 Claudia Minici
Claudia Minici Sabrina Testoni
Sabrina Testoni Emanuel Della-Torre
Emanuel Della-Torre- 1Università Vita-Salute San Raffaele, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Pancreato-Biliary Endoscopy and Endosonography Division, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Division of Pancreatic Surgery, Pancreas Translational and Clinical Research Center, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 4Unit of Immunology, Rheumatology, Allergy and Rare Diseases (UnIRAR), IRCCS San Raffaele Scientific Institute, Milan, Italy
Pancreatic adenocarcinoma is highly infiltrated by B lymphocytes but the relevance of these immune cells in tumor development has been surprisingly overlooked until recently. Based on available evidence from other solid tumors, interaction between B lymphocytes and neoplastic cells is probably not uniformly stimulatory or inhibitory. Although presentation of tumor antigens to T cells and production of antitumor immunoglobulins might intuitively suggest a prominent tumor suppressive activity, specific subsets of B lymphocytes can secrete growth factors for neoplastic cells and immunosuppressive cytokines thus promoting escape from immunosurveillance and cancer progression. Because many of these mechanisms might also be implicated in the development of PDAC, and immune-modulation of B-cell activity is nowadays possible at different levels, determining the role of B-lymphocytes in this lethal cancer becomes of utmost importance to design novel therapeutic strategies. This review aims to discuss the emerging role of B cells in PDAC tumorigenesis, progression, and associated stromal reaction.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignancy with a 5-year survival rate of 5-7% and represents the third leading cause of cancer related mortality (1, 2). Pancreatic ductal adenocarcinoma mainly arises from two types of precursor lesions, pancreatic intraepithelial neoplasia (PanIN) and intraductal papillary mucinous neoplasms (IPMNs) that eventually transform into invasive carcinoma after progression from low to high-grade dysplasia (3, 4). Aside from palliative care or surgery in eligible patients, current treatment options are based on DNA-damaging agents but still offer a life expectancy of less than 40% at three years (5). Failure of available chemotherapy regimens has been traditionally attributed to mechanisms of chemoresistance intrinsic to neoplastic cells and to the characteristic stromal reaction that hampers effective drug delivery into the tumor (6–9).
The immune system plays an equally important role in modulating PDAC progression with opposing influence of tumor promoting and tumor suppressing immune cells (6–9). Specifically, while CD8+ T cells, Th1-type CD4+ T cells, and natural killer (NK) cells bear antitumor activity, regulatory T (Treg) cells, Th2-type CD4+ T cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) have been shown to create a highly immunosuppressive microenvironment that decreases PDAC immunogenicity (7–16). Indeed, cytotoxic T and NK cells infiltrate premalignant IPMN lesions from early stages, but they progressively decrease in number and antitumor activity in parallel with the accumulation of immunosuppressive elements (17).
Pancreatic adenocarcinoma is also highly infiltrated by B lymphocytes but, in contrast to other malignancies, the relevance of these immune cells in tumor development has been surprisingly overlooked until recently. Evidence of B-cells involvement in the progression of solid tumors, in fact, has been appreciated as early as the late 1970s and subsequently explored in a number of non-pancreatic cancers (18–23). In breast, ovarian, and gastric adenocarcinoma, for instance, a prominent B lymphocytic infiltrate correlates with better outcomes (24, 25). Similarly, in non-small cell lung cancer, the presence of tertiary lymphoid structures (TLS) enriched in B cells predicts longer patient survival (26). Yet, B-cell deficiency or depletion reduced disease burden in mouse models of sarcoma, colorectal cancer, melanoma, and squamous carcinomas, suggesting equally relevant tumor promoting properties of the B-cell compartment (27–33). Together, these data support a complex and nuanced interaction between B lymphocytes and neoplastic cells that is probably not uniformly stimulatory or inhibitory across a wide range of cancers. Although presentation of tumor antigens to T cells and production of antitumor immunoglobulins might intuitively suggest a prominent tumor suppressive activity, specific subsets of B lymphocytes can secrete growth factors for neoplastic cells and immunosuppressive cytokines thus promoting escape from immunosurveillance and cancer progression (34–40). The ultimate tumor promoting or tumor suppressing effect of B cells might also be influenced by a number of variables related to the experimental setting, including the B-cell depletion strategy adopted, the mouse model used, and whether the study is focused on peripheral or on tumor-infiltrating B lymphocytes (TIL-B).
Because many of these mechanisms might also be implicated in the development of PDAC, and immune-modulation of B-cell activity is nowadays possible at different levels with a portfolio of targeted treatments, determining the role of B-lymphocytes in this lethal cancer becomes of utmost importance to design novel therapeutic strategies. In the present review we discuss the emerging role of B cells in PDAC tumorigenesis, progression, and stromal reaction. In addition we examine the mechanisms by which B cells might promote as well as inhibit antitumor immunity. To place our work in context, an overview of the available mouse models for studying B cells in PDAC will also be provided as well as hints from targeted B-cell approaches under evaluation in clinical trials.
Mouse Models for Studying B-Lymphocytes in Pancreatic Adenocarcinoma
There are several different genetically modified mouse tumors and xenograft preclinical model systems available to study pancreatic cancer from its pre-neoplastic stage to its metastatic form (41–48). These models use a variety of approaches to target the expression of mutant or endogenous specific genes and, as a result, they develop a broad spectrum of pathologic changes.
In general, as mutations in Kras have been reported in 90% of human PDAC and in more than 95% of PanIN lesions (49–51), most mouse models are based on the alteration of this oncogene. Kras mutations alone are, however, not sufficient to induce PDAC progression to the invasive stage, and different tumor suppressor genes are typically deleted or inactivated to generate metastatic disease including Ink4, p53, Smad4 or TGFβ (52–56). Of note, all these different types of murine models are immunologically intact thus allowing concomitant characterization of the immune response during PDAC progression. Yet, data regarding the composition and functional status of the B-cell infiltrate are available only for KPC, KC mice, and orthotopic models.
The KPC mouse (KrasG12D/+;Trp53R172H/+;Pdx-1-Cre) represents the gold-standard of PDAC preclinical models and is generated by placing a Cre recombinase G12D Kras mutated gene under the control of the pancreas-specific promoter Pdx1 together with silencing of the tumor suppressor p53 gene (57). KPC mice have a median survival of 5 months and faithfully recapitulates human disease in terms of spontaneous progression through PanIN, desmoplastic stromal reaction, and metastatic spread (57). In addition, because Kras expression directly induces Treg cells differentiation, the KPC mouse also offers the opportunity to study the progressive impairment of anti-tumor immune responses that occur in the human counterpart (58, 59). The pancreas of the KPC mouse is strongly infiltrated by B lymphocytes and the proportion of B-cells observed in KPC tumors is comparable to that reported in PDAC patients (60). Of note, B lymphocytes are found not only enmeshed within stromal elements but also clustered in germinal centers-like aggregates, suggesting that the KPC model elicits antigen dependent immune-reactions at disease site (60). Indeed, in analogy to human PDAC, most B-cells infiltrating KPC tumor are memory B-cells, show an activated phenotype, and produce a significant amount of immunoglobulins (60). Lower numbers of regulatory B-cells (Breg) and plasma cells are also observed. Given the analogies with human disease and the capacity to elicit an organized local inflammatory response, the KPC mouse currently represents the best available model to physiologically address the relationship between the B-cell compartment and the various cellular and stromal components of PDAC.
Another related model, the KC mouse (KrasG12D/+;Pdx-1-Cre), expresses only mutant Kras and consequently develops PDAC at a significantly slower rate (61). Mice are histologically normal up to 5 months and do not develop PanIN lesions until after 8 weeks (52). In KC mouse, B cells only modestly infiltrate pancreatic tissue, distribute in proximity to PanIN, and prominently exhibit a B1 and B2 mature phenotype on flow-cytometry. KrasG12D pancreata show diminished frequencies of B2 cells and increased frequencies of B1 cells compared to wild type mice, and their number decreases during carcinogenesis (60, 62). In addition, B1 cells show no to low memory responses, express IgM in greater quantities than IgG, and their B-cell receptors are polyspecific, suggesting a lower immunogenicity of KC lesions compared to KPC mice (60, 63). Because of the latency of tumor formation and the paucity of B-lymphocytic infiltrate, the KC mouse is considered suitable to characterize B-cell contribution to PDAC development rather than PDAC progression. The simultaneous Kras activation and p48/Hif1a deletion (p48-Cre;LSL-KrasG12D;Hif1afl/fl) in the pancreatic epithelium, for instance, has been recently exploited to address B-cell implication in pre-neoplastic PanIN lesions (64). In this model both the percentage and absolute number of B cells were dramatically increased compared with KC and wild type mouse, and, as opposed to the KC model, they did not decrease during carcinogenesis (64). In addition, compared to normal mice, the proportion of CD19hiCD1dhiCD5+ Breg cells was higher both in KrasG12D and KrasG12D;Hif1aKO pancreata, with no significant differences between the two models (64).
A third model to study B-cells in PDAC is the orthotopic model, which is generated by the injection of syngeneic tumor cells derived from primary pancreatic carcinomas of transgenic mice into the pancreas of wild type mice, typically C57BL/6 mice. Orthotopic tumors form rapidly in approximately one month, contain more tumor cells than stromal elements and exhibit significant infiltration by CD45+ leukocytes compared with wild-type mice (52). Yet, due to accelerated disease progression, B-cell infiltration occurs to a far lesser extent and with delayed kinetics than in KPC tumors both in density and proportion, and germinal center (GC)-like structures are typically not observed (60). This evidence suggests that the immunological stimulus provided by tumor cell lines in the orthotopic PDAC model is also less proficient in inducing antibody-secreting plasma cells than KPC mice. Indeed, the activation profile of infiltrating B-cells is much reduced in this model compared to the KPC mouse, and the deposition of immunoglobulins is virtually absent (60). Accordingly, B-cells predominantly bear an IgM transitional and B1 phenotype, with IgMlow memory B cells, Breg cells, plasmablasts, and plasma cells observed to a lesser extent, not significantly different from healthy pancreas (60). Still, although the faster tumor kinetics compared to KPC models probably prevent full B-cell activation, orthotopic tumors seem equally capable of shaping the phenotype of infiltrating B cells as pancreatic B lymphocytes possess a remarkably different activation profile than splenic B cells from the same tumor-bearing mice, including the expression of proinflammatory cytokines and chemokines involved in T-cell recruitment (60).
The relevance of B lymphocytes in orthotopic PDAC models has been recently assessed also by injecting syngeneic pancreatic cancer cell lines into B-cell deficient mice, either μMT or JHT mice (65, 66). The μMT mouse is homozygous for the Ighmtm1Cgn targeted mutation and has no expression of membrane-bound IgM. Similarly, the JHT mouse is homozygous for the Igh-Jtm1Cgn targeted mutation that causes a deletion in the J segment of the Ig heavy-chain locus and prevents the expression of IgM or IgG. The results of both mutations are mouse models that have no mature functional B cells in the bone marrow or periphery due to blocked B-cell differentiation at the precursor stage. These models have both advantages and shortcomings. On the one hand, in fact, orthotopic PDAC models in B-cell deficient mice allow selective evaluation of the contribution of specific B-cell subsets by injecting them intravenously or directly into the recipient pancreas. On the other hand, however, B-cell deficient mice bear intrinsic derangement not only of humoral immune responses but also of cellular immunity, making it possible that intrinsic characteristics of B-cell deficient mice may contribute to orthotopic tumor development, rather than the loss of B cells per se or the effect of a single B-cell subpopulation of interest (60). Finally, the orthotopic model could be also exploited to directly assess the contribution of human B lymphocytes to tumour growth. This model would theoretically imply xenografts of human PDAC cell lines in immunodeficient mice (typically, NOD-scid IL-2 receptor gammanull mice) but we are not aware of studies using this model until the time of writing, and caveats related to the global impairment of the murine immune system remains to be considered. Still, although labor intensive, technically challenging, and expensive, orthotopic xenograft of PDAC represents a valid approach to recapitulate human disease in vivo and to increase the translational relevance of its findings.
Spatial Distribution of B-Lymphocytes in Pancreatic Adenocarcinoma
Spatial compartmentalization of immune cells within the tumor tissue (whether peritumoral, intratumoral, diffuse, or organized in tertiary lymphoid structures) is considered an important determinant of cancer progression (67). In particular, tertiary lymphoid structures are considered crucial for the recruitment of tumor-infiltrating T lymphocytes (TILs), for B-T cells interaction in GCs, and for the activation of tumoricidal cytotoxic responses associated with better clinical outcomes (68–72). Germinal centers are essential also for the development of effective humoral responses whereby naïve B-cells are activated after antigen encountering, undergo affinity maturation under the guidance of follicular helper T (Tfh) cells, and differentiate in antibody producing plasmablasts/plasma cells (73, 74).
In the context of PDAC, B lymphocytic infiltrate have been preferentially observed either scattered within the stromal microenvironment (TILs) or organized in ectopic lymph node-like TLS (75–78) (Figure 1). According to a retrospective analysis of 104 histological PDAC specimens, this bimodal spatial distribution was independently associated with patient survival, suggesting that compartmentalization of B-lymphocytes in the pancreatic tissue reflects opposite immunological commitments (75). Specifically, while TILs interspersed within the tumor stroma were associated with worse prognosis, the presence of intratumoral TLS correlated with longer overall and progression-free survival (75). Accordingly, PDAC specimens rich in TLS showed upregulation of genes related to GC reaction, B-cell activation, and proliferation including activation-induced cytidine deaminase (AICDA), Ki67, and CD27 (75). Conversely, PDAC specimens with prominent TILs infiltrate exhibited increased expression of immunosuppressive genes, including programmed cell death-1 ligand (PDL1) and transforming-growth factor-b (TGF-b) (75). PDAC specimens rich in TLS also showed increased expression of genes related to T-cell activation and proliferation (namely, CD8, IL-2, IL-7, and IL-12), and pancreatic infiltration of cytotoxic CD8+ T cells was observed in orthotopic PDAC models after B-cell depletion (75). Notably, GC reactions occurring in TLS+ PDAC specimens are enriched in class-switched memory B cells, effector memory CD4+ T-cells, and NK cells suggesting that durable immunological memory is associated with a longer survival (78). Similarly, GC reactions in TLS+ tumors express significantly more MHC class I-restricted neoantigens and somatic hypermutations in B-cells, indicating that a more specific humoral immune response also represents an important variable for patient outcome (78).
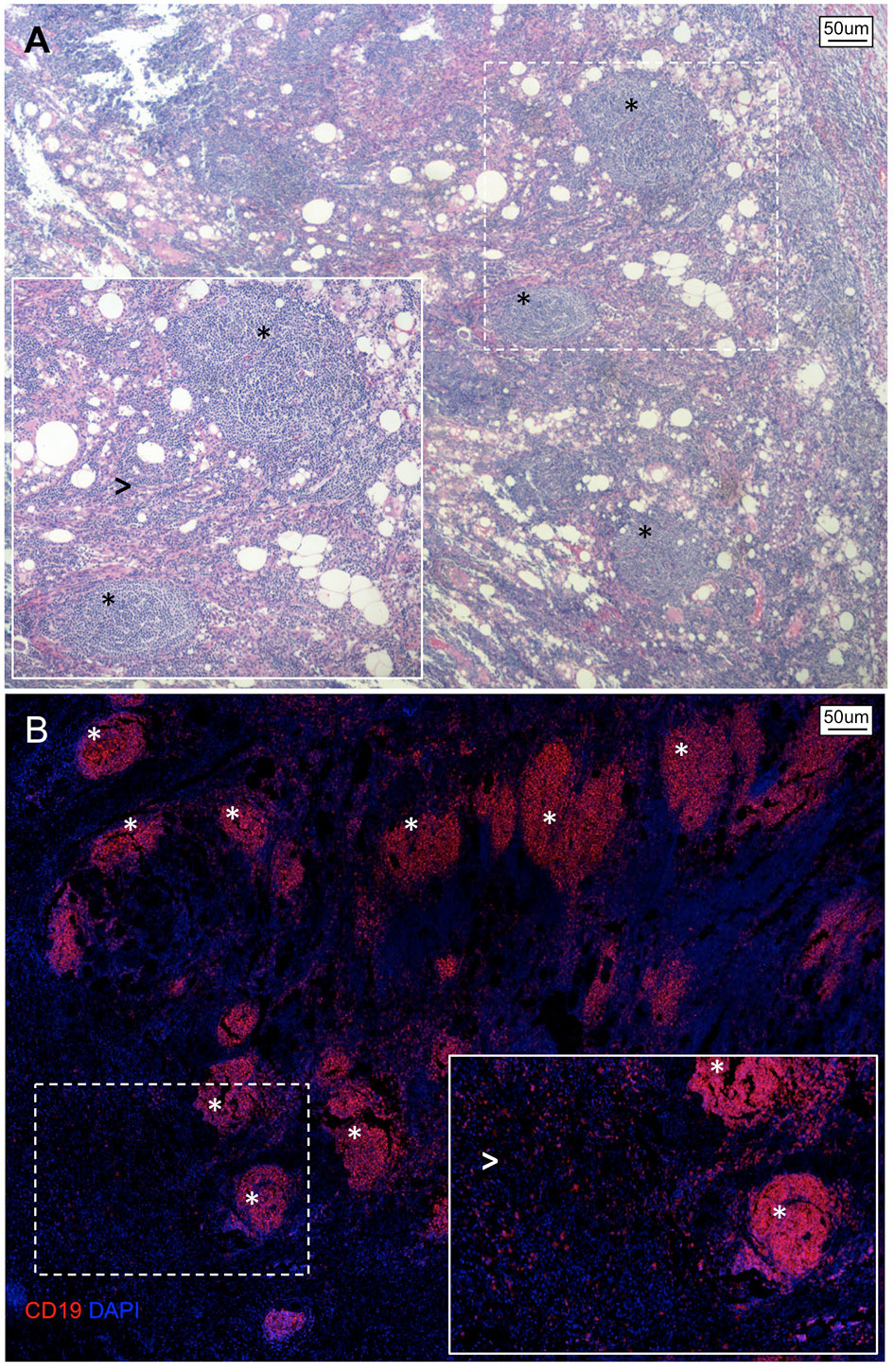
Figure 1 Spatial organization of B lymphocytes in pancreatic adenocarcinoma. (A) Hematoxylin/Eosin stain and (B) immunofluorescence staining of a representative tissue section of PDAC showing an abundant infiltrate of B lymphocytes organized as either multiple lymphoid structures (asterisks) or spread within the neoplastic lesion and tumor stroma (arrowheads).
All together, these results confirm that B-cells bear prognostic relevance in human PDAC and suggest that their ability to influence tumor progression depends on whether they are organized in functionally proficient tertiary lymphoid structures with mature GC reactions (tumor-suppressing) or scattered at the tumor-stroma interface (tumor promoting). Of note, while distribution of B-lymphocytes in PDAC is likely orchestrated by chemotactic factors secreted by neoplastic and/or stromal cells, the dynamics of B-cells compartmentalization during tumor development remain to be fully characterized, and possibly implies antagonizing functions of coexisting TILs and TLS as well as a progressive loss of TLS along with increasing PDAC aggressiveness.
B-Lymphocytes and the Tumorigenesis of Pancreatic Adenocarcinoma
Studies focused on T-cell responses in PDAC have shown that the progression of pre-malignant IPMN lesions to invasive cancer are associated with a consistent modification of the immune landscape whereby cytotoxic tumor-suppressing CD8+ T-cells, Th1 cells, and NK cells are progressively supplanted by immunosuppressive and tumor-promoting Th2 cells and Treg cells during the invasive stage of pancreatic cancer (17, 79).
The role of B-lymphocytes in early stages of PDAC development has been evaluated in few orthotopic mouse models of pancreatic ductal epithelial cells expressing oncogenic Kras (KrasG12D-PDEC) KPC derived cell lines injected into the pancreas of μMT or JHT mice and in organoids (65, 66, 80, 81). Injection of KrasG12D-PDEC or KPC derived cell lines into wild-type pancreas, in fact, typically leads to the accumulation of B-lymphocytes in close proximity to evolving tumor lesions, probably as a consequence of the secretion of the B-cell chemotactic molecule CXCL13 by stromal cells in the context of the growing pancreatic neoplasia (66). Conversely, orthotopic implantation of either KrasG12D-PDEC or KPC derived cell lines in B-cell deficient mice was associated with a significant reduction of tumor lesions compared to wild type mice, and adoptive transfer of B-cells from littermate controls into μMT animals prior to orthotopic injection of neoplastic cells rescued tumor growth together with de novo infiltration of transferred B-cells (66, 80). These findings suggest that the epithelium “in transformation” instructs the recruitment of B-cells and support an essential role of B-lymphocytes in establishing a pro-tumorigenic microenvironment. Accordingly, prevention of early B-cells recruitment in KrasG12D-PDEC orthotopically implanted mice with anti-CXCL13 treatment also resulted in reduced growth of the orthotopic lesions reinforcing possible tumor promoting properties of B-lymphocytes in the initial stages of PDAC development (66). In another study of primary human pancreatic tumors and KC mouse model crossed to Hif1αfl/fl the hypoxia inducible factor-1α (HIF1α) was found to be highly expressed during the pre-invasive stage of PDAC and deletion of HIF1α accelerated PDAC development (61). Elimination of HIF1α resulted in an increased secretion of the B-cell chemoattractants CXCL13 and CCL21 and in intratumoral accumulation of CD19+ B-lymphocytes during early pancreatic neoplasia. On the other hand, depletion of B-cells reduced the progression of PanIN lesions and the development of invasive carcinomas in tumor-bearing mice further supporting the involvement of human B-lymphocytes in PDAC tumorigenesis. Yet, besides this correlative evidence, the exact mechanisms driving B-cell mediated promotion of early tumor development and progression remain elusive.
B-Lymphocytes and the Humoral Response in Pancreatic Adenocarcinoma
Besides functioning primarily as antigen presenting cells and providers of surviving signals to T-cells, B-lymphocytes may contribute to PDAC associated immune response by producing autoantibodies against immunogenic tumor-associated antigens (TAAs). Tumor-associated antigens are proteins expressed by tumor cells capable of activating cellular and humoral immune responses via several mechanisms including, among others, mutations resulting in the formation of neoantigens (82). Pancreatic cancer is, however, a poorly immunogenic tumor due to a low mutational burden and antigenic targets relevant for PDAC progression are currently poorly characterized (83–85).
Yet, in the attempt to identify potentially useful biomarkers, several autoantibodies have been described in patients’ sera (86–89) (Table 1). In a study screening serum IgG against proteins from pancreatic cancer cell lines (CF-PAC-1, MiaPaCa-2, and BxPC-3), two-dimensional gel electrophoresis followed by mass spectrometry identified a number of metabolic enzymes and cytoskeletal proteins as the main target of autoantibodies in PDAC patients (89). Most of these TAAs such as triosephosphateisomerase 1 (TPIS), glucose-6-phosphate-dehydrogenase (G6PD), isocitrate-dehydrogenase (IDHC), keratin 10 (K1C10), cofilin-1 (COF1) and transgelin (TAGL) were also up-regulated in PDAC tissue suggesting an attempt of the immune system to target molecules involved in tumor metabolism and metastasis (89). Additional auto-antigens identified by different proteomic and array approaches include mucin 1 (MUC1), Rad51, insulin, proteins expressed in b-islet cells, phosphoglycerate kinase 1 (PGK1), DEAD-box protein 48 (DDX48), and the tumor suppressor p53, with MUC1, PGK1, and DDX48 having shown some degree of utility for the differential diagnosis of PDAC with chronic pancreatic disorders (86–88, 90, 91). Autoantibodies against PDAC-associated antigens have been, however, observed in up to only 30% of cases and, although their presence suggests that targeted proteins may play a role in carcinogenesis, none has gained sufficient evidence to be exploited for immunotherapeutic purposes (85–89, 91).
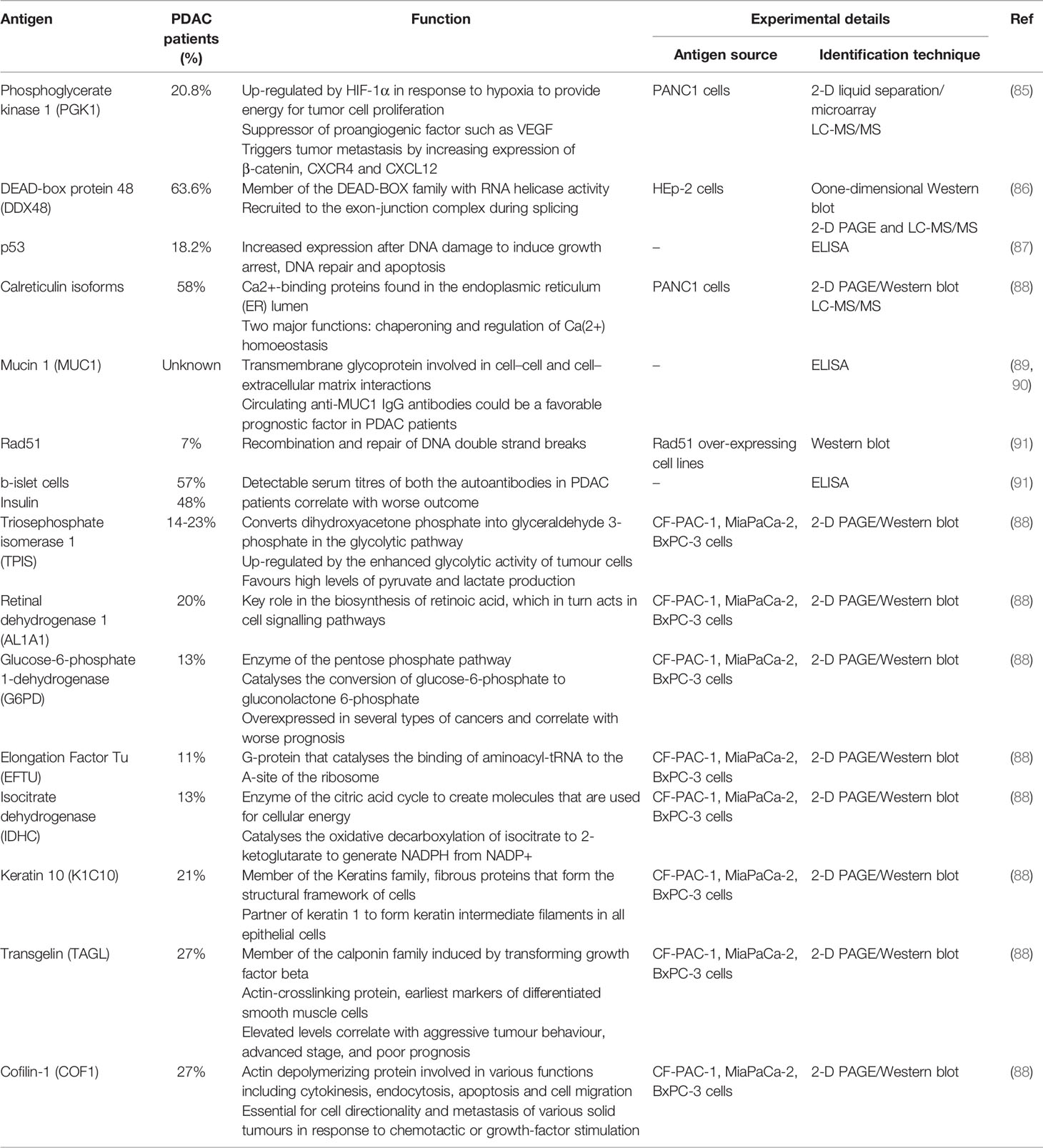
Table 1 Tumour associated antigens in pancreatic adenocarcinoma.
Over and beyond these important aspects pertaining antigen specificity of circulating autoantibodies, humoral response in PDAC patients bears additional features related to IgG subclasses, isotype, and glycosylation profile of secreted immunoglobulins that have relevant prognostic implications. The presence of IgG1+ memory B-cells in TLS+ PDAC specimens, for instance, associates with longer overall survival, while a prominent infiltration of intratumoral IgG4+ plasma cells positively correlates with poor histological grade and worse overall survival, supporting the importance of IgG class-switching for effective anti-tumor humoral immune responses (78, 92). Among the four human IgG subclasses, in fact, IgG1 shows the highest proficiency at triggering immune system components through complement activation, binding of Fc receptors on the surface of effector cells, and engagement of antibody-dependent cell-mediated cytotoxicity (ADCC) (93). On the other hand, IgG4 is considered an “anti-inflammatory” subclass due to its poor ability to fix complement and to activate effector cells (93). Of note, IgG4 antibodies are typically produced in the context of IL-10 and IL-4 driven “modified” Th2 immune responses such as those observed in the microenvironment of PDAC and of other solid tumors (94). Class switching from IgG1 to IgG4 may, therefore, represent an additional immune escape mechanism by which PDAC shapes humoral immunity during its progression to restrain anti-tumor immune responses. Indeed, although overlooked in the setting of pancreatic cancer, this hypothesis has been verified in melanoma patients whereby IgG4 antibodies significantly impaired the potency of tumoricidal IgG1 in vivo in a xenograft mouse model (95). IgE are also immunoglobulins classically associated with Th2 immune responses but, as opposed to IgG4, they have been shown to participate to tumor cells killing in ovarian cancer via ADCC, and their serum concentration correlates with longer overall survival in glioblastoma patients (96, 97). In PDAC patients, IgE levels are significantly increased compared to healthy controls, recognize self antigens, and bear in vitro ADCC activity against pancreatic cancer cells lines but their prognostic relevance has never been assessed in mouse models (98). Indeed, ADCC in PDAC appears markedly attenuated by tumor-derived exosomes that harbor B-cell targets, bind circulating autoantibodies, and exert decoy function against complement-mediated cytotoxicity (99–102).
Finally, as changes in IgG glycoforms have been reported during the progression and metastasis of solid tumors, glycosilation profile of IgG subclasses in PDAC patients has also been evaluated (103–105). Glycosylation is, indeed, one of the most relevant post-translational modifications of human immunoglobulins, and modification of the N-glycan structure in the Fc region of IgG significantly profoundly affect their functions (106). Increased galactosylation and sialylation, for instance, are known to exert anti-inflammatory properties while reduced fucosylation enhances ADCC activity (106). Compared with healthy controls PDAC patients exhibit a significantly increased IgG agalactosylation and a significantly decreased IgG fucosylation and sialylation (103–105). In addition, compared with early stage, advanced PDAC shows reduced galactosylation and increased fucosylation of serum IgG1 confirming the association between IgG glycosylation and tumor progression that has been previously described in gastric and prostate cancers (103–105).
All together, this evidence indicates that PDAC is capable of orienting the anti-tumor humoral response but further studies are needed to unveil the role of different glycoforms and IgG subclasses in cancer progression, possibly in relation with antigen specificity of secreted autoantibodies.
B-Lymphocytes and the Tumor Stroma in Pancreatic Adenocarcinoma
In apparent contrast with PDAC aggressiveness, the majority of the tumor volume is not made of malignant cells, but of a desmoplastic reaction consisting of cancer-associated fibroblasts (CAFs), extracellular matrix (ECM), and immune cells collectively termed as “tumor stroma” (107). Tumor stroma is commonly believed to support PDAC growth by promoting a hypovascular hypoxic microenvironment, escape from immune-surveillance, chemoresistance, and metastatic dissemination (1, 108, 109). Although B-cells represent a salient feature of PDAC microenvironment, their interaction with other stromal elements has not been evaluated until recently.
In a study exploiting co-cultures of B-cells and CAFs from PDAC patients to assess the contribution of B-lymphocytes to the characteristic stromal desmoplasia, unexpected pro-fibrotic properties emerged from functional and transcriptomic analyses (110). In particular, B-lymphocytes purified from PDAC patients (i) induced collagen production by CAFs via soluble factors (ii) up-regulated genes associated with epithelial-to-mesenchymal transition in co-cultured CAFs, and (iii) produced the pro-fibrotic molecule platelet derived growth factor-B (PDGF-B) (110). Circulating plasmablasts were shown to be the most proficient B-cell subset at stimulating collagen production by CAFs, as opposed to naïve B-cells and memory B-cells, and expressed a set of genes implicated in the “activation of fibroblast proliferation” including the collagen genes COL1A1 and COL1A2, insulin-like growth factor-1 (IGF-1), and lysyl oxidase homolog 2 (LOXL2) (110, 111). Of note, plasmablasts are CD19+CD20−CD27+CD38+ cells arising from GCs after affinity maturation from CD20+ naive precursors and differentiate into antibody secreting long-lived plasma cells after homing to inflammatory niches or to the bone marrow (112). They circulate for prolonged periods in the setting of chronic antigen exposure but are infrequently observed in the peripheral blood of healthy individuals (113, 114). Circulating plasmablasts were significantly expanded in the peripheral blood of treatment-naïve PDAC patients compared to healthy individuals and LOXL2 expressing plasmablasts were shown to infiltrate pancreatic lesions (110). LOXL2, on the other hand, is an enzyme that catalyzes the cross-linking of collagen components in the ECM, thus contributing to the overall tensile strength of collagen bundles and, indirectly, to the differentiation of quiescent fibroblasts into activated myofibroblasts through activation of mechanoceptors (111, 115, 116). LOXL2 has been also implicated in carcinogenesis, resistance to gemcitabine, and invasiveness of pancreatic cancer cells, and its inhibition has been shown to reduce tumor volume and metastases in preclinical studies (117).
All together these preliminary evidence demonstrates a direct interaction between B-lymphocytes and CAFs, and suggests an overlooked contribution of B-cells to PDAC pathophysiology via orchestration of stromal activation over and beyond their primary role in humoral response. Whether these pro-fibrotic properties are ultimately tumor promoting or tumor suppressing remains to be fully ascertained as recent in vivo experiences with selective knock out of ECM components in KPC mouse models have raised the possibility that stromal elements does not uniformly promote or restrain PDAC growth, increasing the degree of complexity around tumor-stroma crosstalk (118). Indeed, a slower tumor growth in B cell deficient JH−/− mice orthotopically injected with syngeneic PDAC cell lines was paralleled by a significantly reduced desmoplastic reaction compared to littermate controls, indirectly supporting possible tumorigenic interactions between B-lymphocytes and pancreatic fibroblasts (65).
Tumor Promoting Role of B-Lymphocytes in Pancreatic Adenocarcinoma
B-lymphocytes are generally considered tumor promoting immune cells based on several evidence from a number of solid cancers including prostate cancer (119, 120), liver cancer (121), and breast cancer (122) where they have been shown to sustain tumor growth and metastasis (38). In particular, the tumorigenic role of B-lymphocytes is largely attributed to Breg cells, a B-cell subset with primary immunosuppressive properties (123) that, in addition to Treg cells, hampers antitumor immune response.
Regulatory B-cells represent less than 10% of total circulating B-lymphocytes in healthy subjects and physiologically regulate immune homeostasis mainly, but not exclusively, through the production of the anti-inflammatory cytokine IL-10 (124). Human Breg cells typically express CD24, CD1d, and variable levels of CD27, but subsets with overlapping phenotypes and functions have been identified (125, 126). B-regulatory cells exert their immunosuppressive functions in different ways including (i) induction of CD4+CD25+FOXP3+ Treg cells (27, 38); (ii) inhibition of inflammatory cytokines release by macrophages and monocytes; (iii) inhibition of TH1 responses and TH17 cells differentiation (127, 128); and (iv) promotion of class-switch recombination from pro-inflammatory IgG1 to anti-inflammatory IgG4 antibodies (124, 129).
In PDAC, Breg cells are expanded compared to healthy individuals and their counts in the peripheral blood correlate with tumor stage and poor survival (130). Animal models suggest that Breg cells and PDAC cells engage in mutual activating interactions ultimately leading to tumor progression. In particular, pancreatic cancer cells reportedly induce Breg cells expansion by secreting IL-18 and Breg cells, in turn, support immune escape processes and tumor growth by expressing the programmed cell-death protein-1 ligand (PD-L1) and IL-35 (66, 130–132) (Figure 2).
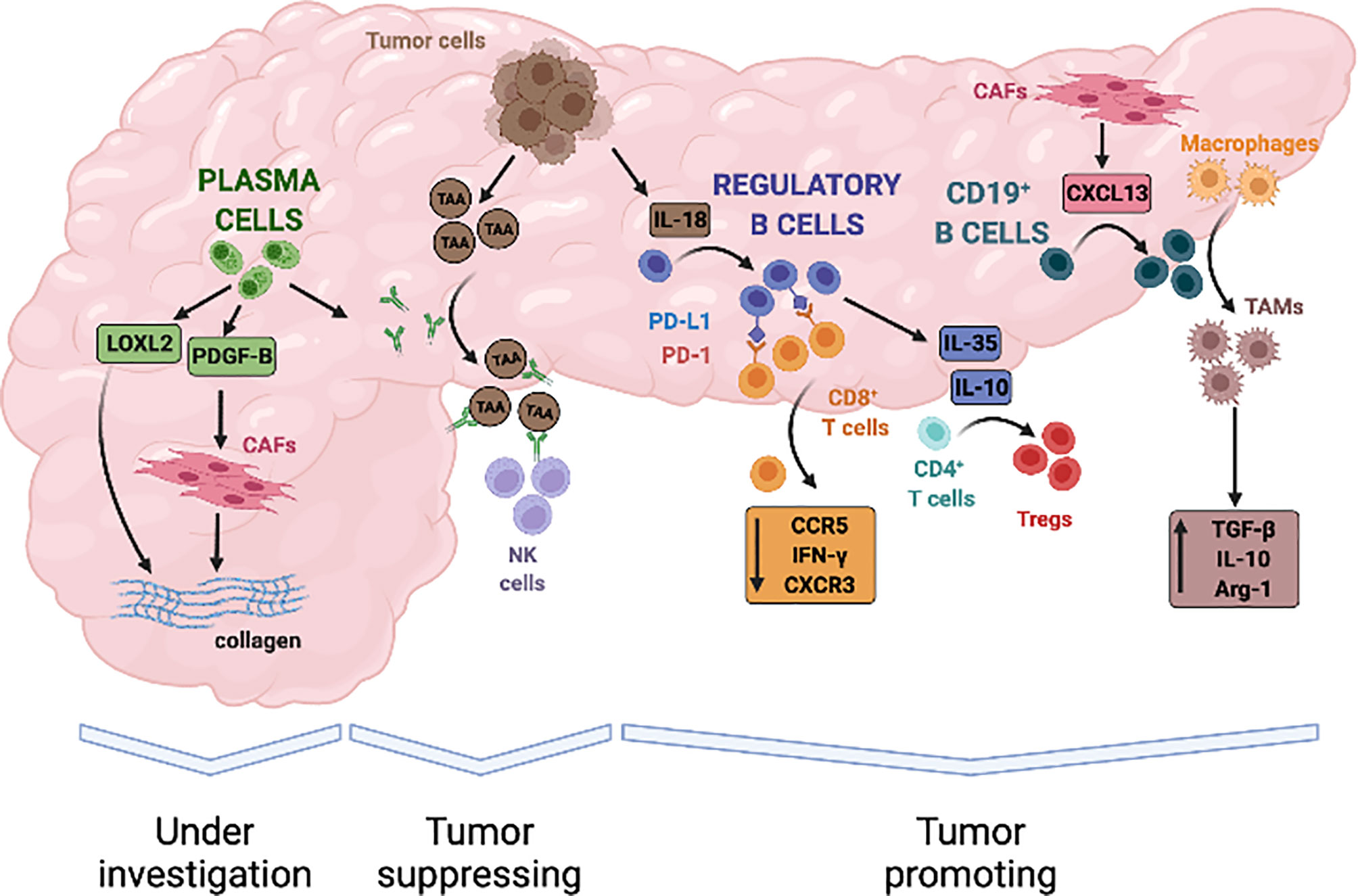
Figure 2 Evidence-based contribution of B lymphocytes to progression of pancreatic adenocarcinoma. B cells infiltrate PDAC in response to the release of local chemokines, such as CXCL13, and can contribute to both tumour promotion and tumour suppression through a variety of mechanisms. Tumour derived IL-18 can induce B lymphocytes with a regulatory phenotype that inhibit anti-tumour cytotoxic T cells responses. B-regulatory cells also secrete immune-modulatory cytokines such as IL-35 and IL-10 that induce T-regulatory cells (Tregs), stimulate tumour proliferation, and promote local angiogenesis. In addition, B-regulatory cells can recruit monocytes to tumour site and foster their differentiation into TGF-β and IL-10 producing M2 macrophages, thus further amplifying immune-evasive strategies and impairing cytotoxic responses. On the other hand, tumour-suppressive activity of B-cells in PDAC has not been clearly established but in-vitro studies suggest that it might depend on the production of autoantibodies against tumour associated antigen (TAA), engagement of natural killer cells (NK cells), and antigen dependent cell-mediated cytotoxicity (ADCC). Finally, plasma cells can sustain activation of the tumour stroma through the secretion of proliferative stimuli for cancer-associated fibroblasts (CAFs), such as PDGFB, and the production of enzymes that regulate extracellular matrix stiffness, such as LOXL2. Whether these properties ultimately exert tumour suppressive or tumour promoting effects is currently under investigation.
PD-L1 is the natural ligand of the T-cell co-inhibitory receptor PD-1 expressed on antigen-presenting cells, and interaction of these two membrane molecules prevents full cytotoxic T-cell activation in physiological condition thus contributing to the removal of auto-reactive T lymphocytes and to the maintenance of immune tolerance (133). PD-L1 is overexpressed in several solid human cancers, including PDAC, and represents an established mechanisms exploited by neoplastic cells to promote tumor escape from immune-surveillance (134, 135). B-regulatory cells from patients with PDAC also express high levels of PD-L1 and in vitro interact with PD-1 expressing CD8+ T cells leading to reduced proliferation and IFN-γ secretion (131, 136) (Figure 2). Combined inhibition of IL-18 and PD-L1/PD-1 pathways with synthetic inhibitors reduces PDAC growth and metastasis formation in orthotopic animal models, reinforcing the in vivo relevance of the crosstalk between Breg and cancer cells for the progression of pancreatic cancer (130).
B regulatory cells - and specifically a subset of CD19+CD1dhiCD5+ Breg cells - can also influence tumor progression via secretion of IL-35 (66, 132). IL-35 is increased in the serum of PDAC patients compared to healthy individuals, and promotes immunosuppression and tumor growth in orthotopic murine models (137). By inducing the phosphorylation of STAT3, in fact, IL-35 causes a reduced expression of the chemotactic receptors CXCR3 and CCR5 as well as of interferon γ (IFN-γ) in CD8+ T cells (137). In addition, IL-35 suppresses endogenous antitumor T-cell responses in vivo by reducing CD4+ effector T cells and by increasing Treg cells in the tumor infiltrate (138). Breg cell derived IL-35 has been also shown to stimulate the proliferation of pancreatic cancer cell lines in vitro therefore harboring direct mitogenic properties (139) (Figure 2). Notably, PDAC growth in IL-35 deficient mice is significantly reduced and accompanied by a robust infiltration of activated cytotoxic CD8+ T cells, especially when combined with anti-PD-1 therapy (138). Conversely, growth deficiency of orthotopic pancreatic neoplasms in μMT mice can be rescued by the reconstitution of IL-35 producing CD1dhiCD5+ B-cells (66).
Finally, activation of the Bruton tyrosine kinase (BTK) in B-cells has been shown to foster reprogramming of tumor infiltrating macrophages towards a M2 phenotype thus impairing CD8+ T-cell anti-tumor responses and supporting PDAC growth (62, 65, 140) (Figure 2). In particular, BTK is activated in CD20+ B lymphocytes infiltrating PDAC compared to peripheral leukocytes and plays an important role in the differentiation of Breg cells and in the recruitment of myeloid cells to tumor site (141, 142). Interestingly, treatment of an orthotopically implanted KrasG12D-PanIN lesions with the BTK inhibitor tirabrutinib reduced the number of intra-tumoral CD1dhiCD5+ Breg cells while only moderately affecting total CD19+ B cells (140). This effect was associated with a decreased production of the immunosuppressive cytokines IL-10 and IL-35 at tissue site, with an increased IFNγ+CD8+ T cells recruitment and, ultimately, with a delayed progression to PDAC (140). Similarly, ibrutinib - another BTK inhibitor - combined with gemcitabine slowed tumor growth of orthotopic PDAC in a T-cell dependent manner (65). BTK inhibition, in fact, reprogrammed macrophages towards a M1 phenotype and restored CD8+ T cell cytotoxicity, thereby improving responsiveness to standard chemotherapy and inhibiting PDAC growth (65). Of note, efficacy of ibrutinib at limiting PDAC growth was previously associated with a reduced stromal reaction and with a decreased infiltrate of BTK expressing mast cells in both transgenic mice and patient-derived xenograft models (143).
Taken together this accumulating evidence suggest that B lymphocytes, specifically Breg cells, bear a spectrum of non-redundant tumor-promoting properties in both early and late PDAC stage, and that most of these properties are tightly linked to modulation of cytotoxic anti-tumor responses. A clinical trial investigating the relevance of targeting B-cells with the BTK inhibitor ibrutinib in metastatic PDAC has been recently completed and results are discussed in the last session of this review (144).
Anti-Tumor Properties of B-Lymphocytes in Pancreatic Adenocarcinoma
As previously discussed most available murine studies point to a tumor-promoting role for B lymphocytes in PDAC (130, 138, 140). Yet, in analogy with other solid cancers, clinical data indicate that tumor-infiltrating B cells represent a positive prognostic factor also in pancreatic cancer, especially when organized in functional lymphoid structures, raising a controversy of crucial relevance for the implementation of effective immunotherapeutic strategies in this lethal tumor (75). In this regard, we are aware of a single study supporting anti-tumor properties of B cells in PDAC (60).
In this study B-cell activation and the effects of B-cell depletion were compared in an injectable orthotopic model of syngeneic KPC derived cell lines and in the KPC mouse, the murine model that most faithfully recapitulates PDAC pathology in terms of stromal reaction and immunological infiltrate. Notably, the authors observed a pronounced B cell infiltrate only in the latter whilst orthotopic tumors exhibited a relative paucity of B and T lymphocytes. B-cells in the KPC mouse also displayed markers of B cell activation (germinal center entry, B cell memory, and plasma cell differentiation) compared to orthotopic tumors and a significant intratumoral immunoglobulin deposition. Furthermore, in contrast to the accepted view of immunosuppressive properties of tumor infiltrating B cells, intratumoral B lymphocytes in both the KPC and orthotopic model up-regulated several pro-inflammatory genes and showed reduced expression of the immunomodulatory IL-35 and IL-10 genes. These results underscore the importance of using relevant preclinical tumor models when assessing B cell function as differences in B cell phenotype may be related to the genetic background of the animal, to the manipulation of orthotopic models, and to differential activation of circulating lymphocytes and of those infiltrating PDAC and secondary lymphoid organs. Indeed, while genetic deletion of B cells in μMT mice resulted in reduced orthotopic tumor growth this was not recapitulated by treatment of orthotopically injected immunocompetent mice with B-cell-depleting anti-CD20 antibody. Delayed tumor growth was also not observed in anti-CD20-treated KPC mice suggesting that B lymphocytes do not favor PDAC progression but rather participate to anti tumor responses as pointed by correlative human studies. Of note, most evidence supporting a tumor promoting role of B cells in PDAC has been obtained in orthotopically injected μMT mice, raising the possibility that reduced tumor progression in these animal models might be, at least in part, related to their genetic background rather than to the lack of B cells. Yet, precise mechanisms implicated in putative PDAC suppressive functions of B lymphocytes remain to be deciphered.
Evidence from Clinical Trials
At the time of writing the RESOLVE study represents the single available clinical trial addressing the utility of targeting B-cells in PDAC. In this randomized, multicenter, double-blind, placebo-controlled, phase III study ibrutinib was added to nab-paclitaxel and gemcitabine as first-line treatment of patients with metastatic PDAC (144).
Ibrutinib is a first-inclass inhibitor of BTK approved for the treatment of various B-cell malignancies where it has been shown to potentiate antitumor activity of standard chemotherapy regimens by decreasing detrimental angiogenesis and desmoplasia as well as by increasing T-cell mediated cytotoxicity (145–148). The rationale for investigating ibrutinib plus nab-paclitaxel and gemcitabine stems from preclinical evidence whereby the combination of these drugs was shown to reduce tumor growth and to increase effector CD8+ T cells activity in an orthotopic model of PDAC (65). In addition, a phase Ib study of PDAC patients treated with ibrutinib before receiving gemcitabine and nabpaclitaxel showed that ibrutinib alone induces immunomodulatory changes in circulating and intramural lymphocytes obtained by endoscopic ultrasound-guided biopsies (149).
Based on these emerging research lines, the RESOLVE trial enrolled 424 patients with metastatic PDAC and randomized them to receive oral ibrutinib (560 mg once daily) (n = 211) or placebo (n = 213) in addition to standard therapy with nab-paclitaxel and gemcitabine. The primary endpoints were overall survival (OS) and investigator-assessed progression-free survival (PFS). With a hazard ratio (HR) of 1.109 (95% confidence interval (CI): 0.903-1.363), the OS of the experimental arm (median = 9.7 months) was not significantly different from the placebo arm (median = 10.8 months) (p value = 0.3225), whilst the PFS was significantly lower in patients treated with ibrutinib (median = 5.3 months) compared to those treated with placebo (median = 6.0 months) (HR = 1.564; 95% CI: 1.277-1.916; p value < 0.0001), suggesting an even potential worse trend.
Owing to the multiple rationales for targeting BTK pathways derived from animal models of PDAC, it is difficult to explain the reasons for the failure of ibrutinib in the RESOLVE trial. Bruton’s tyrosine kinase, in fact, has been implicated the differentiation of many tumor promoting cell lines including Breg cells, M2 macrophages, and mast cells, suggesting non redundant mechanisms equally concurring to immune escape and tumor progression (62, 65, 140, 143). On the one hand it is possible that not all BTK associated pathways identified in animal models are applicable to human disease or consistently inform on the role of B cells in PDAC progression. On the other hand, patient selection in the RESOLVE trial was not based on immunological parameters and we lack information about effective target engagement by ibrutinib, making it impossible to correlate disease outcomes with treatment related perturbations of the B-cell compartment.
Overall, the RESOLVE study does not fully clarify the utility of targeting B cells in pancreatic cancer, but this seminal experience should inform future clinical trials with immune-modulating agents in PDAC on a better patient phenotyping and on an improved study design in order to retrieve useful immunological data for optimizing precision medicine.
Discussion
As current chemotherapeutic regimens - including checkpoint inhibitors - are only marginally effective in prolonging patient survival, there remains an unmet need for novel and innovative approaches for PDAC (150). In the era of personalized cancer medicine B-cells represent an increasingly attractive target for the complex pathophysiology of pancreatic cancer because most available animal models are concordant in pointing at a tumor-promoting role of this overlooked immune cell population. Yet, clinical-pathological evidence indicates that the presence of B-lymphocytes in pancreatic cancer correlate with a better prognosis raising a controversy of central relevance for the implementation of innovative immunotherapeutic strategies.
Looking ahead, as demonstrated by the ibrutinib experience, nonspecific B-cell depletion will very unlikely result in clinical benefit for all PDAC patients. A thorough understanding of the B-cell biology in PDAC will be therefore key to unveil potentially targetable tumor promoting and/or immunosuppressive interactions with neoplastic as well as with other immune and stromal cells. In addition, characterization of B-cell subsets with opposite roles in PDAC progression will instruct patient immunophenotyping and identification of subjects who will most likely benefit from targeted immune modulating therapies.
Author Contributions
All authors contributed to the design of the work, acquisition, analysis and interpretation of data. All authors revised the work critically for important intellectual content and approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Italian Association for Cancer Research (MFAG 2021 ID 26135 to ED-T), and by the Foundation for Research in Rheumatology (FOREUM Career Award 2021 to ED-T).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ryan DP, Hong TS, Bardeesy N. Pancreatic Adenocarcinoma. N Engl J Med (2014) 371:1039–49. doi: 10.1056/NEJMra1404198
2. Rahib L, Fleshman JM, Matrisian LM, Berlin JD. Evaluation of Pancreatic Cancer Clinical Trials and Benchmarks for Clinicallymeaningful Future Trials: A Systematic Review. JAMA Oncol (2016) 2:1209–16. doi: 10.1001/jamaoncol.2016.0585
3. Biankin AV, Kench JG, Dijkman FP, Biankin SA, Henshall SM. Molecular Pathogenesis of Precursor Lesions of Pancreatic Ductal Adenocarcinoma. Pathology (2003) 35:14–24. doi: 10.1080/003130202201472
4. Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, et al. An Illustrated Consensus on the Classification of Pancreatic Intraepithelial Neoplasia and Intraductal Papillary Mucinous Neoplasms. Am J Surg Pathol (2004) 28:977–87. doi: 10.1097/01.pas.0000126675.59108.80
5. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J-L, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med (2018) 379:2395–406. doi: 10.1056/nejmoa1809775
6. Adamska A, Elaskalani O, Emmanouilidi A, Kim M, Abdol Razak NB, Metharom P, et al. Molecular and Cellular Mechanisms of Chemoresistance in Pancreatic Cancer. Adv Biol Regul (2018) 68:77–87. doi: 10.1016/j.jbior.2017.11.007
7. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat Rev Gastroenterol Hepatol (2012) 9:454–67. doi: 10.1038/nrgastro.2012.115
8. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (2009) 324:1457–61. doi: 10.1126/science.1171362
10. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” Versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
11. Granot Z, Jablonska J. Distinct Functions of Neutrophil in Cancer and Its Regulation. Mediators Inflammation (2015) 2015:701067. doi: 10.1155/2015/701067
12. Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual Prognostic Significance of Tumour-Associated Macrophages in Human Pancreatic Adenocarcinoma Treated or Untreated With Chemotherapy. Gut (2016) 65:1710–20. doi: 10.1136/gutjnl-2015-309193
13. Buchholz SM, Goetze RG, Singh SK, Ammer-Herrmenau C, Richards FM, Jodrell DI, et al. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12071978
14. Protti MP, De Monte L. Thymic Stromal Lymphopoietin and Cancer: Th2-Dependent and -Independent Mechanisms. Front Immunol (2020) 11:2088. doi: 10.3389/fimmu.2020.02088
15. De Monte L, Wörmann S, Brunetto E, Heltai S, Magliacane G, Reni M, et al. Basophil Recruitment Into Tumor-Draining Lymph Nodes Correlates With Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res (2016) 76:1792–803. doi: 10.1158/0008-5472.CAN-15-1801-T
16. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T Helper Type 2 Cell Infiltrate Correlates With Cancer-Associated Fibroblast Thymic Stromal Lymphopoietin Production and Reduced Survival in Pancreatic Cancer. J Exp Med (2011) 208:469–78. doi: 10.1084/jem.20101876
17. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the Immune Reaction to Pancreatic Cancer From Inception to Invasion. Cancer Res (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
18. Brodt P, Gordon J. Anti-Tumor Immunity in B Lymphocyte-Deprived Mice. I. Immunity to a Chemically Induced Tumor. J Immunol (1978) 121:359–62.
19. Ioachim HL, Dorsett BH, Paluch E. The Immune Response at the Tumor Site in Lung Carcinoma. Cancer (1976) 38:2296–309. doi: 10.1002/1097-0142(197612)38:6<2296::aid-cncr2820380617>3.0.co;2-1
20. Ruszel KB, Mierzejewska A. [T- and B-Lymphocytes in Bronchial Cancer]. Wiad Lek (1980) 33:951–5.
21. Mandell GL, Fisher RI, Bostick F, Young RC. Ovarian Cancer: A Solid Tumor With Evidence of Normal Cellular Immune Function But Abnormal B Cell Function. Am J Med (1979) 66:621–4. doi: 10.1016/0002-9343(79)91172-0
22. Whitehead RH, Thatcher J, Teasdale C, Roberts GP, Hughes LE. T and B Lymphocytes in Breast Cancer Stage Relationship and Abrogation of T-Lymphocyte Depression by Enzyme Treatment In Vitro. Lancet (London England) (1976) 1:330–3. doi: 10.1016/s0140-6736(76)90085-4
23. House AK, Maley MA. Colorectal Carcinoma in a Rat Model: Suppression of Tumour Development and Altered Host Immune Status Following Treatment With Anti B-Lymphocyte Serum. J Surg Oncol (1986) 32:256–62. doi: 10.1002/jso.2930320416
24. Coronella-Wood JA, Hersh EM. Naturally Occurring B-Cell Responses to Breast Cancer. Cancer Immunol Immunother (2003) 52:715–38. doi: 10.1007/s00262-003-0409-4
25. Milne K, Köbel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, et al. Systematic Analysis of Immune Infiltrates in High-Grade Serous Ovarian Cancer Reveals CD20, FoxP3 and TIA-1 as Positive Prognostic Factors. PloS One (2009) 4:e6412. doi: 10.1371/journal.pone.0006412
26. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B Cells in Tertiary Lymphoid Structures is Associated With a Protective Immunity in Patients With Lung Cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
27. Tadmor T, Zhang Y, Cho H-M, Podack ER, Rosenblatt JD. The Absence of B Lymphocytes Reduces the Number and Function of T-Regulatory Cells and Enhances the Anti-Tumor Response in a Murine Tumor Model. Cancer Immunol Immunother (2011) 60:609–19. doi: 10.1007/s00262-011-0972-z
28. Kim S, Fridlender ZG, Dunn R, Kehry MR, Kapoor V, Blouin A, et al. B-Cell Depletion Using an Anti-CD20 Antibody Augments Antitumor Immune Responses and Immunotherapy in Nonhematopoetic Murine Tumor Models. J Immunother (2008) 31:446–57. doi: 10.1097/CJI.0b013e31816d1d6a
29. Barbera-Guillem E, Nelson MB, Barr B, Nyhus JK, May KFJ, Feng L, et al. B Lymphocyte Pathology in Human Colorectal Cancer. Experimental and Clinical Therapeutic Effects of Partial B Cell Depletion. Cancer Immunol Immunother (2000) 48:541–9. doi: 10.1007/pl00006672
30. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory Effects of B Cells on Antitumor Immunity. Cancer Res (2006) 66:7741–7. doi: 10.1158/0008-5472.CAN-05-3766
31. Perricone MA, Smith KA, Claussen KA, Plog MS, Hempel DM, Roberts BL, et al. Enhanced Efficacy of Melanoma Vaccines in the Absence of B Lymphocytes. J Immunother (2004) 27:273–81. doi: 10.1097/00002371-200407000-00003
32. Oizumi S, Deyev V, Yamazaki K, Schreiber T, Strbo N, Rosenblatt J, et al. Surmounting Tumor-Induced Immune Suppression by Frequent Vaccination or Immunization in the Absence of B Cells. J Immunother (2008) 31:394–401. doi: 10.1097/CJI.0b013e31816bc74d
33. Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B Cells Regulate Macrophage Phenotype and Response to Chemotherapy in Squamous Carcinomas. Cancer Cell (2014) 25:809–21. doi: 10.1016/j.ccr.2014.04.026
34. Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells Are Associated With Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res (2016) 22:3005–15. doi: 10.1158/1078-0432.CCR-15-2762
35. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ Tumor-Infiltrating Lymphocytes Have an Atypical CD27- Memory Phenotype and Together With CD8+ T Cells Promote Favorable Prognosis in Ovarian Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18:3281–92. doi: 10.1158/1078-0432.CCR-12-0234
36. Montfort A, Pearce O, Maniati E, Vincent BG, Bixby L, Böhm S, et al. A Strong B-Cell Response Is Part of the Immune Landscape in Human High-Grade Serous Ovarian Metastases. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23:250–62. doi: 10.1158/1078-0432.CCR-16-0081
37. Balkwill F, Montfort A, Capasso M. B Regulatory Cells in Cancer. Trends Immunol (2013) 34:169–73. doi: 10.1016/j.it.2012.10.007
38. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4+ T Cells to T-Regulatory Cells. Cancer Res (2011) 71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316
39. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, et al. Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely Upon Education From Tumor-Associated B Cells. Cancer Res (2015) 75:3456–65. doi: 10.1158/0008-5472.CAN-14-3077
40. Schrama D, thor Straten P, Fischer WH, McLellan AD, Bröcker EB, Reisfeld RA, et al. Targeting of Lymphotoxin-Alpha to the Tumor Elicits an Efficient Immune Response Associated With Induction of Peripheral Lymphoid-Like Tissue. Immunity (2001) 14:111–21. doi: 10.1016/s1074-7613(01)00094-2
41. Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The Mutant K-Ras Oncogene Causes Pancreatic Periductal Lymphocytic Infiltration and Gastric Mucous Neck Cell Hyperplasia in Transgenic Mice. Cancer Res (2003) 63:2005–9.
42. Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive Pancreatic Neoplasia of Ductal Phenotype Induced by Acinar Cell Targeting of Mutant Kras in Transgenic Mice. Cancer Res (2003) 63:2016–9.
43. Wagner M, Greten FR, Weber CK, Koschnick S, Mattfeldt T, Deppert W, et al. A Murine Tumor Progression Model for Pancreatic Cancer Recapitulating the Genetic Alterations of the Human Disease. Genes Dev (2001) 15:286–93. doi: 10.1101/gad.184701
44. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor Spectrum Analysis in P53-Mutant Mice. Curr Biol (1994) 4:1–7. doi: 10.1016/s0960-9822(00)00002-6
45. Lewis BC, Klimstra DS, Varmus HE. The C-Myc and PyMT Oncogenes Induce Different Tumor Types in a Somatic Mouse Model for Pancreatic Cancer. Genes Dev (2003) 17:3127–38. doi: 10.1101/gad.1140403
46. Means AL, Ray KC, Singh AB, Washington MK, Whitehead RH, Harris RC, et al. Overexpression of Heparin-Binding EGF-Like Growth Factor in Mouse Pancreas Results in Fibrosis and Epithelial Metaplasia. Gastroenterology (2003) 124:1020–36. doi: 10.1053/gast.2003.50150
47. Thayer SP, Di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an Early and Late Mediator of Pancreatic Cancer Tumorigenesis. Nature (2003) 425:851–6. doi: 10.1038/nature02009
48. Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch Signalling Controls Pancreatic Cell Differentiation. Nature (1999) 400:877–81. doi: 10.1038/23716
49. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most Human Carcinomas of the Exocrine Pancreas Contain Mutant C-K-Ras Genes. Cell (1988) 53:549–54. doi: 10.1016/0092-8674(88)90571-5
50. Hilgers W, Kern SE. Molecular Genetic Basis of Pancreatic Adenocarcinoma. Genes Chromosomes Cancer (1999) 26:1–12. doi: 10.1002/(SICI)1098-2264(199909)26:1<1::AID-GCC1>3.0.CO;2-X
51. Wang X, Gao J, Ren Y, Gu J, Du Y, Chen J, et al. Detection of KRAS Gene Mutations in Endoscopic Ultrasound-Guided Fine-Needle Aspiration Biopsy for Improving Pancreatic Cancer Diagnosis. Am J Gastroenterol (2011) 106:2104–11. doi: 10.1038/ajg.2011.281
52. Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse Models of Pancreatic Cancer. World J Gastroenterol (2012) 18:1286–94. doi: 10.3748/wjg.v18.i12.1286
53. Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf Deficiency Cooperate to Produce Metastatic Pancreatic Ductal Adenocarcinoma. Genes Dev (2003) 17:3112–26. doi: 10.1101/gad.1158703
54. Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, et al. Mist1-Kras G12D Knock-in Mice Develop Mixed Differentiation Metastatic Exocrine Pancreatic Carcinoma and Hepatocellular Carcinoma. Cancer Res (2006) 66:242–7. doi: 10.1158/0008-5472.CAN-05-2305
55. Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, et al. Inactivation of Smad4 Accelerates KrasG12D-Mediated Pancreatic Neoplasia. Cancer Res (2007) 67:8121–30. doi: 10.1158/0008-5472.CAN-06-4167
56. Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive Pancreatic Ductal Adenocarcinoma in Mice Caused by Pancreas-Specific Blockade of Transforming Growth Factor-β Signaling in Cooperation With Active Kras Expression. Genes Dev (2006) 20:3147–60. doi: 10.1101/gad.1475506
57. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
58. Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, et al. CD4+ T Lymphocyte Ablation Prevents Pancreatic Carcinogenesis in Mice. Cancer Immunol Res (2014) 2:423–35. doi: 10.1158/2326-6066.CIR-14-0016-T
59. Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, et al. A Central Role for Foxp3+ Regulatory T Cells in K-Ras-Driven Lung Tumorigenesis. PloS One (2009) 4. doi: 10.1371/journal.pone.0005061
60. Spear S, Candido JB, McDermott JR, Ghirelli C, Maniati E, Beers SA, et al. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Front Immunol (2019) 10:542. doi: 10.3389/fimmu.2019.00542
61. Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discovery (2016) 6:256–69. doi: 10.1158/2159-8290.CD-15-0822
62. Schwartz M, Zhang Y, Rosenblatt JD. B Cell Regulation of the Anti-Tumor Response and Role in Carcinogenesis. J Immunother Cancer (2016) 4:40. doi: 10.1186/s40425-016-0145-x
63. Cunningham AF, Flores-Langarica A, Bobat S, Medina CCD, Cook CNL, Ross EA, et al. B1b Cells Recognize Protective Antigens After Natural Infection and Vaccination. Front Immunol (2014) 5:535. doi: 10.3389/fimmu.2014.00535
64. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell (2012) 21:836–47. doi: 10.1016/j.ccr.2012.04.024
65. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discovery (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
66. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discovery (2016) 6:247–55. doi: 10.1158/2159-8290.CD-15-0843
67. Fridman WH, Pagès F, Saut̀s-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
68. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, et al. Occurrence of Tertiary Lymphoid Tissue is Associated With T-Cell Infiltration and Predicts Better Prognosis in Early-Stage Colorectal Cancers. Clin Cancer Res (2014) 20:2147–58. doi: 10.1158/1078-0432.CCR-13-2590
69. Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, et al. Lymphocytic Reaction to Colorectal Cancer is Associated With Longer Survival, Independent of Lymph Node Count, Microsatellite Instability, and CpG Island Methylator Phenotype. Clin Cancer Res (2009) 15:6412–20. doi: 10.1158/1078-0432.CCR-09-1438
70. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-Term Survival for Patients With non-Small-Cell Lung Cancer With Intratumoral Lymphoid Structures. J Clin Oncol (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
71. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique Ectopic Lymph Node-Like Structures Present in Human Primary Colorectal Carcinoma are Identified by Immune Gene Array Profiling. Am J Pathol (2011) 179:37–45. doi: 10.1016/j.ajpath.2011.03.007
72. Cipponi A, Mercier M, Seremet T, Baurain JF, Theáte I, Van Den Oord J, et al. Neogenesis of Lymphoid Structures and Antibody Responses Occur in Human Melanoma Metastases. Cancer Res (2012) 72:3997–4007. doi: 10.1158/0008-5472.CAN-12-1377
73. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC Chemokine Receptor 5 Expression Defines Follicular Homing T Cells With B Cell Helper Function. J Exp Med (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
74. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B Helper T Cells Express CXC Chemokine Receptor 5, Localize to B Cell Follicles, and Support Immunoglobulin Production. J Exp Med (2000) 192:1545–51. doi: 10.1084/jem.192.11.1545
75. Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, et al. Spatial Distribution of B Cells Predicts Prognosis in Human Pancreatic Adenocarcinoma. Oncoimmunology (2016) 5. doi: 10.1080/2162402X.2015.1085147
76. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral Tertiary Lymphoid Organ is a Favourable Prognosticator in Patients With Pancreatic Cancer. Br J Cancer (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
77. Brunner M, Maier K, Rümmele P, Jacobsen A, Merkel S, Benard A, et al. Upregulation of CD20 Positive B-Cells and B-Cell Aggregates in the Tumor Infiltration Zone is Associated With Better Survival of Patients With Pancreatic Ductal Adenocarcinoma. Int J Mol Sci (2020) 21:1–11. doi: 10.3390/ijms21051779
78. Gunderson AJ, Rajamanickam V, Bui C, Bernard B, Pucilowska J, Ballesteros-Merino C, et al. Germinal Center Reactions in Tertiary Lymphoid Structures Associate With Neoantigen Burden, Humoral Immunity and Long-Term Survivorship in Pancreatic Cancer. Oncoimmunology (2021) 10:1–16. doi: 10.1080/2162402X.2021.1900635
79. Roth S, Zamzow K, Gaida MM, Heikenwälder M, Tjaden C, Hinz U, et al. Evolution of the Immune Landscape During Progression of Pancreatic Intraductal Papillary Mucinous Neoplasms to Invasive Cancer. EBioMedicine (2020) 54. doi: 10.1016/j.ebiom.2020.102714
80. Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, et al. Immunoglobulin Gene Rearrangement in B Cell Deficient Mice Generated by Targeted Deletion of the JH Locus. Int Immunol (1993) 5:647–56. doi: 10.1093/intimm/5.6.647
81. Filippini D, Agosto S, Delfino P, Simbolo M, Piro G, Rusev B, et al. Immunoevolution of Mouse Pancreatic Organoid Isografts From Preinvasive to Metastatic Disease. Sci Rep (2019) 9:12286. doi: 10.1038/s41598-019-48663-7
82. Protti MP, Monte LD, Lullo GD. Tumor Antigen-Specific CD4+ T Cells in Cancer Immunity: From Antigen Identification to Tumor Prognosis and Development of Therapeutic Strategies. Tissue Antigens (2014) 83:237–46. doi: 10.1111/tan.12329
83. Biankin AV, Maitra A. Subtyping Pancreatic Cancer. Cancer Cell (2015) 28:411–3. doi: 10.1016/j.ccell.2015.09.020
84. Kabacaoglu D, Ciecielski KJ, Ruess DA, Algül H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front Immunol (2018) 9:1878. doi: 10.3389/fimmu.2018.01878
85. Johnson BA, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res (2017) 23:1656–69. doi: 10.1158/1078-0432.CCR-16-2318
86. Li C, Kim HY, Vuong H, Patwa T, Pal M, Brand RE, et al. The Identification of Auto-Antibodies in Pancreatic Cancer Patient Sera Using a Naturally Fractionated Panc-1 Cell Line. Cancer Biomarkers (2010) 7:25–37. doi: 10.3233/CBM-2010-0145
87. Xia Q, Kong XT, Zhang GA, Hou XJ, Qiang H, Zhong RQ. Proteomics-Based Identification of DEAD-Box Protein 48 as a Novel Autoantigen, a Prospective Serum Marker for Pancreatic Cancer. Biochem Biophys Res Commun (2005) 330:526–32. doi: 10.1016/j.bbrc.2005.02.181
88. Raedle J, Oremek G, Welker M, Roth WK, Caspary WF, Zeuzem S. P53 Autoantibodies in Patients With Pancreatitis and Pancreatic Carcinoma. Pancreas (1996) 13:241–6. doi: 10.1097/00006676-199610000-00005
89. Tomaino B, Cappello P, Capello M, Fredolini C, Ponzetto A, Novarino A, et al. Autoantibody Signature in Human Ductal Pancreatic Adenocarcinoma. J Proteome Res (2007) 6:4025–31. doi: 10.1021/pr070281a
90. Syrigos KN, Konstantoulakis MM, Fyssas I, Katsilambros N, Golematis BC, Svrigos KN, et al. Autoantibodies Against Insulin and Beta-Islet Cells in Pancreatic Adenocarcinoma: A Possible Explanation for Diabetes Mellitus. Int J Cancer (1996) 66:624–6. doi: 10.1002/(SICI)1097-0215(19960529)66:5<624::AID-IJC7>3.0.CO;2-V
91. Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating Anti-MUC1 IgG Antibodies as a Favorable Prognostic Factor for Pancreatic Cancer. Int J Cancer (2003) 103:97–100. doi: 10.1002/ijc.10801
92. Liu Q, Niu Z, Li Y, Wang M, Pan B, Lu Z, et al. Immunoglobulin G4 (IgG4)-Positive Plasma Cell Infiltration is Associated With the Clinicopathologic Traits and Prognosis of Pancreatic Cancer After Curative Resection. Cancer Immunol Immunother (2016) 65:931–40. doi: 10.1007/s00262-016-1853-2
93. Steplewski Z, Sun LK, Shearman CW, Ghrayeb J, Daddona P, Koprowski H. Biological Activity of Human-Mouse IgG1, IgG2, IgG3, and IgG4 Chimeric Monoclonal Antibodies With Antitumor Specificity. Proc Natl Acad Sci U.S.A. (1988) 85:4852–6. doi: 10.1073/pnas.85.13.4852
94. Protti MP, De Monte L. Cross-Talk Within the Tumor Microenvironment Mediates Th2-Type Inflammation in Pancreatic Cancer. Oncoimmunology (2012) 1:89–91. doi: 10.4161/onci.1.1.17939
95. Karagiannis P, Gilbert AE, Josephs DH, Ali N, Dodev T, Saul L, et al. IgG4 Subclass Antibodies Impair Antitumor Immunity in Melanoma. J Clin Invest (2013) 123:1457–74. doi: 10.1172/JCI65579
96. Karagiannis SN, Bracher MG, Hunt J, McCloskey N, Beavil RL, Beavil AJ, et al. IgE-Antibody-Dependent Immunotherapy of Solid Tumors: Cytotoxic and Phagocytic Mechanisms of Eradication of Ovarian Cancer Cells. J Immunol (2007) 179:2832–43. doi: 10.4049/jimmunol.179.5.2832
97. Wrensch M, Wiencke JK, Wiemels J, Miike R, Patoka J, Moghadassi M, et al. Serum IgE, Tumor Epidermal Growth Factor Receptor Expression, and Inherited Polymorphisms Associated With Glioma Survival. Cancer Res (2006) 66:4531–41. doi: 10.1158/0008-5472.CAN-05-4032
98. Fu SL, Pierre J, Smith-Norowitz TA, Hagler M, Bowne W, Pincus MR, et al. Immunoglobulin E Antibodies From Pancreatic Cancer Patients Mediate Antibody-Dependent Cell-Mediated Cytotoxicity Against Pancreatic Cancer Cells. Clin Exp Immunol (2008) 153:401–9. doi: 10.1111/j.1365-2249.2008.03726.x
99. Vykoukal J, Sun N, Aguilar-Bonavides C, Katayama H, Tanaka I, Fahrmann JF, et al. Plasma-Derived Extracellular Vesicle Proteins as a Source of Biomarkers for Lung Adenocarcinoma. Oncotarget (2017) 8:95466–80. doi: 10.18632/oncotarget.20748
100. Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, et al. High Prevalence of Mutant KRAS in Circulating Exosome-Derived DNA From Early-Stage Pancreatic Cancer Patients. Ann Oncol Off J Eur Soc Med Oncol (2017) 28:741–7. doi: 10.1093/annonc/mdx004
101. Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT, et al. Glioblastoma Microvesicles Transport RNA and Proteins That Promote Tumour Growth and Provide Diagnostic Biomarkers. Nat Cell Biol (2008) 10:1470–6. doi: 10.1038/ncb1800
102. Capello M, Vykoukal JV, Katayama H, Bantis LE, Wang H, Kundnani DL, et al. Exosomes Harbor B Cell Targets in Pancreatic Adenocarcinoma and Exert Decoy Function Against Complement-Mediated Cytotoxicity. Nat Commun (2019) 10. doi: 10.1038/s41467-018-08109-6
103. Shih H-C, Chang M-C, Chen C-H, Tsai I-L, Wang S-Y, Kuo Y-P, et al. High Accuracy Differentiating Autoimmune Pancreatitis From Pancreatic Ductal Adenocarcinoma by Immunoglobulin G Glycosylation. Clin Proteomics (2019) 16:1. doi: 10.1186/s12014-018-9221-1
104. Kodar K, Stadlmann J, Klaamas K, Sergeyev B, Kurtenkov O. Immunoglobulin G Fc N-Glycan Profiling in Patients With Gastric Cancer by LC-ESI-MS: Relation to Tumor Progression and Survival. Glycoconj J (2012) 29:57–66. doi: 10.1007/s10719-011-9364-z
105. Kanoh Y, Mashiko T, Danbara M, Takayama Y, Ohtani S, Egawa S, et al. Changes in Serum IgG Oligosaccharide Chains With Prostate Cancer Progression. Anticancer Res (2004) 24:3135–9.
106. Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu Rev Immunol (2007) 25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702
107. Kadaba R, Birke H, Wang J, Hooper S, Andl CD, Di Maggio F, et al. Imbalance of Desmoplastic Stromal Cell Numbers Drives Aggressive Cancer Processes. J Pathol (2013) 230:107–17. doi: 10.1002/path.4172
108. Aroldi F, Bertocchi P, Rosso E, Prochilo T, Zaniboni A. Pancreatic Cancer: Promises and Failures of Target Therapies. Rev Recent Clin Trials (2015) 11:33–8. doi: 10.2174/1574887110666150930122720
109. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The Pancreas Cancer Microenvironment. Clin Cancer Res (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
110. Minici C, Rigamonti E, Lanzillotta M, Monno A, Rovati L, Maehara T, et al. B Lymphocytes Contribute to Stromal Reaction in Pancreatic Ductal Adenocarcinoma. Oncoimmunology (2020) 9:1–10. doi: 10.1080/2162402X.2020.1794359
111. Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B Lymphocytes Directly Contribute to Tissue Fibrosis in Patients With IgG4-Related Disease. J Allergy Clin Immunol (2020) 145:968–981.e14. doi: 10.1016/j.jaci.2019.07.004
112. Lanzillotta M, Della-Torre E, Stone JH. Roles of Plasmablasts and B Cells in IgG4-Related Disease: Implications for Therapy and Early Treatment Outcomes. Curr Top Microbiol Immunol (2017) 401:85–92. doi: 10.1007/82_2016_58
113. Lanzillotta M, Della-Torre E, Milani R, Bozzolo E, Bozzalla-Cassione E, Rovati L, et al. Effects of Glucocorticoids on B-Cell Subpopulations in Patients With IgG4-Related Disease. Clin Exp Rheumatol (2019) 37:S159–66.
114. Lanzillotta M, Della-Torre E, Milani R, Bozzolo E, Bozzalla-Cassione E, Rovati L, et al. Increase of Circulating Memory B Cells After Glucocorticoid-Induced Remission Identifies Patients at Risk of IgG4-Related Disease Relapse. Arthritis Res Ther (2018) 20:1–10. doi: 10.1186/s13075-018-1718-5
115. Kagan HM, Li W. Lysyl Oxidase: Properties, Specificity, and Biological Roles Inside and Outside of the Cell. J Cell Biochem (2003) 88:660–72. doi: 10.1002/jcb.10413
116. Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V, Kulikova M, et al. B-Cell Depletion Attenuates Serological Biomarkers of Fibrosis and Myofibroblast Activation in IgG4-Related Disease. Ann Rheum Dis (2015) 74:2236–43. doi: 10.1136/annrheumdis-2014-205799
117. Tanaka N, Yamada S, Sonohara F, Suenaga M, Hayashi M, Takami H, et al. Clinical Implications of Lysyl Oxidase-Like Protein 2 Expression in Pancreatic Cancer. Sci Rep (2018) 8:1–9. doi: 10.1038/s41598-018-28253-9
118. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
119. Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19+IL-10+ Regulatory B Cells Affect Survival of Tongue Squamous Cell Carcinoma Patients and Induce Resting CD4+ T Cells to CD4+Foxp3+ Regulatory T Cells. Oral Oncol (2016) 53:27–35. doi: 10.1016/j.oraloncology.2015.11.003
120. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-Cell-Derived Lymphotoxin Promotes Castration-Resistant Prostate Cancer. Nature (2010) 464:302–5. doi: 10.1038/nature08782
121. Wang W, Yuan X, Chen H, Xie G, Ma Y, Zheng Y, et al. CD19+CD24hiCD38hi Bregs Involved in Downregulate Helper T Cells and Upregulate Regulatory T Cells in Gastric Cancer. Oncotarget (2015) 6:33486–99. doi: 10.18632/oncotarget.5588
122. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B Regulatory Cells and the Tumor-Promoting Actions of TNF-α During Squamous Carcinogenesis. Proc Natl Acad Sci U.S.A. (2011) 108:10662–7. doi: 10.1073/pnas.1100994108
123. Yuen GJ, Demissie E, Pillai S. B Lymphocytes and Cancer: A Love–Hate Relationship. Trends Cancer (2016) 2:747–57. doi: 10.1016/j.trecan.2016.10.010
124. Candando KM, Lykken JM, Tedder TF. B10 Cell Regulation of Health and Disease. Immunol Rev (2014) 259:259–72. doi: 10.1111/imr.12176
125. Mauri C, Menon M. The Expanding Family of Regulatory B Cells. Int Immunol (2015) 27:479–86. doi: 10.1093/intimm/dxv038
126. Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B Cells in Anti-Tumor Immunity. Int Immunol (2015) 27:521–30. doi: 10.1093/intimm/dxv034
127. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B Cells Exhibit Regulatory Capacity in Healthy Individuals But are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity (2010) 32:129–40. doi: 10.1016/j.immuni.2009.11.009
128. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B Cells Maintain Regulatory T Cells While Limiting TH1 and TH17 Differentiation. Sci Transl Med (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
129. Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-Related Disease. Clin Exp Immunol (2015) 181:191–206. doi: 10.1111/cei.12641
130. Zhao Y, Shen M, Feng Y, He R, Xu X, Xie Y, et al. Regulatory B Cells Induced by Pancreatic Cancer Cell-Derived Interleukin-18 Promote Immune Tolerance via the PD-1/PD-L1 Pathway. Oncotarget (2018) 9:14803–14. doi: 10.18632/oncotarget.22976
131. Tong DN, Guan J, Sun JH, Zhao CY, Chen SG, Zhang ZY, et al. Characterization of B Cell-Mediated PD-1/PD-L1 Interaction in Pancreatic Cancer Patients. Clin Exp Pharmacol Physiol (2020) 47:1342–9. doi: 10.1111/1440-1681.13317
132. Jin P, Ren H, Sun W, Xin W, Zhang H, Hao J. Circulating IL-35 in Pancreatic Ductal Adenocarcinoma Patients. Hum Immunol (2014) 75:29–33. doi: 10.1016/j.humimm.2013.09.018
133. Francisco LM, Sage PT, Sharpe AH. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
134. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2007) 13:2151–7. doi: 10.1158/1078-0432.CCR-06-2746
135. Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
136. Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B Cells are Critical Regulators of Humoral Immunity. Nat Commun (2015) 6:5997. doi: 10.1038/ncomms6997
137. Mirlekar B, Michaud D, Lee SJ, Kren NP, Harris C, Greene K, et al. Bcell-Derived IL35 Drives STAT3-DependentCD8+ T-Cell Exclusion in Pancreatic Cancer. Cancer Immunol Res (2020) 8:292–308. doi: 10.1158/2326-6066.CIR-19-0349
138. Mirlekar B, Michaud D, Searcy R, Greene K, Pylayeva-Gupta Y. IL35 Hinders Endogenous Antitumor T-Cell Immunity and Responsiveness to Immunotherapy in Pancreatic Cancer. Cancer Immunol Res (2018) 6:1014–24. doi: 10.1158/2326-6066.CIR-17-0710
139. Nicholl MB, Ledgewood CL, Chen X, Bai Q, Qin C, Cook KM, et al. IL-35 Promotes Pancreas Cancer Growth Through Enhancement of Proliferation and Inhibition of Apoptosis: Evidence for a Role as an Autocrine Growth Factor. Cytokine (2014) 70:126–33. doi: 10.1016/j.cyto.2014.06.020
140. Das S, Bar-Sagi D. BTK Signaling Drives CD1dhiCD5+ Regulatory B-Cell Differentiation to Promote Pancreatic Carcinogenesis. Oncogene (2019) 38:3316–24. doi: 10.1038/s41388-018-0668-3
141. Schmidt NW, Thieu VT, Mann BA, Ahyi A-NN, Kaplan MH. Bruton’s Tyrosine Kinase is Required for TLR-Induced IL-10 Production. J Immunol (2006) 177:7203–10. doi: 10.4049/jimmunol.177.10.7203
142. Corneth OBJ, de Bruijn MJW, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced Expression of Bruton’s Tyrosine Kinase in B Cells Drives Systemic Autoimmunity by Disrupting T Cell Homeostasis. J Immunol (2016) 197:58–67. doi: 10.4049/jimmunol.1600208
143. Massó-Vallés D, Jauset T, Serrano E, Sodir NM, Pedersen K, Affara NI, et al. Ibrutinib Exerts Potent Antifibrotic and Antitumor Activities in Mouse Models of Pancreatic Adenocarcinoma. Cancer Res (2015) 75:1675–81. doi: 10.1158/0008-5472.CAN-14-2852
144. Tempero M, Oh D-Y, Tabernero J, Reni M, Van Cutsem E, Hendifar A, et al. Ibrutinib in Combination With Nab-Paclitaxel and Gemcitabine for First-Line Treatment of Patients With Metastatic Pancreatic Adenocarcinoma: Phase III RESOLVE Study. Ann Oncol Off J Eur Soc Med Oncol (2021) 32:600–8. doi: 10.1016/j.annonc.2021.01.070
145. Pharmacyclics LLC an AC. IMBRUVICA® Prescribing Information (2019). Available at: https://www.imbruvica.com/files/prescribing-information.pdf.
146. Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib Combined With Bendamustine and Rituximab Compared With Placebo, Bendamustine, and Rituximab for Previously Treated Chronic Lymphocytic Leukaemia or Small Lymphocytic Lymphoma (HELIOS): A Randomised, Double-Blind, Phase 3 Study. Lancet Oncol (2016) 17:200–11. doi: 10.1016/S1470-2045(15)00465-9
147. Brown JR, Barrientos JC, Barr PM, Flinn IW, Burger JA, Tran A, et al. The Bruton Tyrosine Kinase Inhibitor Ibrutinib With Chemoimmunotherapy in Patients With Chronic Lymphocytic Leukemia. Blood (2015) 125:2915–22. doi: 10.1182/blood-2014-09-585869
148. Davids MS, Brander DM, Kim HT, Tyekucheva S, Bsat J, Savell A, et al. Ibrutinib Plus Fludarabine, Cyclophosphamide, and Rituximab as Initial Treatment for Younger Patients With Chronic Lymphocytic Leukaemia: A Single-Arm, Multicentre, Phase 2 Trial. Lancet Haematol (2019) 6:e419–28. doi: 10.1016/S2352-3026(19)30104-8
149. Sinha M, Griffith M, Betts C, Choe G, Sivagnanam S, Cheung A, et al. Immune Modulatory Effects of Ibrutinib in Pancreatic Ductal Adenocarcinoma. Ann Oncol (2019) 30. doi: 10.1093/annonc/mdz155.145
Keywords: B lymphocyte, pancreatic adenocarcinoma, PDAC - pancreatic ductal adenocarcinoma, B cells, fibrosis, cancer associated fibroblast (CAF)
Citation: Minici C, Testoni S and Della-Torre E (2022) B-Lymphocytes in the Pathophysiology of Pancreatic Adenocarcinoma. Front. Immunol. 13:867902. doi: 10.3389/fimmu.2022.867902
Received: 01 February 2022; Accepted: 23 February 2022;
Published: 14 March 2022.
Edited by:
Stefano Ugel, University of Verona, ItalyReviewed by:
Manuela Iezzi, University of Studies G. d’Annunzio Chieti and Pescara, ItalyCarmine Carbone, Agostino Gemelli University Polyclinic (IRCCS), Italy
Copyright © 2022 Minici, Testoni and Della-Torre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanuel Della-Torre, ZGVsbGF0b3JyZS5lbWFudWVsQGhzci5pdA==