 Filippo Annoni1,2†
Filippo Annoni1,2† Federico Moro1†
Federico Moro1† Enrico Caruso1,3
Enrico Caruso1,3 Tommaso Zoerle3,4
Tommaso Zoerle3,4 Fabio Silvio Taccone2
Fabio Silvio Taccone2 Elisa R. Zanier1*
Elisa R. Zanier1*- 1Laboratory of Acute Brain Injury and Therapeutic Strategies, Department of Neuroscience, Mario Negri Institute for Pharmacological Research IRCCS, Milan, Italy
- 2Department of Intensive Care, Erasme Hospital, Free University of Brussels, Anderlecht, Belgium
- 3Neuroscience Intensive Care Unit, Department of Anesthesia and Critical Care, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 4Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
Aneurysmal subarachnoid hemorrhage (SAH) is a substantial cause of mortality and morbidity worldwide. Moreover, survivors after the initial bleeding are often subject to secondary brain injuries and delayed cerebral ischemia, further increasing the risk of a poor outcome. In recent years, the renin–angiotensin system (RAS) has been proposed as a target pathway for therapeutic interventions after brain injury. The RAS is a complex system of biochemical reactions critical for several systemic functions, namely, inflammation, vascular tone, endothelial activation, water balance, fibrosis, and apoptosis. The RAS system is classically divided into a pro-inflammatory axis, mediated by angiotensin (Ang)-II and its specific receptor AT1R, and a counterbalancing system, presented in humans as Ang-(1–7) and its receptor, MasR. Experimental data suggest that upregulation of the Ang-(1–7)/MasR axis might be neuroprotective in numerous pathological conditions, namely, ischemic stroke, cognitive disorders, Parkinson’s disease, and depression. In the presence of SAH, Ang-(1–7)/MasR neuroprotective and modulating properties could help reduce brain damage by acting on neuroinflammation, and through direct vascular and anti-thrombotic effects. Here we review the role of RAS in brain ischemia, with specific focus on SAH and the therapeutic potential of Ang-(1–7).
Introduction
Although aneurysmal subarachnoid hemorrhage (SAH) accounts for only 5% of all strokes, it is a substantial cause of premature death and disability, affecting 10 individuals per 100,000 every year, with a mortality rate of nearly 50% (1–3). Aneurysmal SAH affects younger adults than ischemic stroke and accounts for 27% of all stroke-related years of life lost before age 60 (4), and survivors commonly suffer cognitive and functional impairments (5). Thus SAH is a disease with an important personal and socio-economic impact, for which therapeutic strategies are urgently needed.
Patients surviving the early brain injury related to intracranial bleeding risk re-bleeding, hydrocephalus, seizures, intracranial hypertension, cardiac and pulmonary complications and delayed cerebral ischemia (DCI) (6). Early surgical or endovascular clipping/coiling of the aneurysm can significantly reduce the risk of re-bleeding. Despite preclinical and clinical research efforts (2, 7), strategies to prevent or limit DCI are limited. DCI is a major determinant of poor outcome, with an estimated 30% occurrence rate in SAH survivors (8) and its detection is still a challenge.
Clinical deterioration and/or a new cerebral ischemic lesion detected on cerebral computed tomography (CT-scan) or magnetic resonance imaging (MRI) are essential for the diagnosis of DCI (9). However, these approaches are of limited use in sedated or unconscious patients and offer only a few possible interventions, as cerebral ischemia is already present (10). Therefore additional diagnostic tools, namely, transcranial Doppler, electroencephalography, and cerebral CT-perfusion, have been proposed to optimize the diagnosis of DCI, although their potential is still debated (11).
One of the key determinants of DCI is cerebral vasospasm (CVS), defined as narrowing of the lumen of one or more of the major intracranial arteries (12). However, other mechanisms play a role. Blood extravasation after SAH leads to the exposure of the cerebral tissue to numerous intravascular components, triggering a local inflammatory response (13). This response is further aggravated by the accumulation of free radicals caused by the degradation of cellular components of the clot (14), ultimately generating a self-promoting detrimental loop (15). Microvascular dysfunction and cortical spreading depolarization can also occur in these patients and contribute to the DCI pathogenesis (6).
About two thirds of SAH patients develop CVS within two weeks of the aneurysmal rupture (16) and half of these will also develop DCI (6). These, patients often require intensive care for multimodal neuromonitoring and supportive care to minimize secondary cerebral injuries of systemic origin induced by fluctuations in blood pressure, arterial CO2 concentrations, hemoglobin levels, oxygen demand (i.e., core temperature, adequate sedation) and sodium levels (17).
Besides oral nimodipine, which has been associated with improved neurological outcome though with no significant reduction of CVS (18), treatments have aimed to treat large-vessel spasm (i.e., intravenous and intra-arterial vasodilators; balloon angioplasty), increase cerebral perfusion (i.e., controlled hypertension, milrinone, dobutamine), reduce systemic inflammation (i.e., statins, steroids) (11, 18–20), and maintain brain homeostasis (i.e., temperature control). None of these specifically target the multiple pathophysiological mechanisms involved in DCI.
The Renin–Angiotensin System (RAS)
In recent years, the renin–angiotensin system has been proposed as a possible therapeutic target in different brain-related pathological conditions. In the so-called “classical RAS”, angiotensinogen is transformed by renin into angiotensin (Ang)-I, which is then cleaved into Ang-II by the angiotensin-converting enzyme (ACE); this than exerts its effects through the AT1 receptor (AT1R) or the AT2R subtypes, tuning water and salt homeostasis and modulating inflammation, fibrosis, apoptosis, and vascular tone (21, 22). AT1R stimulation induces vasoconstriction, cell proliferation, protein phosphorylation, sodium retention, fibrosis and oxidative stress, while AT2R activation counterbalances these effects (23). Although Ang-II binds with high affinity to both receptors, their expression widely differs throughout the body (24). The increase in Ang-II production results in a predominant AT1R-mediated effect, namely, vasoconstriction, as shown in the ATHOS trials (25).
Ang-II can also be transformed, by aminopeptidase A (APA), into Ang-III, which can stimulate the AT1R (26), and is then further processed by aminopeptidase N (APN) into Ang-IV, which has high affinity for the AT4R subtype (27). However, an alternative RAS pathway has been flanked and characterized as a counterbalancing system. This pathway has Ang-(1–7) as its main effector, derived either from Ang-II through the action of the monopeptidase ACE2, or through an intermediate transformation of Ang-I into Ang-(1–9) by ACE2 before its final conversion to Ang-(1–7) by ACE (Figure 1). By binding to its specific receptor MasR, Ang-(1–7) exerts a wide range of effects that counteract the pro-inflammatory, pro-apoptotic, pro-fibrotic and vasoconstrictive effects of the AT1R stimulation induced by Ang-II (28).
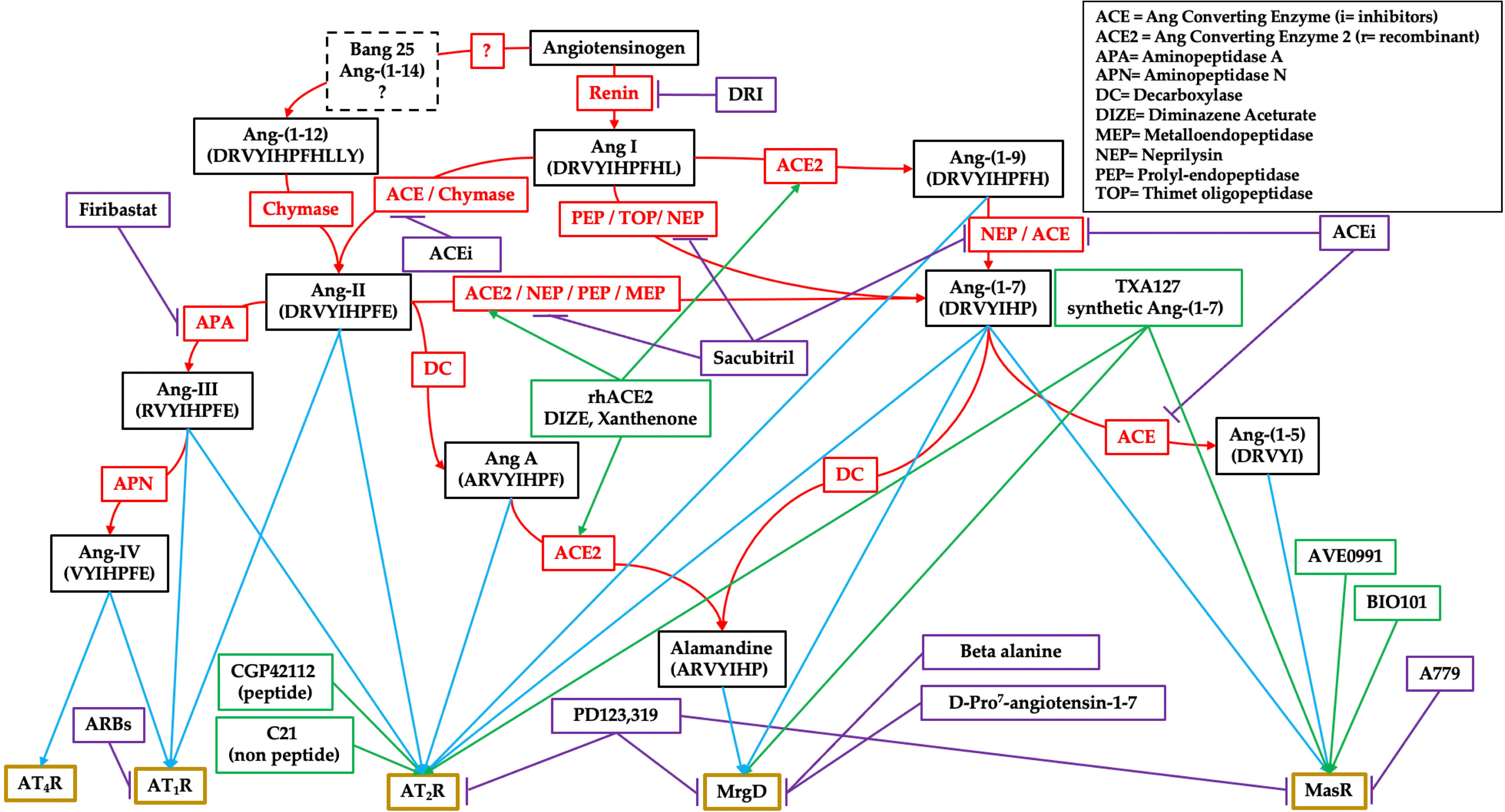
Figure 1 Schematic representation of the renin–angiotensin system (RAS). Black boxes are for the effectors, red ones for enzymes, purple ones for pharmacological inhibitors, green ones for pharmacological stimulators, and gold boxes for receptors.
Ang-(1–7) can be further processed into two biologically active compounds, Ang-(1–5), also capable of binding to MasR, and alamandine, which binds to a specific MasR-related G-protein coupled receptor member D (MrgD), which is active in all the effects produced by this alternative RAS pathway (29, 30). This simplistic representation has led to the misconception of a “positive” RAS as opposed to a “negative” one. However, as suggested by its phylogenesis, RAS should be seen rather as a fine regulatory system, with high complexity, inter-human variability and widespread presence throughout the body (31). RAS is a complex network of interconnected tissue-specific and systemic RAS reactions that finely tune physiological functions.
In recent years several new angiotensins, receptors and enzymes have been discovered and characterized, and a numerous of compounds have been developed with the purpose of stimulating or blocking key components of both RAS pathways in many diseases. To combat hypertension and cardiac failure pharmacological RAS manipulations have aimed to inhibit the overstimulation of AT1R, with selective or unselective blockade (sartans or ACE inhibitors), through renin inhibition (aliskiren) or combined AT1R/neprilysin inhibition (sacubitril) (32). The antihypertensive drug sacubitril can block neprilysin, raising the concentration of natriuretic peptides and, in association with AT1R blockade, preventing the detrimental increase in Ang-II (33).
Given the complexity of RAS interconnections with other systems such as bradykinins, natriuretic peptides and prostaglandins, its manipulation has often produced differing results—as with omapatrilat (a neprilysin blocker combined with an ACE inhibitor), where the beneficial effects of blocking neprilysin are counterbalanced by the risk of angioedema due to the double blockade in the breakdown of bradykinin (34). However, no compound has been developed commercially to specifically target the alternative RAS by stimulating MasR, MrgD or AT2R-related pathways.
This approach has been proposed recently, in the context of the COVID-19 pandemic, as it appears that RAS may become unbalanced during the acute inflammatory response (35), and upregulation of the ACE2/Ang-(1–7)/MasR pathway could re-establish homeostasis within the RAS. Clinical trials evaluating the efficacy of MasR, AT2R agonists, the synthetic form of Ang-(1–7) and recombinant ACE2 in COVID-19, but no data are available about vasculature or cerebral tissues. A synthetic form of Ang-(1–7) has been given to women to treat pre-eclampsia, with vasculature-related endpoints—with some promising results (36).
Despite the large amount of evidence supporting the beneficial role of Ang-(1–7) in various conditions, particularly, cardiovascular disease, cancer, renal failure, and neurological disorders (37), problems related to the peptide’s short half-life and rapid plasmatic metabolism still limit its clinical applications (38).
Given the multifaceted nature of brain damage after SAH, an ideal therapeutic agent in this context should be capable of modulating inflammation, mitigating ischemic injury, and also providing favorable vascular and systemic effects, avoiding hypotension, which could further impair cerebral perfusion. With its favorable local and systemic modulatory effects on most SAH pathophysiological mechanisms, ACE2/Ang-(1–7)/MasR axis manipulation offers a possible new therapeutic target to prevent or mitigate DCI. This article reviews the evidence behind RAS manipulation in acute brain injury, with a focus on ischemia, neuroinflammation, the role of the alternative RAS, and the rationale for the potential use of Ang-(1–7) in the future to prevent DCI in SAH.
Cerebral RAS
Since its first observation over half a century ago (39, 40), the presence of the RAS in the brain and its relationship with systemic physiological functions, such as blood pressure control and water–salt homeostasis, has been extensively investigated. Brain RAS has been implicated in the regulation of arterial blood pressure, body temperature, thirst and hydromineral balance, vasopressin release, adrenocorticotropin synthesis and memory (41, 42). All components of the classical and alternative RAS have been described in the brain (41), namely, MasR, Ang-(1–7), and Ang-(1–9) (43). In particular, renin and angiotensinogen are synthesized mainly by astrocytes, and a small percentage by neurons (44). The presence of AT1R and AT2R has been confirmed in dopaminergic neurons, astrocytes, and glial-cells (45–47), and ACE and ACE2 are expressed in many areas of the mammalian brain (48).
It is still not clear which molecule is the principal effector of the classical pathway in the brain. Ang-III, which is the product of Ang-II cleavage by APA, mediates several RAS pressor effects in the brain (49, 50). For this reason APA inhibitors are being investigated as novel antihypertensive drugs, targeting brain RAS and acting on vasopressin release and sympathetic tone (51). Ang-III is further processed by the APN into Ang-IV, whose receptor (AT4R) has a role in memory (52, 53). Lastly, Ang-(1–7) has also been linked to fine regulation of inflammation, oxidative stress, metabolic homeostasis, angiogenesis, and motor control and cognitive behavior in the brain (37).
RAS in Brain Ischemia
In ischemic stroke, therapeutic manipulation of brain RAS has been investigated to control blood pressure (54) and blockade of the classical RAS with ACE inhibitors and selective AT1R inhibitors (ARBs) has proven more effective than beta-blockers for secondary prevention of stroke (55). The relationship between RAS and stroke has been recently summarized (56, 57). Briefly, AT1R-deficient mice have a larger penumbra area and a smaller area of energy failure than wild-type littermates after middle cerebral artery occlusion (MCAO) (58). It is still not clear whether ARBs have a neuroprotective effect mediated by the decrease in AT1R activation signaling pathways or the AT2R indirect stimulation, but rats immunized with Ang-II peptide vaccine prior to MCAO had less neurodegeneration through suppression of the brain RAS and reduction of oxidative stress (59). In addition, the infarct was smaller, and neurological performance was better in animals given an AT2R agonist (compound 21 or CGP42112) after MCAO (60, 61).
The alternative ACE2/Ang-(1–7)/MasR pathway is also directly involved in the pathophysiology of ischemic stroke, and the expression of both ACE2 and MasR is upregulated after acute stroke in rats. These animals have higher regional and circulating levels of Ang-(1–7), suggesting that this axis could play a key role in the response to brain ischemia (62). There is also experimental evidence of direct neuroprotective effects of Ang-(1–7). Neurological outcome and infarct size improved when Ang-(1–7) was injected in the brain ventriculi of rodents prior to endothelin-1 mediated MCAO (63). Intraventricular Ang-(1–7) also reduced infarct size and inflammation (64) after permanent MCAO in rodents. The concomitant injection of the MasR antagonist A-779 led to similar findings, corroborating a protective action of Ang-(1–7). Interestingly, there were still beneficial effects when Ang-(1–7) was given orally at various times after reperfusion (65). Furthermore, in spontaneously hypertensive rats, prone to hemorrhagic stroke, centrally administered Ang-(1–7) increased the lifespan and improved neurological performance (66). Recently, intranasal administration of AVE 0991, a MasR agonist in an experimental model of SAH was associated with improved neurobehavioral function and a reduction in oxidative stress and neuronal apoptosis (67). These results partially contrast with previous findings in an in vivo model of ischemic stroke, where centrally injected AVE 0991 did not improve functional or histological endpoints, but did protect ischemic neurons in vitro (68).
Targeting ACE2 might also be an important therapeutic option for ischemic stroke (69). Mice overexpressing ACE2 in neurons are protected from ischemic injury through regulation of the NADPH oxidase and endothelial nitric oxide synthase pathways and show a reduction of reactive oxygen species (ROS) after ischemic stroke (70).
The ACE2 increase by central or systemic diminazene aceturate (DIZE) after ischemic stroke improved neurological function and reduced the infarct volume (71). Thus, data suggest that RAS mediates early and delayed consequences after experimental stroke (72, 73) and RAS genetic polymorphisms are thought to be involved in cerebral aneurysm formation and rupture (74), whether these observations can be translated to SAH and/or DCI remains to be shown.
RAS and Neuroinflammation
Even though systemic RAS does not act directly on the central nervous system, tissue-specific RAS is found in the brain and is implicated in several physiological and pathological processes. Local brain RAS may have pro-inflammatory action and pro-fibrotic effects through the activation of the ACE/Ang-II/AT1 axis but also mediates anti-inflammatory effects through the activation of the alternative ACE2/Ang-(1–7)/MasR or AT2R (37).
Microglia are resident brain immune cells responsible for the elimination of microbes, dead cells, protein aggregates, and other dangerous substances or debris, with a key role in orchestrating neuroinflammatory changes (75). Importantly, AT1R and AT2R receptors are present in activated microglia, and RAS stimulation elicits direct activation of microglia cells in vitro and release of pro-inflammatory cytokines through a NF-kB mediated mechanism (76). RAS-mediated microglial inflammation is mainly associated with Nox-derived ROS that can also interfere on intracellular signaling pathways involved in microglial activation, stimulating the release of proinflammatory signals (77).
Together with microglia, astrocytes are important sources of pro- and anti-inflammatory cytokines, they also sustain blood–brain barrier, provide metabolic support to neurons, and maintain synaptic homeostasis (78, 79). Ang-II regulates astroglial functions by inducing astrocyte proliferation and activating intracellular signaling pathways linked to inflammatory status, cellular growth, proliferation and increases in blood–brain barrier permeability, thus favoring the migration of peripheral immune cells into brain tissue (80).
There is growing evidence that RAS can be effectively targeted to reduce neuroinflammation in several brain pathologies, namely, Alzheimer (81–86), Parkinson (87–89), ischemic stroke (90, 91), traumatic brain injury (TBI) (92, 93), hypertension (94, 95) and inflammation (96) (Figure 2). Reduced astro- and micro-gliosis has been reported after blockade of the ACE/Ang-II/AT1R pathway, leading to a reduction in cytokine production and a better functional outcome (81, 83, 86, 90, 93, 96, 97).
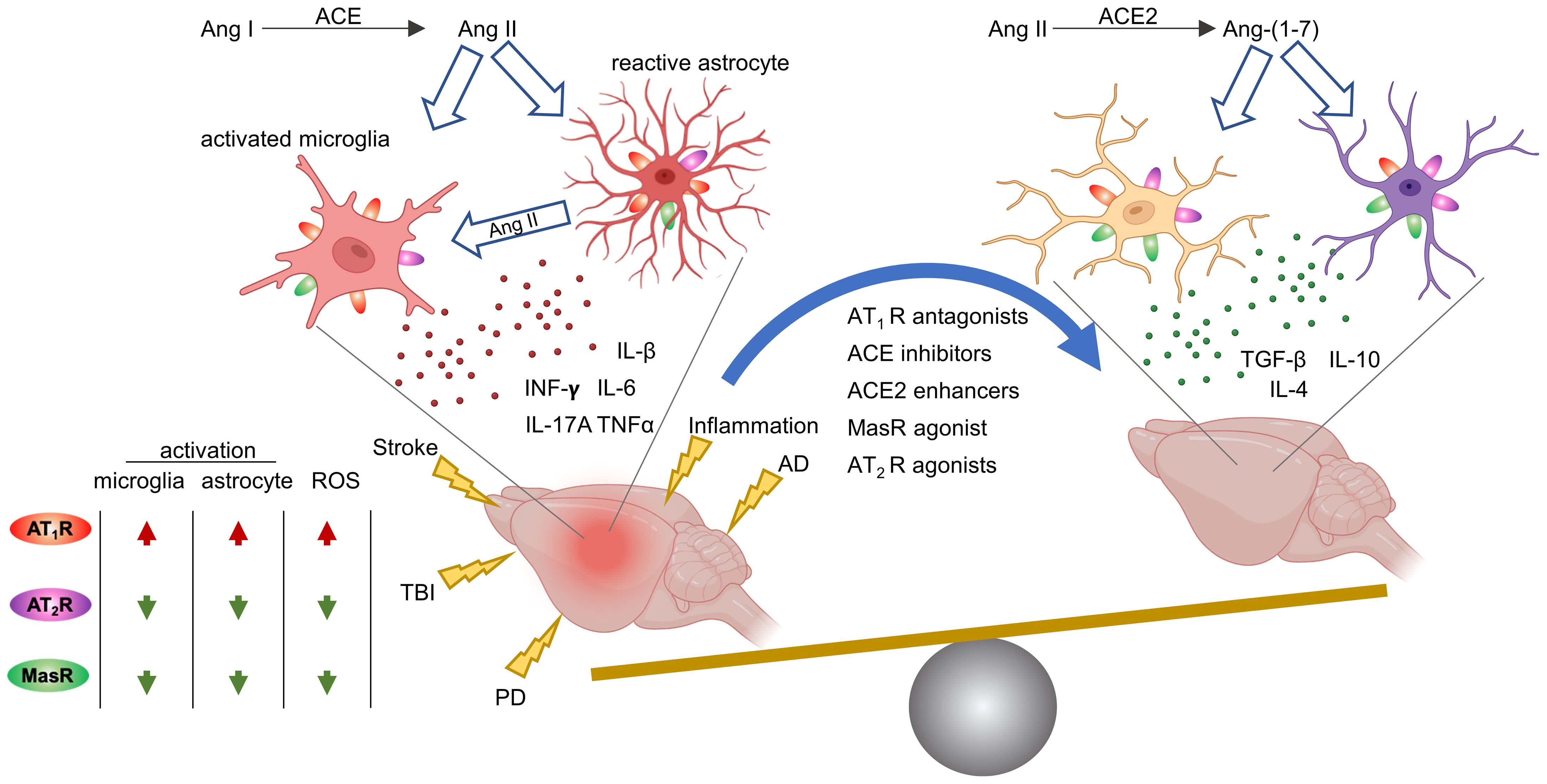
Figure 2 RAS modulation through AT1R, AT2R, MasR counteract neuroinflammation across several brain pathological brain conditions. PD, Parkinson’s disease; AD, Alzheimer’s disease; TBI, traumatic brain injury; ACE, angiotensin-converting enzyme; Ang, angiotensin; ROS, reactive oxygen species. Image created with BioRender.com.
Recent data indicate that vaccination against Ang-II inhibited astrocytic and microglial activation by stimulating basic fibroblast growth factor 2 (FGF2) signaling, and improved cognitive outcome in rats with vascular dementia (59). Neuromodulatory effects mediated by microglia and astrocytes have been reported with ARBs (82, 85, 86, 95, 98–100) and ACE inhibitors (ACEi) (82). Also, AT1R knock down by viral vector, reduced inflammatory mediators and glial activation in hypertensive rats (101). A shift of microglial polarization toward a more protective phenotype has also been observed after either direct stimulation of the AT2R receptor by Compound 21, a potent AT2R agonist (102), or AVE 0991 MasR agonist (103, 104) or ACE2 stimulation (103).
Interestingly, sexual dimorphisms seem implicated in RAS activity on neuroinflammation. Estradiol regulates the expression of AT1R and ACE activity in several peripheral tissues (105) and mediates dopaminergic cell damage in Parkinson’s disease (106). In experimental models of Alzheimer’s disease inhibition of the ACE/Ang-II/AT1R axis by ARBs (98) or ACEi (82, 84) had higher anti-inflammatory effects in the brain of female mice. In line with these observations, the stimulation of estrogen receptor β counteracts the negative effect of Ang-II on microglial polarization (77), highlighting a potential link between low estrogen levels and Ang-II mediated neuroinflammation in microglia. Moreover, positive interactions between the level of estrogen and the expression of AT2R (107, 108) and Ang-(1–7)/MasR (108–111) have been reported at both peripheral and central levels. Female sex hormones upregulate brain ACE2/Ang-(1–7), with a protective role against experimental hypertension in mice by increasing estrogen receptor α and lowering Nox expression in the brain (112, 113). This effect is reversed after either natural or surgical menopause (112). Importantly, estrogen deficiency, as in the perimenstrual phase and menopause, has a role in aneurysm formation and rupture (114), with a higher incidence of SAH in women after the age of 40 (115).
Neuroinflammation has been proposed to contribute to the DCI and poor outcome in SAH patients (15). RAS modulation either by inhibiting ACE/Ang-II/AT1R or stimulating the ACE2/Ang-(1–7)/MasR or AT2R axis has shown anti-neuroinflammatory effects in several brain pathologies (Figure 2) but in vivo studies exploring the possibility of acting on RAS to improve SAH outcome are lacking and are needed in the future.
Ang-(1–7) to Act on Neuroinflammation
The alternative ACE2/Ang-(1–7)/Mas axis exerts effects that appear to be opposite to those of Ang II (116). Like the classical RAS, the receptors of the alternative RAS too are present in microglia and astrocytes (46, 117–121). ACE2/Ang-(1–7)/MasR axis activation either by stimulating ACE2 (103), MasR agonist (104, 122) or with calcitriol (vitamin D) (94) shifts microglial polarization toward a less toxic phenotype. Ang-(1–7) itself can exert direct effects on microglial cells by reducing their activation and the release of pro-inflammatory cytokines, namely, interleukin-1β (IL-1β) and tumor-necrosis factor α (TNF-α), while increasing the anti-inflammatory cytokine interleukin-10 (123).
The option to employ Ang-(1–7) to reduce neuroinflammation has been recently explored in the experimental setting in brain pathologies (Table 1). The effect of Ang-(1–7) on microglial cells is mediated by inhibition of inducible nitric oxide synthase (iNOS) (120) and of the NF-kB pathway (64). Ang-(1–7) also has a positive effect on astrocytes through the regulation of MAP kinase signaling, namely, downstream mediators such as PKCα and MEK (134). Additionally, through inhibition of the MAPK/Nox signaling pathway and by acting on the inflammatory cascade HMGB-1/RAGE/NF-κB/TNF-α, Ang-(1–7) prevented neuronal damage in an experimental model of Parkinson’s disease (125). Ang-(1–7) given subcutaneously 6 h after experimental TBI showed promising anti-inflammatory and neuroprotective properties, with a reduction of astrogliosis and microgliosis, increased neuronal and capillary density, and better cognitive performance one month after TBI (129).
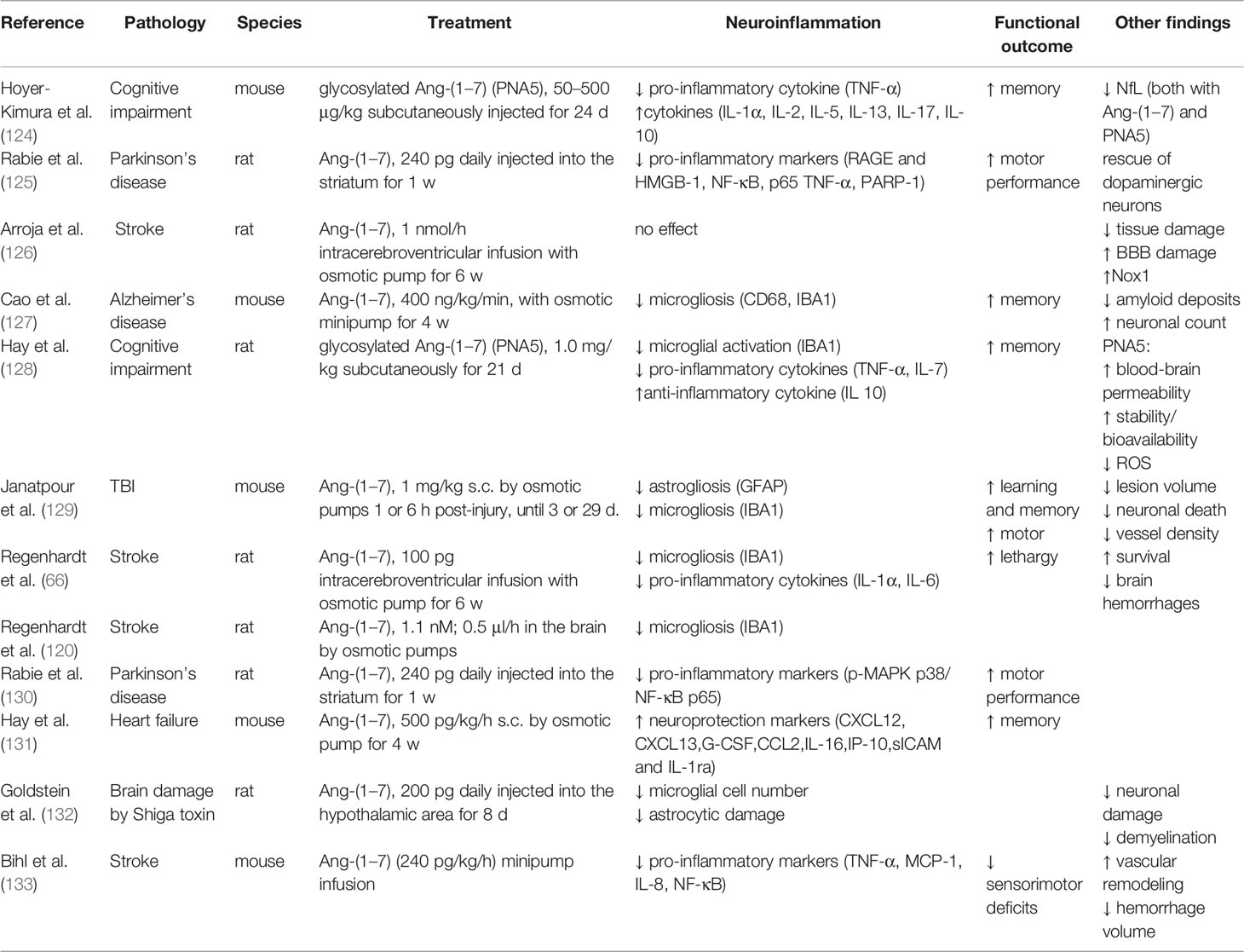
Table 1 In vivo studies showing the anti-inflammatory action of Ang-(1–7) in the brain.
It has been recently proposed that the kinin system, RAS and complement system are closely interconnected and have a major role in regulating vascular tone and inflammation (135). Ang-(1–7), by AT2R-mediated signaling, can counter-regulate blood pressure elevation by stimulating bradykinin production (136) and boosting the bradykinin–NO–cGMP pathway (135). In pathological conditions, particularly SAH (137), over-activation of the complement system induces leukocyte recruitment and extravasation increasing vascular permeability that can lead to tissue edema (135). Thus, Ang-(1–7) given after SAH could restore the balance in vascular permeability (37), mitigating pro-inflammatory mechanisms in brain tissue and blood vessels.
Among pharmacological treatments targeting brain RAS, Ang-(1–7), by stimulating MasR and AT2R, can blunt brain and vascular inflammation and at the same time improve functional outcomes in pathological conditions. Importantly, direct stimulation of one of the MasR (one of the targets of Ang-(1–7)) reduced oxidative stress and neuronal apoptosis in experimental SAH (67). Nevertheless, since Ang-(1–7) might also activate AT2R in tissue there is the possibility that Ang-(1–7) might counteract central and vascular inflammatory processes after SAH.
Potential Beneficial Effect of Ang-(1–7) in SAH
It has recently been proposed that inflammation and oxidative stress may be a common ground for most of the causes of DCI (6, 15). After SAH blood components trigger an inflammatory response in the brain (13), that is further aggravated by the production of free radicals caused by the degradation of red blood cells from the clot (14); this builds up a self-promoting detrimental loop where neuroinflammation causes oxidative stress and oxidative stress aggravates neuroinflammation. As noted above Ang-(1–7) has the potential to block this detrimental loop triggered by SAH, modulating micro- and astro-glial function.
Direct Vascular Activity
Besides its action on neuroinflammation, Ang-(1–7) has other potential beneficial effects in SAH (Figure 3). The vasodilating properties have been described in experimental models (138, 139). In healthy rats, Ang-(1–7) infusion modifies blood flow distribution, increases brain perfusion and vascular conductance, reduces vascular resistance in the brain and increases the cardiac index (140, 141). In situations of unbalanced RAS, such as in diabetic rats, Ang-(1–7) treatment restores carotid blood flow and lowers carotid resistance (142). In Ang-II induced hypertensive mice given intracerebral injections of elastance to increase the incidence of aneurysm, animals treated with Ang-(1–7) had a smaller proportion of ruptured intracranial aneurysms and lower mortality than controls through the MasR dependent pathway (143). The regional impact of Ang-(1–7) in humans has not been clarified yet, but it contrasted Ang-II-induced vasoconstriction in human artery fragments from patients treated for coronary revascularization. Interestingly, this action seems independent from MasR activation, AT2R or endothelium, and involves a direct effect on vascular smooth muscle cells (6). The effects of Ang-(1–7) on vasorelaxant compounds such as NO and prostacyclin have also been linked to the anti-thrombotic action (144–146). The relationship between Ang-(1–7) circulating levels and vascular effects is complex and is affected by comorbidities and concurrent therapies, such as ACEi (147–149).
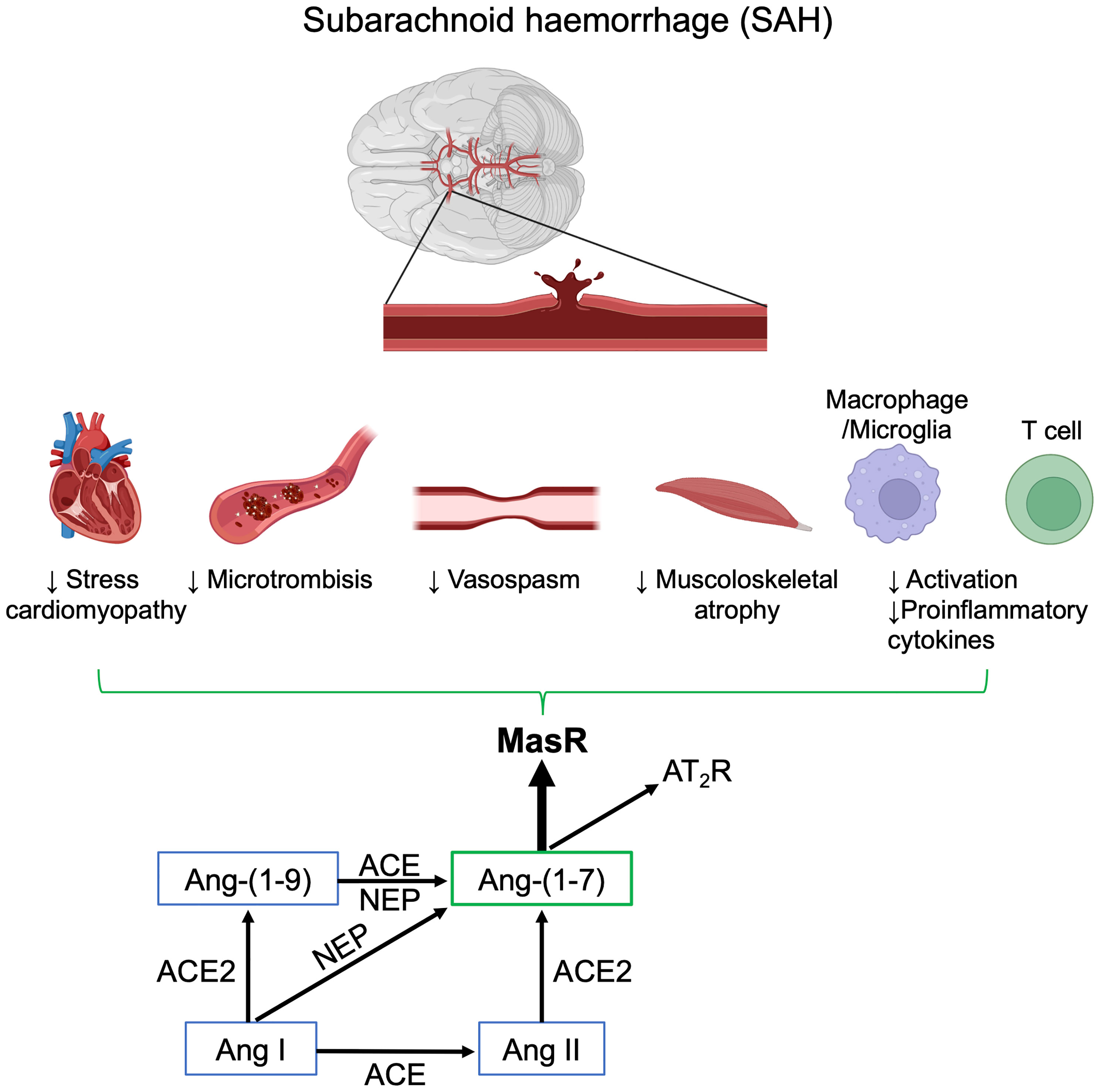
Figure 3 Schematic representation of the positive effects of Ang-(1–7) after subarachnoid hemorrhage (SAH). Image created with BioRender.com.
Anti-Thrombotic Properties
Activation of the coagulation cascade and platelet activation are linked with DCI in SAH patients (150). The presence of microthrombi has been confirmed in two post-mortem studies (151, 152), but the clinical relevance of treatments to reduce thrombus formation is still not clear. Randomized controlled trials so far have found no benefit when aspirin was given to prevent platelet aggregation (153), or have given uncertain responses regarding the use of enoxaparin (154, 155), and guidelines are still based on low-quality evidence (11). In animals the Ang-(1–7)/MasR axis exerts significant antithrombotic effects after both acute and chronic administration (146) and the effect may be mediated by action on NO and prostacyclin release in platelets (144, 145) (Figure 3).
Musculoskeletal Atrophy
SAH patients requiring ICU care are often hospitalized for prolonged periods and suffer to a progressive decrease of muscle mass, with loss of strength and worsening outcome (156, 157). ICU-acquired weakness has been related to later hospital discharge, prolonged mechanical ventilation, and increased mortality (158). RAS has been proposed as a therapeutic target in musculoskeletal diseases, and while the classical RAS has been repeatedly linked with muscle fibrosis (159), Ang-(1–7) exerts a protective role in muscular dystrophy (160). Moreover, Ang-(1–7) prevented Ang-II-related catabolism (161) and muscle fiber shrinkage in an animal model of muscle atrophy due to immobilization (162) (Figure 3).
Stress-Induced Cardiomyopathy
Supranormal blood pressure thresholds (i.e., euvolemic hypertension) are often targeted in severe SAH patients after treatment of an aneurysm, to prevent regional hypoperfusion. Takotsubo syndrome (TTS), or stress-induced cardiomyopathy, is a life-threatening condition associated with SAH, due to increased endogenous and/or iatrogenic catecholamine load (163). In a propensity matched cohort, RAS manipulation using ACEi and ARBs, but not beta blockers, gave survival gain at one year and lower recurrence of the TTS (164). Ang-(1–7) levels were lower than to controls in an animal model of TTS by vagal electrical stimulation (165). The heart might nevertheless benefit from a higher systemic level of Ang-(1–7), as its levels have been associated with less heart failure after myocardial infarction (166), less myocardial swelling (167), and antiarrhythmic effects (168) (Figure 3). Ang-(1–7) infusion as a treatment for TTS still needs to be explored.
Adaptative Immune Cell Modulation
Mas signaling affects macrophage polarization, migration, and macrophage-mediated T-cell activation, all regulated by the alternative Ang-(1–7)/MasR axis (169). At the macrophage level, the lack of Ang-(1–7)-mediated inhibition on MasR results in enhanced T-cell proliferation in vitro co-culture experiments (170–172). In preclinical studies, activating AT1R receptors in T lymphocytes and myeloid cells blunts the polarization of these cells toward pro-inflammatory phenotypes (169) (Figure 3).
Pharmacological Considerations
Ang-(1–7) has a short half-life of ~0.5 h after subcutaneous injection, but it is promptly available and reaches its peak plasma concentration at ~1 h (173). Its bioavailability is even shortened when injected intravenously, as the peptide is rapidly degraded by circulating enzymes, namely, ACE, aminopeptidase A, and DDP3; this makes the development of a commercially distributed drug particularly challenging. In critically ill patients continuous intravenous infusion of Ang-(1–7) would be the safest administration route, achieving a tailored plasmatic increase and allowing for prompt discontinuation in case of hemodynamic instability. Clinically useful data are expected from ongoing clinical trials in severe COVID-19 patients (NCT04332666; NCT04570501; NCT04633772).
To overcome the unfavorable PK/PD profile of the compound, several stabilized forms are currently under investigation, namely, cyclic Ang-(1–7), cyclodextrins-included or bioencapsulated Ang-(1-7), modified amino acids and a new peptide Ang-1–6-O-Ser-Glc-NH2 (PNA5), which offer better brain-penetrating properties than Ang-(1–7). PNA5 given subcutaneously for 24 days reduced the expression of pro-inflammatory cytokines, cognitive impairment, and the plasma level of the axonal damage marker neurofilament light after myocardial infarction (124, 128).
Other pharmacological strategies to boost the alternative RAS include MasR agonists (AVE0991 and BIO101), AT2R agonist (Compound 21), and recombinant ACE2 (rhACE2). Detailed analysis of the characteristics of these treatments is beyond the scope of this review, but it is likely that these strategies will not be biologically equivalent. On the one hand rhACE2 could increase the generation of Ang-(1–7), but on the other it might also lower the overall plasmatic levels of Ang-II, as shown in patients with acute respiratory distress syndrome (174). Their impact after brain injury in patients has not been studied. ACE2 activators, namely, xanthenone and the antiparasitic drug DIZE have a more favorable PK/PD profile than to Ang-(1–7). DIZE appears to stimulate the alternative pathway of the RAS, with favorable cardiovascular, renal, and immune effects (175) but clinical evidence is limited on their effects in the brain. DIZE has been used with off-label in patients, with no toxicity reported (176). However, animal studies show potential drug-related brain toxicity, which appears to be species-dependent (177).
Selective MasR agonism might induce similar biological responses compared to Ang-(1–7), but without the concomitant generation of the derivates of the peptide such as alamandine and Ang-(1–5)—both biologically active. Selective AT2R agonism will not trigger MasR and MrgD cascades, thus only marginally affecting the alternative RAS.
Conclusions
RAS components have numerous effects in the brain, and experimental evidence indicates that inhibiting the ACE/Ang-II/AT1R axis and stimulating the ACE2/Ang-(1-7)/MasR axis have positive effects in ischemic brain injury, particularly stroke. RAS might be important role in SAH and DCI, mainly increasing AT1R mediated oxidative stress and neuroinflammation and by modulating vascular changes that can promote CVS. To our knowledge, the activation of the alternative RAS has not been studied as a strategy to prevent DCI in SAH and only few studies have explored the potential beneficial effects of Ang-(1–7) after SAH, mainly focusing on its vascular effects. Further studies should test the pleiotropic activity of Ang-(1–7) and its potential to counteract Ang-II/AT1R activation to combat DCI.
Author Contributions
FA, FM, and ERZ conceived the study. FA and FM contributed equally to writing the first draft of the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
Partially supported by the Fondazione Cariplo (2019-1632). FA was supported with the mobility fund of the Fonds de Recherche Scientifique (FRS-FNRS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lawton MT, Vates GE. Subarachnoid Hemorrhage. Solomon CG (Ed.). N Engl J Med (2017) 377(3):257–66. doi: 10.1056/NEJMcp1605827
2. Etminan N, Chang H-S, Hackenberg K, de Rooij NK, Vergouwen MDI, Rinkel GJE, et al. Worldwide Incidence of Aneurysmal Subarachnoid Hemorrhage According to Region, Time Period, Blood Pressure, and Smoking Prevalence in the Population: A Systematic Review and Meta-Analysis. JAMA Neurol (2019) 76(5):588. doi: 10.1001/jamaneurol.2019.0006
3. Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G. European Stroke Organization Guidelines for the Management of Intracranial Aneurysms and Subarachnoid Haemorrhage. Cerebrovascular Dis (2013) 35(2):93–112. doi: 10.1159/000346087
4. Johnston SC, Selvin S, Gress DR. The Burden, Trends, and Demographics of Mortality From Subarachnoid Hemorrhage. Neurology (1998) 50(5):1413–8. doi: 10.1212/WNL.50.5.1413
5. Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and Functional Outcome After Aneurysmal Subarachnoid Hemorrhage. Stroke (2010) 41(8):e519–36. doi: 10.1161/STROKEAHA.110.581975
6. Macdonald RL. Delayed Neurological Deterioration After Subarachnoid Haemorrhage. Nat Rev Neurol (2014) 10(1):44–58. doi: 10.1038/nrneurol.2013.246
7. Zoerle T, Ilodigwe DC, Wan H, Lakovic K, Sabri M, Ai J, et al. Pharmacologic Reduction of Angiographic Vasospasm in Experimental Subarachnoid Hemorrhage: Systematic Review and Meta-Analysis. J Cereb Blood Flow Metab (2012) 32(9):1645–58. doi: 10.1038/jcbfm.2012.57
8. van Lieshout JH, Dibué-Adjei M, Cornelius JF, Slotty PJ, Schneider T, Restin T, et al. An Introduction to the Pathophysiology of Aneurysmal Subarachnoid Hemorrhage. Neurosurgical Rev (2018) 41(4):917–30. doi: 10.1007/s10143-017-0827-y
9. Guiraud V, Amor MB, Mas J-L, Touzé E. Triggers of Ischemic Stroke: A Systematic Review. Stroke (2010) 41(11):2669–77. doi: 10.1161/STROKEAHA.110.597443
10. Amodio S, Bouzat P, Robba C, Taccone FS. Rethinking Brain Injury After Subarachnoid Hemorrhage. Crit Care (2020) 24(1):612, s13054-020-03342–2 doi: 10.1186/s13054-020-03342-2
11. Maher M, Schweizer TA, Macdonald RL. Treatment of Spontaneous Subarachnoid Hemorrhage: Guidelines and Gaps. Stroke (2020) 51(4):1326–32. doi: 10.1161/STROKEAHA.119.025997
12. Kassell NF, Sasaki T, Colohan AR, Nazar G. Cerebral Vasospasm Following Aneurysmal Subarachnoid Hemorrhage. Stroke (1985) 16(4):562–72. doi: 10.1161/01.STR.16.4.562
13. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang Q-W. Inflammation in Intracerebral Hemorrhage: From Mechanisms to Clinical Translation. Prog Neurobiol (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
14. Sakamoto M, Takaki E, Yamashita K, Watanabe K, Tabuchi S, Watanabe T, et al. Nonenzymatic Derived Lipid Peroxide, 8-Iso-PGF 2α, Participates in the Pathogenesis of Delayed Cerebral Vasospasm in a Canine SAH Model. Neurol Res (2002) 24(3):301–6. doi: 10.1179/016164102101199783
15. Wu F, Liu Z, Li G, Zhou L, Huang K, Wu Z, et al. Inflammation and Oxidative Stress: Potential Targets for Improving Prognosis After Subarachnoid Hemorrhage. Front Cell Neurosci (2021) 15:3389/fncel.2021.739506. doi: 10.3389/fncel.2021.739506
16. Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. A Clinical Review of Cerebral Vasospasm and Delayed Ischaemia Following Aneurysm Rupture, in: Early Brain Injury or Cerebral Vasospasm (2011). Vienna: Springer Vienna. Available at: 10.1007/978-3-7091-0353-1_1(Accessed 18th September 2021).
17. Taccone FS, De Oliveira Manoel AL, Robba C, Vincent J-L. Use a “GHOST-CAP” in Acute Brain Injury. Crit Care (2020) 24(1):89, s13054-020-2825–2827. doi: 10.1186/s13054-020-2825-7
18. Diringer MN, Bleck TP, Claude Hemphill J, Menon D, Shutter L, Vespa P, et al. Critical Care Management of Patients Following Aneurysmal Subarachnoid Hemorrhage: Recommendations From the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocritical Care (2011) 15(2):211. doi: 10.1007/s12028-011-9605-9
19. Connolly ES, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke (2012) 43(6):1711–37. doi: 10.1161/STR.0b013e3182587839
20. Koyanagi M, Fukuda H, Lo B, Uezato M, Kurosaki Y, Sadamasa N, et al. Effect of Intrathecal Milrinone Injection via Lumbar Catheter on Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. J Neurosurgery (2018) 128(3):717–22. doi: 10.3171/2016.10.JNS162227
21. Aroor AR, DeMarco VG, Jia G, Sun Z, Nistala R, Meininger GA, et al. The Role of Tissue Renin-Angiotensin-Aldosterone System in the Development of Endothelial Dysfunction and Arterial Stiffness. Front Endocrinol (2013) 4:3389/fendo.2013.00161. doi: 10.3389/fendo.2013.00161
22. Kobori H, Nangaku M, Navar LG, Nishiyama A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol Rev (2007) 59(3):251–87. doi: 10.1124/pr.59.3.3
23. Benigni A, Cassis P, Remuzzi G. Angiotensin II Revisited: New Roles in Inflammation, Immunology and Aging. EMBO Mol Med (2010) 2(7):247–57. doi: 10.1002/emmm.201000080
24. Carey RM, Wang Z-Q, Siragy HM. Role of the Angiotensin Type 2 Receptor in the Regulation of Blood Pressure and Renal Function. Hypertension (2000) 35(1):155–63. doi: 10.1161/01.HYP.35.1.155
25. Khanna A, English SW, Wang XS, Ham K, Tumlin J, Szerlip H, et al. Angiotensin II for the Treatment of Vasodilatory Shock. N Engl J Med (2017) 377(5):419–30. doi: 10.1056/NEJMoa1704154
26. Yugandhar VG, Clark MA. Angiotensin III: A Physiological Relevant Peptide of the Renin Angiotensin System. Peptides (2013) 46:26–32. doi: 10.1016/j.peptides.2013.04.014
27. Chai SY, Fernando R, Peck G, Ye S-Y, Mendelsohn FAO, Jenkins TA, et al. What?s New in the Renin-Angiotensin System?: The Angiotensin IV/AT4 Receptor. Cell Mol Life Sci (2004) 61(21):2728–37. doi: 10.1007/s00018-004-4246-1
28. Santos RAS, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-Converting Enzyme 2, Angiotensin-(1–7) and Mas: New Players of the Renin–Angiotensin System. J Endocrinol (2013) 216(2):R1–R17. doi: 10.1530/JOE-12-0341
29. Schleifenbaum J. Alamandine and Its Receptor MrgD Pair Up to Join the Protective Arm of the Renin-Angiotensin System. Front Med (2019) 6:3389/fmed.2019.00107. doi: 10.3389/fmed.2019.00107
30. Villela DC, Passos-Silva DG, Santos RAS. Alamandine: A New Member of the Angiotensin Family. Curr Opin Nephrol Hypertension (2014) 23(2):130–4. doi: 10.1097/01.mnh.0000441052.44406.92
31. Fournier D, Luft FC, Bader M, Ganten D, Andrade-Navarro MA. Emergence and Evolution of the Renin–Angiotensin–Aldosterone System. J Mol Med (2012) 90(5):495–508. doi: 10.1007/s00109-012-0894-z
32. Paz Ocaranza M, Riquelme JA, García L, Jalil JE, Chiong M, Santos RAS, et al. Counter-Regulatory Renin-Angiotensin System in Cardiovascular Disease. Nat Rev Cardiol (2020) 17(2):116–29. doi: 10.1038/s41569-019-0244-8
33. Braunwald E. The Path to an Angiotensin Receptor Antagonist-Neprilysin Inhibitor in the Treatment of Heart Failure. J Am Coll Cardiol (2015) 65(10):1029–41. doi: 10.1016/j.jacc.2015.01.033
34. Campbell DJ. Vasopeptidase Inhibition: A Double-Edged Sword? Hypertension (2003) 41(3):383–9. doi: 10.1161/01.HYP.0000054215.71691.16
35. Pucci F, Annoni F, Dos Santos RAS, Taccone FS, Rooman M. Quantifying Renin-Angiotensin-System Alterations in COVID-19. Cells (2021) 10(10):2755. doi: 10.3390/cells10102755
36. Yamaleyeva LM, Merrill DC, Ebert TJ, Smith TL, Mertz HL, Brosnihan KB. Hemodynamic Responses to Angiotensin-(1-7) in Women in Their Third Trimester of Pregnancy. Hypertension Pregnancy (2014) 33(4):375–88. doi: 10.3109/10641955.2014.911884
38. Touyz RM, Montezano AC. Angiotensin-(1–7) and Vascular Function: The Clinical Context. Hypertension (2018) 71(1):68–9. doi: 10.1161/HYPERTENSIONAHA.117.10406
39. Booth DA. Mechanism of Action of Norepinephrine in Eliciting an Eating Response on Injection Into the Rat Hypothalamus. J Pharmacol Exp Ther (1968) 160(2):336–48.
40. Ganten D, Boucher R, Genest J. Renin Activity in Brain Tissue of Puppies and Adult Dogs. Brain Res (1971) 33(2):557–9. doi: 10.1016/0006-8993(71)90137-5
41. McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, et al. The Brain Renin–Angiotensin System: Location and Physiological Roles. Int J Biochem Cell Biol (2003) 35(6):901–18. doi: 10.1016/S1357-2725(02)00306-0
42. Karamyan VT, Speth RC. Enzymatic Pathways of the Brain Renin–Angiotensin System: Unsolved Problems and Continuing Challenges. Regul Peptides (2007) 143(1–3):15–27. doi: 10.1016/j.regpep.2007.03.006
43. Lombard-Banek C, Yu Z, Swiercz AP, Marvar PJ, Nemes P. A Microanalytical Capillary Electrophoresis Mass Spectrometry Assay for Quantifying Angiotensin Peptides in the Brain. Analytical Bioanalytical Chem (2019) 411(19):4661–71. doi: 10.1007/s00216-019-01771-9
44. Jackson L, Eldahshan W, Fagan S, Ergul A. Within the Brain: The Renin Angiotensin System. Int J Mol Sci (2018) 19(3):876. doi: 10.3390/ijms19030876
45. Lenkei Z, Palkovits M, Corvol P, Llorens-Cortès C. Expression of Angiotensin Type-1 (AT1) and Type-2 (AT2) Receptor mRNAs in the Adult Rat Brain: A Functional Neuroanatomical Review. Front Neuroendocrinol (1997) 18(4):383–439. doi: 10.1006/frne.1997.0155
46. Garrido-Gil P, Rodriguez-Pallares J, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Brain Angiotensin Regulates Iron Homeostasis in Dopaminergic Neurons and Microglial Cells. Exp Neurol (2013) 250:384–96. doi: 10.1016/j.expneurol.2013.10.013
47. Sumners C, Alleyne A, Rodríguez V, Pioquinto DJ, Ludin JA, Kar S, et al. Brain Angiotensin Type-1 and Type-2 Receptors: Cellular Locations Under Normal and Hypertensive Conditions. Hypertension Res (2020) 43(4):281–95. doi: 10.1038/s41440-019-0374-8
48. Harmer D, Gilbert M, Borman R, Clark KL. Quantitative mRNA Expression Profiling of ACE 2, a Novel Homologue of Angiotensin Converting Enzyme. FEBS Letters (2002) 532(1–2):107–10. doi: 10.1016/S0014-5793(02)03640-2
49. Gao J, Marc Y, Iturrioz X, Leroux V, Balavoine F, Llorens-Cortes C. A New Strategy for Treating Hypertension by Blocking the Activity of the Brain Renin–Angiotensin System With Aminopeptidase A Inhibitors. Clin Science (2014) 127(3):135–48. doi: 10.1042/CS20130396
50. Reaux A, Fournie-Zaluski MC, David C, Zini S, Roques BP, Corvol P, et al. Aminopeptidase A Inhibitors as Potential Central Antihypertensive Agents. Proc Natl Acad Sci (1999) 96(23):13415–20. doi: 10.1073/pnas.96.23.13415
51. Llorens-Cortes C, Touyz RM. Evolution of a New Class of Antihypertensive Drugs: Targeting the Brain Renin-Angiotensin System. Hypertension (2020) 75(1):6–15. doi: 10.1161/HYPERTENSIONAHA.119.12675
52. Swanson GN, Hanesworth JM, Sardinia MF, Coleman JKM, Wright JW, Hall KL, et al. Discovery of a Distinct Binding Site for Angiotensin II (3–8), a Putative Angiotensin IV Receptor. Regul Peptides (1992) 40(3):409–19. doi: 10.1016/0167-0115(92)90527-2
53. von Bohlen und Halbach O. Angiotensin IV in the Central Nervous System. Cell Tissue Res (2003) 311(1):1–9. doi: 10.1007/s00441-002-0655-3
54. Kleindorfer DO, Towfighi A, Chaturvedi S, Cockroft KM, Gutierrez J, Lombardi-Hill D, et al. Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke (2021) 2021:52(7). doi: 10.1161/STR.0000000000000375
55. Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular Morbidity and Mortality in the Losartan Intervention For Endpoint Reduction in Hypertension Study (LIFE): A Randomised Trial Against Atenolol. Lancet (London England) (2002) 359(9311):995–1003. doi: 10.1016/S0140-6736(02)08089-3
56. McFall A, Nicklin SA, Work LM. The Counter Regulatory Axis of the Renin Angiotensin System in the Brain and Ischaemic Stroke: Insight From Preclinical Stroke Studies and Therapeutic Potential. Cell Signalling (2020) 76:109809. doi: 10.1016/j.cellsig.2020.109809
57. Arroja MMC, Reid E, McCabe C. Therapeutic Potential of the Renin Angiotensin System in Ischaemic Stroke. Exp Trans Stroke Med (2016) 8(1):8. doi: 10.1186/s13231-016-0022-1
58. Walther T, Olah L, Harms C, Maul B, Bader M, Hörtnagl H, et al. Ischemic Injury in Experimental Stroke Depends on Angiotensin II. FASEB journal: Off Publ Fed Am Societies Exp Biol (2002) 16(2):169–76. doi: 10.1096/fj.01-0601com
59. Wakayama K, Shimamura M, Suzuki J, Watanabe R, Koriyama H, Akazawa H, et al. Angiotensin II Peptide Vaccine Protects Ischemic Brain Through Reducing Oxidative Stress. Stroke (2017) 48(5):1362–8. doi: 10.1161/STROKEAHA.116.016269
60. Joseph JP, Mecca AP, Regenhardt RW, Bennion DM, Rodríguez V, Desland F, et al. The Angiotensin Type 2 Receptor Agonist Compound 21 Elicits Cerebroprotection in Endothelin-1 Induced Ischemic Stroke. Neuropharmacology (2014) 81:134–41. doi: 10.1016/j.neuropharm.2014.01.044
61. Lee S, Brait VH, Arumugam TV, Evans MA, Kim HA, Widdop RE, et al. Neuroprotective Effect of an Angiotensin Receptor Type 2 Agonist Following Cerebral Ischemia In Vitro and In Vivo. Exp Trans Stroke Med (2012) 4(1):16. doi: 10.1186/2040-7378-4-16
62. Lu J, Jiang T, Wu L, Gao L, Wang Y, Zhou F, et al. The Expression of Angiotensin-Converting Enzyme 2–Angiotensin-(1–7)–Mas Receptor Axis are Upregulated After Acute Cerebral Ischemic Stroke in Rats. Neuropeptides (2013) 47(5):289–95. doi: 10.1016/j.npep.2013.09.002
63. Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, et al. Cerebroprotection by Angiotensin-(1-7) in Endothelin-1-Induced Ischaemic Stroke: Angiotensin-(1-7) Cerebroprotection During Stroke. Exp Physiol (2011) 96(10):1084–96. doi: 10.1113/expphysiol.2011.058578
64. Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y. Suppressing Inflammation by Inhibiting the NF-κb Pathway Contributes to the Neuroprotective Effect of Angiotensin-(1-7) in Rats With Permanent Cerebral Ischaemia: Effect of Ang-(1-7) on Neuroinflammation. Br J Pharmacol (2012) 167(7):1520–32. doi: 10.1111/j.1476-5381.2012.02105.x
65. Bennion DM, Jones CH, Donnangelo LL, Graham JT, Isenberg JD, Dang AN, et al. Neuroprotection by Post-Stroke Administration of an Oral Formulation of Angiotensin-(1-7) in Ischaemic Stroke. Exp Physiol (2018) 103(6):916–23. doi: 10.1113/EP086957
66. Regenhardt RW, Bennion DM, Sumners C. Cerebroprotective Action of Angiotensin Peptides in Stroke. Clin Science (2014) 126(3):195–205. doi: 10.1042/CS20130324
67. Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu P, Zhang G, et al. AVE 0991 Attenuates Oxidative Stress and Neuronal Apoptosis via Mas/PKA/CREB/UCP-2 Pathway After Subarachnoid Hemorrhage in Rats. Redox Biol (2019) 20:75–86. doi: 10.1016/j.redox.2018.09.022
68. Lee S, Evans MA, Chu HX, Kim HA, Widdop RE, Drummond GR, et al. Effect of a Selective Mas Receptor Agonist in Cerebral Ischemia In Vitro and In Vivo. Karamyan V (ed.). PloS One (2015) 10(11):e0142087. doi: 10.1371/journal.pone.0142087
69. Peña Silva RA, Heistad DD. Promising Neuroprotective Effects of the Angiotensin-(1-7)-Angiotensin-Converting Enzyme 2-Mas Axis in Stroke: Viewpoint. Exp Physiol (2014) 99(2):342–3. doi: 10.1113/expphysiol.2013.076836
70. Chen J, Zhao Y, Chen S, Wang J, Xiao X, Ma X, et al. Neuronal Over-Expression of ACE2 Protects Brain From Ischemia-Induced Damage. Neuropharmacology (2014) 79:550–8. doi: 10.1016/j.neuropharm.2014.01.004
71. Bennion DM, Haltigan EA, Irwin AJ, Donnangelo LL, Regenhardt RW, Pioquinto DJ, et al. Activation of the Neuroprotective Angiotensin-Converting Enzyme 2 in Rat Ischemic Stroke. Hypertension (2015) 66(1):141–8. doi: 10.1161/HYPERTENSIONAHA.115.05185
72. Fassot C, Lambert G, Gaudet-Lambert E, Friberg P, Elghozi J-L. Beneficial Effect of Renin-Angiotensin System for Maintaining Blood Pressure Control Following Subarachnoid Haemorrhage. Brain Res Bulletin (1999) 50(2):127–32. doi: 10.1016/S0361-9230(99)00089-1
73. Fassot C, Lambert G, Elghozi J, Lambert E. Impact of the Renin-Angiotensin System on Cerebral Perfusion Following Subarachnoid Haemorrhage in the Rat. J Physiol (2001) 535(2):533–40. doi: 10.1111/j.1469-7793.2001.00533.x
74. Griessenauer CJ, Tubbs RS, Foreman PM, Chua MH, Vyas NA, Lipsky RH, et al. Associations of Renin-Angiotensin System Genetic Polymorphisms and Clinical Course After Aneurysmal Subarachnoid Hemorrhage. J Neurosurgery (2017) 126(5):1585–97. doi: 10.3171/2016.4.JNS16409
75. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol (2017) 35(1):441–68. doi: 10.1146/annurev-immunol-051116-052358
76. Shi P, Grobe JL, Desland FA, Zhou G, Shen XZ, Shan Z, et al. Direct Pro-Inflammatory Effects of Prorenin on Microglia. Block ML (Ed.). PloS One (2014) 9(10):e92937. doi: 10.1371/journal.pone.0092937
77. Labandeira-Garcia JL, Rodríguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ. Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front Aging Neurosci (2017) 9:3389/fnagi.2017.00129. doi: 10.3389/fnagi.2017.00129
78. Prat A, Biernacki K, Wosik K, Antel JP. Glial Cell Influence on the Human Blood-Brain Barrier. Glia (2001) 36(2):145–55. doi: 10.1002/glia.1104
79. Bélanger M, Allaman I, Magistretti PJ. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab (2011) 14(6):724–38. doi: 10.1016/j.cmet.2011.08.016
80. O’Connor AT, Clark MA. Astrocytes and the Renin Angiotensin System: Relevance in Disease Pathogenesis. Neurochem Res (2018) 43(7):1297–307. doi: 10.1007/s11064-018-2557-0
81. Drews HJ, Klein R, Lourhmati A, Buadze M, Schaeffeler E, Lang T, et al. Losartan Improves Memory, Neurogenesis and Cell Motility in Transgenic Alzheimer’s Mice. Pharmaceuticals (2021) 14(2):166. doi: 10.3390/ph14020166
82. Cuddy LK, Prokopenko D, Cunningham EP, Brimberry R, Song P, Kirchner R, et al. Aβ-Accelerated Neurodegeneration Caused by Alzheimer’s-Associated ACE Variant R1279Q is Rescued by Angiotensin System Inhibition in Mice. Sci Trans Med (2020) 12(563):eaaz2541. doi: 10.1126/scitranslmed.aaz2541
83. Royea J, Lacalle-Aurioles M, Trigiani LJ, Fermigier A, Hamel E. At2r’s (Angiotensin II Type 2 Receptor’s) Role in Cognitive and Cerebrovascular Deficits in a Mouse Model of Alzheimer Disease. Hypertension (2020) 75(6):1464–74. doi: 10.1161/HYPERTENSIONAHA.119.14431
84. Messiha BAS, Ali MRA, Khattab MM, Abo-Youssef AM. Perindopril Ameliorates Experimental Alzheimer’s Disease Progression: Role of Amyloid β Degradation, Central Estrogen Receptor and Hyperlipidemic-Lipid Raft Signaling. Inflammopharmacology (2020) 28(5):1343–64. doi: 10.1007/s10787-020-00724-4
85. Trigiani LJ, Royea J, Lacalle-Aurioles M, Tong X-K, Hamel E. Pleiotropic Benefits of the Angiotensin Receptor Blocker Candesartan in a Mouse Model of Alzheimer Disease. Hypertension (2018) 72(5):1217–26. doi: 10.1161/HYPERTENSIONAHA.118.11775
86. Torika N, Asraf K, Cohen H, Fleisher-Berkovich S. Intranasal Telmisartan Ameliorates Brain Pathology in Five Familial Alzheimer’s Disease Mice. Brain Behavior Immunity (2017) 64:80–90. doi: 10.1016/j.bbi.2017.04.001
87. Rodriguez-Perez AI, Sucunza D, Pedrosa MA, Garrido-Gil P, Kulisevsky J, Lanciego JL, et al. Angiotensin Type 1 Receptor Antagonists Protect Against Alpha-Synuclein-Induced Neuroinflammation and Dopaminergic Neuron Death. Neurotherapeutics (2018) 15(4):1063–81. doi: 10.1007/s13311-018-0646-z
88. Muñoz A, Garrido-Gil P, Dominguez-Meijide A, Labandeira-Garcia JL. Angiotensin Type 1 Receptor Blockage Reduces L-Dopa-Induced Dyskinesia in the 6-OHDA Model of Parkinson’s Disease. Involvement of Vascular Endothelial Growth Factor and Interleukin-1β. Exp Neurol (2014) 261:720–32. doi: 10.1016/j.expneurol.2014.08.019
89. Sonsalla PK, Coleman C, Wong L-Y, Harris SL, Richardson JR, Gadad BS, et al. The Angiotensin Converting Enzyme Inhibitor Captopril Protects Nigrostriatal Dopamine Neurons in Animal Models of Parkinsonism. Exp Neurol (2013) 250:376–83. doi: 10.1016/j.expneurol.2013.10.014
90. Ahmed HA, Ishrat T, Pillai B, Fouda AY, Sayed MA, Eldahshan W, et al. RAS Modulation Prevents Progressive Cognitive Impairment After Experimental Stroke: A Randomized, Blinded Preclinical Trial. J Neuroinflammation (2018) 15(1):229. doi: 10.1186/s12974-018-1262-x
91. Kono S, Kurata T, Sato K, Omote Y, Hishikawa N, Yamashita T, et al. Neurovascular Protection by Telmisartan via Reducing Neuroinflammation in Stroke-Resistant Spontaneously Hypertensive Rat Brain After Ischemic Stroke. J Stroke Cerebrovascular Dis (2015) 24(3):537–47. doi: 10.1016/j.jstrokecerebrovasdis.2014.09.037
92. Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol (2017) 16:987–1048. doi: 10.1016/S1474-4422(17)30371-X
93. Xiong J, Gao Y, Li X, Li K, Li Q, Shen J, et al. Losartan Treatment Could Improve the Outcome of TBI Mice. Front Neurol (2020) 11:3389/fneur.2020.00992. doi: 10.3389/fneur.2020.00992
94. Cui C, Xu P, Li G, Qiao Y, Han W, Geng C, et al. Vitamin D Receptor Activation Regulates Microglia Polarization and Oxidative Stress in Spontaneously Hypertensive Rats and Angiotensin II-Exposed Microglial Cells: Role of Renin-Angiotensin System. Redox Biol (2019) 26:101295. doi: 10.1016/j.redox.2019.101295
95. Melo MR, Gasparini S, Speretta GF, Silva EF, Rodrigues Pedrino G, Menani JV, et al. Importance of the Commissural Nucleus of the Solitary Tract in Renovascular Hypertension. Hypertension Res (2019) 42(5):587–97. doi: 10.1038/s41440-018-0190-6
96. Salmani H, Hosseini M, Beheshti F, Baghcheghi Y, Sadeghnia HR, Soukhtanloo M, et al. Angiotensin Receptor Blocker, Losartan Ameliorates Neuroinflammation and Behavioral Consequences of Lipopolysaccharide Injection. Life Sci (2018) 203:161–70. doi: 10.1016/j.lfs.2018.04.033
97. Gupta V, Dhull DK, Joshi J, Kaur S, Kumar A. Neuroprotective Potential of Azilsartan Against Cerebral Ischemic Injury: Possible Involvement of Mitochondrial Mechanisms. Neurochemistry Int (2020) 132:104604. doi: 10.1016/j.neuint.2019.104604
98. Scheinman SB, Zaldua S, Dada A, Krochmaliuk K, Dye K, Marottoli FM, et al. Systemic Candesartan Treatment Modulates Behavior, Synaptic Protein Levels, and Neuroinflammation in Female Mice That Express Human Apoe4. Front Neurosci (2021) 15:3389/fnins.2021.628403. doi: 10.3389/fnins.2021.628403
99. Bhat SA, Goel R, Shukla S, Shukla R, Hanif K. Angiotensin Receptor Blockade by Inhibiting Glial Activation Promotes Hippocampal Neurogenesis Via Activation of Wnt/β-Catenin Signaling in Hypertension. Mol Neurobiol (2018) 55(6):5282–98. doi: 10.1007/s12035-017-0754-5
100. Royea J, Zhang L, Tong X-K, Hamel E. Angiotensin IV Receptors Mediate the Cognitive and Cerebrovascular Benefits of Losartan in a Mouse Model of Alzheimer’s Disease. J Neurosci (2017) 37(22):5562–73. doi: 10.1523/JNEUROSCI.0329-17.2017
101. Yu Y, Wei S-G, Weiss RM, Felder RB. Angiotensin II Type 1a Receptors in the Subfornical Organ Modulate Neuroinflammation in the Hypothalamic Paraventricular Nucleus in Heart Failure Rats. Neuroscience (2018) 381:46–58. doi: 10.1016/j.neuroscience.2018.04.012
102. Jackson-Cowan L, Eldahshan W, Dumanli S, Dong G, Jamil S, Abdul Y, et al. Delayed Administration of Angiotensin Receptor (AT2R) Agonist C21 Improves Survival and Preserves Sensorimotor Outcomes in Female Diabetic Rats Post-Stroke Through Modulation of Microglial Activation. Int J Mol Sci (2021) 22(3):1356. doi: 10.3390/ijms22031356
103. Dang R, Yang M, Cui C, Wang C, Zhang W, Geng C, et al. Activation of Angiotensin-Converting Enzyme 2/Angiotensin (1–7)/Mas Receptor Axis Triggers Autophagy and Suppresses Microglia Proinflammatory Polarization via Forkhead Box Class O1 Signaling. Aging Cell (2021) 20(10):e13480. doi: 10.1111/acel.13480
104. Mi X, Cao Y, Li Y, Li Y, Hong J, He J, et al. The Non-Peptide Angiotensin-(1–7) Mimic AVE 0991 Attenuates Delayed Neurocognitive Recovery After Laparotomy by Reducing Neuroinflammation and Restoring Blood-Brain Barrier Integrity in Aged Rats. Front Aging Neurosci (2021) 13:3389/fnagi.2021.624387. doi: 10.3389/fnagi.2021.624387
105. Dean SA, Tan J, O’Brien ER, Leenen FHH. 17β-Estradiol Downregulates Tissue Angiotensin-Converting Enzyme and ANG II Type 1 Receptor in Female Rats. Am J Physiol-Regulatory Integr Comp Physiol (2005) 288(3):R759–66. doi: 10.1152/ajpregu.00595.2004
106. Labandeira-Garcia JL, Rodriguez-Perez AI, Valenzuela R, Costa-Besada MA, Guerra MJ. Menopause and Parkinson’s Disease. Interaction Between Estrogens and Brain Renin-Angiotensin System in Dopaminergic Degeneration. Front Neuroendocrinol (2016) 43:44–59. doi: 10.1016/j.yfrne.2016.09.003
107. Baiardi G, Macova M, Armando I, Ando H, Tyurmin D, Saavedra JM. Estrogen Upregulates Renal Angiotensin II AT1 and AT2 Receptors in the Rat. Regul Peptides (2005) 124(1–3):7–17. doi: 10.1016/j.regpep.2004.06.021
108. Sullivan JC. Sex and the Renin-Angiotensin System: Inequality Between the Sexes in Response to RAS Stimulation and Inhibition. Am J Physiol-Regulatory Integr Comp Physiol (2008) 294(4):R1220–6. doi: 10.1152/ajpregu.00864.2007
109. Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1-7) Receptor Antagonism Equalizes Angiotensin II–Induced Hypertension in Male and Female Spontaneously Hypertensive Rats. Hypertension (2010) 56(4):658–66. doi: 10.1161/HYPERTENSIONAHA.110.153668
110. Brosnihan KB, Neves LAA, Joyner J, Averill DB, Chappell MC, Sarao R, et al. Enhanced Renal Immunocytochemical Expression of ANG-(1-7) and ACE2 During Pregnancy. Hypertension (2003) 42(4):749–53. doi: 10.1161/01.HYP.0000085220.53285.11
111. Neves LAA, Williams AF, Averill DB, Ferrario CM, Walkup MP, Brosnihan KB. Pregnancy Enhances the Angiotensin (Ang)-(1–7) Vasodilator Response in Mesenteric Arteries and Increases the Renal Concentration and Urinary Excretion of Ang-(1–7). Endocrinology (2003) 144(8):3338–43. doi: 10.1210/en.2003-0009
112. Xue B, Zhang Z, Johnson RF, Guo F, Hay M, Johnson AK. Central Endogenous Angiotensin-(1–7) Protects Against Aldosterone/NaCl-Induced Hypertension in Female Rats. Am J Physiology-Heart Circulatory Physiol (2013) 305(5):H699–705. doi: 10.1152/ajpheart.00193.2013
113. Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Estrogen Regulation of the Brain Renin-Angiotensin System in Protection Against Angiotensin II-Induced Sensitization of Hypertension. Am J Physiology-Heart Circulatory Physiol (2014) 307(2):H191–8. doi: 10.1152/ajpheart.01012.2013
114. Tabuchi S. Relationship Between Postmenopausal Estrogen Deficiency and Aneurysmal Subarachnoid Hemorrhage. Behav Neurol (2015) 2015:1–6. doi: 10.1155/2015/720141
115. de Rooij NK, Linn FHH, van der Plas JA, Algra A, Rinkel GJE. Incidence of Subarachnoid Haemorrhage: A Systematic Review With Emphasis on Region, Age, Gender and Time Trends. J Neurol Neurosurgery Psychiatry (2007) 78(12):1365–72. doi: 10.1136/jnnp.2007.117655
116. Gironacci MM, Cerniello FM, Longo Carbajosa NA, Goldstein J, Cerrato BD. Protective Axis of the Renin–Angiotensin System in the Brain. Clin Science (2014) 127(5):295–306. doi: 10.1042/CS20130450
117. Füchtbauer L, Groth-Rasmussen M, Holm TH, Løbner M, Toft-Hansen H, Khorooshi R, et al. Angiotensin II Type 1 Receptor (AT1) Signaling in Astrocytes Regulates Synaptic Degeneration-Induced Leukocyte Entry to the Central Nervous System. Brain Behavior Immunity (2011) 25(5):897–904. doi: 10.1016/j.bbi.2010.09.015
118. Kandalam U, Clark MA. Angiotensin II Activates JAK2/STAT3 Pathway and Induces Interleukin-6 Production in Cultured Rat Brainstem Astrocytes. Regul Peptides (2010) 159(1–3):110–6. doi: 10.1016/j.regpep.2009.09.001
119. Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T. Angiotensin Type 1 Receptor Antagonist Inhibits Lipopolysaccharide-Induced Stimulation of Rat Microglial Cells by Suppressing Nuclear Factor kappaB and Activator Protein-1 Activation. Eur J Neurosci (2008) 27(2):343–51. doi: 10.1111/j.1460-9568.2007.06014.x
120. Regenhardt RW, Desland F, Mecca AP, Pioquinto DJ, Afzal A, Mocco J, et al. Anti-Inflammatory Effects of Angiotensin-(1-7) in Ischemic Stroke. Neuropharmacology (2013) 71:154–63. doi: 10.1016/j.neuropharm.2013.03.025
121. Valenzuela R, Rodriguez-Perez AI, Costa-Besada MA, Rivas-Santisteban R, Garrido-Gil P, Lopez-Lopez A, et al. An ACE2/Mas-Related Receptor MrgE Axis in Dopaminergic Neuron Mitochondria. Redox Biol (2021) 46:102078. doi: 10.1016/j.redox.2021.102078
122. Jiang T, Xue L-J, Yang Y, Wang Q-G, Xue X, Ou Z, et al. AVE0991, a Nonpeptide Analogue of Ang-(1-7), Attenuates Aging-Related Neuroinflammation. Aging (2018) 10(4):645–57. doi: 10.18632/aging.101419
123. Liu M, Shi P, Sumners C. Direct Anti-Inflammatory Effects of Angiotensin-(1-7) on Microglia. J Neurochem (2016) 136(1):163–71. doi: 10.1111/jnc.13386
124. Hoyer-Kimura C, Konhilas JP, Mansour HM, Polt R, Doyle KP, Billheimer D, et al. Neurofilament Light: A Possible Prognostic Biomarker for Treatment of Vascular Contributions to Cognitive Impairment and Dementia. J Neuroinflammation (2021) 18(1):236. doi: 10.1186/s12974-021-02281-1
125. Rabie MA, Abd El Fattah MA, Nassar NN, Abdallah DM, El-Abhar HS. Correlation Between Angiotensin 1–7-Mediated Mas Receptor Expression With Motor Improvement, Activated STAT3/SOCS3 Cascade, and Suppressed HMGB-1/RAGE/NF-κb Signaling in 6-Hydroxydopamine Hemiparkinsonian Rats. Biochem Pharmacol (2020) 171:113681. doi: 10.1016/j.bcp.2019.113681
126. Arroja MMC, Reid E, Roy LA, Vallatos AV, Holmes WM, Nicklin SA, et al. Assessing the Effects of Ang-(1-7) Therapy Following Transient Middle Cerebral Artery Occlusion. Sci Rep (2019) 9(1):3154. doi: 10.1038/s41598-019-39102-8
127. Cao C, Hasegawa Y, Hayashi K, Takemoto Y, Kim-Mitsuyama S. Chronic Angiotensin 1-7 Infusion Prevents Angiotensin-II-Induced Cognitive Dysfunction and Skeletal Muscle Injury in a Mouse Model of Alzheimer’s Disease. J Alzheimer’s Dis: JAD (2019) 69(1):297–309. doi: 10.3233/JAD-181000
128. Hay M, Polt R, Heien ML, Vanderah TW, Largent-Milnes TM, Rodgers K, et al. A Novel Angiotensin-(1-7) Glycosylated Mas Receptor Agonist for Treating Vascular Cognitive Impairment and Inflammation-Related Memory Dysfunction. J Pharmacol Exp Ther (2019) 369(1):9–25. doi: 10.1124/jpet.118.254854
129. Janatpour ZC, Korotcov A, Bosomtwi A, Dardzinski BJ, Symes AJ. Subcutaneous Administration of Angiotensin-(1-7) Improves Recovery After Traumatic Brain Injury in Mice. J Neurotrauma (2019) 36(22):3115–31. doi: 10.1089/neu.2019.6376
130. Rabie MA, Abd El Fattah MA, Nassar NN, El-Abhar HS, Abdallah DM. Angiotensin 1-7 Ameliorates 6-Hydroxydopamine Lesions in Hemiparkinsonian Rats Through Activation of MAS Receptor/PI3K/Akt/BDNF Pathway and Inhibition of Angiotensin II Type-1 Receptor/NF-κb Axis. Biochem Pharmacol (2018) 151:126–34. doi: 10.1016/j.bcp.2018.01.047
131. Hay M, Vanderah TW, Samareh-Jahani F, Constantopoulos E, Uprety AR, Barnes CA, et al. Cognitive Impairment in Heart Failure: A Protective Role for Angiotensin-(1-7). Behav Neurosci (2017) 131(1):99–114. doi: 10.1037/bne0000182
132. Goldstein J, Carden TR, Perez MJ, Taira CA, Höcht C, Gironacci MM. Angiotensin-(1–7) Protects From Brain Damage Induced by Shiga Toxin 2-Producing Enterohemorrhagic Escherichia Coli. Am J Physiol-Regulatory Integr Comp Physiol (2016) 311(6):R1173–85. doi: 10.1152/ajpregu.00467.2015
133. Bihl JC, Zhang C, Zhao Y, Xiao X, Ma X, Chen Y, et al. Angiotensin-(1–7) Counteracts the Effects of Ang II on Vascular Smooth Muscle Cells, Vascular Remodeling and Hemorrhagic Stroke: Role of the NFкB Inflammatory Pathway. Vasc Pharmacol (2015) 73:115–23. doi: 10.1016/j.vph.2015.08.007
134. Moore ED, Kooshki M, Metheny-Barlow LJ, Gallagher PE, Robbins ME. Angiotensin-(1–7) Prevents Radiation-Induced Inflammation in Rat Primary Astrocytes Through Regulation of MAP Kinase Signaling. Free Radical Biol Med (2013) 65:1060–8. doi: 10.1016/j.freeradbiomed.2013.08.183
135. Bekassy Z, Lopatko Fagerström I, Bader M, Karpman D. Crosstalk Between the Renin–Angiotensin, Complement and Kallikrein–Kinin Systems in Inflammation. Nat Rev Immunol (2021) 1–18. doi: 10.1038/s41577-021-00634-8
136. Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, et al. Angiotensin II Type 2 Receptor Overexpression Activates the Vascular Kinin System and Causes Vasodilation. J Clin Invest (1999) 104(7):925–35. doi: 10.1172/JCI7886
137. Holling M, Jeibmann A, Gerss J, Fischer BR, Wassmann H, Paulus W, et al. Prognostic Value of Histopathological Findings in Aneurysmal Subarachnoid Hemorrhage: Clinical Article. J Neurosurgery (2009) 110(3):487–91. doi: 10.3171/2008.8.JNS08789
138. Feterik K, Smith L, Katusic ZS. Angiotensin-(1–7) Causes Endothelium-Dependent Relaxation in Canine Middle Cerebral Artery. Brain Res (2000) 873(1):75–82. doi: 10.1016/S0006-8993(00)02482-3
139. Meng W, Busija DW. Comparative Effects of Angiotensin-(1-7) and Angiotensin II on Piglet Pial Arterioles. Stroke (1993) 24(12):2041–4. doi: 10.1161/01.STR.24.12.2041
140. Sampaio WO, Nascimento AAS, Santos RAS. Systemic and Regional Hemodynamic Effects of Angiotensin-(1–7) in Rats. Am J Physiology-Heart Circulatory Physiol (2003) 284(6):H1985–94. doi: 10.1152/ajpheart.01145.2002
141. Botelho-Santos GA, Sampaio WO, Reudelhuber TL, Bader M, Campagnole-Santos MJ, Souza dos Santos RA. Expression of an Angiotensin-(1-7)-Producing Fusion Protein in Rats Induced Marked Changes in Regional Vascular Resistance. Am J Physiology-Heart Circulatory Physiol (2007) 292(5):H2485–90. doi: 10.1152/ajpheart.01245.2006
142. Pernomian L, Gomes MS, Restini CBA, de Oliveira AM. Mas -Mediated Antioxidant Effects Restore the Functionality of Angiotensin Converting Enzyme 2-Angiotensin-(1–7)- Mas Axis in Diabetic Rat Carotid. BioMed Res Int (2014) 2014:1–20. doi: 10.1155/2014/640329
143. Peña Silva RA, Kung DK, Mitchell IJ, Alenina N, Bader M, Santos RAS, et al. Angiotensin 1–7 Reduces Mortality and Rupture of Intracranial Aneurysms in Mice. Hypertension (2014) 64(2):362–8. doi: 10.1161/HYPERTENSIONAHA.114.03415
144. Fang C, Stavrou E, Schmaier AA, Grobe N, Morris M, Chen A, et al. Angiotensin 1-7 and Mas Decrease Thrombosis in Bdkrb2–/– Mice by Increasing NO and Prostacyclin to Reduce Platelet Spreading and Glycoprotein VI Activation. Blood (2013) 121(15):3023–32. doi: 10.1182/blood-2012-09-459156
145. Fraga-Silva RA, Pinheiro SVB, Gonçalves ACC, Alenina N, Bader M, Souza Santos RA. The Antithrombotic Effect of Angiotensin-(1-7) Involves Mas-Mediated NO Release From Platelets. Mol Med (2008) 14(1–2):28–35. doi: 10.2119/2007-00073.Fraga-Silva
146. Fraga-Silva RA, Costa-Fraga FP, Sousa FBD, Alenina N, Bader M, Sinisterra RD, et al. An Orally Active Formulation of Angiotensin-(1-7) Produces an Antithrombotic Effect. Clinics (2011) 66(5):837–41. doi: 10.1590/S1807-59322011000500021
147. Wilsdorf T, Gainer JV, Murphey LJ, Vaughan DE, Brown NJ. Angiotensin-(1-7) Does Not Affect Vasodilator or TPA Responses to Bradykinin in Human Forearm. Hypertension (2001) 37(4):1136–40. doi: 10.1161/01.HYP.37.4.1136
148. Sasaki S, Higashi Y, Nakagawa K, Matsuura H, Kajiyama G, Oshima T. Effects of Angiotensin-(1-7) on Forearm Circulation in Normotensive Subjects and Patients With Essential Hypertension. Hypertension (2001) 38(1):90–4. doi: 10.1161/01.HYP.38.1.90
149. Ueda S, Masumori-Maemoto S, Wada A, Ishii M, Brosnihan KB, Umemura S. Angiotensin(1–7) Potentiates Bradykinin-Induced Vasodilatation in Man. J Hypertension (2001) 19(11):2001–9. doi: 10.1097/00004872-200111000-00010
150. Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis After Aneurysmal Subarachnoid Hemorrhage: An Additional Explanation for Delayed Cerebral Ischemia. J Cereb Blood Flow Metab (2008) 28(11):1761–70. doi: 10.1038/jcbfm.2008.74
151. Neil-Dwyer G, Lang DA, Doshi B, Gerber C, Smith PWF. Delayed Cerebral Ischaemia: The Pathological Substrate. Acta Neurochirurgica (1994) 131(1–2):137–45. doi: 10.1007/BF01401464
152. Stein SC, Browne KD, Chen X-H, Smith DH, Graham DI. Thromboembolism and Delayed Cerebral Ischemia After Subarachnoid Hemorrhage: An Autopsy Study. Neurosurgery (2006) 59(4):781–8. doi: 10.1227/01.NEU.0000227519.27569.45
153. van den Bergh WM. Randomized Controlled Trial of Acetylsalicylic Acid in Aneurysmal Subarachnoid Hemorrhage: The MASH Study. Stroke (2006) 37(9):2326–30. doi: 10.1161/01.STR.0000236841.16055.0f
154. Siironen J, Juvela S, Varis J, Porras M, Poussa K, Ilveskero S, et al. No Effect of Enoxaparin on Outcome of Aneurysmal Subarachnoid Hemorrhage: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J Neurosurgery (2003) 99(6):953–9. doi: 10.3171/jns.2003.99.6.0953
155. Wurm G, Tomancok B, Nussbaumer K, Adelwöhrer C, Holl K. Reduction of Ischemic Sequelae Following Spontaneous Subarachnoid Hemorrhage: A Double-Blind, Randomized Comparison of Enoxaparin Versus Placebo. Clin Neurol Neurosurgery (2004) 106(2):97–103. doi: 10.1016/j.clineuro.2004.01.006
156. Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, et al. Sarcopenia: Revised European Consensus on Definition and Diagnosis. Age Ageing (2019) 48(1):16–31. doi: 10.1093/ageing/afy169
157. Gonzalez A, Orozco-Aguilar J, Achiardi O, Simon F, Cabello-Verrugio C. SARS-CoV-2/Renin–Angiotensin System: Deciphering the Clues for a Couple With Potentially Harmful Effects on Skeletal Muscle. Int J Mol Sci (2020) 21(21):7904. doi: 10.3390/ijms21217904
158. Lad H, Saumur TM, Herridge MS, dos Santos CC, Mathur S, Batt J, et al. Intensive Care Unit-Acquired Weakness: Not Just Another Muscle Atrophying Condition. Int J Mol Sci (2020) 21(21):7840. doi: 10.3390/ijms21217840
159. Cabello-Verrugio C, Morales MG, Cabrera D, Vio CP, Brandan E. Angiotensin II Receptor Type 1 Blockade Decreases CTGF/CCN2-Mediated Damage and Fibrosis in Normal and Dystrophic Skeletal Muscles. J Cell Mol Med (2012) 16(4):752–64. doi: 10.1111/j.1582-4934.2011.01354.x
160. Acuña MJ, Pessina P, Olguin H, Cabrera D, Vio CP, Bader M, et al. Restoration of Muscle Strength in Dystrophic Muscle by Angiotensin-1-7 Through Inhibition of TGF-β Signalling. Hum Mol Genet (2014) 23(5):1237–49. doi: 10.1093/hmg/ddt514
161. Cisternas F, Morales MG, Meneses C, Simon F, Brandan E, Abrigo J, et al. Angiotensin-(1–7) Decreases Skeletal Muscle Atrophy Induced by Angiotensin II Through a Mas Receptor-Dependent Mechanism. Clin Science (2015) 128(5):307–19. doi: 10.1042/CS20140215
162. Morales MG, Abrigo J, Acuña MJ, Santos RA, Bader M, Brandan E, et al. Angiotensin-(1-7) Attenuates Disuse Skeletal Muscle Atrophy. via Mas receptor Dis Models Mechanisms (2016) 9(4):441–9. doi: 10.1242/dmm.023390
163. Abd TT, Hayek S, Cheng J, Samuels OB, Wittstein IS, Lerakis S. Incidence and Clinical Characteristics of Takotsubo Cardiomyopathy Post-Aneurysmal Subarachnoid Hemorrhage. Int J Cardiol (2014) 176(3):1362–4. doi: 10.1016/j.ijcard.2014.07.279
164. Ghadri J-R, Wittstein IS, Prasad A, Sharkey S, Dote K, Akashi YJ, et al. International Expert Consensus Document on Takotsubo Syndrome (Part II): Diagnostic Workup, Outcome, and Management. Eur Heart J (2018) 39(22):2047–62. doi: 10.1093/eurheartj/ehy077
165. Xi Y, Liu B, Yang L, Kuang C, Guo R. Changes in Levels of Angiotensin II and its Receptors in a Model of Inverted Stress-Induced Cardiomyopathy. Eur J Med Res (2014) 19(1):54. doi: 10.1186/s40001-014-0054-8
166. Loot AE, Roks AJM, Henning RH, Tio RA, Suurmeijer AJH, Boomsma F, et al. Angiotensin-(1–7) Attenuates the Development of Heart Failure After Myocardial Infarction in Rats. Circulation (2002) 105(13):1548–50. doi: 10.1161/01.CIR.0000013847.07035.B9
167. De Mello WC. Angiotensin (1–7) Reduces the Cell Volume of Swollen Cardiac Cells and Decreases the Swelling-Dependent Chloride Current. Implications for Cardiac Arrhythmias and Myocardial Ischemia. Peptides (2010) 31(12):2322–4. doi: 10.1016/j.peptides.2010.08.024
168. Joviano-Santos JV, Santos-Miranda A, Joca HC, Cruz JS, Ferreira AJ. New Insights Into the Elucidation of Angiotensin-(1-7) In Vivo Antiarrhythmic Effects and its Related Cellular Mechanisms: Acute Antiarrhythmic Effects of Angiotensin-(1-7). Exp Physiol (2016) 101(12):1506–16. doi: 10.1113/EP085884
169. Hammer A, Yang G, Friedrich J, Kovacs A, Lee D-H, Grave K, et al. Role of the Receptor Mas in Macrophage-Mediated Inflammation In Vivo. Proc Natl Acad Sci (2016) 113(49):14109–14. doi: 10.1073/pnas.1612668113
170. Gallagher PE. Inhibition of Human Lung Cancer Cell Growth by Angiotensin-(1-7). Carcinogenesis (2004) 25(11):2045–52. doi: 10.1093/carcin/bgh236
171. Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1–7) Reduces Smooth Muscle Growth After Vascular Injury. Hypertension (1999) 33(1):207–11. doi: 10.1161/01.HYP.33.1.207
172. Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1-7) Inhibits Growth of Cardiac Myocytes Through Activation of the Mas Receptor. Am J Physiol Heart Circulatory Physiol (2005) 289(4):H1560–1566. doi: 10.1152/ajpheart.00941.2004
173. Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and Pharmacokinetic Study of Angiotensin-(1-7), an Endogenous Antiangiogenic Hormone. Clin Cancer Res (2009) 15(23):7398–404. doi: 10.1158/1078-0432.CCR-09-1957
174. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, et al. A Pilot Clinical Trial of Recombinant Human Angiotensin-Converting Enzyme 2 in Acute Respiratory Distress Syndrome. Crit Care (2017) 21(1):234. doi: 10.1186/s13054-017-1823-x
175. Qaradakhi T, Gadanec LK, McSweeney KR, Tacey A, Apostolopoulos V, Levinger I, et al. The Potential Actions of Angiotensin-Converting Enzyme II (ACE2) Activator Diminazene Aceturate (DIZE) in Various Diseases. Clin Exp Pharmacol Physiol (2020) 47(5):751–8. doi: 10.1111/1440-1681.13251
176. Peregrine AS, Mamman M. Pharmacology of Diminazene: A Review. Acta Tropica (1993) 54(3–4):185–203. doi: 10.1016/0001-706X(93)90092-P
Keywords: renin–angiotensin system (RAS), delayed cerebral ischemia (DCI), subarachnoid hemorrhage (SAH), anoxic injury, acute brain injury
Citation: Annoni F, Moro F, Caruso E, Zoerle T, Taccone FS and Zanier ER (2022) Angiotensin-(1–7) as a Potential Therapeutic Strategy for Delayed Cerebral Ischemia in Subarachnoid Hemorrhage. Front. Immunol. 13:841692. doi: 10.3389/fimmu.2022.841692
Received: 22 December 2021; Accepted: 04 February 2022;
Published: 09 March 2022.
Edited by:
Yujie Chen, Army Medical University, ChinaReviewed by:
Huijing Xia, Louisiana State University Health Sciences Center, United StatesBaojian Xue, The University of Iowa, United States
Copyright © 2022 Annoni, Moro, Caruso, Zoerle, Taccone and Zanier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa R. Zanier, ZWxpc2EuemFuaWVyQG1hcmlvbmVncmkuaXQ=
†These authors have contributed equally to this work