 Silene M. Silvera-Ruiz1,2
Silene M. Silvera-Ruiz1,2 Corinne Gemperle3
Corinne Gemperle3 Natalia Peano4
Natalia Peano4 Valentina Olivero4
Valentina Olivero4 Adriana Becerra5
Adriana Becerra5 Johannes Häberle3
Johannes Häberle3 Adriana Gruppi1
Adriana Gruppi1 Laura E. Larovere2,6
Laura E. Larovere2,6 Ruben D. Motrich1*
Ruben D. Motrich1*- 1Centro de Investigaciones en Bioquímica Clínica e Inmunología (CIBICI-CONICET), Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba, Argentina
- 2Centro de Estudio de las Metabolopatías Congénitas (CEMECO), Hospital de Niños de la Santísima Trinidad, Facultad de Ciencias Médicas, Universidad Nacional de Córdoba, Córdoba, Argentina
- 3Division of Metabolism and Children’s Research Center, University Children’s Hospital Zurich, Zurich, Switzerland
- 4Fundación para el Progreso de la Medicina, Córdoba, Argentina
- 5División de Enfermedades Metabólicas, Hospital de Niños de la Santísima Trinidad, Córdoba, Argentina
- 6Cátedra de Clínica Pediátrica, Facultad de Ciencias Médicas, Universidad Nacional de Córdoba, Córdoba, Argentina
The hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is a rare autosomal recessive inborn error of the urea cycle caused by mutations in the SLC25A15 gene. Besides the well-known metabolic complications, patients often present intercurrent infections associated with acute hyperammonemia and metabolic decompensation. However, it is currently unknown whether intercurrent infections are associated with immunological alterations besides the known metabolic imbalances. Herein, we describe the case of a 3-years-old girl affected by the HHH syndrome caused by two novel SLC25A15 gene mutations associated with immune phenotypic and functional alterations. She was admitted to the hospital with an episode of recurrent otitis, somnolence, confusion, and lethargy. Laboratory tests revealed severe hyperammonemia, elevated serum levels of liver transaminases, hemostasis alterations, hyperglutaminemia and strikingly increased orotic aciduria. Noteworthy, serum protein electrophoresis showed a reduction in the gamma globulin fraction. Direct sequencing of the SLC25A15 gene revealed two heterozygous non-conservative substitutions in the exon 5: c.649G>A (p.Gly217Arg) and c.706A>G (p.Arg236Gly). In silico analysis indicated that both mutations significantly impair protein structure and function and are consistent with the patient clinical status confirming the diagnosis of HHH syndrome. In addition, the immune analysis revealed reduced levels of serum IgG and striking phenotypic and functional alterations in the T and B cell immune compartments. Our study has identified two non-previously described mutations in the SLC25A15 gene underlying the HHH syndrome. Moreover, we are reporting for the first time functional and phenotypic immunologic alterations in this rare inborn error of metabolism that would render the patient immunocompromised and might be related to the high frequency of intercurrent infections observed in patients bearing urea cycle disorders. Our results point out the importance of a comprehensive analysis to gain further insights into the underlying pathophysiology of the disease that would allow better patient care and quality of life.
Introduction
The mammalian urea cycle provides a pathway for the synthesis of the non-essential amino acid arginine and serves to detoxify ammonia, keeping plasma ammonium concentrations within a narrow range despite ten-fold variations in dietary nitrogen intake (1, 2). The mitochondrial transporter ornithine carrier 1 (ORC1) plays a crucial role in the cycle by transferring cytosolic ornithine into the mitochondrial matrix in exchange for citrulline (3, 4). Mutations in the solute carrier family 25 member 15 (SLC25A15) gene, which encodes for ORC1, are causative of the rare autosomal recessive urea cycle disorder (UCD) called hyperornithinemia–hyperammonemia–homocitrullinuria (HHH) syndrome (OMIM 238970) (1, 5). In affected patients, ORC1 deficiency reduces the rate of the urea cycle leading to hyperammonemia, a typical feature of most UCD (1, 6, 7). A founder effect was reported in the French-Canadian population (1) and then 50 affecting function variants have been already identified in 122 patients worldwide pointing out the diversity of mutations and pan-ethnic distribution (8–10). The disease usually manifests in early infancy or childhood; however, cases of adult onset have also been reported (6, 11, 12). Typical clinical features include lethargy, episodic confusion or coma due to postprandial hyperammonemia, hepatitis-like vomiting, spastic paraplegia, cerebellar ataxia, seizures, failure to thrive, coagulation factor defects, and liver failure (1, 7, 12). Different precipitants such as dietary carelessness, enhanced protein catabolism consequent to dietary over-restriction or infections may trigger an acute deterioration of the metabolic status, which is characterized by potentially life-threatening episodes of hyperammonemia (13), which may lead to cerebral edema, lethargy, anorexia, vomiting, hyperventilation (or hypoventilation), hypothermia, neurologic posturing, coma and death. Interestingly, intercurrent infections are often observed in UCD patients and have been reported as the most frequent precipitant of acute hyperammonemia episodes and metabolic decompensation (13, 14). Furthermore, they are the most dangerous precipitant since the induced inflammation is a catabolic stressor that usually aggravates the patient clinical status significantly increasing morbidity and mortality risks (13, 15–17). Although a considerable advance in the knowledge about the pathophysiology of UCD and their neurologic and metabolic consequences has been achieved during recent years, very scarce data about their putative consequences on the immune system have been reported up to date (12, 17–19). Certainly, it is currently unknown whether the frequently observed intercurrent infections are associated with putative immune alterations, which could render patients susceptible to infections further increasing the severity of the hyperammonemia episodes and thus resulting in higher hospitalization rates and longer hospital stays (13, 17).
Herein, we report the case of a patient with HHH syndrome caused by two novel mutations in the SLC25A15 gene associated with recurrent otitis episodes that precipitated hyperammonemia crises. These non-previously reported mutations consisted of two missense mutations in exon 5 of the SLC25A15 gene, namely c.649G>A (p.Gly217Arg) and c.706A>G (p.Arg236Gly). Interestingly, the patient showed functional and phenotypic immune alterations that could render her immunocompromised and thus explaining her history of recurrent otitis.
Case Description
We present the case of a girl aged 3.4 years, who was admitted to the emergency room on August 5, 2019, for recurrent acute otitis with vomiting, intermittent sensory disturbance, and irritability. She is the second child of healthy non-consanguineous Caucasian parents. She was born at term by spontaneous delivery with a weight of 3500 g and no history of perinatal complications or developmental delay. Interestingly, her parents report that 11 months earlier (September 2018) the child was admitted into another hospital because of a similar episode of acute infectious otitis with somnolence, confusion, and lethargy. When the patient was examined in our hospital, the neurological assessment found her sleepy but reactive to stimuli, with no neurologic deterioration (Glasgow coma score of 15/15), and normal brain computed tomography scan. As shown in Table 1, the patient presented with severe hyperammonemia, increased serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), metabolic alkalosis, and markedly reduced prothrombin activity and prolonged aPTT. Analysis of amino acids in serum revealed increased levels of glutamine. Furthermore, urine analysis revealed strikingly increased orotic acid excretion. Noteworthy, blood cytology showed increased proportions and counts of monocytes and atypical lymphocytes (20) (Table 1), probably a consequence of the ongoing otitis episode. Besides, and remarkably, serum protein electrophoresis revealed low levels of total proteins mainly due to decreased levels of the gamma globulins fraction indicating a possible underproduction of antibodies. Interestingly, reduced levels of IgG and slightly increased levels of IgM were detected (Table 1). Owing to the severe hyperammonemia and increased orotic aciduria, the patient was suspected to suffer from a UCD, more specifically a case of ornithine transcarbamylase deficiency (OTCD) or HHH syndrome. An OTCD was excluded by genetic analysis, which revealed no mutations in the OTC gene. However, direct sequencing of the SLC25A15 gene identified two novel heterozygous mutations in exon 5 of the SLC25A15 gene (ENST00000338625.9): c.649G>A (p.Gly217Arg) and c.706A>G (p.Arg236Gly) (Figure 1A), different from the 50 already reported SLC25A15 gene mutations (5, 8–10), strongly suggesting a case of HHH syndrome.
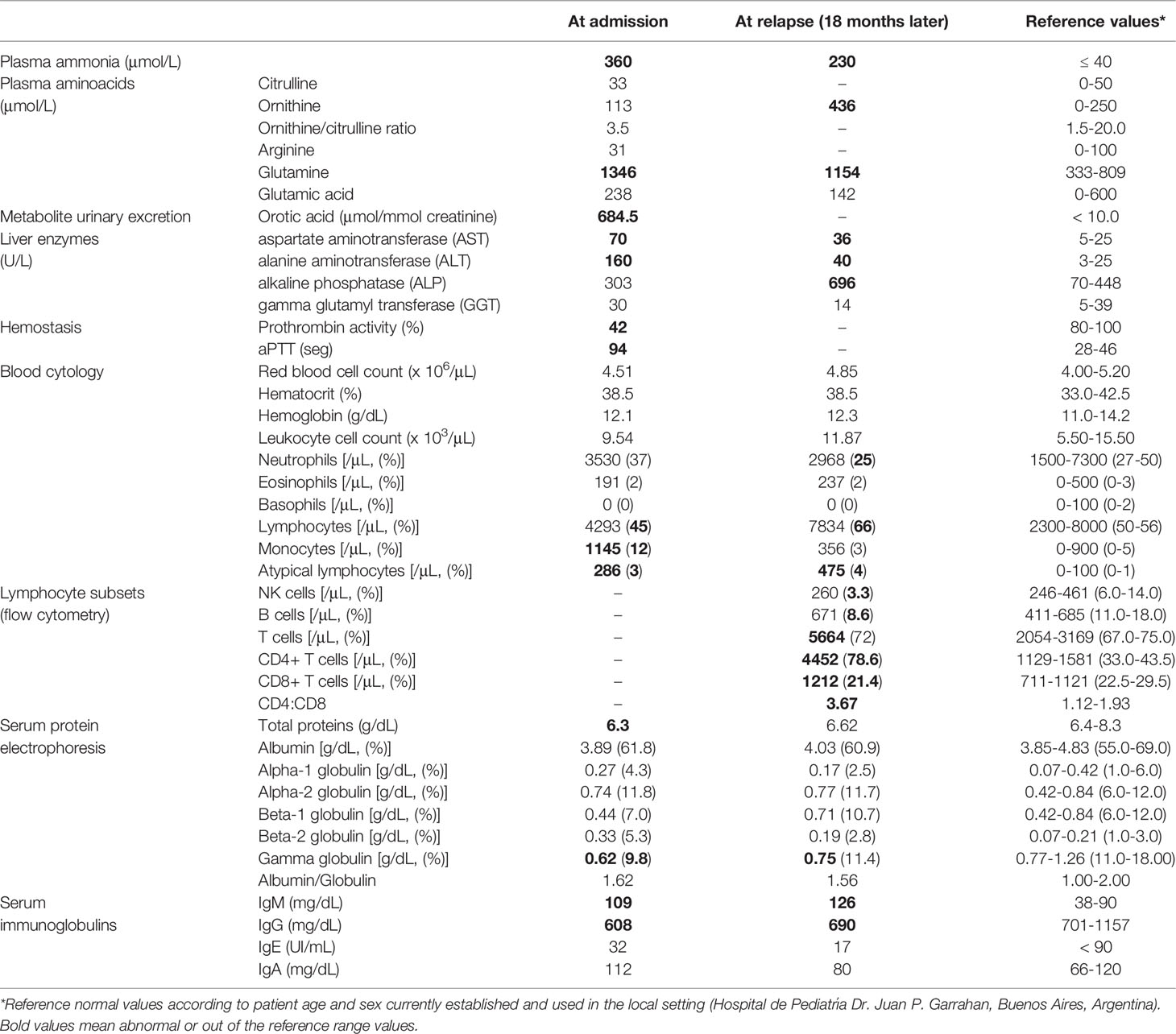
Table 1 Laboratory findings in a patient with HHH syndrome.
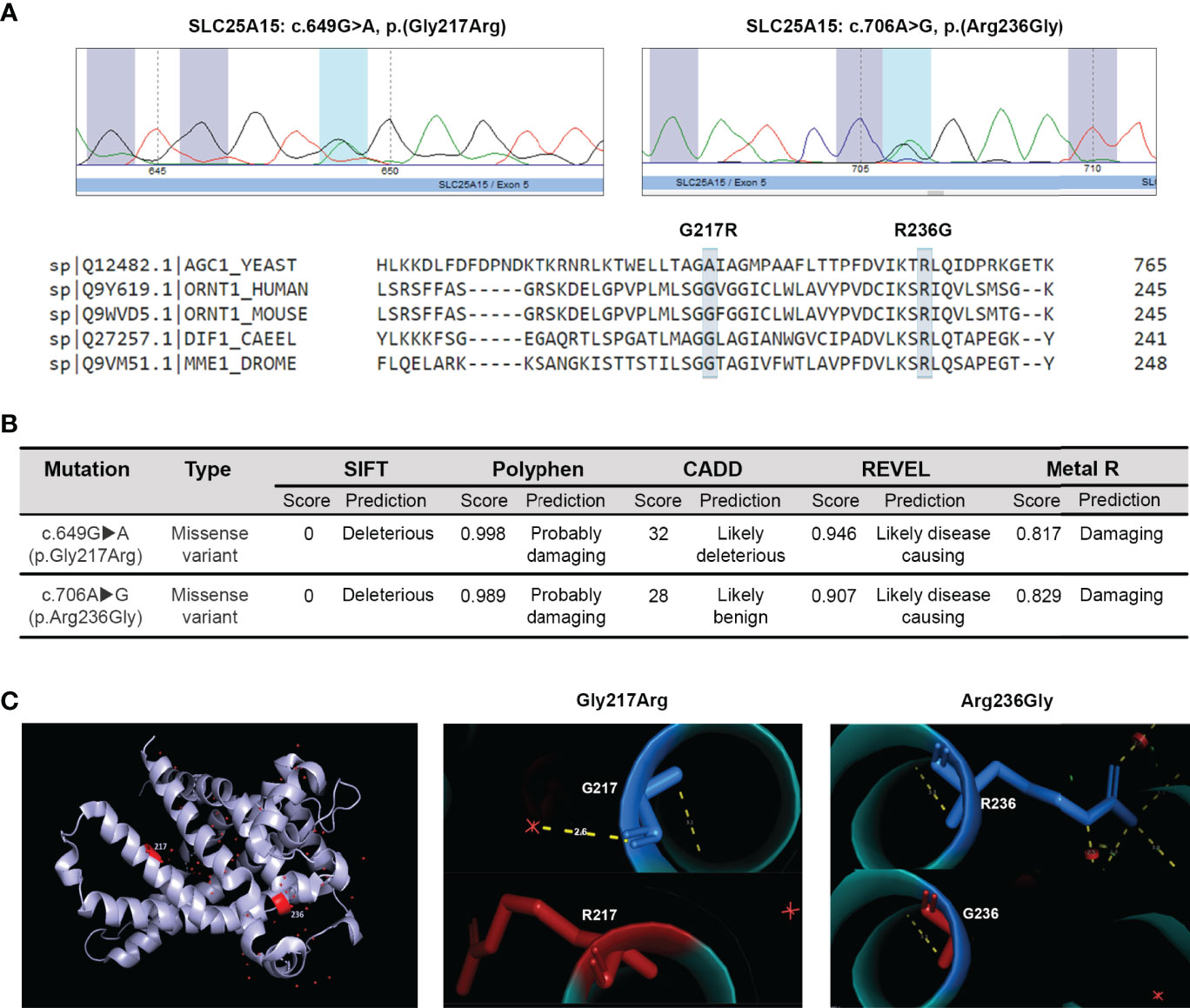
Figure 1 In silico analysis of c.649G>A (p.Gly217Arg) and c.706A>G (p.Arg236Gly) mutations on the ORC1 protein structure and function. (A) Partial electropherograms showing the detected mutations. Multiple alignment of the modified ORC1 aminoacid residues with the protein sequence from different species using the Clustal W2 software. (B) In silico validation of the two SLC25A15 gene mutations identified. (C) Modeling of the tridimensional protein structural changes introduced by the two identified mutations using the human ORC1 homology model. Normal residues are marked in blue (top) and the changes introduced by mutations in red (bottom).
The putative structural and functional consequences of the newly identified SLC25A15 gene missense mutations were assessed in silico. The mitochondrial ORC1 transporter is composed of 301 amino acids with six transmembrane segments. Arginine 217 resides in the fifth transmembrane domain, and Glycine 236 is located in an alpha helix along the substrate translocation pore. As shown in Figure 1A, multiple sequence alignment analysis revealed that G217R and R236G are both non-conservative substitutions in amino acid residues evolutionarily conserved among species, which suggests they are crucial for protein structure and function. Moreover, different bioinformatics prediction tools such as Clustal W (21), SIFT (22), Polyphen (23), CADD (24), REVEL (25) and Metal R (26) indicated that both mutations are highly detrimental for the secondary structure and function of the protein since both substituted amino acids differ in polarity, charge, size, and other biochemical properties (Figure 1B). The majority of the single-residue mutations reported causing the HHH syndrome are non-conservative substitutions that introduce changes in the charge and size of the amino acid side chain (5, 27). Moreover, computer modelling and mapping of both missense mutations using the 3D structural homology model of ORC1 (28) indicated that residue Arg236 is located in the matrix gate area and plays a crucial role in conformational changes which allow substrate translocation (29). Conversely, the residue Gly217 belongs to the Pro-Gly level 1 immediately above the substrate binding region (30), playing the crucial hinge/kink function, which allows conformational changes and the access of the substrate to the ORC1 binding site (Figure 1C). The above reported observations indicate that both mutations have a high likelihood to impair ornithine translocation, coherently with the patient clinical phenotype supporting/confirming a diagnosis of HHH syndrome.
After the initial diagnostic work-up, the patient was treated with intravenous sodium benzoate, sodium phenylacetate, and L-arginine that rapidly normalized plasma ammonia levels in less than 24 hours. Immediately after, the patient was put on a protein restricted diet (1.3 g/kg/day) supplemented with L-carnitine (100 mg/kg/day), L-arginine (100 mg/kg/day), and sodium benzoate (230 mg/kg/day) that resulted in metabolic correction, remarkable clinical and biochemical improvement, without any complications or sequelae. In addition, the otitis episode resolved after amoxicillin treatment (80 mg/kg/day) for 7 days. However, 18 months later (February 2021) the patient relapsed and was re-admitted to the hospital with a similar clinical picture as at first admission: an episode of recurrent otitis associated to irritability, intermittent sensory disturbance, and confusion. Once again, severe hyperammonemia accompanied with increased serum levels of ornithine, glutamine, AST, ALT and also alkaline phosphatase (ALP) were detected. Interestingly, blood cytology revealed increased proportions of lymphocytes and elevated counts of atypical lymphocytes once more (Table 1). Moreover, serum protein electrophoresis consistently revealed reduced levels of the gamma globulins fraction of serum proteins. In addition, reduced levels of IgG and increased levels of IgM were detected again (Table 1). Taking into consideration these data and the patient history, we then performed a comprehensive phenotypic and functional analysis of cellular and humoral adaptive immune compartments once the patient was metabolically compensated and released from the hospital.
Immunocompromised patients usually present a profound immune dysregulation associated with increased morbidity and mortality, which has been characterized by reduced in vitro lymphocyte proliferative capacity in response to mitogens and alterations in the production of pro-inflammatory cytokines (31, 32). Therefore, proliferative responses from peripheral blood mononuclear cell (PBMC) from the patient and sex and age-matched healthy control individuals to well-defined mitogens including concanavalin A (Con A), phytohemagglutinin (PHA), Pokeweed (PKW), candidin, and antigens of Mycobacterium tuberculosis or Trychophyton spp. were assessed. As shown in Figure 2A, significantly reduced proliferation rates in response to most of the mitogens assayed were observed in the patient with respect to controls. When assaying the production of inflammatory cytokines, significantly reduced levels of IFNγ, IL-17A and TNFα secretion were detected in culture supernatants of PBMC from the patient compared with controls (Figure 2B). Conversely, significantly higher levels of the IL-10 were detected in culture supernatants from the patient PBMC stimulated with Con A, PKW, and candidin with respect to controls (Figure 2B). In parallel, we quantified the levels of CD4+ and CD8+ T cells, B cells and NK cells in the patient peripheral blood by flow cytometry. Whereas normal counts of NK and B cells were observed, significantly higher counts of T cells were detected, mainly due to an increase in the CD4+ T cell subset (Table 1). These findings indicated an expansion of that particular T cell population and were in agreement with the increased levels of atypical lymphocytes revealed by blood cytology (Table 1). Considering that, we then evaluated the distribution of CD4+ and CD8+ T cell subsets in the patient under study and sex and age-matched healthy control individuals. CD45RA and CCR7 cell surface markers were used to identify four phenotypically and functionally distinct subsets of CD4+ and CD8+ T-cells: naïve (Naïve: CD45RA+ CCR7+), central memory (CM: CD45RA- CCR7+), effector memory (EM: CD45RA- CCR7-) and terminally differentiated effector memory (TEMRA: CD45RA+ CCR7-) (Figures 3A–C) as previously reported (34–36). Figure 3B shows that, within the CD4+ T-cell compartment, the frequency of naïve and CM T cells were significantly lower in the patient than in controls (p<0.01). In contrast, the patient revealed significantly higher proportions of EM and TEMRA CD4+ T cells than control individuals (p<0.01, Figure 3B). When analyzing the CD8+ T-cell compartment, significantly reduced naïve and increased EM and TEMRA CD8+ T cells were respectively observed in the patient with respect to controls (Figure 3C). These results indicated alterations in the T cell compartment from the patient under study.
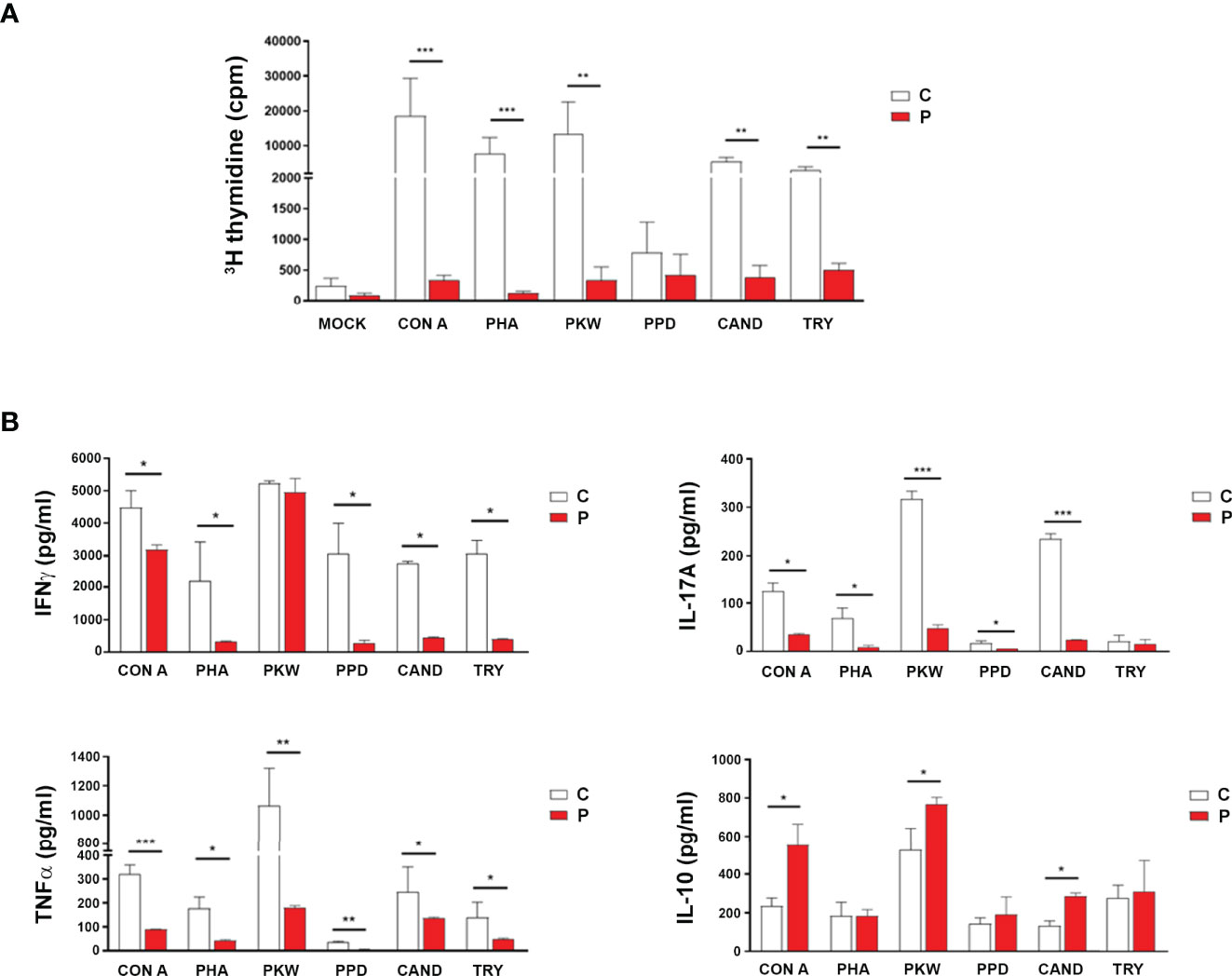
Figure 2 Functional analysis of cellular adaptive immune compartment. (A) Lymphoproliferative responses of peripheral blood mononuclear cells (PBMC) from the patient (P) and control (C) individuals to different mitogenic stimuli [concanavalin A (Con A), phytohemagglutinin (PHA), Pokeweed (PKW), candidin, and antigens of Mycobacterium tuberculosis or Trychophyton spp.] or medium alone (mock). Cells were cultured (3.0x105/well) in quadruplicate for each condition and incubated for 96 h at 37°C/5% CO2 as previously described (33). During the last 18 h of culture, wells were pulsed with 1 µCi of [methyl-3H] thymidine in fresh medium. Cells were harvested onto glass fiber filters and labeled material was counted in a β scintillation counter. Results were expressed as counts per minute (cpm). (B) IFNγ, IL-17A, TNF-α and IL-10 concentrations in PBMCs culture supernatants determined by sandwich ELISA and results expressed as pg/ml. Experiments were performed at least in quadruplicate and repeated twice with similar results. Data are shown as mean ± SEM, the patient and sex and age-matched control individuals (n=6 healthy females, aged 3-7 years old). Red bars correspond to the average values of the patient and white bars correspond to the average values of the controls analyzed. Mann-Whitney test; *p< 0.05, **p< 0.01 and ***p< 0.005.
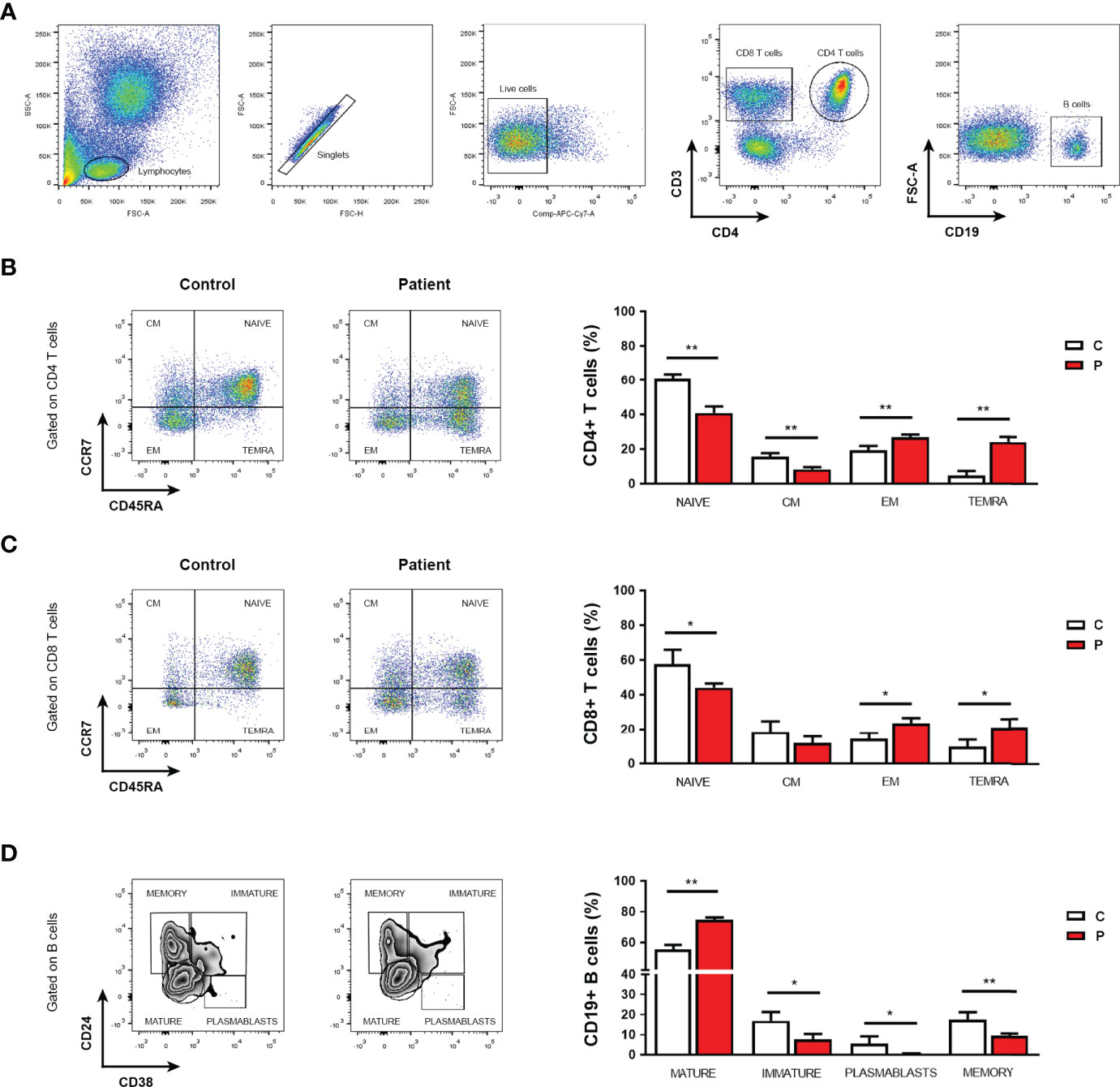
Figure 3 Phenotypic analysis of T and B cells. (A) Flow cytometry gating strategy for the assessment of phenotypic cell markers in live CD4 and CD8 T cells, and B cells from PBMC after red blood cell depletion by lysing buffer treatment. (B, C) Frequencies of naïve T cells, central memory (CM), effector memory (EM), and terminally differentiated effector memory cells (TEMRA) within the CD4+ (B) or CD8+ (C) T cell populations from the patient (P) under study and controls (C). (D) Frequencies of mature, immature and memory B cells, and plasmablasts in peripheral blood from the patient (P) under study and controls (C). Representative flow cytometry dot plots. Experiments were performed in triplicates. Data are shown as mean ± SEM, the patient and sex and age-matched control individuals (n=6 healthy females, aged 3-7 years old). Red bars correspond to the average values of the patient and white bars correspond to the average values of the controls analyzed. Mann-Whitney test; *p< 0.05 and **p< 0.01. The following fluorescent-labeled anti-human antibodies (BioLegend) were used: CD3 (PerCP), CD4 (APC), CD19 (APC), CD45RA (PE-Cy7), CCR7 (PE), CD24 (FITC) and CD38 (PE). Dead cells were excluded using LIVE-DEAD™ fixable (Invitrogen). Data were collected on FACS-CANTO II flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 7.6.2). Proper compensation using Fluorescence Minus One (FMO) controls were used.
As serum protein electrophoresis analyses revealed low levels of gamma globulins, which was further confirmed by the detection of low levels of serum IgG, we then analyzed B cell subsets in peripheral blood by flow cytometry. Although total B cell counts were within reference values, significantly reduced frequencies of plasmablasts, immature and memory B cells were found in the patient with respect to sex and age-matched healthy controls (Figure 3D). Conversely, the patient revealed significantly higher frequencies of mature B cells than control individuals (Figure 3D). These data revealed that the patient had reduced counts of circulating plasmablasts and memory B cells that could be related to the detected hypogammaglobulinemia and low levels of serum IgG but increased IgM. Altogether, the results show alterations in the adaptive T and B cell immune compartments that would render the HHH patient immunocompromised and might explain her history of recurrent otitis episodes.
Discussion
Several inborn errors of metabolism affecting effectors of the urea cycle or UCD have already been described in humans, including the HHH syndrome (12, 18). Although hyperammonemia and related complications are the main and best described consequences of the HHH syndrome and other UCD, little is known about other putative consequences on different tissues and cells such as those from the immune system.
Herein, we are reporting a case of a girl that was admitted to the hospital with an episode of recurrent otitis together with signs and symptoms compatible with a UCD. Due to the clinical presentation, laboratory findings, and local prevalence (37), an OTCD was initially suspected. However, sequencing of the OTC gene revealed no alterations excluding OTCD as the underlying pathology. Based on its prevalence, we then suspected a case of HHH syndrome even though the metabolic triad that usually defines the HHH syndrome, hyperammonemia, hyperornithinemia, and urinary excretion of homocitrulline was not observed in the patient at admission. Nevertheless, genetic analysis revealed two novel heterozygous mutations in trans of the SLC25A15 gene that in silico analysis further indicated to likely impair the functionality of the transporter protein thus confirming a diagnosis of HHH syndrome. These results support previously reported data by Mahmoud et al., 2019, who predicted several possible HHH syndrome causative mutations (38). Interestingly, the R236G mutation is listed in the Single Nucleotide Polymorphism Database (dbSNP; rs142236568) with an allele frequency of 0.02% but it has never been reported as causative of the HHH syndrome or any other disease.
The patient presented hyperammonemic crises after recurrent infectious otitis episodes, which was not surprising since intercurrent infections are commonly observed in UCD patients (13, 15, 16). As it has been shown that the inability to ingest food and medications did not seem to be associated with infection, and cannot readily explain the observed increasing morbidity in UCD patients (13), the pathophysiology of intercurrent infections could be due in part to immune alterations. The assessment of the T cell compartment revealed increased counts of T cells, mainly due to an increase in the CD4+ T helper cell subset. Interestingly, higher levels of effector memory cells and low levels of naïve cells were found in both CD4+ and CD8+ T cell populations. Strikingly, the patient showed significantly increased levels of terminally differentiated effector memory (TEMRA) CD4+ and CD8+ T cells. These results mirrored the dynamics of T cell changes associated with T cell dysfunction typically observed in the elderly and cytomegalovirus infected patients (39–42). Indeed, TEMRA is a hallmark of cellular senescence, including reduced proliferation and defective mitochondrial function (43, 44). In agreement, the patients showed strikingly reduced PBMC proliferative responses to polyclonal stimuli or memory recall antigens associated with reduced secretion of IFNγ, IL-17A, and TNFα, and increased secretion of IL-10. These results indicated an altered lymphocyte proliferation state with reduced ability to produce pro-inflammatory cytokines and enhanced production of immunoregulatory cytokines, a phenotype compatible with immunosuppression (31, 32, 45–47). On the other hand, the patient showed hypogammaglobulinemia and reduced serum IgG but increased IgM levels both at first admission and after relapsing 18 months later. Noteworthy, when assessing B cell subpopulations in peripheral blood, the patient showed strikingly reduced levels of circulating plasmablasts and memory cells but increased frequencies of mature cells suggesting a possible impairment in the activation and differentiation of B cells, and/or an inability to sustain the survival of the former B cell subpopulations. Altogether, these results show alterations in the adaptive T and B cell immune compartments that would render the HHH patient immunocompromised thus explaining recurrent infections.
Although no immune alterations have been reported in HHH syndrome or other UCDs so far, our results suggest they might be present in patients bearing these rare inborn errors of metabolism, which is a still overlooked area in medical research and clinical practice. In fact, our results could be related to the well-known high frequency of intercurrent infections typically observed in these patients. Interestingly, Monaco et al. revealed that the SLC25A15 gene is expressed by most leukocyte subpopulations with plasmablasts and CD4+ T cells showing the highest levels (48, 49). Although the specific implications of defective SLC25A15 gene expression in these cells still need to be unveiled, our results suggest that a defective expression of this gene might alter B and T cell function paving the way for future research in the area. In that regard, argininosuccinate synthetase 1 (ASS1), another component of the urea cycle that when defective causes citrullinemia type I (OMIM# 215700), is also expressed in several organs and cells of the immune system. Interestingly, experimental models have shown that the deficiency of ASS1 leads to abnormal T cell differentiation and function despite normal hepatic expression (50). Moreover, it has been shown that some patients bearing lysinuric protein intolerance, another rare hereditary disorder that secondarily affects the urea cycle and manifests with hyperammonemia, showed decreased levels of serum IgG sub-classes and suboptimal vaccine responses pointing to a B-cell dysfunction (51).
In agreement with our study, these reported data further suggest that in addition to metabolic disturbances, UCD patients may have impaired immune responses. Besides the putative consequences of the deficiency of components of the urea cycle in immune cells, it has been shown that hyperammonemia per se also affects immune cell function. The in vitro exposure to high levels of ammonia impairs neutrophil and dendritic cell phagocytic function and reduces the ability of the latter to induce lymphocyte activation and proliferation (52, 53). Moreover, cirrhotic patients, who are usually hyperammonemic and metabolically decompensated although to a lesser degree than UCD patients, are susceptible to opportunistic infections. Noteworthy, impaired phagocytic neutrophil function, aberrant immunoglobulin glycosylation, and increased exhaust CD8+ T cells have been found in cirrhotic patients indicating a state of immunocompromise associated with hyperammonemia (54–57). A limitation of this study is that the levels of different leukocyte subsets and T and B cell subpopulations from the patient at admission and at different times during her evolution are unknown. In that regard, it would be interesting to assess if correction of hyperammonemia correlates with improvements in T and B cell number and function. In that regard, we believe that the identified immune alterations herein may also be occurring in other metabolic diseases where ammonia is a common factor. Future studies may help confirm the hypothesis that stricter ammonia control is required in these patients, probably much lower than current recommendations (<80 μmol/L) (12), which could improve the overall health status of patients beyond just avoiding neurotoxicity. In addition, although the in silico analyses indicated that both mutations detected would very likely affect the activity of the ORC1 transporter, being compatible with the patient clinical phenotype, the definitive pathogenicity of both mutations should be demonstrated by experimental models in vitro and/or in vivo designed ad hoc.
In conclusion, our study identified two non-previously described mutations in the SLC25A15 gene underlying HHH syndrome. Moreover, we are reporting for the first time functional and phenotypic immunologic alterations in this rare inborn error of metabolism that would render the patient immunocompromised and might be related to the high frequency of intercurrent infections typically observed in UCD patients. Our results highlight the need of a more comprehensive evaluation of patients and warrant further investigation of the putative implications of the HHH syndrome and other UCD on the immune system, which will allow a better understanding of these rare diseases and thus a better patient care and quality of life.
Patient Perspective
The patient has remained stable and with no further complications after adhering to the treatment proposed. Family was satisfied with the improvement in the clinical condition, has been committed to maintain medical care at home and regularly attend to periodic controls.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of the Hospital de Niños de la Santísima Trinidad, Facultad de Ciencias Médicas, Universidad Nacional de Córdoba. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
RM and LL: conceptualization, funding acquisition, resources, supervision, data analysis and curation, writing and editing of the manuscript. SR-R: data acquisition, formal analysis, in silico analysis, validation, writing or the manuscript. CG, NP, VO and JH: methodology, data acquisition. AB: patient care. AG: resources, intellectual content, revision of the manuscript. All authors reviewed the manuscript. All authors significantly contributed to the article and approved the submitted version.
Funding
This work was supported by the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT-FONCyT, grant PICT 2014-2195 and 2019-2451), CONICET (grant PIP0100652) and Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba (Secyt-UNC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
HHH, hyperornithinemia–hyperammonemia–homocitrullinuria; ORC1, ornithine carrier 1; UCD, urea cycle disorder/s; OTCD, ornithine transcarbamylase deficiency; CM, central memory; EM, effector memory; TEMRA, terminally differentiated T effector memory cells that re-express the naïve T cell-associated marker CD45RA.
References
1. Valle D SO. The Hyperornithinemias. In: Beaudet AL SW, Valle D, Scriver CR, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw Hill (2001). p. 1909–64.
2. Brusilow SW HA, Scriver C BA, Sly W, Valle D. Urea Cycle Enzymes. In: The Metabolic and Molecular Bases of Inherited Disease. New York: MacGraw Hill (1995). p. 1187–232.
3. Nakajima M, Ishii S, Mito T, Takeshita K, Takashima S, Takakura H, et al. Clinical, Biochemical and Ultrastructural Study on the Pathogenesis of Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome. Brain Dev (1988) 10(3):181–5. doi: 10.1016/S0387-7604(88)80025-1
4. Hommes FA, Ho CK, Roesel RA, Coryell ME, Gordon BA. Decreased Transport of Ornithine Across the Inner Mitochondrial Membrane as a Cause of Hyperornithinaemia. J Inherit Metab Dis (1982) 5(1):41–7. doi: 10.1007/BF01799753
5. Palmieri F, Scarcia P, Monne M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules (2020) 10(4):655. doi: 10.3390/biom10040655
6. Tessa A, Fiermonte G, Dionisi-Vici C, Paradies E, Baumgartner MR, Chien YH, et al. Identification of Novel Mutations in the SLC25A15 Gene in Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) Syndrome: A Clinical, Molecular, and Functional Study. Hum Mutat (2009) 30(5):741–8. doi: 10.1002/humu.20930
7. Fecarotta S, Parenti G, Vajro P, Zuppaldi A, Della Casa R, Carbone MT, et al. HHH Syndrome (Hyperornithinaemia, Hyperammonaemia, Homocitrullinuria), With Fulminant Hepatitis-Like Presentation. J Inherit Metab Dis (2006) 29(1):186–9. doi: 10.1007/s10545-006-0120-7
8. Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, et al. Hyperornithinaemia-Hyperammonaemia-Homocitrullinuria Syndrome is Caused by Mutations in a Gene Encoding a Mitochondrial Ornithine Transporter. Nat Genet (1999) 22(2):151–8. doi: 10.1038/9658
9. Debray FG, Lambert M, Lemieux B, Soucy JF, Drouin R, Fenyves D, et al. Phenotypic Variability Among Patients With Hyperornithinaemia-Hyperammonaemia-Homocitrullinuria Syndrome Homozygous for the Delf188 Mutation in SLC25A15. J Med Genet (2008) 45(11):759–64. doi: 10.1136/jmg.2008.059097
10. Martinelli D, Diodato D, Ponzi E, Monne M, Boenzi S, Bertini E, et al. The Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome. Orphanet J Rare Dis (2015) 10:29. doi: 10.1186/s13023-015-0242-9
11. Gatfield PD, Taller E, Wolfe DM, Haust MD. Hyperornithinemia, Hyperammonemia, and Homocitrullinuria Associated With Decreased Carbamyl Phosphate Synthetase I Activity. Pediatr Res (1975) 9(5):488–97. doi: 10.1203/00006450-197505000-00006
12. Haberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, et al. Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders: First Revision. J Inherit Metab Dis (2019) 42(6):1192–230. doi: 10.1002/jimd.12100
13. McGuire PJ, Lee HS, Summar ML. Infectious Precipitants of Acute Hyperammonemia are Associated With Indicators of Increased Morbidity in Patients With Urea Cycle Disorders. J Pediatr (2013) 163(6):1705–10 e1. doi: 10.1016/j.jpeds.2013.08.029
14. Singh RH, Rhead WJ, Smith W, Lee B, Sniderman King L, Summar M. Nutritional Management of Urea Cycle Disorders. Crit Care Clin (2005) 21(4 Suppl):S27–35. doi: 10.1016/j.ccc.2005.08.003
15. Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, Symptoms, Frequency and Mortality of 260 Patients With Urea Cycle Disorders From a 21-Year, Multicentre Study of Acute Hyperammonaemic Episodes. Acta Paediatr (2008) 97(10):1420–5. doi: 10.1111/j.1651-2227.2008.00952.x
16. Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, et al. Cross-Sectional Multicenter Study of Patients With Urea Cycle Disorders in the United States. Mol Genet Metab (2008) 94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004
17. Batshaw ML, Tuchman M, Summar M, Seminara J. A Longitudinal Study of Urea Cycle Disorders. Mol Genet Metab (2014) 113(1-2):127–30. doi: 10.1016/j.ymgme.2014.08.001
18. Matsumoto S, Haberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K. Urea Cycle Disorders-Update. J Hum Genet (2019) 64(9):833–47. doi: 10.1038/s10038-019-0614-4
19. Hady-Cohen R, Dragoumi P, Barca D, Plecko B, Lerman-Sagie T, Zafeiriou D. Safety and Recommendations for Vaccinations of Children With Inborn Errors of Metabolism. Eur J Paediatr Neurol (2021) 35:93–9. doi: 10.1016/j.ejpn.2021.10.002
20. van der Meer W, van Gelder W, de Keijzer R, Willems H. The Divergent Morphological Classification of Variant Lymphocytes in Blood Smears. J Clin Pathol (2007) 60(7):838–9. doi: 10.1136/jcp.2005.033787
21. Bao L, Cui Y. Prediction of the Phenotypic Effects of non-Synonymous Single Nucleotide Polymorphisms Using Structural and Evolutionary Information. Bioinformatics (2005) 21(10):2185–90. doi: 10.1093/bioinformatics/bti365
22. Kumar P, Henikoff S, Ng PC. Predicting the Effects of Coding non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat Protoc (2009) 4(7):1073–81. doi: 10.1038/nprot.2009.86
23. Ramensky V, Bork P, Sunyaev S. Human non-Synonymous SNPs: Server and Survey. Nucleic Acids Res (2002) 30(17):3894–900. doi: 10.1093/nar/gkf493
24. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med (2021) 13(1):31. doi: 10.1186/s13073-021-00835-9
25. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet (2016) 99(4):877–85. doi: 10.1016/j.ajhg.2016.08.016
26. Willer CJ, Li Y, Abecasis GR. METAL: Fast and Efficient Meta-Analysis of Genomewide Association Scans. Bioinformatics (2010) 26(17):2190–1. doi: 10.1093/bioinformatics/btq340
27. Monne M, Miniero DV, Daddabbo L, Palmieri L, Porcelli V, Palmieri F. Mitochondrial Transporters for Ornithine and Related Amino Acids: A Review. Amino Acids (2015) 47(9):1763–77. doi: 10.1007/s00726-015-1990-5
28. Monne M, Miniero DV, Daddabbo L, Robinson AJ, Kunji ER, Palmieri F. Substrate Specificity of the Two Mitochondrial Ornithine Carriers can be Swapped by Single Mutation in Substrate Binding Site. J Biol Chem (2012) 287(11):7925–34. doi: 10.1074/jbc.M111.324855
29. Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of Mitochondrial ADP/ATP Carrier in Complex With Carboxyatractyloside. Nature (2003) 426(6962):39–44. doi: 10.1038/nature02056
30. Pierri CL, Palmieri F, De Grassi A. Single-Nucleotide Evolution Quantifies the Importance of Each Site Along the Structure of Mitochondrial Carriers. Cell Mol Life Sci (2014) 71(2):349–64. doi: 10.1007/s00018-013-1389-y
31. Ferguson FG, Wikby A, Maxson P, Olsson J, Johansson B. Immune Parameters in a Longitudinal Study of a Very Old Population of Swedish People: A Comparison Between Survivors and Nonsurvivors. J Gerontol A Biol Sci Med Sci (1995) 50(6):B378–82. doi: 10.1093/gerona/50A.6.B378
32. Ndumbi P, Gilbert L, Tsoukas CM. Comprehensive Evaluation of the Immune Risk Phenotype in Successfully Treated HIV-Infected Individuals. PloS One (2015) 10(2):e0117039. doi: 10.1371/journal.pone.0117039
33. Motrich RD, Breser ML, Molina RI, Tissera A, Olmedo JJ, Rivero VE. Patients With Chronic Prostatitis/Chronic Pelvic Pain Syndrome Show T Helper Type 1 (Th1) and Th17 Self-Reactive Immune Responses Specific to Prostate and Seminal Antigens and Diminished Semen Quality. BJU Int (2020) 126(3):379–87. doi: 10.1111/bju.15117
34. Shirakawa K. Sano M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells (2021) 10(9):2435. doi: 10.3390/cells10092435
35. Tian Y, Babor M, Lane J, Schulten V, Patil VS, Seumois G, et al. Unique Phenotypes and Clonal Expansions of Human CD4 Effector Memory T Cells Re-Expressing CD45RA. Nat Commun (2017) 8(1):1473. doi: 10.1038/s41467-017-01728-5
36. Verma K, Ogonek J, Varanasi PR, Luther S, Bunting I, Thomay K, et al. Human CD8+ CD57- TEMRA Cells: Too Young to be Called "Old". PloS One (2017) 12(5):e0177405. doi: 10.1371/journal.pone.0177405
37. Silvera-Ruiz SM, Arranz JA, Haberle J, Angaroni CJ, Bezard M, Guelbert N, et al. Urea Cycle Disorders in Argentine Patients: Clinical Presentation, Biochemical and Genetic Findings. Orphanet J Rare Dis (2019) 14(1):203. doi: 10.1186/s13023-019-1177-3
38. Mahmoud NA, Ahmed DT, Mohammed ZO, Altyeb FA, Mustafa MI, Hassan MA. The Association Between SLC25A15 Gene Polymorphisms and Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome: Using In Silico Analysis. bioRxiv (2019) 786301. doi: 10.1101/786301
39. Saule P, Trauet J, Dutriez V, Lekeux V, Dessaint JP, Labalette M. Accumulation of Memory T Cells From Childhood to Old Age: Central and Effector Memory Cells in CD4(+) Versus Effector Memory and Terminally Differentiated Memory Cells in CD8(+) Compartment. Mech Ageing Dev (2006) 127(3):274–81. doi: 10.1016/j.mad.2005.11.001
40. Spyridopoulos I, Hoffmann J, Aicher A, Brummendorf TH, Doerr HW, Zeiher AM, et al. Accelerated Telomere Shortening in Leukocyte Subpopulations of Patients With Coronary Heart Disease: Role of Cytomegalovirus Seropositivity. Circulation (2009) 120(14):1364–72. doi: 10.1161/CIRCULATIONAHA.109.854299
41. Weltevrede M, Eilers R, de Melker HE, van Baarle D. Cytomegalovirus Persistence and T-Cell Immunosenescence in People Aged Fifty and Older: A Systematic Review. Exp Gerontol (2016) 77:87–95. doi: 10.1016/j.exger.2016.02.005
42. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function Throughout Life. Immunity (2018) 48(2):202–13. doi: 10.1016/j.immuni.2018.01.007
43. Henson SM, Riddell NE, Akbar AN. Properties of End-Stage Human T Cells Defined by CD45RA Re-Expression. Curr Opin Immunol (2012) 24(4):476–81. doi: 10.1016/j.coi.2012.04.001
44. Mittelbrunn M, Kroemer G. Hallmarks of T Cell Aging. Nat Immunol (2021) 22(6):687–98. doi: 10.1038/s41590-021-00927-z
45. Wong GK, Huissoon AP. T-Cell Abnormalities in Common Variable Immunodeficiency: The Hidden Defect. J Clin Pathol (2016) 69(8):672–6. doi: 10.1136/jclinpath-2015-203351
46. Fenwick C, Joo V, Jacquier P, Noto A, Banga R, Perreau M, et al. T-Cell Exhaustion in HIV Infection. Immunol Rev (2019) 292(1):149–63. doi: 10.1111/imr.12823
47. Hainaut M, Verscheure V, Ducarme M, Schandene L, Levy J, Mascart F. Cellular Immune Responses in Human Immunodeficiency Virus (HIV)-1-Infected Children: Is Immune Restoration by Highly Active Anti-Retroviral Therapy Comparable to non-Progression? Clin Exp Immunol (2011) 165(1):77–84. doi: 10.1111/j.1365-2249.2011.04403.x
48. Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep (2019) 26(6):1627–40.e7. doi: 10.1016/j.celrep.2019.01.041
49. The Human Protein Atlas Project. Stockholm, Sweden: The Knut & Alice Wallenberg Foundation. (2022). SLC25A15. Available at: https://www.proteinatlas.org/ENSG00000102743-SLC25A15/immune+cell [Accessed January 24, 2022].
50. Tarasenko TN, Gomez-Rodriguez J, McGuire PJ. Impaired T Cell Function in Argininosuccinate Synthetase Deficiency. J Leukoc Biol (2015) 97(2):273–8. doi: 10.1189/jlb.1AB0714-365R
51. Lukkarinen M, Parto K, Ruuskanen O, Vainio O, Kayhty H, Olander RM, et al. B and T Cell Immunity in Patients With Lysinuric Protein Intolerance. Clin Exp Immunol (1999) 116(3):430–4. doi: 10.1046/j.1365-2249.1999.00868.x
52. Luo C, Shen G, Liu N, Gong F, Wei X, Yao S, et al. Ammonia Drives Dendritic Cells Into Dysfunction. J Immunol (2014) 193(3):1080–9. doi: 10.4049/jimmunol.1303218
53. Shawcross DL, Wright GA, Stadlbauer V, Hodges SJ, Davies NA, Wheeler-Jones C, et al. Ammonia Impairs Neutrophil Phagocytic Function in Liver Disease. Hepatology (2008) 48(4):1202–12. doi: 10.1002/hep.22474
54. Lebosse F, Gudd C, Tunc E, Singanayagam A, Nathwani R, Triantafyllou E, et al. Cd8(+)T Cells From Patients With Cirrhosis Display a Phenotype That may Contribute to Cirrhosis-Associated Immune Dysfunction. EBioMedicine (2019) 49:258–68. doi: 10.1016/j.ebiom.2019.10.011
55. Bonnel AR, Bunchorntavakul C, Reddy KR. Immune Dysfunction and Infections in Patients With Cirrhosis. Clin Gastroenterol Hepatol (2011) 9(9):727–38. doi: 10.1016/j.cgh.2011.02.031
56. Lamontagne A, Long RE, Comunale MA, Hafner J, Rodemich-Betesh L, Wang M, et al. Altered Functionality of Anti-Bacterial Antibodies in Patients With Chronic Hepatitis C Virus Infection. PloS One (2013) 8(6):e64992. doi: 10.1371/journal.pone.0064992
Keywords: urea cycle defects, HHH syndrome, infection, hyperammonemia, immunodeficiency, T cells, B cells, case report
Citation: Silvera-Ruiz SM, Gemperle C, Peano N, Olivero V, Becerra A, Häberle J, Gruppi A, Larovere LE and Motrich RD (2022) Immune Alterations in a Patient With Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome: A Case Report. Front. Immunol. 13:861516. doi: 10.3389/fimmu.2022.861516
Received: 24 January 2022; Accepted: 02 May 2022;
Published: 27 May 2022.
Edited by:
Anette S. B. Wolff, Haukeland University Hospital, NorwayReviewed by:
Fernando A. Arosa, University of Beira Interior, PortugalCarlo Marya Thomas Marobbio, University of Bari Aldo Moro, Italy
Copyright © 2022 Silvera-Ruiz, Gemperle, Peano, Olivero, Becerra, Häberle, Gruppi, Larovere and Motrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruben D. Motrich, cm1vdHJpY2hAdW5jLmVkdS5hcg==; orcid.org/0000-0002-5772-1285