 Wangko Lundström
Wangko Lundström Rasmus Gustafsson*
Rasmus Gustafsson*- Center for Molecular Medicine, Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden
The role for human herpesvirus (HHV)-6A or HHV-6B in multiple sclerosis (MS) pathogenesis has been controversial. Possibly because the damage of the virus infection may occur before onset of clinical symptoms and because it has been difficult to detect active infection and separate serological responses to HHV-6A or 6B. Recent studies report that in MS patients the serological response against HHV-6A is increased whereas it is decreased against HHV-6B. This effect seems to be even more pronounced in MS patients prior to diagnosis and supports previous studies postulating a predomination for HHV-6A in MS disease and suggests that the infection is important at early stages of the disease. Furthermore, HHV-6A infection interacts with other factors suspected of modulating MS susceptibility and progression such as infection with Epstein-Barr virus (EBV) and Cytomegalovirus (CMV), tobacco smoking, HLA alleles, UV irradiation and vitamin D levels. The multifactorial nature of MS and pathophysiological role for HHV-6A in inflammation and autoimmunity are discussed.
Introduction
Multiple Sclerosis
Multiple Sclerosis (MS) typically presents with alternating bouts of disability and recovery. Over time this pattern often transitions into a more continuous, progressive increase in disability and neurological degeneration. Typical symptoms of MS include sensory and motoric ailments but vary greatly depending on what part of the CNS is targeted. Inflammatory events induce demyelination of neuronal axons reducing signaling capacity that induce disability (1).
The exact pathogenesis of MS is not understood, but epidemiological approaches including twin- and family studies define a complex interplay between genes and environment. For example, concordance rate among monozygotic twins is 25% and 3% for dizygotic twins (2, 3).
Several theories on the pathophysiological origin exist, but autoimmune events, whether primary triggers or secondary drivers, play a central role from disease onset and onwards.
Genetic Risk Factors
Genome wide association studies (GWAS) have provided a fruitful approach in defining genetic associations to susceptibility (4). Prior to the GWAS era the only bona fide genetic risk factors were in the HLA region, underlining autoimmunity as a central player. The associations within the HLA region remain the strongest genetic risk factors. Furthermore, the ~100 loci associated with MS susceptibility outside HLA, almost entirely exist of genes encoding immune regulating proteins, further underlining that interplay (5).
Environmental Risk Factors
Several environmental risk factors have been identified over the years. These include high latitude, Vitamin D deficiency and smoking. High latitude and Vitamin D deficiency are complicated to separate as discrete risk factors. Studies of migrants between low and high prevalence areas suggest the MS risk is affected by such migration (moving to a lower risk area reduces risk and vice versa), indicating the latitude effect may have more to do with sun light exposure than ethnicity (6). Vitamin D is known to regulate T-cells (7), which could explain the mechanics of the effect.
Tobacco smoking is associated with a plethora of disease risks, including inflammatory diseases. MS susceptibility increases in a dose dependent fashion by smoking. Interestingly, a gene-environment interaction seems to occur such that smoking combined with certain HLA genotypes renders larger cumulative risk than the additive risk of both factors by themselves (8).
Herpes Viruses as Risk Factors
Human herpesvirus (HHV)-6A joins a group of herpesviruses suspected of modulating MS susceptibility and progression, most notably Epstein Barr Virus (EBV) and Cytomegalovirus (CMV) (9). HHV-6 (10) belongs to the β-herpesvirus subfamily of Herpesviridae that can establish lifelong latent infections in the host. HHV-6 isolates are classified as two distinct virus species, HHV-6A and 6B (11). Given their high degree homology it has been difficult to separate the viruses serologically. Therefore, in this review, HHV-6 will be used where no distinction has been made. Their genomes are constituted by double stranded DNA and contains hundreds of open reading frames (12). CD46 is a common cellular receptor (13) and CD134 for HHV-6B (14). T cell lines are commonly used for HHV-6 propagation. At one year of age, most individuals have acquired HHV-6 (15, 16). HHV-6A seems the predominant variant in sub-Saharan Africa (17), and HHV-6B in Europe, Japan and USA (18–20). Whereas HHV-6B is the causative agent of exanthema subitum (21), no disease has been clearly linked to HHV-6A but multiple sclerosis (MS) is a candidate (22, 23).
EBV-infection has long been suspected to increase MS risk (24) and EBV transformed B cells and formation of ectopic germinal centers have been seen in MS brains (25). Hence, EBV may be a driver of inflammation and autoimmunity after a primary injury in the CNS. EBV infection is typically asymptomatic during childhood, but generally leads to infectious mononucleosis (IM) in adolescents or adults. The chance (or risk) of avoiding childhood infection is greatest in countries with a high living standard; hence the epidemiological map of IM (implying a person was not exposed to EBV during childhood) correlates with MS prevalence. Furthermore, the risk of several autoimmune diseases (including MS) in developed countries has steadily increased over the last decades whereas the risk for infections has declined. One possible explanation is the “hygiene hypothesis” which proposes that lack of exposure to common pathogens causes increased risk of allergy and autoimmunity. This would explain the link between MS and IM without necessarily providing a direct link between the two (26).
In contrast to EBV, CMV seropositivity protect against MS. As for EBV and HHV-6A (and indeed all herpesviruses), CMV can go into latency after infection. Why CMV seropositivity diverges from EBV and HHV-6A in terms of risk effect is not understood. However, albeit asymptomatic in most infected hosts, CMV occupies large proportions of the adaptive immune system’s cellular antigen specificity (27). It has been speculated that competition between herpes viruses can modulate the immune system’s adaptation, i.e. latent CMV infection could affect the response to subsequent EBV or HHV-6A infection.
In Vitro Diagnosis of HHV-6A or 6B Infection
For in vitro diagnostics correct sample materials and accurate methods for analyses are vital to achieve adequate data for reasonable interpretations. The best indication of active infection is isolation of the virus by inoculating susceptible cells with the biological specimen. Active infection is indicated when cells show signs of infection and/or supported viral replication. However, HHV-6 isolation is very challenging. Alternative approaches include screening plasma for cell free HHV-6A DNA by PCR. Plasma should be used (28) as latent infection may occur in blood cells. However, viral DNA in plasma may origin from latently infected and dying cells, and not necessarily from cell free virions (29, 30). Also, HHV-6 is chromosomally integrated (ciHHV-6) in roughly 1% of the population where all cells in the body contains viral DNA and viral DNA loads are typically high (31). Hence, ciHHV-6 infections may be mistaken for active infections. Also, active HHV-6 infection is highly transient and cell free viral DNA rarely detectable (32), possibly because HHV-6 spreads largely via cell-to-cell contact (33, 34). Therefore, reverse transcription (RT) PCR of HHV-6 mRNAs is more specific. Transcripts of the late genes U31 and U39 were found in 91-96% of samples from children with acute exanthema subitum (caused by HHV-6B), and in non during in the convalescent phase (35). Given that HHV-6A and HHV-6B are distinct species the PCR approach of choice should be able to discriminate between the two viruses (36). Antiviral IgM (37) represent an alternative measure that offers larger time windows. In primary HHV-6B infection virus specific IgM were detected five to seven days after onset of exanthema subitum and lasted for up to two months (38). However, IgM production requires a relatively strong inflammatory response and is not always detectable upon HHV-6 reactivation (39).
Following the natural course of a viral infection and inflammation, the antibody repertoire switches to IgG classes. Children seroconvert upon primary HHV-6 exposure (40), and as most people over two years old has been exposed discrete data on seroprevalence is not very informative. Instead, assessing titers of antiviral antibodies give a hint on how strong a primary infection has been; or how strongly and/or frequently the virus has reactivated. This is seen for infection with Varicella-Zoster virus (VZV) where the antiviral IgG titers increase during the convalescent phase. For CMV reactivation is associated with increased anti-CMV IgG titers (41). Therefore, the antiviral IgG titer seems to reflect the number and/or magnitude of reactivations and serve as a robust proxy indication of HHV-6 infection history.
HHV-6 in MS
HHV-6 in MS Brains and Increased Infection Rates During Relapse
HHV-6 has been associated to MS in numerous reports. A selection of these findings includes increased prevalence of HHV-6 DNA (42), mRNA (43) and protein expression (43–45) in MS plaques compared to in normal appearing white matter. A role for HHV-6 on MS pathogenesis in the brain is supported by increased frequencies and titers of anti-HHV-6 IgG (46, 47) and IgM (48) in cerebrospinal fluids of MS patients compared to controls; and oligoclonal band specificity against HHV-6 (49, 50). In the periphery HHV-6 mRNA and DNA is more frequent in peripheral blood mononuclear cells and serum from MS patients than in controls (51). Even though most studies show a positive association between HHV-6 and MS several conflicting reports exists where no association could be shown. A meta-analysis on the literature published between 1966 and 2009 (52) showed that 60% of the top ranked studies regarding study design, according to pre-determined criteria (53), showed significant differences between MS patients and controls in terms of HHV-6 mRNA, DNA or antibody titers. Active HHV-6 infection is significantly more frequent in MS patients during relapse than during remission (32, 54, 55) and increased titers of serum IgG antibodies against HHV-6 positively associate with relapse risk (56). It is tempting to conclude that these findings support an increased frequency of HHV-6 reactivation as a mechanism of disease activity. However, it is possible that they rather reflect a locally increased general immune activity in the CNS during relapses and that this sets of reactivation of latent viral infections, such as HHV-6, as CNS is a site of latency for HHV-6 from where the virus may reactivate (57).
Role for HHV-6 in MS Onset, and HHV-6A Predomination
Several findings indicate that HHV-6 play a role in MS disease onset and that HHV-6A is predominant over HHV-6B. Serum IgM are detectable in the early events of an infection and increased frequency of anti-HHV-6 IgM have been detected in early stages of MS (58, 59). Marmosets challenged with HHV-6A but not those challenged with HHV-6B gave clinical MS like symptoms and lesions seen by MRI (60). This suggests a role for HHV-6 in disease onset and/or periods of active MS disease and that HHV-6A is predominant over HHV-6B. In humans, this is further supported by that HHV-6A is more neurotropic than HHV-6B and increased cellular immune response against HHV-6A, but not against HHV-6B, has been reported in MS patients compared to controls (22), and increased frequencies of HHV-6A and 6B coinfection in MS patients compared to controls (36). Also, MS serum exhibit increased seroreactivity against HHV-6A compared to HHV-6B infected cells (47) and increased detection of HHV-6A DNA in MS serum and cerebrospinal fluids compared to HHV-6B (61) has been reported.
In our lab we investigated this further in a large clinical material with well characterized MS patients (n=8,742) and closely matched controls (n=7,215) (23) and found that in MS patients the antibody response against HHV-6A was increased (OR=1.55; p=9*10-22) and decreased against HHV-6B (OR=0.74; p=6*10-11), compared to controls. This effect was even more pronounced in samples drawn from MS patients prior to diagnosis, in median 8.3 years (n=478) and closely matched controls (n=476), where people who developed MS later in life exhibited increased anti-HHV-6A antibody levels (OR=2.22; p=2*10-5) compared to controls. No difference was seen for HHV-6B. The serological measurements were performed with a novel bead-based multiplex serology assay that can measure antibodies against the immediate early proteins IE1A (HHV-6A) and IE1B (HHV-6B) encoded by the open reading frame (ORF) U90-U89, the most divergent between the viral species with 62% homology. To conclude, in MS HHV-6 seems important in early stages of the disease, to have a direct effect on the CNS and HHV-6A seems more prominent than HHV-6B. Even though most studies show a positive association the concept is still controversial. Therefore, additional carefully performed studies that can distinguish between HHV-6A and HHV-6B are needed.
Genetic and Environmental Risk Factors for MS, and HHV-6A Infection
To investigate how history of infection (antiviral antibodies) for HHV-6A and HHV-6B correlates with host genetics the serologically characterized MS and control cohort described above were genotyped (23). Most associated single nucleotide polymorphisms (SNP) were found within the HLA region. Interestingly, while 191 SNP were associated with anti-HHV-6A antibody levels only two SNPs were associated with anti-HHV-6B levels. To speculate, as HHV-6 is ubiquitous (15) and as HHV-6B is more common than HHV-6A (18–20) it is possible that HHV-6B has coevolved with the human species to a larger extent than HHV-6A. A similar pattern is seen for CMV, also reaching 100% seroprevalence, and where GWAS data could not be associated to serology (62).
For MS, the major risk HLA haplotype DRB1*15:01 (63) is not associated with anti-HHV-6A levels (23), indicating the serological response could not explain this previously known risk gene. However, presence of DRB1*15:01 in combination with absence of the protective allele HLA-A*02:01 aggravated the MS risk in people with high anti-HHV-6A antibody levels (23). Analogous for EBV, MS patients with DRB1*15:01 and without A*02 had lower serum viral DNA loads compared to those without DRB1*15:01 and with A*02 (24). Other known MS risk factors such as smoking, low ultraviolet radiation and low vitamin D levels all aggravated the MS risk in people with high anti-HHV-6A antibody levels (64). Similar interaction analyses were performed between anti-HHV-6A and anti-EBV (risk factor) and anti-CMV (protective) antibody levels in plasma from people who developed MS 8 years after the sampling date. High anti-HHV-6A levels interacted with high anti-EBV levels and aggravated the MS risk (65). When adding CMV to the model people with high levels of anti-HHV-6A and anti-EBV antibodies, and low levels of anti-CMV antibodies had 15 times higher risk of developing MS later in life compared to people with low levels of IE1A and EBV antibodies and high levels of CMV (66).
Together these data suggests that HHV-6A represents a risk factor by itself and that interaction with other factors suspected of modulating MS disease further impacts the risk for developing MS (Figure 1).
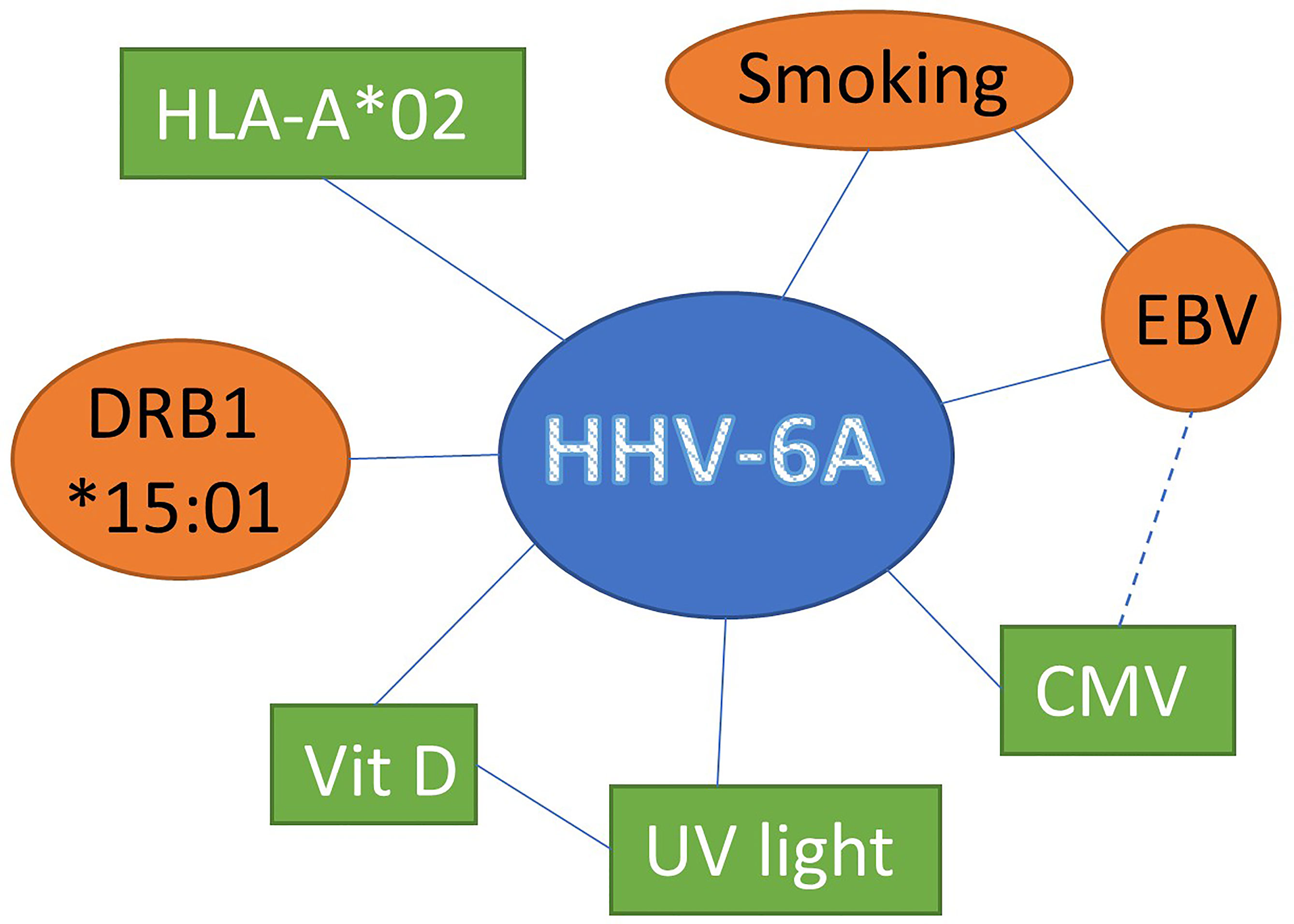
Figure 1 Interaction between HHV-6A and other factors suspected of modulating MS disease susceptibility susceptibility and progression increasing (red circles) or decreasing (green boxes) the risk for MS development.
HHV-6A in MS Pathogenesis
In MS, autoreactive T cells are thought to target the myelin sheath. Antigen presenting cells (APC) such as dendritic cells (DC) are the primary conductors of T cell regulation. DCs mature upon exposure of pathogen-associated molecular patterns (PAMPs) and thereafter can activate T cells. As PAMPs are not present in self-tissue they likely need to be present for breakage of tolerance and autoimmunity. A model has been proposed where APCs need a danger signal in combination with the antigen to elicit an immune response against the antigen (67). An example is the animal model for MS, experimental autoimmune encephalopathy (EAE). Here, the adjuvant provides the immune system with a danger signal to triggering an immune response and the myelin proteins steers the response against myelin. EAE has striking similarities with MS with infiltration of lymphocytes in the CNS and demyelination, as well as clinical symptoms e.g. paralysis (68).
Several mechanisms for HHV-6-induced autoimmunity and myelin breakdown have been suggested. These include molecular mimicry and host cell incorporation in the virion. The molecular mimicry model proposes that the virus encodes for a protein with similar motifs to host cell proteins, and that an immune response directed against this “virus encoded host-like protein” is cross-reactive and also target the host protein that is mimicked. For HHV-6 and MS, molecular mimicry has been shown with cross-reactivity between a peptide encoded by the viral gene U24 and myelin basic protein (MBP) (69). The host cell protein incorporation hypothesis is based on the well-documented notion that the envelop of enveloped viruses origin from the host cell, and therefore, that host cell proteins are present in the viral envelop. When an APC engulfs a virion containing host cell proteins an immune response is triggered by the PAMPs of viral motifs and directed against the host cell proteins contained in the virion (70). Both HHV-6A and HHV-6B have the capacity to infect oligodendrocytes, which is a pre-requisite. But the model suffers from studies where release of cell free virions is not commonly seen during HHV-6 infection of oligodendrocytes (71–73).
Both the molecular mimicry and the host cell incorporation model relies on the idea that HHV-6 virion can trigger the T cell stimulatory capacity of APC. However, we and others have shown that inoculation of DC with both HHV-6A and HHV-6B instead hamper the capacity of DC to activate T cells, both in absence and presence of viral replication, and for both allogenic and autologous T cells (74–79). Hence, HHV-6 induced pathogenesis in MS does not seem to occur through neither of these models.
HHV-6 induces syncytia but also gives semi-lytic infection with dying cells and cell debris. DC exposure to HHV-6A result in cell death and release of high mobility group box 1 (HMGB1) protein (80). This may occur also in the CNS as HHV-6A can induce vigorous apoptosis of a human oligodendrocytes (81). HMGB1 can activate DC in the autoimmune disease systemic lupus erythematosus (82). In MS, HMGB1 has been proposed as an important driver (83) with HMGB1 expression in MS lesions (84) and elevated serum levels of HMGB1 in treatment naïve MS patients, compared to those receiving disease-modifying treatment (85).
Hence, we propose a model for HHV-6A induced pathogenesis where HHV-6A infection and/or reactivation (86) in the CNS gives a primary injury. This in turn gives inflammatory events, possibly driven by HMGB1, with upregulation of metalloproteinase and influx of leukocytes from the periphery which can cause pathological inflammation and subsequent plaque formation. The inflammation is aggravated by EBV infection and tobacco smoking; and hampered if the immune system is occupied by CMV and/or regulated when vitamin D levels are sufficient.
Conclusions
Together, the studies compiled in this review suggest a role for HHV-6A in MS, particularly early in the disease course and/or at MS onset. This is based on findings of increased anti-HHV-6A antibody levels in people who developed MS later in life, compared to people who do not. HHV-6A infection interacts with other risk factors such as carriage of HLA-DRB1*15:01, tobacco smoking, low UV irradiation, low vitamin D levels and EBV infection; and with protective factors such carriage of the protective haplotype HLA-A*02:01 and CMV infection. This shows the multifactorial nature of MS and suggests that accumulated burden of risk factors increases the risk for acquiring the disease. We propose a pathophysiological role for HHV-6A in induction of MS where an infection of the CNS leads to a primary injury and that this in turn leads to inflammatory events and autoimmunity.
Author Contributions
RG conceived this review article. WL and RG contributed to design of the review, wrote the first draft of the manuscript, contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Compston A, Coles A. Multiple Sclerosis. Lancet (2002) 359(9313):1221–31. doi: 10.1016/S0140-6736(02)08220-X
2. Koch-Henriksen N, Stenager E, Bronnum-Hansen H. Studies Based on the Danish Multiple Sclerosis Registry. Scand J Public Health (2011) 39(7 Suppl):180–4. doi: 10.1177/1403494811405097
3. Ebers GC, Bulman DE, Sadovnick AD, Paty DW, Warren S, Hader W, et al. A Population-Based Study of Multiple Sclerosis in Twins. N Engl J Med (1986) 315(26):1638–42. doi: 10.1056/NEJM198612253152603
4. International Multiple Sclerosis Genetics C, Wellcome Trust Case Control C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature (2011) 476(7359):214–9. doi: 10.1038/nature10251
5. Olsson T, Barcellos LF, Alfredsson L. Interactions Between Genetic, Lifestyle and Environmental Risk Factors for Multiple Sclerosis. Nat Rev Neurol (2017) 13(1):25–36. doi: 10.1038/nrneurol.2016.187
6. Simpson S Jr., Blizzard L, Otahal P, van der Mei I, Taylor B. Latitude Is Significantly Associated With the Prevalence of Multiple Sclerosis: A Meta-Analysis. J Neurol Neurosurg Psychiatry (2011) 82(10):1132–41. doi: 10.1136/jnnp.2011.240432
7. Bishop E, Ismailova A, Dimeloe SK, Hewison M, White JH. Vitamin D and Immune Regulation: Antibacterial, Antiviral, Anti-Inflammatory. JBMR Plus (2020). doi: 10.1002/jbm4.10405
8. Hedstrom AK, Sundqvist E, Baarnhielm M, Nordin N, Hillert J, Kockum I, et al. Smoking and Two Human Leukocyte Antigen Genes Interact to Increase the Risk for Multiple Sclerosis. Brain (2011) 134(Pt 3):653–64. doi: 10.1093/brain/awq371
9. Meier UC, Cipian RC, Karimi A, Ramasamy R, Middeldorp JM. Cumulative Roles for Epstein-Barr Virus, Human Endogenous Retroviruses, and Human Herpes Virus-6 in Driving an Inflammatory Cascade Underlying MS Pathogenesis. Front Immunol (2021) 12:757302. doi: 10.3389/fimmu.2021.757302
10. Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, et al. Isolation of a New Virus, HBLV, in Patients With Lymphoproliferative Disorders. Science (1986) 234(4776):596–601. doi: 10.1126/science.2876520
11. Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, et al. Classification of HHV-6A and HHV-6B as Distinct Viruses. Arch Virol (2014) 159(5):863–70. doi: 10.1007/s00705-013-1902-5
12. Finkel Y, Schmiedel D, Tai-Schmiedel J, Nachshon A, Winkler R, Dobesova M, et al. Comprehensive Annotations of Human Herpesvirus 6A and 6B Genomes Reveal Novel and Conserved Genomic Features. Elife (2020) 9. doi: 10.7554/eLife.50960
13. Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. CD46 Is a Cellular Receptor for Human Herpesvirus 6. Cell (1999) 99(7):817–27. doi: 10.1016/s0092-8674(00)81678-5
14. Tang H, Serada S, Kawabata A, Ota M, Hayashi E, Naka T, et al. CD134 Is a Cellular Receptor Specific for Human Herpesvirus-6B Entry. Proc Natl Acad Sci USA (2013) 110(22):9096–9. doi: 10.1073/pnas.1305187110
15. Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, et al. Seroepidemiology of Human Herpesvirus 6 Infection in Normal Children and Adults. J Clin Microbiol (1989) 27(4):651–3. doi: 10.1128/jcm.27.4.651-653.1989
16. Zerr DM, Meier AS, Selke SS, Frenkel LM, Huang ML, Wald A, et al. A Population-Based Study of Primary Human Herpesvirus 6 Infection. N Engl J Med (2005) 352(8):768–76. doi: 10.1056/NEJMoa042207
17. Bates M, Monze M, Bima H, Kapambwe M, Clark D, Kasolo FC, et al. Predominant Human Herpesvirus 6 Variant A Infant Infections in an HIV-1 Endemic Region of Sub-Saharan Africa. J Med Virol (2009) 81(5):779–89. doi: 10.1002/jmv.21455
18. Hall CB, Caserta MT, Schnabel KC, McDermott MP, Lofthus GK, Carnahan JA, et al. Characteristics and Acquisition of Human Herpesvirus (HHV) 7 Infections in Relation to Infection With HHV-6. J Infect Dis (2006) 193(8):1063–9. doi: 10.1086/503434
19. Tanaka-Taya K, Kondo T, Mukai T, Miyoshi H, Yamamoto Y, Okada S, et al. Seroepidemiological Study of Human Herpesvirus-6 and -7 in Children of Different Ages and Detection of These Two Viruses in Throat Swabs by Polymerase Chain Reaction. J Med Virol (1996) 48(1):88–94. doi: 10.1002/(SICI)1096-9071(199601)48:1<88::AID-JMV14>3.0.CO;2-2
20. Ward KN, Andrews NJ, Verity CM, Miller E, Ross EM. Human Herpesviruses-6 and -7 Each Cause Significant Neurological Morbidity in Britain and Ireland. Arch Dis Child (2005) 90(6):619–23. doi: 10.1136/adc.2004.062216
21. Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, et al. Identification of Human Herpesvirus-6 as a Causal Agent for Exanthem Subitum. Lancet (1988) 1(8594):1065–7. doi: 10.1016/s0140-6736(88)91893-4
22. Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S. Increased Lymphoproliferative Response to Human Herpesvirus Type 6A Variant in Multiple Sclerosis Patients. Ann Neurol (2000) 47(3):306–13. doi: 10.1002/1531-8249(200003)47:3<306::AID-ANA5>3.0.CO;2-A
23. Engdahl E, Gustafsson R, Huang J, Bistrom M, Lima Bomfim I, Stridh P, et al. Increased Serological Response Against Human Herpesvirus 6a Is Associated With Risk for Multiple Sclerosis. Front Immunol (2019) 10:2715. doi: 10.3389/fimmu.2019.02715
24. Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, et al. HLA Alleles Modulate EBV Viral Load in Multiple Sclerosis. J Transl Med (2018) 16(1):80. doi: 10.1186/s12967-018-1450-6
25. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr Virus Infection in the Multiple Sclerosis Brain. J Exp Med (2007) 204(12):2899–912. doi: 10.1084/jem.20071030
26. Bach JF. The Effect of Infections on Susceptibility to Autoimmune and Allergic Diseases. N Engl J Med (2002) 347(12):911–20. doi: 10.1056/NEJMra020100
27. Makhani N, Banwell B, Tellier R, Yea C, McGovern S, O’Mahony J, et al. Viral Exposures and MS Outcome in a Prospective Cohort of Children With Acquired Demyelination. Mult Scler (2016) 22(3):385–8. doi: 10.1177/1352458515595876
28. Kainth MK, Caserta MT. Molecular Diagnostic Tests for Human Herpesvirus 6. Pediatr Infect Dis J (2011) 30(7):604–5. doi: 10.1097/INF.0b013e318224947f
29. Caserta MT, Hall CB, Schnabel K, Lofthus G, Marino A, Shelley L, et al. Diagnostic Assays for Active Infection With Human Herpesvirus 6 (HHV-6). J Clin Virol (2010) 48(1):55–7. doi: 10.1016/j.jcv.2010.02.007
30. Achour A, Boutolleau D, Slim A, Agut H, Gautheret-Dejean A. Human Herpesvirus-6 (HHV-6) DNA in Plasma Reflects the Presence of Infected Blood Cells Rather Than Circulating Viral Particles. J Clin Virol (2007) 38(4):280–5. doi: 10.1016/j.jcv.2006.12.019
31. Flamand L, Komaroff AL, Arbuckle JH, Medveczky PG, Ablashi DV. Review, Part 1: Human Herpesvirus-6-Basic Biology, Diagnostic Testing, and Antiviral Efficacy. J Med Virol (2010) 82(9):1560–8. doi: 10.1002/jmv.21839
32. Berti R, Brennan MB, Soldan SS, Ohayon JM, Casareto L, McFarland HF, et al. Increased Detection of Serum HHV-6 DNA Sequences During Multiple Sclerosis (MS) Exacerbations and Correlation With Parameters of MS Disease Progression. J Neurovirol (2002) 8(3):250–6. doi: 10.1080/13550280290049615-1
33. Zhen Z, Bradel-Tretheway B, Sumagin S, Bidlack JM, Dewhurst S. The Human Herpesvirus 6 G Protein-Coupled Receptor Homolog U51 Positively Regulates Virus Replication and Enhances Cell-Cell Fusion In Vitro. J Virol (2005) 79(18):11914–24. doi: 10.1128/JVI.79.18.11914-11924.2005
34. Takemoto M, Imasawa T, Yamanishi K, Mori Y. Role of Dendritic Cells Infected With Human Herpesvirus 6 in Virus Transmission to CD4(+) T Cells. Virology (2009) 385(2):294–302. doi: 10.1016/j.virol.2008.11.049
35. Yoshikawa T, Akimoto S, Nishimura N, Ozaki T, Ihira M, Ohashi M, et al. Evaluation of Active Human Herpesvirus 6 Infection by Reverse Transcription-PCR. J Med Virol (2003) 70(2):267–72. doi: 10.1002/jmv.10388
36. Leibovitch EC, Brunetto GS, Caruso B, Fenton K, Ohayon J, Reich DS, et al. Coinfection of Human Herpesviruses 6A (HHV-6A) and HHV-6B as Demonstrated by Novel Digital Droplet PCR Assay. PloS One (2014) 9(3):e92328. doi: 10.1371/journal.pone.0092328
37. Ortega-Madueno I, Garcia-Montojo M, Dominguez-Mozo MI, Garcia-Martinez A, Arias-Leal AM, Casanova I, et al. Anti-Human Herpesvirus 6A/B IgG Correlates With Relapses and Progression in Multiple Sclerosis. PloS One (2014) 9(8):e104836. doi: 10.1371/journal.pone.0104836
38. Suga S, Yoshikawa T, Asano Y, Nakashima T, Yazaki T, Fukuda M, et al. IgM Neutralizing Antibody Responses to Human Herpesvirus-6 in Patients With Exanthem Subitum or Organ Transplantation. Microbiol Immunol (1992) 36(5):495–506. doi: 10.1111/j.1348-0421.1992.tb02047.x
39. Dzieciatkowski T, Przybylski M, Torosian T, Tomaszewska A, Luczak M. Prevalence of Human Herpesvirus 6 Antibodies and DNA in Allogeneic Stem Cell Transplant Patients: Two-Year Single Centre Experience. Arch Immunol Ther Exp (Warsz) (2008) 56(3):201–6. doi: 10.1007/s00005-008-0021-6
40. Hall CB, Long CE, Schnabel KC, Caserta MT, McIntyre KM, Costanzo MA, et al. Human Herpesvirus-6 Infection in Children. A Prospective Study of Complications and Reactivation. N Engl J Med (1994) 331(7):432–8. doi: 10.1056/NEJM199408183310703
41. Mehta SK, Stowe RP, Feiveson AH, Tyring SK, Pierson DL. Reactivation and Shedding of Cytomegalovirus in Astronauts During Spaceflight. J Infect Dis (2000) 182(6):1761–4. doi: 10.1086/317624
42. Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human Herpesvirus 6 Genome and Antigen in Acute Multiple Sclerosis Lesions. J Infect Dis (2003) 187(9):1365–76. doi: 10.1086/368172
43. Opsahl ML, Kennedy PG. Early and Late HHV-6 Gene Transcripts in Multiple Sclerosis Lesions and Normal Appearing White Matter. Brain (2005) 128(Pt 3):516–27. doi: 10.1093/brain/awh390
44. Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-Associated Expression of Human Herpesvirus 6 in Multiple Sclerosis. Proc Natl Acad Sci USA (1995) 92(16):7440–4. doi: 10.1073/pnas.92.16.7440
45. Friedman JE, Lyons MJ, Cu G, Ablashl DV, Whitman JE, Edgar M, et al. The Association of the Human Herpesvirus-6 and MS. Mult Scler (1999) 5(5):355–62. doi: 10.1177/135245859900500509
46. Derfuss T, Hohlfeld R, Meinl E. Intrathecal Antibody (IgG) Production Against Human Herpesvirus Type 6 Occurs in About 20% of Multiple Sclerosis Patients and Might be Linked to a Polyspecific B-Cell Response. J Neurol (2005) 252(8):968–71. doi: 10.1007/s00415-005-0794-z
47. Virtanen JO, Farkkila M, Multanen J, Uotila L, Jaaskelainen AJ, Vaheri A, et al. Evidence for Human Herpesvirus 6 Variant A Antibodies in Multiple Sclerosis: Diagnostic and Therapeutic Implications. J Neurovirol (2007) 13(4):347–52. doi: 10.1080/13550280701381332
48. Ablashi DV, Lapps W, Kaplan M, Whitman JE, Richert JR, Pearson GR. Human Herpesvirus-6 (HHV-6) Infection in Multiple Sclerosis: A Preliminary Report. Mult Scler (1998) 4(6):490–6. doi: 10.1177/135245859800400606
49. Virtanen JO, Pietilainen-Nicklen J, Uotila L, Farkkila M, Vaheri A, Koskiniemi M. Intrathecal Human Herpesvirus 6 Antibodies in Multiple Sclerosis and Other Demyelinating Diseases Presenting as Oligoclonal Bands in Cerebrospinal Fluid. J Neuroimmunol (2011) 237(1-2):93–7. doi: 10.1016/j.jneuroim.2011.06.012
50. Virtanen JO, Wohler J, Fenton K, Reich DS, Jacobson S. Oligoclonal Bands in Multiple Sclerosis Reactive Against Two Herpesviruses and Association With Magnetic Resonance Imaging Findings. Mult Scler (2014) 20(1):27–34. doi: 10.1177/1352458513490545
51. Chapenko S, Millers A, Nora Z, Logina I, Kukaine R, Murovska M. Correlation Between HHV-6 Reactivation and Multiple Sclerosis Disease Activity. J Med Virol (2003) 69(1):111–7. doi: 10.1002/jmv.10258
52. Voumvourakis KI, Kitsos DK, Tsiodras S, Petrikkos G, Stamboulis E. Human Herpesvirus 6 Infection as a Trigger of Multiple Sclerosis. Mayo Clin Proc (2010) 85(11):1023–30. doi: 10.4065/mcp.2010.0350
53. Moore FG, Wolfson C. Human Herpes Virus 6 and Multiple Sclerosis. Acta Neurol Scand (2002) 106(2):63–83. doi: 10.1034/j.1600-0404.2002.01251.x
54. Alvarez-Lafuente R, De las Heras V, Bartolome M, Picazo JJ, Arroyo R. Relapsing-Remitting Multiple Sclerosis and Human Herpesvirus 6 Active Infection. Arch Neurol (2004) 61(10):1523–7. doi: 10.1001/archneur.61.10.1523
55. Alvarez-Lafuente R, Garcia-Montojo M, De las Heras V, Bartolome M, Arroyo R. Clinical Parameters and HHV-6 Active Replication in Relapsing-Remitting Multiple Sclerosis Patients. J Clin Virol (2006) 37(Suppl 1):S24–6. doi: 10.1016/S1386-6532(06)70007-5
56. Simpson S Jr., Taylor B, Dwyer DE, Taylor J, Blizzard L, Ponsonby AL, et al. Anti-HHV-6 IgG Titer Significantly Predicts Subsequent Relapse Risk in Multiple Sclerosis. Mult Scler (2012) 18(6):799–806. doi: 10.1177/1352458511428081
57. Handley G, Hasbun R, Okhuysen P. Human Herpesvirus 6 and Central Nervous System Disease in Oncology Patients: A Retrospective Case Series and Literature Review. J Clin Virol (2021) 136:104740. doi: 10.1016/j.jcv.2021.104740
58. Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, et al. Association of Human Herpes Virus 6 (HHV-6) With Multiple Sclerosis: Increased IgM Response to HHV-6 Early Antigen and Detection of Serum HHV-6 DNA. Nat Med (1997) 3(12):1394–7. doi: 10.1038/nm1297-1394
59. Villoslada P, Juste C, Tintore M, Llorenc V, Codina G, Pozo-Rosich P, et al. The Immune Response Against Herpesvirus is More Prominent in the Early Stages of MS. Neurology (2003) 60(12):1944–8. doi: 10.1212/01.wnl.0000069461.53733.f7
60. Leibovitch E, Wohler JE, Cummings Macri SM, Motanic K, Harberts E, Gaitan MI, et al. Novel Marmoset (Callithrix Jacchus) Model of Human Herpesvirus 6A and 6B Infections: Immunologic, Virologic and Radiologic Characterization. PloS Pathog (2013) 9(1):e1003138. doi: 10.1371/journal.ppat.1003138
61. Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland HF, et al. Tissue Distribution and Variant Characterization of Human Herpesvirus (HHV)-6: Increased Prevalence of HHV-6A in Patients With Multiple Sclerosis. J Infect Dis (2000) 182(5):1321–5. doi: 10.1086/315893
62. Kuparinen T, Seppala I, Jylhava J, Marttila S, Aittoniemi J, Kettunen J, et al. Genome-Wide Association Study Does Not Reveal Major Genetic Determinants for Anti-Cytomegalovirus Antibody Response. Genes Immun (2012) 13(2):184–90. doi: 10.1038/gene.2011.71
63. Jersild C, Fog T, Hansen GS, Thomsen M, Svejgaard A, Dupont B. Histocompatibility Determinants in Multiple Sclerosis, With Special Reference to Clinical Course. Lancet (1973) 2(7840):1221–5. doi: 10.1016/s0140-6736(73)90970-7
64. Wu J, Engdahl E, Gustafsson R, Fogdell-Hahn A, Waterboer T, Hillert J, et al. High Antibody Levels Against Human Herpesvirus-6A Interact With Lifestyle Factors in Multiple Sclerosis Development. Mult Scler (2021). doi: 10.1177/13524585211022011
65. Bistrom M, Jons D, Engdahl E, Gustafsson R, Huang J, Brenner N, et al. Epstein-Barr Virus Infection After Adolescence and Human Herpesvirus 6A as Risk Factors for Multiple Sclerosis. Eur J Neurol (2021) 28(2):579–86. doi: 10.1111/ene.14597
66. Grut V, Bistrom M, Salzer J, Stridh P, Jons D, Gustafsson R, et al. Cytomegalovirus Seropositivity Is Associated With Reduced Risk of Multiple Sclerosis-a Presymptomatic Case-Control Study. Eur J Neurol (2021) 28(9):3072–9. doi: 10.1111/ene.14961
67. Matzinger P. The Danger Model: A Renewed Sense of Self. Science (2002) 296(5566):301–5. doi: 10.1126/science.1071059
68. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms Regulating Regional Localization of Inflammation During CNS Autoimmunity. Immunol Rev (2012) 248(1):205–15. doi: 10.1111/j.1600-065X.2012.01126.x
69. Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-Reactivity With Myelin Basic Protein and Human Herpesvirus-6 in Multiple Sclerosis. Ann Neurol (2003) 53(2):189–97. doi: 10.1002/ana.10425
70. Dunn N, Kharlamova N, Fogdell-Hahn A. The Role of Herpesvirus 6A and 6B in Multiple Sclerosis and Epilepsy. Scand J Immunol (2020) 92(6):e12984. doi: 10.1111/sji.12984
71. Albright AV, Lavi E, Black JB, Goldberg S, O’Connor MJ, Gonzalez-Scarano F. The Effect of Human Herpesvirus-6 (HHV-6) on Cultured Human Neural Cells: Oligodendrocytes and Microglia. J Neurovirol (1998) 4(5):486–94. doi: 10.3109/13550289809113493
72. Mock DJ, Strathmann F, Blumberg BM, Mayer-Proschel M. Infection of Murine Oligodendroglial Precursor Cells With Human Herpesvirus 6 (HHV-6)–Establishment of a Murine In Vitro Model. J Clin Virol (2006) 37(Suppl 1):S17–23. doi: 10.1016/S1386-6532(06)70006-3
73. Ahlqvist J, Fotheringham J, Akhyani N, Yao K, Fogdell-Hahn A, Jacobson S. Differential Tropism of Human Herpesvirus 6 (HHV-6) Variants and Induction of Latency by HHV-6A in Oligodendrocytes. J Neurovirol (2005) 11(4):384–94. doi: 10.1080/13550280591002379
74. Gustafsson R, Svensson M, Fogdell-Hahn A. Modulatory Effects on Dendritic Cells by Human Herpesvirus 6. Front Microbiol (2015) 6:388. doi: 10.3389/fmicb.2015.00388
75. Asada H, Yalcin S, Balachandra K, Higashi K, Yamanishi K. Establishment of Titration System for Human Herpesvirus 6 and Evaluation of Neutralizing Antibody Response to the Virus. J Clin Microbiol (1989) 27(10):2204–7. doi: 10.1128/jcm.27.10.2204-2207.1989
76. Kakimoto M, Hasegawa A, Fujita S, Yasukawa M. Phenotypic and Functional Alterations of Dendritic Cells Induced by Human Herpesvirus 6 Infection. J Virol (2002) 76(20):10338–45. doi: 10.1128/jvi.76.20.10338-10345.2002
77. Smith AP, Paolucci C, Di Lullo G, Burastero SE, Santoro F, Lusso P. Viral Replication-Independent Blockade of Dendritic Cell Maturation and Interleukin-12 Production by Human Herpesvirus 6. J Virol (2005) 79(5):2807–13. doi: 10.1128/JVI.79.5.2807-2813.2005
78. Niiya H, Lei J, Guo Y, Azuma T, Yakushijin Y, Sakai I, et al. Human Herpesvirus 6 Impairs Differentiation of Monocytes to Dendritic Cells. Exp Hematol (2006) 34(5):642–53. doi: 10.1016/j.exphem.2006.02.001
79. Gustafsson RK, Engdahl EE, Hammarfjord O, Adikari SB, Lourda M, Klingstrom J, et al. Human Herpesvirus 6A Partially Suppresses Functional Properties of DC Without Viral Replication. PloS One (2013) 8(3):e58122. doi: 10.1371/journal.pone.0058122
80. Gustafsson R. Human Herpesvirus 6a Induces Dendritic Cell Death and HMGB1 Release Without Virus Replication. Pathogens (2021) 10(1). doi: 10.3390/pathogens10010057
81. Gardell JL, Dazin P, Islar J, Menge T, Genain CP, Lalive PH. Apoptotic Effects of Human Herpesvirus-6A on Glia and Neurons as Potential Triggers for Central Nervous System Autoimmunity. J Clin Virol (2006) 37 Suppl 1:S11–6. doi: 10.1016/S1386-6532(06)70005-1
82. Song X, Zhang H, Zhao Y, Lin Y, Tang Q, Zhou X, et al. HMGB1 Activates Myeloid Dendritic Cells by Up-Regulating mTOR Pathway in Systemic Lupus Erythematosus. Front Med (Lausanne) (2021) 8:636188. doi: 10.3389/fmed.2021.636188
83. Paudel YN, Angelopoulou E, C BK, Piperi C, Othman I. High Mobility Group Box 1 (HMGB1) Protein in Multiple Sclerosis (MS): Mechanisms and Therapeutic Potential. Life Sci (2019) 238:116924. doi: 10.1016/j.lfs.2019.116924
84. Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, Khademi M, et al. Pivotal Advance: HMGB1 Expression in Active Lesions of Human and Experimental Multiple Sclerosis. J Leukoc Biol (2008) 84(5):1248–55. doi: 10.1189/jlb.1207844
85. Sternberg Z, Sternberg D, Chichelli T, Drake A, Patel N, Kolb C, et al. High-Mobility Group Box 1 in Multiple Sclerosis. Immunol Res (2016) 64(2):385–91. doi: 10.1007/s12026-015-8673-x
Keywords: human herpesvirus 6A, HHV-6A, HHV-6, infection, multiple sclerosis, environment, epidemiology, risk factor
Citation: Lundström W and Gustafsson R (2022) Human Herpesvirus 6A Is a Risk Factor for Multiple Sclerosis. Front. Immunol. 13:840753. doi: 10.3389/fimmu.2022.840753
Received: 21 December 2021; Accepted: 20 January 2022;
Published: 10 February 2022.
Edited by:
Per Höllsberg, Aarhus University, DenmarkReviewed by:
Simone Agostini, Fondazione Don Carlo Gnocchi Onlus (IRCCS), ItalyCopyright © 2022 Lundström and Gustafsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rasmus Gustafsson, UmFzbXVzLkd1c3RhZnNzb25Aa2kuc2U=