 Ganggui Zhu
Ganggui Zhu Xiaoyu Wang
Xiaoyu Wang Luxi Chen
Luxi Chen Cameron Lenahan4,5
Cameron Lenahan4,5 Zaixiang Fu
Zaixiang Fu Yuanjian Fang
Yuanjian Fang- 1Department of Neurosurgery, Hangzhou First People’s Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Medical Genetics, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 4Center for Neuroscience Research, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 5Department of Biomedical Science, Burrell College of Osteopathic Medicine, Las Cruces, NM, United States
Stroke is the second leading cause of global death and is characterized by high rates of mortality and disability. Oxidative stress is accompanied by other pathological processes that together lead to secondary brain damage in stroke. As the major component of the brain, glial cells play an important role in normal brain development and pathological injury processes. Multiple connections exist in the pathophysiological changes of reactive oxygen species (ROS) metabolism and glia cell activation. Astrocytes and microglia are rapidly activated after stroke, generating large amounts of ROS via mitochondrial and NADPH oxidase pathways, causing oxidative damage to the glial cells themselves and neurons. Meanwhile, ROS cause alterations in glial cell morphology and function, and mediate their role in pathological processes, such as neuroinflammation, excitotoxicity, and blood-brain barrier damage. In contrast, glial cells protect the Central Nervous System (CNS) from oxidative damage by synthesizing antioxidants and regulating the Nuclear factor E2-related factor 2 (Nrf2) pathway, among others. Although numerous previous studies have focused on the immune function of glial cells, little attention has been paid to the role of glial cells in oxidative stress. In this paper, we discuss the adverse consequences of ROS production and oxidative-antioxidant imbalance after stroke. In addition, we further describe the biological role of glial cells in oxidative stress after stroke, and we describe potential therapeutic tools based on glia cells.
Introduction
Accounting for 9% of all deaths worldwide, stroke is the second leading cause of death following ischemic heart diseases (1). Hemorrhagic strokes caused by rupture of blood vessels in the cerebrum and ischemic strokes caused by occlusion of cerebral arteries constitute the two subtypes of stroke; the latter accounted for approximately 87% of all strokes (2). The high disability rate and poor prognosis that accompany stroke have greatly increased individual and societal burden. Therefore, stroke has become a major global public health and economic concern (3).
Ischemia-reperfusion and the release of damage associated molecular patterns (DAMPs) after stroke induce a series of pathological changes, including oxidative and nitrative stress, neuroinflammation, excitotoxicity, and apoptosis (4). Oxidative stress describes the state of imbalance between the systemic performance of reactive oxygen species and the ability of biological systems to detoxify active media or repair damage (5). Excess ROS interact with several of the pathological mechanisms described above and collectively mediates secondary brain injury after stroke (6–8).
Glial cells serve as the main supporting cells of the CNS. Recent studies have found that they are involved in the regulation of oxidative stress in the CNS. On one hand, Astrocytes and microglia are responsible for the imbalance of redox status after stroke (9). Glial cells have the potential to release large amounts of free radicals through various pathways (10, 11). The generated ROS promote the activation of inflammatory pathways in astrocytes (12), interfere with their transport of glutamate (13), and affect homeostasis of the intra- and extracellular microenvironment. Microglia are involved in the disruption of the blood-brain barrier after stroke and affect the post-stroke neural repair process (14). Conversely, astrocytes maintain high intracellular antioxidant concentrations to protect themselves from oxidative damage after stroke (15). Glutathione is the main antioxidant synthesized by glial cells and plays an important role in warding off oxidative damage (16). The intracellular Nrf2-ARE pathway is the main antioxidant pathway that synergistically regulates the redox status of glial cells and neurons (17).
Previous studies have mostly focused on the effects of oxidative damage on neurons within the brain, but few studies have explored the regulation of glial cells in oxidative stress after stroke. From the perspective of glial cells, this article aims to explain its influence on the occurrence and development of oxidative stress after stroke and to explore its interaction with other pathological mechanisms to provide new insight for future stroke treatments.
The Role of ROS Under Physiological Conditions
As oxidants, physiological doses of ROS play an active role in various aspects of the body’s functional activities. From one hand, ROS contribute to the support of immune functions, such as enhancing macrophage phagocytosis and activating T cells (18). For another, ROS are engaged in the regulation of vascular cell proliferation and apoptosis. NO can maintain blood flow homeostasis in blood vessels (19). ROS promote the release of various germinal factors during wound repair and facilitate the repair process (20). During neurodevelopment, ROS is essential for synaptic plasticity and memory formation (21). In addition, researchers have found that ROS are also essential in muscle cell development and muscle remodeling (22). In conclusion, ROS can act as signaling molecules and are widely involved in maintaining homeostasis, metabolism, growth and differentiation in vivo.
Oxidative Stress Generated by Glial Cells
Mitochondria-Derived Oxidative Stress in Glial Cells
Mitochondrial metabolism in glial cells plays an important role in the regulation of redox status and biometabolism (see Figure 1) (10). Under physiological conditions, electrons released from the electron transport chain (ETC) react with O2 to produce superoxide .Free complex I in astrocytes is the major site of mitochondrial ROS (mROS) production (23). The metabolic state of the mitochondria determines the rate of production. The inhibition of respiration and the reduction in mitochondrial membrane potential following ischemia or hypoxia each induce electron leakage, which increases the output rate of reactive oxygen species significantly (24). Edward et al. (25) found that in a rat model of cerebral ischemia, succinate drove the rapid release of superoxide from complex I during reperfusion. Furthermore,higher ROS concentrations may result in prolonged activation of the mitochondrial permeability transition pore (MPTP) and inner membrane anion channel (IMAC). The opening of the above channels alters the changes in intra- and inter- mitochondrial redox homeostasis, leading to a surge of ROS. This ROS-induced ROS release (RIRR) mechanism highlights the mitochondria as the main source of ROS production (26). Superoxide dismutase(SOD) is present in large quantities in the mitochondria of glial cells, and it breaks down superoxide into oxygen and hydrogen peroxide (27). Catalase (CAT) and glutathione peroxidase (GPx) are also involved in the degradation of free radicals (28).
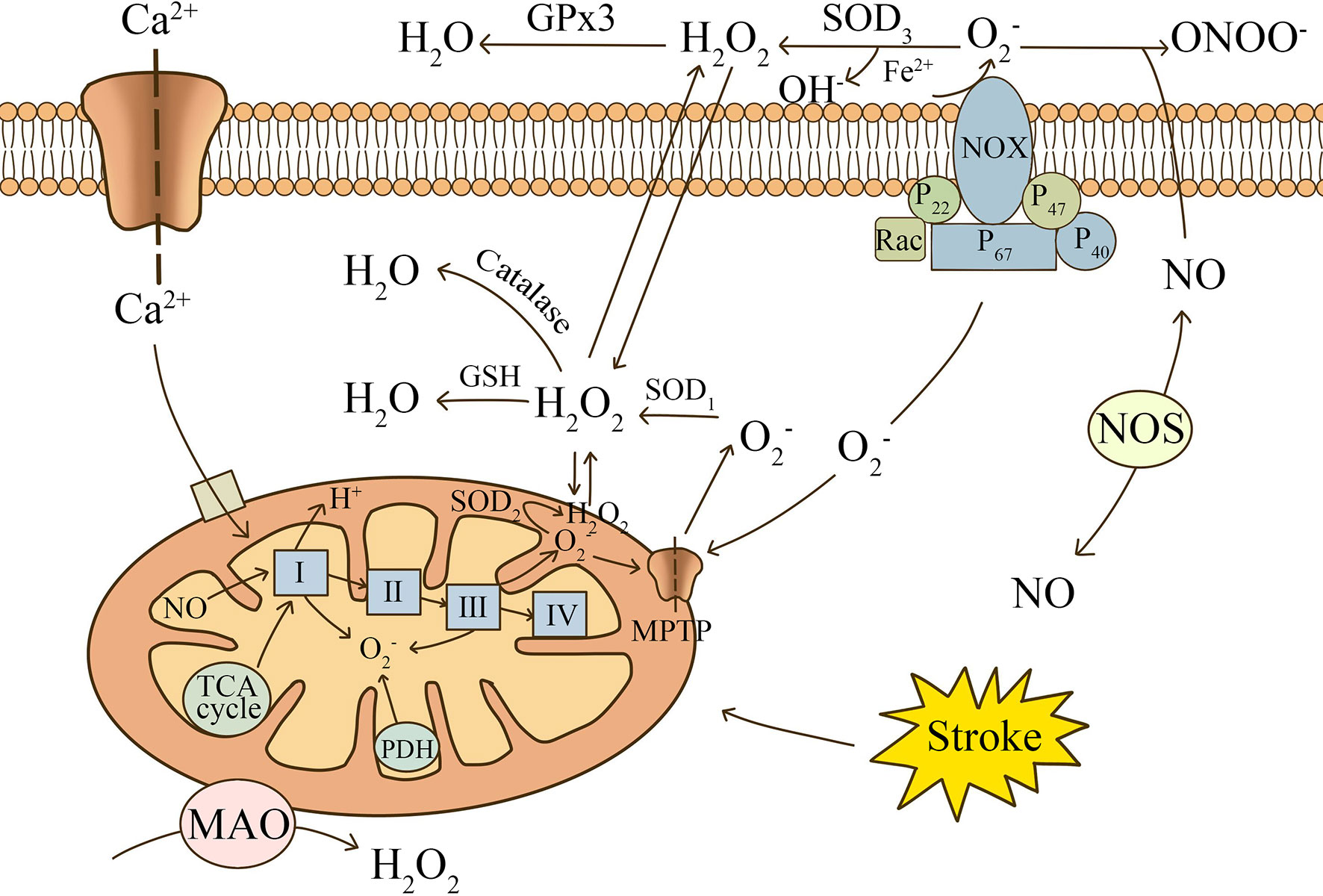
Figure 1 The main mechanisms of ROS production after stroke. Electrons released from the electron transport chain (ETC) react with O2 to produce superoxide. Mitochondrial complexes, oxidases in the matrix, and NADPH oxidase are involved in the release of ROS. Ca2+, NO, and succinate produced by the TCA cycle are important contributing factors. is catalyzed by SOD to produce H2O2, which can be decomposed into H2O and O2. NO can combine with to form ONOO-, causing nitrosative stress.
NADPH-Derived Oxidative Stress in Glial Cells
The NOX family, especially NOX2 and NOX4, is highly expressed in glial cells (29–31). NADPH oxidase generates considerably more ROS than mitochondria and can release ROS out of the cell (32). Therefore, it is considered to be a key factor in the abnormal elevation of ROS after stroke. Translocation of the functional subunits, p47phox and p67phox, and the small GTPase, Rac1, is required for the activation of NOX2 (33). Toll-like receptors (TLRs) are important receptors on the glial cell surface that promote NOX activation. TLRs mediate phosphorylation of NOX functional subunits and oxidase activation mainly through MyD88-dependent and IL-1 receptor-associated kinase 4 (IRAK4)-dependent pathways (34, 35). The remaining surface receptors, including Complement receptor 3, Ionotropic P2X and metabotropic P2Y purinergic receptors, activate NOX mainly through the p38MAPK and ERK pathways (36, 37). Upon activation, NOX2 carries cytoplasmic electrons across the cell membrane and reduces extracellular molecular oxygen to . The released rapidly produces H2O2 with the participation of SOD3. H2O2 is highly lipid soluble and produces OH- through the Fenton reaction in the presence of iron, resulting in oxidative damage to most biological components (38). It is worth mentioning that mitochondria cross over with the NADPH oxidase pathway, increasing the likelihood of mitochondrial uncoupling, thus initiating a secondary surge of mitochondrial ROS (39).
RNS Produced in Glial Cells
In addition to the oxygen radicals and derivatives that can be created after stroke as portrayed above, reactive nitrogen species (RNS) is also introduced in biological oxidation reactions. In the early stages of ischemia, NO released from endothelial NOS (eNOS) is usually considered to have a neuroprotective effect. Researchers have found that low doses of NO can cause vasodilation and improve blood supply to the penumbra region (40). In addition, Irina et al. found that eNOS-deficient mice had a higher likelihood of microcirculatory failure and death occurring after subarachnoid hemorrhage (41). However, studies have shown that glial cells release large amounts of NO under pathological conditions mainly through inducible NOS (iNOS) (42, 43). Lipopolysaccharide (LPS) and iron ions were found to induce iNOS expression in astrocytes and promote NO production (44, 45). Excess NO reacts with to form a strongly oxidizing peroxynitrite (ONOO-), leading to nitrosative stress (46). Mice experiencing transient or permanent cerebral ischemia have reduced infarct size and more preserved neurological function by knocking down iNOS gene expression or using NOS inhibitors (47, 48).Therefore, RNS may be a therapeutic target for the treatment of post-stroke ischemic injury.
Oxidative Stress-Associated Pathological Processes After Stroke
Oxidative Stress and Neuroinflammation
After cerebral ischemia, inflammatory reactions are initiated within the occluded blood vessels. Numerous cells, including astrocytes, are involved in the inflammatory process. ROS trigger nuclear transcription factors, such as nuclear factor kappa B (NF-κB), interferon regulatory factor 1, hypoxia-inducible factor 1, STAT3, activator protein 1 (AP-1), and p53, triggering the release of pro-inflammatory mediators (49). Interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and IL-6, are the main inflammatory factors that trigger and exacerbate the inflammatory response after stroke (50). Cell injury or necrosis release DAMPs, including high-mobility group box 1 (HMGB1), heat shock proteins (Hsps), and peroxiredoxin. DAMPs support excessive ROS production by activating NOX2 and NOX4 in inflammatory cells through pattern recognition receptors, CR3 and TLR4 (31). It is worth mentioning that microglia, driven by inflammatory factors, exhibit greater ROS production than neutrophils after stroke (51). Nuclear translocation of NF-κB plays a critical role in inflammatory cell-mediated responses to neuroinflammation and other injuries (52). Junghyung et al. (53) found that mROS regulates LPS-induced pro-inflammatory responses in microglia through the pathways of NF-κB and MAPK. In summary, oxidative stress and neuroinflammation are closely related, and together they contribute to the development of post-ischemic cerebral histopathological damage.
Oxidative Stress and Excitotoxicity
Glutamate, the main neurotransmitter of the CNS, plays an essential role in message transmission, axon guidance, synaptic plasticity, and neuronal growth and development (54). After ischemia, free radicals are involved in the extracellular release of glutamate from nerve cells (55). Accumulation of glutamate in the extracellular space causes excitotoxicity (56). Glutamate can alter the osmotic gradient of the cell membrane, leading to excessive influx of water and Cl- into the cell, causing neuronal swelling. In addition, glutamate activates receptors such as NMDA and AMPA, and the impaired Na+/K+ATPase function promotes the inward flow of calcium ions (57). Excess calcium ions accumulate in the mitochondrial matrix, impairing the mitochondrial electron transport chain and increasing ROS production (58). Furthermore, high extracellular glutamate concentrations inhibit cysteine transport into the C6 glia cells (59). As a raw material for glutathione (GSH) synthesis, a decrease in cystine implies a reduced antioxidant capacity, making neurons inevitably vulnerable to oxidative damage. Overall, the accumulation of glutamate, with abnormal changes in Ca2+ concentration, is very closely related to the increase in ROS after stroke.
Oxidative Stress and Apoptosis
Programmed cell death, or apoptosis for short, is a strictly controlled process used to remove harmful cells in order to maintain cellular homeostasis (60). Excess free radicals can induce apoptosis by causing oxidative damage from lipid exposure, abnormal protein folding, and DNA breakage from multiple angles. Most of the peroxide-mediated apoptosis involves the activation of cysteinases. Additionally, H2O2 can induce increased mitochondrial membrane permeability, chromatin aggregation, and ultimately apoptosis by binding to death receptors and stimulating endoplasmic reticulum stress (60). When exposed to higher doses of H2O2, the tumor suppressor protein, p53, activates the transcription of pro-apoptotic genes (Bcl-2, Noxa) intracellularly and stimulates pro-apoptotic factors (Fas, DR-x) extracellularly to regulate apoptosis (61). Oxidative stress also upregulates the expression of MAPK and NF-KB pathways (62), and downregulates PI3K/Akt expression to trigger apoptosis (63). Additionally, the ROS-induced target protein, JNK, activates the expression of members of the Bcl family of pro-apoptotic proteins (64). The internal and external pathways of apoptosis are coordinated, as they both ultimately activate caspase proteins, which together play a role in causing programmed cell death (65).
Astrocyte in Stroke
Reactive Astrogliosis and Glial Scar in Oxidative Stress
The normal functions of astrocytes in the maintenance of the CNS have been well documented (9, 66). In the absence of cerebral blood flow, astrocytes swell, proliferate rapidly, and migrate, and is coupled with increased cell retention of glial fibrillary acidic protein (GFAP) and vimentin (Vim), which signals the onset of reactive astrogliosis (67). A growing number of studies report that certain oxidative stress molecules, such as NO, H2O2, are important in the induction of reactive astrogliosis and glial scar formation (see Figure 2) (68). Amita et al. (69) observed that GFAP and VIM expression was upregulated in astrocytes after exposure to H2O2, and that the intervention of curcumin served to mitigate this tendency. Swarnkaret et al. (70) found that increased ROS upregulated GFAP expression in C6 astrocytes after mitochondrial inhibition with rotenone. Several researchers have shown increased expression and secretion of chondroitin sulfate proteoglycan (CSPG) in an in vitro reactive astrocyte model they designed, which was shown to have an inhibitory effect on neurite growth in axon guidance site assays (71). Excessive astrocyte proliferation combined with pericytes is involved in the formation of glial scar, which is thought to act as a mechanical barrier to the recovery of neurological function (72).Nrf2 is a major antioxidant pathway in astrocytes, and researchers found that a significant reduction in the number of reactive astrocytes in the CA1 region of the hippocampus was related to activation of the Nrf2 pathway following ischemia and hypoxia (73). Liu et al. found that mice lacking Nrf2 had reactive glial proliferation after permanent distal middle cerebral artery occlusion and elevated expression of the water regulatory protein aquaporin 4 levels, and significantly larger infarct size compared to controls (74).Sustained attenuation of reactive glial proliferation in astrocytes and microglia is highly correlated with Nrf2, providing late neuroprotection to ischemic and hypoxic neurons (73). In summary, redox status influences the state of reactive astrocytes and glial scar formation, in which Nrf2 plays an important role.
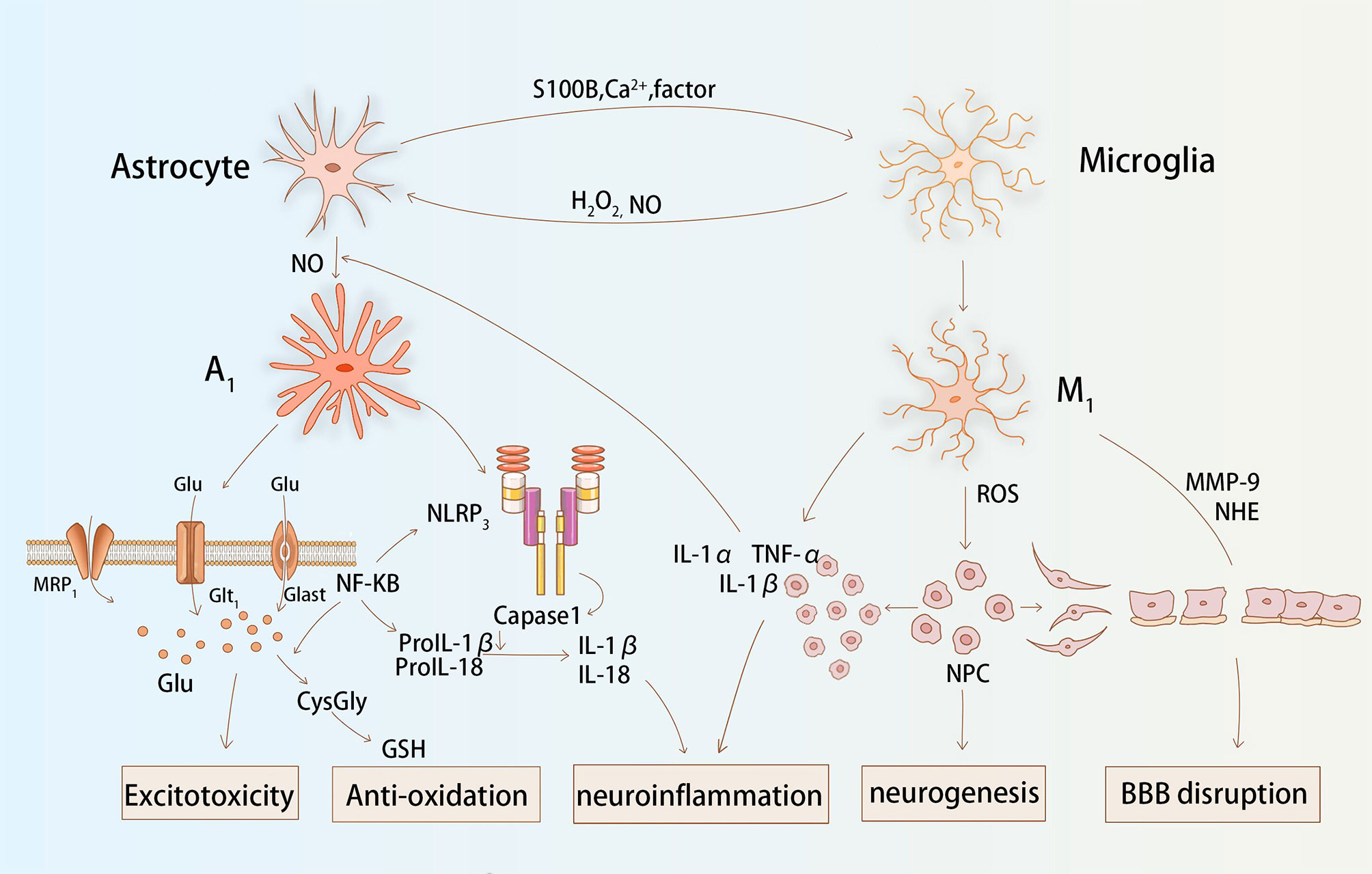
Figure 2 Astrocytes and microglia interact and function together during stroke. Astrocytes release Ca2+, S100B, and cytokines that affect the redox state of microglia. The production of H2O2 and NO by microglia facilitates the activation process of astroglial. Glial cells affect glutamate transport, inflammatory factor secretion, matrix protease activation, and antioxidant production via ROS, which together cause post-stroke excitotoxicity, neuroinflammation, BBB destruction, and neurogenesis.
Astrocytic Oxidative Stress and Neuroinflammation in Stroke
It has been found that glial cells are frequently present in both oxidative stress and inflammatory processes in CNS. ROS activate reactive astrocytes and microglia, stimulate multiple inflammatory signaling pathways, and consistently release large amounts of inflammatory factors (75). NLRP3 inflammasome is the protein complex that has received the most attention in the NLRP subfamily. It has been noted that mROS is involved in ethanol-induced NLRP3 inflammasome activation in astrocytes and that it recruits procaspase 1 (76). It is hydrolyzed to caspase 1 to promote the release of IL-1β and IL-18, which ultimately induces inflammation. Not coincidentally, NADPH oxidase-derived H2O2 is involved in the induction of the inflammatory cascade reaction that occurs in astrocytes with LPS/IFNγ, causing glial cells to express different cytokines, namely IL-1, IL-6, and TNF (77). In in vitro experiments, researchers pre-treated cultured primary astrocytes with oxygen and glucose deprivation. They found that astrocytes secreted significantly higher levels of IL-1β and TNF-α when re-exposed to temporal oxidative stress (78). Moreover, glial cells exposed to inflammation also release ROS to increase the level of oxidative stress. Studies have shown that stimulation of astrocytes with TNF-α and IFN-γ leads to mild upregulation of IL-6 and iNOS and the production of harmful free radicals (79). IL-1 is also involved in the activation of iNOS and stimulates the release of high levels of NO. Nrf2 is a major antioxidant gene in astrocytes. Researchers found that knockout Nrf2 mice secreted more TNF-α, IL-1β, IL-6, and MMP9 via the NF-κB pathway in astrocytes than in controls (80).During the later stages of stroke, astrocytes also secrete factors, such as transforming growth factor-beta (TGF-β), that exert an active anti-inflammatory effect (81).
Overall, ROS can influence the inflammatory response of glial cells. Similarly, inflammatory factors can regulate the redox status of glial cells. The interaction between glial cell ROS and neuroinflammation is a common pathological basis for several CNS diseases.
Astrocytic Oxidative Stress and Excitotoxicity in Stroke
Astrocytes indirectly influence redox levels by regulating extracellular glutamate concentrations mainly through two specific transporter proteins (GLAST: glutamate/aspartate transporter protein and GLT-1: glutamate transporter protein 1) (82). In experiments using GLAST/GLT-1 knockout animals, slower glutamate clearance rates were found in the knockout animals compared to controls, and neuronal deformation was also observed in the hippocampal CA1 region (83). After conducting in vitro experiments, researchers found that GLAST/GLT-1 redistribution occurred in astrocytes after oxygen and glucose deprivation, causing a consequent impairment of glutamate transport (84). The accumulation of extracellular glutamate stimulates Ca2+ channels to stay open for an extended duration, resulting in a large influx of Ca2+. NMDA receptors or AMPA receptors are activated by Ca2+, and generate large amounts of ROS (57).
Relatively, the transport of glutamate by astrocytes is also conditioned by the redox state of the CNS (84, 85). In a rat model of cerebral hemorrhage, researchers observed a decrease in GLAST and GLT-1 expression at 6 hours and 72 hours, respectively, which was accompanied by a massive amount of ROS production (86). Antioxidants such as hydrogen sulphide (H2S) maintain normal glutamate metabolism by enhancing glutamate uptake by astrocytes pre-treated with H2O2 (87). In the study by Matos et al. (88), Aβ inhibited GLAST and GLT-1 in a non-competitive manner to reduce glutamate intake, with a cascade of oxidative stress and MAPK signaling pathways involved. In astrocytes, ROS is also involved in the secretion of glutamate. A study found that ROS production in rat hippocampal astrocytes cultured with ethanol stimulated glial cell cytostasis and increased glutamate secretion, leading to hippocampal neurotoxicity (89). Paralleled with ROS, the RNS plays a similar role. The project by Tomoaki et al. confirms that there is a prominent relationship between NO production and enhanced cytokine-induced calcium-dependent glutamate release in astrocytes cultured in vitro (90). Conversely, the effects of the remaining free radical components on glutamate transport and metabolism in astrocytes have seldomly been reported and still warrants exploration.
Astrocytic Oxidative Stress and Synthetic GSH in Stroke
Glutathione, as an important oxidative detoxifier in the body, plays an important role in protecting cells from ROS. GSH exerts its antioxidant effect when oxidized by hydrogen peroxide and lipid peroxides to Glutathione disulfide (GSSG) (91). Astrocytes appear to be important in GSH metabolism as they can deliver more GSH compared to microglia, oligodendrocytes, and neurons (92). Researchers found that in mice with brain hemorrhage, heme induced peroxynitrite production, causing oxidative damage. This damaging effect was shown to be associated with GSH depletion in astrocytes, and the toxic response was attenuated by exogenous GSH supplementation (93). Guo et al. (94) showed that the AMPK-PGC-1α signaling axis positively stimulates γ-glutamylcysteine synthetase expression in astrocytes, and it increases the rate of GSH synthesis. The antioxidant effects of dehydroascorbic acid (DHA) are widely known, and studies have found that DHA increases the rate of glutathione production in primary astrocytes, thereby protecting astrocytes from H2O2-induced cytotoxicity (95). GSH synthesis is also regulated by the inflammatory response, and In vitro studies have demonstrated that IL-1β stimulates astrocyte upregulation of GSH synthesis via the NF-κB pathway (96). Besides, astrocytes also release GSH precursors (CysGly) that can be used for de novo synthesis of GSH in neurons, indirectly increasing oxidative defense capacity (97). Members of the multidrug resistance protein (MRP) family, mainly MRP1, reportedly mediate the extracellular release of astrocytic GSH and similarly transmit the efflux of the strongly oxidative GSSG (98). It has been demonstrated that the antioxidant, alpha-tocotrienol (TCT), may exert neuroprotective effects after stroke by inhibiting MRP1-mediated GSSG release (99). Johannes et al. (100) showed that high concentrations of MK571 (MRP1 inhibitor) could only achieve 63% inhibition of GSH release from astrocytes. This suggests that there are other protein channels involved in GSH transport and that other MRP family members are candidates (92).
Astrocytic Oxidative Stress and Nrf2-ARE Pathway in Stroke
The Nrf2-ARE signaling axis is an essential endogenous antioxidant pathway (101). Nrf2 is an inducible transcription factor that promotes the expression of cytoprotective proteins. ARE is an antioxidant response element present in a variety of functional enzymes, such as HO-1, NADPH-quinone oxidoreductase (NQO1), glutathione synthase and MRP1 (see Figure 3) (102). Astrocytes have a powerful capacity to cope with oxidative harm and are resistant to oxidative stress because the Nrf2-ARE pathway is extremely stable within them (103). A study using mixed primary cultures showed that tert-butylhydroquinone (tBHQ)-induced ARE activation effects were highly selective for astrocyte subpopulations, not neurons (104). Furthermore, in an in vitro model of cerebral ischemia, increased expression of Nrf2 by tBHQ prompted astrocytes to secrete the anti-inflammatory cytokine, IL-10 (78). Another experiment using sub-toxic doses of H2O2 to induce Nrf2 activation also demonstrated that astrocytes are critical loci (105). In some models of permanent cerebral ischemia(pMCAO), Nrf2 and protein expression levels of HO-1 and SOD were upregulated in the ischemic cortex within 24 hours of the intervention (106). Cytoprotective proteins downstream of Nrf2, such as HO-1 and NQO1, were significantly increased in peri-infarct tissue samples ranging from 1.8 to 3.6-fold shortly after ischemia, but not in Nrf2- mice (107). Xie et al. found that the use of Leonurine increased the expression level of Nrf-2 mRNA, and decreased the level of ROS in pMCAO of male rats (108). Dimethyl fumarate (DMF) specifically activates Nrf2 and exerts neuroprotective effects against ischemic brain injury via the Nrf2/HO-1 pathway. Researchers found that DMF significantly reduced the area of brain swelling, the incidence of neurological deficits, and the quantity of activated microglia and macrophages (109, 110). Moreover, Nrf2 is also involved in regulating the metabolism of hemoglobin and iron, and plays a role in avoiding neurological damage after intracerebral hemorrhage (111). In a Parkinson’s disease(PD) mouse model, Nrf2 knockout mice are more prone to PD-like damage, and this toxic response is mitigated by overexpression of Nrf2 in astrocytes (112). Using transgenic mice overexpressing Nrf2 crossed with amyotrophic lateral sclerosis mice, the researchers found that mice with the specific gene had a delayed onset of disease and longer duration of survival (113). The Nrf2 pathway is therefore an effective and promising therapeutic target in the regulation of oxidative and reductive homeostasis, whether in neurodegenerative disease or in stroke.
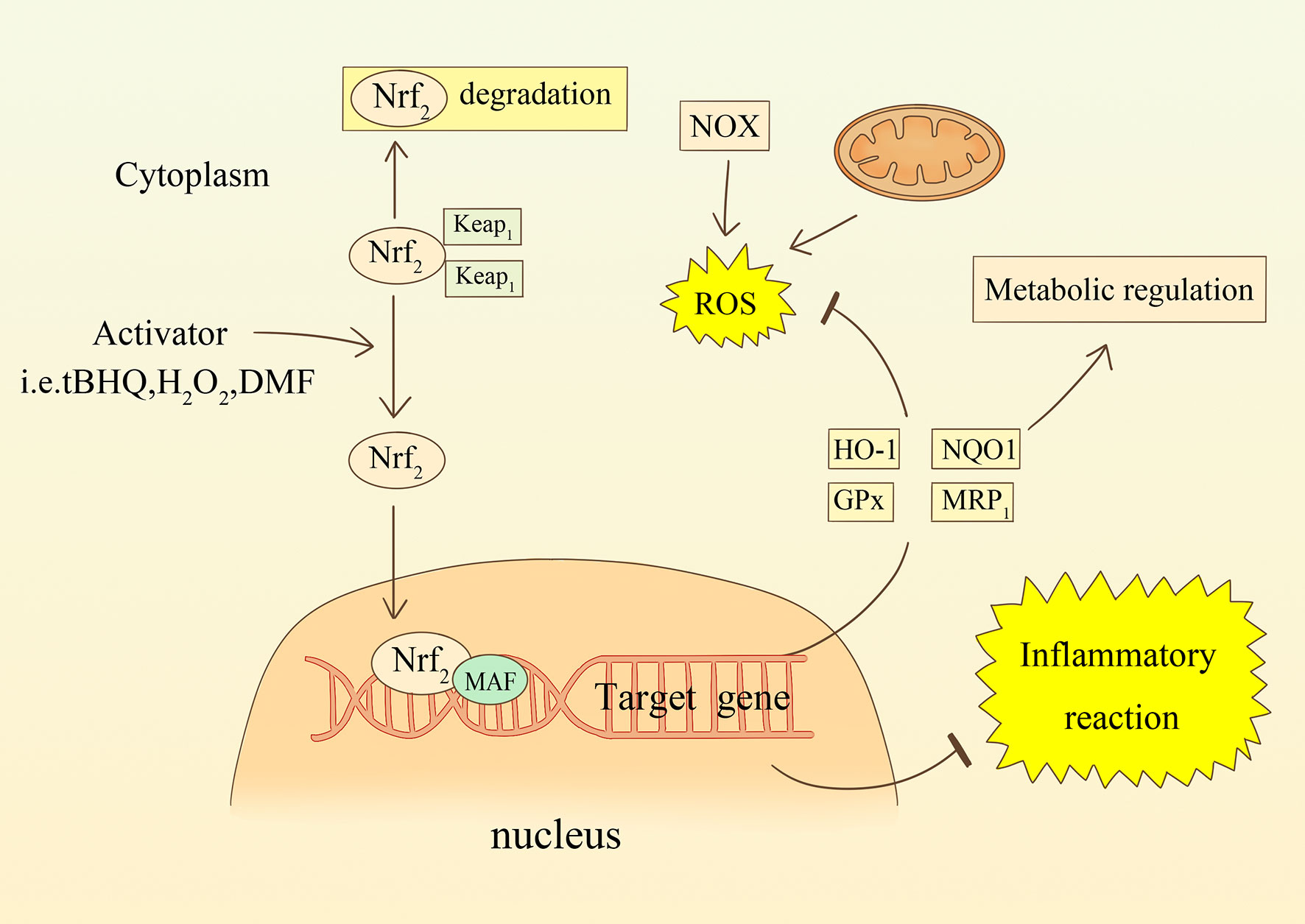
Figure 3 The relationship between oxidative stress and the Nrf2 signaling pathway in astrocytes. ROS or compounds stimulate Nrf2 transcription into the nucleus and participate in the transcription of target genes. increased synthesis of enzymes such as HO-1, NQO1, GPx, and MRP1 counteract oxidative stress and regulate metabolism. Nrf2 is also involved in regulating inflammatory responses.
Microglia in Stroke
Microglia Activation After Stroke
Microglia are the resident immune cells of the CNS. They are rapidly activated and migrate to the lesion area in response to harmful stimuli, such as infection, stroke, trauma, tumors, etc. Activation of microglia is a key part of the cerebral defense system. Microglial activation can be detected in the ischemic core and in the peri-infarct area 24 hours after cerebral ischemia (114). To identify activated microglia more precisely, Iba1, IB4, F4/80, CD11b, and CD68 have been defined as surface markers of activated cells, and increased CD11b expression corresponds to the severity of microglial activation (115). The researchers induced high CD11b expression in mouse BV-2 microglia by stimulating them with bacterial LPS and other harmful molecules, such as IL-1β, IL-12, and double-stranded RNA (poly(IC)), but this effect was blocked by antioxidants, suggesting that ROS are involved in microglial activation (116). Another study also corroborated that inhibition of ROS significantly prevented cell proliferation and microglial activation (117). According to Palwinder’s description, microglial activation due to the pro-inflammatory factor, IL-1β, was inhibited in primary rat glial cultures when an NADPH oxidase inhibitor (apocynin) was introduced. When NADPH oxidase activator, such as arachidonic acid, was introduced into the system, the rate of microglia proliferation was accelerated (118). The above evidence suggests that ROS, mainly hydrogen peroxide from NADPH oxidase, may be a key player in the activation and proliferation of microglia after stroke.
Oxidative Stress and Neuroinflammation in Microglia After Stroke
Different stimulating factors, periods of action, and environmental variation stimulate microglia to undergo polarization into different phenotypes: M1 (classical pro-inflammation) and M2 (alternative protection) (119, 120). INOS and arginase-1 are often considered accurate markers for the characterization of M1 and M2 microglia, respectively (121). Siddharama et al. (77) found that NADPH oxidase in rat microglia was rapidly activated by LPS and IFNγ and that it released large amounts of ROS, which induced iNOS and NO production. However, when they transfected microglia with a mutant form of the p47 phox subunit of NADPH oxidase, the expression of both ROS and iNOS was inhibited. This suggests that the pro-inflammatory response of microglia requires the involvement of functional NADPH oxidase and that ROS is an important mediator in the activation of MAPK and NF-κB. Furthermore, the researchers found that H2O2 was the ROS that promoted iNOS expression in LPS-pre-treated microglia, although this effect could not be triggered when H2O2 was applied alone (122). In in vivo and in vitro stroke models, it was found that NOX2-mediated oxidative stress in microglia induces the onset of inflammatory responses after ischemia-reperfusion. Inhibition of NADPH oxidase using apocynin resulted in significant inhibition of IL-6 release levels from microglia (123). IL-4 is commonly used to induce activation of M2 microglia and expression of arginase. Rotenone inhibits IL-4-induced activation of the M2 phenotype in microglia and expression of arginase, as described by Annette et al. (124). Furthermore, they found that IL-4 reduced LPS-stimulated IL-6 and TNF-α secretion, and that the addition of rotenone attenuated this effect. By specifically knocking out p47 phox subunit, the researchers discovered that microglia from p47 phox-/- mice overexpressed IL-4 and arginase 1 compared to wild-type mice, a phenomenon also verified in mice treated with NADPH oxidase inhibitors (125). Nrf2 plays a role in facilitating the transition of microglia to M2. The researchers found that in a model of Parkinson’s disease, Nrf2-deficient mice exhibited an increased M1 phenotype, as well as a decreased M2 phenotype (126). We can conclude that in the presence of ROS, microglia may tend to polarize toward M1 and reduce the activation of M2 to exert their vital role in inflammation.
Notably, in recent years, M2 microglia exhibit three subtypes, M2a, M2b, and M2c, depending on the period of stimulation (127).The role of ROS in regulating these three subtypes in ischemia should be further investigated.
Oxidative Stress and Neurogenesis in Microglia After Stroke
Although stroke results in massive neuronal death, a mounting number of studies are finding compensatory neurogenesis as an important mechanism for neural repair and maintaining brain plasticity after injury (128, 129). The subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) of the lateral ventricles are commonly believed to be areas of neurogenesis (130). The aggregation of chemotactic microglia to neurogenic ecological sites is fundamental in exerting their function. Lelli et al. (33) found that NOX2 mediates these processes through colony-stimulating factor 1 receptor (CSF-1R) and vascular endothelial growth factor receptor 1 (VEGFR1). Estrada et al. (131) found that exogenous addition of H2O2 promoted neural progenitor cell (NPC) differentiation and increased the number of oligodendrocytes. Furthermore, microglia remove redundant neurons through phagocytosis. Wakselman et al. (132) demonstrated that CD11b integrin and DAP12 immunoreceptors regulate ROS production in microglia and mediate apoptosis of neurons in the developing hippocampal region. Another study demonstrated that microglia produce and subsequently, physiologically reject Purkinje fibers (133). However, the effect of microglia on neurogenesis is biphasic. After ischemia, Nrf2 levels were significantly reduced in pyridoxine-deficient mice, and neurotrophic factor (BDNF) showed the same trend. This suggests that oxidative stress reduces the regenerative capacity of neurons in the hippocampal region by reducing the secretion of trophic factors (134). In an experiment with co-culture of NPCs and microglia, researchers found that microglia-derived ROS inhibited NPC proliferation via glycogen synthase 3, thereby affecting neural recovery in mice with Parkinson’s disease (135). This was associated with inhibition of the wnt/β-catenin signaling pathway. The secretion of IL-1β by microglia in another experiment activated the p53 gene in NPC by promoting the production of ROS, which stalled the NPC cell cycle and hindered their differentiation process (136). Besides, NO released from microglia exhibits an antiproliferative effect on NPC in the SVZ region (137). Compared with the control group, the release of NO was significantly reduced and the proliferation of NPC was remarkably enhanced in the knockdown iNOS medium. Indeed, the specific relationship between microglia-derived ROS and neurogenesis remains unclear, which requires more research in the future.
Oxidative Stress and Blood Brain Barrier in Microglia After Stroke
The blood-brain barrier (BBB) is a physical barrier formed by endothelial cells (EC) relying on tight junctions (TJ), and is essential for maintaining CNS homeostasis. After stroke, BBB disruption leads to inflammatory cell infiltration and release of toxic factors, increasing disability and mortality. There is evidence that ROS released from activated microglia inevitably damage the BBB (138). In the early stages, ROS induce EC death and drive BBB destruction. EC structures can be maintained via inhibition of ROS through apocynin (14). The researchers found that NOX4 was hyper-expressed in microglia in the ICH model, resulting in remarkable downregulation of multiple TJ protein, including ZO-1, occludin, and claudin-5 (139). After knockdown of NOX4, tight junction proteins were upregulated, brain edema was significantly improved, and BBB integrity was restored. At later stages of BBB disruption, ROS activated MMP-9, which degrades collagen and laminin in the basement membrane and disrupts basement membrane integrity. Usatyuk et al. (140) found that ROS cause lipid peroxidation, leading to an increase in 4-hydroxynonenal (NHE). NHE alters BBB permeability by modulating intercellular adhesion, but also diffuses to distant compartments to impair cellular function. Besides, after stroke, iNOS expression is increased in M1 microglia. Both NO and peroxynitrite can lead to BBB destruction (141). The use of NOS inhibitors prevents MMP activation and attenuates BBB damage. Not specifically, ROS can induce chemotaxis in leukocytes to migrate to CNS, and their released products further degrade the BBB (142). In conclusion, microglia-derived ROS have a very important role in regulating the integrity of the BBB, and will likely be a new direction for future treatment.
Glial Cell-Related Neural Repair After Stroke
A growing number of studies have demonstrated that the adult CNS remains plastic after damage. Glial cells are important players in many processes of neural repair or regeneration. First, glial cells, especially M2 microglia secrete the anti-inflammatory factor IL-10, which counteracts the development of neuroinflammation after stroke and protects endothelial cells from oxidative stress injury (143). IL-4 downregulates the release of ROS or RNS, thereby limiting BBB infiltration (144). Secondly, glial cells can promote stroke repair by promoting angiogenesis and BBB repair. ROS promotes the release of vascular growth factor (VEGF) (145). VEGF, administered late after stroke, promotes angiogenesis in ischemic strokes (146). Although MMP-9, which has a destructive effect in the early phase, is upregulated in the peri-infarct cortex 7-14 days after stroke and shows a angiogenesis-promoting function (147). Moreover, sonic hedgehog released from astrocytes promotes the upregulation of tight junction proteins between capillary endothelial cells to promote the restoration of BBB integrity (148). Thirdly, glial cells play an important role in promoting synaptogenesis and plasticity. Glial cell-derived cholesterol serves as a raw material for synaptogenesis, promotes synaptogenesis and regulates myelin formation after stroke (149). Studies have reported that various molecules secreted by glial cells, such as TNF-α, thrombospondin, hevin have the ability to repair synaptic dysfunction after stroke (150). Beyond that, microglia and astrocytes secrete various nutrients and growth factors such as BDNF, nerve growth factor (NGF), and basic fibroblast growth factor (bFGF), which are useful for neural repair and plasticity (151, 152). In fact, the role of ROS and oxidative stress on brain tissue repair after stroke is still poorly understood, and most of the current studies focus on the aspects that promote brain injury; therefore, further studies on the specific mechanisms of ROS and oxidative stress and cell repair after stroke are needed in the future.
Crosstalk Between Astrocytes and Microglia in Oxidative Stress After Stroke
Both microglia and astrocytes are active participants in the post-stroke pathological process. On one hand, microglia respond to injury earlier than astrocytes and promote astrocyte activation. The surge of ROS after stroke promotes the release of multiple inflammatory mediators from microglia (53). IL-1α and TNF-α induce astrocyte activation and promote their proliferation (153). IL-1β mediates the release of NO from astrocytes, which induces neurotoxicity (154). IL-1β also inhibits the ability of astrocytes to transport glutamate, inducing excitotoxicity in neurons (155). In addition, in Parkinson’s disease, H2O2 produced by microglia can act as a direct signal to regulate astrocyte proliferation, which is associated with STAT1/3 phosphorylation (156). In a model of chronic neuroinflammation, the P2Y6 receptor is involved in the release of NO from microglia, and it promotes astrocytic apoptosis to prevent excessive proliferation (157). Conversely, astrocytes are also involved in regulating the redox status of microglia. The Ca2+ modulated protein, S100B, is released by astrocytes. At physiological concentrations, S100B acts as a trophic factor and exerts neuroprotective effects (158). At higher concentrations, researchers found that S100B stimulates ROS release from microglia and activates p38 and JNK kinases, which mediate NO production (159). Furthermore, Min et al. (160) found that astrocytes produce a soluble heat-intolerant factor that regulates the redox state of microglia. They found that the level of ROS was significantly reduced after treating microglia with astrocyte culture conditioned medium for 6 and 12 hours. This inhibitory effect was associated with nuclear translocation of Nrf2 and promoted the expression of the antioxidant enzyme, HO-1, in an ARE-dependent manner. Moreover, astrocytes contribute to the regulation of microglial cell function. H2O2-exposed astrocytes activate the STAT6-COX2 pathway, which regulates the release of TNF-α from microglia through the release of PGE2 and PGI2 (161). Most of the current research exploring crosstalk between microglia and astrocytes has focused on the interaction of immune function, but further studies on the role of oxidative stress in each are necessary.
Potential Therapeutic Targets
It is widely recognized that oxidative stress mechanisms following stroke is an important cause of secondary brain damage. Therefore, inhibition of ROS/RNS production and enhancement of its scavenging efficiency are currently considered viable antioxidant strategies. Table 1 summarizes the relevant drugs and their main mechanisms of action.

Table 1 Approaches to post-stroke oxidative stress.
Inhibition of ROS-Producing Enzymes
The role of NADPH oxidase in mediating oxidative stress after stroke is well established. Apocynin inhibits NADPH oxidase by inhibiting the migration of the p47 phox subunit to the membrane, thus exerting its antioxidant capacity (162). Apocynin-treated microglia exhibit reduced NADPH oxidase activity, decreased ROS production, and reduced necrosis of SH-SY5Y neurons (163). The study demonstrated that after 24 hours of ischemia-reperfusion, mice in the Apocynin-treated group had approximately 50% less infarct area than the wild group (164). Besides, Apocynin significantly inhibits the development of the inflammatory cascade response and decreases the level of apoptosis (165). It is worth mentioning that Apocynin can also act as a non-specific free radical scavenger to reduce oxidative stress (166). Meanwhile, melatonin, an endogenous antioxidant, prevents the complete assembly of NADPH oxidase in microglia. It inhibits the production of superoxide and its derivatives, which may be related to the PI3K/Akt signalling pathway (167).
Scavenge ROS
Edaravone is a synthetic free radical scavenger whose conferring neuroprotective effects have been demonstrated in animal studies (196). In vitro, researchers have demonstrated that edaravone protects neurovascular unit components, including neurons, glial cells, and endothelial cells, from oxidative cell death (168–170). In a randomized clinical trial for acute ischemic stroke, the edaravone treatment group showed higher neurological outcomes at day 90 post-onset (171). However, whether edaravone can modulate inflammation is controversial, and this warrants further exploration in the future.
Melatonin is a neuroendocrine hormone that protects normal cell function by scavenging free radicals, enhancing antioxidants, and inhibiting inflammation (172). We found that ROS and superoxide production were reduced and DNA damage was significantly alleviated in C6 astrocytes treated with melatonin (172, 173). Additionally, melatonin has been demonstrated as a targeted mitochondrial protector, reducing mROS formation (174).
Furthermore, a multitude of compounds exist that exert antioxidant effects and protect the nervous system, such as Sirtuin 1 (175), Ebselen (176), Resveratrol (177), and Sulforaphane (178).
Endogenous Antioxidants
Vitamin C (ascorbic acid) scavenges free radicals produced by normal cellular metabolic processes and also down-regulates the activity of NADPH oxidase, reducing the generation of ROS (179). Nevertheless, fewer studies have been conducted to clarify whether the antioxidant properties of the vitamin are related to glial cells. Hypoxia-inducible factor 1 (HIF-1) plays an integral role in inducing cells to build tolerance to hypoxia (197). Following ischemia, HIF-1 and many of its target genes, such as erythropoietin (EPO) and VEGF, are increased and exert neuroprotective effects (198). Deferoxamine is an inducer of HIF-1 and its mediated antioxidant effect may be related to the inhibition of the Fenton reaction by chelating Fe2+ (180). Clinical trials conducted by Mònica et al. confirmed the effectiveness and safety of desferrioxamine in the treatment of ischemic stroke (181).Neuroglobin protein (ngb1) is a member of the bead protein family and its initiation process is regulated by transcription factors, such as HIF-1 and VEGF (182). Animal experiments have demonstrated that sub-nanomolar concentrations of Ngb1 prevent ROS accumulation, reduced antioxidant enzymes, and reduced expression of apoptotic proteins in astrocytes after exposure to H2O2 (183).
Mitochondria-Targeted Protective Agents
Mitoquinone (MitoQ), a mitochondria-specific antioxidant, has the advantage of being able to penetrate the mitochondrial phospholipid layer and accumulate rapidly, thereby eliminating ROS at the source (184). The researchers observed that carbon nanoparticles C3, through photosensitizing effects and depolarization of mitochondrial membrane potential, caused astrocytes to produce large amounts of ROS (174). This effect was significantly inhibited when glial cells were protected by MitoQ. Furthermore, it has been observed in ICH models that MitoQ inhibits M1 microglia activation and promotes the M2 phenotype shift, which also contributes to the reduction of brain edema and BBB destruction (185). Mice treated with 4 mg/kg MitoQ after TBI exhibited increased activity of multiple antioxidant enzymes (including SOD and GPx) and attenuation of neurological deficits, an effect likely mediated through activation of the Nrf2/ARE pathway (186). A recent clinical study showed that high oral ingestion of Mitoquinone improved endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral arterial disease (187). Despite its powerful and rapid antioxidant capacity, the safe dose range of MitoQ has not been defined (188).
Epigenetic Factors (microRNA)
The intron DNA encodes the production of MicroRNA, which binds to mRNA, and either accelerates, slows, or blocks the translation process of mRNA, thereby regulating different aspects of the pathophysiological process of stroke (199). Studies have shown that treatment with miR-424 mice after 24 hours of acute cerebral ischemia exhibited increased levels of SOD, MnSOD, and Nrf2 expression (189). MiR-424 also limits the development of cerebral infarct volume by inhibiting microglial activation and inflammatory factor production. Besides, miR-152-3p agomirs (190) and miR-652 (191) also showed different levels of antioxidant capacity. A certain miRNA expressed only in the CNS, such as miR-124, can reduce neuronal death after MCAO by affecting the activation of the M2 phenotype in microglia (192, 193). Finally, miR-421-3p (194) and miR-181a (195) could also protect neurological function by modulating the post-stroke inflammatory response. These studies suggest that the use of miRNAs offers new insights into mitigating neurological impairment and promoting brain recovery after stroke.
Conclusion and Perspectives
In summary, the redox status of the central nervous system influences the secondary damage and repair of brain tissue after stroke. Glial cells play a key role in maintaining the balance of oxidation and antioxidation in the CNS under physiological or pathological conditions. On one hand, neuroglia can act as a major source of ROS, causing oxidative damage and mediating secondary damage, such as neuroinflammation, excitotoxicity, and blood-brain barrier disruption. On the other hand, ROS affect the phenotype of glial cells, such as activating astrocytes and promoting polarization of microglia. Thus, ROS is an important mediator for glial cells to perform their functions. Glial cells are involved in the neural repair process after stroke, including limiting the spread of diseased tissue, enhancing antioxidant capacity, and participating in neurogenesis. In addition, there are associations between various glial cells that jointly mediate the pathophysiological processes after stroke. Recently, studies on antioxidant therapy have started to focus on the role of glial cells. These studies have been conducted from the perspective of inhibiting ROS production or accelerating ROS clearance, and some progress has been made. However, considering the different roles played by glial cells at various times, studies should be individually designed. Future studies can further explore the specific mechanisms of glial cell-mediated oxidative stress and the functional differences between different glial cell types, which would be very promising targets in the treatment of CNS diseases.
Author Contributions
GZ and XW were responsible for the study concept, design, and writing of the first draft. LC, CL, ZF, YF, and WY participated in the literature collection. All authors agreed to approve the final manuscript.
Funding
This study was supported by financial assistance from the Department of Neurosurgery, Hangzhou First People’s Hospital, Zhejiang University School of Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boursin P, Paternotte S, Dercy B, Sabben C, Maier B. [Semantics, Epidemiology and Semiology of Stroke]. Soins (2018) 63(828):24–7. doi: 10.1016/j.soin.2018.06.008
2. Barthels D, Das H. Current Advances in Ischemic Stroke Research and Therapies. Biochim Biophys Acta Mol Basis Dis (2020) 1866(4):165260. doi: 10.1016/j.bbadis.2018.09.012
3. Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, et al. Global and Regional Burden of Stroke During 1990-2010: Findings From the Global Burden of Disease Study 2010. Lancet (2014) 383(9913):245–54. doi: 10.1016/S0140-6736(13)61953-4
4. Belur PK, Chang JJ, He S, Emanuel BA, Mack WJ. Emerging Experimental Therapies for Intracerebral Hemorrhage: Targeting Mechanisms of Secondary Brain Injury. Neurosurg Focus (2013) 34(5):E9. doi: 10.3171/2013.2.FOCUS1317
5. Islam MT. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol Res (2017) 39(1):73–82. doi: 10.1080/01616412.2016.1251711
6. Malik AR, Willnow TE. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int J Mol Sci (2019) 20(22):5671. doi: 10.3390/ijms20225671
7. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
8. Oakes SA, Papa FR. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
9. Pekny M, Wilhelmsson U, Pekna M. The Dual Role of Astrocyte Activation and Reactive Gliosis. Neurosci Lett (2014) 565:30–8. doi: 10.1016/j.neulet.2013.12.071
10. Vicente-Gutierrez C, Bonora N, Bobo-Jimenez V, Jimenez-Blasco D, Lopez-Fabuel I, Fernandez E, et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat Metab (2019) 1(2):201–11. doi: 10.1038/s42255-018-0031-6
11. Nayernia Z, Jaquet V, Krause KH. New Insights on NOX Enzymes in the Central Nervous System. Antioxid Redox Signal (2014) 20(17):2815–37. doi: 10.1089/ars.2013.5703
12. Alfonso-Loeches S, Urena-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C. Role of Mitochondria ROS Generation in Ethanol-Induced NLRP3 Inflammasome Activation and Cell Death in Astroglial Cells. Front Cell Neurosci (2014) 8:216. doi: 10.3389/fncel.2014.00216
13. Dal-Cim T, Poluceno GG, Lanznaster D, de Oliveira KA, Nedel CB, Tasca CI. Guanosine Prevents Oxidative Damage and Glutamate Uptake Impairment Induced by Oxygen/Glucose Deprivation in Cortical Astrocyte Cultures: Involvement of A1 and A2A Adenosine Receptors and PI3K, MEK, and PKC Pathways. Purinergic Signal (2019) 15(4):465–76. doi: 10.1007/s11302-019-09679-w
14. Kacimi R, Giffard RG, Yenari MA. Endotoxin-Activated Microglia Injure Brain Derived Endothelial Cells via NF-KappaB, JAK-STAT and JNK Stress Kinase Pathways. J Inflamm (Lond) (2011) 8:7. doi: 10.1186/1476-9255-8-7
15. Becerra-Calixto A, Cardona-Gomez GP. The Role of Astrocytes in Neuroprotection After Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci (2017) 10:88. doi: 10.3389/fnmol.2017.00088
16. Dringen R. Metabolism and Functions of Glutathione in Brain. Prog Neurobiol (2000) 62(6):649–71. doi: 10.1016/s0301-0082(99)00060-x
17. Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, et al. Controlled Enzymatic Production of Astrocytic Hydrogen Peroxide Protects Neurons From Oxidative Stress via an Nrf2-Independent Pathway. Proc Natl Acad Sci U S A (2010) 107(40):17385–90. doi: 10.1073/pnas.1003996107
18. Nathan C, Cunningham-Bussel A. Beyond Oxidative Stress: An Immunologist's Guide to Reactive Oxygen Species. Nat Rev Immunol (2013) 13(5):349–61. doi: 10.1038/nri3423
19. Tejero J, Shiva S, Gladwin MT. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol Rev (2019) 99(1):311–79. doi: 10.1152/physrev.00036.2017
20. Andre-Levigne D, Modarressi A, Pepper MS, Pittet-Cuenod B. Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair. Int J Mol Sci (2017) 18(10):2149. doi: 10.3390/ijms18102149
21. Massaad CA, Klann E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxid Redox Signal (2011) 14(10):2013–54. doi: 10.1089/ars.2010.3208
22. Yang S, Lian G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol Cell Biochem (2020) 467(1-2):1–12. doi: 10.1007/s11010-019-03667-9
23. Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, et al. Complex I Assembly Into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc Natl Acad Sci U S A (2016) 113(46):13063–8. doi: 10.1073/pnas.1613701113
24. Andrabi SS, Parvez S, Tabassum H. Ischemic Stroke and Mitochondria: Mechanisms and Targets. Protoplasma (2020) 257(2):335–43. doi: 10.1007/s00709-019-01439-2
25. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury Through Mitochondrial ROS. Nature (2014) 515(7527):431–5. doi: 10.1038/nature13909
26. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
27. Ma X, He P, Sun P, Han P. Lipoic Acid: An Immunomodulator That Attenuates Glycinin-Induced Anaphylactic Reactions in a Rat Model. J Agric Food Chem (2010) 58(8):5086–92. doi: 10.1021/jf904403u
28. Wang Y, Branicky R, Noe A, Hekimi S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J Cell Biol (2018) 217(6):1915–28. doi: 10.1083/jcb.201708007
29. Devanney NA, Stewart AN, Gensel JC. Microglia and Macrophage Metabolism in CNS Injury and Disease: The Role of Immunometabolism in Neurodegeneration and Neurotrauma. Exp Neurol (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
30. Li H, Wang Y, Feng D, Liu Y, Xu M, Gao A, et al. Alterations in the Time Course of Expression of the Nox Family in the Brain in a Rat Experimental Cerebral Ischemia and Reperfusion Model: Effects of Melatonin. J Pineal Res (2014) 57(1):110–9. doi: 10.1111/jpi.12148
31. Simpson DSA, Oliver PL. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel) (2020) 9(8):743. doi: 10.3390/antiox9080743
32. Brown GC, Borutaite V. There is No Evidence That Mitochondria Are the Main Source of Reactive Oxygen Species in Mammalian Cells. Mitochondrion (2012) 12(1):1–4. doi: 10.1016/j.mito.2011.02.001
33. Haslund-Vinding J, McBean G, Jaquet V, Vilhardt F. NADPH Oxidases in Oxidant Production by Microglia: Activating Receptors, Pharmacology and Association With Disease. Br J Pharmacol (2017) 174(12):1733–49. doi: 10.1111/bph.13425
34. Pacquelet S, Johnson JL, Ellis BA, Brzezinska AA, Lane WS, Munafo DB, et al. Cross-Talk Between IRAK-4 and the NADPH Oxidase. Biochem J (2007) 403(3):451–61. doi: 10.1042/BJ20061184
35. Qu Z, Chen Y, Luo ZH, Shen XL, Hu YJ. 7-Methoxyflavanone Alleviates Neuroinflammation in Lipopolysaccharide-Stimulated Microglial Cells by Inhibiting TLR4/MyD88/MAPK Signalling and Activating the Nrf2/NQO-1 Pathway. J Pharm Pharmacol (2020) 72(3):385–95. doi: 10.1111/jphp.13219
36. Zhou H, Liao J, Aloor J, Nie H, Wilson BC, Fessler MB, et al. CD11b/CD18 (Mac-1) is a Novel Surface Receptor for Extracellular Double-Stranded RNA to Mediate Cellular Inflammatory Responses. J Immunol (2013) 190(1):115–25. doi: 10.4049/jimmunol.1202136
37. Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carri MT, Cozzolino M, et al. The NADPH Oxidase Pathway is Dysregulated by the P2X7 Receptor in the SOD1-G93A Microglia Model of Amyotrophic Lateral Sclerosis. J Immunol (2013) 190(10):5187–95. doi: 10.4049/jimmunol.1203262
38. Armogida M, Nistico R, Mercuri NB. Therapeutic Potential of Targeting Hydrogen Peroxide Metabolism in the Treatment of Brain Ischaemia. Br J Pharmacol (2012) 166(4):1211–24. doi: 10.1111/j.1476-5381.2012.01912.x
39. Fukai T, Ushio-Fukai M. Cross-Talk Between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells (2020) 9(8):1849. doi: 10.3390/cells9081849
40. Bulhak AA, Jung C, Ostenson CG, Lundberg JO, Sjoquist PO, Pernow J. PPAR-Alpha Activation Protects the Type 2 Diabetic Myocardium Against Ischemia-Reperfusion Injury: Involvement of the PI3-Kinase/Akt and NO Pathway. Am J Physiol Heart Circ Physiol (2009) 296(3):H719–27. doi: 10.1152/ajpheart.00394.2008
41. Lenz IJ, Plesnila N, Terpolilli NA. Role of Endothelial Nitric Oxide Synthase for Early Brain Injury After Subarachnoid Hemorrhage in Mice. J Cereb Blood Flow Metab (2021) 41(7):1669–81. doi: 10.1177/0271678X20973787
42. Shen J, Ma S, Chan P, Lee W, Fung PC, Cheung RT, et al. Nitric Oxide Down-Regulates Caveolin-1 Expression in Rat Brains During Focal Cerebral Ischemia and Reperfusion Injury. J Neurochem (2006) 96(4):1078–89. doi: 10.1111/j.1471-4159.2005.03589.x
43. Liu H, Li J, Zhao F, Wang H, Qu Y, Mu D. Nitric Oxide Synthase in Hypoxic or Ischemic Brain Injury. Rev Neurosci (2015) 26(1):105–17. doi: 10.1515/revneuro-2014-0041
44. Jomova K, Valko M. Advances in Metal-Induced Oxidative Stress and Human Disease. Toxicology (2011) 283(2-3):65–87. doi: 10.1016/j.tox.2011.03.001
45. Moriyama M, Fujitsuka S, Kawabe K, Takano K, Nakamura Y. Zinc Potentiates Lipopolysaccharide-Induced Nitric Oxide Production in Cultured Primary Rat Astrocytes. Neurochem Res (2018) 43(2):363–74. doi: 10.1007/s11064-017-2431-5
46. Radi R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc Natl Acad Sci U S A (2018) 115(23):5839–48. doi: 10.1073/pnas.1804932115
47. Zheng L, Ding J, Wang J, Zhou C, Zhang W. Effects and Mechanism of Action of Inducible Nitric Oxide Synthase on Apoptosis in a Rat Model of Cerebral Ischemia-Reperfusion Injury. Anat Rec (Hoboken) (2016) 299(2):246–55. doi: 10.1002/ar.23295
48. Kim DW, Im SH, Kim JY, Kim DE, Oh GT, Jeong SW. Decreased Brain Edema After Collagenase-Induced Intracerebral Hemorrhage in Mice Lacking the Inducible Nitric Oxide Synthase Gene. Laboratory investigation. J Neurosurg (2009) 111(5):995–1000. doi: 10.3171/2009.3.JNS081285
49. Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation After Ischemic Stroke: The Role of Leukocytes and Glial Cells. Exp Neurobiol (2016) 25(5):241–51. doi: 10.5607/en.2016.25.5.241
50. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
51. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional Differences Between Microglia and Monocytes After Ischemic Stroke. J Neuroinflamm (2015) 12:106. doi: 10.1186/s12974-015-0329-1
52. Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte Crosstalk in CNS Inflammation. Neuron (2020) 108(4):608–22. doi: 10.1016/j.neuron.2020.08.012
53. Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, et al. Mitochondrial ROS Govern the LPS-Induced Pro-Inflammatory Response in Microglia Cells by Regulating MAPK and NF-kappaB Pathways. Neurosci Lett (2015) 584:191–6. doi: 10.1016/j.neulet.2014.10.016
54. Fern R, Matute C. Glutamate Receptors and White Matter Stroke. Neurosci Lett (2019) 694:86–92. doi: 10.1016/j.neulet.2018.11.031
55. Wang Z, Wei X, Liu K, Zhang X, Yang F, Zhang H, et al. NOX2 Deficiency Ameliorates Cerebral Injury Through Reduction of Complexin II-Mediated Glutamate Excitotoxicity in Experimental Stroke. Free Radic Biol Med (2013) 65:942–51. doi: 10.1016/j.freeradbiomed.2013.08.166
56. Choi DW. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front Neurosci (2020) 14:579953. doi: 10.3389/fnins.2020.579953
57. Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol Disord Drug Targets (2018) 17(9):689–95. doi: 10.2174/1871527317666180627120501
58. Nicholls DG. Mitochondrial Calcium Function and Dysfunction in the Central Nervous System. Biochim Biophys Acta (2009) 1787(11):1416–24. doi: 10.1016/j.bbabio.2009.03.010
59. Kato S, Negishi K, Mawatari K, Kuo CH. A Mechanism for Glutamate Toxicity in the C6 Glioma Cells Involving Inhibition of Cystine Uptake Leading to Glutathione Depletion. Neuroscience (1992) 48(4):903–14. doi: 10.1016/0306-4522(92)90278-A
60. Redza-Dutordoir M, Averill-Bates DA. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim Biophys Acta (2016) 1863(12):2977–92. doi: 10.1016/j.bbamcr.2016.09.012
61. Yoshida K, Miki Y. The Cell Death Machinery Governed by the P53 Tumor Suppressor in Response to DNA Damage. Cancer Sci (2010) 101(4):831–5. doi: 10.1111/j.1349-7006.2009.01488.x
62. Han Y, Li X, Yan M, Yang M, Wang S, Pan J, et al. Oxidative Damage Induces Apoptosis and Promotes Calcification in Disc Cartilage Endplate Cell Through ROS/MAPK/NF-kappaB Pathway: Implications for Disc Degeneration. Biochem Biophys Res Commun (2019) 516(3):1026–32. doi: 10.1016/j.bbrc.2017.03.111
63. Li D, Ni S, Miao KS, Zhuang C. PI3K/Akt and Caspase Pathways Mediate Oxidative Stress-Induced Chondrocyte Apoptosis. Cell Stress Chaperones (2019) 24(1):195–202. doi: 10.1007/s12192-018-0956-4
64. Sinha K, Das J, Pal PB, Sil PC. Oxidative Stress: The Mitochondria-Dependent and Mitochondria-Independent Pathways of Apoptosis. Arch Toxicol (2013) 87(7):1157–80. doi: 10.1007/s00204-013-1034-4
65. Fujikawa DG. The Role of Excitotoxic Programmed Necrosis in Acute Brain Injury. Comput Struct Biotechnol J (2015) 13:212–21. doi: 10.1016/j.csbj.2015.03.004
66. Misra UK, Singh SK, Kalita J, Kumar A. Astrocyte Activation Following Nitrous Oxide Exposure is Related to Oxidative Stress and Glutamate Excitotoxicity. Brain Res (2020) 1730:146645. doi: 10.1016/j.brainres.2020.146645
67. Nedergaard M, Dirnagl U. Role of Glial Cells in Cerebral Ischemia. Glia (2005) 50(4):281–6. doi: 10.1002/glia.20205
68. Swanson RA, Ying W, Kauppinen TM. Astrocyte Influences on Ischemic Neuronal Death. Curr Mol Med (2004) 4(2):193–205. doi: 10.2174/1566524043479185
69. Daverey A, Agrawal SK. Curcumin Alleviates Oxidative Stress and Mitochondrial Dysfunction in Astrocytes. Neuroscience (2016) 333:92–103. doi: 10.1016/j.neuroscience.2016.07.012
70. Swarnkar S, Singh S, Goswami P, Mathur R, Patro IK, Nath C. Astrocyte Activation: A Key Step in Rotenone Induced Cytotoxicity and DNA Damage. Neurochem Res (2012) 37(10):2178–89. doi: 10.1007/s11064-012-0841-y
71. Yu P, Wang H, Katagiri Y, Geller HM. An In Vitro Model of Reactive Astrogliosis and its Effect on Neuronal Growth. Methods Mol Biol (2012) 814:327–40. doi: 10.1007/978-1-61779-452-0_21
72. Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A Pericyte Origin of Spinal Cord Scar Tissue. Science (2011) 333(6039):238–42. doi: 10.1126/science.1203165
73. Liu L, Vollmer MK, Kelly MG, Fernandez VM, Fernandez TG, Kim H, et al. Reactive Gliosis Contributes to Nrf2-Dependent Neuroprotection by Pretreatment With Dimethyl Fumarate or Korean Red Ginseng Against Hypoxic-Ischemia: Focus on Hippocampal Injury. Mol Neurobiol (2020) 57(1):105–17. doi: 10.1007/s12035-019-01760-0
74. Liu L, Kelly MG, Wierzbicki EL, Escober-Nario IC, Vollmer MK, Dore S. Nrf2 Plays an Essential Role in Long-Term Brain Damage and Neuroprotection of Korean Red Ginseng in a Permanent Cerebral Ischemia Model. Antioxidants (Basel) (2019) 8(8):273. doi: 10.3390/antiox8080273
75. Jensen CJ, Massie A, De Keyser J. Immune Players in the CNS: The Astrocyte. J Neuroimmune Pharmacol (2013) 8(4):824–39. doi: 10.1007/s11481-013-9480-6
76. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci (2014) 15(2):84–97. doi: 10.1038/nrn3638
77. Pawate S, Shen Q, Fan F, Bhat NR. Redox Regulation of Glial Inflammatory Response to Lipopolysaccharide and Interferongamma. J Neurosci Res (2004) 77(4):540–51. doi: 10.1002/jnr.20180
78. Segev-Amzaleg N, Trudler D, Frenkel D. Preconditioning to Mild Oxidative Stress Mediates Astroglial Neuroprotection in an IL-10-Dependent Manner. Brain Behav Immun (2013) 30:176–85. doi: 10.1016/j.bbi.2012.12.016
79. Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential Activation of Astrocytes by Innate and Adaptive Immune Stimuli. Glia (2005) 49(3):360–74. doi: 10.1002/glia.20117
80. Pan H, Wang H, Wang X, Zhu L, Mao L. The Absence of Nrf2 Enhances NF-kappaB-Dependent Inflammation Following Scratch Injury in Mouse Primary Cultured Astrocytes. Mediators Inflamm (2012) 2012:217580. doi: 10.1155/2012/217580
81. Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic Transforming Growth Factor-Beta Signaling Reduces Subacute Neuroinflammation After Stroke in Mice. Glia (2014) 62(8):1227–40. doi: 10.1002/glia.22675
82. Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance Between Glutamate Uptake and Release. Cells (2019) 8(2):184. doi: 10.3390/cells8020184
83. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and Exacerbation of Brain Injury in Mice Lacking the Glutamate Transporter GLT-1. Science (1997) 276(5319):1699–702. doi: 10.1126/science.276.5319.1699
84. Gouix E, Buisson A, Nieoullon A, Kerkerian-Le Goff L, Tauskela JS, Blondeau N, et al. Oxygen Glucose Deprivation-Induced Astrocyte Dysfunction Provokes Neuronal Death Through Oxidative Stress. Pharmacol Res (2014) 87:8–17. doi: 10.1016/j.phrs.2014.06.002
85. Rao SD, Yin HZ, Weiss JH. Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons. J Neurosci (2003) 23(7):2627–33. doi: 10.1523/JNEUROSCI.23-07-02627.2003
86. Neves JD, Vizuete AF, Nicola F, Da Re C, Rodrigues AF, Schmitz F, et al. Glial Glutamate Transporters Expression, Glutamate Uptake, and Oxidative Stress in an Experimental Rat Model of Intracerebral Hemorrhage. Neurochem Int (2018) 116:13–21. doi: 10.1016/j.neuint.2018.03.003
87. Lu M, Hu LF, Hu G, Bian JS. Hydrogen Sulfide Protects Astrocytes Against H(2)O(2)-Induced Neural Injury via Enhancing Glutamate Uptake. Free Radic Biol Med (2008) 45(12):1705–13. doi: 10.1016/j.freeradbiomed.2008.09.014
88. Matos M, Augusto E, Oliveira CR, Agostinho P. Amyloid-Beta Peptide Decreases Glutamate Uptake in Cultured Astrocytes: Involvement of Oxidative Stress and Mitogen-Activated Protein Kinase Cascades. Neuroscience (2008) 156(4):898–910. doi: 10.1016/j.neuroscience.2008.08.022
89. Salazar M, Pariente JA, Salido GM, Gonzalez A. Ethanol Induces Glutamate Secretion by Ca2+ Mobilization and ROS Generation in Rat Hippocampal Astrocytes. Neurochem Int (2008) 52(6):1061–7. doi: 10.1016/j.neuint.2007.11.001
90. Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H. Cytokine-Induced Enhancement of Calcium-Dependent Glutamate Release From Astrocytes Mediated by Nitric Oxide. Neurosci Lett (2008) 432(3):232–6. doi: 10.1016/j.neulet.2007.12.047
91. Lu SC. Glutathione Synthesis. Biochim Biophys Acta (2013) 1830(5):3143–53. doi: 10.1016/j.bbagen.2012.09.008
92. Hirrlinger J, Schulz JB, Dringen R. Glutathione Release From Cultured Brain Cells: Multidrug Resistance Protein 1 Mediates the Release of GSH From Rat Astroglial Cells. J Neurosci Res (2002) 69(3):318–26. doi: 10.1002/jnr.10308
93. Laird MD, Wakade C, Alleyne CH Jr., Dhandapani KM. Hemin-Induced Necroptosis Involves Glutathione Depletion in Mouse Astrocytes. Free Radic Biol Med (2008) 45(8):1103–14. doi: 10.1016/j.freeradbiomed.2008.07.003
94. Guo X, Jiang Q, Tuccitto A, Chan D, Alqawlaq S, Won GJ, et al. The AMPK-PGC-1alpha Signaling Axis Regulates the Astrocyte Glutathione System to Protect Against Oxidative and Metabolic Injury. Neurobiol Dis (2018) 113:59–69. doi: 10.1016/j.nbd.2018.02.004
95. Kim EJ, Park YG, Baik EJ, Jung SJ, Won R, Nahm TS, et al. Dehydroascorbic Acid Prevents Oxidative Cell Death Through a Glutathione Pathway in Primary Astrocytes. J Neurosci Res (2005) 79(5):670–9. doi: 10.1002/jnr.20384
96. He Y, Jackman NA, Thorn TL, Vought VE, Hewett SJ. Interleukin-1beta Protects Astrocytes Against Oxidant-Induced Injury via an NF-kappaB-Dependent Upregulation of Glutathione Synthesis. Glia (2015) 63(9):1568–80. doi: 10.1002/glia.22828
97. Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Munch G. Effect of Nrf2 Activators on Release of Glutathione, Cysteinylglycine and Homocysteine by Human U373 Astroglial Cells. Redox Biol (2013) 1:441–5. doi: 10.1016/j.redox.2013.08.006
98. Minich T, Riemer J, Schulz JB, Wielinga P, Wijnholds J, Dringen R. The Multidrug Resistance Protein 1 (Mrp1), But Not Mrp5, Mediates Export of Glutathione and Glutathione Disulfide From Brain Astrocytes. J Neurochem (2006) 97(2):373–84. doi: 10.1111/j.1471-4159.2006.03737.x
99. Park HA, Kubicki N, Gnyawali S, Chan YC, Roy S, Khanna S, et al. Natural Vitamin E Alpha-Tocotrienol Protects Against Ischemic Stroke by Induction of Multidrug Resistance-Associated Protein 1. Stroke (2011) 42(8):2308–14. doi: 10.1161/STROKEAHA.110.608547
100. Steinmeier J, Dringen R. Exposure of Cultured Astrocytes to Menadione Triggers Rapid Radical Formation, Glutathione Oxidation and Mrp1-Mediated Export of Glutathione Disulfide. Neurochem Res (2019) 44(5):1167–81. doi: 10.1007/s11064-019-02760-1
101. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 Signaling in Oxidative and Reductive Stress. Biochim Biophys Acta Mol Cell Res (2018) 1865(5):721–33. doi: 10.1016/j.bbamcr.2018.02.010
102. Xu X, Zhang Y, Li W, Miao H, Zhang H, Zhou Y, et al. Wogonin Reverses Multi-Drug Resistance of Human Myelogenous Leukemia K562/A02 Cells via Downregulation of MRP1 Expression by Inhibiting Nrf2/ARE Signaling Pathway. Biochem Pharmacol (2014) 92(2):220–34. doi: 10.1016/j.bcp.2014.09.008
103. Vargas MR, Johnson JA. The Nrf2-ARE Cytoprotective Pathway in Astrocytes. Expert Rev Mol Med (2009) 11:e17. doi: 10.1017/S1462399409001094
104. Kraft AD, Johnson DA, Johnson JA. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by Tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons Against Oxidative Insult. J Neurosci (2004) 24(5):1101–12. doi: 10.1523/JNEUROSCI.3817-03.2004
105. Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, et al. Mild Oxidative Stress Activates Nrf2 in Astrocytes, Which Contributes to Neuroprotective Ischemic Preconditioning. Proc Natl Acad Sci U S A (2011) 108(1):E1–2; author reply E3-4. doi: 10.1073/pnas.1015229108
106. Chang CY, Kuan YH, Li JR, Chen WY, Ou YC, Pan HC, et al. Docosahexaenoic Acid Reduces Cellular Inflammatory Response Following Permanent Focal Cerebral Ischemia in Rats. J Nutr Biochem (2013) 24(12):2127–37. doi: 10.1016/j.jnutbio.2013.08.004
107. Liu L, Vollmer MK, Fernandez VM, Dweik Y, Kim H, Dore S. Korean Red Ginseng Pretreatment Protects Against Long-Term Sensorimotor Deficits After Ischemic Stroke Likely Through Nrf2. Front Cell Neurosci (2018) 12:74. doi: 10.3389/fncel.2018.00074
108. Xie YZ, Zhang XJ, Zhang C, Yang Y, He JN, Chen YX. Protective Effects of Leonurine Against Ischemic Stroke in Mice by Activating Nuclear Factor Erythroid 2-Related Factor 2 Pathway. CNS Neurosci Ther (2019) 25(9):1006–17. doi: 10.1111/cns.13146
109. Yao Y, Miao W, Liu Z, Han W, Shi K, Shen Y, et al. Dimethyl Fumarate and Monomethyl Fumarate Promote Post-Ischemic Recovery in Mice. Transl Stroke Res (2016) 7(6):535–47. doi: 10.1007/s12975-016-0496-0
110. Lin R, Cai J, Kostuk EW, Rosenwasser R, Iacovitti L. Fumarate Modulates the Immune/Inflammatory Response and Rescues Nerve Cells and Neurological Function After Stroke in Rats. J Neuroinflamm (2016) 13(1):269. doi: 10.1186/s12974-016-0733-1
111. Kasai S, Mimura J, Ozaki T, Itoh K. Emerging Regulatory Role of Nrf2 in Iron, Heme, and Hemoglobin Metabolism in Physiology and Disease. Front Vet Sci (2018) 5:242. doi: 10.3389/fvets.2018.00242
112. Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, et al. Nrf2-Mediated Neuroprotection in the MPTP Mouse Model of Parkinson's Disease: Critical Role for the Astrocyte. Proc Natl Acad Sci U S A (2009) 106(8):2933–8. doi: 10.1073/pnas.0813361106
113. Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 Activation in Astrocytes Protects Against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J Neurosci (2008) 28(50):13574–81. doi: 10.1523/JNEUROSCI.4099-08.2008
114. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull (2019) 35(5):921–33. doi: 10.1007/s12264-019-00388-3
115. Morrison HW, Filosa JA. A Quantitative Spatiotemporal Analysis of Microglia Morphology During Ischemic Stroke and Reperfusion. J Neuroinflamm (2013) 10:4. doi: 10.1186/1742-2094-10-4
116. Roy A, Jana A, Yatish K, Freidt MB, Fung YK, Martinson JA, et al. Reactive Oxygen Species Up-Regulate CD11b in Microglia via Nitric Oxide: Implications for Neurodegenerative Diseases. Free Radic Biol Med (2008) 45(5):686–99. doi: 10.1016/j.freeradbiomed.2008.05.026
117. Zou CG, Zhao YS, Gao SY, Li SD, Cao XZ, Zhang M, et al. Homocysteine Promotes Proliferation and Activation of Microglia. Neurobiol Aging (2010) 31(12):2069–79. doi: 10.1016/j.neurobiolaging.2008.11.007
118. Mander PK, Jekabsone A, Brown GC. Microglia Proliferation is Regulated by Hydrogen Peroxide From NADPH Oxidase. J Immunol (2006) 176(2):1046–52. doi: 10.4049/jimmunol.176.2.1046
119. Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM. Microglia and Macrophages Differentially Modulate Cell Death After Brain Injury Caused by Oxygen-Glucose Deprivation in Organotypic Brain Slices. Glia (2013) 61(5):813–24. doi: 10.1002/glia.22478
120. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
121. Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J Neuroinflamm (2014) 11:98. doi: 10.1186/1742-2094-11-98
122. Eguchi H, Fujiwara N, Sakiyama H, Yoshihara D, Suzuki K. Hydrogen Peroxide Enhances LPS-Induced Nitric Oxide Production via the Expression of Interferon Beta in BV-2 Microglial Cells. Neurosci Lett (2011) 494(1):29–33. doi: 10.1016/j.neulet.2011.02.047
123. Liu H, Wei X, Kong L, Liu X, Cheng L, Yan S, et al. NOD2 is Involved in the Inflammatory Response After Cerebral Ischemia-Reperfusion Injury and Triggers NADPH Oxidase 2-Derived Reactive Oxygen Species. Int J Biol Sci (2015) 11(5):525–35. doi: 10.7150/ijbs.10927
124. Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC, et al. Effects of Mitochondrial Dysfunction on the Immunological Properties of Microglia. J Neuroinflamm (2010) 7:45. doi: 10.1186/1742-2094-7-45
125. Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH Oxidase Promotes Alternative and Anti-Inflammatory Microglial Activation During Neuroinflammation. J Neurochem (2012) 120(2):292–301. doi: 10.1111/j.1471-4159.2011.07572.x
126. Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 Regulates Microglial Dynamics and Neuroinflammation in Experimental Parkinson's Disease. Glia (2010) 58(5):588–98. doi: 10.1002/glia.20947
127. Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, et al. Characterization of Phenotype Markers and Neuronotoxic Potential of Polarised Primary Microglia In Vitro. Brain Behav Immun (2013) 32:70–85. doi: 10.1016/j.bbi.2013.02.005
128. Lin R, Cai J, Nathan C, Wei X, Schleidt S, Rosenwasser R, et al. Neurogenesis Is Enhanced by Stroke in Multiple New Stem Cell Niches Along the Ventricular System at Sites of High BBB Permeability. Neurobiol Dis (2015) 74:229–39. doi: 10.1016/j.nbd.2014.11.016
129. Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O. Neurogenesis and Inflammation After Ischemic Stroke: What is Known and Where We Go From Here. J Cereb Blood Flow Metab (2014) 34(10):1573–84. doi: 10.1038/jcbfm.2014.130
130. Zhao C, Deng W, Gage FH. Mechanisms and Functional Implications of Adult Neurogenesis. Cell (2008) 132(4):645–60. doi: 10.1016/j.cell.2008.01.033
131. Perez Estrada C, Covacu R, Sankavaram SR, Svensson M, Brundin L. Oxidative Stress Increases Neurogenesis and Oligodendrogenesis in Adult Neural Progenitor Cells. Stem Cells Dev (2014) 23(19):2311–27. doi: 10.1089/scd.2013.0452
132. Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental Neuronal Death in Hippocampus Requires the Microglial CD11b Integrin and DAP12 Immunoreceptor. J Neurosci (2008) 28(32):8138–43. doi: 10.1523/JNEUROSCI.1006-08.2008
133. Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia Promote the Death of Developing Purkinje Cells. Neuron (2004) 41(4):535–47. doi: 10.1016/S0896-6273(04)00069-8
134. Jung HY, Kim W, Hahn KR, Kang MS, Kim TH, Kwon HJ, et al. Pyridoxine Deficiency Exacerbates Neuronal Damage After Ischemia by Increasing Oxidative Stress and Reduces Proliferating Cells and Neuroblasts in the Gerbil Hippocampus. Int J Mol Sci (2020) 21(15):5551. doi: 10.3390/ijms21155551
135. L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Deleidi M, et al. Plasticity of Subventricular Zone Neuroprogenitors in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Mouse Model of Parkinson's Disease Involves Cross Talk Between Inflammatory and Wnt/beta-Catenin Signaling Pathways: Functional Consequences for Neuroprotection and Repair. J Neurosci (2012) 32(6):2062–85. doi: 10.1523/JNEUROSCI.5259-11.2012
136. Guadagno J, Swan P, Shaikh R, Cregan SP. Microglia-Derived IL-1beta Triggers P53-Mediated Cell Cycle Arrest and Apoptosis in Neural Precursor Cells. Cell Death Dis (2015) 6:e1779. doi: 10.1038/cddis.2015.151
137. Carreira BP, Morte MI, Santos AI, Lourenco AS, Ambrosio AF, Carvalho CM, et al. Nitric Oxide From Inflammatory Origin Impairs Neural Stem Cell Proliferation by Inhibiting Epidermal Growth Factor Receptor Signaling. Front Cell Neurosci (2014) 8:343. doi: 10.3389/fncel.2014.00343
138. Rojo AI, McBean G, Cindric M, Egea J, Lopez MG, Rada P, et al. Redox Control of Microglial Function: Molecular Mechanisms and Functional Significance. Antioxid Redox Signal (2014) 21(12):1766–801. doi: 10.1089/ars.2013.5745
139. Xie J, Hong E, Ding B, Jiang W, Zheng S, Xie Z, et al. Inhibition of NOX4/ROS Suppresses Neuronal and Blood-Brain Barrier Injury by Attenuating Oxidative Stress After Intracerebral Hemorrhage. Front Cell Neurosci (2020) 14:578060. doi: 10.3389/fncel.2020.578060
140. Usatyuk PV, Parinandi NL, Natarajan V. Redox Regulation of 4-Hydroxy-2-Nonenal-Mediated Endothelial Barrier Dysfunction by Focal Adhesion, Adherens, and Tight Junction Proteins. J Biol Chem (2006) 281(46):35554–66. doi: 10.1074/jbc.M607305200
141. Ronaldson PT, Davis TP. Regulation of Blood-Brain Barrier Integrity by Microglia in Health and Disease: A Therapeutic Opportunity. J Cereb Blood Flow Metab (2020) 40(1_suppl):S6–S24. doi: 10.1177/0271678X20951995
142. Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of Ischemic Brain Damage. Neuropharmacology (2008) 55(3):310–8. doi: 10.1016/j.neuropharm.2008.01.005
143. Garcia JM, Stillings SA, Leclerc JL, Phillips H, Edwards NJ, Robicsek SA, et al. Role of Interleukin-10 in Acute Brain Injuries. Front Neurol (2017) 8:244. doi: 10.3389/fneur.2017.00244
144. Kang R, Gamdzyk M, Lenahan C, Tang J, Tan S, Zhang JH. The Dual Role of Microglia in Blood-Brain Barrier Dysfunction After Stroke. Curr Neuropharmacol (2020) 18(12):1237–49. doi: 10.2174/1570159X18666200529150907
145. Ushio-Fukai M. VEGF Signaling Through NADPH Oxidase-Derived ROS. Antioxid Redox Signal (2007) 9(6):731–9. doi: 10.1089/ars.2007.1556
146. Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, et al. VEGF Enhances Angiogenesis and Promotes Blood-Brain Barrier Leakage in the Ischemic Brain. J Clin Invest (2000) 106(7):829–38. doi: 10.1172/JCI9369
147. Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, et al. Role of Matrix Metalloproteinases in Delayed Cortical Responses After Stroke. Nat Med (2006) 12(4):441–5. doi: 10.1038/nm1387
148. Xing G, Zhao T, Zhang X, Li H, Li X, Cui P, et al. Astrocytic Sonic Hedgehog Alleviates Intracerebral Hemorrhagic Brain Injury via Modulation of Blood-Brain Barrier Integrity. Front Cell Neurosci (2020) 14:575690. doi: 10.3389/fncel.2020.575690
149. Kiray H, Lindsay SL, Hosseinzadeh S, Barnett SC. The Multifaceted Role of Astrocytes in Regulating Myelination. Exp Neurol (2016) 283(Pt B):541–9. doi: 10.1016/j.expneurol.2016.03.009
150. Yamagata K. Astrocyte-Induced Synapse Formation and Ischemic Stroke. J Neurosci Res (2021) 99(5):1401–13. doi: 10.1002/jnr.24807
151. Bejot Y, Prigent-Tessier A, Cachia C, Giroud M, Mossiat C, Bertrand N, et al. Time-Dependent Contribution of non Neuronal Cells to BDNF Production After Ischemic Stroke in Rats. Neurochem Int (2011) 58(1):102–11. doi: 10.1016/j.neuint.2010.10.019
152. Zhang H, Xiao J, Hu Z, Xie M, Wang W, He D. Blocking Transient Receptor Potential Vanilloid 2 Channel in Astrocytes Enhances Astrocyte-Mediated Neuroprotection After Oxygen-Glucose Deprivation and Reoxygenation. Eur J Neurosci (2016) 44(7):2493–503. doi: 10.1111/ejn.13352
153. Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity (2017) 46(6):957–67. doi: 10.1016/j.immuni.2017.06.006
154. Chao CC, Hu S, Sheng WS, Bu D, Bukrinsky MI, Peterson PK. Cytokine-Stimulated Astrocytes Damage Human Neurons via a Nitric Oxide Mechanism. Glia (1996) 16(3):276–84. doi: 10.1002/(SICI)1098-1136(199603)16:3<276::AID-GLIA10>3.0.CO;2-X
155. Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine Effects on Glutamate Uptake by Human Astrocytes. Neuroimmunomodulation (2000) 7(3):153–9. doi: 10.1159/000026433
156. Hou L, Zhou X, Zhang C, Wang K, Liu X, Che Y, et al. NADPH Oxidase-Derived H2O2 Mediates the Regulatory Effects of Microglia on Astrogliosis in Experimental Models of Parkinson's Disease. Redox Biol (2017) 12:162–70. doi: 10.1016/j.redox.2017.02.016
157. Quintas C, Pinho D, Pereira C, Saraiva L, Goncalves J, Queiroz G. Microglia P2Y(6) Receptors Mediate Nitric Oxide Release and Astrocyte Apoptosis. J Neuroinflamm (2014) 11:141. doi: 10.1186/s12974-014-0141-3
158. Zhao Y, Rempe DA. Targeting Astrocytes for Stroke Therapy. Neurotherapeutics (2010) 7(4):439–51. doi: 10.1016/j.nurt.2010.07.004
159. Adami C, Bianchi R, Pula G, Donato R. S100B-Stimulated NO Production by BV-2 Microglia is Independent of RAGE Transducing Activity But Dependent on RAGE Extracellular Domain. Biochim Biophys Acta (2004) 1742(1-3):169–77. doi: 10.1016/j.bbamcr.2004.09.008
160. Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes Induce Hemeoxygenase-1 Expression in Microglia: A Feasible Mechanism for Preventing Excessive Brain Inflammation. J Neurosci (2006) 26(6):1880–7. doi: 10.1523/JNEUROSCI.3696-05.2006
161. Park SJ, Lee JH, Kim HY, Choi YH, Park JS, Suh YH, et al. Astrocytes, But Not Microglia, Rapidly Sense H(2)O(2)via STAT6 Phosphorylation, Resulting in Cyclooxygenase-2 Expression and Prostaglandin Release. J Immunol (2012) 188(10):5132–41. doi: 10.4049/jimmunol.1101600
162. Touyz RM. Apocynin, NADPH Oxidase, and Vascular Cells: A Complex Matter. Hypertension (2008) 51(2):172–4. doi: 10.1161/HYPERTENSIONAHA.107.103200
163. Hur J, Lee P, Kim MJ, Kim Y, Cho YW. Ischemia-Activated Microglia Induces Neuronal Injury via Activation of Gp91phox NADPH Oxidase. Biochem Biophys Res Commun (2010) 391(3):1526–30. doi: 10.1016/j.bbrc.2009.12.114
164. Chen H, Song YS, Chan PH. Inhibition of NADPH Oxidase is Neuroprotective After Ischemia-Reperfusion. J Cereb Blood Flow Metab (2009) 29(7):1262–72. doi: 10.1038/jcbfm.2009.47
165. Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea S. Modulation of NADPH Oxidase Activation in Cerebral Ischemia/Reperfusion Injury in Rats. Brain Res (2011) 1372:92–102. doi: 10.1016/j.brainres.2010.11.088
166. Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, et al. Apocynin is Not an Inhibitor of Vascular NADPH Oxidases But an Antioxidant. Hypertension (2008) 51(2):211–7. doi: 10.1161/HYPERTENSIONAHA.107.100214
167. Zhou J, Zhang S, Zhao X, Wei T. Melatonin Impairs NADPH Oxidase Assembly and Decreases Superoxide Anion Production in Microglia Exposed to Amyloid-Beta1-42. J Pineal Res (2008) 45(2):157–65. doi: 10.1111/j.1600-079X.2008.00570.x
168. Higashi Y. Edaravone for the Treatment of Acute Cerebral Infarction: Role of Endothelium-Derived Nitric Oxide and Oxidative Stress. Expert Opin Pharmacother (2009) 10(2):323–31. doi: 10.1517/14656560802636888
169. Evren V, Apaydin M, Khalilnezhad A, Erbas O, Taskiran D. Protective Effect of Edaravone Against Manganese-Induced Toxicity in Cultured Rat Astrocytes. Environ Toxicol Pharmacol (2015) 40(2):563–7. doi: 10.1016/j.etap.2015.08.010
170. Banno M, Mizuno T, Kato H, Zhang G, Kawanokuchi J, Wang J, et al. The Radical Scavenger Edaravone Prevents Oxidative Neurotoxicity Induced by Peroxynitrite and Activated Microglia. Neuropharmacology (2005) 48(2):283–90. doi: 10.1016/j.neuropharm.2004.10.002
171. Sharma P, Sinha M, Shukla R, Garg RK, Verma R, Singh MK. A Randomized Controlled Clinical Trial to Compare the Safety and Efficacy of Edaravone in Acute Ischemic Stroke. Ann Indian Acad Neurol (2011) 14(2):103–6. doi: 10.4103/0972-2327.82794
172. Tordjman S, Chokron S, Delorme R, Charrier A, Bellissant E, Jaafari N, et al. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr Neuropharmacol (2017) 15(3):434–43. doi: 10.2174/1570159X14666161228122115
173. Goswami P, Gupta S, Joshi N, Sharma S, Singh S. Astrocyte Activation and Neurotoxicity: A Study in Different Rat Brain Regions and in Rat C6 Astroglial Cells. Environ Toxicol Pharmacol (2015) 40(1):122–39. doi: 10.1016/j.etap.2015.06.001
174. Jou MJ. Pathophysiological and Pharmacological Implications of Mitochondria-Targeted Reactive Oxygen Species Generation in Astrocytes. Adv Drug Deliv Rev (2008) 60(13-14):1512–26. doi: 10.1016/j.addr.2008.06.004
175. Cheng Y, Takeuchi H, Sonobe Y, Jin S, Wang Y, Horiuchi H, et al. Sirtuin 1 Attenuates Oxidative Stress via Upregulation of Superoxide Dismutase 2 and Catalase in Astrocytes. J Neuroimmunol (2014) 269(1-2):38–43. doi: 10.1016/j.jneuroim.2014.02.001
176. Gabryel B, Malecki A. Ebselen Attenuates Oxidative Stress in Ischemic Astrocytes Depleted of Glutathione. Comparison With Glutathione Precursors. Pharmacol Rep (2006) 58(3):381–92.
177. Quincozes-Santos A, Bobermin LD, Latini A, Wajner M, Souza DO, Goncalves CA, et al. Resveratrol Protects C6 Astrocyte Cell Line Against Hydrogen Peroxide-Induced Oxidative Stress Through Heme Oxygenase 1. PloS One (2013) 8(5):e64372. doi: 10.1371/journal.pone.0064372
178. Danilov CA, Chandrasekaran K, Racz J, Soane L, Zielke C, Fiskum G. Sulforaphane Protects Astrocytes Against Oxidative Stress and Delayed Death Caused by Oxygen and Glucose Deprivation. Glia (2009) 57(6):645–56. doi: 10.1002/glia.20793
179. Ulker S, McKeown PP, Bayraktutan U. Vitamins Reverse Endothelial Dysfunction Through Regulation of eNOS and NAD(P)H Oxidase Activities. Hypertension (2003) 41(3):534–9. doi: 10.1161/01.HYP.0000057421.28533.37
180. Xie P, Yang L, Talaiti A, Wu JJ, Yu J, Yu T, et al. Deferoxamine-Activated Hypoxia-Inducible Factor-1 Restores Cardioprotective Effects of Sevoflurane Postconditioning in Diabetic Rats. Acta Physiol (Oxf) (2017) 221(2):98–114. doi: 10.1111/apha.12874
181. Millan M, DeGregorio-Rocasolano N, Perez de la Ossa N, Reverte S, Costa J, Giner P, et al. Targeting Pro-Oxidant Iron With Deferoxamine as a Treatment for Ischemic Stroke: Safety and Optimal Dose Selection in a Randomized Clinical Trial. Antioxidants (Basel) (2021) 10(8):1270. doi: 10.3390/antiox10081270
182. Cabezas R, Baez-Jurado E, Hidalgo-Lanussa O, Echeverria V, Ashrad GM, Sahebkar A, et al. Growth Factors and Neuroglobin in Astrocyte Protection Against Neurodegeneration and Oxidative Stress. Mol Neurobiol (2019) 56(4):2339–51. doi: 10.1007/s12035-018-1203-9
183. Amri F, Ghouili I, Amri M, Carrier A, Masmoudi-Kouki O. Neuroglobin Protects Astroglial Cells From Hydrogen Peroxide-Induced Oxidative Stress and Apoptotic Cell Death. J Neurochem (2017) 140(1):151–69. doi: 10.1111/jnc.13876
184. James AM, Cocheme HM, Smith RA, Murphy MP. Interactions of Mitochondria-Targeted and Untargeted Ubiquinones With the Mitochondrial Respiratory Chain and Reactive Oxygen Species. Implications for the Use of Exogenous Ubiquinones as Therapies and Experimental Tools. J Biol Chem (2005) 280(22):21295–312. doi: 10.1074/jbc.M501527200
185. Chen W, Guo C, Huang S, Jia Z, Wang J, Zhong J, et al. MitoQ Attenuates Brain Damage by Polarizing Microglia Towards the M2 Phenotype Through Inhibition of the NLRP3 Inflammasome After ICH. Pharmacol Res (2020) 161:105122. doi: 10.1016/j.phrs.2020.105122
186. Zhou J, Wang H, Shen R, Fang J, Yang Y, Dai W, et al. Mitochondrial-Targeted Antioxidant MitoQ Provides Neuroprotection and Reduces Neuronal Apoptosis in Experimental Traumatic Brain Injury Possibly via the Nrf2-ARE Pathway. Am J Transl Res (2018) 10(6):1887–99.
187. Park SY, Pekas EJ, Headid RJ 3rd, Son WM, Wooden TK, Song J, et al. Acute Mitochondrial Antioxidant Intake Improves Endothelial Function, Antioxidant Enzyme Activity, and Exercise Tolerance in Patients With Peripheral Artery Disease. Am J Physiol Heart Circ Physiol (2020) 319(2):H456–67. doi: 10.1152/ajpheart.00235.2020
188. Jou MJ, Peng TI, Yu PZ, Jou SB, Reiter RJ, Chen JY, et al. Melatonin Protects Against Common Deletion of Mitochondrial DNA-Augmented Mitochondrial Oxidative Stress and Apoptosis. J Pineal Res (2007) 43(4):389–403. doi: 10.1111/j.1600-079X.2007.00490.x
189. Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, et al. MicroRNA-424 Protects Against Focal Cerebral Ischemia and Reperfusion Injury in Mice by Suppressing Oxidative Stress. Stroke (2015) 46(2):513–9. doi: 10.1161/STROKEAHA.114.007482
190. Zhang A, Qian Y, Qian J. MicroRNA-152-3p Protects Neurons From Oxygen-Glucose-Deprivation/Reoxygenation-Induced Injury Through Upregulation of Nrf2/ARE Antioxidant Signaling by Targeting PSD-93. Biochem Biophys Res Commun (2019) 517(1):69–76. doi: 10.1016/j.bbrc.2019.07.012
191. Zuo ML, Wang AP, Song GL, Yang ZB. miR-652 Protects Rats From Cerebral Ischemia/Reperfusion Oxidative Stress Injury by Directly Targeting NOX2. BioMed Pharmacother (2020) 124:109860. doi: 10.1016/j.biopha.2020.109860
192. Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. MiRNA-124 Induces Neuroprotection and Functional Improvement After Focal Cerebral Ischemia. Biomaterials (2016) 91:151–65. doi: 10.1016/j.biomaterials.2016.03.025
193. Liu X, Feng Z, Du L, Huang Y, Ge J, Deng Y, et al. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int J Mol Sci (2019) 21(1):120. doi: 10.3390/ijms21010120
194. Zheng L, Tang X, Lu M, Sun S, Xie S, Cai J, et al. microRNA-421-3p Prevents Inflammatory Response in Cerebral Ischemia/Reperfusion Injury Through Targeting M6a Reader YTHDF1 to Inhibit P65 mRNA Translation. Int Immunopharmacol (2020) 88:106937. doi: 10.1016/j.intimp.2020.106937
195. Xu LJ, Ouyang YB, Xiong X, Stary CM, Giffard RG. Post-Stroke Treatment With miR-181 Antagomir Reduces Injury and Improves Long-Term Behavioral Recovery in Mice After Focal Cerebral Ischemia. Exp Neurol (2015) 264:1–7. doi: 10.1016/j.expneurol.2014.11.007
196. Watanabe T, Tanaka M, Watanabe K, Takamatsu Y, Tobe A. [Research and Development of the Free Radical Scavenger Edaravone as a Neuroprotectant]. Yakugaku Zasshi (2004) 124(3):99–111. doi: 10.1248/yakushi.124.99
197. Yang J, Liu C, Du X, Liu M, Ji X, Du H, et al. Hypoxia Inducible Factor 1alpha Plays a Key Role in Remote Ischemic Preconditioning Against Stroke by Modulating Inflammatory Responses in Rats. J Am Heart Assoc (2018) 7(5):e007589. doi: 10.1161/JAHA.117.007589
198. Jones NM, Bergeron M. Hypoxic Preconditioning Induces Changes in HIF-1 Target Genes in Neonatal Rat Brain. J Cereb Blood Flow Metab (2001) 21(9):1105–14. doi: 10.1097/00004647-200109000-00008
Keywords: stroke, oxidative stress, astrocyte, microglia, therapies
Citation: Zhu G, Wang X, Chen L, Lenahan C, Fu Z, Fang Y and Yu W (2022) Crosstalk Between the Oxidative Stress and Glia Cells After Stroke: From Mechanism to Therapies. Front. Immunol. 13:852416. doi: 10.3389/fimmu.2022.852416
Received: 11 January 2022; Accepted: 08 February 2022;
Published: 25 February 2022.
Edited by:
Yujie Chen, Army Medical University, ChinaReviewed by:
Zhen-Ni Guo, First Affiliated Hospital of Jilin University, ChinaWeixiang Chen, General Hospital of Xinjiang Military Region, China
Kirill Kiselyov, University of Pittsburgh, United States
Copyright © 2022 Zhu, Wang, Chen, Lenahan, Fu, Fang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanjian Fang, c2FuZG1hbjA1MDZAemp1LmVkdS5jbg==; Wenhua Yu, eXdoNjk5QHpqdS5lZHUuY24=
†These authors have contributed equally to this work