 Tariq Al Farsi1
Tariq Al Farsi1 Khwater Ahmed1
Khwater Ahmed1 Jalila Alshekaili2
Jalila Alshekaili2 Mahmood Al Kindi2
Mahmood Al Kindi2 Matthew Cook3,4,5
Matthew Cook3,4,5 Aliya Al-Hosni6Zainab Ansari7
Aliya Al-Hosni6Zainab Ansari7 Iman Nasr7
Iman Nasr7 Nashat Al Sukaiti1*
Nashat Al Sukaiti1*- 1Department of Pediatric Allergy and Clinical Immunology, The Royal Hospital, Muscat, Oman
- 2Department of Microbiology and Immunology, Sultan Qaboos University Hospital, Muscat, Oman
- 3Department of Immunology and Infectious Disease, John Curtin School of Medical Research, Australian National University, Canberra, NSW, Australia
- 4Translational Research Unit, Department of Immunology, The Canberra Hospital, Canberra, NSW, Australia
- 5Centre for Personalized Immunology (National Health and Medical Research Council (NHMRC) Centre of Research Excellence), John Curtin School of Medical Research, Australian National University, Canberra, NSW, Australia
- 6Molecular Genetics, National Genetics Center, Muscat, Oman
- 7Department of Adult Allergy and Clinical Immunology, The Royal Hospital, Muscat, Oman
Background: Inborn errors of immunity (IEIs) are being recognized as an important cause of morbidity and mortality in communities with a high frequency of consanguinity, such as Oman, and thus recessively inherited conditions. Various monogenic causes of IEI have been recently discovered; however, the disease phenotype may be variable and does not always include infection at presentation, leading to a delay in diagnosis and a poor outcome. It is now well recognized that immune dysregulation manifestations are observed in a significant proportion of patients with IEI and occasionally precede infection.
Methods: Here, we retrospectively report the epidemiological, clinical, immunological, and molecular findings and outcomes from 239 patients with IEI who were diagnosed and managed at the Royal Hospital, Oman, from January 2010 to October 2021.
Results: The estimated annual cumulative mean incidence of IEI was 25.5 per 100,000 Omani live births with a total prevalence of 15.5 per 100,000 Omani population. Both the high incidence and prevalence are attributed to the high rate of consanguinity (78.2%). Defects affecting cellular and humoral immunity including severe combined immunodeficiency (SCID), combined immunodeficiency (CID), and CID with syndromic features were the predominant defects in IEI (36%). Immune dysregulation was a prominent manifestation and occurred in approximately a third of all patients with IEI (32%), with a mean age of onset of 81 months and a mean diagnostic delay of 50.8 months. The largest percentage of patients who showed such clinical signs were in the category of diseases of immune dysregulation (41%), followed by predominantly antibody deficiency (18%). The overall mortality rate in our cohort was 25.1%, with higher death rates seen in CID including SCID and diseases of immune dysregulation.
Conclusion: Immune dysregulation is a frequent manifestation of Omani patients with IEI. Early detection through raising awareness of signs of IEI including those of immune dysregulation and implementation of newborn screening programs will result in early intervention and improved overall outcome.
Introduction
Inborn errors of immunity (IEIs) are a heterogeneous group of more than 400 primary immunodeficiency (PID) disorders, which are classified into 10 groups based on the involved pathophysiology, clinical phenotype, and genotype (1, 2). Prevalence studies based on the given clinical diagnosis have been conducted worldwide. Using disease registries, the prevalence is estimated to range from 1:8,500 to 1:100,000 people (3), according to the ethnic group and method used.
In a historical cohort study performed over a 31-year period using a public administrative healthcare database in the USA, the incidence rate of IEI was approximately 10.3/100,000 person-years, with predominantly antibody deficiency (PAD) accounting for 78% of all cases compared to 10.5% for combined immunodeficiency (CID) including severe combined immunodeficiency (SCID) (4). However, Asian registries especially those of Arabian gulf countries reported an IEI prevalence (per 100,000 children) of 20.2 in Kuwait (5), 4.7 in Qatar (6), and 7.2 in Saudi Arabia (7), with a predominance of CIDs affecting cellular and humoral immunity. In Oman, a previous report on PID among a pediatric cohort described a prevalence of 7/100,000 with an estimated incidence of 5.0/100,000. The main encountered IEIs were phagocytic defects (35%), followed by PAD (20.7%) and CID (17.8%) (8). In contrast, a study from our center demonstrated a higher incidence of SCID among an Omani pediatric population (4/100,000) compared to the earlier published data (9). Such epidemiological discrepancies in Oman can suggest underreporting of IEI in such retrospective reports.
A previous report from Oman described recurrent and severe infections as the only main clinical presentation of IEI (8). Infection is the cornerstone of the 10 warning signs of PID first proposed by the Jeffrey Modell Foundation in 1993 as a screening tool. However, with the expansion of knowledge on IEI, it is gradually becoming clearer that immune dysregulation is a major manifestation of this disease group. Indeed, later studies have demonstrated inadequate sensitivity of the initial 10 warning signs especially in infant and pediatric populations and have discussed the need for adjustment (10–12). In line with this, Thalhammer et al. (13) recommended adding immune dysregulation and syndromic features to the list of IEI warning signs to avoid diagnostic delay, which is associated with poorer health outcomes.
This study aimed to collect information on the frequency and manifestation of non-infectious phenotypes in the current extended cohort of pediatric and adult patients with IEI in Oman. Additionally, we provide more recent data on the incidence, prevalence, molecular diagnosis, and outcome of IEI in the Omani population.
Methods
Data were extracted from electronic medical records by International Statistical Classification of Diseases (ICD)-10 codes (D80–D89) and were filled into Google forms and then an Excel sheet. A manual selection of patients was also performed, as some patients were not linked to ICD-10 codes. Further analysis was performed via Excel and GraphPad Prism-9 (GraphPad Software, Inc., San Diego, CA, USA). The results are expressed as means and medians with ranges and frequency (%) for continuous and categorical variables, respectively. The study was approved by the Research and Ethics Review and Approval Committee in the Ministry of Health, Sultanate of Oman.
Patients
We performed an analysis of a cohort of 239 children and adults with IEI, who were seen and managed at the Royal Hospital—the main governmental tertiary hospital in Oman—from January 2010 to October 2021. The epidemiological details included the annual incidence per Omani live birth and the prevalence among the Omani population. The cumulative incidence was calculated by defining the mean of the yearly new diagnoses of IEI from the annual population of Omani live births obtained from local registries for the last 11 years. Given that most of our cohort were from the pediatric age group, the Omani live birth group was considered a reasonable population at risk to extrapolate a relatively true incidence. However, prevalence included all patients with IEI who were diagnosed from the Omani population over the last 11 years. Additionally, other information such as demographics (sex and age), family history, and clinical manifestations of infection, immune dysregulation, and malignancy, as well as molecular diagnosis, long-term events, and outcomes were collected into an electronic database. The adopted diagnostic criteria were based on The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of IEI (14). The classification and subclassification of IEI were based on the Human Inborn Errors of Immunity: 2019 Update of the International Union of Immunological Societies (IUIS) Phenotypical Classification (1, 2).
Age Categories and International Union of Immunological Societies (IUIS) Classification
The age of onset and age of diagnosis were grouped into the following eight categories: 0–2, 3–5, 6–10, 11–15, 16–20, 21–30, 31–40, and >40 years. The phenotypic and genotypic classification was based on the main categories of IEI described in the IUIS (1, 2). Autoinflammatory disorders and syndromes of familial hemophagocytic lymphohistocytosis were excluded from the analysis due to the unavailability of patient’s details. Unclassified IEI was defined when the clinical phenotype and genotype did not fit the above classes. Diseases with possible contributions to IEI and those that were yet to be further studied were highlighted.
Clinical Presentation, Disease Manifestation, and IEI Complications
Clinical presentations were divided into infection, immune dysregulation, malignancy, or asymptomatic. Detailed infectious and non-infectious disease manifestations and complications are listed. We performed a focused analysis on immunodysregulatory disorders to reveal specific characteristics with IEI classification, phenotype, genotype, diagnostic delay, and outcome.
Laboratory Evaluation
The laboratory evaluation included full blood count (hemoglobin level and total lymphocyte, neutrophil, eosinophil, and platelet count), basic lymphocyte subsets [T, B, and Natural killer (NK) cell count] and total serum immunoglobulin levels (IgG, IgA, and IgM, IgE). Moreover, we also utilized a specific antibody panel for tetanus, diphtheria toxin, Haemophilus influenzae type B, pneumococcal antibodies, hepatitis B, measles, mumps, rubella and varicella, and dihydrorhodamine (DHR) assay for neutrophil function, a functional complement assay for classical and alternative hemolytic pathways and a CD11/CD18 expression assay for adhesion defect when clinically indicated.
Genotype
Molecular diagnosis was obtained through whole-exome sequencing, Sanger sequencing, targeted mutation analysis, fluorescent in situ hybridization (FISH), or microarray. Confirmed genetic diagnosis was determined when patient had a positive pathogenic or likely pathogenic genetic change that could explain the clinical phenotype. A note was made on the proportion of positive mutations according to the results of a commercial lab, National Genetic Centre (NGC), in Oman or via the help of external expert research laboratories. The novelty of the mutation detected was confirmed by reviewing the query interface on the ClinVar database. Genetic workup for patients with Predominantly Antibody Deficiency (PAD) (IUIS class III) was not frequently requested due to limited resources.
Long-Term Events and Outcome
The patients’ clinical outcomes were determined after reviewing the medical record system, noting long-term infectious and non-infectious manifestations. We also reported on the treatment options received, and information on the recent status was defined as follows: alive and well, alive with complications, need for Intensive Care Unit (ICU) admission, defaulted patients, mortality, and the contributions to mortality.
Results
The Sultanate of Oman is the second largest territory in the Arabian Peninsula with an area of 120,000 square miles, a total coastal border of 3,165 km, and a total population of 4,471,148 in 2020. The geographical location has led to open access to immigration and trading from various ancient civilizations in Asia and North Africa. Oman has two major tertiary referral centers for IEI, and one bone marrow transplant unit, which is limited to four beds. The Bacillus Calmette–Guerin (BCG) vaccine is a routine immunization at birth for all newborns. There is currently no newborn screening program for T- and B-cell deficiencies, and referrals from primary physician to tertiary units are based on clinical suspicion and previous family history of IEI. Most patients were referred from government primary or secondary hospitals (68.6%). This is the first report to describe our IEI cohort over the last 11 years.
Patient Demographics
Two hundred thirty-nine (n = 239) patients were diagnosed and managed at the Royal Hospital over a period of 11 years (January 2010–October 2021). The estimated annual cumulative mean incidence of IEI was 25.5 (range: 20.0–29.8) per 100,000 Omani live births, and combined with the Middle East and North Africa region (MENA) Omani cohort (15), the total prevalence of IEI was 15.5/100,000 Omani population (Table 1). There were slightly more boys in the registry (n = 139, 58.1%, and M/F: 1.4), and 194 (81.2%) patients were under 18 years of age. More than two-thirds of the cohort (n = 187, 78.2%) were born to consanguineous parents. Among the relatives of patients with IEI, a positive family history of recurrent infections was detected in 100 patients (41.8%) compared to a positive family history immune dysregulation (n = 38, 15.9%) and a positive family history of malignancy (n = 17, 7.1%). Regardless of the age of disease onset, there was a diagnostic delay. The mean and median age differences between the onset of symptoms and diagnosis were 41.7 and 15 months (range: 1–453), respectively. Details of the age of onset and diagnosis for Omani patients with IEI are shown in Table 2.
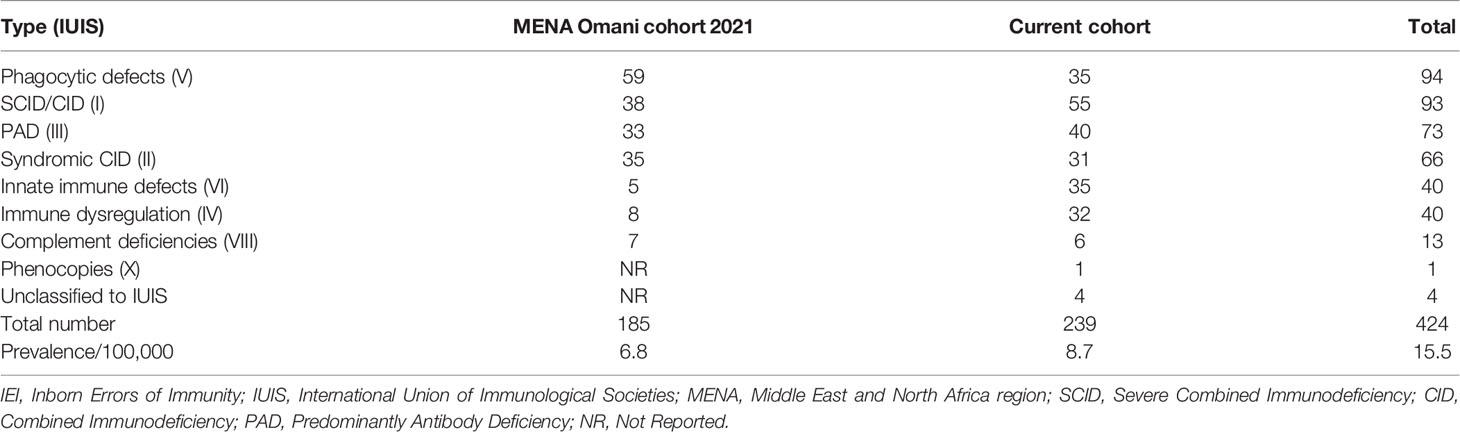
Table 1 Combined data showing the prevalence of IEI in Omani patients according to IUIS classification.
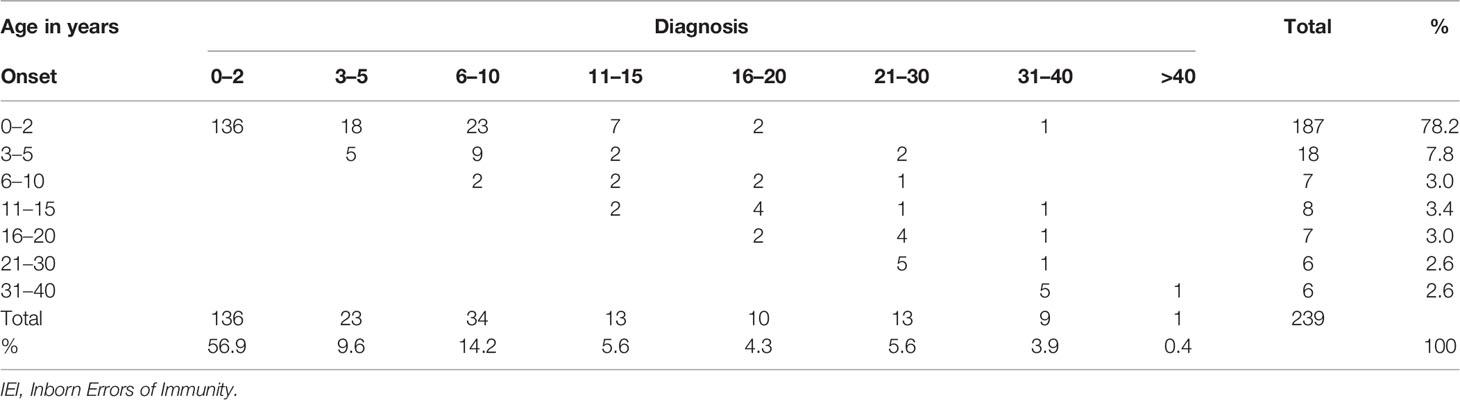
Table 2 Distribution of patients with IEI in the categories of age according to onset and diagnosis.
Clinical Phenotype
Most patients presented with infection (n = 219, 91.6%) at clinical presentation, among whom 40 of them (16.7%) also had symptoms of immune dysregulation. Five patients presented with malignancy (2%), and the remainder were asymptomatic (n = 5, 2%) at clinical presentation as they were familial.
Infection
The mean age of infection episode onset was 42.6 months (range: 1–480). The documented infectious etiology varied but mainly included Gram-negative (n = 143, 61.6%) and Gram-positive (n = 130, 56%) bacterial infections; this was followed by viral infections including the common viruses (n = 114, 49.1%), cytomegalovirus (n = 29, 12.5%), Epstein–Barr virus, (n = 16, 6.9%), and adenovirus (n = 13, 5.6%); fungal infections, including non-filamentous (n = 37, 15.9%), filamentous (n = 29, 12.5%), and dimorphic fungi (n = 13, 5.6%); and mycobacterial infections with Mycobacterium bovis, including BCG strain (n = 41, 17.7%), Mycobacterium tuberculosis (n = 3, 1.3%), and atypical mycobacterium (n = 3, 1.3%). Of note, most patients (n = 231, 96.7%) received BCG vaccination at birth before diagnosis. The lowest reported infection etiology was protozoal agents (n = 5, 2.2%). In regard to the frequency of infectious etiology among patients with infections (n = 219), approximately half (n = 106, 48.4%) had up to two etiologies, a third (n = 64, 29.2%) had three etiologies, and a fifth have four or more etiologies (n = 49, 22.3%). The most commonly reported infection site was the respiratory tract (41.1%), followed by gastrointestinal (12.2%), cutaneous (8.5%), bacteremia and septicemia (8.4%), and other infections. The frequency of clinical presentations is grouped under infections, and the involved sites are illustrated in Figure 1.
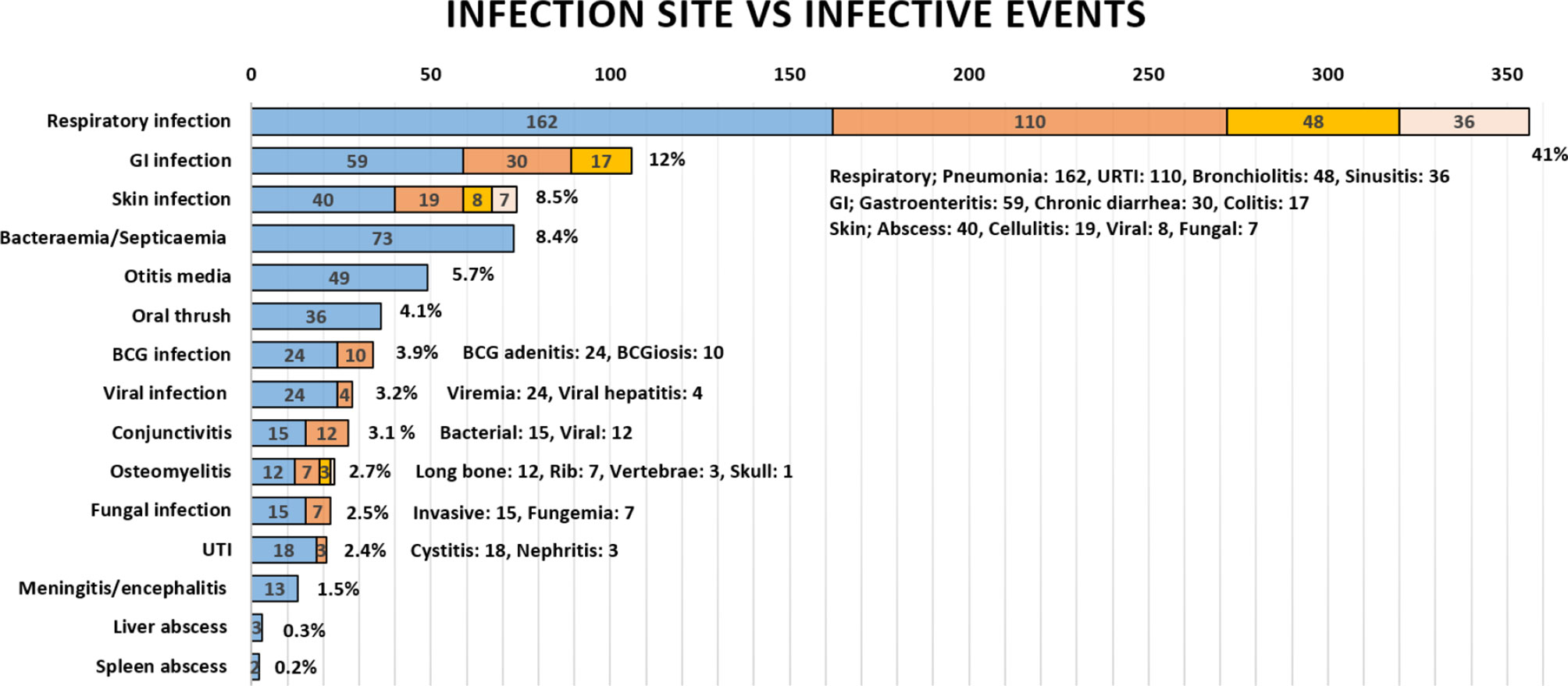
Figure 1 The frequency of infections in Omani patients with IEI according to infection site. GI, gastrointestinal; BCG, Bacillus Calmette–Guerin; UTI, urinary tract infection; URTI, upper respiratory tract infection.
Immunodysregulation
Immunodysregulatory manifestations were prominent in 77 patients (32.2%), among whom 27 patients (35%) died and 5 patients received hematopoietic stem cell transplantation (HSCT), as it was clinically indicated for their phenotype and genotype (DCLRE1C, RAB27A, LYST, LRBA, and XIAP). The mean and median age of onset of immune dysregulation was 81 and 25 (range: 1–480) months, respectively. The mean and median diagnostic delay in patients with immunodysregulatory manifestations was 50.8 and 19 months, respectively. Thirty-two patients were categorized as IEI with immunodysregulation (13.4%) according to IUIS classification, among whom the genotype of 21 patients (8.8%) was identified. The recorded immunodysregulatory manifestations included Omenn’s syndrome, recurrent oral ulcers, autoimmune cytopenia, lymphoproliferation, autoimmune enteropathy, autoimmune thyroid disease, autoimmune nephropathy, diabetes mellitus type 1, autoimmune arthritis, systemic lupus erythematosus, hemophagocytic lymphohistiocytosis, interstitial lung disease, autoimmune eye disease, and others. The features of the genotype, phenotype, and outcomes of patients with immune dysregulation are detailed in Table 3. Patients with non-infective lymphoproliferation were found to have splenomegaly (n = 32, 13.4%), hepatomegaly (n = 29, 12.1%), generalized (n = 25, 10.5%), and localized lymphadenopathy (n = 13, 5.4%).
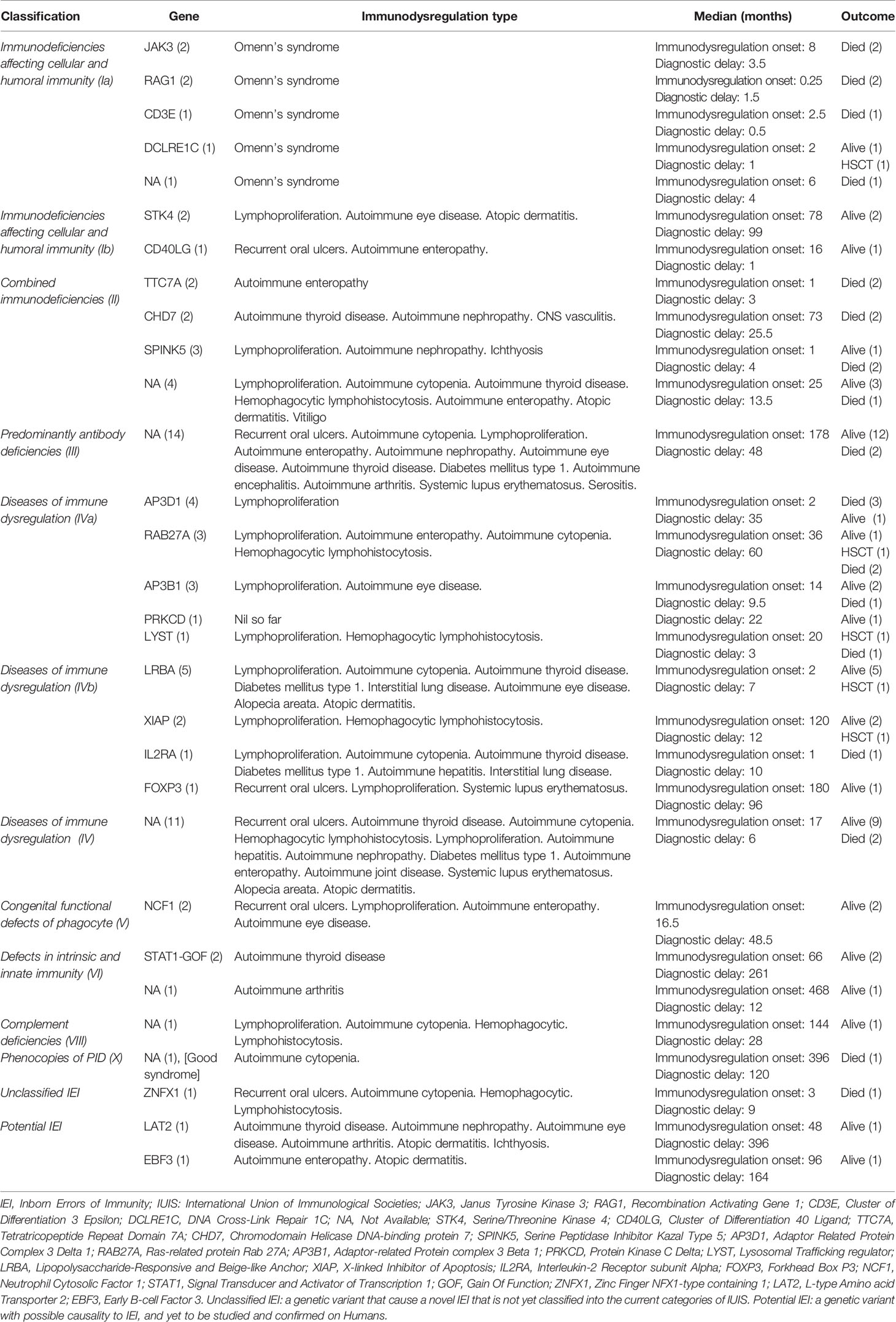
Table 3 Summary describes the immunodysregulatory manifestations of patients with IEI according to IUIS category, genotype, phenotype, and outcome.
Malignancy
The mean and median age of malignancy episode onset was 54 and 36 months, respectively (range: 12–114). A malignant process was reported as Hodgkin’s lymphoma (n = 3, 1.3%), non-Hodgkin’s lymphoma (n = 3, 1.3%), and other malignancies (n = 2, 0.8%).
Immunological Evaluation
In more than a third of the patients, the basic immune workup revealed T-cell lymphopenia (n = 88, 37.9%) and B-cell lymphopenia (n = 70, 30.1%). Other observed cell line abnormalities excluding autoimmune cytopenia included anemia (n = 70, 30.1%), neutropenia (n = 45, 19.4%), thrombocytopenia (n = 34, 14.7%), and eosinophilia (n = 5, 2.2%). Various serum immunoglobulin abnormalities were detected including hypogammaglobulinemia (n = 55, 23.7%), hypergammaglobulinemia (n = 30, 12.9%), hyper-IgE (n = 28, 12.1%), dysgammaglobulinemia (n = 26, 11.2%), agammaglobulinemia (n = 18, 7.8%), specific antibody deficiency (n = 7, 3%), IgG subclass deficiency (n = 6, 2.6%), IgA deficiency (n = 2, 0.9%), and hyper-IgM (n = 2, 0.9%). Other reported immunological findings were abnormal DHR123 assay (n = 33, 14.2%), abnormal complement number and function assay (n = 6, 2.6%), and abnormal expression of CD11/CD18 on flow cytometry (n = 1, 0.4%).
Genotype
Genetic diagnosis was confirmed in 120 patients (50%), with a mean and median age of 59 and 21 months, respectively (range: 0.5–492). However, the median age (in months) of a delay in obtaining a genotype was 69 for class VI, 26.5 for class V, 21.5 for class III, 14 for class IV, 13.5 for class II, and 6 for class I. Homozygosity was paramount, as 90% of all genotypes were autosomal recessive in inheritance (n = 108). In addition to the known mutations associated with the described groups of IEI, we reported three mutations in genes described to cause IEI that are not yet classified into IUIS [LIFR (16), DIAPH1 (17), ZNFX1 (18)]. Additionally, there were two homozygous variants [LAT2 (19) and EBF3 (20)] in genes implicated in immune function in mice that require further immunological functional studies to support a disease phenotype–genotype correlation in humans (Table 4). The modalities used for confirming the molecular diagnosis were whole-exome sequencing and next-generation sequencing (n = 70), Sanger sequencing as targeted mutation analysis (n = 38), and FISH and microarray (n = 12). We identified the genotype using a commercial lab (n = 92, 38.5%), the NGC in Oman (n = 20, 8.4%), and through external expert research laboratories (n = 8, 3.3%). The percentage of familial cases among IUIS classes was 67.6% for class V, 61.8% for class I, 46% for class II, 33.3% for class IV, 16.7% for class VI, and 7.5% for class III. No genetic diagnosis was obtained after a patient’s death.
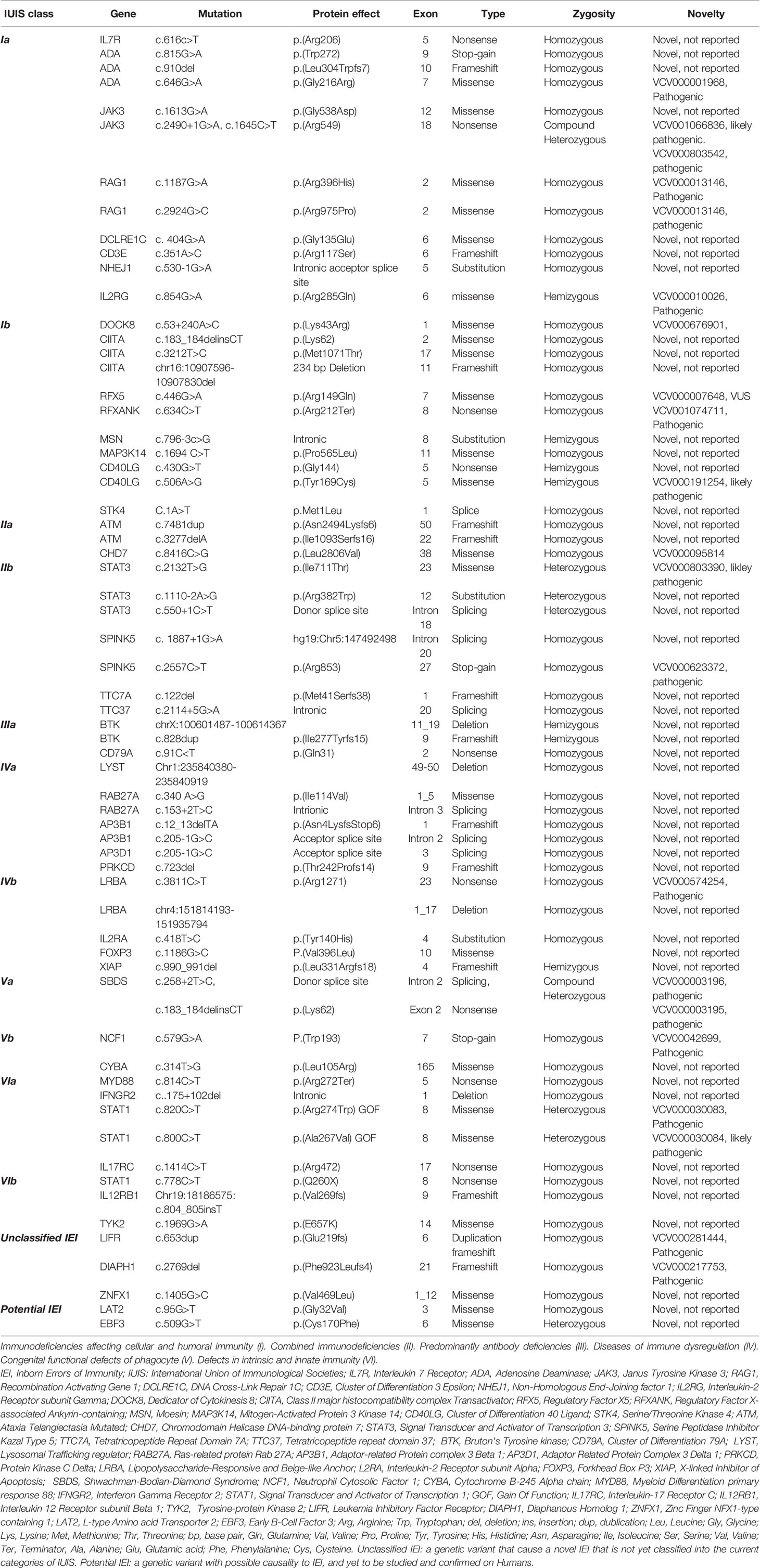
Table 4 Summary of the identified genotype of IEI cohort shown in categories of IUIS, unclassified IEI, and potentially disease-causing variants.
Long-Term Events and Outcome
One hundred thirty-one (54.8%) patients failed to gain adequate weight during their early life. Chronic lung disease constituted a major complication of IEI. Bronchiectasis (n = 62, 25.9%), bronchial asthma (n = 27, 11.3%), and interstitial lung disease (ILD; n = 12, 5%) were the most commonly reported lung complications. End-organ damage (n = 54, 22.6%) was recorded for the brain (n = 14, 5.9%), hearing (n = 12, 5%), liver (n = 8, 3.3%), bone marrow (n = 6, 2.5%), kidney (n = 6, 2.5%), skin (n = 4, 1.5%), and eye (n = 4, 1.5%). In comparison to the overall median age of diagnostic delay (15 months), further delay was noted for patients with the following: bronchiectasis, 60 (2–453); combined end-organ damage, 32.5 (1–204); and bronchial asthma, 28.5 (1–396); and a lesser delay was observed for patients with ILD, 8.5 (2–84) months. Long-term enteropathy (n = 23, 9.6%) was noted as one of the infectious and non-infections complications that contributed to morbidity.
Management embraced frequent interaction with patients and their families. Extensive counseling was provided on nutrition, infection prevention, recognition of warning signs, adherence to action plans, and disease-specific treatment options. Proper assessment and follow-up of an individual clinical course for each patient were implemented. Most patients were evaluated by multidisciplinary teams for disease monitoring, complication prevention, and management. Management strategies included prophylactic antimicrobials (n = 142, 59.4%), intravenous immunoglobulin (IVIG) reconstitution therapy (n = 98, 41%), HSCT (n = 24, 10%), targeted immune therapy (n = 14, 5.9%), and biological therapy (n = 9, 3.8%). One patient with complete 22q11.2 deletion syndrome received thymic transplantation. At the time of the study, 92 patients (38.5%) were alive and well, and 66 patients (27.6%) were alive but with complications and morbidity. Seventy-six patients (31.8%) had been admitted to the ICU at least once, and 21 patients defaulted (8.8%). The management of immune dysregulation involved high-dose immunoglobulin therapy and topical and/or systemic immunosuppressive therapy with corticosteroids, interleukin-1, and interleukin-6 receptor antagonists, calcineurin inhibitors, mammalian Target of Rapamycin (mTOR) inhibitors, Janus kinase (JAK) inhibitors, and anti-CD20 monoclonal antibody. All patients with Lipopolysaccharide-Responsive Beige-Like Anchor Protein (LRBA) deficiency received abatacept.
The mean and median age of transplant was 32 and 19 months, respectively (range: 5–144). Among the 24 patients who received HSCT, 14 (58.3%) were reported to be in remission, 3 (12.5%) demonstrated rejection, and 2 (8.3%) had a relapse of original disease. Five patients are currently undergoing HSCT, with no conclusion of outcome at the time of this report. The donor source was a matched sibling in 12 patients (50%), haploidentical in 8 patients (33.3%), and a matched family donor in 4 patients (16.7%). More than half of the patients who received HSCT (n = 14/24, 58.3%) were transplanted in centers abroad, while 10 patients (41.7%) received HSCT in Oman.
The overall mortality rate was 25.1% (n = 60), with a mean and median age of 60 and 14 months, respectively (range: 1–408). The survival curve for patients with IEI is illustrated in Figure 2. Immunodeficiencies affecting cellular and humoral immunity (I) had the worst probability of survival at 44%, followed by syndromic CID (II) and immune dysregulation (IV) at approximately 68%. The causes of death included severe infection (n = 51, 21.3%), end-organ disease (n = 27, 11.3%), immunodysregulation complications (n = 13, 5.4%), do-not-resuscitate (DNR) order (n = 12, 5%), HSCT-related (n = 6, 2.5%), malignancy (n = 3, 1.2%), and others (n = 2, 0.8%). The bulk of the mortality was reported in class I (n = 20, 58.8%), followed by class II (n = 15, 30%), and class IV (n = 10, 37%). The mortality in class I was due to severe infection in 95%, uncontrolled immune dysregulation in 30%, end-organ damage in 30%, HSCT-related in 15%, and DNR order in 15%. The mortality in class II was due to severe infection in 93%, end-organ damage in 53%, HSCT-related in 40%, uncontrolled immune dysregulation in 26.7%, and DNR order in 6.7%. The mortality in class IV was due to uncontrolled immune dysregulation in 100%, severe infection in 60%, end-organ damage in 50%, HSCT-related in 10%, and DNR order in 10%.
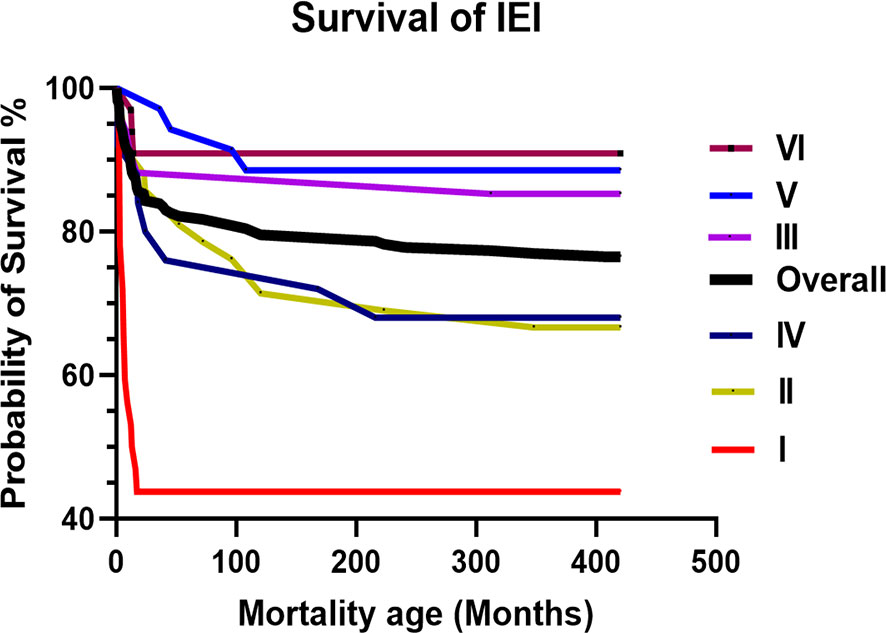
Figure 2 The overall and IUIS class-specific survival curve for Omani patients with IEI. Immunodeficiencies affecting cellular and humoral immunity (I). Combined immunodeficiencies (II). Predominantly antibody deficiencies (III). Diseases of immune dysregulation (IV). Congenital functional defects of phagocyte (V). Defects in intrinsic and innate immunity (VI). IEI, Inborn Errors of Immunity; IUIS, International Union of Immunological Societies.
Discussion
The IUIS PID classification committee classified IEI into 10 major groups based on clinical and laboratory findings along with molecular diagnosis. The current study is the first to characterize 239 pediatric and adult patients with IEI in Oman and provides up-to-date detailed clinical and genetic data. The present results support a higher prevalence and incidence of IEI in such highly consanguineous community and poor survival. Early prevention and early management through screening programs for the classical T- and B-cell deficiencies as well as incorporating immunodysregulation as a warning sign for IEI are considered to represent effective approaches to improve survival.
In this study, we analyzed the demographic, clinical, and molecular data and outcomes of 239 patients diagnosed with IEI at the Royal Hospital. In line with the previously reported high prevalence of consanguineous marriage rate in Oman (49%) (21), greater than two-thirds of the cohort had a history of consanguinity. Therefore, one would expect to see more autosomal recessive and almost equal female-to-male distribution. However, this cohort had slightly more boys to girls (male/female: 58.1% vs. 41.9%), which is similar to the ESID registry (56% vs. 44%) (15).This finding may be because males are more commonly affected with X-linked diseases in addition to the autosomal recessive disease that affect both sexes in a similar proportion.
According to a previous Omani study, Al-Tamemi et al. (8) described an IEI prevalence of 7/100,000, while the current cohort reports a prevalence of 8.7/100,000. The overall calculated combined prevalence of this cohort and the Omani cohort included in the recent MENA report in 2021 (15) was 15.5 per 100,000 Omani population (Table 1). This emphasizes the importance of establishing a detailed national IEI registry in Oman.
Similar to previously published literature, infection was found to be the predominant clinical presentation, followed by immune dysregulation and then malignancy. We also observed that symptomatic recurrent bacterial infections were the most common clinical presentation with the respiratory tract being the predominant site of infection. Failure to thrive, chronic lung diseases, infective lymphadenopathy, and enteropathy were major complications of IEI in our group. This is in agreement with previous studies conducted in Qatar, Kuwait, and Egypt (5, 6, 22).
Interestingly, we found that immune dysregulation was prominent and occurred in approximately one-third of the patients (32%), which was a higher rate than that reported by previous studies from France (26%), Kuwait (20%), and ESID registries (18%) (23, 24). During the last 25 years, immune dysregulation was frequently reported in patients who suffer from IEI, and this association prompted further studies that led to discovery of new monogenic disorders, improvement in the knowledge of the pathogenesis of autoimmunity, and introduction of targeted treatments. The largest percentage of patients who showed clinical signs of immune dysregulation was in the category of diseases of immune dysregulation (41%), followed by PAD (18%) (Table 3). As depicted by Costagliola et al. (25), failure to eliminate self-reactive B cells, T-cell dysfunction, and reduced number and function of regulatory T cells (Tregs) are the main contributing factors in the development of immune dysregulation in such patients. A previous literature review showed that, regardless of what aspect of PID has been studied, early diagnosis reduces healthcare consumption and leads to better health outcomes in nearly all cases (26). Unfortunately, 35% of those who manifested with immune dysregulation died at a mean age of 50.8 months compared to the overall mean age of 41.7 months for the entire cohort. However, a mean difference of 9 months of delay is not statistically significant, highlighting the possibility of other factors such as uncontrolled immune dysregulation, ongoing end-organ damage, and/or treatment-related immunosuppression and effect of toxicity.
Another relevant finding was that the prevalence of both CID and SCID exceeded the frequency of congenital functional phagocyte defects in this cohort. A similar pattern was observed by Barbouche et al. (27) in Egypt, but was in contrast to other studies from the ESID registry (13), Iran (28), Kuwait (5), Tunisia (27), and Turkey (29), where other types of IEI were dominant. One possibility of the higher prevalence of these groups of immunodeficiencies in the current study compared to previous reported findings in the country (8) is related to the direct access and referral of different healthcare facilities to the Royal Hospital, which is considered the largest tertiary hospital in Oman. However, late recognition and referral hamper the appropriate care needed and often increase morbidity and mortality at peripheral hospitals.
We also found that 77% (n = 184) of this cohort had an attempt made toward obtaining molecular confirmation of their underlying IEI, with 50% (n = 120/239) having their genetic defect identified. Compared to the global molecular diagnosis rate of 13.2% (3) this higher percentage could be attributed to the higher rate of consanguinity in our cohort. The method of detection varies, but almost 90% were identified using whole-exome sequencing/next-generation sequencing and Sanger sequencing/targeted mutation analysis. The establishment of a molecular diagnosis has enabled the support of the suspected diagnosis and its expected spectrum of different IEIs. Moreover, molecular characterization guides the treating physician to establish a proper targeted and personalized clinical management, minimizing complications and allowing the provision of evidence-based genetic counseling.
Long-term follow-up of patients revealed that IVIG replacement therapy was conducted in 41% of patients, which is concordant with other cohorts from Germany (47%) (30) and Kuwait (58%) (5). HSCT was achieved in 10% of patients with IEI, among whom more than 56% underwent remission. The mortality rate in our cohort was 25.1% (n = 60), with a mean age of 60 months (range: 1–408). This is comparable to other reports of 26% in Kuwait (5) and 18.7% in Iran (28). A higher mortality rate was seen in patients with SCID/CID due to the delayed diagnosis. Similarly, patients with immune dysregulation had a mortality rate that was comparable to CID probably due to delayed diagnosis. Overall, the mean delayed diagnosis for all patients was 41.7 months, while it was 50 months for those with immune dysregulation. The lack of awareness that such patients is among the spectrum of IEI that has also contributed to the delay in diagnosis. As most of patients with IEI (SCID, CID and patients with immune dysregulation) are relatively asymptomatic at birth, in the absence of a proactive screening program such as a newborn screen program for T- and B-cell deficiencies, the presence of disease can easily be missed until manifestation. Indeed, the newborn screen program has been shown to be sensitive in detection by the new finding that Saudi Arabia has a SCID incidence of 1/2,906 live births (31) compared to the previous retrospectively reported 5.39/100,000 (7). The lack of adequate HSCT units in Oman that can urgently accommodate a sufficient number of patients also contributed in management delay, as most patients were transplanted abroad (58.3%). To overcome the aforementioned obstacles, our prospective proposition is to implement a national newborn screening program, recommend expanding the HSCT service in the country, establish a national IEI registry, and increase the awareness toward the phenotype and genotype of IEI in Oman.
Conclusion
This study is the largest in the country and addresses important factors that lead to poor outcome in patients with IEI. Our findings acknowledge the need to include immune dysregulation as one of the warning signs of primary immunodeficiency to avoid delayed diagnosis of patients with non-infectious manifestations. The higher mortality rate seen in patients with SCID/CID highlights the urgent need for proactive detection through the establishment of a national IEI newborn screening program. Finally, the development of a national IEI registry is a priority, as it will help to unify the national efforts and develop effective short- and long-term strategies. Addressing the above issues will pave the way for better disease ascertainment, phenotype characterization, early diagnosis, and early intervention, thereby improving the likelihood of positive health outcomes for Omani patients with IEI.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
TF: Conception of idea, research design, data collection, data management, analysis, report writing, and intellectual input. KA: Data collection and data management. JA: Critical reviewing and report writing. MK: Critical reviewing and report writing. MC: Critical reviewing. AH: Reviewing genotype. ZA: Data collection and data management. IN: Data collection and data management. NS: Conception of idea, research design, data collection, data management, analysis, report writing, and intellectual input. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41(3):666–79. doi: 10.1007/s10875-021-00980-1
3. Abolhassani H, Azizi G, Sharifi L, Yazdani R, Mohsenzadegan M, Delavari S, et al. Global Systematic Review of Primary Immunodeficiency Registries. Expert Rev Clin Immunol (2020) 16(7):717–32. doi: 10.1080/1744666X.2020.1801422
4. Joshi AY, Iyer VN, Hagan JB, St. Sauver JL, Boyce TG. Incidence and Temporal Trends of Primary Immunodeficiency: A Population-Based Cohort Study. Mayo Clin Proc (2009) 84(1):16–22. doi: 10.4065/84.1.16
5. Al-Herz W, Al-Ahmad M, Al-Khabaz A, Husain A, Sadek A, Othman Y. The Kuwait National Primary Immunodeficiency Registry 2004-2018. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01754
6. Ehlayel MS, Bener A, Laban MA. Primary Immunodeficiency Diseases in Children: 15 Year Experience in a Tertiary Care Medical Center in Qatar. J Clin Immunol (2013) 33(2):317–24. doi: 10.1007/s10875-012-9812-y
7. Al-Saud B, Al-Mousa H, Al Gazlan S, Al-Ghonaium A, Arnaout R, Al-Seraihy A, et al. Primary Immunodeficiency Diseases in Saudi Arabia: A Tertiary Care Hospital Experience Over a Period of Three Years (2010-2013). J Clin Immunol (2015) 35(7):651–60. doi: 10.1007/s10875-015-0197-6
8. Al-Tamemi S, Naseem SUR, Al-Siyabi N, El-Nour I, Al-Rawas A, Dennison D. Primary Immunodeficiency Diseases in Oman: 10-Year Experience in a Tertiary Care Hospital. J Clin Immunol (2016) 36(8):785–92. doi: 10.1007/s10875-016-0337-7
9. Al Sukaiti N, Ahmed K, Alshekaili J, Al Kindi M, Cook MC, Farsi T. A Decade Experience on Severe Combined Immunodeficiency Phenotype in Oman, Bridging to Newborn Screening. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.623199
10. Arkwright PD, Gennery AR. Ten Warning Signs of Primary Immunodeficiency: A New Paradigm Is Needed for the 21st Century. Ann N Y Acad Sci (2011) 1238(1):7–14. doi: 10.1111/j.1749-6632.2011.06206.x
11. Lankisch P, Schiffner J, Ghosh S, Babor F, Borkhardt A, Laws HJ. The Duesseldorf Warning Signs for Primary Immunodeficiency: Is It Time to Change the Rules? J Clin Immunol (2015) 35(3):273–9. doi: 10.1007/s10875-015-0149-1
12. Bjelac JA, Yonkof JR, Fernandez J. Differing Performance of the Warning Signs for Immunodeficiency in the Diagnosis of Pediatric Versus Adult Patients in a Two-Center Tertiary Referral Population. J Clin Immunol (2019) 39(1):90–8. doi: 10.1007/s10875-018-0582-z
13. Thalhammer J, Kindle G, Nieters A, Rusch S, Seppänen MRJ, Fischer A, et al. Initial Presenting Manifestations in 16,486 Patients With Inborn Errors of Immunity Include Infections and Noninfectious Manifestations. J Allergy Clin Immunol (2021) 148(5):1332–41.e5. doi: 10.1016/j.jaci.2021.04.015
14. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract (2019) 7(6):1763–70. doi: 10.1016/j.jaip.2019.02.004
15. Aghamohammadi A, Rezaei N, Yazdani R, Delavari S, Kutukculer N, Topyildiz E, et al. Consensus Middle East and North Africa Registry on Inborn Errors of Immunity. J Clin Immunol (2021) 41(6):1339–51. doi: 10.1007/s10875-021-01053-z
16. Van De Maele K, Smulders C, Ecury-Goossen G, Rosina-Angelista I, Redeker E, Van Haelst M. Stüve-Wiedemann Syndrome: Recurrent Neonatal Infections Caused by Impairment of JAK/STAT 3 Pathway. Clin Dysmorphol (2019) 28(2):57–62. doi: 10.1097/MCD.0000000000000255
17. Kaustio M, Nayebzadeh N, Hinttala R, Tapiainen T, Åström P, Mamia K, et al. Loss of DIAPH1 Causes SCBMS, Combined Immunodeficiency, and Mitochondrial Dysfunction. J Allergy Clin Immunol (2021) 148(2):599–611. doi: 10.1016/j.jaci.2020.12.656
18. Alawbathani S, Westenberger A, Ordonez-Herrera N, Al-Hilali M, Al Hebby H, Al Abbas F, et al. Biallelic ZNFX1 Variants Are Associated With a Spectrum of Immunohematological Abnormalities. Clin Genet (2022) 101(2):247–54. doi: 10.1111/cge.14081
19. Whittaker GC, Orr SJ, Quigley L, Hughes L, Francischetti IMB, Zhang W, et al. The Linker for Activation of B Cells (LAB)/Non-T Cell Activation Linker (NTAL) Regulates Triggering Receptor Expressed on Myeloid Cells (TREM)-2 Signaling and Macrophage Inflammatory Responses Independently of the Linker for Activation of T Cells. J Biol Chem (2010) 285(5):2976–85. doi: 10.1074/jbc.M109.038398
20. Warg LA, Oakes JL, Burton R, Neidermyer AJ, Rutledge HR, Groshong S, et al. The Role of the E2F1 Transcription Factor in the Innate Immune Response to Systemic LPS. Am J Physiol Lung Cell Mol Physiol (2012) 303(5):396–9. doi: 10.1152/ajplung.00369.2011
21. Mazharul Islam M. Consanguineous Marriage in Oman: Understanding the Community Awareness About Congenital Effects of and Attitude Towards Consanguineous Marriage. Ann Hum Biol (2017) 44(3):273–86. doi: 10.1080/03014460.2016.1224385
22. Reda SM, Afifi HM, Amine MM. Primary Immunodeficiency Diseases in Egyptian Children: A Single-Center Study. J Clin Immunol (2009) 29(3):343–51. doi: 10.1007/s10875-008-9260-x
23. Massaad MJ, Zainal M, Al-Herz W. Frequency and Manifestations of Autoimmunity Among Children Registered in the Kuwait National Primary Immunodeficiency Registry. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01119
24. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, Adoue D, et al. Autoimmune and Inflammatory Manifestations Occur Frequently in Patients With Primary Immunodeficiencies. J Allergy Clin Immunol (2017) 140(5):1388–93.e8. doi: 10.1016/J.JACI.2016.12.978
25. Costagliola G, Cappelli S, Consolini R. Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. J Clin Med (2021) 10(20):3–4. doi: 10.3390/jcm10204729
26. Elsink K, van Montfrans JM, van Gijn ME, Blom M, van Hagen PM, Kuijpers TW, et al. Cost and Impact of Early Diagnosis in Primary Immunodeficiency Disease: A Literature Review. Clin Immunol (2020) 213:6–9. doi: 10.1016/j.clim.2020.108359
27. Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary Immunodeficiencies in Highly Consanguineous North African Populations. Ann N Y Acad Sci (2011) 1238(1):42–52. doi: 10.1111/j.1749-6632.2011.06260.x
28. Aghamohammadi A, Mohammadinejad P, Abolhassani H, Mirminachi B, Movahedi M, Gharagozlou M, et al. Primary Immunodeficiency Disorders in Iran: Update and New Insights From the Third Report of the National Registry. J Clin Immunol (2014) 34(4):478–90. doi: 10.1007/s10875-014-0001-z
29. Sanal O, Tezcan I. Thirty Years of Primary Immunodeficiencies in Turkey. Ann N Y Acad Sci (2011) 1238(1):15–23. doi: 10.1111/j.1749-6632.2011.06242.x
30. Gathmann B, Goldacker S, Klima M, Belohradsky BH, Notheis G, Ehl S, et al. The German National Registry for Primary Immunodeficiencies (PID). Clin Exp Immunol (2013) 173(2):372–80. doi: 10.1111/cei.12105
31. Al-Mousa H, Al-Dakheel G, Jabr A, Elbadaoui F, Abouelhoda M, Baig M, et al. High Incidence of Severe Combined Immunodeficiency Disease in Saudi Arabia Detected Through Combined T Cell Receptor Excision Circle and Next Generation Sequencing of Newborn Dried Blood Spots. Front Immunol (2018) 9(APR). doi: 10.3389/fimmu.2018.00782
Keywords: inborn errors of immunity, immunodeficiency, immune dysregulation, phenotype, genotype, children, adults, Omani
Citation: Al Farsi T, Ahmed K, Alshekaili J, Al Kindi M, Cook M, Al-Hosni A, Ansari Z, Nasr I and Al Sukaiti N (2022) Immune Dysregulation in Monogenic Inborn Errors of Immunity in Oman: Over A Decade of Experience From a Single Tertiary Center. Front. Immunol. 13:849694. doi: 10.3389/fimmu.2022.849694
Received: 06 January 2022; Accepted: 24 February 2022;
Published: 06 April 2022.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Vassilios Lougaris, University of Brescia, ItalySaul Oswaldo Lugo Reyes, National Institute of Pediatrics, Mexico
Copyright © 2022 Al Farsi, Ahmed, Alshekaili, Al Kindi, Cook, Al-Hosni, Ansari, Nasr and Al Sukaiti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nashat Al Sukaiti, bmFzaGF0YWxzdWthaXRpQHlhaG9vLmNvbQ==