 Lisa Hirahara†
Lisa Hirahara† Kaoru Takase-Minegishi†
Kaoru Takase-Minegishi† Yohei Kirino*
Yohei Kirino* Yuki Iizuka-IribeYutaro Soejima
Yuki Iizuka-IribeYutaro Soejima Ryusuke Yoshimi
Ryusuke Yoshimi Hideaki Nakajima
Hideaki Nakajima- Department of Stem Cell and Immune Regulation, Yokohama City University Graduate School of Medicine, Yokohama, Japan
Behçet’s disease (BD) is a systemic inflammatory disease characterized by recurrent oral ulcers, genital ulcers, cutaneous inflammation, and uveitis. In addition, other potentially life-threatening lesions may occur in the intestinal tract, blood vessels, and central nervous system. This heterogeneity of the BD phenotype hampers development of a targeted treatment strategy. The pathogenesis of BD is not fully elucidated, but it is likely that genetically susceptible people develop BD in response to environmental factors, such as microbiome factors. Genetic analyses have identified various BD susceptibility loci that function in HLA-antigen presentation pathways, Th1 and Th17 cells, and autoinflammation related to monocytes/macrophages, or that increase levels of pro-inflammatory cytokines, reduce levels of anti-inflammatory cytokines, or act in dysfunctional mucous barriers. Our functional analyses have revealed that impairment of M2 monocyte/macrophage-mediated anti-inflammatory function through IL-10 is crucial to BD pathogenesis. We, therefore, propose that BD is an M1-dominant disease. In this review, we describe the roles of monocytes and macrophages in BD and consider the potential of these cells as therapeutic targets.
Introduction
Behçet’s disease (BD) is a systemic inflammatory disease initially reported in 1937 by Hulusi Behçet, a prominent Turkish dermatologist. The disease is epidemiologically characterized by a high incidence along the ancient Silk Route, and Professor Shigeaki Ohno has named the disease “Silk Road Disease” (1). The typical manifestations of BD are recurrent oral ulcers, genital ulcers, cutaneous inflammation, and uveitis. However, other potentially life-threatening lesions may occur in the intestinal tract, blood vessels, and central nervous system and this heterogeneity results in the disease being considered a syndrome (2). There are currently no disease-specific antibodies or biomarkers for BD; therefore, diagnosis is made solely on clinical symptoms. The most important symptom is recurrent oral ulcers, which is seen in more than 95% of Japanese patients and is mandatory for diagnosis according to the International Study Group criteria (3, 4). The pathogenesis of BD is not fully determined, but it is likely that people who are genetically susceptible to the disease may develop BD as a response to environmental factors, such as microbiome factors. HLA-B*51 is the most widely known BD susceptibility gene, with an odds ratio of 5.9 for the development of BD, but its allele frequency is about 20% in the Japanese population and is, therefore, not a disease-causative locus (5). Genetic analyses have identified various susceptibility loci involved in HLA-antigen presentation pathways, Th1 and Th17 cells, pro-inflammatory cytokine regulation, dysfunctional mucous barriers, and autoinflammation related to monocytes/macrophages, the main focus of this review. Our research goal is to categorize the complexity of BD and to establish treatment strategies that are tailored to individual patients. In this respect, macrophages and monocytes could be important targets. In this review, we discuss the roles of monocytes and macrophages based on the results of a recent literature survey and how monocyte-macrophages may be considered as therapeutic targets of BD.
Elucidation of Monocyte Function
Neutrophil Hyperchemotaxis and Monocytes in BD
Early studies of the involvement of monocytes and macrophages in BD focused on their function as part of the pathogenesis of enhanced neutrophil chemotaxis in response to environmental factors. Aberrant neutrophil chemotaxis in BD was reported in 1975 and colchicine was found to exert a therapeutic effect by inhibiting neutrophil migration (6). Monocyte involvement in the mechanism of neutrophil chemotaxis was then investigated. In a report from 1993, monocytes from patients with active BD displayed increased secretion of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-8 (7). Then, in 1995, increased levels of the soluble monocyte-activation marker, CD14, were reported in the sera of BD patients, and monocyte culture supernatants from BD patients were shown to significantly enhance neutrophil adhesion to endothelial cells (8). These results indicated that monocytes in BD patients are highly activated and involved in chronic inflammation by continuous production of pro-inflammatory cytokines. Histopathological examination then revealed that the cells infiltrating oral ulcer sites in BD patients were lymphocytes and monocytes/macrophages (9). Furthermore, mononuclear cells consisting of CD4+ T cells and monocytes were demonstrated to infiltrate the periphery of small blood vessels at the site of the pathergy reaction, suggesting that monocytes are the essential driver of inflammation in BD (10).
Activation of the Innate Immune System via Toll-Like Receptors
Involvement of monocytes in the pathogenesis of BD was further strengthened by the discovery of the pattern recognition mechanism of Toll-like receptors (TLRs). TLRs are a defense mechanism against pathogens-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). Microbes have long been assumed to be an external factor triggering BD inflammation. In fact, Hulusi Behçet reported that BD is induced by herpes virus infection, and herpes simplex-induced animal models elicit BD-like symptoms (11). Herpes viruses replicate in monocytes and can be detected by TLR2 and TLR9, which are highly expressed in monocytes.
Oral commensal bacteria are another candidate risk factor for BD. It is hypothesized that streptococci are particularly prevalent in BD because oral ulcers often worsen after dental treatment (12). Heat shock protein (HSP) 65 produced by these bacteria is homologous to human HSP60, and hyper-reactivity of lymphocytes to HSP has been reported in BD patients. Ten human TLRs have been identified and peptidoglycan (PTG) is a ligand for TLR2 and lipopolysaccharide (LPS) is a ligand for TLR4. Furthermore, HSP60 can bind to several PAMPs and induces cytokine production via TLR2 and TLR4 signaling, indicating that TLR2 and TLR4 are involved in the pathogenesis of BD (13).
In 2008, we reported the overexpression of TLR4 in peripheral blood mononuclear cells (PBMCs) of BD patients (14). Subsequently, it was reported that TLR2/TLR4 expression was also increased in monocytes from BD patients (15). In 2013, upregulated expression of TLR2/TLR4 was found in macrophages isolated from BD patients, and that TLR2/TLR4-mediated IL-1β was upregulated in patients with active uveitis when stimulated with peptidoglycan/LPS (16). These results indicate that monocytes are involved in BD pathogenesis in part by activating the innate immune system against external stimuli via the TLR pathway. Concordant with these observations, a targeted resequencing of innate-immune genes in Japanese and Turkish populations identified low-frequency TLR4 variants associated with BD, supporting the hypothesis of innate immune system activation through TLRs in BD (17).
Functions of Monocytes and Macrophages Revealed by Genetic Studies
Since 2010, genome-wide association studies (GWAS) and other large-scale genetics studies have been conducted to explore BD. These investigations confirmed that the HLA region has the highest association with BD development. HLA-B*51, A*26, B*15, B*27, and B*57 were identified as disease-susceptibility alleles, and A*03 and B*49 were identified as disease-protective alleles (18). In addition to HLA genes, nearly 20 disease-susceptibility loci were identified, including IL10, IL23R, and ERAP1, which contributed to the proposal of the disease concept of “MHC class-I-opathy”, similar to spondyloarthritis (Table 1) (19–21, 23). BD-related loci that may affect monocyte and macrophage function include IL10, CCR1–CCR3, MEFV, IL1B, IRF8, and most recently, IFNGR1 (27, 28). The pathways associated with BD are summarized in Figure 1.

Table 1 Genome-wide significant disease susceptibility loci for BD.
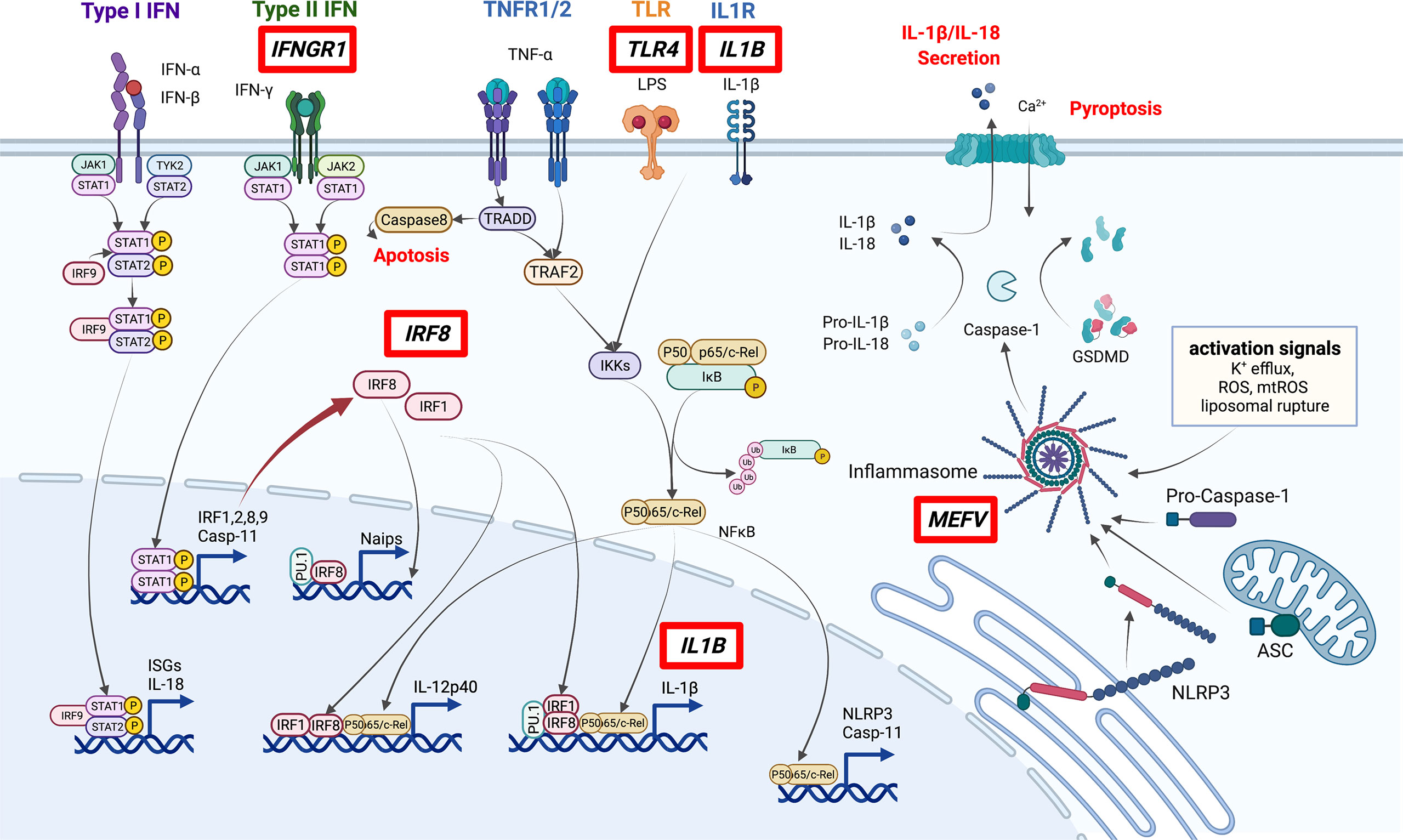
Figure 1 Pathways involved in the pathogenesis of BD. Genes discovered by GWAS to be associated with BD (red boxes) include several involved in macrophage inflammation. In particular, IRF8 and MEFV are important in the regulation of STAT signaling and inflammasome activation. ISGs: Interferon-stimulated genes. This figure was created with BioRender.com.
Elucidation and Interpretation of CCR1 Function
An important finding in understanding the function of monocytes and macrophages in the pathogenesis of BD was identification of the CCR1–CCR3 locus. CCR1 encodes CC-motif chemokine receptor1(CCR1), which is highly expressed on monocytes/macrophages, whereas CCR3 is highly expressed on eosinophils, basophils, and T cells. The ligands for CCR1 are macrophage inflammatory protein-1 alpha (MIP-1α), regulated on activation normal T expressed and secreted protein (RANTES), and monocyte chemoattractant protein 3 (MCP3). Upon detection of these chemokines, monocytes migrate to the site of high chemokine production (29). GWAS identified a locus tagged with SNP rs7616215 located in the 3′ non-cording region of CCR1, and functional analysis revealed that risk allele T was associated with reduced expression of CCR1. Furthermore, the migratory ability of monocytes in response to MIP-1α was lower in individuals with risk allele T (23). These results indicate that monocyte chemotaxis is reduced in patients with BD, in contrast to increased monocyte infiltration of lesions. Inconsistent with our GWAS findings, systematic expression quantitative trait analysis with various cell subsets indicated that rs7616215 affects CCR3 more than CCR1, resulting in higher CCR3 expression (30). However, a recent large GWAS on canker sores, a refractory form of which is considered a “Behçet-spectrum disorder” (31), confirmed association between CCR1-CCR3, and the Genotype-Tissue expression of CCR1 and CCR3 were down-regulated and up-regulated, respectively, indicating that the locus has a binary effect on CCR1 and CCR3 expression (32). In addition, a recent single-cell whole-genome expression quantitative trait analysis (33) showed consistent results that the risk allele T is associated with decreased expression of CCR1 and increased expression of CCR3 in classical monocytes as well as neutrophils and plasmacytoid dendritic cells (Supplementary Figure 2).
Abnormal Function of Polarized Macrophages in the Context of IL10
Macrophage polarization has attracted much attention in recent years. M1 macrophages secrete pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, which are therapeutic targets of BD, while M2 macrophages secrete anti-inflammatory cytokine represented by IL-10 (34). M2 can be further classified into four subtypes, M2a, M2b, M2c and M2d. Each subtype is characterized by cell surface markers, secreted cytokines and function, but all subtypes secrete IL-10 (35). IL-10 receptor deficiency is known to cause severe inflammation in skin and intestines, symptoms that partially overlap with BD (36). GWAS showed association of BD with IL10 intronic variant, rs1518111, in both Japanese and Turkish populations (20, 21). IL10 mRNA and protein levels were reduced in monocytes of individuals with risk alleles compared with those in individuals without risk alleles (21). As mentioned earlier, CCR1–CCR3 SNPs are associated with BD, and reduced expression of CCR1 is associated with disease risk.
We addressed how decreased monocyte migration ability revealed by GWAS is related to the pathogenesis of BD by analyzing the polarization of M1 and M2 macrophages (37). Expression of IL10 and CCR1 was generally higher in M2 than M1 macrophages, and CCR1 expression in M1 macrophages was higher in BD patients than in healthy controls. We also found significant infiltration of M1 macrophages in erythema nodosum lesions of BD patients. CCR1 SNP, rs7616215, is a BD-risk allele associated with reduced M2 migration in response to MIP-1α. These results suggest that M2 macrophage infiltration capacity is lower in BD than in healthy subjects, and that fewer M2 cells and BD-associated SNPs may result in lower levels of IL-10, resulting in M1-predominant inflammation in BD. As mentioned above, recent quantitative trait analysis of single cell expression revealed that CCR1 has a role in the migration of various immune cells, and that not only monocytes but also neutrophils and acquired immune responses are affected by this CCR1-CCR3 locus (Supplementary Figure 2). The crosstalk between polarized macrophages and neutrophils and acquired immune cells is not fully understood, but the interaction may be important for inflammation in BD.
In line with our findings, macrophages collected from the peritoneal fluid of herpes simplex virus-induced BD model mice were predominantly M1 (38). Macrophages collected from healthy individuals treated with serum from BD patients promoted differentiation into M1 macrophages, while differentiation into M2 macrophages was suppressed (39). We also reported that the production of heme oxygenase 1, an anti-inflammatory heme degrading enzyme, is high in M2 macrophages but reduced in BD patients in response to TLR4 stimulation (14). Together these results support the hypothesis of impaired M2 function in BD (Figure 2).
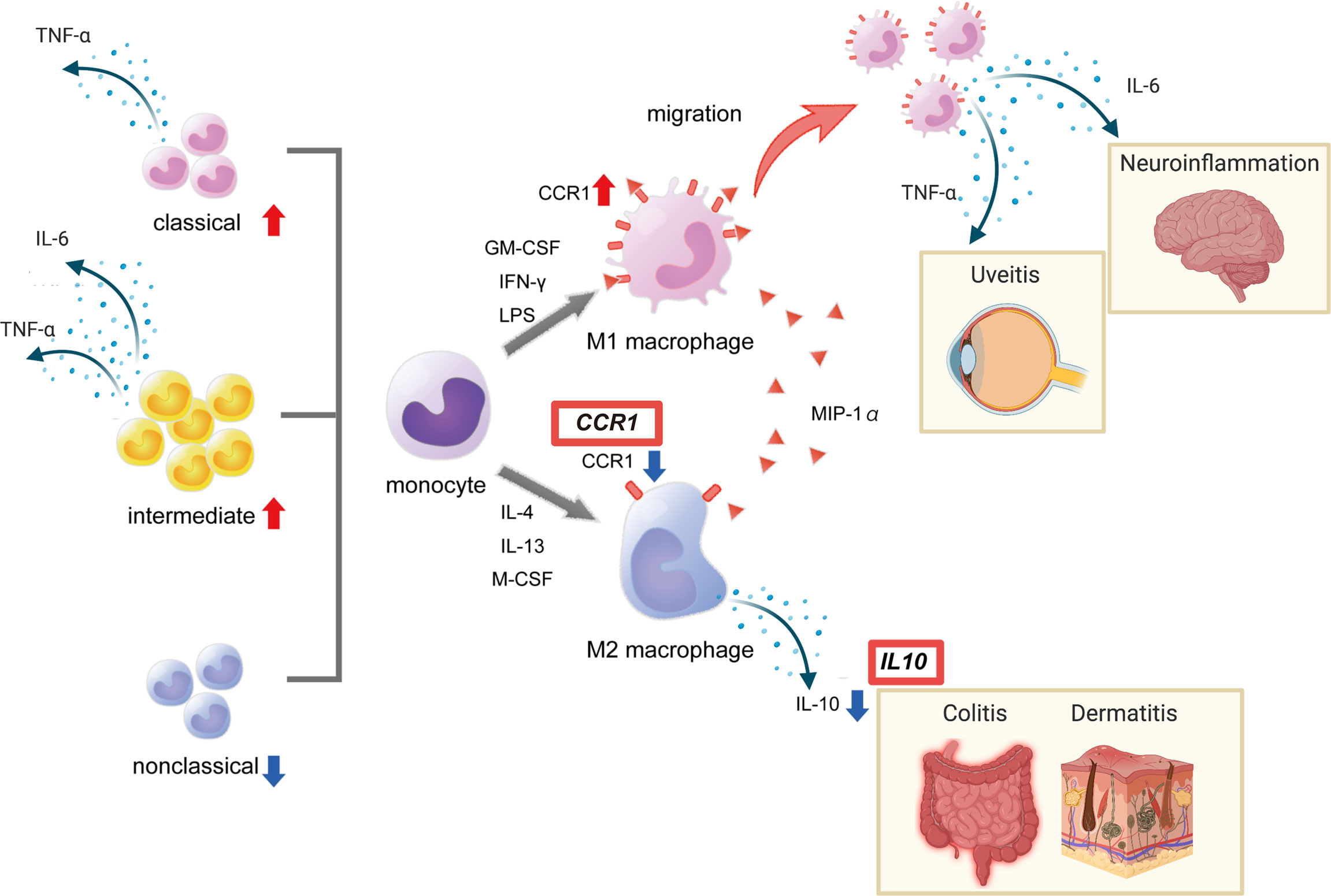
Figure 2 Red boxes show disease susceptibility genes for BD associated with this pathway. This figure was created with BioRender.com.
Importantly, anti-inflammatory M2 macrophages may not be consistently anti-inflammatory. We previously tested whether established M1 macrophages could be converted to M2 macrophages in human primary cells (37). For this purpose, M1 and M2 macrophages that underwent 9 days of in vitro polarization in response to GM-CSF or M-CSF were cultured for another 9 days in the presence of the same or other cytokines. Treatment with M-CSF restored the expression of IL10 (Figure 3). Conversely, the expression of IL10 mRNA was decreased by GM-CSF in M2 macrophages. IL6 mRNA also showed changes in this experiment. These results suggest that the macrophage phenotype is partially interchangeable between M1 and M2, depending on situational factors, including cytokines.
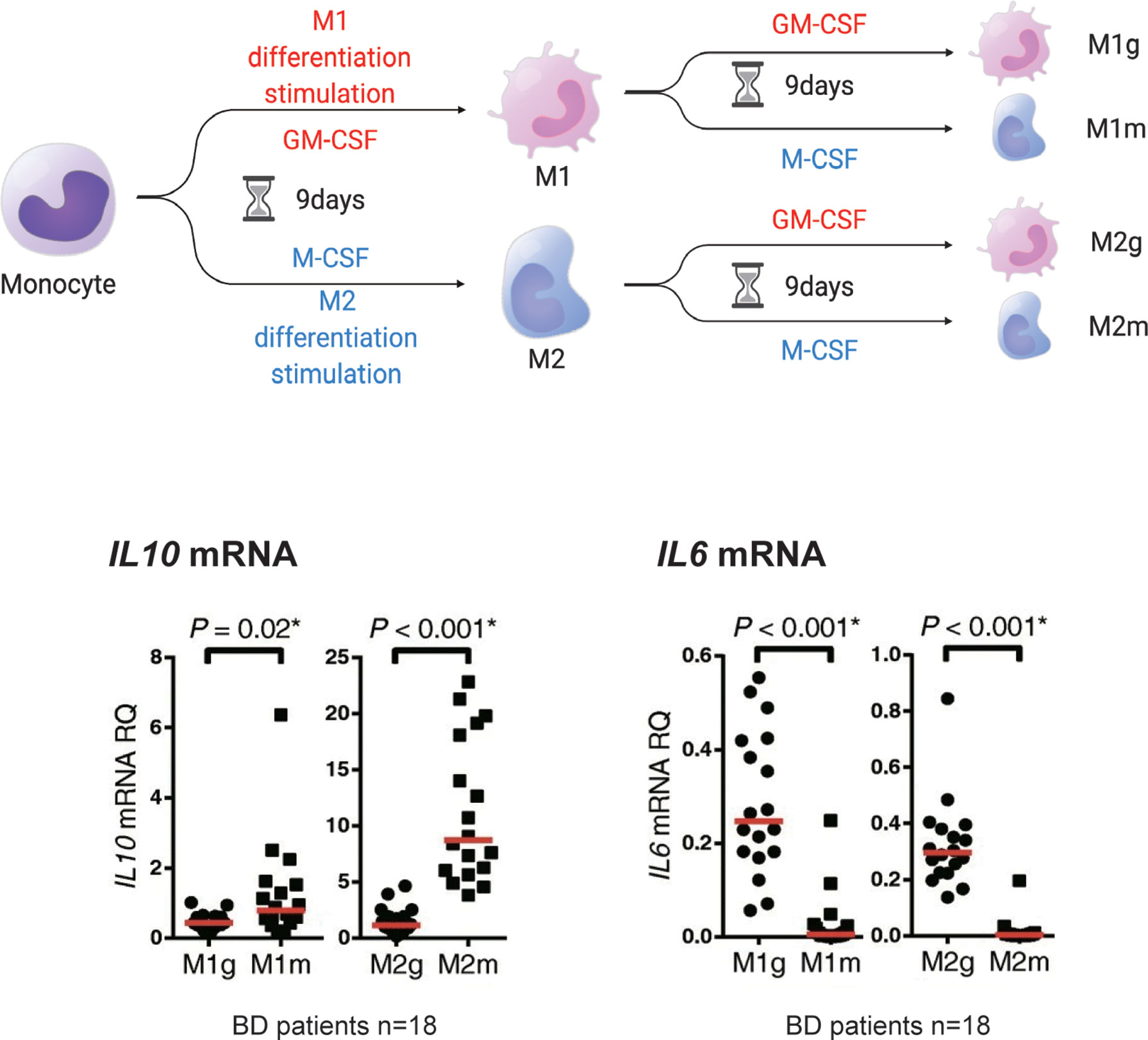
Figure 3 M1 and M2 macrophages may be interchangeable. Monocytes (mono) treated in vitro with GM-CSF (M1g and M2g) or M-CSF (M1m and M2m) for 9 days developed an M1 or M2 macrophage-like phenotype (defined here as M1 or M2 stimulation). However, the M2 macrophages stimulated with “M2” or “M1” stimulation for another 9 days (the first M1 stimulus followed by M2 stimulus is defined as “M2g” and the second M1 stimulus followed by M2 stimulus is defined as “M1m”) maintained or decreased IL10 mRNA production. IL6 mRNA production was reduced by M2 stimulation of M1 macrophages. This figure is a modified version of Figure 4 a-e published in Nakano et al. (37). The figure is modified and distributed under the Creative Commons Attribution 4.0 International License (CCBY4.0, http://creativecommons.org/licenses/by/4.0/). This figure was partially created by BioRender.com.
Aberrant Monocyte Subsets in BD Patients
Interestingly, several recent reports have shown that not only macrophages but also monocyte subsets are imbalanced in BD. Based on the expression of molecules on the cell surface, monocytes can be classified into three categories: classical (CD14++CD16-), intermediate (CD14++CD16+), and non-classical (CD14+CD16++) (40). These three subtypes are also considered to be functionally distinct. Classical monocyte represents the majority of circulating monocytes. In general, classical monocytes have a pro-inflammatory and phagocytic phenotype. They express high level of CCR2, migrate in response to CCL2(MCP-1) stimulation and are involved in initiation of inflammation. In contrast, non-classical monocytes that patrol the vascular endothelium to survey for damage. Particularly in the area of vascular diseases, they have been focused on as intravascular scavenger that remove cellular debris from blood vessels (41, 42). They produce less reactive oxygen species (ROS) and cytokines in response to stimulation with cell surface TLRs, but produce pro-inflammatory cytokines in response to stimulation with viruses and immune complexes containing nucleic acids (43). Although the functions of intermediate monocytes are not fully understood, they are involved in ROS production and highly express genes related to antigen presentation, cytokine production, apoptosis control, T cell proliferation and cell differentiation (44, 45).
Increased numbers of classical monocytes and decreased numbers of nonclassical monocytes have been reported in BD patients with uveitis (46). In addition, monocytes from BD patients and healthy controls differ in TLR expression by subset, with classical monocytes expressing TLR2, TLR4, and TLR5, nonclassical monocytes expressing TLR2 and TLR5, and intermediate monocytes expressing TLR2 (47). BD patients have increased numbers of intermediate monocytes and decreased numbers of nonclassical monocytes compared with healthy controls. The results showed that classical and intermediate monocytes overproduce TNF-α and intermediate monocytes overproduce IL-6 (48). At present, little is known about monocyte subsets in BD patients. However, there is also a theory that non-classical monocytes can differentiate into M2 macrophages; hopefully future research will answer these questions.
IL1A/IL1B and MEFV
Immunochip analysis in Turkish, Japanese, and Iranian subjects revealed association of IL1A–IL1B and IRF8 loci with BD. Functional analysis revealed that risk SNPs in the IL1A–IL1B region were associated with high levels of IL1β expression (27). Targeted-resequencing of innate-immune genes in a Turkish population identified association of BD with MEFV M694V, a known disease-causing variant of familial Mediterranean fever. MEFV encodes a pyrin, which forms pyrin-inflammasomes in response to toxins produced by Clostridium difficile (17, 49). A pyrin ligand β2-microglobulin (β2MG) induces pyrin inflammasome formation, while the caspase-1p20 subunit produced by the pyrin inflammasome inhibits the pyrin-β2MG interaction in neutrophils (50). The M694V mutation weakens the inhibitory effect of caspase-1p20 on the pyrin-β2MG interaction (50). Inflammasomes are composed of pattern recognition receptors, such as NOD-like receptors, and an adaptor protein, pro-caspase 1 (51). When the inflammasome is formed, pro-caspase 1 becomes activated caspase 1, which cleaves pro-IL-1β into active IL-1β (52). Caspase 1 also cleaves gasdermin D, the N-terminus of which forms membrane pores, leading to pyroptosis (53). This pathway is called the canonical pathway. In addition to this process, there is the noncanonical pathway that releases IL-1β/IL-18 and causes pyroptosis via caspase 11 (54). Together, these findings indicate that excessive production of IL-1β by activated inflammasomes is likely to be involved in the pathogenesis of BD.
Conclusions from studies using cells from BD patients have been controversial. In monocyte-derived macrophages from BD patients, TLR2 and TLR4 expression and IL-1β/ROS production were upregulated and IL-1β production was suppressed by inhibition of the NLRP3 inflammasome (55). In PBMCs from BD patients, production of NLRP3 inflammasomes and IL-1β in response to LPS stimulation was increased compared with that in healthy controls (56). The expression of NLRP3, caspase 1 and gasdermin D was also significantly higher in the intestinal tissues of BD patients (57). Meanwhile, no enhancement of the caspase 1 pathway was observed in dendritic cells of BD patients (58). The serum level of gasdermin D was lower in BD patients used as disease controls for adult-onset Still’s disease (59). The differences in these conclusions might be affected by different cell types and patients’ backgrounds. Focusing on the relationship between risk alleles and monocyte/macrophage polarity may provide additional autoinflammatory insights into BD.
IRF8
IRF8 is one of the nine members of the interferon regulatory factor (IRF) family (60). In IRF8-deficient mice, monocytic phagocyte differentiation is inhibited and abnormal differentiation into neutrophils occurs, indicating that IRF8 regulates the differentiation of progenitor cells into monocytic phagocytes (61, 62). IRF8 is also involved in the formation of M1 macrophages (63).
IRFs also function in signaling by TLRs and IFN receptors. IRF8 is expressed in macrophages and is responsible for induction of interferon production; IRF8 regulates transcription by forming a complex with IRF1 and transcription factors such as AP1 and PU.1 (64–67). Stimulation of myeloid cells with LPS results in increased expression of IRF8, which then binds to sites on the genome that are different from its steady-state binding site (68). The activity of IRF8/PU.1 is required for the regulation of IL18 expression in macrophages in mice (69). Furthermore, IRF8-PU.1 forms a complex with IRF1 and increases the promoter activity of IL1β (70). IRF8 is also required to activate the NLRC inflammasome (71).
IRF8 is involved in polarization of T-cells. In antigen-presenting cells (APCs), IFN-γ binding to IFNGR1/2 transactivates IRF8 expression through a STAT1-mediated pathway (72). IRF8 binds to the promoter region of IL12p40 and induces the production of IL-12 p40 from APCs, which promotes differentiation into Th1 cells (73, 74). IL-12p40 is a subunit of IL-12, but it is also a subunit of IL-23, which induces Th17 cell differentiation. In addition, IRF8 regulates IL27 and TGFβ expression in APCs. IRF8 expression decreases IL-27 production and activates TGFβ, thereby enhancing the induction of Th17 cell differentiation (75).
The association between IRF8 and BD has been reported in Chinese populations in addition to that described in the aforementioned Immunochip analysis (76). Functional analysis showed increased IRF8 mRNA expression and IFN-γ production and decreased production of IL-10 in the risk allele group, indicating higher macrophage differentiation ability in BD (76).
IFNGR1
A large genetic association study involving 9,444 individuals from seven diverse populations was recently performed and IFNGR1 and LNCAROD/DKK1 were identified as new BD susceptibility loci (28). IFNGR1 expression was increased in CD14+ monocytes 2 hours after LPS stimulation in the presence of the risk allele (28). IFNGR1 is a receptor for IFN-γ, and when IFN-γ binds to IFNGR1, it regulates the transcription of genes that contain gamma-activating sequences (GAS) in the promoter region via the STAT1 pathway (77). In addition to activating macrophages, IFN-γ has many other functions, including controlling Th cell polarity, enhancing antigen presentation, leukocyte homing and cell adhesion (78). Of note, IFNGR1 polymorphisms affect the immune response to mycobacterium and Helicobacter pylori, which are assumed to be pathogens associated with BD (79, 80). Patients with complete loss of IFNGR1 are repeatedly infected with mycobacterium and develop disseminated Bacille Calmette-Guerin (BCG) and die from BCG inoculation (81, 82). Furthermore, IFNGR1-mediated transactivation of caspase 11 has been reported in mice, indicating that IFNGR1 may be involved in the noncanonical pathway of inflammasome formation (83). These results strengthen the hypothesis of an abnormal innate immune response to external stimuli (including pathogens) in BD pathogenesis. The discovery of the involvement of IGNGR1 and STAT1 pathways may support the use of JAK inhibitors for BD.
Serum Cytokine Levels in BD
To clarify the role of cytokines, especially those produced by monocytes and macrophages, in BD pathogenesis, we systematically searched PubMed, the Cochrane Central Register of Controlled Trials, and the Web of Science Core Collection (up to November 30, 2021) for literature comparing cytokine levels in healthy controls and BD patients. Search formulae are presented in Supplementary Text 1. Differences in cytokine levels between BD and control subjects were calculated as the standardized mean difference (SMD) with confidence intervals (CIs). Pooled analyses were performed using the generic inverse variance method with a random effects model. Heterogeneity was indicated by I2, where 0% meant no heterogeneity and 100% meant the strongest heterogeneity. Review Manager version 5.4 software (Cochrane, London, UK) was used to draw paired forest plots.
Among 2,429 candidate articles, we identified 26 eligible studies (84–95). The quality of the original studies was assessed using the Newcastle-Ottawa Quality Assessment Scale for case-control study design (96). Characteristics of the included studies are summarized in Supplementary Table 1. The Newcastle-Ottawa Scale score of the included studies was good (Supplementary Table 2).
Proinflammatory cytokines play crucial roles in the initiation and perpetuation of disease. Levels of IL-6 and TNF-α are associated with disease activity according to several studies (91, 94). Serum IL-6 levels were measured in 15 cohorts involving 594 BD patients and 479 controls. The meta-analysis showed that IL-6 levels were significantly higher in BD patients than in controls (SMD = 3.20, 95% CI: 2.14–4.26, I2 = 97%, p < 0.001) (Figure 4A). Similarly, serum TNF-α levels were measured in 14 cohorts involving 552 BD patients and 413 controls, and were significantly increased in BD patients compared with controls (SMD = 3.19, 95% CI: 2.22–4.16, I2 = 97%, p < 0.001) (Figure 4B). Subgroup analysis revealed that IL-6 and TNF-α levels were increased more in active BD than in inactive BD (Supplementary Figure 1). Serum IL-1β levels were studied in only four cohorts involving 191 BD patients and 128 controls. IL-1β levels were increased in BD patients compared with controls (SMD = 1.67, 95% CI: 0.18–3.16, I2 = 97%, p < 0.001).
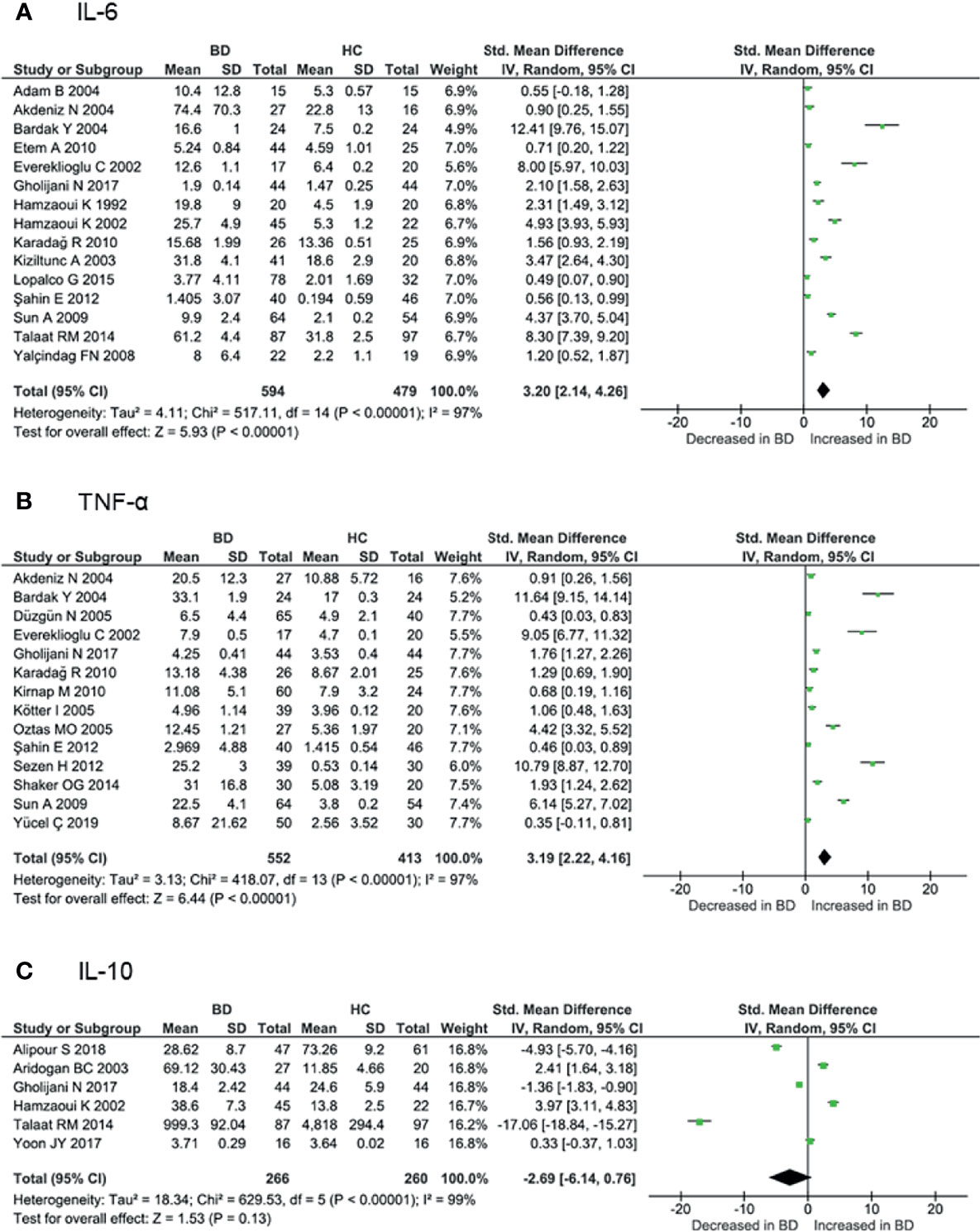
Figure 4 Forest plot of the standardized mean difference of serum cytokine levels in BD patients compared with controls. (A) IL-6, (B) TNF-α, (C) IL-10.
As noted above, IL-10 confers an anti-inflammatory effect involved in the pathogenesis of various autoimmune diseases. Serum IL-10 levels were measured in six cohorts involving 266 BD patients and 260 controls. Some studies have shown that IL-10 is increased in inflamed tissues of BD patients (92, 93). However, serum levels have been reported to be both increased and decreased in BD, and the meta-analysis showed that IL-10 levels were not higher in BD patients compared with controls (SMD = -2.69, 95% CI: -6.14–0.76, I2 = 99%, p < 0.001) (Figure 4C). Although GWAS have shown decreased IL10 expression associated with BD (20, 21), various disease activities included in the analyses may have influenced the results (Supplementary Table 1). Subgroup analysis revealed that IL-10 levels were higher in active BD than in inactive BD (92, 93).
Involvement of Monocytes With Current Treatment Strategies
Colchicine inhibits the polymerization of microtubules and is an established treatment for BD. Inhibition of microtubule polymerization prevents NLRP3 from approaching the adopter molecule apoptosis-associated speck-like protein containing a CARD (ASC) and suppresses activation of the NLRP3 inflammasome (97). Colchicine is also associated with increased numbers of nonclassical monocytes, indicating that it may affect monocyte polarization (98). GWAS indicate abnormal inflammasome activation in monocytes, supporting the use of colchicine as an anchor drug for BD.
Anti-TNF-α antibodies have been used to treat uveitis, and intestinal, neurological, and vascular lesions of BD (99). TNF-α receptors include TNFR1 and TNFR2. TNFR1 has the death domain, Tumor necrosis factor receptor type 1-associated DEATH domain (TRADD), and TRADD-mediated aggregation of induced Fas-associated protein with death domain (FADD)and receptor-interacting protein (RIP) activates caspase-8, leading to apoptosis (100). In contrast, TNFR2 does not have a death domain and activation of the TRAF2-mediated pathway leads to the activation of NFκB (101). Haploinsufficiency of A20, encoded by the TNFAIP3 gene, causes prolonged activation of the NFκB pathway and BD like symptoms (102–105). Anti-TNF antibodies and monocytes have been studied in response to rheumatoid arthritis treatment. Serum monocyte counts are decreased in patients who are responsive to anti-TNF therapy (106) and an early decrease in circulating monocyte count is a predictor of maintenance of remission (107). Anti-TNF antibodies also affect the polarity of macrophages and monocytes. A psoriasis study showed that anti-TNF therapy inhibited the polarity of M1 macrophages in an IRF1- and STAT1-independent manner (108). Another report of inflammatory bowel disease showed that infliximab infusion caused a decrease in monocyte count, which was more pronounced in classical and intermediate monocytes (109).
In 2019, Apremilast produced successful results in a phase 3 trial for refractory oral ulcers in BD (110). Apremilast is an inhibitor of phosphodiesterase (PDE)4, which regulates signal transduction of the intracellular second messengers, cyclic AMP (cAMP) and cyclic GMP (cGMP). The 11 gene families that comprise the PDE superfamily differ in function, primary structure, affinity for cAMP and cGMP, and regulatory mechanisms. Most cells express more than one PDE family member, but the degree of expression varies among tissues and cells (111). PED4 is cAMP-specific and is distributed throughout the body, but is expressed predominantly on T cells, monocytes, macrophages, neutrophils, dendritic cells, and eosinophils (112). In vivo stimulation of CD14+ monocytes with LPS and apremilast increased the expression of the SOCS3 gene, which then up-regulated the enhanced IL-10 and IL-6 expression, and decreased IFN-γ expression (113), thus apremilast may correct the genetically driven IL-10 impairment in M2 macrophages of BD patients. Similarly, sub-analysis of a phase 3 trial showed serum IFN-γ levels in the apremilast group were significantly lower than in the placebo group at 12 weeks (114).
Future Perspectives
Elucidation and interpretation of the function of disease susceptibility loci discovered by GWAS have led to significant progress in understanding the pathogenesis of BD. The pathogenesis of BD cannot be described by an abnormality in a single immune process but is undoubtedly a complex interplay of multiple immune processes. Monocytes/macrophages are responsible for initiating the inflammatory response and directing the subsequent activation of the acquired immune system. Considering that current therapies target monocytes, monocytes are promising targets for new agents, especially those that can alter the polarity of macrophages and monocytes.
The current unmet medical needs of BD are residual disease activity in some patients using current therapies, lack of treatment goals, and personalization of treatment regimens. As seen in the cytokine meta-analysis, the heterogeneity of BD symptoms makes the study of BD difficult. The classification of disease type is necessary to inform therapeutic strategies. We and others have analyzed the phenotypic subtypes of BD and have described five subtypes (115, 116). A disadvantage of categorizing the disease into clinical subtypes is the reduction in patient number for each subtype, especially as BD is a rare disease with a regional bias. A large international cohort to increase the number of participants is, therefore, necessary. In addition to symptom subtypes, there may also be genetic and immunological subsets. IL-17 inhibitors and JAK inhibitors were recently shown to be effective for BD (117, 118). By identifying subsets with predominant monocyte/macrophage activity, subsets with predominant Th17 activity, etc., it may be possible to identify patient groups for which inhibition of specific cells is predicted to be useful. We suggest that further subtype analysis and elucidation of immunological pathogenesis will contribute to personalized medicine in BD.
Author Contributions
LH, KT-M, and YK designed the research. LH, KT-M, YK, YI-I, YS, and RY conducted the research, statistical analysis, and interpretation of the data. LH, KT-M, and YK drafted the manuscript. YI-I, YS, RY, and HN were involved in writing the article or revising it critically for intellectual content. LH, KT-M, and YK had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a Japanese Society for the Promotion of Science Grant-in-Aid for Scientific Research # 19H03700 (YK), and the Japan College of Rheumatology Novartis Pharma Grants for Basic Research 2020. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
Conflict of Interest
YK reports personal fees from Amgen, and grant support from Nippon Shinyaku, Tanabe-Mitsubishi, and Chugai.
The authors declare that this study received funding from a Japanese Society for the Promotion of Science Grant-in-Aid for Scientific Research #19H03700 (YK), and the Japan College of Rheumatology Novartis Pharma Grants for Basic Research 2020 (YK). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Jeremy Allen, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.852297/full#supplementary-material
References
1. Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M. Close Association of HLA-Bw51 With Behcet’s Disease [Research Support, Non-U.S. Gov’t] Arch Ophthalmol (1982) 100(9):1455–8. doi: 10.1001/archopht.1982.01030040433013
2. Yazici H, Seyahi E, Hatemi G, Yazici Y. Behcet Syndrome: A Contemporary View. Nat Rev Rheumatol (2018) 14(2):107–19. doi: 10.1038/nrrheum.2017.208
3. Ideguchi H, Suda A, Takeno M, Ueda A, Ohno S, Ishigatsubo Y. Behcet Disease: Evolution of Clinical Manifestations. Med (Baltimore) (2011) 90(2):125–32. doi: 10.1097/MD.0b013e318211bf28
4. International Study Group for Behcet’s Disease. Criteria for Diagnosis of Behcet’s Disease. Lancet (1990) 335(8697):1078–80. doi: 10.1016/0140-6736(90)92643-V
5. de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the Risk of Behcet’s Disease: A Systematic Review and Meta-Analysis of Case-Control Genetic Association Studies. Arthritis Rheum (2009) 61(10):1287–96. doi: 10.1002/art.24642
6. Matsumura N, Mizushima Y. Leucocyte Movement and Colchicine Treatment in Behcet’s Disease. Lancet (1975) 2(7939):813. doi: 10.1016/s0140-6736(75)80031-6
7. Mege JL, Dilsen N, Sanguedolce V, Gul A, Bongrand P, Roux H, et al. Overproduction of Monocyte Derived Tumor Necrosis Factor Alpha, Interleukin (IL) 6, IL-8 and Increased Neutrophil Superoxide Generation in Behcet’s Disease. A Comparative Study With Familial Mediterranean Fever and Healthy Subjects. J Rheumatol (1993) 20(9):1544–9.
8. Sahin S, Lawrence R, Direskeneli H, Hamuryudan V, Yazici H, Akoglu T. Monocyte Activity in Behcet’s Disease. Br J Rheumatol (1996) 35(5):424–9. doi: 10.1093/rheumatology/35.5.424
9. Muller W, Lehner T. Quantitative Electron Microscopy Microscopical Analysis of Leukocyte Infiltration in Oral Ulcers of Behcet’s Syndrome. Br J Dermatol (1982) 106(5):535–44. doi: 10.1111/j.1365-2133.1982.tb04556.x
10. Gül A, Esin S, Dilsen N, Konice M, Wigzell H, Biberfeld P. Immunohistology of Skin Pathergy Reaction in Behçet’s Disease. Br J Dermatol (1995) 132(6):901–7. doi: 10.1111/j.1365-2133.1995.tb16946.x
11. Sohn S, Lee ES, Bang D, Lee S. Behcet’s Disease-Like Symptoms Induced by the Herpes Simplex Virus in ICR Mice [Comparative StudyResearch Support, Non-U.S. Gov’t]. Eur J Dermatol (1998) 8(1):21–3.
12. Kaneko F, Oyama N, Yanagihori H, Isogai E, Yokota K, Oguma K. The Role of Streptococcal Hypersensitivity in the Pathogenesis of Behcet’s Disease [Review]. Eur J Dermatol (2008) 18(5):489–98. doi: 10.1684/ejd.2008.0484
13. Dabbagh F, Haghighi AB, Ghasemi Y. Behcet’s Disease: From Heat Shock Proteins to Infections. Asian Biomed (2014) 8(2):139–55. doi: 10.5372/1905-7415.0802.274
14. Kirino Y, Takeno M, Watanabe R, Murakami S, Kobayashi M, Ideguchi H, et al. Association of Reduced Heme Oxygenase-1 With Excessive Toll-Like Receptor 4 Expression in Peripheral Blood Mononuclear Cells in Behcet’s Disease [Research Support, Non-U. S Gov’t] Arthritis Res Ther (2008) 10(1):R16. doi: 10.1186/ar2367
15. Rucinski M, Zok A, Guidolin D, De Caro R, Malendowicz LK. Expression of Precerebellins in Cultured Rat Calvaria Osteoblast-Like Cells. Int J Mol Med (2008) 22(4):553–8.
16. Liang L, Tan X, Zhou Q, Zhu Y, Tian Y, Yu H, et al. IL-1beta Triggered by Peptidoglycan and Lipopolysaccharide Through TLR2/4 and ROS-NLRP3 Inflammasome-Dependent Pathways is Involved in Ocular Behcet’s Disease [Comparative Study Research Support, Non-U.S. Gov’t]. Invest Ophthalmol Vis Sci (2013) 54(1):402–14. doi: 10.1167/iovs.12-11047
17. Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Targeted Resequencing Implicates the Familial Mediterranean Fever Gene MEFV and the Toll-Like Receptor 4 Gene TLR4 in Behcet Disease [Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci USA (2013) 110(20):8134–9. doi: 10.1073/pnas.1306352110
18. Ombrello MJ, Kirino Y, de Bakker PI, Gul A, Kastner DL, Remmers EF. Behcet Disease-Associated MHC Class I Residues Implicate Antigen Binding and Regulation of Cell-Mediated Cytotoxicity. Proc Natl Acad Sci USA (2014) 111(24):8867–72. doi: 10.1073/pnas.1406575111
19. Meguro A, Inoko H, Ota M, Katsuyama Y, Oka A, Okada E, et al. Genetics of Behcet Disease Inside and Outside the MHC [Research Support, Non-U.S. Gov’t]. Ann Rheum Diseases (2010) 69(4):747–54. doi: 10.1136/ard.2009.108571
20. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-Wide Association Studies Identify IL23R-IL12RB2 and IL10 as Behcet’s Disease Susceptibility Loci [Comment Research Support, Non-U.S. Gov’t]. Nat Genet (2010) 42(8):703–6. doi: 10.1038/ng.624
21. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-Wide Association Study Identifies Variants in the MHC Class I, IL10, and IL23R-IL12RB2 Regions Associated With Behcet’s Disease [Research Support, N.I.H., ExtramuralResearch Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t]. Nat Genet (2010) 42(8):698–702. doi: 10.1038/ng.625
22. Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a Susceptibility Locus in STAT4 for Behcet’s Disease in Han Chinese in a Genome-Wide Association Study [Research Support, Non-U.S. Gov’t]. Arthritis Rheum (2012) 64(12):4104–13. doi: 10.1002/art.37708
23. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-Wide Association Analysis Identifies New Susceptibility Loci for Behcet’s Disease and Epistasis Between HLA-B*51 and ERAP1. Nat Genet (2013) 45(2):202–7. doi: 10.1038/ng.2520
24. Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, et al. Genome-Wide Association Study Identifies GIMAP as a Novel Susceptibility Locus for Behcet’s Disease [Research Support, Non-U.S. Gov’t] Ann Rheum Diseases (2013) 72(9):1510–6. doi: 10.1136/annrheumdis-2011-200288
25. Xavier JM, Shahram F, Sousa I, Davatchi F, Matos M, Abdollahi BS, et al. FUT2: Filling the Gap Between Genes and Environment in Behcet’s Disease? Ann Rheum Dis (2015) 74(3):618–24. doi: 10.1136/annrheumdis-2013-204475
26. Kappen JH, Medina-Gomez C, van Hagen PM, Stolk L, Estrada K, Rivadeneira F, et al. Genome-Wide Association Study in an Admixed Case Series Reveals IL12A as a New Candidate in Behcet Disease [Research Support, Non-U.S. Gov’t]. PloS One (2015) 10(3):e0119085. doi: 10.1371/journal.pone.0119085
27. Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense Genotyping of Immune-Related Loci Implicates Host Responses to Microbial Exposure in Behcet’s Disease Susceptibility. Nat Genet (2017) 49(3):438–43. doi: 10.1038/ng.3786
28. Ortiz Fernandez L, Coit P, Yilmaz V, Yentur SP, Alibaz-Oner F, Aksu K, et al. Genetic Association of a Gain-Of-Function IFNGR1 Polymorphism and the Intergenic Region LNCAROD/DKK1 With Behcet’s Disease [Research Support, N.I.H., Extramural]. Arthritis Rheumatol (2021) 73(7):1244–52. doi: 10.1002/art.41637
29. Kaufmann A, Salentin R, Gemsa D, Sprenger H. Increase of CCR1 and CCR5 Expression and Enhanced Functional Response to MIP-1 Alpha During Differentiation of Human Monocytes to Macrophages. J Leukoc Biol (2001) 69(2):248–52. doi: 10.1189/jlb.69.2.248
30. Ishigaki K, Kochi Y, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S, et al. Polygenic Burdens on Cell-Specific Pathways Underlie the Risk of Rheumatoid Arthritis. Nat Genet (2017) 49(7):1120–5. doi: 10.1038/ng.3885
31. Manthiram K, Preite S, Dedeoglu F, Demir S, Ozen S, Edwards KM, et al. Common Genetic Susceptibility Loci Link PFAPA Syndrome, Behcet’s Disease, and Recurrent Aphthous Stomatitis [Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci USA (2020) 117(25):14405–11. doi: 10.1073/pnas.2002051117
32. Dudding T, Haworth S, Lind PA, Sathirapongsasuti JF, Me Research T, Tung JY, et al. Genome Wide Analysis for Mouth Ulcers Identifies Associations at Immune Regulatory Loci. Nat Commun (2019) 10(1):1052. doi: 10.1038/s41467-019-08923-6
33. Ota M, Nagafuchi Y, Hatano H, Ishigaki K, Terao C, Takeshima Y, et al. Dynamic Landscape of Immune Cell-Specific Gene Regulation in Immune-Mediated Diseases [Research Support, Non-U.S. Gov’t]. Cell (2021) 184(11):3006–21.e17. doi: 10.1016/j.cell.2021.03.056
34. Peng H, Xian D, Liu J, Pan S, Tang R, Zhong J. Regulating the Polarization of Macrophages: A Promising Approach to Vascular Dermatosis. J Immunol Res (2020) 2020:8148272. doi: 10.1155/2020/8148272
35. Yao Y, Xu XH, Jin L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front Immunol (2019) 10:792. doi: 10.3389/fimmu.2019.00792
36. Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. New Engl J Med (2009) 361(21):2033–45. doi: 10.1056/nejmoa0907206
37. Nakano H, Kirino Y, Takeno M, Higashitani K, Nagai H, Yoshimi R, et al. GWAS-Identified CCR1 and IL10 Loci Contribute to M1 Macrophage-Predominant Inflammation in Behcet’s Disease. Arthritis Res Ther (2018) 20(1):124. doi: 10.1186/s13075-018-1613-0
38. Anower AKMM, Shim JA, Choi B, Kwon HJ, Sohn S. The Role of Classical and Alternative Macrophages in the Immunopathogenesis of Herpes Simplex Virus-Induced Inflammation in a Mouse Model. J Dermatol Sci (2014) 73(3):198–208. doi: 10.1016/j.jdermsci.2013.11.001
39. Shi J, Wu XH, Liu JJ, Chen H, Zheng WJ. Aberrant M1 Polarization of Macrophages in Behcet’s Disease. Arthritis Rheumatol (2019) 71.
40. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of Monocytes and Dendritic Cells in Blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
41. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
42. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler Thromb Vasc Biol (2015) 35(6):1306–16. doi: 10.1161/ATVBAHA.114.304650
43. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim Monocytes Patrol and Sense Nucleic Acids and Viruses via TLR7 and TLR8 Receptors. Immunity (2010) 33(3):375–86. doi: 10.1016/j.immuni.2010.08.012
44. Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. Monocyte Subsets: Phenotypes and Function in Tuberculosis Infection. Front Immunol (2018) 9:1726. doi: 10.3389/fimmu.2018.01726
45. Gren ST, Rasmussen TB, Janciauskiene S, Hakansson K, Gerwien JG, Grip O. A Single-Cell Gene-Expression Profile Reveals Inter-Cellular Heterogeneity Within Human Monocyte Subsets. PloS One (2015) 10(12):e0144351. doi: 10.1371/journal.pone.0144351
46. Esen F, TÜRkyilmaz Ö, Aykut H, DİReskenelİ H, Denİ ZG, OĞUz H. Influence of İnterferon Alfa-2a Treatment on Monocyte Subsets in Patients With Uveitis. Turkish J Immunol (2020) 8(2):50–6. doi: 10.25002/tji.2020.1261
47. van der Houwen TB, Dik WA, Goeijenbier M, Hayat M, Nagtzaam NMA, van Hagen M, et al. Leukocyte Toll-Like Receptor Expression in Pathergy Positive and Negative Behcet’s Disease Patients. Rheumatol (Oxford) (2020) 59(12):3971–9. doi: 10.1093/rheumatology/keaa251
48. Li C, Liu J, Yu X, Li L, Wang Z, Shi J, et al. Aberrant Monocyte Subsets in Patients With Behcet’s Disease. Clin Immunol (2021) 225:108683. doi: 10.1016/j.clim.2021.108683
49. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate Immune Sensing of Bacterial Modifications of Rho GTPases by the Pyrin Inflammasome. Nature (2014) 513(7517):237–41. doi: 10.1038/nature13449
50. Samukawa S, Yoshimi R, Kirino Y, Nakajima H. The PRY/SPRY Domain of Pyrin/TRIM20 Interacts With Beta2-Microglobulin to Promote Inflammasome Formation. Sci Rep (2021) 11(1):23613. doi: 10.1038/s41598-021-03073-6
51. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and Reacting to Microbes Through the Inflammasomes. Nat Immunol (2012) 13(4):325–32. doi: 10.1038/ni.2231
52. Martinon F, Burns K, Tschopp J. The Inflammasome. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
53. Fink SL, Cookson BT. Caspase-1-Dependent Pore Formation During Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
54. Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
55. Liang L, Tan X, Zhou Q, Zhu Y, Tian Y, Yu H, et al. IL-1β Triggered by Peptidoglycan and Lipopolysaccharide Through TLR2/4 and ROS-NLRP3 Inflammasome–Dependent Pathways Is Involved in Ocular Behçet’s Disease. Invest Opthalmol Visual Sci (2013) 54(1):402. doi: 10.1167/iovs.12-11047
56. Kim EH, Park M-J, Park S, Lee E-S. Increased Expression of the NLRP3 Inflammasome Components in Patients With Behçet’s Disease. J Inflamm (2015) 12(1):41. doi: 10.1186/s12950-015-0086-z
57. Hou C-C, Ma H-F, Ye J-F, Luo D, Bao H-F, Guan J-L. Plasma Exosomes Derived From Patients With Intestinal Behçet’s Syndrome Induce Intestinal Epithelial Cell Pyroptosis. Clin Rheumatol (2021) 40(10):4143–55. doi: 10.1007/s10067-021-05755-y
58. Ture-Ozdemir F, Tulunay A, Elbasi MO, Tatli I, Maurer AM, Mumcu G, et al. Pro-Inflammatory Cytokine and Caspase-1 Responses to Pattern Recognition Receptor Activation of Neutrophils and Dendritic Cells in Behcet’s Disease. Rheumatol (Oxford) (2013) 52(5):800–5. doi: 10.1093/rheumatology/kes399
59. Nagai H, Kirino Y, Nakano H, Kunishita Y, Henmi R, Szymanski AM, et al. Elevated Serum Gasdermin D N-Terminal Implicates Monocyte and Macrophage Pyroptosis in Adult-Onset Still’s Disease. Rheumatol (Oxford) (2021) 60(8):3888–95. doi: 10.1093/rheumatology/keaa814
60. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front Immunol (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
61. Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, et al. IRF8 Inhibits C/Ebpα Activity to Restrain Mononuclear Phagocyte Progenitors From Differentiating Into Neutrophils. Nat Commun (2014) 5(1):4978. doi: 10.1038/ncomms5978
62. Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP Directs Bipotential Myeloid Progenitor Cells to Differentiate Into Mature Macrophages. Immunity (2000) 13(2):155–65. doi: 10.1016/s1074-7613(00)00016-9
63. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The Impact of Interferon-Regulatory Factors to Macrophage Differentiation and Polarization Into M1 and M2. Immunobiology (2018) 223(1):101–11. doi: 10.1016/j.imbio.2017.10.005
64. Bovolenta C, Driggers PH, Marks MS, Medin JA, Politis AD, Vogel SN, et al. Molecular Interactions Between Interferon Consensus Sequence Binding Protein and Members of the Interferon Regulatory Factor Family. Proc Natl Acad Sci (1994) 91(11):5046–50. doi: 10.1073/pnas.91.11.5046
65. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, et al. A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1–IRF Complexes. Science (2012) 338(6109):975–80. doi: 10.1126/science.1228309
66. McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, et al. Targeted Disruption of the PU.1 Gene Results in Multiple Hematopoietic Abnormalities. EMBO J (1996) 15(20):5647–58. doi: 10.1002/j.1460-2075.1996.tb00949.x
67. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of Transcription Factor PU.1 in the Development of Multiple Hematopoietic Lineages. Science (1994) 265(5178):1573–7. doi: 10.1126/science.8079170
68. Mancino A, Termanini A, Barozzi I, Ghisletti S, Ostuni R, Prosperini E, et al. A Dual Cis-Regulatory Code Links IRF8 to Constitutive and Inducible Gene Expression in Macrophages. Genes Dev (2015) 29(4):394–408. doi: 10.1101/gad.257592.114
69. Kim YM, Im JY, Han SH, Kang HS, Choi I. IFN-Gamma Up-Regulates IL-18 Gene Expression via IFN Consensus Sequence-Binding Protein and Activator Protein-1 Elements in Macrophages. J Immunol (2000) 165(6):3198–205. doi: 10.4049/jimmunol.165.6.3198
70. Marecki S, Fenton MJ. PU.1/Interferon Regulatory Factor Interactions; Mechanisms of Transcriptional Regulation. Cell Biochem Biophys (2000) 33(2):127–48. doi: 10.1385/cbb:33:2:127
71. Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell (2018) 173(4):920–933.e13. doi: 10.1016/j.cell.2018.02.055
72. Kanno Y, Kozak CA, Schindler C, Driggers PH, Ennist DL, Gleason SL, et al. The Genomic Structure of the Murine ICSBP Gene Reveals the Presence of the Gamma Interferon-Responsive Element, to Which an ISGF3 Alpha Subunit (or Similar) Molecule Binds. Mol Cell Biol (1993) 13(7):3951–63. doi: 10.1128/mcb.13.7.3951-3963.1993
73. Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. An IFN-Gamma-Inducible Transcription Factor, IFN Consensus Sequence Binding Protein (ICSBP), Stimulates IL-12 P40 Expression in Macrophages. J Immunol (2000) 165(1):271–9. doi: 10.4049/jimmunol.165.1.271
74. Giese NA, Gabriele L, Doherty TM, Klinman DM, Tadesse-Heath L, Contursi C, et al. Interferon (IFN) Consensus Sequence-Binding Protein, a Transcription Factor of the IFN Regulatory Factor Family, Regulates Immune Responses In Vivo Through Control of Interleukin 12 Expression. J Exp Med (1997) 186(9):1535–46. doi: 10.1084/jem.186.9.1535
75. Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey A, Xiong H, et al. The Transcription Factor IRF8 Activates Integrin-Mediated TGF-β Signaling and Promotes Neuroinflammation. Immunity (2014) 40(2):187–98. doi: 10.1016/j.immuni.2013.11.022
76. Jiang Y, Wang H, Yu H, Li L, Xu D, Hou S, et al. Two Genetic Variations in the IRF8 Region are Associated With Behcet’s Disease in Han Chinese. Sci Rep (2016) 6:19651. doi: 10.1038/srep19651
77. van de Wetering D, de Paus RA, van Dissel JT, van de Vosse E. Functional Analysis of Naturally Occurring Amino Acid Substitutions in Human IFN-Gammar1. Mol Immunol (2010) 47(5):1023–30. doi: 10.1016/j.molimm.2009.11.016
78. Boehm U, Klamp T, Groot M, Howard JC. Cellular Responses to Interferon-Gamma. Annu Rev Immunol (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
79. Wu S, Wang Y, Zhang M, Wang M, He JQ. Genetic Variants in IFNG and IFNGR1 and Tuberculosis Susceptibility. Cytokine (2019) 123:154775. doi: 10.1016/j.cyto.2019.154775
80. Zhang Y, Dong Q, Tian L, Zhang S, Zuo N, Zhang S, et al. Risk Factors for Recurrence of Helicobacter Pylori Infection After Successful Eradication in Chinese Children: A Prospective, Nested Case-Control Study. Helicobacter (2020) 25(5):e12749. doi: 10.1111/hel.12749
81. Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile J-F, Newport M, et al. Interferon-γ –Receptor Deficiency in an Infant With Fatal Bacille Calmette–Guérin Infection. New Engl J Med (1996) 335(26):1956–62. doi: 10.1056/nejm199612263352604
82. Jouanguy E, Lamhamedi-Cherradi S, Altare F, Fondanèche MC, Tuerlinckx D, Blanche S, et al. Partial Interferon-Gamma Receptor 1 Deficiency in a Child With Tuberculoid Bacillus Calmette-Guérin Infection and a Sibling With Clinical Tuberculosis. J Clin Invest (1997) 100(11):2658–64. doi: 10.1172/jci119810
83. Aachoui Y, Kajiwara Y, Irina, Mao D, Jenny, Coers J, et al. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense Against a Cytosol-Invasive Bacterium. Cell Host Microbe (2015) 18(3):320–32. doi: 10.1016/j.chom.2015.07.016
84. Alipour S, Nouri M, Khabbazi A, Samadi N, Babaloo Z, Abolhasani S, et al. Hypermethylation of IL-10 Gene is Responsible for its Low mRNA Expression in Behcet’s Disease [Clinical TrialResearch Support, Non-U.S. Gov’t]. J Cell Biochem (2018) 119(8):6614–22. doi: 10.1002/jcb.26809
85. Gholijani N, Ataollahi MR, Samiei A, Aflaki E, Shenavandeh S, Kamali-Sarvestani E. An Elevated Pro-Inflammatory Cytokines Profile in Behcet’s Disease: A Multiplex Analysis. Immunol Lett (2017) 186:46–51. doi: 10.1016/j.imlet.2016.12.001
86. Talaat RM, Ashour ME, Bassyouni IH, Raouf AA. Polymorphisms of Interleukin 6 and Interleukin 10 in Egyptian People With Behcet’s Disease [Research Support, Non-U.S. Gov’t] Immunobiol (2014) 219(8):573–82. doi: 10.1016/j.imbio.2014.03.004
87. Sun A, Wang YP, Chia JS, Liu BY, Chiang CP. Treatment With Levamisole and Colchicine can Result in a Significant Reduction of IL-6, IL-8 or TNF-Alpha Level in Patients With Mucocutaneous Type of Behcet’s Disease [Comparative Study Controlled Clinical Trial]. J Oral Pathol Med: Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol (2009) 38(5):401–5. doi: 10.1111/j.1600-0714.2009.00774.x
88. Akdeniz N, Esrefoglu M, Keles MS, Karakuzu A, Atasoy M. Serum Interleukin-2, Interleukin-6, Tumour Necrosis Factor-Alpha and Nitric Oxide Levels in Patients With Behcet’s Disease [Comparative Study]. Ann Acad Med Singapore (2004) 33(5):596–9.
89. Bardak Y, Aridogan BC. The Demonstration of Serum Interleukin 6-8, Tumor Necrosis Factor-Alpha, Complement, and Immunoglobulin Levels in Behcet’s Disease With Ocular Involvement. Ocular Immunol Inflamm (2004) 12(1):53–8. doi: 10.1076/ocii.12.1.53.28062
90. Adam B, Calikoglu E. Serum Interleukin-6, Procalcitonin and C-Reactive Protein Levels in Subjects With Active Behcet’s Disease [Comparative Study]. J Eur Acad Dermatol Venereol: JEADV (2004) 18(3):318–20. doi: 10.1111/j.1468-3083.2004.00907.x
91. Duzgun N, Ayaslioglu E, Tutkak H, Aydintug OT. Cytokine Inhibitors: Soluble Tumor Necrosis Factor Receptor 1 and Interleukin-1 Receptor Antagonist in Behcet’s Disease. Rheumatol Int (2005) 25(1):1–5. doi: 10.1007/s00296-003-0400-6
92. Aridogan BC, Yildirim M, Baysal V, Inaloz HS, Baz K, Kaya S. Serum Levels of IL-4, IL-10, IL-12, IL-13 and IFN-Gamma in Behcet’s Disease. J Dermatol (2003) 30(8):602–7. doi: 10.1111/j.1346-8138.2003.tb00442.x
93. Hamzaoui K, Hamzaoui A, Guemira F, Bessioud M, Hamza M, Ayed K. Cytokine Profile in Behcet’s Disease Patients. Relationship With Disease Activity. Scand J Rheumatol (2002) 31(4):205–10. doi: 10.1080/030097402320318387
94. Evereklioglu C, Er H, Turkoz Y, Cekmen M. Serum Levels of TNF-Alpha, sIL-2r, IL-6, and IL-8 are Increased and Associated With Elevated Lipid Peroxidation in Patients With Behcet’s Disease. Mediators Inflamm (2002) 11(2):87–93. doi: 10.1080/09629350220131935
95. Hamzaoui K, Hamzaoui A, Kahan A, Hamza M, Chabbou A, Ayed K. Interleukin-6 in Peripheral Blood and Inflammatory Sites in Behcet’s Disease. Mediators Inflamm (1992) 1(4):281–5. doi: 10.1155/S0962935192000437
96. Stang A. Critical Evaluation of the Newcastle-Ottawa Scale for the Assessment of the Quality of Nonrandomized Studies in Meta-Analyses. Eur J Epidemiol (2010) 25(9):603–5. doi: 10.1007/s10654-010-9491-z
97. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-Driven Spatial Arrangement of Mitochondria Promotes Activation of the NLRP3 Inflammasome. Nat Immunol (2013) 14(5):454–60. doi: 10.1038/ni.2550
98. Gazzito Del Padre TC, Belem J, de Aguiar MF, Torquato HFV, Paredes-Gamero EJ, Abdulahad WH, et al. Distribution of Monocytes Subpopulations in the Peripheral Blood From Patients With Behcet’s Disease - Impact of Disease Status and Colchicine Use. Clin Immunol (2021) 231:108854. doi: 10.1016/j.clim.2021.108854
99. Hatemi G, Christensen R, Bang D, Bodaghi B, Celik AF, Fortune F, et al. Update of the EULAR Recommendations for the Management of Behçet’s Syndrome. Ann Rheumatic Dis (2018) 2018:annrheumdis-201. doi: 10.1136/annrheumdis-2018-213225
100. Micheau O, Tschopp J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell (2003) 114(2):181–90. doi: 10.1016/s0092-8674(03)00521-x
101. Cabal-Hierro L, Rodriguez M, Artime N, Iglesias J, Ugarte L, Prado MA, et al. TRAF-Mediated Modulation of NF-kB AND JNK Activation by TNFR2. Cell Signal (2014) 26(12):2658–66. doi: 10.1016/j.cellsig.2014.08.011
102. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-Of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48(1):67–73. doi: 10.1038/ng.3459
103. Ohnishi H, Kawamoto N, Seishima M, Ohara O, Fukao T. A Japanese Family Case With Juvenile Onset Behcet’s Disease Caused by TNFAIP3 Mutation. Allergol Int (2017) 66(1):146–8. doi: 10.1016/j.alit.2016.06.006
104. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 Haploinsufficiency (HA20): Clinical Phenotypes and Disease Course of Patients With a Newly Recognised NF-kB-Mediated Autoinflammatory Disease. Ann Rheumatic Dis (2018) 77(5):728–35. doi: 10.1136/annrheumdis-2017-212403
105. Tsuchida N, Kirino Y, Soejima Y, Onodera M, Arai K, Tamura E, et al. Haploinsufficiency of A20 Caused by a Novel Nonsense Variant or Entire Deletion of TNFAIP3 is Clinically Distinct From Behçet’s Disease. Arthritis Res Ther (2019) 21(1):137. doi: 10.1186/s13075-019-1928-5
106. Chara L, Sánchez-Atrio A, Pérez A, Cuende E, Albarrán F, Turrión A, et al. Monocyte Populations as Markers of Response to Adalimumab Plus MTX in Rheumatoid Arthritis. Arthritis Res Ther (2012) 14(4):R175. doi: 10.1186/ar3928
107. Shipa MRA, Amarnani R, Yeoh SA, Mainuddin MD, Ehrenstein MR. Early Reduction in Circulating Monocyte Count Predicts Maintenance of Remission in Patients With Rheumatoid Arthritis Treated With Anti-TNF Therapy. Ann Rheum Dis (2021) 80(12):1628–9. doi: 10.1136/annrheumdis-2021-220642
108. Lin SH, Chuang HY, Ho JC, Lee CH, Hsiao CC. Treatment With TNF-Alpha Inhibitor Rectifies M1 Macrophage Polarization From Blood CD14+ Monocytes in Patients With Psoriasis Independent of STAT1 and IRF-1 Activation. J Dermatol Sci (2018) 91(3):276–84. doi: 10.1016/j.jdermsci.2018.05.009
109. Slevin SM, Dennedy MC, Connaughton EP, Ribeiro A, Ceredig R, Griffin MD, et al. Infliximab Selectively Modulates the Circulating Blood Monocyte Repertoire in Crohn’s Disease. Inflamm Bowel Dis (2016) 22(12):2863–78. doi: 10.1097/mib.0000000000000964
110. Hatemi G, Mahr A, Ishigatsubo Y, Song YW, Takeno M, Kim D, et al. Trial of Apremilast for Oral Ulcers in Behcet’s Syndrome [Clinical Trial, Phase III Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t]. New Engl J Med (2019) 381(20):1918–28. doi: 10.1056/NEJMoa1816594
111. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat Rev Drug Discov (2014) 13(4):290–314. doi: 10.1038/nrd4228
112. Li H, Zuo J, Tang W. Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front Pharmacol (2018) 9:1048. doi: 10.3389/fphar.2018.01048
113. Schafer PH, Parton A, Capone L, Cedzik D, Brady H, Evans JF, et al. Apremilast is a Selective PDE4 Inhibitor With Regulatory Effects on Innate Immunity. Cell Signal (2014) 26(9):2016–29. doi: 10.1016/j.cellsig.2014.05.014
114. Deeks ED. Apremilast: A Review in Oral Ulcers of Behcet’s Disease. Drugs (2020) 80(2):181–8. doi: 10.1007/s40265-019-01253-3
115. Soejima Y, Kirino Y, Takeno M, Kurosawa M, Takeuchi M, Yoshimi R, et al. Changes in the Proportion of Clinical Clusters Contribute to the Phenotypic Evolution of Behçet’s Disease in Japan. Arthritis Res Ther (2021) 23(1):49. doi: 10.1186/s13075-020-02406-6
116. Zou J, Luo J-F, Shen Y, Cai J-F, Guan J-L. Cluster Analysis of Phenotypes of Patients With Behçet’s Syndrome: A Large Cohort Study From a Referral Center in China. Arthritis Res Ther (2021) 23(1):45. doi: 10.1186/s13075-021-02429-7
117. Liu J, Hou Y, Sun L, Li C, Li L, Zhao Y, et al. A Pilot Study of Tofacitinib for Refractory Behcet’s Syndrome. Ann Rheum Dis (2020) 79(11):1517–20. doi: 10.1136/annrheumdis-2020-217307
Keywords: polarization, genetics, innate immunity, macrophages, monocytes, Behcet’s disease
Citation: Hirahara L, Takase-Minegishi K, Kirino Y, Iizuka-Iribe Y, Soejima Y, Yoshimi R and Nakajima H (2022) The Roles of Monocytes and Macrophages in Behçet’s Disease With Focus on M1 and M2 Polarization. Front. Immunol. 13:852297. doi: 10.3389/fimmu.2022.852297
Received: 11 January 2022; Accepted: 22 February 2022;
Published: 11 March 2022.
Edited by:
Atsushi Kawakami, Nagasaki University, JapanReviewed by:
Yuko Kaneko, Keio University School of Medicine, JapanYudong Liu, Chinese Academy of Medical Sciences, China
Kei Ikeda, Chiba University Hospital, Japan
Copyright © 2022 Hirahara, Takase-Minegishi, Kirino, Iizuka-Iribe, Soejima, Yoshimi and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yohei Kirino, a2lyaW5vQHlva29oYW1hLWN1LmFjLmpw
†These authors have contributed equally to this work and share first authorship