 Wen Xu
Wen Xu Yi Huang
Yi Huang- 1Neurology Department, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
- 2Wuxi School of Medicine, Jiangnan University, Wuxi, China
Cell death is a necessary event in multi-cellular organisms to maintain homeostasis by eliminating unrequired or damaged cells. Currently, there are many forms of cell death, and several of them, such as necroptosis, pyroptosis and ferroptosis, even apoptosis trigger an inflammatory response by releasing damage-associated molecular patterns (DAMPs), which are involved in the pathogenesis of a variety of human inflammatory diseases, including autoimmunity disease, diabetes, Alzheimer’s disease and cancer. Therefore, the occurrence of inflammatory cell death must be strictly regulated. Recently, increasing studies suggest that phosphorylation plays a critical role in inflammatory cell death. In this review, we will summarize current knowledge of the regulatory role of phosphorylation in inflammatory cell death and also discuss the promising treatment strategy for inflammatory diseases by targeting related protein kinases that mediate phosphorylation or phosphatases that mediate dephosphorylation.
Introduction
Cell death is a process common to all creatures. Early classified cell death based on morphology into apoptosis, autophagy, and necrosis. According to the latest recommendations by the Nomenclature Committee on Cell Death (NCCD) in 2018, there are two types of cell death named programmed cell death (PCD) and non-PCD (1, 2). Non-PCD is referred as accidental cell death (ACD), Which is stimulated by many external factors, such as infection, toxins and physical damage that cause morphological changes, for instance cytoplasmic swelling, rupture of the plasma membrane, and subsequent loss of cell contents, there is no severe chromatin condensation, but with random degradation of DNA (3). ACD is common in ischemia, tumor, and some form of neurodegeneration disease. It is often thought of as a passive process (4, 5). PCD can be regulated by drug or genetic interventions. PCD is divided into apoptotic type (apoptosis) and non-apoptotic type (such as ferroptosis, necroptosis, pyroptosis) (6–9). Apoptosis maintains cell membrane integrity and occurs in a caspase-dependent manner. In contrast, the main characteristics of non-apoptotic cell death are membrane rupture and caspase independence. PCD is a necessary event in multi-cellular organisms to maintain homeostasis by eliminating unrequired or damaged cells. However, some types of cell death, for example necroptosis, pyroptosis and ferroptosis, even apoptosis trigger an inflammatory response by releasing damage-associated molecular patterns (DAMPs), including HMGB1, IL-1α and IL-6, both of which are involved in the development of different kinds of inflammatory diseases, such as diabetes, Alzheimer’s disease, cancer. and even COVID-19 (10–15). Therefore, it is necessary to strictly control the inflammatory activation secondary to cell death and further explore the comprehensive mechanism of its activation.
Phosphorylation and dephosphorylation are the most common and widely studied post-translational modifications in eukaryotes (16). Reversible protein phosphorylation occurs mainly at serine, threonine and tyrosine and is mediated by kinases and phosphatases. Protein phosphorylation and dephosphorylation have been shown to effectively modulate immune responses. In recent years, increasing studies suggest that phosphorylation plays a critical role in regulating inflammatory cell death. For example, MK2 prevent TNF-induced apoptosis and necroptosis by directly phosphorylation of RIPK1 S321 residue (17). TBK1 and IKKϵ were also found to inhibit TNF-induced cell death via inducing the kinase RIPK1 phosphorylation (18). In addition, the kinases IKKα/β, TAK1, PLK1 and AMPK, and even the phosphatases Ppm1b and PTEN were shown to regulate inflammatory cell death through phosphorylation and dephosphorylation (19–24). In this review, we have outlined inflammatory cell death and phosphorylation, including apoptosis, pyroptosis, necroptosis and ferroptosis (Table 1). Moreover, we discuss the potential treatment strategy for diseases associated with cell death by targeting related protein kinases that mediate phosphorylation or phosphatases that mediate dephosphorylation.
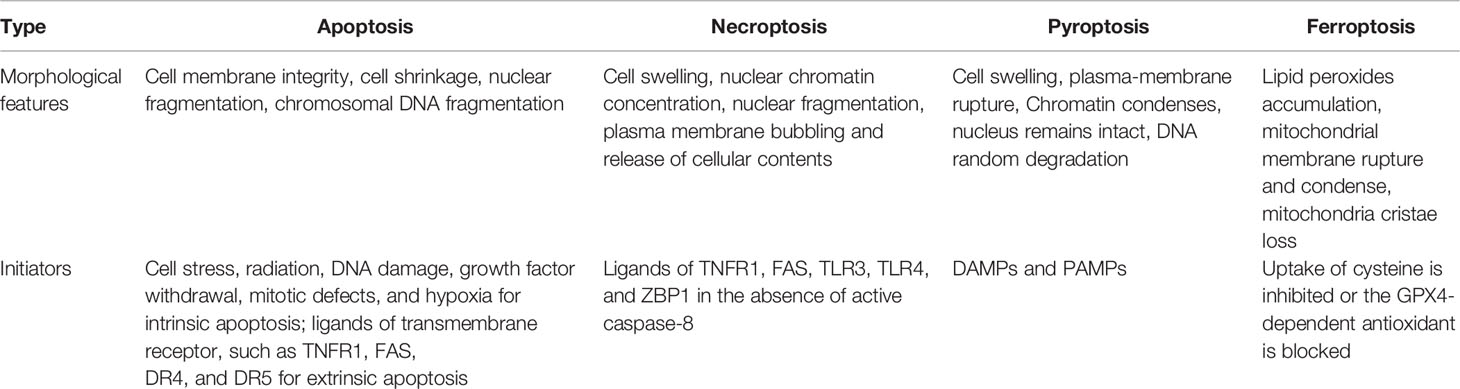
Table 1 Comparison of the main features of different cell death.
Cell Death and Inflammation
Cell death can promote inflammation by releasing so-called endogenous DAMPs that promote the activation of dendritic cells, macrophages and other sentinel cells of the innate immune system (25). Excessive cell death can also directly and indirectly trigger an inflammatory response, leading to persistent inflammation (26).
Inflammatory activation signaling pathways is one of the pioneer innate immune responses. Acute inflammation is a defense response, usually caused by detecting extracellular stimulation and mediated by cytokines, chemokines, and other second messengers. Priming signals mainly consist of DAMPs and pathogen-associated molecular patterns (PAMPs), which activate cellular receptors on or within individual cells in exposed tissue (27). Thus, signaling molecules induce and regulate part and systemic inflammation, cell proliferation, and tissue repairmen (28). Cytokines amplify inflammation, increase the production of inflammatory mediators, coordinating part tissue and systemic immune responses.
Resolution of acute inflammation is critical to restoring homeostasis if danger or damage is handled. Failure to deal with the inflammatory response can lead to chronic inflammation. Amplifying the cycle to ensure adequate acute inflammatory responses, promote excessive inflammation and a long-lasting immune responses, leading to sustained tissue damage and ultimately to chronic inflammatory disease (29, 30).
During necroptosis, receptors downstream of TNF receptor 1 (TNFR1) interact, and activation of serine/threonine protein kinase 3 (RIPK3), which promotes the release of inflammatory cytokines and chemokines (31). Necroptosis causes early infiltration of the plasma membrane, and it is highly pro-inflammatory. Response to DAMPs activates pyroptosis, and sterile, while inflammation induced by sterile DAMPs is the underlying cause of inflammatory disease, which is activated after activation of pro-inflammatory caspases to a series of PAMPs and DAMPs (8, 32). Activated caspases can cleave a variety of substrates, including nucleases and gasdermin D, which promote DNA disruption, during both classical and non-classical pyroptosis processes. Once cracked, the N-terminal part of gasdermin D binds to the lipid membrane to forms oligomer pores. Due to increased osmotic pressure, cell swelling and cell membranes rupture, followed by the release of DAMPs, including ATP, HMGB1 and mitochondrial DNA (33, 34).
In classical pyroptosis processes, activated caspase 1 directly cleaves pro-IL-1β and pro-IL-18, and is released into the extracellular space through the pyroptoptic pores (35). Although the non-classical caspase cannot cleave pro-IL-1β and Pro-IL-18 directly, non-classical caspase cleavage of the gasdermin D can also lead to NLRP3 activation. Subsequent activation of caspase-1 depends on IL-1 β and IL-18 maturation (36).
Cell death plays an important role in cancer therapy, many anti-cancer therapies eliminate cancer cells by using modulate cell death pathways. Necroptosis can serve as an alternative mode of cell death that can potentially eliminate cancer cells and prevent tumorigenesis (37). Evasion of the apoptotic cell death program can facilitate tumor initiation and maintenance as well as promote therapeutic resistance (38).Pyroptosis has emerged as a key form of cancer that serve as a promising target for enhancing tumor immunity. It has been shown that the key pyroptotic proteins GSDMB and GSDME/DFNA5 can be cleaved by granzymes, especially granzymes A and B, respectively, to induce pyroptosis in cancer cells (39, 40). Triggering ferroptosis may prove to be a very effective means of selectively target and overcome iron-addicted and treat resistant tumor cells (41).
Necroptosis and Phosphorylation
Necroptosis was previously thought to be accidental death due to cytotoxic over damage caused by a variety of routine molecular events (42, 43). But now necroptosis was thought the best characterized programmed and regulated cell death, including cell swelling, nuclear chromatin condensation, nuclear fragmentation, plasma membrane bubbling and release of cellular contents (1, 44).
The necroptosis pathway is initiated by numerous specific death receptors, including Z-nucleic acid binding protein 1 (ZBP1) (45), interferon receptors (IFNRs), tumor necrosis factor receptor (TNFR), and Toll-like receptors (TLRs) (46–48). The key molecules of necroptosis are receptor interacting protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain like pseudokinase(MLKL) (49). Many studies have shown that RIPK1 and RIPK3 form a RIP1-RIP3 complex (necrosome),which can be phosphorylated mutually (50, 51). MLKL oligomerize and translocate to plasma membrane, forming a RIPK1/RIPK3/MLKL complex to induce cell expansion and necroptosis (52, 53). MLKL is composed of N-terminal helical bundle domain (HBD) and a C-terminal kinase-like domain, and HBD is critical for the oligomerization of MLKL (54). MLKL oligomerization leads to dysfunction of plasma membrane ion channel, resulting in ion imbalance between internal and external compartments of the plasma membrane and ultimately leading to necroptosis (55). When caspase-8 is inhibited by the apoptosis inhibitor zVAD-fmk or other drugs, RIPK1 binds to RIPK3 to form necrosome. The kinase activity of RIPK3, the oligomerization of MLKL, and the formation of necrosome are considered to be important in the necroptosis processes (51, 53, 56).
Tumor necrosis factor receptor 1 (TNFR1) has been reported induces the expression of many genes that regulate inflammation in necroptosis (57). However, under certain conditions, during TNF-induced cell death, TNF-α is required to bind to TNFR1 on the cell membrane and recruit a range of proteins within the cell to form different complexes (58–60). Among them, complex I includes TNFR-associated death domain (TRADD), TRAF2, RIPK1, cylindromatosis, the cellular inhibitor of apoptosis protein 1 (cIAP1), and the ubiquitin complex (59).
The IκB kinase (IKK) complex consists of a nuclear factor-kappa B essential modulator (NEMO, also known as IKKγ) and two catalytic subunits (IKKα and IKKβ). It plays an important role in mediating immune inflammatory responses and promoting cell survival and tumorigenesis. NEMO recruits IKKα/IKKβ, leading to IKK-mediated rapid selective phosphorylation of IkBα. IkBα activates NF-kB and upregulates genes that encode pro-survival and pro-inflammatory molecules (61). IKKα/β is recruited by NEMO into complex I, which phosphorylates RIPK1 at Ser25 and inhibits RIPK1 kinase-dependent cell death (62, 63) (Table 2).

Table 2 Clinical Trials Targeting cell death -Related Kinases or Phosphatases.
The complex II were consist of TRADD, RIPK1, FAS-associated death domain protein(FADD) and caspase 8 (64). Caspase 8 inactivates RIPK1 and RIPK3 through proteolytic cleavage. When caspase 8 is inhibited, the necrosome is formed, and RIPK1 and RIPK3 were phosphorylated and activated (65). MLKL mediates necroptosis by translocate to the plasma membrane and inducing rupture of plasma membrane. MLKL activation occurs in the necrosome, which is composed of MLKL, RIPK3 and RIPK1 (66, 67). In this necrosome complex, RIPK3 phosphorylates the activation ring of MLKL, promoting conformational changes and the formation of MLKL oligomers. It is found that the phosphorylation of Ser345 is important to RIPK3-mediated necroptosis, It is prove that Ser345 is not required to phosphorylate the interaction between RIPK3 and MLKL in the necrosome, but is critical for MLKL translocation, accumulation in the plasma membrane, and following necroptosis (68).
RIPK3 is an RHIM-domain kinase, it forms complexes with other RHIM-domain proteins, such as RIPK1, ZBP1, and TRIF under activation of necroptosis (67). RIPK3 is phosphorylated at many sites in the necrosome, after Ser227 of RIPK3 was autophosphorylation, it formed hydrogen bonds with Ser404 of MLKL, triggering MLKL to recruit necrosome, resulting in MLKL phosphorylation and induces necroptosis (51, 69). In addition to IKK and IKKβ, TANK-bound kinase 1 (TBK1) is recruited to complex I, which regulates RIPK1 phosphorylation (60, 70). TBK1/IKKϵ recruits complex I through the interactions with NEMO, NAP1, and TANK, then phosphorylated RIPK1 is following inhibited by RIPK1 kinase-dependent cell death. Compare to IKKα/β, TBK1/IKKϵ-mediated RIPK1 phosphorylation does not require TAK1 activation, suggesting that TBK1/IKKϵ phosphorylates and regulates RIPK1 in a manner independent of NF-κB signaling (60, 70). More recently, three separate experiments have proved that RIPK1-dependent cell death is controlled by phosphorylation of RIPK1-ser321 and Ser336 by MK2. In response to TNF signaling, TAK1 phosphorylates p38, which then activates MK2 (17, 71, 72). Active MK2 phosphorylates RIPK1 and it recruited by complex I, which inhibits the formation of complex II b by blocking the interaction of complex II b with FADD. Although TAK1 indirectly phosphorylates RIPK1 through activating IKKα/β and MK2, TAK1 has also been proved to directly phosphorylate RIPK1 (73). RIPK1 autophosphorylation leads to its enzymatic activation, which activates its cytotoxic function. It is shown that Ser14/15, Ser20, Ser161 and Ser166 were autophosphorylation sites in RIPK1 (74). Furthermore, knockdown of L929 cells by reconstructed RIPK1 in RIPK1 K45A/S161E or D138N/S161E mutants is enough to induce RIPK1-dependent necroptosis and the formation of necrosome. These results suggest that RIPK1 autophosphorylation at Ser161 is crucial in RIPK1-dependent cell death (75). RIPK3 is phosphorylated at many sites in the necrosome, in these sites, the autophosphorylation of RIPK3 on Ser227 bind with of the MLKL on Ser404 and formed the necrosome, which phosphorylates MLKL and induces necroptosis (51, 76). Ppm1b was separated by mass spectrometry as the binding partner of RIPK3, which dephosphorylated RIPK3 (77). Consistent with this result, Ppm1b KO mice showed higher mortality in TNF-induced SIRS and increased phosphorylation of RIPK3, suggesting that Ppm1b negatively regulates RIPK3 dephosphorylation under physiological conditions (20).
Parkin was identified as a RIPK3 E3 ligase (78). During the processes of necroptosis, AMP-activated protein kinase (AMPK) is activated by RIPKs and following phosphorylates Parkin’s Ser9. Phosphorylated Parkin causes polyubiquitination of the K33 link on Lys197, Lys302, and Lys364 of RIPK3, and inhibit the formation of necrosome (78). Mass spectrometry showed that MLKL and phosphoglycerate mutase family 5 (PGAM5) are downstream molecules of RIPK3 (79). The N-terminal of MLKL contains 4HB and the C-terminal contains a pseudokinase domain. In case of RIPK3 activation, MLKL is recruited into the necrosome through its interaction with RIPK3 and following phosphorylated on Thr357 and Ser358 in the pseudokinase domain. This phosphorylation of RIPK3 by MLKL leads to a conformational change in the 4HB domain, leading to oligomerization of MLKL (80–82).
MER receptor kinases (TAM), TYRO3 and AXL were identified as MLKL kinase. TAMs phosphorylate MLKL on Tyr376 in response to necroptosis stress and promotes MLKL oligomerization (83). The loss of TAMs attenuated necroptosis but did not alter the phosphorylation of RIPK1, RIPK3, or MLKL. It is suggesting that TAM phosphorylation of MLKL may be the final modification step of necroptosis.
Bcl-2 has been considered as a negative regulator of MLKL. Bcl-2 binds to MLKL and interferes with RIPK3-mediated MLKL phosphorylation and MLKL oligomerization (84). Beclin1 was considered as an inhibitory member of necrosome. Beclin1 binds to necrosome by interacting with MLKL during necroptosis. The interaction between Beclin1 and MLKL requires MLKL phosphorylation of RIPK3, suggesting that Beclin1 is the final barrier to necroptosiss (85).
MLKL exhibits a similar phosphorylation dependence through RIPK3-dependent signaling prior to pore formation and subsequent necroptosis. Therefore, the phosphorylation of pore-forming proteins may be a common mechanism of cells regulate pore formation through death effector proteins (53) (Figure 1).
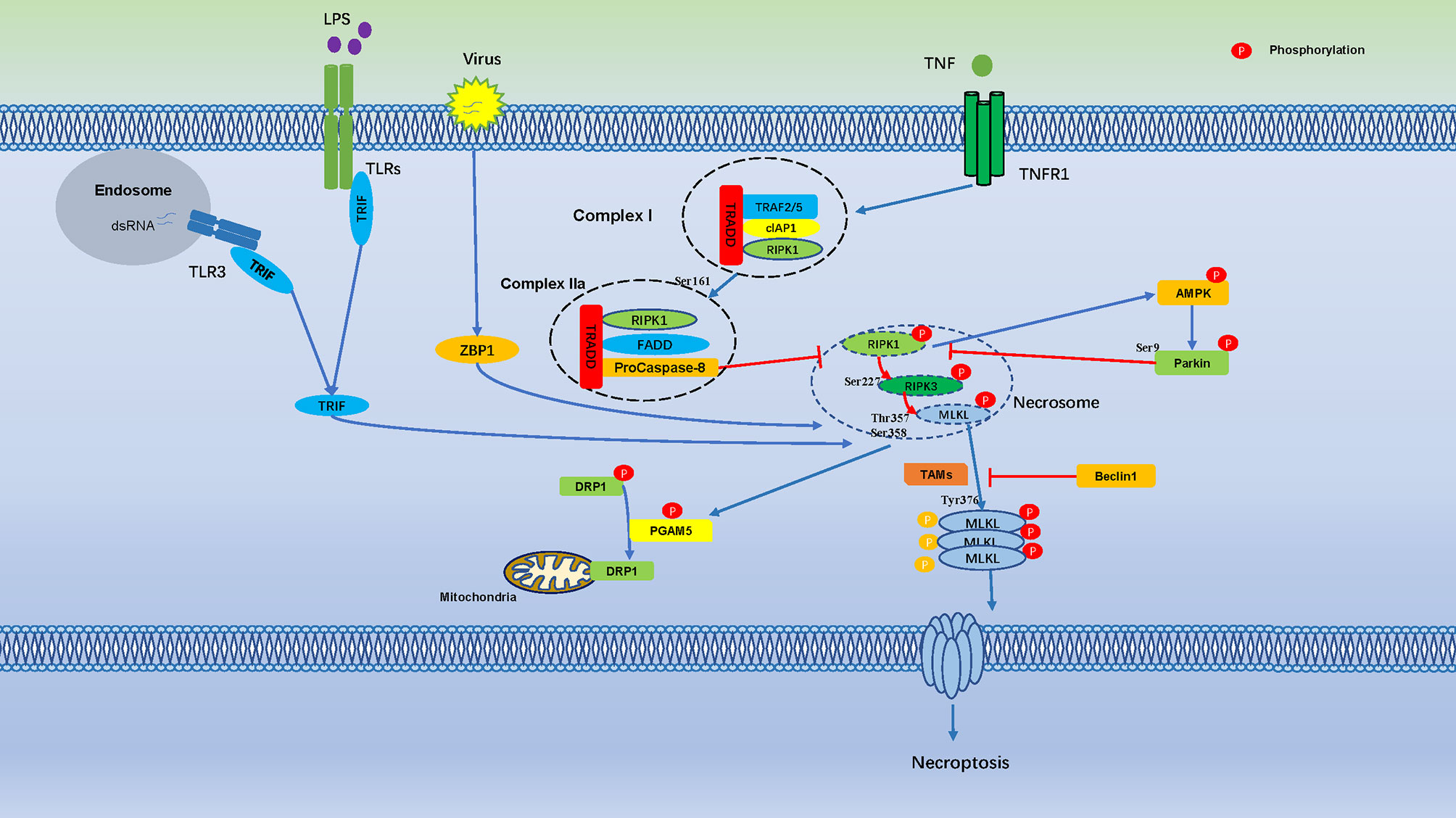
Figure 1 Cell Necroptosis is Regulated by Numerous Phosphorylation Events. Phosphorylation regulates cell necroptosis through numerous phosphorylation events, TNF bind to TNFR1 on the cell membrane and recruit a series of proteins to form complex I, which is includes TNFR-associated death domain (TRADD), RIPK1, TRAF2, the cellular inhibitor of apoptosis protein 1 (cIAP1). Autophosphorylation of RIPK1 leads to its enzymatic activation, Ser161 of RIPK1 is an important autophosphorylation sites, which triggers RIPK3 phosphorylation, the phosphorylation of Ser227 of RIPK3 triggers MLKL recruitment to the necrosome, which phosphorylates MLKL in Ser358, Thr357, and induces necroptosis. AMPK is activated by RIPKs and subsequently phosphorylates Parkin’s Ser9, which is inhibits phosphorylation of RIPK3. Necrosome promotes phosphorylation of PGAM5, which induces dephosphorylation of DRP1.
Apoptosis and Phosphorylation
Apoptosis is a process of cell suicide triggered by foreign factor, It’s the spontaneous, orderly death of cells controlled by certain genes, one highly regulated programmed cell death (PCD) (86), The characteristics of apoptosis are cell becoming small and fine cytoplasmic density increased, mitochondrial membrane permeability changed, cytochrome C released into the cytoplasm, the nucleus shrinks, and eventually forms apoptosome, which are then engulfed by phagocytes (87, 88). The regulation of apoptosis is mediated by two pathways: intrinsic and extrinsic apoptosis signaling pathways. Caspase plays the key role in many apoptotic regulatory pathways (46, 86, 89).
The Intrinsic Pathway
The intrinsic pathway is caused by many of non-receptor-mediated stimuli, such as cellular stress, radiation, DNA damage, and hypoxia (90–92). In the presence of dATP, cytochrome C released into the cytoplasm and binds with apoptosis-related factors to form a polymer, and caspase-9 binds with it to form an apoptosome, when apoptosome activate, and then activates other caspases to induce apoptosis (93). After receiving these endogenous stimuli signals, the pro-apoptotic proteins BAX and BAK form pores on the mitochondrial membrane, destroy the mitochondrial membrane potential, and induce the release of the cytochrome C and apoptotic inducer into cytoplasm (90, 94). Cytochrome C binds to apoptotic peptidase activator 1 and pro-caspase-9, to form the apoptosome. The process of apoptosome formation is essential for the cleavage and activation of pro-caspase-9. Activated caspase-9 cleaves pro-caspase-3 and pro-caspase-7, activated caspase-3 and -7 ultimately initiate apoptosis (15, 95, 96).
The Extrinsic Apoptosis Pathway
The extrinsic apoptosis pathway is triggered by many transmembrane receptors, such as Fas/FasL, death receptor 4 (DR4), death receptor 5 (DR5) and TNF-1/TNF-α, they are typical molecular model signals that caused to apoptosis (46, 97). Stimulated by ligands, these receptors recruit appropriate adaptors to synthesis of a protein complex called a death-inducing signaling complex (DISC) (98). These receptors and ligands activate procaspase by binding to the caspase-8/caspase-10 precursor protein through the Fas-associated DD protein (FADD), then FADD recruits pro-caspase-8 through the death effect domain and induces pro-Caspase-8 activation through its own proteolytic cleavage (68, 99). Activated caspase-8 cleaves and activates effector caspases, including caspase-3 and caspase-7, following activate the downstream effector molecules then leads to apoptosis (68, 99).
Apoptosis signals lead to activation of Mst through phosphorylation and proteolytic cleavage. Apoptotic stimuli result in Mst autophosphorylation and activate caspases. The autophosphorylated Mst is active, and phosphorylates substrates amplify the apoptosis signals, then increasing caspase activation. Futhermore, active caspases cleaved the regulatory domain of the Mst carboxy-terminal. This releases the active amino-terminal kinase domain that migrate to the nucleus, where they phosphorylate transcription factors and histones (100).
HSP27 inhibits ASK1 cell death signaling by phosphorylation of PKD1. After ischemia, PKD1 is phosphorylate, but without HSP27, unable to inhibit ASK1 activity and resulting in cell death (101).
Phosphorylation plays critical role in apoptosis, although the extent to which proteolysis and phosphorylation pathways interact during PCD remains unclear. Using a quantitative proteomic platform integrates phosphorylation sites into protein topologies, a set of over 500 apoptosis phosphorylation events has been identified and shown to be rich on cleaved proteins and clustered around caspase protein hydrolysis sites. Meanwhile, it was found that caspase-cleavage can expose new phosphorylation sites. Conversely, phosphorylation at the +3 position directly promote proteolysis of caspase-8 substrate. Tt is revealed a form of functional communication between phosphorylation and caspase proteolytic pathways, leading to increased protein cleavage rates and the emergence of new phosphorylation sites (102).
After DNA damage, p53 is phosphorylated on Ser-15 and Ser-20, stimulating p53 to bind to promoter regions of genes subset. If DNA damage is severe, TP53INP1 forms a protein complex with Ser-46 kinase HIPK2 and PKCδ, resulting in phosphorylation of p53 on Ser-46, and induction of p53AIP1 gene transcription and cell apoptosis (103). It is suggested that TP53INP1 induces phosphorylation of p53 protein on Ser-46 and apoptosis after DNA damage (Figure 2).
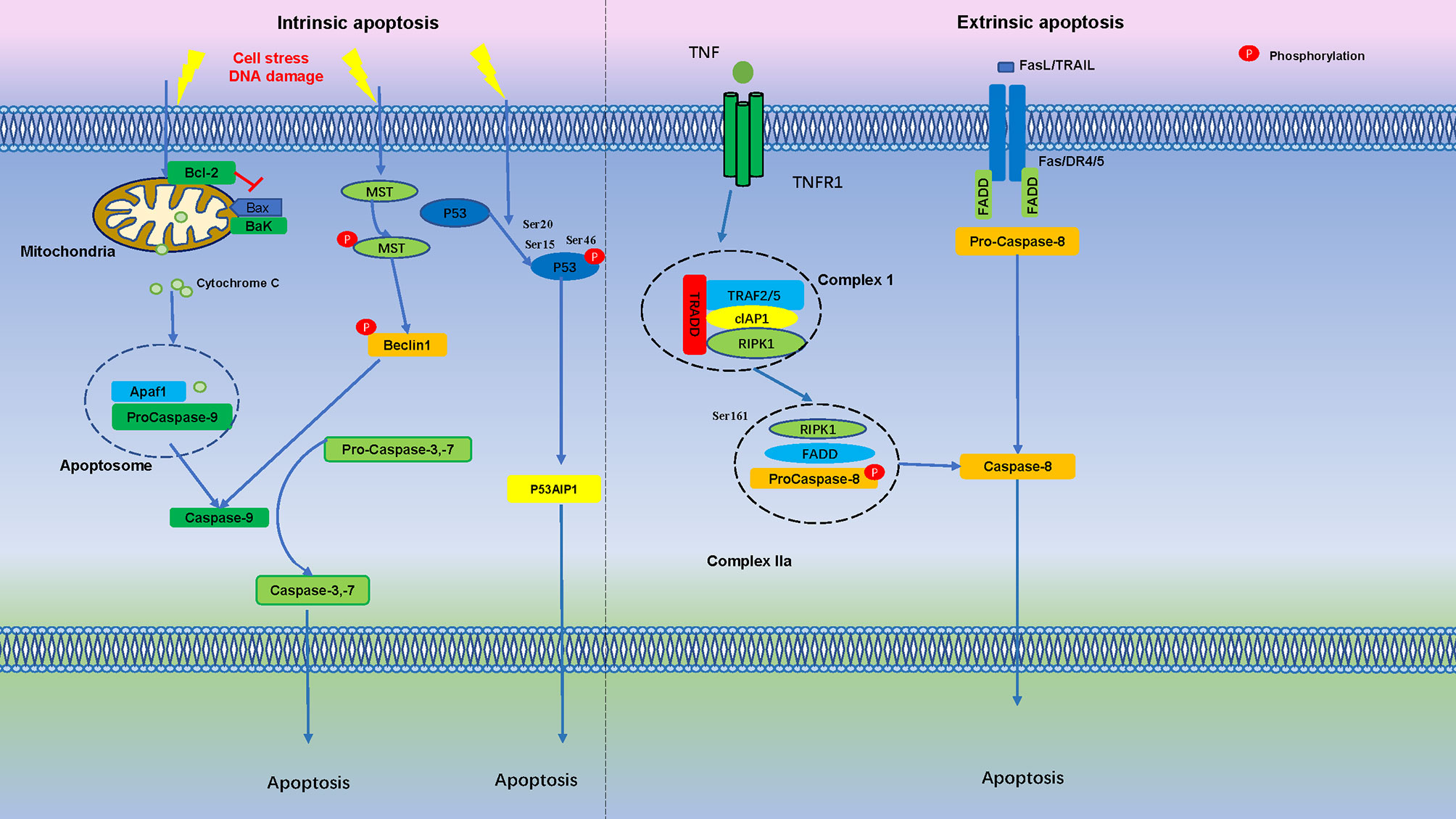
Figure 2 Apoptosis is Regulated by Numerous Phosphorylation Events. In the intrinsic apoptosis pathway, cell stress and DNA damage promotes mitochondria release cytochrome C into the cytoplasm, binds with Apaf1 and pro-caspase-9 to form an apoptosome. Apoptotic stimuli lead to MST autophosphorylation and triggers Beclin1 phosphorylation, which activate caspases. After initial DNA damage, p53 is phosphorylated at Ser15 and Ser20, if DNA damage is severe, phosphorylation of p53 protein at Ser-46 forms TP53INP1 and induce cell apoptosis. In the extrinsic apoptosis pathway, Fas/FasL, death receptor 4 (DR4), death receptor 5 (DR5) and TNFR1 bind to their ligands to induce pro-caspase-8 activation, which then cleaves and activates effector caspases, such as caspase-3 and caspase-7, further activation of downstream effector molecules then leads to apoptosis.
Pyroptosis and Phosphorylation
Pyroptosis is a newly discovered PCD in recent years. It is a main non-specific defense mechanism in the body, which plays an irreplaceable role in antagonizing the invasion of external pathogens and sensing the endogenous danger signals. In 2001, Cookson et al. (104) have found that there was a mode of death dependent on caspase-1 activity, which was different from apoptosis dependent on caspase-3 activity, and they defined cell pyroptosis as caspase-1 dependent cell death in the first time. Shao et al. have found that pyroptosis can be triggered by the activation of caspase-4/5/11 by intracellular LPS, and the activated caspase-4/5/11 ultimately induces pyroptosis through the cleavage of Gasdermin family proteins (105, 106). Therefore, they defined pyrotopsis as PCD mediated by the Gasdermin family.
Pyroptosis with some characteristics of cell necroptosis and apoptosis on the morphology. When cell pyroptosis occurs, the nucleus condensation, chromatin DNA random fracture degradation, Numerous pores appear in the cell membrane, Cell membranes lose their ability to regulate the movement of substances in and out, cell loss of ion balance, both inside and outside osmotic swelling and membrane rupture occurred, releasing active substances such as cell content, stimulate the body The immune response recruits more inflammatory cells and amplifies the inflammatory response (107, 108).When microorganisms infect host cells externally or internally, the pattern recognition receptor (PRR) located in the cytoplasm is associated with PAMP and DAMP recognize and bind to the corresponding ligand to assemble and form the intracellular multiprotein complex, which activates inflammatory caspase-1 and caspase-4/5/11 further cleavage of GSDMD protein punch holes on the cell membrane and promoted the occurrence of pyroptosis. At the same time, inflammasome acts on downstream molecules to promote the maturation of inflammatory cytokines (such as IL-1β, IL-18, IL-6, etc.), chemokines, adhesion molecules, and release them into the extracellular domain through the ruptured cell membrane, recruit and activate more inflammatory cells and amplify local and systemic inflammatory responses (109).The activation pathway of pyroptosis was divided into two groups: the classical cell pyroptosis pathway activated by inflammasome caspase-1 and the non-classical pyroptosis pathway activated by LPS.
Classical Cell Pyroptosis Pathway
The activation of classical cell pyroptosis pathway mainly depends on PRRs receiving danger signal molecule stimulation, recruiting pro-caspase1 to assemble and form inflammasome through ASC, activating caspase-1 molecule to further cleaves downstream GSDMD protein and promoting pyroptosis (110).
Non-Classical Pyroptosis Pathway
Unlike the classical pathway, the non-classical pathway depends on caspase-4/5/11 activation. In response to intracellular LPS stimulation, mouse caspase-11 and its human counterpart caspase-4/5 can bind directly to conserve structure lipid A of LPS and be activated. The activated caspase-4/5/11 further cleaves GSDMD protein and promotes cell pyroptosis (111, 112). GSDMD protein exists in cytoplasm and can be stimulated by caspase-1 activated in classical and caspase-4/5/11 activated in non-classical pyroptosis pathway. GSDMD protein is divided into lipophilic N-terminal domain and hydrophilic C-terminal domain at specific sites. They are connected by a joint region with a protease cleavage site (113). The N-terminal domain bind to the biofilm and aggregate in the inner side of the biofilm to form pores, where water molecules invade cells and trigger cell pyroptosis (114–116).
So far, evidence of phosphorylation only exists at Thr8 and Thr6 of human GSDMA and GSDME, both phosphorylation sites can prevent Gasdermin oligomerization and pore formation (117). Serine threonine kinase Polo-like kinase 1 (PLK1) mediates phosphorylation of GSDMA, but it is unclear if it phosphorylates Thr6 in GSDME (117, 118). GSDMD-NT, GSDMA-NT and GSDMA3-NT, can bind to membrane lipids only when phosphorylated, membrane lipid composition and phosphorylation modification were identified as another control mechanism for pyroptosis (119). In addition, TGF-β -activated kinase (TAK) 1 and PTEN has been found to control inflammasome activation and pyroptosis by regulating NLRP3 phosphorylation (19, 120) (Figure 3).
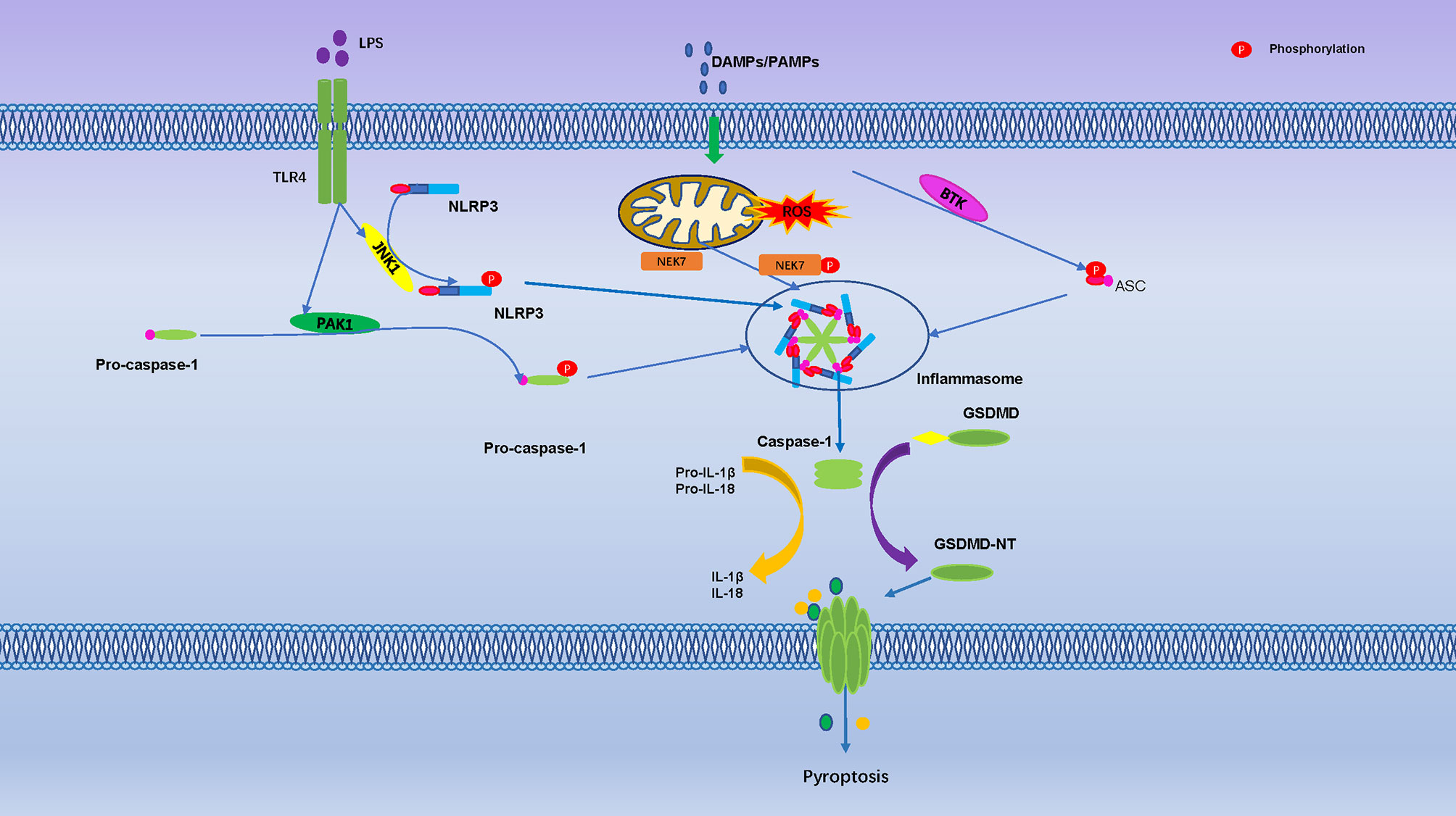
Figure 3 Pyroptosis is Regulated by Numerous Phosphorylation Events. The toll-like receptor 4 (TLR-4) activate Jun N terminal kinase 1(JNK1), which primes NLRP3 by direct phosphorylation at S198. The PAK1 promote phosphorylation of pro-caspase-1. Mitosis A-Related Kinase 7 (NEK7) and Bruton’s tyrosine kinase (BTK) promote NLRP3 activation through direct interaction. They are regulated activation of the NLRP3 inflammasome, cleavage of GSDMD protein punch holes on the cell membrane and promoted the occurrence of pyroptosis.
Ferroptosis and Phosphorylation
It is have found that iron metabolism disorder and consequent iron overload are strongly linked to the occurrence and development of tumors. In addition, the presence of iron, especially iron divalent greatly aggravated the lipid peroxidation of saturated fatty acids (FAs) in human body. During n mitochondrial iron related oxidative phosphorylation, cells produce reactive oxygen species (ROS) and ATP. Overload ROS levels can lead to oxidative stress reactions that directly or indirectly destroy proteins, lipids and other macromolecules, leading to cell damage or death (121). Ferroptosis is recognized as non-apoptotic mediated cell death, usually caused by iron-dependent lipid peroxidation (122, 123). Ferroptosis characterized by the accumulation of lipid peroxides and lethal ROS, which is distinct from other types of cell death in morphologically, genetically, and biochemistry and is controlled by integrated oxidation and antioxidant systems. Morphologically, cells that undergo ferroptosis usually show typical necrotic morphological changes, such as cytoplasmic organelle swelling and rupture of plasma membrane, it is different from traditional apoptosis cells, which are characterized by blistering and contracting of plasma membrane, fragmentation and marginalization of chromatin, production of apoptosome, and cytoskeleton destruction (124).
Ferroptosis is an ROS-dependent form of cell death, associated with excess iron accumulation and lipid peroxidation, and changes in specific genes involved in regulating iron homeostasis and lipid peroxidation metabolism (125). Ferroptosis can be prevented by the use of iron chelators, while the provision of exogenous iron (such as ammonium ferric citrate) enhances ferroptosis (126). Glutathione-GPX4, FSP1-coenzyme Q10 (CoQ 10) and GTP cyclohydrolase-1- (GCH1-) tetrahydrobiotrexate (BH4) pathway have been confirmed to be involved in the regulation of ferroptosis (9, 127).
The phosphorylation typically occurs at the serine, tyrosine, and threonine residues of the target proteins (128). Energy stress causes cell death, which is related to the induction of reactive oxygen species (ROS) (129). Since lipid peroxidation is the main characteristic of ferroptosis, theoretically energy stress should promote ferroptosis. However, Lee et al. shown that glucose starvation inhibited erastin-induced ferroptosis in mouse embryonic fibroblasts, and this effect can also be observed in cystine depletion-, RSL3-, and GPX4-induced ferroptosis (129). AMPK is a key sensor of cellular energy status, and glucose starvation leads to phosphorylation and activation of AMPK (130). AMPK mediated the inhibitory effect of glucose starvation on ferroptosis by accelerating the phosphorylation of ACC and inhibiting lipid biosynthesis containing PUFA (129). Cancer cells with high AMPK are resistant to ferroptosis, and the inactivation of AMPK makes these cells sensitive to ferroptosis (129), it suggests that AMPK negatively regulates ferroptosis. During ferroptosis, BECN1 was identified as a novel SLC7A11/system Xc−binding protein that inhibit activity of type 1 ferroptosis inducers (such as erastin, sulfasalazine) (131, 132). Erastin and sulfasalazine activate AMPK as ferroptosis inducers, and AMPK phosphorylates BECN1 on Ser90/93/96, thereby promoting the formation of the BECN1-SLC7A11 complex (131). The interaction between BECN1 and SLC7A11 directly inhibit system Xc to promoting ferroptosis. Knockout of BECN1 could inhibited ferroptosis induced by system Xc inhibitors but not by RSL3, FIN56, or sulfoximine. Overexpression of BECN1 or application of the BECN1-activating peptide Tat-Beclin 1 promoted cancer cell ferroptosis (131). Although autophagy machinery are involved in ferroptosis, and BECN1 plays an important role in autophagy (133, 134). The downregulation of BECN1 cannot affect the formation of lipidated microtubule-associated protein 1 light chain 3 (MAP1LC3B) in ferroptosis (134). It suggests that, within the presence of certain stimuli, BECN1 bind to different partners to play different roles in autophagy and ferroptosis.
The Toll-like receptor 4- (TLR4-) nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway activates the expression of multiple proinflammatory cytokine genes, and play an important role in inflammatory disorders (135, 136). Bone marrow-derived macrophages (BMDMs) pretreat with erastin obviously attenuates the expression of proinflammatory cytokines induced by LPS treatment, these effects are mediated by inhibition of the phosphorylation of IKKβ, and the phosphorylation and degradation of IκBα and NF-κB, consequently caused to inhibit the development of sepsis (136). Ferroptosis was detected in intestinal epithelial cells in patients with ulcerative colitis and colitis mice, it is indicate that NF-κBp65 phosphorylation suppresses ER stress-mediated ferroptosis in intestinal epithelial cells alleviates ulcerative colitis (137). Moreover, folic acid induced kidney damage was alleviated by FG-4592, which increased phosphorylated of AKT and GSK-3 β, and activated NFE2-related factor 2 (Nrf2) to inhibit ferroptosis (138). Protein kinase C (PKC) mediated phosphorylation of HSPB1 prevents erastin-induced ferroptosis by reducing lipid peroxidation (139) (Figure 4).
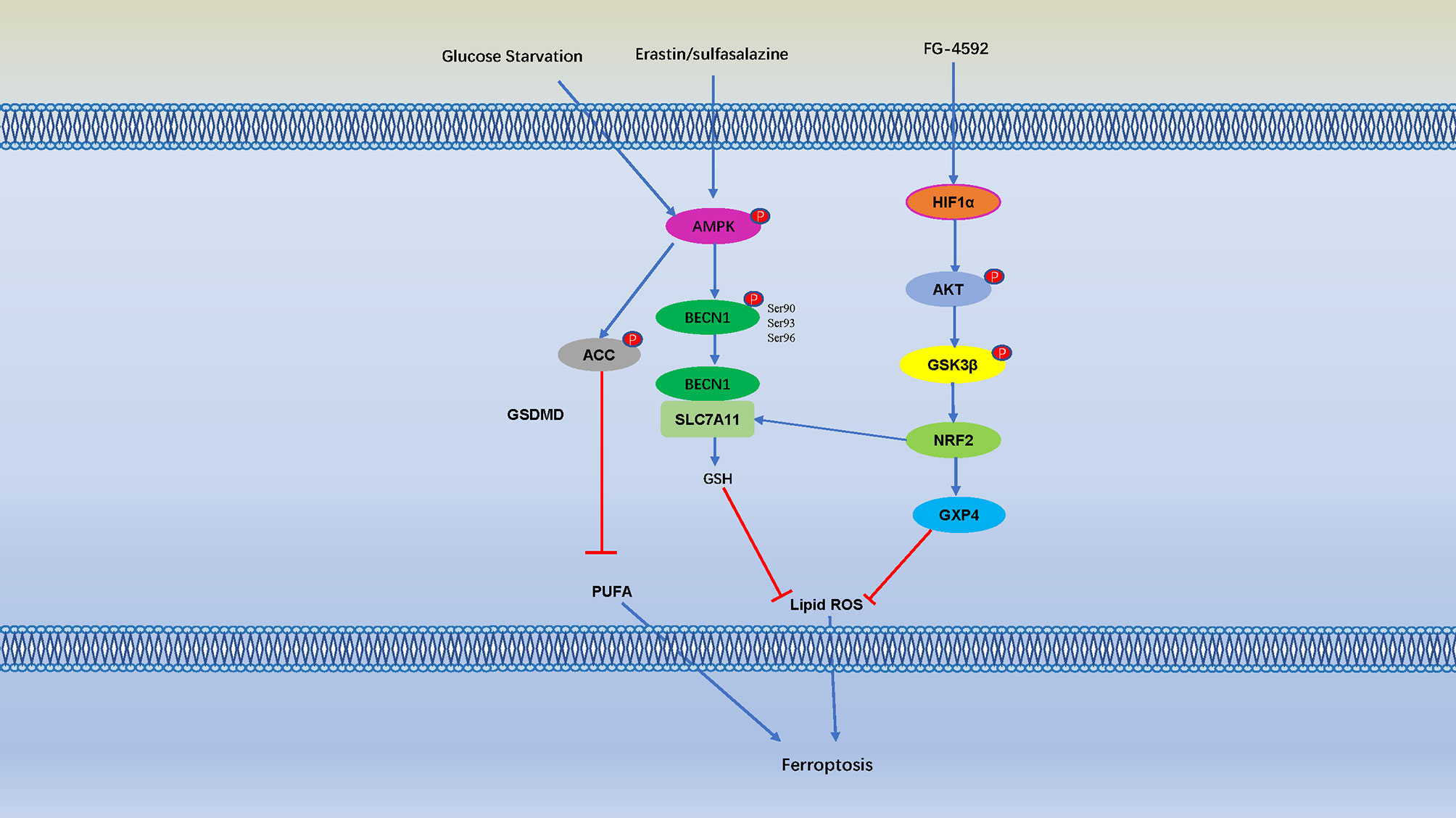
Figure 4 Cell Ferroptosis is Regulated by Numerous Phosphorylation Events. AMP-activated protein kinase (AMPK) is a key sensor of cellular energy status, and glucose starvation leads to phosphorylation and activation of AMPK, AMPK mediated phosphorylation of Acetyl-CoA carboxylase (ACC) and inhibiting PUFA-containing lipid biosynthesis and negatively regulates ferroptosis. Erastin and sulfasalazine also activate AMPK phosphorylation, and AMPK phosphorylates BECN1 at Ser90/93/96, thereby promoting the formation of the BECN1-SLC7A11 complex (131). The interaction between BECN1 and SLC7A11 promote GSH, thereby inhibit lipid ROS and negatively regulates ferroptosis. Moreover, folic acid induced kidney damage was alleviated by pretreatment of FG-4592, which increased phosphorylated of protein kinase B (AKT) and glycogen synthase kinase 3β (GSK-3 β) and activated NFE2-related factor 2 (Nrf2) to inhibit ferroptosis.
Targeting Phosphorylation for Treatment of Cell Death-Associated Inflammatory Disease
Protein kinases and phosphatases are highly regarded as very important drug targets, and now it is known that protein phosphorylation can be involved in regulating inflammatory cell death, which plays an important role in multiple diseases. Since many types of cell death are regulated by protein phosphorylation, targeted phosphorylation can be used to treat many inflammatory cell death-related diseases (Table 2).
Necrosulfonamide is a very specific and potent inhibitor of MLKL, which inhibits MLKL-mediated necrosis by blocking its N-terminal CC domain. It inhibits necroptosis downstream of RIPK3 activation. Necrosulfonamide efficiently blocks necroptosis in human cells (140).
Metformin, as an AMPK activator, which inhibits the proliferation, migration, and activation of rheumatoid arthritis (RA)- fibroblast-like synoviocytes (FLS). Metformin treatment inhibited the expression of pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β, and increased the expression of p-AMPK-α1, protein kinase A α1 (PKA-α1), and hyaluronic acid and proteoglycan associated protein 1 (HAPLN1). The increased expression of HAPLN1 in RA-FLS may be beneficial for joint protection (141) (Table 3).
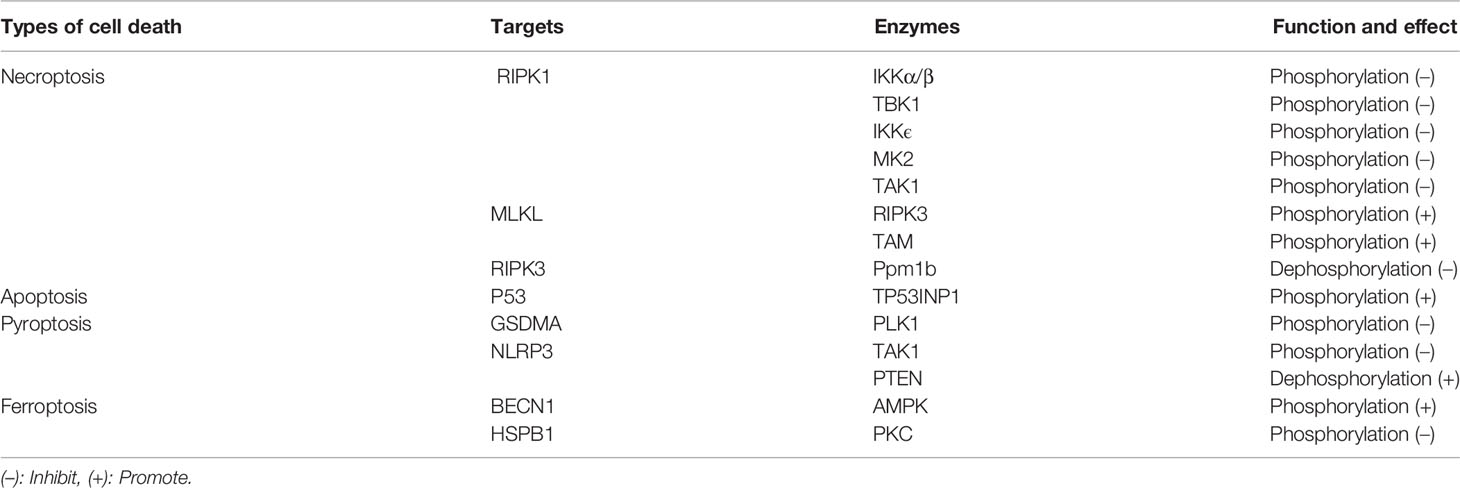
Table 3 Key kinases and phosphatases that regulate inflammatory cell death.
Concluding Remarks and Future Perspectives
Cell death is a common process in all creatures, which is also involved in the development of various inflammatory diseases. Phosphorylation of proteins that regulate cell death is associated with a variety of inflammatory diseases, such as diabetes, Alzheimer’s disease, cancer, and so on, as mentioned above. In the past decade, there have been rapid advances in the understanding of how cell death regulates disease progression, including apoptosis, pyroptosis, necroptosis and ferroptosis, all of which are regulated by protein phosphorylation. Targeting the phosphorylation of MLKL, RIPK3, AMPK, BECN1 and other proteins can directly regulate cell death, which can then be used to treat cell death-related diseases by targeting these phosphorylated proteins. Understanding the relationship between inflammatory cell death and protein phosphorylation will not only improve our understanding of inflammation and cell death, but will also provide new targets and therapeutic strategies to inflammatory cell death-related diseases (Figure 5).
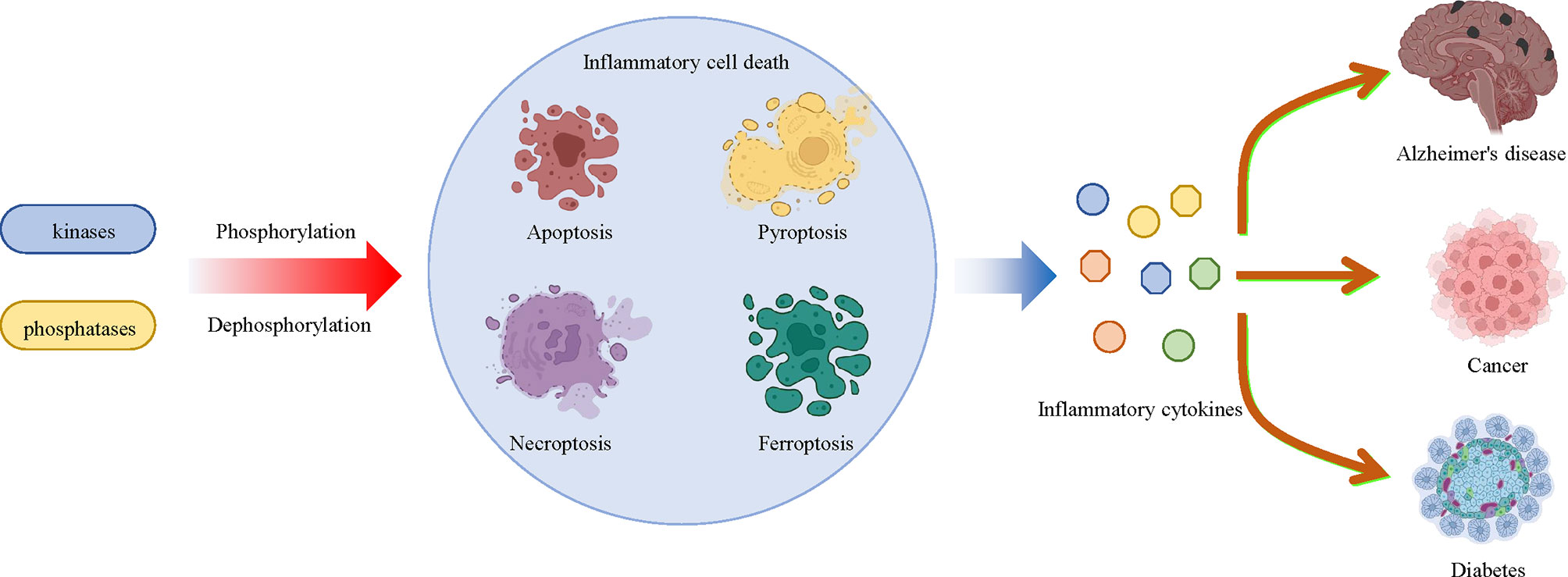
Figure 5 Multiple kinases and phosphatases regulate inflammatory cell death and related diseases through phosphorylation and dephosphorylation modifications. Multiple protein kinases that mediate phosphorylation and phosphatases that mediate dephosphorylation affect the pathologic development of a large number of inflammatory diseases, including diabetes, Alzheimer’s disease and cancer by regulating inflammatory cell death, such as apoptosis, pyroptosis, necroptosis and ferroptosis, and inflammatory cytokine release.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by the Natural Science Foundation of Anhui province (1808085QH244), The Fundamental Research Funds for the Central Universities (WK2070000191, WK9110000037), and the fellowship of China National Postdoctoral Program for Innovative Talents (BX20200325).
References
1. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
2. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
3. Weerasinghe P, Buja LM. Oncosis: An Important Non-Apoptotic Mode of Cell Death. Exp Mol Pathol (2012) 93(3):302–8. doi: 10.1016/j.yexmp.2012.09.018
4. Fuchs Y, Steller H. Programmed Cell Death in Animal Development and Disease. Cell (2011) 147(4):742–58. doi: 10.1016/j.cell.2011.10.033
5. Syntichaki P, Tavernarakis N. Death by Necrosis. Uncontrollable Catastrophe, or Is There Order Behind the Chaos? EMBO Rep (2002) 3(7):604–9. doi: 10.1093/embo-reports/kvf138
6. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in Development, Inflammation and Disease. Nat Rev Mol Cell Biol (2017) 18(2):127–36. doi: 10.1038/nrm.2016.149
7. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
8. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
9. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: Machinery and Regulation. Autophagy (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
10. Bi M, Du X, Jiao Q, Chen X, Jiang H. Expanding the Role of Proteasome Homeostasis in Parkinson's Disease: Beyond Protein Breakdown. Cell Death Dis (2021) 12(2):154. doi: 10.1038/s41419-021-03441-0
11. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis (2020) 11(11):1013. doi: 10.1038/s41419-020-03221-2
12. Vora SM, Lieberman J, Wu H. Inflammasome Activation at the Crux of Severe COVID-19. Nat Rev Immunol (2021) 21(11):694–703. doi: 10.1038/s41577-021-00588-x
13. Xu C, Wu J, Wu Y, Ren Z, Yao Y, Chen G, et al. TNF-α-Dependent Neuronal Necroptosis Regulated in Alzheimer's Disease by Coordination of RIPK1-P62 Complex With Autophagic UVRAG. Theranostics (2021) 11(19):9452–69. doi: 10.7150/thno.62376
14. Chee SEJ, Solito E. The Impact of Ageing on the CNS Immune Response in Alzheimer's Disease. Front Immunol (2021) 12:738511. doi: 10.3389/fimmu.2021.738511
15. Huang Y, Xu W, Zhou R. NLRP3 Inflammasome Activation and Cell Death. Cell Mol Immunol (2021) 18(9):2114–27. doi: 10.1038/s41423-021-00740-6
16. Cohen P. The Origins of Protein Phosphorylation. Nat Cell Biol (2002) 4(5):E127–30. doi: 10.1038/ncb0502-e127
17. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell (2017) 66(5):698–710.e5. doi: 10.1016/j.molcel.2017.05.003
18. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon Prevent TNF-Induced Cell Death by RIPK1 Phosphorylation. Nat Cell Biol (2018) 20(12):1389–99. doi: 10.1038/s41556-018-0229-6
19. Huang Y, Wang H, Hao Y, Lin H, Dong M, Ye J, et al. Myeloid PTEN Promotes Chemotherapy-Induced NLRP3-Inflammasome Activation and Antitumour Immunity. Nat Cell Biol (2020) 22(6):716–27. doi: 10.1038/s41556-020-0510-3
20. Chen W, Wu J, Li L, Zhang Z, Ren J, Liang Y, et al. Ppm1b Negatively Regulates Necroptosis Through Dephosphorylating Rip3. Nat Cell Biol (2015) 17(4):434–44. doi: 10.1038/ncb3120
21. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol (2018) 28(15):2388–99 e5. doi: 10.1016/j.cub.2018.05.094
22. Liccardi G, Ramos Garcia L, Tenev T, Annibaldi A, Legrand AJ, Robertson D, et al. RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Mol Cell (2019) 73(3):413–28 e7. doi: 10.1016/j.molcel.2018.11.010
23. Podder B, Gutta C, Rozanc J, Gerlach E, Feoktistova M, Panayotova-Dimitrova D, et al. TAK1 Suppresses RIPK1-Dependent Cell Death and is Associated With Disease Progression in Melanoma. Cell Death Differ (2019) 26(12):2520–34. doi: 10.1038/s41418-019-0315-8
24. Koppe C, Verheugd P, Gautheron J, Reisinger F, Kreggenwinkel K, Roderburg C, et al. IkappaB Kinasealpha/Beta Control Biliary Homeostasis and Hepatocarcinogenesis in Mice by Phosphorylating the Cell-Death Mediator Receptor-Interacting Protein Kinase 1. Hepatology (2016) 64(4):1217–31. doi: 10.1002/hep.28723
25. Martin SJ. Cell Death and Inflammation: The Case for IL-1 Family Cytokines as the Canonical DAMPs of the Immune System. FEBS J (2016) 283(14):2599–615. doi: 10.1111/febs.13775
26. Anderton H, Wicks IP, Silke J. Cell Death in Chronic Inflammation: Breaking the Cycle to Treat Rheumatic Disease. Nat Rev Rheumatol (2020) 16(9):496–513. doi: 10.1038/s41584-020-0455-8
27. Chen GY, Nuñez G. Sterile Inflammation: Sensing and Reacting to Damage. Nat Rev Immunol (2010) 10(12):826–37. doi: 10.1038/nri2873
28. Arango Duque G, Descoteaux A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front Immunol (2014) 5:491. doi: 10.3389/fimmu.2014.00491
29. Gilroy D, De Maeyer R. New Insights Into the Resolution of Inflammation. Semin Immunol (2015) 27(3):161–8. doi: 10.1016/j.smim.2015.05.003
30. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune network (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
31. Orozco SL, Daniels BP, Yatim N, Messmer MN, Quarato G, Chen-Harris H, et al. RIPK3 Activation Leads to Cytokine Synthesis That Continues After Loss of Cell Membrane Integrity. Cell Rep (2019) 28(9):2275–87.e5. doi: 10.1016/j.celrep.2019.07.077
32. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-Induced Pyroptosis is an Innate Immune Effector Mechanism Against Intracellular Bacteria. Nat Immunol (2010) 11(12):1136–42. doi: 10.1038/ni.1960
33. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
34. Frank D, Vince JE. Pyroptosis Versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death And Differ (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
35. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM Structure of the Gasdermin A3 Membrane Pore. Nature (2018) 557(7703):62–7. doi: 10.1038/s41586-018-0058-6
36. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
37. Linkermann A, Green DR. Necroptosis. N Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
38. Carneiro BA, El-Deiry WS. Targeting Apoptosis in Cancer Therapy. Nat Rev Clin Oncol (2020) 17(7):395–417. doi: 10.1038/s41571-020-0341-y
39. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (2020) 368(6494). doi: 10.1126/science.aaz7548
40. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
41. Koren E, Fuchs Y. Modes of Regulated Cell Death in Cancer. Cancer Discovery (2021) 11(2):245–65. doi: 10.1158/2159-8290.Cd-20-0789
42. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
43. Pasparakis M, Vandenabeele P. Necroptosis and Its Role in Inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
44. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and Tumor Progression. Trends Cancer (2021) 8(1):21–27. doi: 10.1016/j.trecan.2021.09.003
45. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell (2020) 180(6):1115–29.e13. doi: 10.1016/j.cell.2020.02.050
46. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: A Crucial Pathogenic Mediator of Human Disease. JCI Insight (2019) 8(12):e128834. doi: 10.1172/jci.insight.128834
47. Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, et al. BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1β Maturation in Macrophages. Cell Rep (2018) 25(9):2354–68.e5. doi: 10.1016/j.celrep.2018.10.087
48. Wu YH, Kuo WC, Wu YJ, Yang KT, Chen ST, Jiang ST, et al. Participation of C-FLIP in NLRP3 and AIM2 Inflammasome Activation. Cell Death differ (2014) 21(3):451–61. doi: 10.1038/cdd.2013.165
49. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma Membrane Translocation of Trimerized MLKL Protein is Required for TNF-Induced Necroptosis. Nat Cell Biol (2014) 16(1):55–65. doi: 10.1038/ncb2883
50. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 Mediates the Embryonic Lethality of Caspase-8-Deficient Mice. Nature (2011) 471(7338):368–72. doi: 10.1038/nature09857
51. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed Lineage Kinase Domain-Like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
52. Garnish SE, Meng Y, Koide A, Sandow JJ, Denbaum E, Jacobsen AV, et al. Conformational Interconversion of MLKL and Disengagement From RIPK3 Precede Cell Death by Necroptosis. Nat Commun (2021) 12(1):2211. doi: 10.1038/s41467-021-22400-z
53. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed Lineage Kinase Domain-Like Protein MLKL Causes Necrotic Membrane Disruption Upon Phosphorylation by RIP3. Mol Cell (2014) 54(1):133–46. doi: 10.1016/j.molcel.2014.03.003
54. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the Pseudokinase MLKL Unleashes the Four-Helix Bundle Domain to Induce Membrane Localization and Necroptotic Cell Death. Proc Natl Acad Sci USA (2014) 111(42):15072–7. doi: 10.1073/pnas.1408987111
55. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of Mixed Lineage Kinase Domain-Like Protein to Plasma Membrane Leads to Necrotic Cell Death. Cell Res (2014) 24(1):105–21. doi: 10.1038/cr.2013.171
56. Zhou W, Yuan J. SnapShot: Necroptosis. Cell (2014) 158(2):464–.e1. doi: 10.1016/j.cell.2014.06.041
57. Liu S, Wang X, Li Y, Xu L, Yu X, Ge L, et al. Necroptosis Mediates TNF-Induced Toxicity of Hippocampal Neurons. BioMed Res Int (2014) 2014:290182. doi: 10.1155/2014/290182
58. Micheau O, Tschopp J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell (2003) 114(2):181–90. doi: 10.1016/s0092-8674(03)00521-x
59. Amin P, Florez M, Najafov A, Pan H, Geng J, Ofengeim D, et al. Regulation of a Distinct Activated RIPK1 Intermediate Bridging Complex I and Complex II in Tnfα-Mediated Apoptosis. Proc Natl Acad Sci USA (2018) 115(26):E5944–e53. doi: 10.1073/pnas.1806973115
60. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and Ikkϵ Prevent TNF-Induced Cell Death by RIPK1 Phosphorylation. Nat Cell Biol (2018) 20(12):1389–99. doi: 10.1038/s41556-018-0229-6
61. Silke J, Brink R. Regulation of TNFRSF and Innate Immune Signalling Complexes by TRAFs and cIAPs. Cell Death Differ (2010) 17(1):35–45. doi: 10.1038/cdd.2009.114
62. Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-κb-Independent Role of Ikkα/Ikkβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death During TNF Signaling. Mol Cell (2015) 60(1):63–76. doi: 10.1016/j.molcel.2015.07.032
63. Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 Phosphorylation Inhibits RIPK1 Kinase-Dependent Cell Death in Models of Infection and Inflammation. Nat Commun (2019) 10(1):1729. doi: 10.1038/s41467-019-09690-0
64. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a Signaling Platform That Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell (2011) 43(3):432–48. doi: 10.1016/j.molcel.2011.06.006
65. Wu W, Liu P, Li J. Necroptosis: An Emerging Form of Programmed Cell Death. Crit Rev oncol/hematol (2012) 82(3):249–58. doi: 10.1016/j.critrevonc.2011.08.004
66. Sun L, Wang X. A New Kind of Cell Suicide: Mechanisms and Functions of Programmed Necrosis. Trends Biochem Sci (2014) 39(12):587–93. doi: 10.1016/j.tibs.2014.10.003
67. Seo J, Kim MW, Bae KH, Lee SC, Song J, Lee EW. The Roles of Ubiquitination in Extrinsic Cell Death Pathways and Its Implications for Therapeutics. Biochem Pharmacol (2019) 162:21–40. doi: 10.1016/j.bcp.2018.11.012
68. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, et al. Characterization of RIPK3-Mediated Phosphorylation of the Activation Loop of MLKL During Necroptosis. Cell Death Differ (2016) 23(1):76–88. doi: 10.1038/cdd.2015.70
69. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity (2013) 39(3):443–53. doi: 10.1016/j.immuni.2013.06.018
70. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation During Development and in Aging. Cell (2018) 174(6):1477–91.e19. doi: 10.1016/j.cell.2018.07.041
71. Menon MB, Gropengießer J, Fischer J, Novikova L, Deuretzbacher A, Lafera J, et al. P38(MAPK)/MK2-Dependent Phosphorylation Controls Cytotoxic RIPK1 Signalling in Inflammation and Infection. Nat Cell Biol (2017) 19(10):1248–59. doi: 10.1038/ncb3614
72. Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, et al. MK2 Phosphorylation of RIPK1 Regulates TNF-Mediated Cell Death. Nat Cell Biol (2017) 19(10):1237–47. doi: 10.1038/ncb3608
73. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat Commun (2017) 8(1):359. doi: 10.1038/s41467-017-00406-w
74. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat Chem Biol (2008) 4(5):313–21. doi: 10.1038/nchembio.83
75. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 Autophosphorylation is Promoted by Mitochondrial ROS and is Essential for RIP3 Recruitment Into Necrosome. Nat Commun (2017) 8:14329. doi: 10.1038/ncomms14329
76. Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural Insights Into RIP3-Mediated Necroptotic Signaling. Cell Rep (2013) 5(1):70–8. doi: 10.1016/j.celrep.2013.08.044
77. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The Ubiquitin-Modifying Enzyme A20 Restricts Ubiquitination of the Kinase RIPK3 and Protects Cells From Necroptosis. Nat Immunol (2015) 16(6):618–27. doi: 10.1038/ni.3172
78. Lee SB, Kim JJ, Han SA, Fan Y, Guo LS, Aziz K, et al. The AMPK-Parkin Axis Negatively Regulates Necroptosis and Tumorigenesis by Inhibiting the Necrosome. Nat Cell Biol (2019) 21(8):940–51. doi: 10.1038/s41556-019-0356-8
79. Wang Z, Jiang H, Chen S, Du F, Wang X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell (2012) 148(1-2):228–43. doi: 10.1016/j.cell.2011.11.030
80. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational Switching of the Pseudokinase Domain Promotes Human MLKL Tetramerization and Cell Death by Necroptosis. Nat Commun (2018) 9(1):2422. doi: 10.1038/s41467-018-04714-7
81. Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A Plug Release Mechanism for Membrane Permeation by MLKL. Struct (London Engl 1993) (2014) 22(10):1489–500. doi: 10.1016/j.str.2014.07.014
82. Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C, et al. Necroptosis Signalling is Tuned by Phosphorylation of MLKL Residues Outside the Pseudokinase Domain Activation Loop. Biochem J (2015) 471(2):255–65. doi: 10.1042/bj20150678
83. Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol Cell (2019) 75(3):457–68.e4. doi: 10.1016/j.molcel.2019.05.022
84. Shi CS, Kehrl JH. Bcl-2 Regulates Pyroptosis and Necroptosis by Targeting BH3-Like Domains in GSDMD and MLKL. Cell Death Discovery (2019) 5:151. doi: 10.1038/s41420-019-0230-2
85. Seo J, Seong D, Nam YW, Hwang CH, Lee SR, Lee CS, et al. Beclin 1 Functions as a Negative Modulator of MLKL Oligomerisation by Integrating Into the Necrosome Complex. Cell Death differ (2020) 27(11):3065–81. doi: 10.1038/s41418-020-0561-9
86. Chinnaiyan AM. The Apoptosome: Heart and Soul of the Cell Death Machine. Neoplasia (1999) 1(1):5–15. doi: 10.1038/sj.neo.7900003
88. Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential Role of the Mitochondrial Apoptosis-Inducing Factor in Programmed Cell Death. Nature (2001) 410(6828):549–54. doi: 10.1038/35069004
89. Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity (2020) 52(6):978–93.e6. doi: 10.1016/j.immuni.2020.04.002
90. Fleten KG, Flørenes VA, Prasmickaite L, Hill O, Sykora J, Mælandsmo GM, et al. hvTRA, a Novel TRAIL Receptor Agonist, Induces Apoptosis and Sustained Growth Retardation in Melanoma. Cell Death Discov (2016) 2:16081. doi: 10.1038/cddiscovery.2016.81
91. Wajant H. The Fas Signaling Pathway: More Than a Paradigm. Science (2002) 296(5573):1635–6. doi: 10.1126/science.1071553
92. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell (2001) 104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9
93. Bonora M, Pinton P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front Oncol (2014) 4:302. doi: 10.3389/fonc.2014.00302
94. Kallenberger SM, Beaudouin J, Claus J, Fischer C, Sorger PK, Legewie S, et al. Intra- and Interdimeric Caspase-8 Self-Cleavage Controls Strength and Timing of CD95-Induced Apoptosis. Sci Signaling (2014) 7(316):ra23. doi: 10.1126/scisignal.2004738
95. Oberst A, Pop C, Tremblay AG, Blais V, Denault JB, Salvesen GS, et al. Inducible Dimerization and Inducible Cleavage Reveal a Requirement for Both Processes in Caspase-8 Activation. J Biol Chem (2010) 285(22):16632–42. doi: 10.1074/jbc.M109.095083
96. Hague A, Paraskeva C. Apoptosis and Disease: A Matter of Cell Fate. Cell Death Differ (2004) 11(12):1366–72. doi: 10.1038/sj.cdd.4401497
97. Dhuriya YK, Sharma D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J Neuroinflamm (2018) 15(1):199. doi: 10.1186/s12974-018-1235-0
98. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 Complexes With RIP3 to Mediate Virus-Induced Programmed Necrosis That is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe (2012) 11(3):290–7. doi: 10.1016/j.chom.2012.01.016
99. Vanden Berghe T, Hassannia B, Vandenabeele P. An Outline of Necrosome Triggers. Cell Mol Life Sci (2016) 73(11-12):2137–52. doi: 10.1007/s00018-016-2189-y
100. Radu M, Chernoff J. The DeMSTification of Mammalian Ste20 Kinases. Curr Biol (2009) 19(10):R421–5. doi: 10.1016/j.cub.2009.04.022
101. Stetler RA, Gao Y, Zhang L, Weng Z, Zhang F, Hu X, et al. Phosphorylation of HSP27 by Protein Kinase D is Essential for Mediating Neuroprotection Against Ischemic Neuronal Injury. J Neurosci (2012) 32(8):2667–82. doi: 10.1523/jneurosci.5169-11.2012
102. Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF. Functional Interplay Between Caspase Cleavage and Phosphorylation Sculpts the Apoptotic Proteome. Cell (2012) 150(2):426–40. doi: 10.1016/j.cell.2012.05.040
103. Shahbazi J, Lock R, Liu T. Tumor Protein 53-Induced Nuclear Protein 1 Enhances P53 Function and Represses Tumorigenesis. Front Genet (2013) 4:80. doi: 10.3389/fgene.2013.00080
104. Cookson BT, Brennan MA. Pro-Inflammatory Programmed Cell Death. Trends Microbiol (2001) 9(3):113–4. doi: 10.1016/s0966-842x(00)01936-3
105. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
106. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
107. Jorgensen I, Rayamajhi M, Miao EA. Programmed Cell Death as a Defence Against Infection. Nat Rev Immunol (2017) 17(3):151–64. doi: 10.1038/nri.2016.147
108. McKenzie BA, Dixit VM, Power C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci (2020) 43(1):55–73. doi: 10.1016/j.tins.2019.11.005
109. Jorgensen I, Miao EA. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265(1):130–42. doi: 10.1111/imr.12287
110. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
111. Thurston TL, Matthews SA, Jennings E, Alix E, Shao F, Shenoy AR, et al. Growth Inhibition of Cytosolic Salmonella by Caspase-1 and Caspase-11 Precedes Host Cell Death. Nat Commun (2016) 7:13292. doi: 10.1038/ncomms13292
112. Yang Q, Stevenson HL, Scott MJ, Ismail N. Type I Interferon Contributes to Noncanonical Inflammasome Activation, Mediates Immunopathology, and Impairs Protective Immunity During Fatal Infection With Lipopolysaccharide-Negative Ehrlichiae. Am J Pathol (2015) 185(2):446–61. doi: 10.1016/j.ajpath.2014.10.005
113. Broz P, Pelegrín P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
114. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep (2019) 27(12):3646–56.e5. doi: 10.1016/j.celrep.2019.05.065
115. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-Inflammatory Effects by Promoting Neutrophil Death. Cell Rep (2018) 22(11):2924–36. doi: 10.1016/j.celrep.2018.02.067
116. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
117. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation During Apoptosis and Inflammasome Activation. Nat Commun (2019) 10(1):1689. doi: 10.1038/s41467-019-09397-2
118. Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, et al. The Plk1-Dependent Phosphoproteome of the Early Mitotic Spindle. Mol Cell Proteomics MCP (2011) 10(1):M110.004457. doi: 10.1074/mcp.M110.004457
119. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
120. Gong YN, Wang X, Wang J, Yang Z, Li S, Yang J, et al. Chemical Probing Reveals Insights Into the Signaling Mechanism of Inflammasome Activation. Cell Res (2010) 20(12):1289–305. doi: 10.1038/cr.2010.135
121. Ray PD, Huang BW, Tsuji Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell Signalling (2012) 24(5):981–90. doi: 10.1016/j.cellsig.2012.01.008
122. Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and Cancer: Recent Insights. Ann N Y Acad Sci (2016) 1368(1):149–61. doi: 10.1111/nyas.13008
123. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem Sci (2016) 41(3):274–86. doi: 10.1016/j.tibs.2015.11.012
124. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
125. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: Mechanisms and Links With Diseases. Signal Transduction Targeted Ther (2021) 6(1):49. doi: 10.1038/s41392-020-00428-9
126. Doll S, Conrad M. Iron and Ferroptosis: A Still Ill-Defined Liaison. IUBMB Life (2017) 69(6):423–34. doi: 10.1002/iub.1616
127. Wei X, Yi X, Zhu XH, Jiang DS. Posttranslational Modifications in Ferroptosis. Oxid Med Cell Longevity (2020) 2020:8832043. doi: 10.1155/2020/8832043
128. Chen L, Liu S, Tao Y. Regulating Tumor Suppressor Genes: Post-Translational Modifications. Signal Transduction Targeted Ther (2020) 5(1):90. doi: 10.1038/s41392-020-0196-9
129. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat Cell Biol (2020) 22(2):225–34. doi: 10.1038/s41556-020-0461-8
130. Hardie DG, Ross FA, Hawley SA. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat Rev Mol Cell Biol (2012) 13(4):251–62. doi: 10.1038/nrm3311
131. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(-) Activity. Curr Biol (2018) 28(15):2388–99.e5. doi: 10.1016/j.cub.2018.05.094
132. Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a New Driver of Ferroptosis. Autophagy (2018) 14(12):2173–5. doi: 10.1080/15548627.2018.1513758
133. Wang Y, Peng X, Zhang M, Jia Y, Yu B, Tian J. Revisiting Tumors and the Cardiovascular System: Mechanistic Intersections and Divergences in Ferroptosis. Oxid Med Cell Longevity (2020) 2020:9738143. doi: 10.1155/2020/9738143
134. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a Type of Autophagy-Dependent Cell Death. Semin Cancer Biol (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
135. Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W, et al. The Emerging Role of Toll-Like Receptor 4 in Myocardial Inflammation. Cell Death Dis (2016) 7(5):e2234. doi: 10.1038/cddis.2016.140
136. Oh BM, Lee SJ, Park GL, Hwang YS, Lim J, Park ES, et al. Erastin Inhibits Septic Shock and Inflammatory Gene Expression via Suppression of the NF-κb Pathway. J Clin Med (2019) 8(12):2210. doi: 10.3390/jcm8122210
137. Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang J, et al. Ferroptosis Involves in Intestinal Epithelial Cell Death in Ulcerative Colitis. Cell Death Dis (2020) 11(2):86. doi: 10.1038/s41419-020-2299-1
138. Li X, Zou Y, Xing J, Fu YY, Wang KY, Wan PZ, et al. Pretreatment With Roxadustat (FG-4592) Attenuates Folic Acid-Induced Kidney Injury Through Antiferroptosis via Akt/GSK-3β/Nrf2 Pathway. Oxid Med Cell Longevity (2020) 2020:6286984. doi: 10.1155/2020/6286984
139. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene (2015) 34(45):5617–25. doi: 10.1038/onc.2015.32
140. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione Peroxidase 4-Regulated Neutrophil Ferroptosis Induces Systemic Autoimmunity. Nat Immunol (2021) 22(9):1107–17. doi: 10.1038/s41590-021-00993-3
Keywords: phosphorylation, pyroptosis, apoptosis, necroptosis, ferroptosis
Citation: Xu W and Huang Y (2022) Regulation of Inflammatory Cell Death by Phosphorylation. Front. Immunol. 13:851169. doi: 10.3389/fimmu.2022.851169
Received: 09 January 2022; Accepted: 09 February 2022;
Published: 01 March 2022.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaReviewed by:
Atsushi Koike, Osaka University of Pharmaceutical Sciences, JapanShankargouda Patil, Jazan University, Saudi Arabia
Divakar Sharma, University of Delhi, India
Copyright © 2022 Xu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Huang, SFk1MjdAamlhbmduYW4uZWR1LmNu