 Jared L. Casteel1Kasey R. Keever1,2
Jared L. Casteel1Kasey R. Keever1,2 Christopher L. Ardell1David L. Williams2,3Detao Gao4
Christopher L. Ardell1David L. Williams2,3Detao Gao4 Eugene A. Podrez4Tatiana V. Byzova5
Eugene A. Podrez4Tatiana V. Byzova5 Valentin P. Yakubenko1,2*
Valentin P. Yakubenko1,2*- 1Department of Biomedical Sciences, Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 2Center of Excellence in Inflammation, Infectious Disease and Immunity, Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 3Department of Surgery, Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 4Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, United States
- 5Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, United States
Oxidation of polyunsaturated fatty acids contributes to different aspects of the inflammatory response due to the variety of products generated. Specifically, the oxidation of DHA produces the end-product, carboxyethylpyrrole (CEP), which forms a covalent adduct with proteins via an ϵ-amino group of lysines. Previously, we found that CEP formation is dramatically increased in inflamed tissue and CEP-modified albumin and fibrinogen became ligands for αDβ2 (CD11d/CD18) and αMβ2 (CD11b/CD18) integrins. In this study, we evaluated the effect of extracellular matrix (ECM) modification with CEP on the adhesive properties of M1-polarized macrophages, particularly during chronic inflammation. Using digested atherosclerotic lesions and in vitro oxidation assays, we demonstrated the ability of ECM proteins to form adducts with CEP, particularly, DHA oxidation leads to the formation of CEP adducts with collagen IV and laminin, but not with collagen I. Using integrin αDβ2-transfected HEK293 cells, WT and mouse M1-polarized macrophages, we revealed that CEP-modified proteins support stronger cell adhesion and spreading when compared with natural ECM ligands such as collagen IV, laminin, and fibrinogen. Integrin αDβ2 is critical for M1 macrophage adhesion to CEP. Based on biolayer interferometry results, the isolated αD I-domain demonstrates markedly higher binding affinity to CEP compared to the “natural” αDβ2 ligand fibrinogen. Finally, the presence of CEP-modified proteins in a 3D fibrin matrix significantly increased M1 macrophage retention. Therefore, CEP modification converts ECM proteins to αDβ2-recognition ligands by changing a positively charged lysine to negatively charged CEP, which increases M1 macrophage adhesion to ECM and promotes macrophage retention during detrimental inflammation, autoimmunity, and chronic inflammation.
Introduction
Low-grade chronic inflammation is a key component in the development of metabolic and autoimmune diseases such as diabetes, atherosclerosis, obesity, and rheumatoid arthritis (1–3). A critical step in the progression of the disease is the accumulation of classically activated pro-inflammatory (M1-like) macrophages in the extracellular matrix (ECM) of inflamed tissues (4, 5). The failure of pro-inflammatory macrophages to emigrate from the inflamed tissue leads to excessive leukocyte recruitment, metabolic imbalance, and damage of healthy tissue, which all together promote the development of chronic inflammation (1, 6). The mechanism and key components of macrophage retention within the site of inflammation are not yet well understood, although this information can improve the treatment of a variety of different pathologies. Enhanced adhesion that prevents macrophage migration is a potential central mechanism of chronic inflammation development. The cell adhesion force is regulated by the density of adhesive receptors, ligand availability, and receptor-ligand affinity (7). The major adhesive receptors on macrophages are integrins. β1 integrins (α1β1, α2β1, α3β1, etc.) have a moderate expression on macrophages (8, 9) and support adhesion/migration to major ECM proteins, such as collagens and laminins. The role of high-density expressed β2 integrins (αMβ2, αDβ2, etc.) in macrophage migration/retention in tissue is thought to be limited by lack of binding to major ECM proteins (10, 11).
Nevertheless, we recently demonstrated that macrophage receptor integrin αDβ2 (CD11d/CD18) contributes to the development of atherosclerosis and diabetes due to the generation of strong adhesion of pro-inflammatory macrophages to the inflamed ECM, thereby promoting the retention of macrophages at the site of chronic inflammation (12). Integrin αDβ2 is the most recently discovered leukocyte integrin (13, 14), that contributes to an inflammatory response (12, 15–17) and infection in different pathological conditions (18–21). Interestingly, integrin αDβ2 is upregulated on pro-inflammatory macrophages, which makes it potentially important for macrophage retention (12). However, the critical αDβ2 ligand in the inflamed tissue is not yet defined.
We recently discovered a potential mechanism that could explain the presence of inflammation-specific ligands in the ECM for integrin αDβ2. We revealed that during acute inflammation, the oxidation of docosahexaenoic acid (DHA) leads to the generation of the end-product carboxyethylpyrrole (CEP), which forms an adduct with proteins via an ϵ-amino group of lysines (22, 23). In vitro analysis with BSA demonstrated that 10-20 lysines can be modified by CEP in one protein molecule. Notably, positively charged lysines are substituted to the pyrroles with exposed negatively charged carboxyl groups. (Figure 1). Based on our recent data, a carboxyl group in the CEP structure is specifically responsible for the binding to integrins αDβ2 and αMβ2 (23). Accordingly, CEP-modification significantly changes the surface charge of the targeted protein and its binding properties.
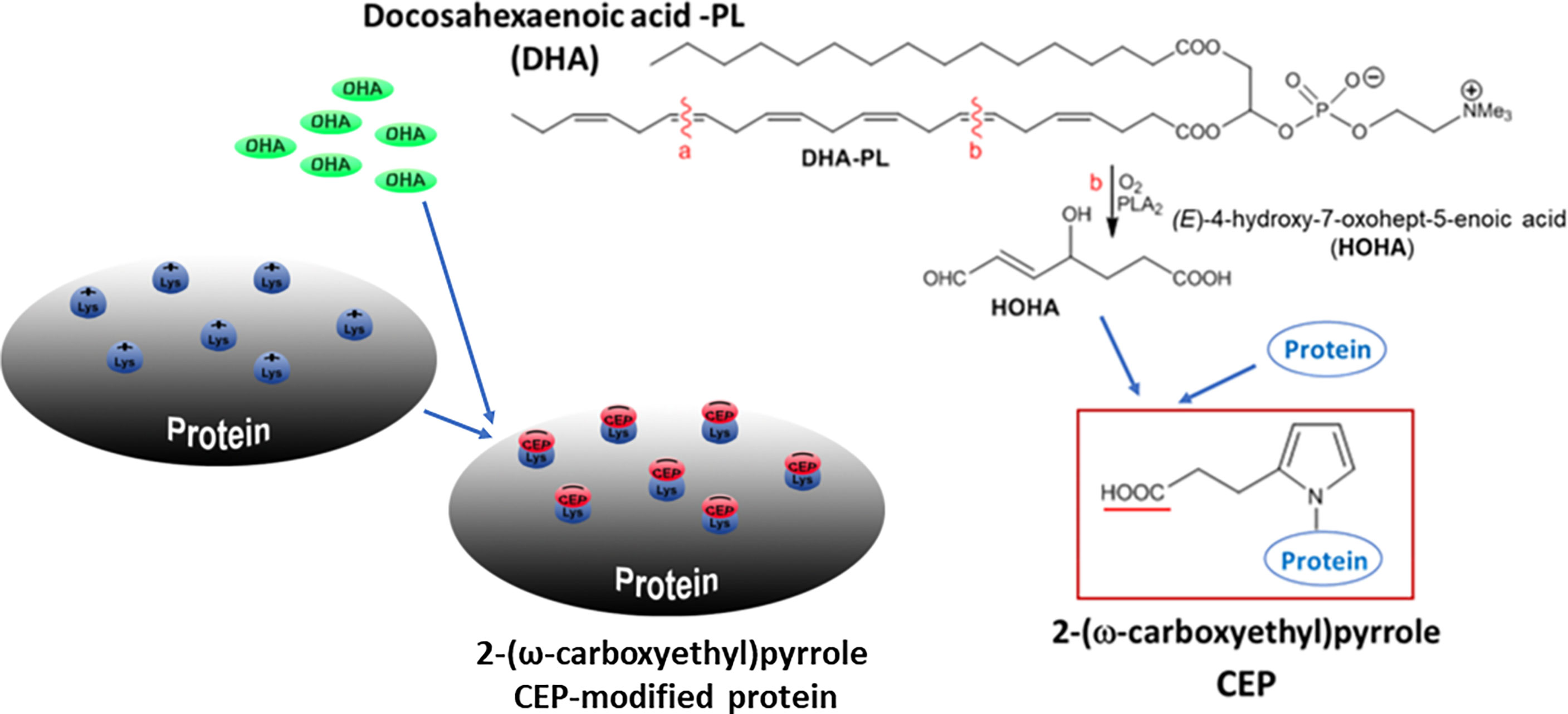
Figure 1 Schematic representation of CEP formation. PLA2-catalyzed hydrolysis of DHA generates 4-hydroxy-7-oxo-hept-5-eonate (HOHA), which, in turn, produces 2-ω-Carboxyethylpyrrole (CEP)-protein derivatives through condensation with the primary amino groups of protein lysyl residues. A positively charged lysine is modified by pyrrole with a negatively charged carboxyl group. Multiple lysines can be modified by CEP in one protein molecule.
In our previous study, we found that during peritoneal inflammation at least two proteins are modified with CEP in the peritoneal cavity - fibrinogen and albumin. Most importantly, we found that CEP-modified proteins support the migration of non-polarized macrophages to the site of acute inflammation due to interaction with macrophage integrins αDβ2 and αMβ2 (23). Integrins αDβ2 and αMβ2 are adhesive receptors, which interact with many ligands and transduce a signal to the actin cytoskeleton that regulates cell adhesion, migration, and other cell responses (24, 25). αDβ2 possesses a high homology and similar ligand-binding specificity to αMβ2 (11); however, these two integrins demonstrate different roles in chronic inflammatory diseases. As discussed above, αD supports the development of atherosclerosis (12) and diabetes (26), while integrin αM protects against these metabolic diseases (27, 28). Apparently, the different surface densities of these integrins on primary monocytes (29) and distinct subsets of macrophages (12, 18, 26), determine their diverse roles in cell migration and chronic inflammation. Interestingly, the analysis of αDβ2 and αMβ2 binding to CEP demonstrated that αD has a 5-folds higher affinity to CEP (23), which may have a physiological importance for macrophage adhesion.
The goal of the current project is to evaluate how the modification of ECM proteins with CEP affects their binding properties and the strength of macrophage adhesion, which is critical for macrophage retention during chronic inflammation.
Material and Methods
Reagents and Antibodies
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) and Thermo Fisher Scientific (Waltham, MA). Recombinant mouse IFNγ, CCL2, and CCL5 were purchased from Invitrogen Corporation (Carlsbad, CA). Human fibrinogen and thrombin were obtained from Enzyme Research Laboratories (South Bend, IN). Collagen IV, collagen I, and laminin were purchased from Corning (Corning, NY). CEP-BSA (21:1 molar ratio) and CEP-Fg (29:1 molar ratio) were prepared through a Paar-Knorr pyrrole synthesis with (9H-fluoren-9-ylmethyl ester 4,7-dioxo-heptanoic acid) DOHA-Fm as described previously (28). Anti-CD68 mAb was from eBioscience. Polyclonal antibody against the αD I-domain was described previously (11). The antibody recognizes both human and mouse αD I-domains and has no cross-reactivity with recombinant human and mouse αM, αX, and αL I-domains. The antibody was isolated from rabbit serum by affinity chromatography using αD I-domain-Sepharose. Anti-macrophage antibody F4/80 was from eBioscience (San Diego, CA). A polyclonal antibody against CEP and monoclonal IgM anti-CEP antibody were described previously (30, 31). Blocking IgG anti-CEP antibody (Clone 3C9) was generated in Dr. Tatiana Byzova’s laboratory (23).
Animals
Wild type (C57BL/6J, stock # 000664) and integrin αD-deficient (B6.129S7-Itgadtm1Bll/J, stock # 005258) mice were bought from Jackson Laboratory (Bar Harbor, ME). αD-deficient have been backcrossed to C57BL/6 for at least ten generations. All procedures were performed according to animal protocols approved by East Tennessee State University IACUC.
Generation of Classically Activated (M1) Macrophages
Notably, the terms M1 and M2 macrophages are artificial and do not represent all variabilities of macrophage subsets in vivo. We used the term M1 in our manuscript to refer to in vitro polarized macrophages activated by IFN-γ which serve as a model for pro-inflammatory macrophages in vivo. Therefore, peritoneal macrophages from 8-12-week-old mice (WT and ) were harvested by lavage of the peritoneal cavity with 5 ml of sterile PBS 3 days after intraperitoneal (IP) injection of 4% thioglycollate (TG; 0.5ml). The cells were washed twice with PBS and resuspended in RPMI media supplemented with 10% FBS, 0.1 mg/ml streptomycin, and 0.1 unit/ml penicillin. The cell suspension was transferred into 100mm Petri dishes and incubated for 2h at 37°C in humidified air containing a 5% CO2 atmosphere. Nonadherent cells were washed out with PBS, and the adherent macrophages were replenished with complete RPMI media. The macrophages were differentiated to M1 phenotypes by treatment with recombinant mouse interferon-γ (IFN-γ) (100 U/ml, Thermo Fisher) for 4 days. Medium with IFN-γ was changed every 2 days or as required. Macrophages were dissociated from the plates using 5mM EDTA in PBS and used for the experiments thereafter.
Generation of αDβ2-Transfected HEK293 Cells
The HEK293 cells stably expressing human integrin αDβ2 were generated as described previously (11) and maintained in DMEM/F-12 (Invitrogen) supplemented with 10% FBS, 2 mM glutamine, 15 mM HEPES, 0.1 mg/ml streptomycin, and 0.1 unit/ml penicillin.
Isolation of Recombinant αD I-Domain in an Active Conformation
The construct for the αD I-domain was generated and recombinant protein was isolated as described in our previous paper (23). Briefly, αD in the active conformation (Pro128-Lys314) was inserted in the pET15b vector, expressed in E. Coli as a His-tag fusion protein, and purified using affinity chromatography on Ni-chelating agarose (Qiagen Inc., Valencia, CA).
Flow Cytometry Analysis
Flow cytometry analysis was performed to assess the expression of integrin αD on mouse peritoneal macrophages and αDβ2-transfected HEK293 cells. Cells were harvested and pre-incubated with 4% normal goat serum for 30 min at 4°C, then 2x106 cells were incubated with a specific antibody for 30 min at 4°C. Non-conjugated antibodies required additional incubation with Alexa 488 or Alexa 647-donkey anti-mouse IgG (at a 1:1000 dilution) for 30 min at 4°C. Finally, the cells were washed and analyzed using a Fortessa X-20 (Becton Dickinson).
Cell Adhesion Assay
The adhesion assay was performed as described previously (23) with modifications. Briefly, 96-well plates (Immulon 2HB, Cambridge, MA) were coated with different concentrations of fibrinogen, collagen IV, collagen I, or laminin for 3 h at 37°C. The wells were post-coated with 0.5% polyvinyl alcohol for 1 h at room temperature. Mouse peritoneal macrophages or HEK293 cells transfected with integrin αDβ2 were labeled with 10 µM Calcein AM (Molecular Probes, Eugene, OR) for 30 min at 37°C and washed with HBSS and resuspended in the same medium at a concentration of 1 × 106 cells/mL. αDβ2-transfected HEK 293 cells were also supplemented with 2mM MnCl2. Aliquots (50 µL) of the labeled cells were added to each well. After 30 minutes of incubation at 37°C in a 5% CO2 humidified atmosphere, the nonadherent cells were removed by washing with PBS. The fluorescence was measured in a Synergy H1 fluorescence plate reader (BioTek, Winooski, VT), and the number of adherent cells was determined from a labeled control.
Cell Spreading Assay
Glass coverslips were coated with fibrinogen (20 µg/ml), Collagen IV (5 μg/ml) or CEP-BSA (750 μM) for 3 hours at 37°C and post-coated with 0.5% polyvinylpyrrolidone for 1 hour at 37°C. To determine cell spreading, inflammatory macrophages isolated from the WT and mice were allowed to adhere to coverslips for 1 hour at 37°C. Macrophages were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with Phalloidin-iFluor 555 and DAPI. Fluorescent images were obtained with EVOS FL auto cell fluorescent imaging system using 20x objective with multi-frame automatic setup (9-fields). The cell areas were calculated using Imaris 8.0 software.
Migration of Macrophages in 3D Fibrin Gel
Migration assay was performed as described previously (26). Polarized M1 macrophages were labeled with PKH26 red fluorescent dye. Cell migration assay was performed for 48 hours at 37°C in 5% CO2 in a sterile condition. An equal number of WT macrophages was evaluated by cytospin of mixed cells before the experiment and at the starting point before migration. Labeled WT (1.5x105) activated macrophages were plated on the membranes of transwell inserts with a pore size of 8 μm and 6.5 mm in diameter (Costar, Corning, NY) precoated with fibrinogen (Fg). Fibrin gel (100 µl/sample) was generated by mixing 0.75mg/ml Fg containing 1% FBS and 1% P/S and 0.5 U/ml thrombin. In some samples 9 µM CEP was incorporated into the gel during polymerization. 30 nM of MCP-1 was added on top of the gel to initiate the migration. Migrating cells were detected by Leica Confocal microscope (Leica-TCS SP8), and the results were analyzed and reconstructed using IMARIS 8.0 software.
Biolayer Interferometry
The binding parameters of the interaction between the αD I-domain with CEP or fibrinogen were determined using an Octet K2 instrument (FortéBio, Sartorius Group). CEP-BSA or fibrinogen was immobilized on the Amine Reactive Second-generation (AR2G) biosensor using the amine coupling kit according to the manufacturer’s protocol. Different concentrations of the active forms of αD I-domain (50-1000 nM) were applied in the mobile phase, and the association between the immobilized and flowing proteins was detected. Experiments were performed in 20 mm HEPES, 150 mm NaCl, 1 mM CaCl2, 1 mM MgCl2, 0.02% (v/v) Tween-20, 0.02% BSA, pH 7.5. The protein-coated biosensors were regenerated with 25 mm NaOH and 2M NaCl. Analyses of the binding kinetics were performed using DataAnalysis HT11.0 software (ForteBio). The value of the equilibrium dissociation constant (KD) was obtained by fitting a plot of response at equilibrium (Req) against the concentration.
Immunostaining
Cryosections (10 μm) of aorta roots (from WT and ApoE-/- mice after five weeks on a western diet) were warmed to room temperature for 30 minutes prior to immunofluorescence staining. Tissue sections were fixed in ice-cold acetone for 10 minutes followed by permeabilizing with 0.2% Tween-20 for 10 min to increase the signal of intracellular binding sites. Tissue sections were washed in PBS and incubated with SuperBlock (PBS) Blocking buffer (Thermo Scientific, Rockford, IL, USA) for 45 min to block nonspecific binding. Tissues were then incubated at 4°C overnight with the primary antibody (rabbit polyclonal anti-CEP and rat anti-mouse CD68 (macrophage marker). After washing several times with PBS, the sections were incubated with Alexa Fluor 488-conjugated donkey anti-rabbit IgG and Alexa Fluor 568-conjugated donkey anti-rat for 1 hour at room temperature. The sections were subsequently washed and sealed. The tissue sections were examined by Leica Confocal microscope (Leica-TCS SP8). Control sections without the primary antibody were also performed at the same time.
Analysis of CEP Formation in Atherosclerotic Lesions of ApoE-/- Mice by Western Blot
Male ApoE-deficient mice were kept on a Western diet for 16 weeks. After that, ApoE-/- and the same age WT C57BL/6 mice (as the control) were euthanized, their vascular system was perfused with 10 ml PBS, and the aortas were cleaned from the outer layers of connective tissue and removed. The isolated aortas were digested with Collagenase type I, type XI, Hyaluronidase, and DHAase I according to the published protocol (12). The cells were separated by centrifugation. The non-cellular supernatant was high speed centrifugated to exclude aggregates and cell traces, and used for Western Blot analysis.
The same concentration of proteins isolated from ApoE-/- and WT aortas was loaded on SDS-electrophoresis analyzed by 7% SDS-PAGE electrophoresis and Western blotting. The Immobilon-P membranes (Millipore) were incubated with rabbit polyclonal anti-CEP antibody, followed by incubation with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase and developed using enhanced SuperSignal Chemiluminescent Substrate (Pierce).
Generation of CEP-Modified ECM Proteins
The evaluation of CEP-modified proteins was performed as described previously (23). Collagen I, collagen IV, and laminin were coated on a 96-well plate at a concentration of 20 μg/ml overnight at 4°C. Wells were post-coated with 0.5% polyvinyl alcohol for 1 h at 37°C. 20 μM DHA in 20mM Hepes, 150 mM NaCl, 0.01% H2O2, and 1mM CaCl2 were added to the wells and incubated in an oxygen-free environment (under argon) for 18 hours at 37°C. After incubation, the plate was washed out with PBS supplemented with 0.05% Tween-20 3 times and incubated with anti-CEP polyclonal antibody (0.9 μg/ml) for 2 hours at 37°C. After washing, wells were incubated with goat-anti-rabbit HRP conjugated antibody for 1 h at 37°C and the binding was developed using TMB-ELISA substrate solution (Pierce). The result was detected by a plate reader using a wavelength of 450 nm.
Statistical Analysis
Statistical analyses were performed using a Student’s t-test or Student’s paired t-tests where indicated in the text using SigmaPlot 13. A value of p<0.05 was considered significant. Error bars represent the SEM of experiments.
Results
DHA Oxidation Generates CEP Adducts With ECM Proteins in Atherosclerotic Lesions and In Vitro
We and others have demonstrated the presence of CEP in different inflammatory tissues using immunohistochemistry (23, 30–34). Particularly, anti-CEP antibody detected strong immunostaining in the sections of atherosclerotic lesions of ApoE-/- mice on a Western diet (Figure 2A). However, the direct formation of CEP-protein adducts was shown previously only in the peritoneal cavity, but not in the tissue (23). To demonstrate the link between CEP immunostaining and the formation of CEP-modified proteins in the inflamed tissue during chronic inflammation, we evaluated CEP-protein adduct formation in atherosclerotic lesions (Figure 2B). Male ApoE-deficient mice were kept on a Western diet for 16 weeks (n=3). After that, ApoE-/- and the same age WT C57BL/6 mice (as the control) were euthanized, their vascular system was perfused and aortas were removed. The isolated aortas were digested according to the published protocol (12) and cells were separated by centrifugation. The non-cellular supernatant was high speed centrifugated to exclude aggregates and cell traces, and used for Western Blot analysis.
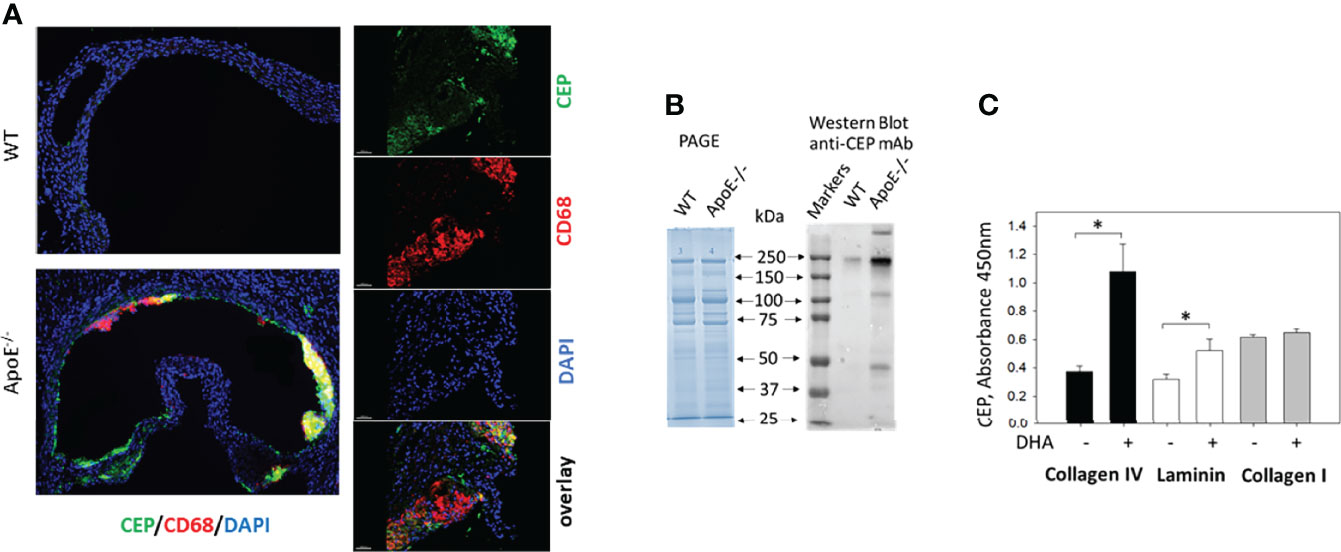
Figure 2 CEP forms adducts with ECM proteins during inflammation and oxidation of DHA. (A) CEP deposition in atherosclerotic lesions. Aortic sinuses were isolated from the ApoE-deficient mice after five weeks on a western diet. The sections were prepared and immunostained with anti-CEP (green) and anti-CD68 (red) antibodies. The crosssection of entire aorta roots (right) and 200x magnified inlet (left) are shown (scale bar 200 μm). (B) Electrophoresis (PAGE) and Western blot analysis with anti-CEP antibody of digested aortas. Aortas of ApoE-/- and WT mice were digested, cells were separated and the non-cellular contest was analyzed by electrophoresis with Coomassie Blue and Western Blot (7% PAGE) with anti-CEP antibody (representative experiment from 4 performed). (C) Generation of CEP-modified ECM proteins. Collagen IV, collagen I, and laminin were coated on a 96-well plate and incubated in the presence (or absence) of 2 μM DHA in 20 mM HEPES, 150 mM NaCl, 0.01% H2O2, and 1mM CaCl2 in an oxygen-free environment (under argon atmosphere) for 18 hours at 37°C. After incubation, the plate was incubated with an anti-CEP polyclonal antibody (0.9 μg/ml) followed by goat-anti-rabbit HRP conjugated antibody. The binding was developed using a TMB-ELISA substrate solution (Pierce). The result was detected by plate reader using wavelength 450 nm. Statistical analyses were performed using Student’s paired t-tests from 3 independent experiments. *P < 0.05.
The same concentration of proteins isolated from ApoE-/- and WT aortas was loaded on SDS-electrophoresis (Figure 2B, left panel) and analyzed using an anti-CEP antibody (Figure 2B, right panel). We found a dramatic difference in anti-CEP staining of proteins isolated from the atherosclerotic lesions of ApoE-deficient and WT mice. While only one weak band was spotted in the sample isolated from WT mice, several intensive bands were detected in the ApoE-/- mice samples that represent several CEP-modified proteins. Notably, the faded CEP signal from the WT mice corresponds to the concept that the natural oxidation of PUFA in a healthy organism can lead to a low level of adduct formation. We detected such signal previously in commercial fibrinogen that we used in our experiments as a control (23).
To confirm the ability of CEP to form the covalent adduct with ECM proteins, we tested the formation of CEP modification in collagen I, laminin, and collagen IV using in vitro DHA oxidation assay that we developed previously (23). ECM proteins were incubated with DHA and 0.01% H2O2 for 18 h at 37°C. After incubation, CEP formation was detected by ELISA with an anti-CEP antibody (Figure 2C). We found that DHA oxidation led to the formation of CEP-collagen IV adduct and CEP-laminin adduct, while collagen I was not modified.
These experiments clearly demonstrated that ECM proteins are modified by CEP, and inflamed tissue (e.g. atherosclerotic lesions) can be a source of specific adhesive substrate for macrophages.
M1 Macrophages Demonstrate Stronger Adhesion and Spreading to CEP-Modified Proteins
Pro-inflammatory, M1-like macrophages, are the major source of inflammatory stimuli during chronic inflammation. We tested how protein modification with CEP affects M1 macrophage adhesion to ECM proteins such as collagen IV, laminin, and fibrinogen (which leaks from blood to the ECM through the damaged endothelial monolayer during inflammation). Different concentrations of collagen IV, laminin, CEP-modified BSA, and BSA alone as a control were immobilized on the 96-well plate and tested as substrates for M1 macrophage adhesion (Figure 3A). We found that the adhesion of pro-inflammatory macrophages to CEP-modified BSA dramatically overreached the adhesion of macrophages to natural ECM ligands. Notably, BSA alone does not support the adhesion of macrophages. In a separate experiment, we compared the adhesion of fibrinogen-modified by CEP and native fibrinogen. Clearly, CEP modification dramatically increased macrophage adhesion to fibrinogen (Figure 3B).
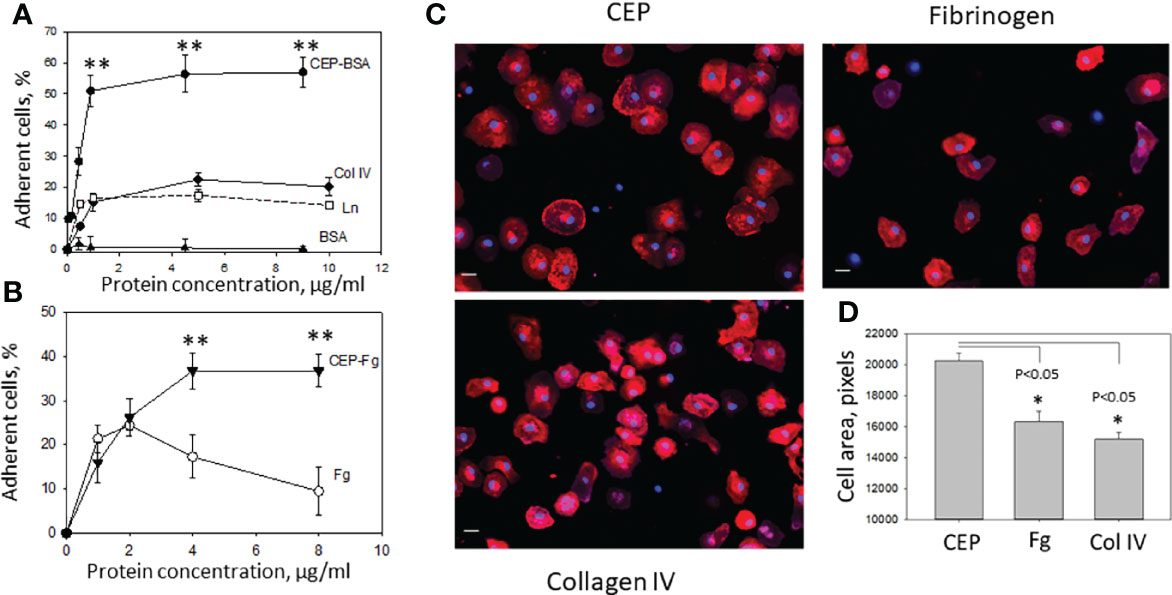
Figure 3 Adhesion of M1-polarized macrophages to CEP and ECM proteins. (A) Different concentrations of CEP-BSA, collagen IV (Col IV), laminin (Ln) and BSA were immobilized on a 96-well plate for 3 h at 37°C. Fluorescently labeled macrophages were added to the wells and cell adhesion was determined after 30 min in a fluorescence plate reader. CEP-BSA is presented based on the concentration of BSA. 1µg BSA contains 250 nM CEP. A representative experiment from 4 performed. Data are presented as mean ± SEM **p < 0.01. (B) Different concentrations of CEP-fibrinogen (CEP-Fg) and fibrinogen (Fg) were immobilized and tested in an adhesion assay as described above. A summary result from 3 experiments. CEP-Fg is shown based on the concentration of Fg. 1µg Fg contains 87 nM CEP Data are presented as mean ± SEM **p < 0.01. (C) Macrophages were allowed to adhere to glass coverslips coated with fibrinogen, Collagen IV or CEP-BSA for 1 hour at 37°C. Macrophages were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with Alexa Fluor 555-conjugated phalloidin and DAPI. Fluorescent images were obtained with EVOS FL auto cell fluorescent imaging system using 20x objective with mosaic 9-fields setup. (D) The cell area was calculated using Imaris 8.0 software. Data are presented as mean ± SEM *p < 0.05.
To verify these results macrophage spreading on CEP, collagen IV, and fibrinogen was evaluated. Cells were labeled with phalloidin-PE, examined by fluorescent microscopy, and analyzed using the ImarisCell module (Imaris 8.0). Corresponding to the adhesion data, the results demonstrate stronger spreading to CEP with larger cell areas and focal adhesion complexes (Figures 3C, D).
The Presence of CEP in a 3D Matrix Inhibits M1 Macrophage Migration
To evaluate the effect of CEP modification in ECM on cell motility we performed a 3D migration assay by comparing M1 macrophage migration in a polymerized fibrin matrix and polymerized fibrin matrix supplemented with 9 μM CEP. Based on our previous result the migration of M1 macrophages in a fibrin matrix is lower compared to non-activated or M2 macrophages (26). The goal of this experiment was to evaluate the additional effect of CEP on this migration. Cells were plated on one side of the gel and migration was initiated by CCL2 and CCL5 added to the other side of the gel (Figure 4A). We found that migration of M1 macrophages was dramatically reduced in fibrin gel supplemented with CEP (Figures 4B–D), which corresponds to strong adhesion events and promotion of macrophage retention at the site of chronic inflammation.
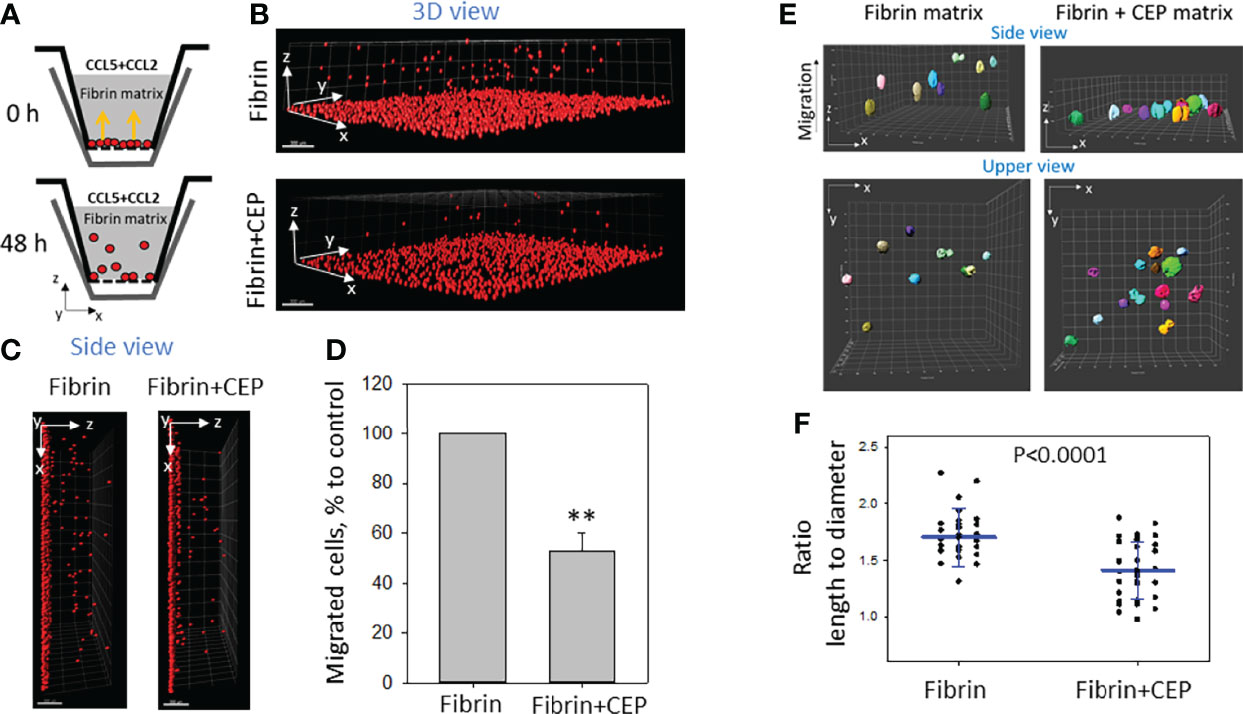
Figure 4 The presence of CEP in the matrix inhibits the 3D migration of M1 macrophages. (A) Sketch diagram of the migrating cells in Boyden transwell chamber. Before migration (upper panel) and after 48h migration (lower panel). (B) WT M1 macrophages were labeled with red PKH26 fluorescent dye. Labeled WT (1.5x105) activated macrophages were plated on the membranes of transwell inserts (Boyden chamber) with a pore size of 8 μm and 6.5 mm in diameter (Costar, Corning, NY) precoated with fibrinogen (Fg). Fibrin gel (100 µl/sample) was generated by mixing 0.75mg/ml Fg containing 1% FBS and 1% P/S and 0.5 U/ml thrombin with or without 9uM CEP. Migration of macrophages was stimulated by 30 nM CCL2 and 100nM CCL5 added to the top of the gel. After 48 hours, migrating cells were detected by a Leica Confocal microscope (B, C). (D) The results were analyzed by IMARIS 8.0 software, and statistical analyses were performed using Student’s paired t-tests (n = 5 per group). Scale bar= 300 μm. Data are presented as mean ± SEM. **P < 0.01. (E) The morphology of non-labeled macrophages was evaluated after gel staining with CytoPainet Phalloidinin-iFluor 555 in fibrin matrix (left panel) and Fibrin matrix supplemented with CEP (right panel). The ratio length/diameter was calculated using the ImarisCell module (IMARIS 8.0). (F) Statistical analyses were performed using Student’s t-tests, n = 30 (fibrin gel) 36 (fibrin+CEP gel). P < 0.0001.
To more critically evaluate these results, we tested the morphology of macrophages in a 3D matrix supplemented with CEP. 3D gels were generated using polymerized fibrin and polymerized fibrin/9 uM CEP. The same macrophages were incorporated into both matrixes during polymerization and migration was stimulated with a gradient of CCL2. After 18 hours, the cells inside the gel (at least 80 µM from the gel surface) were analyzed using the confocal microscope. We found that cells in the fibrin matrix present a more elongated shape when compared with the more rounded cells in the matrix with supplemented CEP (Figure 4E) The difference between the length-to-diameter ratio was extremely significant (P<0.0001, n=30-35/group) (Figure 4F). According to published data, elongated cell morphology in a 3D matrix corresponds to migrating cells.
Therefore, our results demonstrate that modification with CEP affects M1 macrophage adhesion, spreading, and migration-dependent morphology.
Integrin αDβ2 Is Critical for M1 Macrophage Adhesion and Spreading to CEP-Modified Proteins
The major adhesive receptors for CEP on macrophages are integrins αDβ2 and αMβ2. Integrin αD is upregulated on M1 macrophages (12), (Figure 5A), while αM maintains the same level of expression. This difference determines the distinct roles of these β2 integrins in inflammation. While αMβ2 supports macrophage migration and demonstrates some protective function during chronic inflammation (27, 35), integrin αDβ2 inhibits M1 macrophage migration and contributes to the development of chronic inflammation (12, 26). We hypothesized that integrin αDβ2 is critical for M1 macrophage adhesion to CEP-modified proteins. To test this hypothesis, we compared the adhesion of M1 macrophages isolated from the WT and αD-deficient mice to CEP. The adhesion assay demonstrated a dramatic reduction in macrophage adhesion in αD-deficient mice (Figure 5B). Moreover, macrophage spreading to CEP was significantly affected in M1 macrophages, the total cell areas were reduced as well as cell shapes were irregular (Figures 5C, D).
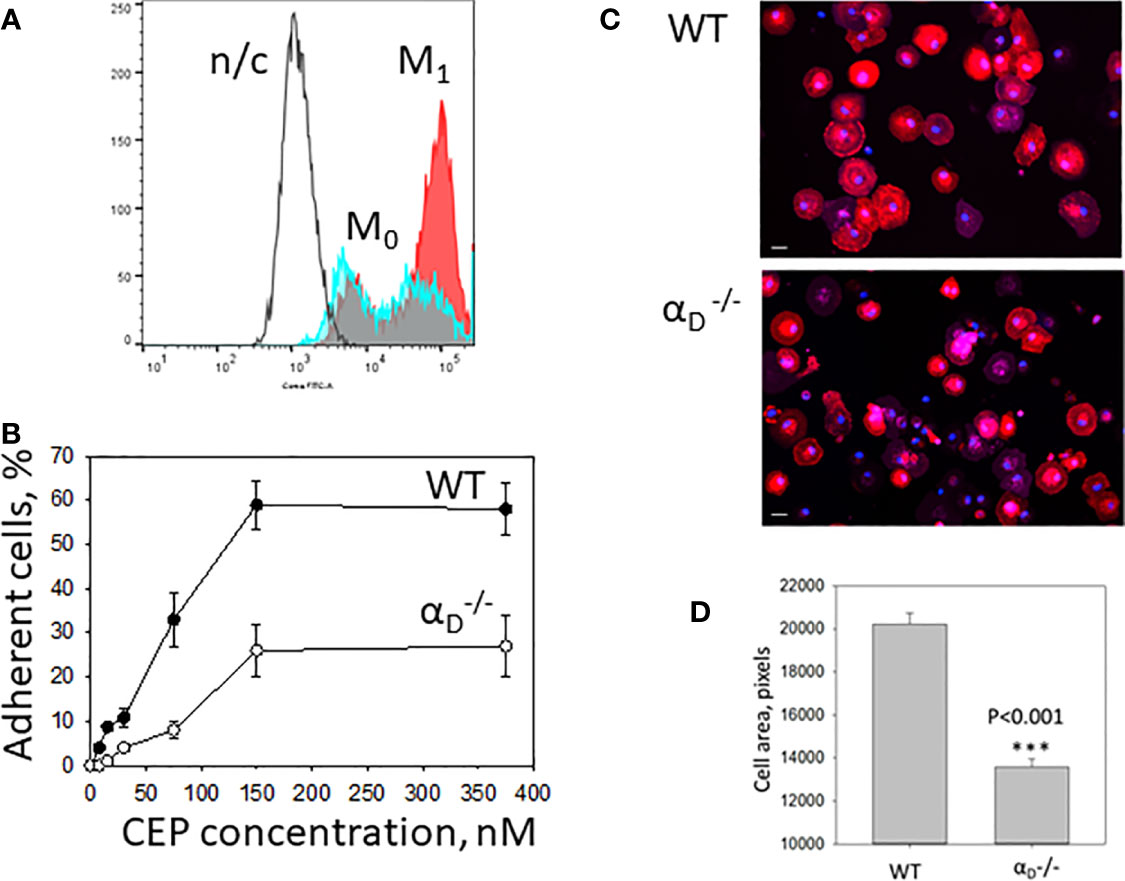
Figure 5 Integrin αDβ2 is critical for M1 macrophage adhesion to CEP-modified proteins. (A) The expression of integrin αD is upregulated on M1 macrophages. Peritoneal macrophages were isolated and plated for 4 days in the presence of IFNg in complete RPMI media (Red). M1 macrophages were collected using cell dissociation buffer and stain with anti-αD polyclonal antibody, followed by staining with donkey-anti-rabbit Alexa 647secondary antibody. Freshly isolated peritoneal macrophages were used as a control (M0)(Blue). Integrin αD expression was tested using flow cytometry (Fortessa X-20). N/c-negative control. (B) Adhesion of WT and M1 macrophages to CEP-BSA. Isolated M1 macrophages were fluorescently labeled with Calcein AM and added to the well coated with CEP-BSA. cell adhesion was determined after 30 min in a fluorescence plate reader. (C) Macrophages were allowed to adhere to glass coverslips coated with CEP-BSA for 1 hour at 37°C. After fixation with 2% paraformaldehyde and permeabilization with 0.1% Triton X-100, cells were incubated with CytoPainet Phalloidinin-iFluor 555 and DAPI. Fluorescent images were obtained with EVOS FL auto cell fluorescent imaging system using 20x objective with mosaic 9-fields setup. (D) The cell area was calculated using Imaris 8.0 software. Data are presented as mean ± SEM ***p < 0.001.
Therefore, our data demonstrate that αD-deficiency significantly reduced adhesion and spreading of M1 macrophages on CEP, which confirmed the critical role of αDβ2 in macrophage adhesion and potential retention in CEP-modified substrate.
The Binding Properties of CEP Exceed the Binding Properties of Natural αDβ2 Ligands
Macrophages are a complex system with a variety of adhesive receptors that have overlapping ligand binding properties. To verify the role of CEP as a strong αDβ2 ligand, we tested αDβ2-transfected HEK293 cells (Figure 6A). HEK293 cells do not express any receptors that interact with CEP such as integrin αMβ2, CD36, or TLRs (23, 34). Adhesion of αDβ2-transfected HEK293 cells was tested to CEP-BSA versus BSA and CEP-fibrinogen versus fibrinogen. As we expected αDβ2-cells do not adhere to BSA, but demonstrate the strong concentration-dependent adhesion to CEP-BSA. (Figure 6B, upper).
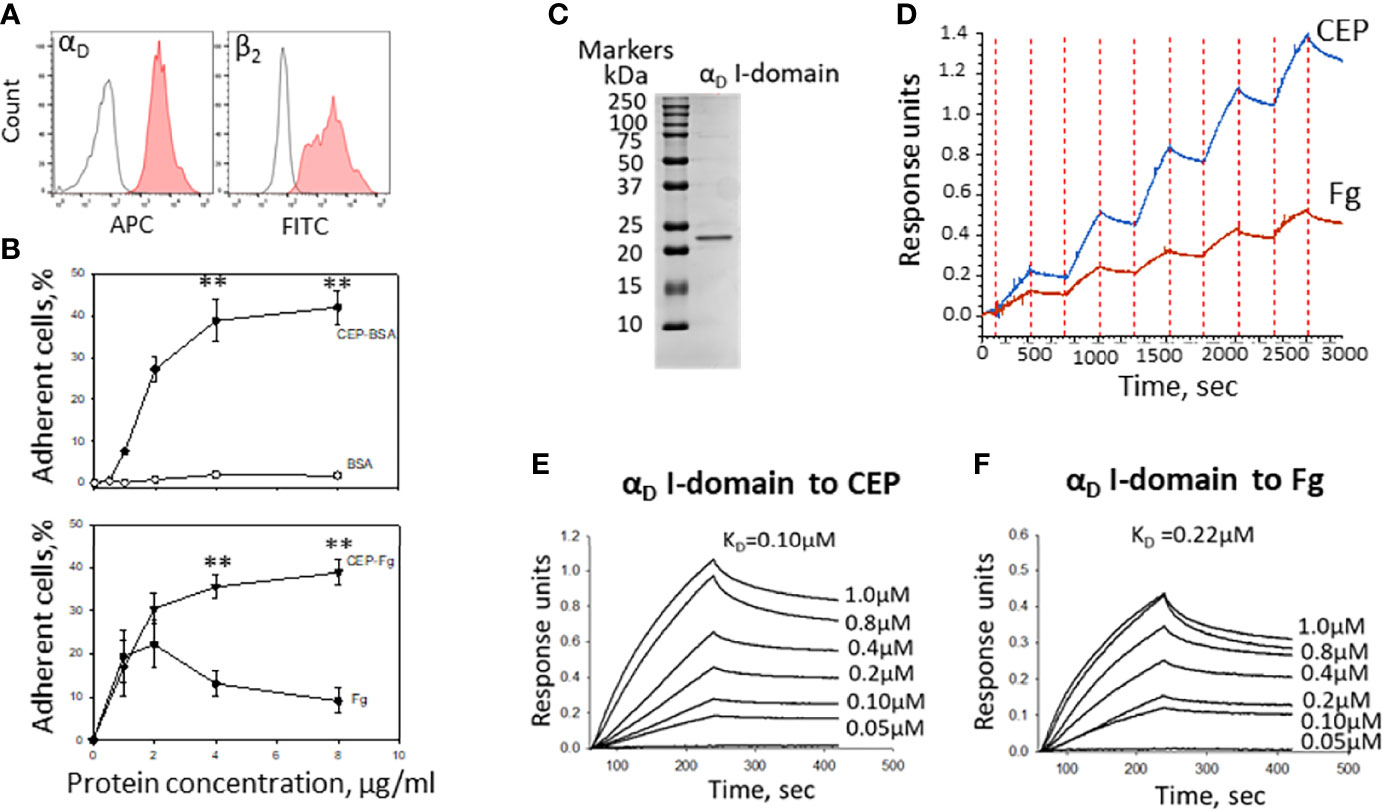
Figure 6 The binding properties of CEP exceed the binding properties of natural αDβ2 ligands. (A) The expression of integrins αD and β2 on the surface of HEK293-transfected cells. After isolation, cells were stained with anti-αD polyclonal antibody and anti-β2 monoclonal antibody, followed by staining with donkey-anti-rabbit Alexa 647 and donkey-anti-mouse Alexa 488 secondary antibodies. Control is shown in open histogram. (B) Different concentrations of CEP-BSA and BSA (Upper panel) or CEP-fibrinogen (CEP-Fg) and Fg (lower panel) were immobilized on a 96-well plate for 3 h at 37°C. Fluorescently labeled αDβ2 HEK293-transfected cells were added to the wells and cell adhesion was determined after 30 min in a fluorescence plate reader. A summary result from 4-6 experiments. Data are presented as mean ± SEM **p < 0.01. (C) Isolated αD I-domain binds to CEP in biolayer interferometry assays. SDS-PAGE of generated I-domain. αD active (D122-K314) I-domain was isolated from soluble fractions of (E) coli lysates and purified using affinity chromatography. The protein purity was assessed by SDS-PAGE on 4-15% gradient gel under non-reducing conditions followed by staining with Coomassie Blue. Representative profiles of the biolayer interferometry responses for αD binding to the immobilized CEP-BSA and Fibrinogen (Fg). (D) Single cycle kinetic of αD I-domain binding (concentrations ranging from 24-750 nM) to CEP-BSA and Fg coupled to AR2G biosensor. (F) Steady state analysis of αD I-domain binding to CEP (E) and Fg (F) (concentrations ranging from 50-1000 nM). The KD of binding was calculated using DataAnalysis HT11.0 software (ForteBio).
Fibrinogen is a natural ligand for integrin αDβ2 and a common element of the ECM during inflammation. Markedly, CEP modification significantly improves the adhesive properties of fibrinogen to αDβ2-HEK293 transfected cells similar to the result, which was obtained for M1 macrophages (Figure 3B). Notably, it has been shown previously that cell adhesion to fibrinogen achieved maximum at fibrinogen concentration 2-3 µg/ml and is significantly reduced at higher concentrations (10, 36). The mechanism is not known but may depend on the surface-induced lateral aggregation of fibrinogen (37). This result demonstrated that CEP modification in different proteins provides a similar augmentation of cell adhesive properties.
To evaluate a difference in the binding of αDβ2 to CEP and fibrinogen, we isolated αDβ2 ligand-binding motif – I-domain (Figure 6C) and examined its binding properties using BioLayer Interferometry. CEP and fibrinogen were immobilized on amino coupling biosensors and tested with different concentrations of αD I-domain using two approaches such as kinetic titration (single cycle kinetics) (Figure 6D) and equilibrium analysis (steady-state analysis) (Figures 6E, F). The binding affinity was analyzed using DataAnalysis HT11.0 software (ForteBio). We found that the affinity of αD binding to CEP (KD=1.0x10-7M) is 2-fold stronger than the αD interaction with fibrinogen (KD=2.2x10-7).
Therefore, αDβ2 generates a more powerful adhesion to CEP-modified proteins due to a higher affinity mediated by a negative charge of a carboxyl group in CEP’s structure. This result suggests the unique role of CEP-modified ECM proteins in macrophage adhesion that generates strong αDβ2-dependent adhesion via CEP that can be critical for macrophage retention.
Discussion
In our previous project, we found that CEP, the product of DHA oxidation, is a ligand for integrins αMβ2 and αDβ2 (23). In the current study, we tested how the modification of the ECM proteins with CEP affects macrophage adhesion. Now we found that 1) Polyunsaturated fatty acid oxidation generates the formation of adducts between CEP and ECM proteins in vitro. 2) The development of atherosclerosis promotes a CEP formation in proteins within the atherosclerotic lesions. 3) Pro-inflammatory M1 macrophages demonstrate a stronger adhesion to CEP-modified proteins compared to natural ECM ligands. 4) M1 macrophage adhesion to CEP inhibits cell migration. 5) The adhesion of M1 macrophages to CEP-modified proteins is driven by integrin αDβ2.
The previous experiments using immunostaining with the anti-CEP antibody demonstrated the deposition of CEP in tissue during inflammation including myocardial infarction (38), peritoneal inflammation (23), atherosclerosis (38), and macular degeneration (32, 39). Our published data revealed that neutrophil-mediated oxidation led to the formation of CEP-fibrinogen and CEP-BSA adducts (23). We hypothesize that PUFA oxidation can lead to the modification of different ECM proteins that contain an exposed lysine on a protein’s surface. We do expect some limitations, which can depend on special tertiary structures, post-translational modifications, or cross-linkage through lysines. Our concern was primarily based on the absence of CEP adduct formation with the E-fragment of fibrin (23), which has a strong triple-coiled helix structure. (The triple-coiled helix is also a major component of laminin and collagen fibrils). In addition to a triple-coiled structure, collagens IV, V, and VI contain flexible globular domains, which are absent in collagens I and III (40). In our current experiments, we found that in vitro oxidation of DHA generates CEP-collagen IV adduct formation, while collagen I is not modified by CEP, which confirms our hypothesis regarding the potential role of the globular domains in CEP adducts formation.
Previously we demonstrated that non-activated peritoneal macrophages can bind CEP via αMβ2 and αDβ2. However, we did not compare the level of macrophage adhesion to natural adhesive ligands and CEP-modified ligands. Moreover, we did not test the effect of CEP modification on the adhesion and migration of M1 macrophages, which is critical for the development of inflammatory response. In this paper, we are particularly focused on integrin αDβ2 since it is upregulated on pro-inflammatory M1-like macrophages and our previous data demonstrated that the αD I-domain showed 5 folds stronger affinity to CEP compared to the αM I domain (23, 41). This difference most likely is mediated by clusters of positively charged amino acids on the surface of the αD I-domain. The increased binding activity to negatively charged ligands was described previously for integrin αXβ2 which has a significantly lower expression on macrophages but has a high level of homology with integrin αDβ2 (42).
In our previous project, we found that αDβ2 on the surface of pro-inflammatory M1 macrophages contributed to the retention of macrophages at the site of atherosclerotic lesions due to strong cell adhesion (12). It has been shown that αDβ2 is a multiligand protein that can bind to ICAM-3, VCAM-1, fibrinogen, fibronectin, and vitronectin (11). However, these proteins are cell surface receptors or plasma proteins, which are sporadic in the interstitium, while the major ECM proteins such as collagens and laminins do not support αDβ2 -mediated adhesion (26). This raises the question regarding the potential ECM ligands for αDβ2 -mediated macrophage retention. The modification of collagen IV and laminin with CEP (Figure 2C) demonstrates the protein modification in ECM and resolves this issue. This mechanism presents the formation of the universal ligand for αDβ2-mediated adhesion. Notably, in our previous project, we found that a carboxyl group in CEP’s structure is critical for integrin binding since another product of DHA oxidation, ethylpyrole, has a similar structure, but does not have a carboxyl group and loses the ability to interact with integrins αMβ2 and αDβ2 (23). Therefore, the positively charged lysines in different proteins can be modified with negatively charged CEP, which converts these proteins to the αDβ2-dependent substrate.
We found that adhesion and spreading of M1 macrophages to the CEP-modified proteins is much stronger compared to native collagen IV, laminin, or fibrinogen (Figure 3). The adhesion to collagens and laminins is mediated via β1 integrins such as α1β1, α2β2, etc. The level of β1 integrins is lower on macrophages compared to β2 integrins (8, 9). Therefore, the switch of binding from classical collagen/β1-dependent to CEP-modified collagen/β2-dependent increased cell adhesion. Moreover, we demonstrated that M1 macrophage adhesion to CEP is integrin αDβ2-dependent since it is dramatically reduced in αD-deficient macrophages (Figure 5B). Notably, αD-deficiency does not affect the expression of other major adhesive receptors on the macrophage surface (26). We verified this result on several systems including αDβ2-transfected cells and isolated αD I-domain. αDβ2-transfected cells generated lower adhesion compared to M1 macrophages (Figures 5B, 6B) since the expression of αDβ2 on these cells is lower (Figures 5A, 6A). Nevertheless, these cells demonstrate a specific effect of integrin αDβ2 on cell adhesion. The results of the adhesion assay are confirmed by BioLayer Interferometry, which measures the direct protein-protein interaction between αD I-domain and CEP or fibrinogen.
I-domain is a region of αDβ2 that contains most of the ligand-binding sites for β2 integrins (43). We previously showed that binding sites for CEP and fibrinogen are located within the I-domain (23). In our earlier study regarding the ligand binding properties of integrin αDβ2, we compared αD I-domain binding to all previously identified ligands including fibrinogen, fibronectin, ICAM-3, VCAM-1, plasminogen, vitronectin, and CYR61 using the surface plasmon resonance (Biacore 3000) (11). These studies demonstrated a KD for αD I-domain/fibrinogen binding 7.6x10-7 M. In another project we tested αD I-domain binding to CEP by Biacore and calculated constant dissociation for this binding as KD 1.8x10-7 M (23). However, we did not compare αD I-domain binding to fibrinogen and CEP in the same experiment. Previously, the Biacore results for αD binding to fibrinogen and to CEP were obtained from different αD I-domain isolations, using different ligand densities on sensor chips and were performed several years apart (11, 23). Therefore, it was critical to analyze these interactions in one experiment to verify this result. Based on Biolayer Interfereometry results, which were performed on one αD I-domain isolation using the same I-domain concentrations, we found that the calculated affinity of αD I-domain/CEP interaction (1.0x10-7M) is 2 folds stronger compared to the αD I-domain/fibrinogen binding (2.2x10-7M). These results are slightly different from our previous Biocare results. We explain this difference by the difference in the experimental platforms between surface plasmon resonance (Biocore) and biolayer interferometry (Octet). Nevertheless, both methods provides a similar result and demonstrates the stronger binding of αD I-domain to CEP compared to fibrinogen.
All αD ligands analyzed by Biacore previously (11) showed a KD for αD I-domain binding in the range 0.2-8x10-6 M. The strongest binding was found for αD I-domain interaction with the rare ECM protein CYR61 (CCN1) with the KD 2.9x10-7 M. This affinity is still weaker than αD I-domain/CEP binding that we detected. Most importantly, the binding to “classical” αDβ2 ligands such as ICAM-3 (1.89x10-6 M), VCAM-1 (8.13x10-6 M), and fibrinogen (0.76x10-6 M) (11) are significantly less compared to CEP. These data together demonstrate that CEP modification generates preferential ligands for αDβ2-mediated macrophage adhesion during inflammation. Therefore, inflammation and oxidation generate the improved conditions for αDβ2-dependent macrophage adhesion.
The theory of mesenchymal cell migration postulates that optimal migration occurs with an intermediate level of receptor expression and substrate density, while the high density of receptors and corresponding substrates generate very strong adhesion, preventing cell motility (7, 26, 44–46).
Previously we found that non-activated macrophages demonstrated an improved migration in a CEP-supplemented matrix (23). Non-activated macrophages express an intermediate level of αDβ2 and αMβ2 integrins (26). We and others have shown experimentally that intermediate levels of integrin expression support cell migration (7, 26, 47, 48).
We propose that upregulation of αDβ2 on M1 macrophages and modification of ECM proteins with CEP promotes the conditions for the formation of an excessive number of receptor-ligand pairs with a high binding affinity. Accordingly, this high level of macrophage adhesion can prevent macrophage migration. In agreement with this concept, we found that migration of M1 macrophages in a 3D fibrin matrix supplemented with CEP is significantly reduced compared to migration in a pure fibrin matrix. Interestingly, we found that the morphology of M1 macrophages in a 3D matrix is affected by the presence of CEP (Figure 4D). Particularly, we revealed, that M1 macrophages in CEP supplemented matrix have a more rounded shape compared to macrophages in fibrin gel. It has been shown previously that migrated macrophages have an elongated shape and accordingly a greater length-to-diameter ratio (49, 50). The length-to-diameter ratio of macrophages in a CEP supplemented matrix is statistically significantly lower compared to control, which is in agreement with the reduced macrophage migration that we detected (Figure 4A).
To summarize, we propose a potential mechanism that affects macrophage adhesion and migration in inflamed tissue. Oxidation of DHA, which is a common structural component of cell membranes, leads to the formation of adducts between ECM proteins and CEP. Experimental data demonstrated that 20-30 modifications can occur in one protein molecule (23, 30, 32, 51). These modifications substitute a lysine-mediated positive charge for a CEP-mediated negative charge, which affects the conformation and ligand-binding properties of modified proteins. CEP is recognized by integrins αMβ2 and αDβ2. Accordingly, CEP modification can transform random proteins in integrin-related ligands. Freshly differentiated non-activated macrophages possess an intermediate level of αMβ2 and αDβ2 expression that supports CEP-mediated migration to the site of inflammation. Polarized pro-inflammatory (M1-like) macrophages express a high level of αDβ2, generating a high adhesion to CEP-modified proteins in the inflamed tissue that prevents M1 macrophage migration and leads to the development of chronic inflammation. The high affinity between αDβ2 and CEP additionally intensifies the potential effect. This model proposes that targeting the binding between αDβ2 and CEP can provide benefits against macrophage retention in the inflamed tissue and the development of chronic inflammation during metabolic and autoimmune diseases.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by East Tennessee State University IACUC.
Author Contributions
JC performed the experiments, analyzed the data, and edited the manuscript; KK, DG, CA, performed the experiments and analyzed the data; DW, EP, TB analyzed the data and edited the manuscript; VY designed the research, performed the experiments, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
These studies were supported by the American Heart Association 20AIREA35150018 (VY); and partially supported by the National Institutes of Health grants R15HL157836 (VY), R01GM119197 (DW), R01GM083016 (DW), R01HL071625 (TB), R01HL145536 (TB) and by the National Institute of Health grant C06RR0306551 for East Tennessee State University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CEP, carboxyethylpyrrole; ECM, Extracellular matrix; EDTA, Ethylenediaminetetraacetic acid; FACS, Fluorescence-activated cell sorting; IFNγ, interferon-γ; CCL2, C-C motif chemokine ligand 2 (MCP-1); CCL5, C-C motif chemokine ligand 5 (RANTES); TG, thioglycollate; WT, wide type; 2D, 2 dimensional; 3D, 3 dimensional.
References
1. Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J Immunol Res (2018) 2018:8917804. doi: 10.1155/2018/8917804
2. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and Macrophage Immunometabolism in Atherosclerosis. Semin Immunopathol (2018) 40(2):203–14. doi: 10.1007/s00281-017-0656-7
3. Ross EA, Devitt A, Johnson JR. Macrophages: The Good, the Bad, and the Gluttony. Front Immunol (2021) 12:708186. doi: 10.3389/fimmu.2021.708186
4. Koltsova EK, Hedrick CC, Ley K. Myeloid Cells in Atherosclerosis: A Delicate Balance of Anti-Inflammatory and Proinflammatory Mechanisms. Curr Opin Lipidol (2013) 24(5):371–80. doi: 10.1097/MOL.0b013e328363d298
5. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
6. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
7. Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-Ligand Binding Properties Govern Cell Migration Speed Through Cell-Substratum Adhesiveness. Nature (1997) 385:537–40. doi: 10.1038/385537a0
8. Ammon C, Meyer SP, Schwarzfischer L, Krause SW, Andreesen R, Kreutz M. Comparative Analysis of Integrin Expression on Monocyte-Derived Macrophages and Monocyte-Derived Dendritic Cells. Immunology (2000) 100(3):364–9. doi: 10.1046/j.1365-2567.2000.00056.x
9. Ou Z, Dolmatova E, Lassegue B, Griendling KK. β1- and β2-Integrins: Central Players in Regulating Vascular Permeability and Leukocyte Recruitment During Acute Inflammation. Am J Physiol Heart Circ Physiol (2021) 320(2):H734–H9. doi: 10.1152/ajpheart.00518.2020
10. Yakubenko VP, Lishko VK, Lam SC-T, Ugarova TP. A Molecular Basis for Integrin αMβ2 in Ligand Binding Promiscuity. J Biol Chem (2002) 277:48635–42. doi: 10.1074/jbc.M208877200
11. Yakubenko VP, Yadav SP, Ugarova TP. Integrin αDβ2, an Adhesion Receptor Up-Regulated on Macrophage Foam Cells, Exhibits Multiligand-Binding Properties. Blood (2006) 107:1643–50. doi: 10.1182/blood-2005-06-2509
12. Aziz MH, Cui K, Das M, Brown KE, Ardell CL, Febbraio M, et al. The Upregulation of Integrin αDβ2 (CD11d/CD18) on Inflammatory Macrophages Promotes Macrophage Retention in Vascular Lesions and Development of Atherosclerosis. J Immunol (2017) 198(12):4855–67. doi: 10.4049/jimmunol.1602175
13. Danilenko DM, Rossito PV, van der Vieren M, Le Tring H, Mc Donagh SP, Affolter VK, et al. A Novel Canine Leukointegrin, Alpha D Beta 2, is Expressed by Specific Macrophage Subpopulations in Tissue and a Minot CD8+ Lymphocyte Subpopulation in Peripheral Blood. J Immunol (1995) 155:35–44.
14. Van der Vieren M, Le Trong H, St.John T, Staunton DE, Gallatin WM. A Novel Leukointegrin, αDβ2, Binds Preferentially to ICAM-3. Immunity (1995) 3:683–90. doi: 10.1016/1074-7613(95)90058-6
15. Bao F, Brown A, Dekaban GA, Omana V, Weaver LC. CD11d Integrin Blockade Reduces the Systemic Inflammatory Response Syndrome After Spinal Cord Injury. Exp Neurol (2011) 231(2):272–83. doi: 10.1016/j.expneurol.2011.07.001
16. Thomas AP, Dunn TN, Oort PJ, Grino M, Adams SH. Inflammatory Phenotyping Identifies CD11d as a Gene Markedly Induced in White Adipose Tissue in Obese Rodents and Women. J Nutr (2011) 141(6):1172–80. doi: 10.3945/jn.110.127068
17. Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, et al. CD11d Integrin Blockade Reduces the Systemic Inflammatory Response Syndrome After Traumatic Brain Injury in Rats. Exp Neurol (2015) 271:409–22. doi: 10.1016/j.expneurol.2015.07.003
18. Miyazaki Y, Bunting M, Stafforini DM, Harris ES, McIntyre TM, Prescott SM, et al. Integrin αDβ2 Is Dynamically Expressed by Inflamed Macrophages and Alters the Natural History of Lethal Systemic Infections. J Immunol (2008) 180(1):590–600. doi: 10.4049/jimmunol.180.1.590
19. de Azevedo-Quintanilha IG, Vieira-de-Abreu A, Ferreira AC, Nascimento DO, Siqueira AM, Campbell RA, et al. Integrin αDβ2 (CD11d/CD18) Mediates Experimental Malaria-Associated Acute Respiratory Distress Syndrome (MA-ARDS). Malar J (2016) 15(1):393. doi: 10.1186/s12936-016-1447-7
20. Nascimento DO, Vieira-de-Abreu A, Arcanjo AF, Bozza PT, Zimmerman GA, Castro-Faria-Neto HC. Integrin αDβ2 (CD11d/CD18) Modulates Leukocyte Accumulation, Pathogen Clearance, and Pyroptosis in Experimental Salmonella Typhimurium Infection. Front Immunol (2018) 9:1128. doi: 10.3389/fimmu.2018.01128
21. Bailey WP, Cui K, Ardell CL, Keever KR, Singh S, Rodriguez-Gil DJ, et al. The Expression of Integrin αDβ2 (CD11d/CD18) on Neutrophils Orchestrates the Defense Mechanism Against Endotoxemia and Sepsis. J Leukoc Biol (2021) 109(5):877–90. doi: 10.1002/JLB.3HI0820-529RR.
22. Yakubenko VP, Byzova TV. Biological and Pathophysiological Roles of End-Products of DHA Oxidation. Biochim Biophys Acta (2017) 1862(4):407–15. doi: 10.1016/j.bbalip.2016.09.022
23. Yakubenko VP, Cui K, Ardell CL, Brown KE, West XZ, Gao D, et al. Oxidative Modifications of Extracellular Matrix Promote the Second Wave of Inflammation via β2 Integrins. Blood (2018) 132(1):78–88. doi: 10.1182/blood-2017-10-810176
24. Blythe EN, Weaver LC, Brown A, Dekaban GA. Beta2 Integrin CD11d/CD18: From Expression to an Emerging Role in Staged Leukocyte Migration. Front Immunol (2021) 12:775447. doi: 10.3389/fimmu.2021.775447
25. Shi C, Simon DI. Integrin Signals, Transcription Factors, and Monocyte Differentiation. Trends Cardiovasc Med (2006) 16(5):146–52. doi: 10.1016/j.tcm.2006.03.002
26. Cui K, Ardell CL, Podolnikova NP, Yakubenko VP. Distinct Migratory Properties of M1, M2, and Resident Macrophages Are Regulated by αDβ2 and αMβ2 Integrin-Mediated Adhesion. Front Immunol (2018) 9:2650. doi: 10.3389/fimmu.2018.02650
27. Szpak D, Izem L, Verbovetskiy D, Soloviev DA, Yakubenko VP, Pluskota E. Alphambeta2 Is Antiatherogenic in Female But Not Male Mice. J Immunol (2018) 200(7):2426–38. doi: 10.4049/jimmunol.1700313
28. Wolf D, Bukosza N, Engel D, Poggi M, Jehle F, Anto MN, et al. Inflammation, But Not Recruitment, of Adipose Tissue Macrophages Requires Signalling Through Mac-1 (CD11b/CD18) in Diet-Induced Obesity (DIO). Thromb Haemost (2017) 117(2):325–38. doi: 10.1160/TH16-07-0553
29. Miyazaki Y, Vieira-de-Abreu A, Harris ES, Shah AM, Weyrich AS, Castro-Faria-Neto HC, et al. Integrin αDβ2 (CD11d/CD18) Is Expressed by Human Circulating and Tissue Myeloid Leukocytes and Mediates Inflammatory Signaling. PloS One (2014) 9(11):e112770. doi: 10.1371/journal.pone.0112770
30. Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, et al. Carboxyethylpyrrole Protein Adducts and Autoantibodies, Biomarkers for Age-Related Macular Degeneration. J Biol Chem (2003) 278(43):42027–35. doi: 10.1074/jbc.M305460200
31. West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, et al. Oxidative Stress Induces Angiogenesis by Activating TLR2 With Novel Endogenous Ligands. Nature (2010) 467(7318):972–6. doi: 10.1038/nature09421
32. Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, et al. Carboxyethylpyrrole Oxidative Protein Modifications Stimulate Neovascularization: Implications for Age-Related Macular Degeneration. Proc Natl Acad Sci USA (2006) 103(36):13480–4. doi: 10.1073/pnas.0601552103
33. Hoff HF, O'Neil J, Wu Z, Hoppe G, Salomon RL. Phospholipid Hydroxyalkenals: Biological and Chemical Properties of Specific Oxidized Lipids Present in Atherosclerotic Lesions. Arterioscler Thromb Vasc Biol (2003) 23(2):275–82. doi: 10.1161/01.ATV.0000051407.42536.73
34. Kim YW, Yakubenko VP, West XZ, Gugiu GB, Renganathan K, Biswas S, et al. Receptor-Mediated Mechanism Controlling Tissue Levels of Bioactive Lipid Oxidation Products. Circ Res (2015) 117(4):321–32. doi: 10.1161/CIRCRESAHA.117.305925
35. Wolf D, Anto-Michel N, Blankenbach H, Wiedemann A, Buscher K, Hohmann JD, et al. A Ligand-Specific Blockade of the Integrin Mac-1 Selectively Targets Pathologic Inflammation While Maintaining Protective Host-Defense. Nat Commun (2018) 9(1):525. doi: 10.1038/s41467-018-02896-8
36. Lishko VK, Burke T, Ugarova T. Antiadhesive Effect of Fibrinogen: A Safeguard for Thrombus Stability. Blood (2007) 109(4):1541–9. doi: 10.1182/blood-2006-05-022764
37. Yermolenko IS, Gorkun OV, Fuhrmann A, Podolnikova NP, Lishko VK, Oshkadyerov SP, et al. The Assembly of Nonadhesive Fibrinogen Matrices Depends on the alphaC Regions of the Fibrinogen Molecule. J Biol Chem (2012) 287(50):41979–90. doi: 10.1074/jbc.M112.410696
38. Kerr BA, Ma L, West XZ, Ding L, Malinin NL, Weber ME, et al. Interference With Akt Signaling Protects Against Myocardial Infarction and Death by Limiting the Consequences of Oxidative Stress. Sci Signal (2013) 6(287):ra67. doi: 10.1126/scisignal.2003948
39. Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, et al. Oxidative Damage-Induced Inflammation Initiates Age-Related Macular Degeneration. Nat Med (2008) 14(2):194–8. doi: 10.1038/nm1709
40. Shoulders MD, Raines RT. Collagen Structure and Stability. Annu Rev Biochem (2009) 78:929–58. doi: 10.1146/annurev.biochem.77.032207.120833
41. Cui K, Podolnikova NP, Bailey W, Szmuc E, Podrez EA, Byzova TV, et al. Inhibition of Integrin αDβ2-Mediated Macrophage Adhesion to End-Product of DHA Oxidation Prevents Macrophage Accumulation During Inflammation. J Biol Chem (2019) 294(39):14370–82. doi: 10.1074/jbc.RA119.009590
42. Vorup-Jensen T, Carman CV, Shimaoka M, Schuck P, Svitel J, Springer TA. Exposure of Acidic Residues as a Danger Signal for Recognition of Fibrinogen and Other Macromolecules by Integrin αXβ2. Proc Natl Acad Sci USA (2005) 102(5):1614–9. doi: 10.1073/pnas.0409057102
43. Luo BH, Carman CV, Springer TA. Structural Basis of Integrin Regulation and Signaling. Annu Rev Immunol (2007) 25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618
44. Wiesner C, Le-Cabec V, El AK, Maridonneau-Parini I, Linder S. Podosomes in Space: Macrophage Migration and Matrix Degradation in 2D and 3D Settings. Cell Adh Migr (2014) 8(3):179–91. doi: 10.4161/cam.28116
45. DiMilla PA, Barbee K, Lauffenburger DA. Mathematical Model for the Effects of Adhesion and Mechanics on Cell Migration Speed. Biophys J (1991) 60:15–37. doi: 10.1016/S0006-3495(91)82027-6
46. DiMilla PA, Stone JA, Quinn JA, Albelda SM, Lauffenburger DA. Maximal Migration of Human Smooth Muscle Cells on Fibronectin and Type IV Collagen Occurs at an Intermediate Attachment Strength. J Cell Biol (1993) 122:729–37. doi: 10.1083/jcb.122.3.729
47. Lishko VK, Yakubenko VP, Ugarova TP. The Interplay Between Integrins αMβ2 and α5β1 During Cell Migration to Fibronectin. Exp Cell Res (2003) 283:116–26. doi: 10.1016/S0014-4827(02)00024-1
48. Yakubenko VP, Belevych N, Mishchuk D, Schurin A, Lam SC, Ugarova TP. The Role of Integrin Alpha D Beta2 (CD11d/CD18) in Monocyte/Macrophage Migration. Exp Cell Res (2008) 314:2569–78. doi: 10.1016/j.yexcr.2008.05.016
49. Cougoule C, Van GE, Le C, Lafouresse F, Dupre L, Mehraj V, et al. Blood Leukocytes and Macrophages of Various Phenotypes Have Distinct Abilities to Form Podosomes and to Migrate in 3D Environments. Eur J Cell Biol (2012) 91(11-12):938–49. doi: 10.1016/j.ejcb.2012.07.002
50. McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of Macrophage Phenotype by Cell Shape. Proc Natl Acad Sci USA (2013) 110(43):17253–8. doi: 10.1073/pnas.1308887110
Keywords: macrophage, adhesion, migration, integrin αDβ2, CD11d/CD18, carboxyethylpyrrole (CEP), ECM, inflammation
Citation: Casteel JL, Keever KR, Ardell CL, Williams DL, Gao D, Podrez EA, Byzova TV and Yakubenko VP (2022) Modification of Extracellular Matrix by the Product of DHA Oxidation Switches Macrophage Adhesion Patterns and Promotes Retention of Macrophages During Chronic Inflammation. Front. Immunol. 13:867082. doi: 10.3389/fimmu.2022.867082
Received: 31 January 2022; Accepted: 27 April 2022;
Published: 26 May 2022.
Edited by:
Zhichao Fan, UCONN Health, United StatesReviewed by:
Rongrong Liu, Northwestern University, United StatesDennis Wolf, University of Freiburg, Germany
Craig T. Lefort, Rhode Island Hospital, United States
Copyright © 2022 Casteel, Keever, Ardell, Williams, Gao, Podrez, Byzova and Yakubenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentin P. Yakubenko, eWFrdWJlbmtvQGV0c3UuZWR1, orcid.org/0000-0002-0242-3061