both genes are inactivated and the cell eventually dies under the influence of synthetic death (158). However, with the discovery and application of BRCA gene mutations, high-grade serous ovarian cancer (159, 160), advanced prostate cancer (161), and pancreatic cancer (162) have been found to potentially benefit from PARP inhibitor therapy. The earliest clinically used PARP inhibitor is rucaparib in combination with the chemotherapeutic agent, temozolomide (163, 164).
With the discovery of synthetic death, rucaparib has also been shown to treat metastatic prostate cancer patients with BRCA mutations (165). After treatment with rucaparib, olaparib was also widely promoted. Trials showed that 63% of breast cancer patients with BRCA1 or BRCA2 germline mutations could benefit from olaparib treatment (166). Niraparib and talazoparib have also been used to treat patients with BRCA germline or systemic mutations (167, 168).
It took more than a decade for the first PARP inhibitor to be FDA approved through the discovery of a relationship between PARP inhibitors and BRCA synthetic death. Currently, PARP inhibitors are still widely investigated, with several clinical trials underway to be approved for cancer treatment.
RCD in Immunotherapy
Immunogenic Cell Death (ICD) and Tumor Microenvironment
When tumor cells die under the influence of external stimulation (i.e., chemotherapy and radiotherapy), they change from non-immunogenic to immunogenic and mediate anti-tumor immune response after tumor cell death. This phenomenon is known as immunogenic cell death (ICD) (17). The classification of ICDs is unique, where necrosis occurs only if cell death caused by pathogen invasion takes place. However, if the immune cells encounter the target cells infected by pathogens and cause the latter to die, effector T cells directly lead to cell death. Thus, in the latter case, pathogens are only the inducing factors of the target cells’ death. The immunological properties of ICD are mediated by damage-associated molecular patterns (DAMPs), which are endogenous molecules released during cell death (169). Theoretically, many intracellular substances, including cytokines and intracellular matrix, fall into the DAMP category. DAMPs, which are also called ICD-associated DAMPs, produced in the body during chemotherapy mainly include surface-exposed calreticulin (CRT), secreted ATP, released high mobility group protein B1 (HMGB1), and heat shock protein (HSP70, HSP90, etc.) (17, 118, 170). Unlike the various programmed cell deaths mentioned above, ICD is a form of cell death that occurs during chemotherapy and can increase the effectiveness of cancer treatment through cellular immunogenicity. Therefore, DAMPs can be used to enhance the effect of chemotherapy through amplifying the effect of ICD.
High Mobility Group Protein 1 (HMGB1)
When ICD occurs, HMGB1 is released from cells. The release of HMGB1 involves in it crossing the nuclear membrane and the plasma membrane to complete the transfer of HMGB1 from the nucleus to the cytoplasm and finally to the extracellular space. Extracellular HMGB1 binds to PRRs expressed on the surface of bone marrow cells, advanced glycosylation end-product-specific receptor (AGER or RAGE), and toll-like receptor 4 (TLR4), which activate corresponding signaling pathways and promote immune response (171). HMGB1 has been confirmed to be a tumor suppressor. Kang et al. showed that HMGB1 in the pancreas highly sensitized newborn mice to carcinogenic K-Ras-driven precancerous lesions and promoted tumor metastasis and invasion (172). Other experiments have confirmed that HMGB1 released by GSDME-mediated pyroptotic epithelial cells can be involved in colitis-related colorectal cancer lesions (173). However, HMGB1 still plays a role in promoting cancer development. A study has shown that HMGB1 regulates VEGF-D to mediate the formation of cancer blood vessels, thus promoting cancer (174).
Calreticulin (CRT)
When ICD occurs in tumor cells, CRT is exposed to the cell membrane surface and acts as an “Eat-ME” signal. This promotes dendritic cells (DCs) to engulf dead or dying tumor cells and promotes the maturation and function of DCs (175). Under stress, CRT translocates from the endoplasmic reticulum and is exposed to the membrane surface, releasing the “Eat-me” signal to activate the immune response (176). Drugs known to induce the transfer of CRT from the intracellular space to the cell membrane include anthracyclines and oxaliplatin (177). The adverse effects of mutations in the regulatory genes of CRT on ICD are of interest. Liu et al. have shown that cancer cells secrete soluble mutant CRT into the tumor microenvironment, which binds to the CRT receptor on DC cells and prevents DC cells from contacting cancer cells, thus blocking the progression of ICD (178). By using the retention using selective hooks system (RUSH) to observe the transport of CRT, Liu et al. found that CRT produced by exon 9 mutation of the CALR gene can inhibit the phagocytosis of dendritic cells (DCS) to dying cancer cells, thus reducing the effectiveness of tumor immunotherapy by chemotherapeutic drugs or PD-1 blockade (179).
ATP
ATP is released mainly in autophagy-dependent mode during ICD and is released in the form of vesicles via the ANNexin channel. ICD is activated by the release of a “Find-me” signal via the purinergic receptor P2RY2, which is picked up by DC progenitors and macrophages. ATP released from the cell is also involved in mediating the development of proinflammatory cytokines, activating the formation of the casp1-dependent NLRP3 inflammasome and secretion of mature IL-1β and IL-18 (180, 181). This process is like the typical inflammasome pathway of pyroptosis. Therefore, the ICD process is believed to involve pyroptosis.
iDAMPs
Recent studies have shown that DAMPs not only activate the immunogenicity of the body against cancer cells, but also many immunosuppressive DAMPs have been discovered successively, including HSP60 and adenosine (182). There is an intricate balance between immune stimulation and inhibition by DAMPs. Striking this balance is important to help some drugs that do not have an intrinsic ability to induce ICD to produce greater therapeutic efficacy. A new perspective, which categorizes immunosuppressive DAMPs as iDAMP, suggests that by blocking their effects, it is possible to transform the nature of some chemotherapeutic drugs and improve their efficacy (183). Unfortunately, there has not been much experimental confirmation of this novel idea. Recent studies have shown that gemcitabine can be transformed from being a non-ICD-mediated chemotherapeutic agent to an ICD-mediated chemotherapeutic agent through the COX-2/PGE2 pathway, which mediates a large number of CD8+ T cells into tumor tissues and enhances the anticancer efficacy of gemcitabine (184). This experiment also demonstrated that the immunosuppressive function of iDAMP is a physiological response to drug-induced cell death, which helps chemotherapeutic drugs better mediate ICD.
Application of Nano-Vesicle Carrier Combined With Chemotherapy Drugs
Activating the anti-tumor immune response of T cells by triggering ICD is a conventional approach for the application of ICD in tumor therapy. Many conventional clinical trials based on this approach have been initiated. The current problems include low drug delivery efficiency and avoidance of immunosuppression in the tumor microenvironment. To solve these problems, the integration of chemotherapeutic drugs and polyethylene glycol photosensitizers with nano-platforms seems to be the best solution. Zhou et al. used a tumor microenvironment-activatable prodrug vesicle for cancer chemoimmunotherapy to treat cancer. Their study showed that oxaliplatin and PEGylated photosensitizers could be integrated into the same nanoplatform to effectively improve the efficiency of drug delivery and inhibit tumor immune escape by blocking CD47 (185). Another study showed that integrated ph-responsive nanovesicles(pRNVs)/2-(1-hexyloxyethyl)-2-devinyl pyropheophorbide–A(HPHH)/indoximod (IND) could produce significant anti-tumor effects in melanoma (186). It was found that HPHH-mediated photodynamic therapy (PDT) that produces singlet oxygen, combined with PRNV to induce ICD, promoted dendritic cell (DC) recruitment and increased immune response stimulation. Meanwhile, IND regulates the tumor microenvironment by promoting the development of CD8+ T cells. ICD and tumor-infiltrating T lymphocytes are severely impaired by elevated ROS levels in the tumor microenvironment. Therefore, the regulation of extracellular ROS levels is essential to reverse the immunosuppressive environment. Based on the reactivity between pRNV and ROS, Deng et al. (187) targeted the ROS removal in the tumor microenvironment by anchoring pRNV on tumor ECM, alleviated immunosuppressant ICD caused by specific chemotherapy, and extended the survival time of T cells.
In addition to anthracycline-based drugs, some of the commonly used chemotherapeutic drugs have been found to promote ICD, including bleomycin, bortezomib, cyclophosphamide, actinomycin D, and teniposide. Some drugs can only promote the secretion of CRT like bortezomib, others can promote the secretion of CRT and HMGB1 like Teniposide, while most of the drugs can induce a variety of DAMPs (181). In addition, we found that many drugs, including cyclophosphamide and teniposide, can also enhance anti-PD-1 and anti-CTLA-4 therapies. PD-1 and CTLA-4 are protein receptors located on the surface of T cells for immune regulation, The combination of PD-L1 and CD80/86 on the surface of tumors can shut down the function of T cells and prevent tumor cell death (188, 189). The PD1 ligand, PD-L1, is highly expressed in several cancers; hence, the role of PD1 in cancer immune evasion has been well established. Inhibition of PD-1 and PD-L1 interactions enhances T cell responses in vitro and mediates preclinical antitumor activity. This is known as immune checkpoint blockade therapy (190). Our study demonstrated the efficacy of anti-PD-1 and anti-CTLA-4 therapies in renal cancer (191). Unfortunately, widespread resistance has been found against this treatment; therefore, we believe that the appropriate combination of these ICD-promoting agents with immune checkpoint blockade therapy could provide a new therapeutic potential for this approach.
Xie et al. prepared a phenol-based ICD inducer for tumor cells in vitro by combining doxorubicin (DOX), phenolic manganese dioxide nanoreactor, ferric iron, and polyethylene glycol polyphenol (MDP NPs) through metal phenol coordination assembly (192). They found that MDP NPs enhanced DOX-mediated ROS-dependent cell death and accelerated ICD induction. Subsequently, MDP NPs successively lead to the enhancement of tumor-associated antigens, maturation of dendritic cells, and ultimately enhancement of tumor-specific T cell infiltration. MDP and NPs can also effectively recruit macrophages, thus improving the tumor response to PD-1 checkpoint blocking immunotherapy. This eventually resulted in a significant anti-tumor immune response.
In these studies, we found that PDT and nanoparticles are an important means of using ICD to treat cancer. However, PDT has some limitations, including a reduction in the efficiency of ICD in the hypoxic tumor microenvironment. Technology based on both PDT and nano-platforms can perfectly compensate for each other’s shortcomings without compromising their own efficacy, which renders this combined therapeutic approach high efficiency, high targeting and personalization.
NETosis
Neutrophils play a key role in immunity. Neutrophils usually perform their functions by directly phagocytosing pathogens and secreting cytotoxic enzymes to produce neutrophil extracellular traps (NETs) (193, 194). Brinkmann et al. first discovered the NETs and their function in immunity in 2004 (195). The main structure of NETs is reticular DNA formed by depolymerized chromatin and is surrounded by a variety of nuclear proteins, including histone granular proteins and cytoplasmic proteins (196). Brinkmann et al. found that the released DNA can capture and neutralize pathogens. This process is called NETosis.
NET release begins with the activation of neutrophils, where neutrophil surface receptors bind to ligands. Studies have shown that neutrophils lacking surface receptors do not develop NETosis (197). G protein-coupled receptor ligand (GPCR), interleukin-1(IL-1), tumor necrosis factor (TNF) and Fc receptor can induce NETosis (198–200). In addition, activation of Nod-like receptor protein 3, some bacterial toxins, and ROS can also induce NETosis instead of binding to neutrophil surface receptors (201–203). There is also a theory that injury or infection induces the body’s stress state, which activates oxidative stress and causes histone citrullination by NAPDH, producing a high amount of ROS (204). The formation of NETs depends on chromatin desorption, nuclear membrane degradation, and cell lysis by peptidyl-arginine deiminase 4-mediated citrullination (conversion of arginine to citrulline). It is a type of epigenetic histone modification (205). Many studies have confirmed that PAD4 is a marker for NETosis, and the loss of PAD4 causes NETosis to fail to initiate (206, 207). Degradation of the nuclear membrane is driven by neutrophil elastase (NE), which receives a superior signal transfer to the nucleus, resulting in the rupture of the nuclear membrane (208). It is exciting that the latest research results show that NE plays a role in the destruction of nuclear membrane mechanisms, which are closely related to pyroptosis. ROS promotes NE release into the cytoplasm. NE released from granules can shear GSDMD and release GSDMD-NT, destroying the nuclear membrane and cell membrane and causing neutrophil lysis. The production of GSDMD-NT is the main reason for NET secretion into the extracellular domain (209). This study not only revealed the mechanism of NET secretion, but also provided evidence that the GSDM family plays a role in a variety of different cell death pathways as cell death executors (Figure 2).
The role of neutrophils in tumor progression is controversial because neutrophils have both pro-tumor and anti-tumor properties (210). The neutrophil chromatin release affects tumor growth, angiogenesis, metastasis, and immunosuppression (211). Many studies have found large concentrations of neutrophils and high expression of PAD4 in cancer tissues, including Lewis lung cancer and Ewing’s sarcoma (212, 213). Simultaneously, NETs allow detached tumor cells to attach to other tissues, promoting cancer metastasis, and the by-products of NET decomposition can also cause immunosuppression (210, 214). Some studies have also found that NETosis can cause post-tumor thrombosis, further aggravating tumor damage e (211). Plasma DNA, neutrophil counts, and NET biomarkers have been suggested as diagnostic tools for assessing the propensity for thrombosis (215).
Many of the above findings suggest that research on tumor-induced NETosis should focus on targeting NETs that may benefit cancer patients. As research progresses, NETs have shifted from being initially considered a defense against serious infectious diseases to negatively impacting the body during cancer by promoting deadly processes such as thrombosis as well as systemic inflammation and cancer recurrence. With this in mind, NETs could provide excellent targets for future anticancer therapies.
Conclusion
Cells can quickly disintegrate and die when exposed to extremely harsh environmental conditions, which is called accidental cell death (ACD). Minor exogenous or endogenous disturbances promote adaptive stress, thus restoring intracellular homeostasis. When the stress response fails to restore homeostasis, one or more signaling cascades are activated in the cell, contributing to the regulation of cell death.
There are many types of RCDSs with different mechanisms (Table 3). However, there is a very close relationship between many types of RCD. Although the application of RCD in cancer is mainly to promote the process of death, there is still a part of RCD that promotes the effect of cancer; hence, we cannot induce cell death for cancer treatment. As a bridge between different RCDSs, the caspase protein family, the Bcl-2 family, and the GSDM family are involved in most RCD processes and are representative markers of RCD. This also proves the connectivity between different RCDs from the side, which is also a breakthrough in the application of RCD in cancer treatment research. Currently, we have a better understanding of apoptosis, and we have learned about RCTs including necroptosis, pyroptosis, and ferroptosis, which remain to be understood, particularly the role of these processes in the development or treatment of cancer. The role of cell death in the tumor microenvironment is unique. From immunogenic cell death to immune checkpoint inhibition, various discoveries continue to remind us of the significance of cell death in cancer treatment. In addition to these common classic RCDs, many newly discovered types of RCDs are worth discussing. We believe that the widespread discovery of various non-caspase-dependent cell death means that our understanding of cell death is further advanced. The application of PARP inhibitors to improve the efficacy of chemotherapy is worth advocating. Various forms of death, including ferroptosis, play a double-role in cancer cells. Therefore, exploring the causes and mechanisms of their different outcomes in cancer patients is an issue that needs to be addressed.
Currently, we need to understand more about cell death, which will not only provide new ideas for current cancer treatment, but also provide more resources for cancer chemotherapeutic drugs, immunotherapy drugs, and targeted drugs.
Author Contributions
GW conceived the idea of the manuscript. QL and XC undertook the initial research. XQ was involved in writing and plotting. QW reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The PhD Start-up Fund of Liaoning Province from GW (2021-BS-209, Liaoning Province, 30000 CNY). Dalian Young Science and Technology Star from GW (2021RQ010 Dalian, Liaoning Province, 100000CNY).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of Precision Cancer Medicine: Evolution of the Treatment Paradigm. Cancer Treat Rev (2020) 86:102019. doi: 10.1016/j.ctrv.2020.102019
3. Strasser A, Vaux DL. Cell Death in the Origin and Treatment of Cancer. Mol Cell (2020) 78:1045–54. doi: 10.1016/j.molcel.2020.05.014
4. Vossenkamper A, Warnes G. Flow Cytometry Reveals the Nature of Oncotic Cells. Int J Mol Sci (2019) 20:4379. doi: 10.3390/ijms20184379
5. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The Molecular Machinery of Regulated Cell Death. Cell Res (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
6. D’Arcy MS. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol Int (2019) 43:582–92. doi: 10.1002/cbin.11137
7. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
8. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, Autophagy, Necroptosis, and Cancer Metastasis. Mol Cancer (2015) 14:48. doi: 10.1186/s12943-015-0321-5
9. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The Role of Necroptosis in Cancer Biology and Therapy. Mol Cancer (2019) 18:100. doi: 10.1186/s12943-019-1029-8
10. Koren E, Fuchs Y. Modes of Regulated Cell Death in Cancer. Cancer Discovery (2021) 11:245–65. doi: 10.1158/2159-8290.CD-20-0789
11. Sevinsky CJ, Khan F, Kokabee L, Darehshouri A, Maddipati KR, Conklin DS. NDRG1 Regulates Neutral Lipid Metabolism in Breast Cancer Cells. Breast Cancer Res (2018) 20:55. doi: 10.1186/s13058-018-0980-4
12. Wettersten HI, Aboud OA, Lara PN, Weiss RH. Metabolic Reprogramming in Clear Cell Renal Cell Carcinoma. Nat Rev Nephrol (2017) 13:410–9. doi: 10.1038/nrneph.2017.59
13. Cheng C, Geng F, Cheng X, Guo D. Lipid Metabolism Reprogramming and its Potential Targets in Cancer. Cancer Commun (Lond) (2018) 38:27. doi: 10.1186/s40880-018-0301-4
14. Gao L, Xu Z, Huang Z, Tang Y, Yang D, Huang J, et al. CPI-613 Rewires Lipid Metabolism to Enhance Pancreatic Cancer Apoptosis via the AMPK-ACC Signaling. J Exp Clin Cancer Res (2020) 39:73. doi: 10.1186/s13046-020-01579-x
15. Huang C, Freter C. Lipid Metabolism, Apoptosis and Cancer Therapy. Int J Mol Sci (2015) 16:924–49. doi: 10.3390/ijms16010924
16. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
17. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic Cell Death in Cancer Therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
18. Majno G, Joris I. Apoptosis, Oncosis, and Necrosis. An overview of cell death. Am J Pathol (1995) 146:3–15.
19. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol Rev (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
21. Kerr JF, Wyllie AH, Currie AR. Apoptosis: A Basic Biological Phenomenon With Wide-Ranging Implications in Tissue Kinetics. Br J Cancer (1972) 26:239–57. doi: 10.1038/bjc.1972.33
22. Shi Y. Mechanisms of Caspase Activation and Inhibition During Apoptosis. Mol Cell (2002) 9:459–70. doi: 10.1016/S1097-2765(02)00482-3
23. Laubach V, Kaufmann R, Bernd A, Kippenberger S, Zöller N. Extrinsic or Intrinsic Apoptosis by Curcumin and Light: Still a Mystery. Int J Mol Sci (2019) 20:E905. doi: 10.3390/ijms20040905
24. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome C and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell (1997) 91:479–89. doi: 10.1016/S0092-8674(00)80434-1
25. Sairanen T, Szepesi R, Karjalainen-Lindsberg M-L, Saksi J, Paetau A, Lindsberg PJ. Neuronal Caspase-3 and PARP-1 Correlate Differentially With Apoptosis and Necrosis in Ischemic Human Stroke. Acta Neuropathol (2009) 118:541–52. doi: 10.1007/s00401-009-0559-3
26. Creagh EM. Caspase Crosstalk: Integration of Apoptotic and Innate Immune Signalling Pathways. Trends Immunol (2014) 35:631–40. doi: 10.1016/j.it.2014.10.004
27. Kesavardhana S, Malireddi RKS, Kanneganti T-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
28. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol (2017) 24:507–14.e4. doi: 10.1016/j.chembiol.2017.03.009
29. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
30. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
31. Zheng M, Karki R, Vogel P, Kanneganti T-D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell (2020) 181:674–87.e13. doi: 10.1016/j.cell.2020.03.040
32. Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, et al. Caspase-8 is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
33. Bruckheimer EM, Cho SH, Sarkiss M, Herrmann J, McDonnell TJ. The Bcl-2 Gene Family and Apoptosis. Adv Biochem Eng Biotechnol (1998) 62:75–105. doi: 10.1007/BFb0102306
34. Gottlieb TM, Oren M. P53 and Apoptosis. Semin Cancer Biol (1998) 8:359–68. doi: 10.1006/scbi.1998.0098
35. Herbst RS, Frankel SR. Oblimersen Sodium (Genasense Bcl-2 Antisense Oligonucleotide): A Rational Therapeutic to Enhance Apoptosis in Therapy of Lung Cancer. Clin Cancer Res (2004) 10:4245s–8s. doi: 10.1158/1078-0432.CCR-040018
36. Abou-Nassar K, Brown JR. Novel Agents for the Treatment of Chronic Lymphocytic Leukemia. Clin Adv Hematol Oncol (2010) 8(12):886–95.
37. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature (2005) 435:677–81. doi: 10.1038/nature03579
38. Albershardt TC, Salerni BL, Soderquist RS, Bates DJP, Pletnev AA, Kisselev AF, et al. Multiple BH3 Mimetics Antagonize Antiapoptotic MCL1 Protein by Inducing the Endoplasmic Reticulum Stress Response and Up-Regulating BH3-Only Protein NOXA. J Biol Chem (2011) 286:24882–95. doi: 10.1074/jbc.M111.255828
39. Ocker M, Neureiter D, Lueders M, Zopf S, Ganslmayer M, Hahn EG, et al. Variants of Bcl-2 Specific siRNA for Silencing Antiapoptotic Bcl-2 in Pancreatic Cancer. Gut (2005) 54:1298–308. doi: 10.1136/gut.2004.056192
40. Wu X, Liu X, Sengupta J, Bu Y, Yi F, Wang C, et al. Silencing of Bmi-1 Gene by RNA Interference Enhances Sensitivity to Doxorubicin in Breast Cancer Cells. Indian J Exp Biol (2011) 49(2):105–12.
41. Pekarsky Y, Balatti V, Croce CM. BCL2 and miR-15/16: From Gene Discovery to Treatment. Cell Death Differ (2018) 25:21–6. doi: 10.1038/cdd.2017.159
42. Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a P53-Mediated Activity During Tumour Suppression. Nature (2015) 520:57–62. doi: 10.1038/nature14344
43. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-Mediated Neuroinflammation in CNS Diseases. Nat Rev Neurosci (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
44. Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-Regulated Necroptosis as a Target for Multifaceted Disease Therapy (Review). Int J Mol Med (2019) 44:771–86. doi: 10.3892/ijmm.2019.4244
45. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in Development and Diseases. Genes Dev (2018) 32:327–40. doi: 10.1101/gad.312561.118
46. Legarda D, Justus SJ, Ang RL, Rikhi N, Li W, Moran TM, et al. CYLD Proteolysis Protects Macrophages From TNF-Mediated Auto-Necroptosis Induced by LPS and Licensed by Type I IFN. Cell Rep (2016) 15:2449–61. doi: 10.1016/j.celrep.2016.05.032
47. Silke J, Rickard JA, Gerlic M. The Diverse Role of RIP Kinases in Necroptosis and Inflammation. Nat Immunol (2015) 16:689–97. doi: 10.1038/ni.3206
48. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat Commun (2020) 11:3151. doi: 10.1038/s41467-020-16887-1
49. Christofferson DE, Li Y, Yuan J. Control of Life-or-Death Decisions by RIP1 Kinase. Annu Rev Physiol (2014) 76:129–50. doi: 10.1146/annurev-physiol-021113-170259
50. Peltzer N, Darding M, Walczak H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol (2016) 26:445–61. doi: 10.1016/j.tcb.2016.01.006
51. Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by Caspase-8 is Crucial for Limiting Apoptosis and Necroptosis. Nature (2019) 574:428–31. doi: 10.1038/s41586-019-1548-x
52. Koo G-B, Morgan MJ, Lee D-G, Kim W-J, Yoon J-H, Koo JS, et al. Methylation-Dependent Loss of RIP3 Expression in Cancer Represses Programmed Necrosis in Response to Chemotherapeutics. Cell Res (2015) 25:707–25. doi: 10.1038/cr.2015.56
53. Stoll G, Ma Y, Yang H, Kepp O, Zitvogel L, Kroemer G. Pro-Necrotic Molecules Impact Local Immunosurveillance in Human Breast Cancer. Oncoimmunology (2017) 6:e1299302. doi: 10.1080/2162402X.2017.1299302
54. Wang Q, Chen W, Xu X, Li B, He W, Padilla MT, et al. RIP1 Potentiates BPDE-Induced Transformation in Human Bronchial Epithelial Cells Through Catalase-Mediated Suppression of Excessive Reactive Oxygen Species. Carcinogenesis (2013) 34:2119–28. doi: 10.1093/carcin/bgt143
55. McCormick KD, Ghosh A, Trivedi S, Wang L, Coyne CB, Ferris RL, et al. Innate Immune Signaling Through Differential RIPK1 Expression Promote Tumor Progression in Head and Neck Squamous Cell Carcinoma. Carcinogenesis (2016) 37:522–9. doi: 10.1093/carcin/bgw032
56. Ke H, Augustine CK, Gandham VD, Jin JY, Tyler DS, Akiyama SK, et al. CYLD Inhibits Melanoma Growth and Progression Through Suppression of the JNK/AP-1 and β1-Integrin Signaling Pathways. J Invest Dermatol (2013) 133:221–9. doi: 10.1038/jid.2012.253
57. Wu W, Zhu H, Fu Y, Shen W, Xu J, Miao K, et al. Clinical Significance of Down-Regulated Cylindromatosis Gene in Chronic Lymphocytic Leukemia. Leuk Lymphoma (2014) 55:588–94. doi: 10.3109/10428194.2013.809077
58. Ertao Z, Jianhui C, Kang W, Zhijun Y, Hui W, Chuangqi C, et al. Prognostic Value of Mixed Lineage Kinase Domain-Like Protein Expression in the Survival of Patients With Gastric Caner. Tumour Biol (2016) 37:13679–85. doi: 10.1007/s13277-016-5229-1
59. He L, Peng K, Liu Y, Xiong J, Zhu F-F. Low Expression of Mixed Lineage Kinase Domain-Like Protein is Associated With Poor Prognosis in Ovarian Cancer Patients. Onco Targets Ther (2013) 6:1539–43. doi: 10.2147/OTT.S52805
60. Ruan J, Mei L, Zhu Q, Shi G, Wang H. Mixed Lineage Kinase Domain-Like Protein Is a Prognostic Biomarker for Cervical Squamous Cell Cancer. Int J Clin Exp Pathol (2015) 8(11):15035–8.
62. Valentijn AJ, Zouq N, Gilmore AP. Anoikis. Biochem Soc Trans (2004) 32:421–5. doi: 10.1042/bst0320421
63. Cao Z, Livas T, Kyprianou N. Anoikis and EMT: Lethal “Liaisons” During Cancer Progression. Crit Rev Oncog (2016) 21:155–68. doi: 10.1615/CritRevOncog.2016016955
64. Bouillet P, Strasser A. BH3-Only Proteins - Evolutionarily Conserved Proapoptotic Bcl-2 Family Members Essential for Initiating Programmed Cell Death. J Cell Sci (2002) 115:1567–74. doi: 10.1242/jcs.115.8.1567
65. Valentijn AJ, Gilmore AP. Translocation of Full-Length Bid to Mitochondria During Anoikis. J Biol Chem (2004) 279:32848–57. doi: 10.1074/jbc.M313375200
66. Paoli P, Giannoni E, Chiarugi P. Anoikis Molecular Pathways and its Role in Cancer Progression. Biochim Biophys Acta (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
67. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Surveillance of Tumor Metastasis by Liver Natural Killer Cells. Nat Med (2001) 7:94–100. doi: 10.1038/83416
68. Sakamoto S, Kyprianou N. Targeting Anoikis Resistance in Prostate Cancer Metastasis. Mol Aspects Med (2010) 31:205–14. doi: 10.1016/j.mam.2010.02.001
69. Ye G, Yang Q, Lei X, Zhu X, Li F, He J, et al. Nuclear MYH9-Induced CTNNB1 Transcription, Targeted by Staurosporin, Promotes Gastric Cancer Cell Anoikis Resistance and Metastasis. Theranostics (2020) 10:7545–60. doi: 10.7150/thno.46001
70. Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming Anoikis–Pathways to Anchorage-Independent Growth in Cancer. J Cell Sci (2011) 124:3189–97. doi: 10.1242/jcs.072165
71. Lock R, Debnath J. Extracellular Matrix Regulation of Autophagy. Curr Opin Cell Biol (2008) 20:583–8. doi: 10.1016/j.ceb.2008.05.002
72. Desgrosellier JS, Cheresh DA. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat Rev Cancer (2010) 10:9–22. doi: 10.1038/nrc2748
73. Hamidi H, Ivaska J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat Rev Cancer (2018) 18:533–48. doi: 10.1038/s41568-018-0038-z
74. Caswell PT, Vadrevu S, Norman JC. Integrins: Masters and Slaves of Endocytic Transport. Nat Rev Mol Cell Biol (2009) 10:843–53. doi: 10.1038/nrm2799
75. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT. Cell Plasticity and Metastasis. Cancer Metastasis Rev (2016) 35:645–54. doi: 10.1007/s10555-016-9648-7
76. Levy JMM, Towers CG, Thorburn A. Targeting Autophagy in Cancer. Nat Rev Cancer (2017) 17:528–42. doi: 10.1038/nrc.2017.53
77. Peng Y-F, Shi Y-H, Ding Z-B, Ke A-W, Gu C-Y, Hui B, et al. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy (2013) 9:2056–68. doi: 10.4161/auto.26398
78. Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of Autophagy During Extracellular Matrix Detachment Promotes Cell Survival. Mol Biol Cell (2008) 19:797–806. doi: 10.1091/mbc.e07-10-1092
79. Wang Y-N, Zeng Z-L, Lu J, Wang Y, Liu Z-X, He M-M, et al. CPT1A-Mediated Fatty Acid Oxidation Promotes Colorectal Cancer Cell Metastasis by Inhibiting Anoikis. Oncogene (2018) 37:6025–40. doi: 10.1038/s41388-018-0384-z
80. Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, et al. The PLAG1-GDH1 Axis Promotes Anoikis Resistance and Tumor Metastasis Through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol Cell (2018) 69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025
81. Jin L, Chun J, Pan C, Alesi GN, Li D, Magliocca KR, et al. Phosphorylation-Mediated Activation of LDHA Promotes Cancer Cell Invasion and Tumour Metastasis. Oncogene (2017) 36:3797–806. doi: 10.1038/onc.2017.6
82. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
83. Xia S, Hollingsworth LR, Wu H. Mechanism and Regulation of Gasdermin-Mediated Cell Death. Cold Spring Harb Perspect Biol (2020) 12:a036400. doi: 10.1101/cshperspect.a036400
84. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
85. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A New Frontier in Cancer. Biomed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
86. Frank D, Vince JE. Pyroptosis Versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
87. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The Inflammasome: A Caspase-1-Activation Platform That Regulates Immune Responses and Disease Pathogenesis. Nat Immunol (2009) 10:241–7. doi: 10.1038/ni.1703
88. Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The NLRC4 Inflammasome Receptors for Bacterial Flagellin and Type III Secretion Apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
89. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 Proteins Regulate Caspase Activation in Response to Foreign Cytoplasmic DNA. Sci Am Assoc Adv Science (2009) 323:1057–60. doi: 10.1126/science.1169841
90. Davis BK, Wen H, Ting JP-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
91. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
92. Rathinam VAK, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
93. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense Against Infection. Curr Opin Microbiol (2013) 16:319–26. doi: 10.1016/j.mib.2013.04.004
94. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
95. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 Self-Cleavage is an Intrinsic Mechanism to Terminate Inflammasome Activity. J Exp Med (2018) 215:827–40. doi: 10.1084/jem.20172222
96. Man SM, Karki R, Kanneganti T-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol Rev (2017) 277:61–75. doi: 10.1111/imr.12534
97. Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep (2017) 21:3846–59. doi: 10.1016/j.celrep.2017.12.018
98. Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R, et al. A PLK1 Kinase Inhibitor Enhances the Chemosensitivity of Cisplatin by Inducing Pyroptosis in Oesophageal Squamous Cell Carcinoma. EBioMedicine (2019) 41:244–55. doi: 10.1016/j.ebiom.2019.02.012
99. Martín A, Odajima J, Hunt SL, Dubus P, Ortega S, Malumbres M, et al. Cdk2 is Dispensable for Cell Cycle Inhibition and Tumor Suppression Mediated by P27(Kip1) and P21(Cip1). Cancer Cell (2005) 7:591–8. doi: 10.1016/j.ccr.2005.05.006
100. Oakes V, Wang W, Harrington B, Lee WJ, Beamish H, Chia KM, et al. Cyclin A/Cdk2 Regulates Cdh1 and Claspin During Late S/G2 Phase of the Cell Cycle. Cell Cycle Taylor Francis (2014) 13:3302–11. doi: 10.4161/15384101.2014.949111
101. Gopinathan L, Tan SLW, Padmakumar VC, Coppola V, Tessarollo L, Kaldis P. Loss of Cdk2 and Cyclin A2 Impairs Cell Proliferation and Tumorigenesis. Cancer Res (2014) 74:3870–9. doi: 10.1158/0008-5472.CAN-13-3440
102. Yam CH, Fung TK, Poon RYC. Cyclin A in Cell Cycle Control and Cancer. Cell Mol Life Sci (2002) 59:1317–26. doi: 10.1007/s00018-002-8510-y
103. Pizato N, Luzete BC, Kiffer LFMV, Corrêa LH, de Oliveira Santos I, Assumpção JAF, et al. Omega-3 Docosahexaenoic Acid Induces Pyroptosis Cell Death in Triple-Negative Breast Cancer Cells. Sci Rep (2018) 8:1952. doi: 10.1038/s41598-018-20422-0
104. Magna M, Pisetsky DS. The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases. Mol Med BioMed Central (2014) 20:138–46. doi: 10.2119/molmed.2013.00164
105. Hou J, Zhao R, Xia W, Chang C-W, You Y, Hsu J-M, et al. PD-L1-Mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
106. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
107. Ashrafizadeh M, Mohammadinejad R, Tavakol S, Ahmadi Z, Roomiani S, Katebi M. Autophagy, Anoikis, Ferroptosis, Necroptosis, and Endoplasmic Reticulum Stress: Potential Applications in Melanoma Therapy. J Cell Physiol (2019) 234:19471–9. doi: 10.1002/jcp.28740
108. Liu J, Xia X, Huang P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol Ther (2020) 28:2358–66. doi: 10.1016/j.ymthe.2020.08.021
109. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J Hematol Oncol (2019) 12:34. doi: 10.1186/s13045-019-0720-y
110. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
111. Lipper CH, Stofleth JT, Bai F, Sohn Y-S, Roy S, Mittler R, et al. Redox-Dependent Gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A (2019) 116:19924–9. doi: 10.1073/pnas.1908271116
112. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature (2007) 447:864–8. doi: 10.1038/nature05859
113. Frazer DM, Anderson GJ. The Regulation of Iron Transport. Biofactors (2014) 40(2):206–14. doi: 10.1002/biof.1148
114. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene (2015) 34:5617–25. doi: 10.1038/onc.2015.32
115. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
116. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
117. Wu G, Wang Q, Xu Y, Li Q, Cheng L. A New Survival Model Based on Ferroptosis-Related Genes for Prognostic Prediction in Clear Cell Renal Cell Carcinoma. Aging (Albany NY) (2020) 12:14933–48. doi: 10.18632/aging.103553
118. Garg AD, Agostinis P. Cell Death and Immunity in Cancer: From Danger Signals to Mimicry of Pathogen Defense Responses. Immunol Rev (2017) 280:126–48. doi: 10.1111/imr.12574
119. Taguchi K, Yamamoto M. The KEAP1-NRF2 System in Cancer. Front Oncol (2017) 7:85. doi: 10.3389/fonc.2017.00085
120. Sajadimajd S, Khazaei M. Oxidative Stress and Cancer: The Role of Nrf2. Curr Cancer Drug Targets (2018) 18:538–57. doi: 10.2174/1568009617666171002144228
121. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-Keap1-NRF2 Pathway Protects Against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology (2016) 63:173–84. doi: 10.1002/hep.28251
122. Yang B, Chen Y, Shi J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem Rev (2019) 119:4881–985. doi: 10.1021/acs.chemrev.8b00626
123. Jeschke J, O’Hagan HM, Zhang W, Vatapalli R, Calmon MF, Danilova L, et al. Frequent Inactivation of Cysteine Dioxygenase Type 1 Contributes to Survival of Breast Cancer Cells and Resistance to Anthracyclines. Clin Cancer Res (2013) 19:3201–11. doi: 10.1158/1078-0432.CCR-12-3751
124. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is Induced Following Siramesine and Lapatinib Treatment of Breast Cancer Cells. Cell Death Dis (2016) 7:e2307. doi: 10.1038/cddis.2016.208
125. Deckers IAG, Schouten LJ, Van Neste L, van Vlodrop IJH, Soetekouw PMMB, Baldewijns MMLL, et al. Promoter Methylation of CDO1 Identifies Clear-Cell Renal Cell Cancer Patients With Poor Survival Outcome. Clin Cancer Res (2015) 21:3492–500. doi: 10.1158/1078-0432.CCR-14-2049
126. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
127. Che X, Qi X, Xu Y, Wang Q, Wu G. Using Genomic and Transcriptome Analyses to Identify the Role of the Oxidative Stress Pathway in Renal Clear Cell Carcinoma and Its Potential Therapeutic Significance. Oxid Med Cell Longev (2021) 2021:5561124. doi: 10.1155/2021/5561124
128. Liu M-R, Zhu W-T, Pei D-S. System Xc-: A Key Regulatory Target of Ferroptosis in Cancer. Invest New Drugs (2021) 39:1123–31. doi: 10.1007/s10637-021-01070-0
129. Cao JY, Dixon SJ. Mechanisms of Ferroptosis. Cell Mol Life Sci (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
130. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The Ferroptosis Inducer Erastin Enhances Sensitivity of Acute Myeloid Leukemia Cells to Chemotherapeutic Agents. Mol Cell Oncol (2015) 2:e1054549. doi: 10.1080/23723556.2015.1054549
131. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib Induces Cisplatin-Resistant non-Small Cell Lung Cancer Cell Ferroptosis Through Inhibition of the Nrf2/xCT Pathway. Oncol Lett (2020) 19:323–33. doi: 10.3892/ol.2019.11066
132. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther (2020) 13:5429–41. doi: 10.2147/OTT.S254995
133. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol (2019) 26:623–33.e9. doi: 10.1016/j.chembiol.2019.01.008
134. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal Transduct Target Ther (2020) 5:87. doi: 10.1038/s41392-020-0187-x
135. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal Cell Carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi: 10.1038/nrdp.2017.9
136. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
137. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G Facilitates Sorafenib Resistance Through Inhibition of Ferroptosis. Hepatology (2016) 64:488–500. doi: 10.1002/hep.28574
138. Ursini F, Maiorino M. Lipid Peroxidation and Ferroptosis: The Role of GSH and Gpx4. Free Radic Biol Med (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
139. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
140. Yangyun W, Guowei S, Shufen S, Jie Y, Rui Y, Yu R. Everolimus Accelerates Erastin and RSL3-Induced Ferroptosis in Renal Cell Carcinoma. Gene (2022) 809:145992. doi: 10.1016/j.gene.2021.145992
141. Ghoochani A, Hsu E-C, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res (2021) 81:1583–94. doi: 10.1158/0008-5472.CAN-20-3477
142. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat Chem Biol (2016) 12:497–503. doi: 10.1038/nchembio.2079
143. Sun Y, Berleth N, Wu W, Schlütermann D, Deitersen J, Stuhldreier F, et al. Fin56-Induced Ferroptosis is Supported by Autophagy-Mediated GPX4 Degradation and Functions Synergistically With mTOR Inhibition to Kill Bladder Cancer Cells. Cell Death Dis (2021) 12:1028. doi: 10.1038/s41419-021-04306-2
144. Zhang X, Guo Y, Li H, Han L. FIN56, a Novel Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization in a TFEB-Dependent Manner in Glioblastoma. J Cancer (2021) 12:6610–9. doi: 10.7150/jca.58500
145. Fatokun AA, Dawson VL, Dawson TM. Parthanatos: Mitochondrial-Linked Mechanisms and Therapeutic Opportunities. Br J Pharmacol (2014) 171:2000–16. doi: 10.1111/bph.12416
146. Grignani G, Merlini A, Sangiolo D, D’Ambrosio L, Pignochino Y. Delving Into PARP Inhibition From Bench to Bedside and Back. Pharmacol Ther (2020) 206:107446. doi: 10.1016/j.pharmthera.2019.107446
147. Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
148. Lonskaya I, Potaman VN, Shlyakhtenko LS, Oussatcheva EA, Lyubchenko YL, Soldatenkov VA. Regulation of Poly(ADP-Ribose) Polymerase-1 by DNA Structure-Specific Binding. J Biol Chem (2005) 280:17076–83. doi: 10.1074/jbc.M413483200
149. Bürkle A, Virág L. Poly(ADP-Ribose): PARadigms and PARadoxes. Mol Aspects Med (2013) 34:1046–65. doi: 10.1016/j.mam.2012.12.010
150. Zhong H, Song R, Pang Q, Liu Y, Zhuang J, Chen Y, et al. Propofol Inhibits Parthanatos via ROS-ER-Calcium-Mitochondria Signal Pathway In Vivo and Vitro. Cell Death Dis (2018) 9:932. doi: 10.1038/s41419-018-0996-9
151. Koehler RC, Dawson VL, Dawson TM. Targeting Parthanatos in Ischemic Stroke. Front Neurol (2021) 12:662034. doi: 10.3389/fneur.2021.662034
152. Wang X, Ge P. Parthanatos in the Pathogenesis of Nervous System Diseases. Neuroscience (2020) 449:241–50. doi: 10.1016/j.neuroscience.2020.09.049
153. Alemasova EE, Lavrik OI. Poly(ADP-Ribosyl)Ation by PARP1: Reaction Mechanism and Regulatory Proteins. Nucleic Acids Res (2019) 47:3811–27. doi: 10.1093/nar/gkz120
154. Piskunova TS, Yurova MN, Ovsyannikov AI, Semenchenko AV, Zabezhinski MA, Popovich IG, et al. Deficiency in Poly(ADP-Ribose) Polymerase-1 (PARP-1) Accelerates Aging and Spontaneous Carcinogenesis in Mice. Curr Gerontol Geriatr Res (2008) 2008:754190. doi: 10.1155/2008/754190
155. Toprani SM. DNA Damage and Repair Scenario in Ameloblastoma. Oral Oncol (2020) 108:104804. doi: 10.1016/j.oraloncology.2020.104804
156. Lord CJ, Ashworth A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
157. O’Neil NJ, Bailey ML, Hieter P. Synthetic Lethality and Cancer. Nat Rev Genet (2017) 18:613–23. doi: 10.1038/nrg.2017.47
158. Faraoni I, Graziani G. Role of BRCA Mutations in Cancer Treatment With Poly(ADP-Ribose) Polymerase (PARP) Inhibitors. Cancers (Basel) (2018) 10:487. doi: 10.3390/cancers10120487
159. Hodgson DR, Dougherty BA, Lai Z, Fielding A, Grinsted L, Spencer S, et al. Candidate Biomarkers of PARP Inhibitor Sensitivity in Ovarian Cancer Beyond the BRCA Genes. Br J Cancer (2018) 119:1401–9. doi: 10.1038/s41416-018-0274-8
160. George A, Kaye S, Banerjee S. Delivering Widespread BRCA Testing and PARP Inhibition to Patients With Ovarian Cancer. Nat Rev Clin Oncol (2017) 14:284–96. doi: 10.1038/nrclinonc.2016.191
161. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
162. Gout J, Perkhofer L, Morawe M, Arnold F, Ihle M, Biber S, et al. Synergistic Targeting and Resistance to PARP Inhibition in DNA Damage Repair-Deficient Pancreatic Cancer. Gut (2021) 70:743–60. doi: 10.1136/gutjnl-2019-319970
163. Daniel RA, Rozanska AL, Mulligan EA, Drew Y, Thomas HD, Castelbuono DJ, et al. Central Nervous System Penetration and Enhancement of Temozolomide Activity in Childhood Medulloblastoma Models by Poly(ADP-Ribose) Polymerase Inhibitor AG-014699. Br J Cancer (2010) 103:1588–96. doi: 10.1038/sj.bjc.6605946
164. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates With SLFN11 Expression and Demonstrates Synergy With Temozolomide in Small Cell Lung Cancer. Clin Cancer Res (2017) 23:523–35. doi: 10.1158/1078-0432.CCR-16-1040
165. Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J Clin Oncol (2020) 38:3763–72. doi: 10.1200/JCO.20.01035
166. Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients With a Germline BRCA Mutation. N Engl J Med (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
167. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med (2016) 375:2154–64. doi: 10.1056/NEJMoa1611310
168. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, et al. Talazoparib in Patients With Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
169. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
170. Krysko O, Løve Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many Faces of DAMPs in Cancer Therapy. Cell Death Dis (2013) 4:e631. doi: 10.1038/cddis.2013.156
171. Wang S, Zhang Y. HMGB1 in Inflammation and Cancer. J Hematol Oncol (2020) 13:116. doi: 10.1186/s13045-020-00950-x
172. Kang R, Xie Y, Zhang Q, Hou W, Jiang Q, Zhu S, et al. Intracellular HMGB1 as a Novel Tumor Suppressor of Pancreatic Cancer. Cell Res (2017) 27:916–32. doi: 10.1038/cr.2017.51
173. Tan G, Huang C, Chen J, Zhi F. HMGB1 Released From GSDME-Mediated Pyroptotic Epithelial Cells Participates in the Tumorigenesis of Colitis-Associated Colorectal Cancer Through the ERK1/2 Pathway. J Hematol Oncol (2020) 13:149. doi: 10.1186/s13045-020-00985-0
174. Da W, Zhang J, Zhang R, Zhu J. Curcumin Inhibits the Lymphangiogenesis of Gastric Cancer Cells by Inhibiton of HMGB1/VEGF-D Signaling. Int J Immunopathol Pharmacol (2019) 33:2058738419861600. doi: 10.1177/2058738419861600
175. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat Rev Cancer (2012) 12:860–75. doi: 10.1038/nrc3380
176. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med (2007) 13:54–61. doi: 10.1038/nm1523
177. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic Death of Colon Cancer Cells Treated With Oxaliplatin. Oncogene (2010) 29:482–91. doi: 10.1038/onc.2009.356
178. Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, et al. Immunosuppression by Mutated Calreticulin Released From Malignant Cells. Mol Cell (2020) 77:748–60.e9. doi: 10.1016/j.molcel.2019.11.004
179. Liu P, Zhao L, Kroemer G, Kepp O. Secreted Calreticulin Mutants Subvert Anticancer Immunosurveillance. Oncoimmunology (2020) 9:1708126. doi: 10.1080/2162402X.2019.1708126
180. Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic Cell Death in Cancer Therapy: Present and Emerging Inducers. J Cell Mol Med (2019) 23:4854–65. doi: 10.1111/jcmm.14356
181. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of Immunogenic Cell Death and its Relevance for Cancer Therapy. Cell Death Dis (2020) 11:1013. doi: 10.1038/s41419-020-03221-2
182. Gong T, Liu L, Jiang W, Zhou R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
183. Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, et al. PGE2 Induced in and Released by Dying Cells Functions as an Inhibitory DAMP. Proc Natl Acad Sci U S A (2016) 113:3844–9. doi: 10.1073/pnas.1602023113
184. Hayashi K, Nikolos F, Lee YC, Jain A, Tsouko E, Gao H, et al. Tipping the Immunostimulatory and Inhibitory DAMP Balance to Harness Immunogenic Cell Death. Nat Commun (2020) 11:6299. doi: 10.1038/s41467-020-19970-9
185. Zhou F, Feng B, Yu H, Wang D, Wang T, Ma Y, et al. Tumor Microenvironment-Activatable Prodrug Vesicles for Nanoenabled Cancer Chemoimmunotherapy Combining Immunogenic Cell Death Induction and CD47 Blockade. Adv Mater (2019) 31:e1805888. doi: 10.1002/adma.201805888
186. Yang W, Zhang F, Deng H, Lin L, Wang S, Kang F, et al. Smart Nanovesicle-Mediated Immunogenic Cell Death Through Tumor Microenvironment Modulation for Effective Photodynamic Immunotherapy. ACS Nano (2020) 14:620–31. doi: 10.1021/acsnano.9b07212
187. Deng H, Yang W, Zhou Z, Tian R, Lin L, Ma Y, et al. Targeted Scavenging of Extracellular ROS Relieves Suppressive Immunogenic Cell Death. Nat Commun (2020) 11:4951. doi: 10.1038/s41467-020-18745-6
188. Chamoto K, Al-Habsi M, Honjo T. Role of PD-1 in Immunity and Diseases. Curr Top Microbiol Immunol (2017) 410:75–97. doi: 10.1007/82_2017_67
189. Pai C-CS, Simons DM, Lu X, Evans M, Wei J, Wang Y-H, et al. Tumor-Conditional Anti-CTLA4 Uncouples Antitumor Efficacy From Immunotherapy-Related Toxicity. J Clin Invest (2019) 129:349–63. doi: 10.1172/JCI123391
190. Mikami S, Mizuno R, Kondo T, Shinohara N, Nonomura N, Ozono S, et al. Clinical Significance of Programmed Death-1 and Programmed Death-Ligand 1 Expression in the Tumor Microenvironment of Clear Cell Renal Cell Carcinoma. Cancer Sci (2019) 110:1820–8. doi: 10.1111/cas.14019
191. Che X, Qi X, Xu Y, Wang Q, Wu G. Genomic and Transcriptome Analysis to Identify the Role of the mTOR Pathway in Kidney Renal Clear Cell Carcinoma and Its Potential Therapeutic Significance. Oxid Med Cell Longev (2021) 2021:6613151. doi: 10.1155/2021/6613151
192. Xie L, Wang G, Sang W, Li J, Zhang Z, Li W, et al. Phenolic Immunogenic Cell Death Nanoinducer for Sensitizing Tumor to PD-1 Checkpoint Blockade Immunotherapy. Biomaterials (2021) 269:120638. doi: 10.1016/j.biomaterials.2020.120638
193. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
194. Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells (2020) 9:915. doi: 10.3390/cells9040915
195. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
196. Tan C, Aziz M, Wang P. The Vitals of NETs. J Leukoc Biol (2021) 110:797–808. doi: 10.1002/JLB.3RU0620-375R
197. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-Induced NETosis is a Dynamic Process Involving Neutrophil Multitasking In Vivo. Nat Med (2012) 18:1386–93. doi: 10.1038/nm.2847
198. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient Neutrophil Extracellular Trap Induction Requires Mobilization of Both Intracellular and Extracellular Calcium Pools and is Modulated by Cyclosporine a. PloS One (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
199. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PloS One (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
200. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized Integrin Engagement and Chemokine Activation is Crucial in Neutrophil Extracellular Trap-Mediated Sterile Inflammation. Blood (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
201. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
202. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
203. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
204. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc Natl Acad Sci U S A (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
205. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis Proceeds by Cytoskeleton and Endomembrane Disassembly and PAD4-Mediated Chromatin Decondensation and Nuclear Envelope Rupture. Proc Natl Acad Sci U S A (2020) 117:7326–37. doi: 10.1073/pnas.1909546117
206. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
207. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-Mediated Neutrophil Extracellular Trap Formation is Not Required for Immunity Against Influenza Infection. PloS One (2011) 6:e22043. doi: 10.1371/journal.pone.0022043
208. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191:677–91. doi: 10.1083/jcb.201006052
209. Chen KW, Demarco B, Broz P. Beyond Inflammasomes: Emerging Function of Gasdermins During Apoptosis and NETosis. EMBO J (2020) 39:e103397. doi: 10.15252/embj.2019103397
210. Demers M, Wagner DD. NETosis: A New Factor in Tumor Progression and Cancer-Associated Thrombosis. Semin Thromb Hemost (2014) 40:277–83. doi: 10.1055/s-0034-1370765
211. Demers M, Wagner DD. Neutrophil Extracellular Traps: A New Link to Cancer-Associated Thrombosis and Potential Implications for Tumor Progression. Oncoimmunology (2013) 2:e22946. doi: 10.4161/onci.22946
212. Ho-Tin-Noé B, Carbo C, Demers M, Cifuni SM, Goerge T, Wagner DD. Innate Immune Cells Induce Hemorrhage in Tumors During Thrombocytopenia. Am J Pathol (2009) 175:1699–708. doi: 10.2353/ajpath.2009.090460
213. Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R, et al. A Proposed Role for Neutrophil Extracellular Traps in Cancer Immunoediting. Front Immunol (2013) 4:48. doi: 10.3389/fimmu.2013.00048
214. Decker AS, Pylaeva E, Brenzel A, Spyra I, Droege F, Hussain T, et al. Prognostic Role of Blood NETosis in the Progression of Head and Neck Cancer. Cells (2019) 8:946. doi: 10.3390/cells8090946
Keywords: regulatory cell death (RCD), immunotherapy, tumor microenvironment, caspase, GSDM, PARP, ECM, DAMPs
Citation: Qi X, Li Q, Che X, Wang Q and Wu G (2022) Application of Regulatory Cell Death in Cancer: Based on Targeted Therapy and Immunotherapy. Front. Immunol. 13:837293. doi: 10.3389/fimmu.2022.837293
Received: 16 December 2021; Accepted: 21 February 2022;
Published: 10 March 2022.
Edited by:
Zong Sheng Guo, Roswell Park Comprehensive Cancer Center, United StatesReviewed by:
Stephen Hatfield, Northeastern University, United StatesGaocai Li, Huazhong University of Science and Technology, China
Copyright © 2022 Qi, Li, Che, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangzhen Wu, d3VndWFuZzA2MTNAaG90bWFpbC5jb20=; Qifei Wang, d2FuZ3FpZmVpNjAwOEBob3RtYWlsLmNvbQ==