 Timothy C. Frommeyer1†
Timothy C. Frommeyer1† Michael M. Gilbert1†
Michael M. Gilbert1† Garrett V. Brittain1†
Garrett V. Brittain1† Tongfan Wu1†
Tongfan Wu1† Trang Q. Nguyen1†
Trang Q. Nguyen1† Craig A. Rohan1,2,3
Craig A. Rohan1,2,3 Jeffrey B. Travers1,2,3*
Jeffrey B. Travers1,2,3*- 1Department of Pharmacology and Toxicology, Boonshoft School of Medicine at Wright State University, Dayton, OH, United States
- 2Department of Dermatology, Boonshoft School of Medicine at Wright State University, Dayton, OH, United States
- 3Department of Medicine, Dayton Veterans Administration Medical Center, Dayton, OH, United States
Ultraviolet B radiation (UVB) has profound effects on human skin that results in a broad spectrum of immunological local and systemic responses and is the major cause of skin carcinogenesis. One important area of study in photobiology is how UVB is translated into effector signals. As the skin is exposed to UVB light, subcellular microvesicle particles (MVP), a subtype of bioactive extracellular vesicles, are released causing a variety of local and systemic immunological effects. In this review, we highlight keratinocyte MVP release in keratinocytes in response to UVB. Specifically, Platelet-activating factor receptor agonists generated by UVB result in MVP released from keratinocytes. The downstream effects of MVP release include the ability of these subcellular particles to transport agents including the glycerophosphocholine-derived lipid mediator Platelet-activating factor (PAF). Moreover, even though UVB is only absorbed in the epidermis, it appears that PAF release from MVPs also mediates systemic immunosuppression and enhances tumor growth and metastasis. Tumor cells expressing PAF receptors can use this mechanism to evade chemotherapy responses, leading to treatment resistance for advanced cancers such as melanoma. Furthermore, novel pharmacological agents provide greater insight into the UVB-induced immune response pathway and a potential target for pharmacological intervention. This review outlines the need to more clearly elucidate the mechanism linking UVB-irradiation with the cutaneous immune response and its pathological manifestations. An improved understanding of this process can result in new insights and treatment strategies for UVB-related disorders from carcinogenesis to photosensitivity.
Introduction
Ultraviolet B (UVB; 290-320nm) radiation found in sunlight is essential for the production of vitamin D in humans (1). However, prolonged exposure can lead to a myriad of pathologic effects including erythema, photoaging, inflammatory responses, and skin cancer (2–4). Within the epidermis, UVB is able to damage DNA in various cell types in the skin, especially the keratinocyte (5–7). The UVB rays induce formation of cyclobutane pyrimidine dimers which have the potential to be propagated to subsequent cellular populations, which may result in pro-carcinogenic changes (8, 9). UVB also has the ability to act as a pro-oxidative stressor, generating various immunoregulatory mediators including prostaglandin E2, serotonin, histamines, interleukin (IL)-6, IL-10, tumor necrosis factor-alpha (TNFα) and the lipid mediator Platelet-activating factor (PAF) (10–12). UVB-induced production of these bioactive molecules results in acute inflammation, erythema, cancer, and degenerative aging (13, 14). Moreover, at high doses, UVB-irradiation and subsequent release of PAF and other agents such as TNFα can lead to systemic effects including fever, malaise, and immunosuppression (15, 16). PAF exerts a variety of effects through the G-protein coupled transmembrane Platelet-activating factor receptor (PAFR), which is expressed on multiple cell types including keratinocytes (17, 18). Cutaneous UVB exposure and activation of keratinocytic PAFR results in similar signaling pathways, which suggests that many UVB-induced effects are mediated via PAFR or modified by associated PAFR activation (19, 20). Since UVB absorption is limited to the epidermis, bioactive agents that leave the skin are thought to act as effectors for UVB responses.
Platelet-activating factor (PAF) is the term denoting a family of glycerophosphocholine (GPC)-derived lipid mediators implicated in a number of pathologic processes including skin carcinogenesis, liver disease, and allergic rhinitis (16, 21, 22). Although PAF application results in pro-inflammatory processes similar to the acute effects of UVB radiation, it can also produce immunosuppressive effects via upregulation of regulatory T cells (10, 16, 17, 23). The synthesis of PAF is associated with several different pathways including the remodeling and de novo pathways (24, 25). Of particular interest is the formation of PAFR agonists in response to increased reactive oxygen species (ROS) resulting in non-enzymatic formation of oxidized GPC (ox-GPC) with PAFR agonistic activities (26–28). This has prompted the notion that UVB-irradiation results in activation of the PAF system. The link between UVB, PAFR signaling, and photobiology is further implicated by PAFR activation in the early acute response of UVB as well as UVB-mediated systemic immunosuppression (10, 16, 17, 23, 29, 30). Thus, the PAF system plays an integral role in dermal pathophysiology and skin response to environmental stressors (31). After generation, PAF resides in cellular membranes where it can act upon itself or neighboring cells through juxtacrine signaling (32, 33). It should be noted that once generated, PAF/ox-GPCs are rapidly metabolized by removal of the sn-2 short-chained fatty acid by acetyl hydrolases. However, certain cell types such as keratinocytes have demonstrated the ability of PAF to exert its effects some distance away from the host cell (34). Previous studies by our group have shown that UVB-irradiated keratinocytes can induce the production of subcellular microvesicle particles (MVP), which contains PAFR agonist activity (34, 35).
Microvesicle particles are small membrane-bound particles that are shed from the plasma membrane of various cell types including keratinocytes (36). Their release is largely dependent on the action of the lipid enzyme acid sphingomyelinase (aSMase) (37, 38). Also named microvesicles and microparticles, MVPs are thought to provide a cellular mechanism to transport a variety of bioactive substances including proteins, lipids, cytokines, and nucleic acids (39, 40). Furthermore, their integral role within cell-to-cell signaling has suggested their mediation of pathogenic processes (41). Emerging literature suggests that UVB-generated ox-GPC PAFR agonists activate keratinocytic PAFR, which translocates aSMase to trigger MVP release from the plasma membrane (42). However, the exact mechanism linking UVB-irradiation with the cutaneous immune response and its pathological manifestations needs to be further elucidated. In this review, we highlight the potential role of UVB-generated MVPs and subsequent release of PAF in cutaneous immune responses.
Background
Cutaneous Microenvironment
Skin with subcutaneous fat (called the hypodermis) is the largest and most exposed organ of the human body. As such, it acts as a physical barrier, which leaves it vulnerable to a myriad of potentially harmful agents including bacteria, viruses and other environmental contaminants. In addition, the skin has important functions in regulating body temperature, preventing dehydration, and protecting against ultraviolet (UV) radiation (43). Cutaneous anatomy consists of an outer epidermal layer and an inner dermal layer that are joined by a basement membrane as well as an underlying layer of subcutaneous fat and connective tissue (44, 45). The epidermal layer is made up of five distinct layers that consist primarily of keratinocytes, which are bioactive cells that are highly proliferative and secrete a variety of cytokines (46). As keratinocytes detach from the basal layer of the epidermis, they stop dividing and undergo a final differentiation process known as cornification (44, 47). Melanocytes, Langerhans cells (LC), and Merkel cells are other cell types in the epidermis that are involved in melanin synthesis, immunoregulation and sensory functions respectively (46). The underlying dermis supplies nutrients to the epidermis and consists of two layers: the upper papillary layer and the lower reticular layer. The papillary dermis consists of densely packed fibroblasts whereas the reticular layer has lesser numbers of cells such as fibroblasts but contains more densely packed collagen and sits just above the subcutaneous fat (44). Various immune cells reside in the dermis which provide routine immune surveillance; these include macrophages, dendritic cells, T cells, mast cells, neutrophils and eosinophils. Inflammation and immune responses lead to significant proliferation of dermal immune cell populations (48, 49). The underlying hypodermis is a layer of subcutaneous tissue, composed of blood vessels and adipocytes that provide storage of lipids and fatty acids. Adipocytes help generate a number of bioactive lipid mediators and peptide hormones that partake in a number of dermal biological processes including cell signaling, inflammation, and regulation of local and systemic effects (50, 51). The connection between the local and systemic pathways are based on both internal and external variables including serotonergic, melatoninergic, and cholinergic pathways (52). The skin responds distinctly depending on the type of stressor and amount of exposure, which results in specific physiologic responses and subsequent signaling pathways. Of particular interest is the effect of UVB radiation on the skin and its effects on keratinocytes.
Ultraviolet radiation is implicated in a number of cutaneous pathologies and is divided into three distinct wavelengths: UVA (320-400 nm), UVB (290-320 nm) and UVC (100 -290 nm). Accounting for only 0.3% of the total light, UVB is absorbed by the epidermis, and can directly damage DNA by forming photoproducts including cyclobutane pyrimidine dimers and 6-4 photoproducts (53). In addition, UVB exposure results in the upregulation and release of various factors from keratinocytes including growth factors, antimicrobial peptides (human β-defensin-2, -3, ribonuclease-7 and psoriasin (SA100A7)) and cytokines (IL-1α, IL-1β, IL-6, IL-8, granulocyte colony stimulating factor [G-CSF], macrophage-CSF, interferon gamma [INF-γ], platelet-derived growth factor (PDGF) as well as the increased accumulation of reactive oxygen species (ROS) (54, 55). UVB also has additional roles in immune function by upregulating toll-like receptors and can disrupt the delicate skin microbiome (56, 57). One such consequence of prolonged UV-exposure is immune suppression, which includes inhibition of antigen presentation, induction of leukocyte apoptosis, and generation of immunosuppressive cytokines and (16). Additionally, UV-induced immunosuppression has been implicated to act through several molecular including DNA and membrane lipids, tryptophan in skin cells, and trans-urocanic acid (UCA) as well as through the depletion of other molecules including nicotinamide adenine dinucleotide (NAD) levels in keratinocytes (58). Moreover, UVB appears to inhibit immune reactions in an antigen-specific fashion, suppressing primary immune reactions. This is evidenced by suppressed contact hypersensitivity and delayed type hypersensitivity (DTH) reactions after UVB-irradiation, which show a diminished T-cell mediated immune response and generation of antigen-specific tolerance and desensitization (59–61). Extensive UVB damage alters vitamin D uptake, induces wrinkling of the skin, promotes photoaging, and can result in skin carcinogenesis as well as immunosuppression (14, 58, 62, 63). Simultaneously, vitamin D has been shown to have photoprotective effects against UV damage, which include diminishing the production of free radicals and attenuating DNA repair (64). As a result, the mechanisms by which UVB-mediated damage has a wide spectrum of effects in the epidermis and beyond are becoming increasingly important. Interestingly, UV radiation is able to upregulate local neuroendocrine axes through locally induced cytokines, corticotropin-releasing hormones, urocortin’s, proopiomelanocortin-peptides, and enkephalins. These local compounds can induce systemic effects such as the activation of central hypothalamic-pituitary-adrenal axis as well as immunosuppression (65). Of note, UVB enhances the production of a variety of bioactive lipids including PAF and eicosanoids such as PGE2, which appear to play an important role in UV-induced immunosuppression, inflammation, and carcinogenesis (26, 66).
The Platelet-Activating Factor Family
In 1972, the term Platelet-activating Factor was first utilized in reference to a released product of IgE-induced basophil activation which results in subsequent platelet aggregation in rabbit models (67). Since its initial discovery, further investigations have indicated that PAF asserts a multitude of physiological and pathophysiological effects through binding with a specific G-protein-coupled receptor, PAF-R, extensively expressed by many immune and epithelial cell types (68). Despite new understandings of the functional significance, Benveniste and colleague’s cognomination persisted. The PAF family has been implicated in a variety of conditions ranging from malignancies to neurological conditions. PAF-related mechanisms have also been described in asthma and of interest to our group, UVB-mediated responses (42). Interestingly, PAF’s proposed acute and chronic impacts appear contradictory, the former pro-inflammatory and the latter immunosuppressive as will be discussed further (31).
Throughout the body, PAF production can be stimulated by a variety of cell types. Through a variety of mechanisms, eosinophils, neutrophils, macrophages, monocytes, basophils, and mast cells all play a role in PAF synthesis (69). The biosynthesis of PAF has two separate pathways: a remodeling pathway and a de novo pathway (Figure 1). An interesting facet of the remodeling pathway of PAF production includes its influences from cell-specific inflammatory tracks. Upon cellular stimulation, increased intracellular calcium levels activate Ca-dependent MAPK kinase which phosphorylates phospholipase A2 (PLA2). PLA2 then deacylates alkylacyl-glycerophosphocholine to produce lyso-PAF and the unsaturated sn-2 fatty acid, often arachidonate. Lyso-PAF (1-alkyl-sn-glycero-3-phosphocholine) is further acetylated by CoA lyso PAF acetyltransferase (LPCAT2) forming PAF (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) and Coenzyme A. The terminal enzyme in PAF biosynthesis, LPCAT2, is also highly expressed in inflammatory cells (16, 42, 70). One example of this includes its role in macrophages. Following lipopolysaccharides (LPS) binding to TLR4 on macrophages, protein kinase 2 activates MAPK which phosphorylates LPCAT2 resulting in a rapid surge in PAF. In addition, the PAFR, once activated, results in the activation of protein kinase Cα which phosphorylates LPCAT2, modeling a positive feed forward loop for PAF formation (16, 31, 69). Moreover, the de novo model of PAF production, unlike its counterpart, is not influenced by inflammatory pathways but plays an important role in baseline PAF levels due to constitutive activation. This pathway has three primary steps. Initially, 1-alkyl-2-lyso-sn-glycero-3-P is acetylated by CoA-alkyl-lysoglycero-P acetyltransferase forming 1-alkyl-2-acetyl-sn-glycero-3-P. Next, this intermediate undergoes dephosphorylation via an alkylacylglycerol-P phosphohydrolase forming 1-alkyl-2-lyso-sn-glycerol. Lastly, this glycerol molecule receives a phosphate group and choline by CDP-choline alkylacetylglycerol cholinephosphotransferase (25, 71, 72).
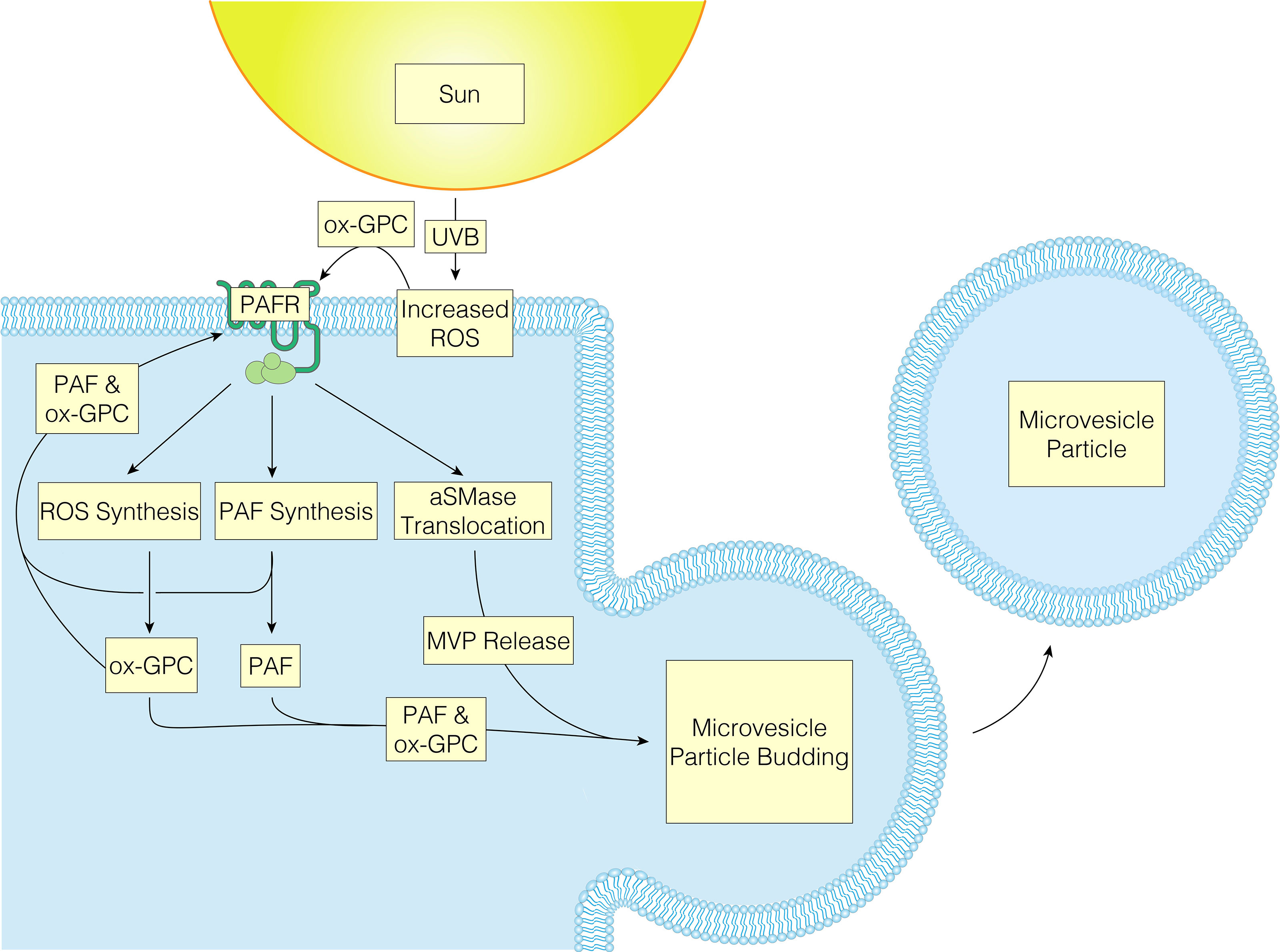
Figure 1 PAF biosynthesis pathways.
The metabolism of PAF is highly regulated by the PAF acetylhydrolase (PAF-AH) subclass of phospholipase A2 enzymes (73). By hydrolyzing the short-chained fatty acid moiety (e.g., acetyl group) at the sn-2 position of PAF and related GPC, these signaling molecules form the biologically inactive lyso-PAF. The PAF-AH family members PAF-AH type I, PAF-AH type II, and plasma PAF-AH are pertinent to PAF degradation (31, 74). While these enzymes were originally named due to their role in PAF metabolism, they play a multitude of physiological and pathophysiological roles which are further discussed in the literature (73). The intracellular PAH-AH type II heterotetrameric has also been found in both sebaceous glands and epidermal keratinocytes (73, 75). Due to the rapid degradation and tight regulation of PAF and its implication in a vast number of processes, there is a need to better understand the mechanisms which allow for the systemic effects of PAF. Keratinocyte-derived MVPs are a major point of interest for this role.
Microvesicle Particles
UVB-mediated bioactive product release can take place in a variety of ways, including through the release of MVPs. MVPs are extracellular vesicles that are formed from outward protrusion of the plasma membrane when there is an increase in intracellular calcium (76). Ranging from 100 – 1,000 nm, MVPs are important for cellular signaling due to them carrying a variety of different proteins, lipids, and mRNA. MVPs can be shed during physiological conditions including cell growth but also are increased in a variety of pathological processes ranging from hypoxia to oxidative stress (40, 77). Many cytokines including TNFα and cell damage can release MVP (38). They are present in a range of physiologic and pathologic processes throughout the body (77–79). Of particular interest are MVPs generated by a keratinocyte cell line through budding from the plasma membrane which contains PAFR agonist activity (80, 81). Both MVPs and PAF are released due to activation of PAFR. In fact, MVP production and activation can be triggered by a great variety of mechanisms, including acute alcohol poisoning (35, 82).
Throughout the human body, extracellular vesicles like MVPs and smaller exosomes frequent matrices of the system. UVB has been demonstrated to induce the release of both of these particles (34, 83). While MVPs are often parceled with exosomes, each are separate entities expressing different characteristics. Exosome formation occurs via reverse budding of multivesicular bodies prior to cellular release (84, 85). This process allows for delivery of exosome contents with plasma membrane fusion (85). On the other hand, MVPs are contrived from external budding and fission of cellular plasma membrane which allows for an additional function in MVPs of cargo delivery into the extracellular environments (86). The variance in formation of each of these extracellular vesicles results in discrepancy in sizes as well. Exosomes typically range from 30nm – 150nm with less deviance from mean values (84–86). This range is smaller than the previously mentioned range for MVPs and has a lower peak value. Due to the external plasma membrane invagination formation mechanisms of MVPs, cytosolic elements are accrued secondary to proximity to the plasma membrane. In exosomes, contiguity with intraluminal vesicles play a greater significance in free exosome content (85, 87).
While the exact mechanisms of keratinocyte-derived MVP generation and release are unclear, much research has been done on the biogenesis of MVPs (34). The production of MVPs has three elemental steps. To begin, membrane and lipid proteins are reorganized into distinct groups within the plasma membrane. Next, these microdomains aid in various pathways attracting cargo. Lastly, in conjunction with other machinery, membrane budding and fission is promoted (88). The initiation of these processes is highly unlikely to be secondary to spontaneous intrinsic property changes of the phospholipid bilayer but rather unclear emerging external mechanisms (86). In particular, MVP generation from diverse stimuli involve the enzyme acid sphingomyelinase (aSMase) appears to play an important role in this process (42). Furthermore, MVP cargo, which is dependent upon parent cell archetype, is preferentially gathered through a variety of possible mechanisms- most notably ARF6 of the GTPase family (89). Downstream effects of MVPs are secondary to their respective cargo, highlighting the importance of these pathways. Of interest, keratinocyte-formed MVPs contain a myriad of bioactive agents and molecules including PAF and ox-GPC, which have implications in systemic immune responses (34). Of interest, protein cytokines found in UVB-generated MVP contain lesser numbers of classic pro-inflammatory cytokines (e.g., TNF-alpha), but increased numbers of anti-inflammatory cytokines such as IL-1 receptor antagonist, in comparison to baseline (unstimulated) MVP (37).
As previously discussed, MVPs functions are dependent upon the appropriate intercellular messaging and respective cargo. As more research is needed on keratinocyte-derived MVPs uptake at their respective target cells, we will only briefly cover this topic. Parent cells of MVP, by nature of their biosynthesis pathway, additionally impact the plasma membrane content of this extracellular vesicle. The adoption of plasma membrane from origin cells is believed to play a role in MVP intercellular communication (88). With this in mind, certain MVPs transmit signals by direct physical contact between membrane-associated ligands and cell surface receptors. One such example is the production of chemokine (C-C motif) ligand 3 (CCL3), chemokine (C-C motif) ligand 7 (CCL7) and IL-24 in T-cell-derived MVPs co-cultured with in-vitro keratinocytes (90, 91). Indirect mechanisms of MVP function have also been observed. For instance, MVP release of cargo into extracellular milieu near the vicinity of target cells has been shown to stimulate cell surface receptors (92). Furthermore, MVPs can merge with beneficiary cells’ plasma membranes, sending internal cargo into the recipient cells’ cytoplasm with the additional potential of membrane property transference (93). Finally, select MVPs are absorbed into their destination cells through non-selective macropinocytosis or endocytosis (94).
UVB Activation of MVPs
UVB radiation (290-320 nm) results in a pro-oxidative stress that exerts significant effects within the skin. One of which is the generation of ROS, which modify biologically active agents, such as lipids (28). GPC is an integral structural lipid found in all cellular membranes and functions as a precursor of bioactive PAF. Due to the presence of bisallylic double bonds, esterified polyunsaturated fatty acids are vulnerable to oxidation from ROS attack of the hydrogen donors. This results in the introduction of oxy functions to the chain of carbon atoms, rearranges bonds, fragments carbon-carbon bonds by β-scission, which can all give rise to a multitude of lipid reaction products (27, 28). In particular are a series of phospholipids with oxidatively fragmented sn-2 acyl residues with a terminal methyl group or ω-oxy function. Potent PAF agonists (ox-GPC) are generated if the oxidatively modified sn-2 acyl residue is a 1-alkyl GPC (26–28). There are multiple literature reports that link UVB generation of ROS with subsequent activation of the PAF system (10, 16, 17, 23, 29, 30). Early research into these findings was based on a knowledge of how UV response affects the cell membrane rather than the nucleus (95). Additional evidence found overexpressing the PAFR enhances PAF synthesis by keratinocytes in response to UVB radiation, while administering a PAFR antagonist inhibits PAF synthesis in UVB radiated keratinocytes (96). Furthermore, Yao and colleagues discovered that a PAFR antagonist inhibited UVB-mediated skin inflammation and TNF-alpha synthesis in the photosensitive Xpa-/- mice (29). Interestingly, it has also been found that PAFR expression is necessary for UVB-induced activation and subsequent migration of mast cells (97). Thus, UVB acts as a pro-oxidative stressor that generates ox-GPC and subsequent activation of PAFR through its PAFR agonist activity. Of importance, ox-GPC activation of the PAFR can generate enzymatic PAF synthesis, thus forming a positive feedback loop (31).
UVB radiation and PAFR activation of keratinocytes have been noted to result in multiple similar signaling pathways, implying that some UVB-induced effects are mediated via PAFR or modified by associated PAFR activation (19, 20). There are several cutaneous cell types that express PAFR including keratinocytes, mast cells, monocytes, granulocytes, and B cells (30, 97–102). The actions of PAF and its agonists are mediated through the PAFR, a unique G-protein-coupled seven transmembrane receptor (GPCR), which when activated results in several intracellular signaling pathways (68). These include mitogen kinase (both ERK and P38), JNK, and can also indirectly activate the EGFR (80). While coupled to G proteins Gi, Gq, and G12/13, PAFR also controls the production of inositol 1,4,5-triphosphate (IP3) and calcium mobilization in GPCRs, while suppressing forskolin-stimulated cAMP synthesis (103). Emerging literature supports the theory that UVB produces ox-GPC PAFR agonists, which then act upon PAFR-positive keratinocytes, resulting in PAFR activation. As a result of PAFR activation, more PAF is produced enzymatically as well as more ROS and subsequent ox-GPC. Finally, PAFR activation induces aSMase to translocate to the plasma membrane causing MVP to be generated and released from the keratinocyte plasma membrane. As the PAF/ox-GPC will be retained in the plasma membranes this allows PAFR agonist to be transported in the MVP to additional locations (Figure 2) (42). Of interest, studies of the crystal structure of the PAFR have revealed one of the protein loops appears draped over the central area of this GPCR (100). This novel putative conformational change suggests that PAFR agonists interact with the receptor optimally from the plasma membrane rather from the exterior of the cell. Hence, it is possible that the PAFR agonists in the MVP when are incorporated into the target cell could be in a pharmacologically active form. In sum, a feed-forward loop is created because PAFR activation results in continual release of PAF from MVPs and subsequent activation of PAFR.
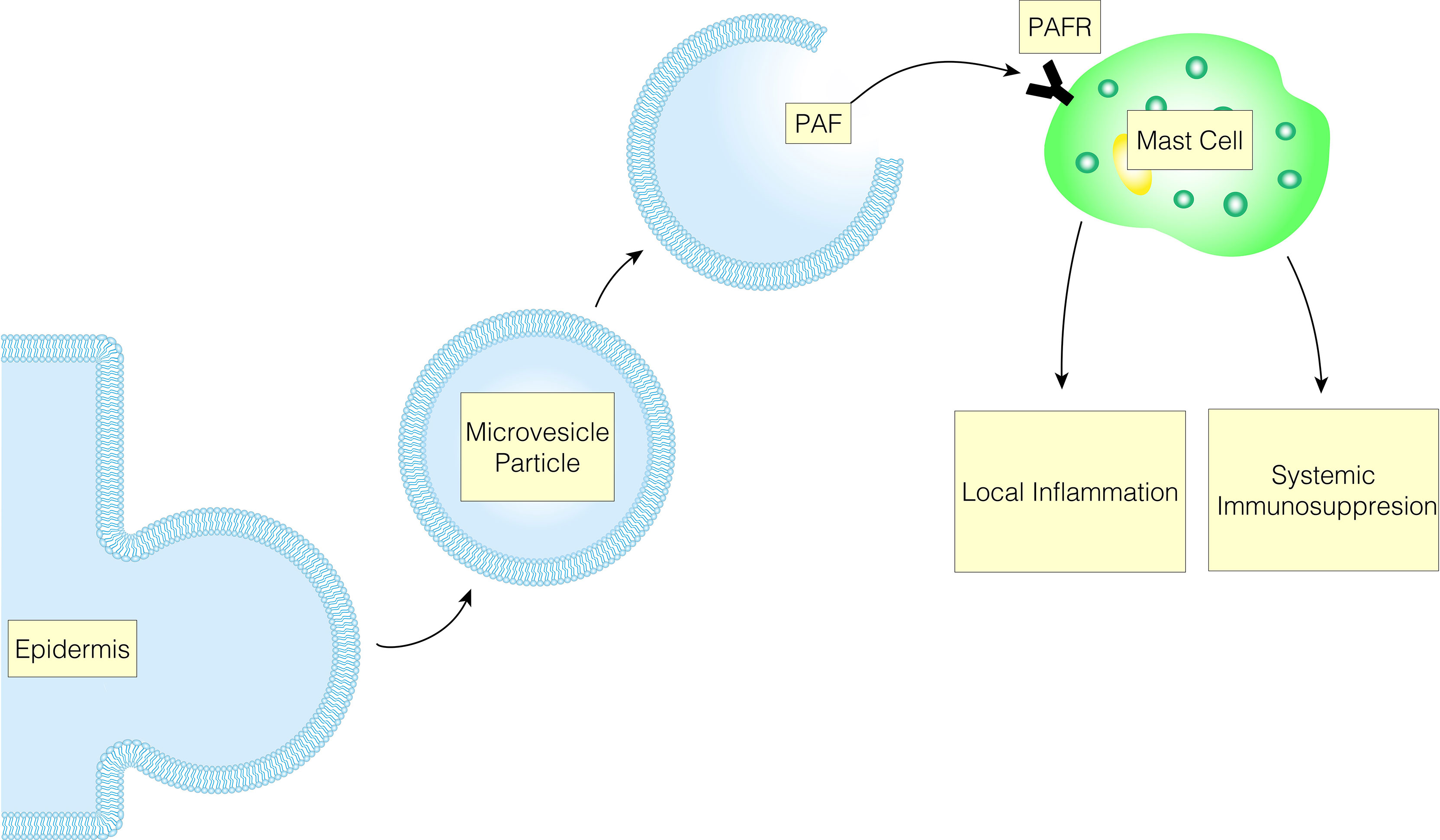
Figure 2 UVB oxidized-GPC results in PAFR activation, causing MVP to be released from the keratinocyte plasma membrane, allowing PAFR agonist to be transported to additional locations.
As mentioned previously, a common lipid pathway causing MVP release is the stimulus-mediated translocation of the enzyme aSMase from lysosomes to plasma membranes (38, 42, 104). Importantly, activation of PAFR has been shown to generate membrane translocation of aSMase as well as boost its enzymatic activity (105, 106). Multiple lines of evidence support aSMase serving as the effector for PAFR-mediated MVP release. For instance, our group found that both application of the metabolically stable PAFR agonist carbamyl-PAF (CPAF) as well as UVB exposure led to increased aSMase enzymatic activity in HaCaT keratinocytes (37). In addition, after UVB irradiation, application of the aSMase inhibitor imipramine inhibited MVP generation in HaCaT keratinocyte cells, human skin explants, and murine skin. Of note, multiple agents including many tricyclic anti-depressants act as functional inhibitors of aSMase (FIASM) (107).These findings were further substantiated by the use of PAFR-KO (Ptafr-/-) and aSMase-KO (Smpd1-/-) mice, which corroborated functions of PAFR and aSMase in UVB-mediated MVP release in skin (37). Of note, application of the phorbol ester phorbol-12-myristate -13-acetate (TPA) results in MVP generation in PAFR-deficient but not in aSMase-deficient hosts. Finally, topical treatment with the aSMase product C2 ceramide also resulted in amplified MVP levels in mice, including aSMase-deficient animals. Ultimately, UVB-induced production of MVPs relies on a process that involves both PAFR signaling and aSMase.
There are many stressors and pathways that can generate MVP. However, our group was the first to demonstrate the ability of UVB to induce PAF-mediated MVP formation and release (35). Using a keratinocyte-derived cell line (HaCaT), UVB irradiation resulted in increased levels of MVP generation. Furthermore, application of the non-metabolizable PAFR agonist CPAF resulted in MVP formation in a dose-dependent manner. In addition, to test if the PAF system was involved in UVB-mediated MVP generation, we exposed UVB to PAFR-positive and PAFR-negative cell lines (KBP and KBM, respectively). UVB-irradiation generated MVP formation in only the PAFR-positive cell line, while no differences in MVP levels were seen in the PAFR-negative cells. Similar findings were noted using PAFR-deficient mice. These findings demonstrate PAF’s involvement in UVB-induced MVP formation, which adds to the extensive literature supporting the role of the PAF system in mediated UVB-induced acute inflammation and delayed immunosuppression (19, 27, 66). It is also known that UVB-induced PAF agonists involve ROS that can be blocked by antioxidants (18, 27, 66). Our group found that UVB-mediated MVP generation involves ROS-induced PAF agonists, as evidenced by the ability of antioxidants to block MVP production (35). More specifically, pretreatment of HaCaT cells with N-acetylcysteine and vitamin C resulted in hampered UVB-induced MVP release while CPAF-mediated MVP release was unaffected. Furthermore, the UVB fluences required to produce MVP in keratinocytes are quite high, with a two- to three-fold increase in the minimum erythema dosage required to detect quantifiable MVP within the skin (108). While addressing this phenomenon, our group demonstrated that pretreatment of epithelial skin cells with PAFR agonist can synergize with low fluences of UVB to produce high levels of MVP. This suggests that MVP could play a role in combinatorial pathologic processes involving UVB, which is important since synergistic responses from multiple agents are likely more common than presently recognized (108).
PAF persists in the cellular membrane after production where it can operate on itself or nearby cells via juxtacrine signaling (32, 33). Additionally, studies have shown that in certain cell types, such as keratinocytes, PAF can exert its effects some distance away from the host cell (34). This phenomenon is likely the result of MVPs, which are thought to provide PAF protection from degradation by PAF-AH as opposed to being free or protein-bound within tissue fluids (42). By extension, the advantage of this arrangement is the protection of metabolically unstable compounds from enzymatic degradation. Thus, UVB-induced systemic effects may be mediated in part by MVPs produced by epithelial cells.
MVP-Mediated Local Inflammation
UVB damage causes both local and systemic inflammation and immunosuppression. While considering the cutaneous response to environmental stressors, there are a number of cytokines and cells signaling pathways that are mediated through T-cells, antigen-presenting cells (APCs), mast cells, fibroblasts, and keratinocytes. Consequently, the release of many cytokines including TNF-alpha, IL-6, IL-8, IL-10, and others leading to a range of pathological processes (11, 109). The aforementioned effector cells and cytokines create a complex interaction that initially generates an acute and local reaction that may eventually progress to a systemic response. It appears that this UVB-induced systemic response is in part mediated by MVPs, which act on these effector cells and amplify the UVB-damage. The end result is an MVP-mediated inflammation and immunosuppression that may give rise to a number of pathological processes.
As aforementioned, UVB produces ox-GPC PAFR agonists, which then activate PAFR in PAFR-positive keratinocytes. More PAF is created enzymatically as a result of PAFR activation along with more ROS and hence more ox-GPC. Moreover, its activation also causes aSMase to translocate, leading to MVP release. The epidermis then releases MVP, which contains PAF and ox-GPC (42). Locally, a variety of downstream effects are then generated as previously mentioned (11, 109). Recently, MVP release has been found to be associated with the production of the pro-inflammatory cytokine IL-8. Studies done by Bhadri and colleagues explored the possible link between the pro-inflammatory cytokine IL-8 and MVP. The authors demonstrated that pretreatment of HaCaT keratinocytes with PAFR agonists synergizes with low fluences of UBV to produce increased levels of IL-8 and MVPs (110). Further illustrating this linkage, application of the FIASM imipramine blocked both MVP and IL-8 release following UVB treatment (110). Furthermore, the ability of IL-8 to activate and attract neutrophils suggests that it may play a role in the acute UVB response. This is highlighted by the report that treatment with an IL-8 neutralizing antibody, which reduced UVB-induced synthesis of fibroblast neprilysin and matrix metalloproteinase 1 (MMP-1) in keratinocyte-fibroblast cocultures (94). Interestingly, the role of PAF in local immunosuppression is still under debate due to Sahu and colleagues showing that UVB-mediated LC depletion is not mediated though PAFR (111). Rather, UVB-exposed mouse skin treated with the contact allergen DNFB (2,4-dinitro-1-fluorobenzene) resulted in a significant inhibition of contact hypersensitive response in both WT and PAFR knockout mice. Additionally, the presence of LC was reduced in both types of mice compared to their sham irradiated controls (111). Thus, it appears that MVP-mediated PAF release caused by UVB needs further research to establish a causal role in the local immune response.
The immunological responses to UVB are characterized by both local and systemic inflammation and immunosuppression. These phenomena are not mutually exclusive as many acute mediators serve to bridge the local and systemic responses. The connection between these two systems is illustrated by the effect of PAF on keratinocytes with the mast cell playing an effector role (Figure 3). UVB-induced damage mediated through PAF on keratinocytes decreases DNA repair mechanisms by decreasing the expression of response elements such as ataxia telangiectasia and rad3 related protein (ATR) (112). Additionally, the secretion of PAF by keratinocytes increases IL-10, activates COX-2 and decreases delayed type hypersensitivity reactions (102, 113). Furthermore, PAF release from keratinocytes promotes mast cell migration into the lymph nodes and subsequent systemic effects. Moreover, PAF stimulation of mast cells upregulates the epigenome to increase the expression of DNMT1/3b and p300, and decreases the expression of HDAC2. Ultimately, this increases the responsiveness of mast cells to CXCR4 agonist, in which mast cells will release IL-10 and histamine (112, 114, 115). Further, this relationship is supported by UV radiation not suppressing the contact hypersensitivity (CHS) response in mast-cell deficient mice, but when these mice are reconstituted with normal bone marrow-derived mast cells, UV-induced suppression of CHS is restored (115).
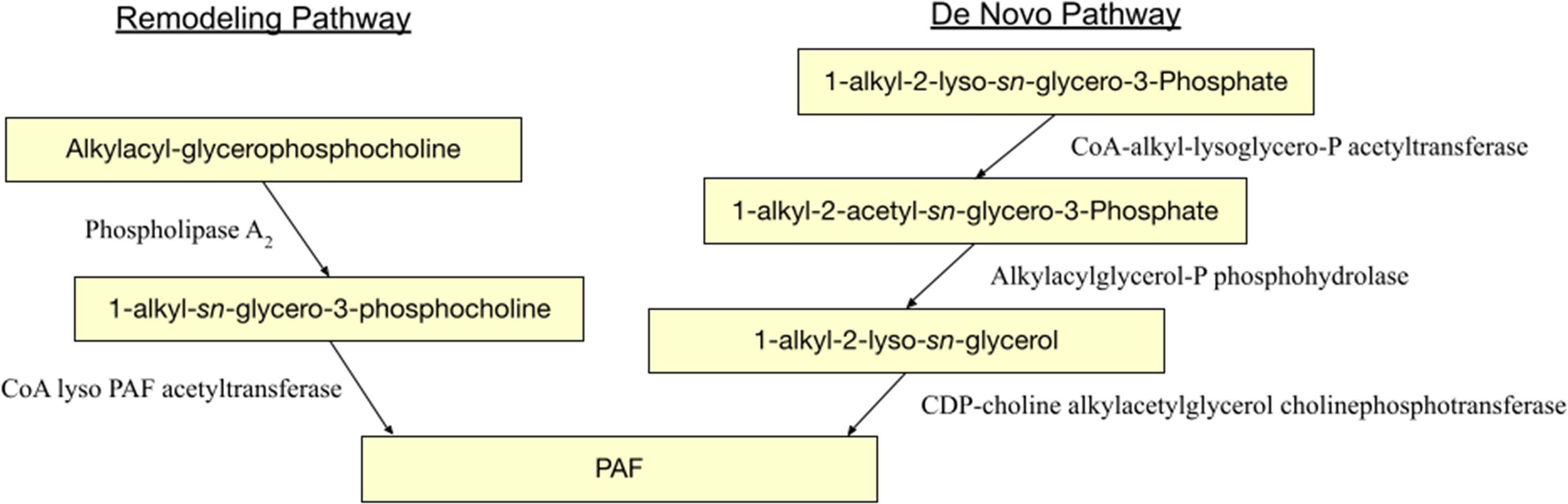
Figure 3 UVB-induced MVP release leads to mast cell PAFR activation, which results in immunological responses characterized by both local and systemic inflammation and immunosuppression.
The role of LC UV-induced PAF-mediated local inflammation is still being established, but there appears to be a relationship between PAF-induced LC migration, consequently leading to immunosuppression (116). In particular, UV irradiation induces expression of RANK-L in keratinocytes which stimulates the migration of LC to the lymph nodes where they activate immunological Treg cells, decreasing the DTH, CHS, and tumor immunity (117–120). PAF-induced PGE2 may be implicated in the process by upregulation of RANK-L on keratinocytes since inhibiting PGE2 binding to its receptor EP4 with specific receptor antagonists inhibits RANK-L upregulation, impairs UV-induced Treg activation, and inhibits UV-induced CHS suppression (121). PAF is also known to induce regulatory dendritic cells through several cytokines including PGE2 and IL-10 (122). These examples help illustrate the growing picture linking UV exposure, MVP-mediated PAF release, and subsequent local and systemic immunosuppression.
The relationship between UVB-mediated MVP release in systemic cutaneous disease is still evolving, but current literature has shown the PAF system induces inflammation in a variety of skin pathologies. For example, PAF can be found to be increased in inflammatory lesions of psoriasis favoring Th17 development. More specifically, this appears to occur through PAF-mediated increase in the expression of IL-1ß, IL-5, and IL-23 in LC and keratinocyte, subsequently leading to Th17 development (123–125). Other illustrations of the detection of the PAF in cutaneous disease states are in bullous pemphigoid, sunburn, cold urticaria and in burn injuries (66, 126–128). However, it is unclear in these pathogenic states if the PAF measured is truly contributory or serves as a “bystander effect” of the concomitant inflammation. In sum, there is a need to use specific pharmacologic and genetic tools to expand on this body of knowledge, particularly MVP’s role in UVB-induced cutaneous inflammation, immunosuppression, and systemic cutaneous disease.
Delayed Immunosuppression and Blocking of Tumor Immunity
Tumorigenesis/Metastasis in Melanoma
Exposure to UVB radiation is a major risk factor in developing skin cancer, especially melanoma. UVB induces oxidative stress and generates lipid mediators such as PAF and Ox-GPC (129). These factors signal inflammatory cytokines that lead to systemic immunosuppression, which leads to tumor augmentation in experimental murine models by two-fold (130). Not only are MVPs released by UVB-induced PAFs, Lima et al. demonstrated that malignant melanoma cells (B16F10) produce a large quantity of microvesicles in vitro that mediate immunosuppression and tumor progression (131). TGF-beta is an important immunoregulatory factor produced by phagocytes; B16F10-produced MVP upregulates this production, leading to changes in the tumor microenvironment promoting tumor dissemination (131). Moreover, it is noteworthy that antioxidants attenuate the effect of UVB-irradiation induced melanoma growth, suggesting that reactive oxygen species (ROS) leading to PAF release could play an important role in tumor growth. Likewise, UVB radiation activates tumorigenesis through the PAF-PAFR pathway. While PAFR are not found in melanocytes, the release of PAFR agonist due to environmental stressors, such as UVB, activates a cascade of downstream effects that leads to tumorigenesis and metastasis. Overexpression of PAFR has been shown to lead to acanthosis and increased numbers of dermal melanocytes, fibroblasts, mast cells, filaggrin and endothelial cells (132, 133). Interestingly, PAFR transgenic mice spontaneously developed hyperpigmentation on the ears, tails, and external genital area and melanocytic tumors in the dermis with age (132). It is likely that keratinocyte hyperplasia stimulated this growth of dermal melanocytes directly or indirectly. Of note, topical use of PAFR antagonist, WEB 2086, inhibited keratinocyte proliferation in both transgenic mice and control mice, suggesting that PAFR inhibition may mediate cellular hyperplasia and tumor growth (132). However, the phenotype of the PAFR transgenic mouse could likely be due to inappropriate expression of the PAFR using a widespread actin promoter. Sahu and colleagues found the mechanism which leads to melanoma tumor progression is due to the systemic expression and activation of PAFR; and that whether the tumor cells themselves expressed PAFR or not does not impact the tumor development, suggesting that PAFR-mediated anti-tumoral immunity effects are involved (130). Support for this finding relies on several mechanisms. One, PAF-induced release of inflammatory cytokines, such as IL-10, Treg, IL-1, and TNF-alpha lead to systemic immunosuppression and tumor metastasis (130, 134). Two, UVB-induced immunosuppression can be blocked by PAFR-antagonists, therefore attenuating the effect on tumorigenesis (135). Three, administration of a systemic PAFR agonist, carbamoyl PAF (CPAF), will increase tumor growth in wildtype mice or mice injected with PAFR onto host cells. However, CPAF failed to augment the tumor size in PAFR -/- mice (130). Lastly, PAFR acetylhydrolase (PAFR-AH), an enzyme that breaks down PAF, will decrease tumor vascularization and growth (133). These observations suggest that systemic PAF/PAFR expression and activation contribute to carcinogenesis, and inhibition of these reactions may provide insight to cancer treatment.
Melanoma is the most lethal skin cancer due to its high likelihood to metastasize and ability to evade therapy (136). The PAF-PAFR pathway has been implicated for the promotion of metastasis in melanoma (134, 137, 138). An inflammatory tumor microenvironment leads to proliferation, invasion, and angiogenesis of tumors which may metastasize. Matrix metalloproteinase-2 (MMP-2) is a main contributor to cancer cell migration, which can be stimulated by PAFR (139). Melnikova and their group studied melanoma metastasis to the lung and found that these cell lines all expressed PAFR to a varying degree, although the extent of metastasis does not correspond to the level of PAFR expressed (5). PAF upregulates multiple pathways that lead to cell migration and metastasis (137, 138). Importantly, PAF-induced expression of pro-MMP-2 was related to PAF-induced cAMP response element (CREB) and activating transcription factor (ATF-1) phosphorylation, and addition of a PAFR antagonist prevented this stimulation (137). It is thought that PAFR-induced CREB phosphorylation in more aggressive melanoma is through two intermediate signaling proteins, PKA and p38 MAPK. In addition, incubation of melanoma cell lines with the PAF analog, CPAF, increased phosphorylation of CREB (138). Another study looking at lung metastasis relating to melanoma also found that PAF enhances metastasis (134). In Im and colleagues’ experiments, pulmonary metastasis from B16F10 melanoma were significantly increased with addition of dose- and time-dependent PAF. Furthermore, administration of PAF-R antagonists reduced the pulmonary tumor colonization (134). Additionally, IL-1 and TNF-alpha were thought to be at least partially mediated by PAF generation, which can cause metastasis (134). Another important mechanism for metastasis is the cooperation between PAR-1 and PAFR to regulate the expression of MCAM/MUC18 (melanoma cellular adhesion molecules) (138). Taken together, metastatic melanoma carries higher expression of PAF/PAFR and increases migration through mechanisms modulated by the inflammatory tumor microenvironment.
Angiogenesis is an important step in tumor metastasis. Platelets and factors in the coagulation cascade, especially thrombin, are stimulated by PAFR and play crucial roles in vascularization that leads to tumor growth and metastasis (137, 138). Notably, PAF/PAFR modulate tumor cell adhesion to endothelial cells, angiogenesis, tumor growth, and metastasis (137). Inhibition of neoangiogenesis by inhibiting PAF could play a part in mediating metastasis. Indeed, mice bearing B16F10/PAF-AH tumors survived significantly longer than those bearing B16F10/neo tumors (133). Thus, it is clear that the PAF/PAFR systemic activation is implicated in tumorigenesis, which further highlights the need to understand how UVB-induced MVPs mediate this process. Finally, metastatic melanoma has especially poor prognosis due to failure of response to chemotherapeutics and radiation therapy. Therefore, understanding mechanisms for metastasis and tumor survival will provide insight to better treatment in melanoma.
Chemotherapeutics
Environmental stressors, such as UVB radiation and cigarette smoke, could help tumors such as melanomas escape antitumor immunity. Similarly, chemotherapy and radiation therapy increase PAFR expression in tumors as a protective response through generating reactive oxygen species (ROS). ROS activates host-immunity and increases glycerophosphocholine (GPC) species that can act as PAFR agonists (129, 130, 140). The release of PAF subsequently augments cytokine production through the NF-kB pathway, assisting the evasion of chemotherapy. The NF-κB pathway leads to the loss of E-cadherin, contributing to melanoma invasion and resistance to apoptosis (136). Another study showed that cancer therapy activated PAF-R systems augment the cytokine production, specifically IL-8 and TNF-alpha through the NF-κB pathway (141). Cyclooxygenase-2 (COX-2) is a major promoter in immune suppression in melanoma, through its ability to stimulate angiogenesis, inhibit apoptosis, increase cellular proliferation and increase cellular invasiveness, enhance immunosuppression, and produce mutagens (136). PAF is deducted to be a component of oxidative stress and these downstream pathways, and manipulation of PAF/PAFR gene expression, provides an understanding of tumor cell reactions to ROS and chemotherapy.
Cell repopulation is the phenomenon where tumor cells that survive chemotherapy or radiation therapy will undergo accelerated proliferation (142). Thus, finding the mediator that allows tumor cell survival will improve cancer treatment outcomes. Treatment with cisplatin increased PAFR gene expression and accumulation of its products in SKmel37 (melanoma cells) in vitro that express PAFR (143). Blocking the release of PAF induced by chemotherapy or the PAF systemic effect may enhance cancer treatment. Indeed, Onuchic et al. found a combination therapy with cisplatin and WEB2086, a PAFR-antagonist, in melanoma-bearing mice showed slowing tumor progression and increasing tumor regression (143). It is conceivable that PAF release protects tumor cells. Another example in the literature was done by Sahu et al. with the focus on ROS and COX-2 inhibition. Both chemotherapy and radiation therapy induce the release of PAF and PAF-like ox-GPC due to oxidative stress and generation of ROS. In a murine model of melanoma, addition of antioxidant regimen reduced the formation of PAFR agonist activity, therefore preventing the augmentation of tumor growth (130). Moreover, COX-2 inhibitors contribute to antitumor immunity by downregulating IL-10 and Tregs that modulate the systemic effect of PAFR-mediated tumor growth (130). Inhibiting effects of PAFR will block immunosuppression and photocarcinogenesis as well as accelerating DNA repair. Therefore, PAF induced immunosuppression may promote survival response in tumor cells (144). Finally, PAF antagonists may work synergistically with other agents that reduce immune suppression, such as 5-HT2A receptor antagonists. Evidence shows that combination of PAF and 5-HT2A receptor antagonists blocks skin cancer induction in UV-irradiated mice (145). These antagonists first prevent UV-induced damage in the skin from apoptosis after a single exposure to UV radiation, then prevent cytokine release and immune suppression (145). In conclusion, these results provide compelling evidence that potentially future chemotherapeutics could be created by manipulating the PAF/PAFR-mediated responses.
Prosurvival Response in Tumor Cells - Anti-Apoptosis
Most chemotherapeutic agents exert their therapeutic effects through programmed cell death. It is understood that some cancer treatment failures can be attributed to tumor cells’ prosurvival response mechanisms that escape the apoptotic activities induced by chemotherapy. Chemotherapy and radiation therapy cause oxidative stress and induce PAF/PAF-like molecules release. PAFR-activated modification of cell microenvironment and upregulation of anti-apoptotic molecules favor tumor growth (146, 147). Specifically, PAF increases the expression of mRNA and protein synthesis of anti-apoptotic factors and inhibits caspase activities (147). Tumor-derived microvesicle particles are another source of anti-apoptosis endorsed by cancer cells to evade cell death (148). Chemotherapeutic agents induce cell death by increasing mitochondrial enzyme caspases. De Olivera and colleagues found that combination of dacarbazine (DTIC) and WEB 2170 (PAFR antagonist) significantly improves survival of B16F10 melanoma-bearing mice compared to either agent used alone (146). Using a PAFR antagonist alone, however, did substantially delay tumor growth (146). PAF molecules are found to inhibit the activities of caspase-3, caspase-8, and caspase-9, as well as cell death induced by etoposide, a common chemotherapy agent (147). These findings suggest that tumor cells release PAF/PAF-like molecules to prolong tumor survival and escape treatment desired cell death.
While abundant reports provide evidence that PAF leads to anti-apoptotic activities, some have found that the opposite could also happen. PAF may produce pro-apoptotic response to chemotherapy as well as anti-apoptotic response depending on which agent is being used (149). This finding complicates our understanding of the effects PAF has on chemotherapeutics and survival of tumor cells. The differences in PAF effect on chemotherapy may be due to varying pathways that PAF can induce. Depending on how the cancer medication works and what it targets, PAF may augment or attenuate the efficacy of chemotherapeutics. PAFR, for example, can enhance apoptosis induced by etoposide and mitomycin C, but not by other agents such as C2 ceramide or tumor necrosis factor related apoptosis-induced ligand (TRAIL) (149).. In sum, further research is needed to study the specifics of how PAF/PAFR affects cancer treatment and tumor cell survival response.
Mechanistic Insight Through Pharmacological Interventions
The downstream effects mediated by UVB-induced MVP release have been implicated in both local and systemic pathological processes, including inflammatory responses, immunosuppression, and blocking of tumor immunity. As outlined, the mechanism involves UVB-generated ox-PAFR agonists and subsequent MVP generation, which then transports several bioactive molecules such as PAF both locally and systemically. There are a number of pharmacological agents that may modulate this pathway including aSMase inhibitors, antioxidants, COX-2 inhibitors, and PAFR modulators (agonists and antagonists). These agents may serve as potential pharmaceutical strategies to mitigate the pathological consequences seen from UVB exposure.
aSMase Inhibitors
MVP and ceramide release are dependent upon the lipid enzyme aSMase (38, 150). Thus, as evidenced by multiple lines of evidence, aSMase inhibitors pharmacologically inhibit the effects of MVPs and PAF. The literature demonstrating the utility of aSMase inhibitors within the skin is limited. However, there are many models supporting their value in other tissues and organ systems. For example, the mechanisms driving formation of hepatic steatosis are due in part to the activation of aSMase and the production of ceramide in response to ethanol consumption. Liangpunsakul and colleagues found that aSMase inhibitors, such as imipramine, may serve as a therapeutic target for alcohol-induced hepatic steatosis by inhibiting the release of ceramides by aSMase (151). In addition, transfusion-related acute lung injury (TRALI) is mediated by ceramide-mediated endothelial barrier dysfunction, in which extracellular vesicles (EVs) may be required for transport from platelets to endothelial cells. McVey and colleagues reported that blockage of aSMase reduced the formation of EVs and may present as a promising strategy for TRALI prevention (150). By increasing vascular permeability, PAF is a known mediator of pulmonary edema in acute lung injury. This is due in part through the activation of aSMase (152). Yang and collaborators found that pharmacological inhibition of the aSMase pathway blocked the PAF-induced increase in caveolin-1 and endothelial nitric oxide synthase, suggesting aSMase inhibitors as a novel mechanism to regulate vascular permeability (152). Importantly, Chauhan and colleagues explored the role of PAFR signaling in MVP release and the underlying mechanisms using non-small cell lung cancer cell lines. They found that aSMase inhibition significantly blocked MVP release, highlighting the utility of modulating the aSMase and PAFR pathways for targeted therapies in lung cancer cells (153). Lastly, the Sahu research group determined the significance of PAFR in chemotherapy-mediated MVP generation in human pancreatic cancer cells and found the inhibition of aSMase blocked the generation of MVP (154). The aforementioned studies demonstrate the utility of aSMase inhibition in prevention of various pathologies within targeted tissues.
While sparse, there is some literature highlighting the usefulness of aSMase inhibitors within the dermis. Nakatsuji and collaborators found that C.acnes Christie, Atkins, Munch-Peterson (CAMP) factor may hijack host aSMase to increase bacterial virulence in order to impair and invade host cells (155). Similarly, Ma and colleagues demonstrated how S.aureus α-toxin activates aSMase in macrophages and precipitates the release of ceramides, which activate the inflammasome and mediate the generation and release of cytokines (156). Of note, a-toxin has been reported to be a potent stimulus for enzymatic PAF production (151). These findings suggest that inhibition of aSMase may block cutaneous microbial-driven pathogenesis. Furthermore, UV irradiation is known to impart a variety of negative pathological consequences within the skin. Appelqvist and colleagues explored the initial signaling during UV-induced damage in human keratinocytes by investigating apoptosis induction and lysosomal exocytosis. The authors found that the addition of anti-aSMase reduced the activation of caspase-8, which plays a central role in the execution-phase of cellular apoptosis (157). Likewise, UV irradiation stimulates the generation of ceramide through the de novo synthesis and hydrolysis of sphingomyelin. Kim and collaborators found that UV-induced intracellular ceramide may activate matrix metalloproteinase-1 (MMP-1) expression in dermal fibroblasts via JAK1/STAT-1 pathway (158). The findings from these two reports suggest that the targeted modulation of ceramide signaling and aSMase may offer a novel therapeutic approach to allay the risk of UV-mediated cutaneous pathology. Similarly to aSMase inhibitors, antioxidants have shown utility in the prevention of MVP release.
Antioxidants and PAFR Antagonists
The PAF/PAFR signaling pathway has been shown to be exploited by UVB-induced oxidation and PAFR-agonists. This allows for the therapeutic potential of pharmacological agents including antioxidants and PAFR antagonists. Antioxidants may diminish the effects of UVB-induced ROS, which generate PAF agonists and trigger MVP release (34). There are multiple lines of evidence exhibiting the ability of antioxidants to block UVB-mediated PAFR agonist formation and UVB-MVP release (27, 34, 35, 66, 130). Moreover, the antioxidants N-acetyl cysteine, 1,1,3,3-tetramethyl-2-thiourea, and vitamins C and E completely suppressed PAF synthesis in cultured keratinocytes and human skin (159, 160). Furthermore, Sahu and colleagues demonstrated that pretreatment with antioxidants could block PAFR-dependent tumor growth (129). Antioxidants also restricted gefitinib and erlotinib ROS generation and subsequent PAFR activation, which highlights how modulation of PAFR signaling can modify the cellular responses of targeted cancer therapies (153). Lastly, pretreatment with PAFR antagonists and the antioxidant vitamin E resulted in inhibition of UVB-induced TNF-alpha production, suggesting that epidermal PAF-R may be a pharmacological target for UVB in skin (19).
As aforementioned, PAF-R antagonist intervention has the potential to reduce the negative effects of PAFR agonist signaling. Early in vitro literature showed pretreatment of PAFR positive keratinocytes with PAFR antagonists Web 2086 and A-85783 reduced UVB-induced TNF-alpha production. Zhang and collaborators provided additional support of the role of PAFR antagonist in the skin, demonstrating that sebaceous glands express PAFRs and PAFR antagonism can suppress COX-2 production in sebaceous glands (161). Interestingly, PAFR activity has been inhibited by pharmacological inhibitors of protein kinase C (PKC), implying that PKC mediates some PAFR actions (162). Inasmuch the subsequent release of MVP particles leads to immunomodulatory effects, the role of PAFR antagonists have also been shown to alter systemic immune effects (163). Additionally, PAFR antagonists have reduced experimental tumor repopulation both in vitro and in vivo (142, 164). These studies further illustrate the utility of PAFR antagonists as a pharmacological agent aimed at modulating the PAF/PAFR system and subsequent MVP release. Unfortunately, selective PAFR antagonists are not commercially available for clinical application for the targeting of MVP’s.
Selective COX-2 Inhibitors
Nonsteroidal anti-inflammatory drugs (NSAIDs) are commercially accessible drugs utilized across the globe for anti-inflammatory, antipyretic, and analgesic use. Through inhibition of cyclooxygenase (COX) enzymes in the arachidonic acid metabolism pathway, these pharmaceutical agents decrease prostaglandin and thromboxane levels. Within this group of medications, COX-1 and COX-2 isoforms are inhibited to various degrees of selection by pharmaceutical agents. Of interest, our lab has shown the activation of epidermal PAFR leads to increased PAF, eicosanoids, COX-2, its product PGE2, arachidonic acid, IL-6, and IL-8. Furthermore, our lab’s findings with HaCaT and KB keratinocytes demonstrate that PAFR is sufficient to induce epidermal COX-2 production (165) Additionally, ultraviolet radiation, along with CPAF induced COX-2 activation, and subsequent PGE2 and IL-10 production, has been found to be inhibited by PAFR and COX-2 antagonists, further highlighting the linkage between PAF and its downstream effects with COX-2 (10, 17).
Due to the increased levels of COX-2 and prostaglandins in cutaneous malignancies, pharmacological intervention with COX-2 inhibitors for skin cancer has been examined in clinical trials and systematic reviews. In 2010, a double-blinded placebo-controlled trial on patients with pre-existing actinic keratoses found fewer incidences of squamous cell carcinomas and basal cell carcinomas in patients taking 200 mg of celecoxib twice daily (161). It has been hypothesized that these changes are linked in part to the impact of COX-2 inhibitors on UVB-based systemic immune suppression (42, 166). In particular with melanomas, COX-2 has been implicated as an important pro-immunosuppressive agent. COX-2 also enhances tumor-induced melanoma angiogenesis by increasing vascular endothelial growth factor (VEGF) through phosphoinositide 3-kinase (PI3K)/protein kinase C (PKC) mechanisms. Furthermore, there is emerging evidence correlating COX-2 with Breslow thickness and metastasis, indicating COX-2 roles in prognostic outcomes (136). One mechanism of interest is the role of COX-2 inhibitors on Treg cell depletion in PAFR pathways thus inhibiting tumor growth (42, 129). The woven nature of COX-2 and melanoma implicates roles for COX-2 inhibitors possibly impacting downstream effects of MVPs. Nonetheless, selective COX-2 inhibitors have associated cardiovascular risks which must be taken into account when considering a broad application in skin cancer (167, 168). In conclusion, targeted therapy of COX-2 offers a prospective mechanism to inhibit PAFR activation and MVP release.
Conclusion
In conclusion, accumulating evidence has implicated the PAF family of mediators in UV cutaneous responses. Recent studies have suggested that their ability to generate and travel in MVP could provide a mechanism by which a highly potent yet metabolically labile family of lipids may leave the epidermis and thus impact the host. Moreover, it is likely that other agents can utilize subcellular particles such as MVP or exosomes to travel from the skin. These new areas of study have tremendous therapeutic implications, especially given the ability of aSMase inhibitors including FIASM agents such as imipramine and other FIASMs to potently block this pathway. The use of these pharmacologic tools can provide important insights into the roles of this pathway in UV as well as other environmental cutaneous insults.
Author Contributions
TF, MG, GB, TW, and TN contributed equally to this work and wrote the first draft; JT and CR helped conceptualize the work and edited and provided further input. All authors contributed to the article and approved the submitted version.
Funding
This research was supported in part by grants from the National Institutes of Health R01 HL062996 (JT), R01 ES031087 (JT and CR) and Veteran’s Administration Merit Award 5I01BX000853 (JT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
UVB, Ultraviolet B; MVP, microvesicle particles; PAF, platelet-activating factor; PAFR, Platelet-activating factor receptor; GPC, glycerophosphocholine; PAF-AH, PAF acetylhydrolase; aSMase, acid sphingomyelinase.
References
1. Leitenberger J, Jacobe HT, Cruz PD Jr. Photoimmunology–illuminating the Immune System Through Photobiology. Semin Immunopathol (2007) 29(1):65–70. doi: 10.1007/s00281-007-0063-6
2. Murphy GM. Ultraviolet Radiation and Immunosuppression. Br J Dermatol (2009) 161(Suppl 3):90–5. doi: 10.1111/j.1365-2133.2009.09455.x
3. Soehnge H, Ouhtit A, Ananthaswamy ON. Mechanisms of Induction of Skin Cancer by UV Radiation. Front Biosci (1997) 2:d538–51. doi: 10.2741/a211
4. Narayanan DL, Saladi RN, Fox JL. Ultraviolet Radiation and Skin Cancer. Int J Dermatol (2010) 49(9):978–86. doi: 10.1111/j.1365-4632.2010.04474.x
5. Melnikova VO, Ananthaswamy HN. Cellular and Molecular Events Leading to the Development of Skin Cancer. Mutat Res (2005) 571(1-2):91–106. doi: 10.1016/j.mrfmmm.2004.11.015
6. Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M, et al. UV-Induced Skin Damage. Toxicology (2003) 189(1-2):21–39. doi: 10.1016/S0300-483X(03)00150-1
7. Nishigori C. Cellular Aspects of Photocarcinogenesis. Photochem Photobiol Sci (2006) 5(2):208–14. doi: 10.1039/B507471A
8. Krtolica A, Campisi J. Cancer and Aging: A Model for the Cancer Promoting Effects of the Aging Stroma. Int J Biochem Cell Biol (2002) 34(11):1401–14. doi: 10.1016/S1357-2725(02)00053-5
9. Campisi J. Cancer and Ageing: Rival Demons? Nat Rev Cancer (2003) 3(5):339–49. doi: 10.1038/nrc1073
10. Walterscheid JP, Ullrich SE, Nghiem DX. Platelet-Activating Factor, a Molecular Sensor for Cellular Damage, Activates Systemic Immune Suppression. J Exp Med (2002) 195(2):171–9. doi: 10.1084/jem.20011450
11. Ullrich SE. Mechanisms Underlying UV-Induced Immune Suppression. Mutat Res (2005) 571(1-2):185–205. doi: 10.1016/j.mrfmmm.2004.06.059
12. Shreedhar V, Giese T, Sung VW, Ullrich SE. A Cytokine Cascade Including Prostaglandin E2, IL-4, and IL-10 is Responsible for UV-Induced Systemic Immune Suppression. J Immunol (1998) 160(8):3783–9.
13. Kochevar IE, Taylor CR, Krutmann J. Chapter 90. In: Goldsmith LA, editor. Fundamentals of Cutaneous Photobiology and Photoimmunology, in Fitzpatrick’s Dermatology in General Medicine, 8e. New York, NY: The McGraw-Hill Companies (2012).
14. D’Orazio J, Jarrett S, Amaro-Orti A, Scott T. UV Radiation and the Skin. Int J Mol Sci (2013) 14(6):12222–48. doi: 10.3390/ijms140612222
15. Ansel JC, Luger TA, Green I. Fever and Increased Serum IL-1 Activity as a Systemic Manifestation of Acute Phototoxicity in New Zealand White Rabbits. J Invest Dermatol (1987) 89(1):32–7. doi: 10.1111/1523-1747.ep12580362
16. Damiani E, Ullrich SE. Understanding the Connection Between Platelet-Activating Factor, a UV-Induced Lipid Mediator of Inflammation, Immune Suppression and Skin Cancer. Prog Lipid Res (2016) 63:14–27. doi: 10.1016/j.plipres.2016.03.004
17. Zhang Q, Yao Y, Konger RL, Sinn AL, Cai S, Pollok KE, et al. UVB Radiation-Mediated Inhibition of Contact Hypersensitivity Reactions is Dependent on the Platelet-Activating Factor System. J Invest Dermatol (2008) 128(7):1780–7. doi: 10.1038/sj.jid.5701251
18. Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, et al. Ultraviolet B Radiation Generated Platelet-Activating Factor Receptor Agonist Formation Involves EGF-R-Mediated Reactive Oxygen Species. J Immunol (2009) 182(5):2842–8. doi: 10.4049/jimmunol.0802689
19. Dy LC, Pei Y, Travers JB. Augmentation of Ultraviolet B Radiation-Induced Tumor Necrosis Factor Production by the Epidermal Platelet-Activating Factor Receptor. J Biol Chem (1999) 274(38):26917–21. doi: 10.1074/jbc.274.38.26917
20. Travers JB, Edenberg HJ, Zhang Q, Al-Hassani M, Yi Q, Baskaran S, et al. Augmentation of UVB Radiation-Mediated Early Gene Expression by the Epidermal Platelet-Activating Factor Receptor. J Invest Dermatol (2008) 128(2):455–60. doi: 10.1038/sj.jid.5701083
21. Detopoulou P, Nomikos T, Fragopoulou E, Antonopoulou S. Association of PAF and its Metabolic Enzymes With GGT and the Fatty Liver Index in Healthy Volunteers. Curr Vasc Pharmacol (2021) 19(6):663–72. doi: 10.2174/1570161119666210628125239
22. Muñoz-Cano RM, Casas-Saucedo R, Valero Santiago A, Bobolea I, Ribó P, Mullol J. Platelet-Activating Factor (PAF) in Allergic Rhinitis: Clinical and Therapeutic Implications. J Clin Med (2019) 8(9):1338. doi: 10.3390/jcm8091338
23. Bernard JJ, Gallo RL, Krutmann J. Photoimmunology: How Ultraviolet Radiation Affects the Immune System. Nat Rev Immunol (2019) 19(11):688–701. doi: 10.1038/s41577-019-0185-9
24. Shimizu T. Lipid Mediators in Health and Disease: Enzymes and Receptors as Therapeutic Targets for the Regulation of Immunity and Inflammation. Annu Rev Pharmacol Toxicol (2009) 49:123–50. doi: 10.1146/annurev.pharmtox.011008.145616
25. Kita Y, Shindou H, Shimizu T. Cytosolic Phospholipase A(2) and Lysophospholipid Acyltransferases. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(6):838–45. doi: 10.1016/j.bbalip.2018.08.006
26. Marathe GK, Prescott SM, Zimmerman GA, McIntyre TM. Oxidized LDL Contains Inflammatory PAF-Like Phospholipids. Trends Cardiovasc Med (2001) 11(3-4):139–42. doi: 10.1016/S1050-1738(01)00100-1
27. Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, et al. Ultraviolet B Radiation Generates Platelet-Activating Factor-Like Phospholipids Underlying Cutaneous Damage. J Biol Chem (2005) 280(42):35448–57. doi: 10.1074/jbc.M503811200
28. Konger RL, Marathe GK, Yao Y, Zhang Q, Travers JB. Oxidized Glycerophosphocholines as Biologically Active Mediators for Ultraviolet Radiation-Mediated Effects. Prostaglandins Other Lipid Mediat (2008) 87(1-4):1–8. doi: 10.1016/j.prostaglandins.2008.04.002
29. Yao Y, Harrison KA, Al-Hassani M, Murphy RC, Rezania S, Konger RL, et al. Platelet-Activating Factor Receptor Agonists Mediate Xeroderma Pigmentosum A Photosensitivity. J Biol Chem (2012) 287(12):9311–21. doi: 10.1074/jbc.M111.332395
30. Ocana JA, Romer E, Sahu R, Pawelzik SC, FitzGerald GA, Kaplan MH, et al. Platelet-Activating Factor-Induced Reduction in Contact Hypersensitivity Responses Is Mediated by Mast Cells via Cyclooxygenase-2-Dependent Mechanisms. J Immunol (2018) 200(12):4004–11. doi: 10.4049/jimmunol.1701145
31. Travers JB. Platelet-Activating Factor as an Effector for Environmental Stressors. Handb Exp Pharmacol (2020) 259:185–203. doi: 10.1007/164_2019_218
32. Silva AR, de Assis EF, Caiado LF, Marathe GK, Bozza MT, McIntyre TM, et al. Monocyte Chemoattractant Protein-1 and 5-Lipoxygenase Products Recruit Leukocytes in Response to Platelet-Activating Factor-Like Lipids in Oxidized Low-Density Lipoprotein. J Immunol (2002) 168(8):4112–20. doi: 10.4049/jimmunol.168.8.4112
33. Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The Platelet-Activating Factor Signaling System and its Regulators in Syndromes of Inflammation and Thrombosis. Crit Care Med (2002) 30(5 Suppl):S294–301. doi: 10.1097/00003246-200205001-00020
34. Fahy K, Liu L, Rapp CM, Borchers C, Bihl JC, Chen Y, et al. UVB-Generated Microvesicle Particles: A Novel Pathway by Which a Skin-Specific Stimulus Could Exert Systemic Effects. Photochem Photobiol (2017) 93(4):937–42. doi: 10.1111/php.12703
35. Bihl JC, Rapp CM, Chen Y, Travers JB. UVB Generates Microvesicle Particle Release in Part Due to Platelet-Activating Factor Signaling. Photochem Photobiol (2016) 92(3):503–6. doi: 10.1111/php.12577
36. Mause SF, Weber C. Microparticles: Protagonists of a Novel Communication Network for Intercellular Information Exchange. Circ Res (2010) 107(9):1047–57. doi: 10.1161/CIRCRESAHA.110.226456
37. Liu L, Awoyemi AA, Fahy KE, Thapa P, Borchers C, Wu BY, et al. Keratinocyte-Derived Microvesicle Particles Mediate Ultraviolet B Radiation-Induced Systemic Immunosuppression. J Clin Invest (2021) 131(10):e144963. doi: 10.1172/JCI144963
38. Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, et al. Acid Sphingomyelinase Activity Triggers Microparticle Release From Glial Cells. EMBO J (2009) 28(8):1043–54. doi: 10.1038/emboj.2009.45
39. Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/Microvesicles as a Mechanism of Cell-to-Cell Communication. Kidney Int (2010) 78(9):838–48. doi: 10.1038/ki.2010.278
40. Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-Derived Microvesicles: Important and Underappreciated Mediators of Cell-to-Cell Communication. Leukemia (2006) 20(9):1487–95. doi: 10.1038/sj.leu.2404296
41. Xiao X, Ma X, Liu L, Wang J, Bi K, Liu Y, et al. Cellular Membrane Microparticles: Potential Targets of Combinational Therapy for Vascular Disease. Curr Vasc Pharmacol (2015) 13(4):449–58. doi: 10.2174/1570161112666141014145440
42. Travers JB, Rohan JG, Sahu RP. New Insights Into the Pathologic Roles of the Platelet-Activating Factor System. Front Endocrinol (Lausanne) (2021) 12:624132. doi: 10.3389/fendo.2021.624132
44. Gruber F, Kremslehner C, Eckhart L, Tschachler E. Cell Aging and Cellular Senescence in Skin Aging - Recent Advances in Fibroblast and Keratinocyte Biology. Exp Gerontol (2020) 130:110780. doi: 10.1016/j.exger.2019.110780
45. Gordon R. Skin Cancer: An Overview of Epidemiology and Risk Factors. Semin Oncol Nurs (2013) 29(3):160–9. doi: 10.1016/j.soncn.2013.06.002
46. Nguyen AV, Soulika AM. The Dynamics of the Skin’s Immune System. Int J Mol Sci (2019) 20(8):1811. doi: 10.3390/ijms20081811
47. Eckhart L, Zeeuwen P. The Skin Barrier: Epidermis vs Environment. Exp Dermatol (2018) 27(8):805–6. doi: 10.1111/exd.13731
48. Kish DD, Li X, Fairchild RL. CD8 T Cells Producing IL-17 and IFN-Gamma Initiate the Innate Immune Response Required for Responses to Antigen Skin Challenge. J Immunol (2009) 182(10):5949–59. doi: 10.4049/jimmunol.0802830
49. Friedmann PS, Strickland I, Memon AA, Johnson PM. Early Time Course of Recruitment of Immune Surveillance in Human Skin After Chemical Provocation. Clin Exp Immunol (1993) 91(3):351–6. doi:10.1111/j.1365-2249.1993.tb05908.x
50. Falcão-Pires I, Castro-Chaves P, Miranda-Silva D, Lourenço AP, Leite-Moreira AF. Physiological, Pathological and Potential Therapeutic Roles of Adipokines. Drug Discov Today (2012) 17(15-16):880–9. doi: 10.1016/j.drudis.2012.04.007
51. Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Inflammatory Lipid Mediators in Adipocyte Function and Obesity. Nat Rev Endocrinol (2010) 6(2):71–82. doi: 10.1038/nrendo.2009.264
52. Zmijewski M, Skobowiat C, Zbytek B, Slominski R, Steketee J. Sensing the Environment: Regulation of Local and Global Homeostasis by the Skin Neuroendocrine System. Adv Anatomy Embryol Cell Biol (2012) 212:1–115. doi: 10.1007/978-3-642-19683-6_1
53. Berneburg M, Plettenberg H, Krutmann J. Photoaging of Human Skin. Photodermatol Photoimmunol Photomed (2000) 16(6):239–44. doi: 10.1034/j.1600-0781.2000.160601.x
54. Gläser R, Navid F, Schuller W, Jantschitsch C, Harder J, Schröder JM, et al. UV-B Radiation Induces the Expression of Antimicrobial Peptides in Human Keratinocytes In Vitro and In Vivo. J Allergy Clin Immunol (2009) 123(5):1117–23. doi: 10.1016/j.jaci.2009.01.043
55. Heck DE, Vetrano AM, Mariano TM, Laskin JD. UVB Light Stimulates Production of Reactive Oxygen Species: Unexpected Role for Catalase. J Biol Chem (2003) 278(25):22432–6. doi: 10.1074/jbc.C300048200
56. Johnson KE, Wulff BC, Oberyszyn TM, Wilgus TA. Ultraviolet Light Exposure Stimulates HMGB1 Release by Keratinocytes. Arch Dermatol Res (2013) 305(9):805–15. doi: 10.1007/s00403-013-1401-2
57. Burns EM, Ahmed H, Isedeh PN, Kohli I, Van DerPol W, Shaheen A, et al. Ultraviolet Radiation, Both UVA and UVB, Influences the Composition of the Skin Microbiome. Exp Dermatol (2019) 28(2):136–41. doi: 10.1111/exd.13854
58. Hart PH, Gorman S, Finlay-Jones JJ. Modulation of the Immune System by UV Radiation: More Than Just the Effects of Vitamin D? Nat Rev Immunol (2011) 11(9):584–96. doi: 10.1038/nri3045
59. Schwarz T. Mechanisms of UV-Induced Immunosuppression. Keio J Med (2005) 54(4):165–71. doi: 10.2302/kjm.54.165
60. Nghiem DX, Kazimi N, Clydesdale G, Ananthaswamy HN, Kripke ML, Ullrich SE. Ultraviolet a Radiation Suppresses an Established Immune Response: Implications for Sunscreen Design. J Invest Dermatol (2001) 117(5):1193–9. doi: 10.1046/j.0022-202x.2001.01503.x
61. Moyal DD, Fourtanier AM. Broad-Spectrum Sunscreens Provide Better Protection From the Suppression of the Elicitation Phase of Delayed-Type Hypersensitivity Response in Humans. J Invest Dermatol (2001) 117(5):1186–92. doi: 10.1046/j.0022-202x.2001.01545.x
62. Bikle DD. Protective Actions of Vitamin D in UVB Induced Skin Cancer. Photochem Photobiol Sci (2012) 11(12):1808–16. doi: 10.1039/c2pp25251a
63. Imokawa G. Mechanism of UVB-Induced Wrinkling of the Skin: Paracrine Cytokine Linkage Between Keratinocytes and Fibroblasts Leading to the Stimulation of Elastase. J Investig Dermatol Symp Proc (2009) 14(1):36–43. doi: 10.1038/jidsymp.2009.11
64. Slominski AT, Chaiprasongsuk A, Janjetovic Z, Kim T-K, Stefan J, Slominski RM, et al. Photoprotective Properties of Vitamin D and Lumisterol Hydroxyderivatives. Cell Biochem Biophysics (2020) 78(2):165–80. doi: 10.1007/s12013-020-00913-6
65. Slominski AT, Zmijewski MA, Plonka PM, Szaflarski JP, Paus R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology (2018) 159(5):1992–2007. doi: 10.1210/en.2017-03230
66. Travers JB, Berry D, Yao Y, Yi Q, Konger RL, Travers JB. Ultraviolet B Radiation of Human Skin Generates Platelet-Activating Factor Receptor Agonists. Photochem Photobiol (2010) 86(4):949–54. doi: 10.1111/j.1751-1097.2010.00743.x
67. Benveniste J, Henson PM, Cochrane CG. Leukocyte-Dependent Histamine Release From Rabbit Platelets. The Role of IgE, Basophils, and a Platelet-Activating Factor. J Exp Med (1972) 136(6):1356–77. doi: 10.1084/jem.136.6.1356
68. Ishii S, Nagase T, Shimizu T. Platelet-Activating Factor Receptor. Prostaglandins Other Lipid Mediators (2002) 68-69:599–609. doi: 10.1016/S0090-6980(02)00058-8
69. Gill P, Jindal NL, Jagdis A, Vadas P. Platelets in the Immune Response: Revisiting Platelet-Activating Factor in Anaphylaxis. J Allergy Clin Immunol (2015) 135(6):1424–32. doi: 10.1016/j.jaci.2015.04.019
70. Abate W, Alrammah H, Kiernan M, Tonks AJ, Jackson SK. Lysophosphatidylcholine Acyltransferase 2 (LPCAT2) Co-Localises With TLR4 and Regulates Macrophage Inflammatory Gene Expression in Response to LPS. Sci Rep (2020) 10(1):10355. doi: 10.1038/s41598-020-67000-x
71. Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-Activating Factor and Related Lipid Mediators. Annu Rev Biochem (2000) 69:419–45. doi: 10.1146/annurev.biochem.69.1.419
72. Snyder F. Platelet-Activating Factor: The Biosynthetic and Catabolic Enzymes. Biochem J (1995) 305(Pt 3):689–705. doi: 10.1042/bj3050689
73. Kono N, Arai H. Platelet-Activating Factor Acetylhydrolases: An Overview and Update. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids (2019) 1864(6):922–31. doi: 10.1016/j.bbalip.2018.07.006
74. Karasawa K, Inoue K. Chapter One - Overview of PAF-Degrading Enzymes, in The Enzymes. Inoue K, Stafforini DM, Tamanoi F, editors. Cambridge, MA: Academic Press (2015) p. 1–22.
75. Marques M, Pei Y, Southall MD, Johnston JM, Arai H, Aoki J, et al. Identification of Platelet-Activating Factor Acetylhydrolase II in Human Skin. J Invest Dermatol (2002) 119(4):913–9. doi: 10.1046/j.1523-1747.2002.01859.x
76. Pasquet JM, Dachary-Prigent J, Nurden AT. Calcium Influx is a Determining Factor of Calpain Activation and Microparticle Formation in Platelets. Eur J Biochem (1996) 239(3):647–54. doi: 10.1111/j.1432-1033.1996.0647u.x
77. Ståhl AL, Arvidsson I, Johansson KE, Chromek M, Rebetz J, Loos S, et al. A Novel Mechanism of Bacterial Toxin Transfer Within Host Blood Cell-Derived Microvesicles. PloS Pathog (2015) 11(2):e1004619. doi:10.1371/journal.ppat.1004619
78. Kahn R, Mossberg M, Ståhl AL, Johansson K, Lopatko Lindman I, Heijl C, et al. Microvesicle Transfer of Kinin B1-Receptors is a Novel Inflammatory Mechanism in Vasculitis. Kidney Int (2017) 91(1):96–105. doi: 10.1016/j.kint.2016.09.023
79. Abid Hussein MN, Böing AN, Sturk A, Hau CM, Nieuwland R. Inhibition of Microparticle Release Triggers Endothelial Cell Apoptosis and Detachment. Thromb Haemost (2007) 98(5):1096–107. doi:10.1160/th05-04-0231
80. Liu L, Fahy KE, Awoyemi AA, Thapa P, Kelly LE, Chen J, et al. Thermal Burn Injury Generates Bioactive Microvesicles: Evidence for a Novel Transport Mechanism for the Lipid Mediator Platelet-Activating Factor (PAF) That Involves Subcellular Particles and the PAF Receptor. J Immunol (2020) 205(1):193–201. doi: 10.4049/jimmunol.1901393
81. Liu L, Rapp CM, Zheng S, Travers JB. 627 UVB-Generated Microvesicle Particles Mediate Systemic Immunosuppression. J Invest Dermatol (2020) 140(7):S85. doi: 10.1016/j.jid.2020.03.638
82. Awoyemi AA, Borchers C, Liu L, Chen Y, Rapp CM, Brewer CA, et al. Acute Ethanol Exposure Stimulates Microvesicle Particle Generation in Keratinocytes. Toxicol Lett (2022) 355:100–5. doi: 10.1016/j.toxlet.2021.11.008
83. Wang J, Pothana K, Chen S, Sawant H, Travers JB, Bihl J, et al. Ultraviolet B Irradiation Alters the Level and miR Contents of Exosomes Released by Keratinocytes in Diabetic Condition. Photochem Photobiol (2021). doi: 10.1111/php.13583
84. Pitt JM, Kroemer G, Zitvogel L. Extracellular Vesicles: Masters of Intercellular Communication and Potential Clinical Interventions. J Clin Invest (2016) 126(4):1139–43. doi: 10.1172/JCI87316
85. McBride JD, Rodriguez-Menocal L, Badiavas EV. Extracellular Vesicles as Biomarkers and Therapeutics in Dermatology: A Focus on Exosomes. J Invest Dermatol (2017) 137(8):1622–9. doi: 10.1016/j.jid.2017.04.021
86. Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: Mediators of Extracellular Communication During Cancer Progression. J Cell Sci (2010) 123(Pt 10):1603–11. doi: 10.1242/jcs.064386
87. Riazifar M, Pone EJ, Lötvall J, Zhao W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu Rev Pharmacol Toxicol (2017) 57:125–54. doi: 10.1146/annurev-pharmtox-061616-030146
88. van Niel G, D’Angelo G, Raposo G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
89. Tricarico C, Clancy J, D’Souza-Schorey C. Biology and Biogenesis of Shed Microvesicles. Small GTPases (2017) 8(4):220–32. doi: 10.1080/21541248.2016.1215283
90. Leroyer AS, Rautou P-E, Silvestre J-S, Castier Y, Lesèche G, Devue C, et al. CD40 Ligand+ Microparticles From Human Atherosclerotic Plaques Stimulate Endothelial Proliferation and Angiogenesis: A Potential Mechanism for Intraplaque Neovascularization. J Am Coll Cardiol (2008) 52(16):1302–11. doi: 10.1016/j.jacc.2008.07.032
91. Shefler I, Pasmanik-Chor M, Kidron D, Mekori YA, Hershko AY. T Cell-Derived Microvesicles Induce Mast Cell Production of IL-24: Relevance to Inflammatory Skin Diseases. J Allergy Clin Immunol (2014) 133(1):217–24.e1-3. doi: 10.1016/j.jaci.2013.04.035
92. Taverna S, Ghersi G, Ginestra A, Rigogliuso S, Pecorella S, Alaimo G, et al. Shedding of Membrane Vesicles Mediates Fibroblast Growth Factor-2 Release From Cells *. J Biol Chem (2003) 278(51):51911–9. doi: 10.1074/jbc.M304192200
93. Obregon C, Rothen-Rutishauser B, Gitahi SK, Gehr P, Nicod LP. Exovesicles From Human Activated Dendritic Cells Fuse With Resting Dendritic Cells, Allowing Them to Present Alloantigens. Am J Pathol (2006) 169(6):2127–36. doi: 10.2353/ajpath.2006.060453
94. Faille D, El-Assaad F, Mitchell AJ, Alessi M-C, Chimini G, Fusai T, et al. Endocytosis and Intracellular Processing of Platelet Microparticles by Brain Endothelial Cells. J Cell Mol Med (2012) 16(8):1731–8. doi: 10.1111/j.1582-4934.2011.01434.x
95. Simon MM, Aragane Y, Schwarz A, Luger TA, Schwarz T. UVB Light Induces Nuclear Factor Kappa B (NF Kappa B) Activity Independently From Chromosomal DNA Damage in Cell-Free Cytosolic Extracts. J Invest Dermatol (1994) 102(4):422–7. doi: 10.1111/1523-1747.ep12372194
96. Barber LA, Spandau DF, Rathman SC, Murphy RC, Johnson CA, Kelley SW, et al. Expression of the Platelet-Activating Factor Receptor Results in Enhanced Ultraviolet B Radiation-Induced Apoptosis in a Human Epidermal Cell Line. J Biol Chem (1998) 273(30):18891–7. doi: 10.1074/jbc.273.30.18891
97. Chacón-Salinas R, Chen L, Chávez-Blanco AD, Limón-Flores AY, Ma Y, Ullrich SE. An Essential Role for Platelet-Activating Factor in Activating Mast Cell Migration Following Ultraviolet Irradiation. J Leukoc Biol (2014) 95(1):139–48. doi: 10.1189/jlb.0811409
98. Wardlaw AJ, Moqbel R, Cromwell O, Kay AB. Platelet-Activating Factor. A Potent Chemotactic and Chemokinetic Factor for Human Eosinophils. J Clin Invest (1986) 78(6):1701–6. doi: 10.1172/JCI112765
99. Håkansson L, Venge P. Inhibition of Neutrophil and Eosinophil Chemotactic Responses to PAF by the PAF-Antagonists WEB-2086, L-652,731, and SRI-63441. J Leukoc Biol (1990) 47(5):449–56. doi: 10.1002/jlb.47.5.449
100. Travers JB, Li Q, Kniss DA, Fertel RH. Identification of Functional Platelet-Activating Factor Receptors in Raji Lymphoblasts. J Immunol (1989) 143(11):3708–13.
101. Travers JB, Shaw JE, Li Q, Kniss DA, Fertel RH. Evidence for Platelet-Activating Factor Receptors in Several B Lymphoblastoid Cell Lines. Life Sci (1991) 49(23):1755–60. doi: 10.1016/0024-3205(91)90318-6
102. Travers JB, et al. Identification of Functional Platelet-Activating Factor Receptors on Human Keratinocytes. J Invest Dermatol (1995) 105(6):816–23. doi: 10.1111/1523-1747.ep12326581
103. Honda Z, Ishii S, Shimizu T. Platelet-Activating Factor Receptor. J Biochem (2002) 131(6):773–9. doi: 10.1093/oxfordjournals.jbchem.a003164
104. Record M, Silvente-Poirot S, Poirot M, Wakelam MJO. Extracellular Vesicles: Lipids as Key Components of Their Biogenesis and Functions. J Lipid Res (2018) 59(8):1316–24. doi: 10.1194/jlr.E086173
105. Göggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, et al. PAF-Mediated Pulmonary Edema: A New Role for Acid Sphingomyelinase and Ceramide. Nat Med (2004) 10(2):155–60. doi: 10.1038/nm977
106. Lang PA, Kempe DS, Tanneur V, Eisele K, Klarl BA, Myssina S, et al. Stimulation of Erythrocyte Ceramide Formation by Platelet-Activating Factor. J Cell Sci (2005) 118(Pt 6):1233–43. doi: 10.1242/jcs.01730
107. Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, et al. Identification of Novel Functional Inhibitors of Acid Sphingomyelinase. PloS One (2011) 6(8):e23852. doi: 10.1371/journal.pone.0023852
108. Thapa P, Bhadri S, Borchers C, Liu L, Chen Y, Rapp CM, et al. Low UVB Fluences Augment Microvesicle Particle Generation in Keratinocytes. Photochem Photobiol (2021) 98(1):248–53. doi: 10.1111/php.13495
109. Ullrich SE, Byrne SN. The Immunologic Revolution: Photoimmunology. J Invest Dermatol (2012) 132(3 Pt 2):896–905. doi: 10.1038/jid.2011.405
110. Bhadri S, Thapa P, Chen Y, Rapp CM, Travers JB. Evidence for Microvesicle Particles in UVB-Mediated IL-8 Generation in Keratinocytes. J Clin Invest Dermatol (2021) 9(2):10.13188/2373-1044.1000076. doi: 10.13188/2373-1044.1000076
111. Sahu RP, Yao Y, Konger RL, Travers JB. Platelet-Activating Factor Does Not Mediate UVB-Induced Local Immune Suppression. Photochem Photobiol (2012) 88(2):490–3. doi: 10.1111/j.1751-1097.2011.01071.x
112. Puebla-Osorio N, Damiani E, Bover L, Ullrich SE. Platelet-Activating Factor Induces Cell Cycle Arrest and Disrupts the DNA Damage Response in Mast Cells. Cell Death Dis (2015) 6(5):e1745. doi: 10.1038/cddis.2015.115
113. Vieyra-Garcia PA, Wolf P. From Early Immunomodulatory Triggers to Immunosuppressive Outcome: Therapeutic Implications of the Complex Interplay Between the Wavebands of Sunlight and the Skin. Front Med (2018) 5:232–2. doi: 10.3389/fmed.2018.00232
114. Damiani E, Puebla-Osorio N, Gorbea E, Ullrich SE. Platelet-Activating Factor Induces Epigenetic Modifications in Human Mast Cells. J Invest Dermatol (2015) 135(12):3034–40. doi: 10.1038/jid.2015.336
115. Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay-Jones JJ. Dermal Mast Cells Determine Susceptibility to Ultraviolet B-Induced Systemic Suppression of Contact Hypersensitivity Responses in Mice. J Exp Med (1998) 187(12):2045–53. doi: 10.1084/jem.187.12.2045
116. Schwarz A, Noordegraaf M, Maeda A, Torii K, Clausen BE, Schwarz T. Langerhans Cells are Required for UVR-Induced Immunosuppression. J Invest Dermatol (2010) 130(5):1419–27. doi: 10.1038/jid.2009.429
117. Fukunaga A, Khaskhely NM, Ma Y, Sreevidya CS, Taguchi K, Nishigori C, et al. Langerhans Cells Serve as Immunoregulatory Cells by Activating NKT Cells. J Immunol (2010) 185(8):4633–40. doi: 10.4049/jimmunol.1000246
118. Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE. Immune Suppression and Skin Cancer Development: Regulation by NKT Cells. Nat Immunol (2000) 1(6):521–5. doi: 10.1038/82782
119. Schwarz T. 25 Years of UV-Induced Immunosuppression Mediated by T Cells-From Disregarded T Suppressor Cells to Highly Respected Regulatory T Cells. Photochem Photobiol (2008) 84(1):10–8. doi: 10.1111/j.1751-1097.2007.00223.x
120. Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL Controls Regulatory T-Cell Numbers via Activation of Dendritic Cells. Nat Med (2006) 12(12):1372–9. doi: 10.1038/nm1518
121. Soontrapa K, Honda T, Sakata D, Yao C, Hirata T, Hori S, et al. Prostaglandin E2–Prostoglandin E Receptor Subtype 4 (EP4) Signaling Mediates UV Irradiation-Induced Systemic Immunosuppression. Proc Natl Acad Sci (2011) 108(16):6668–73. doi: 10.1073/pnas.1018625108
122. Koga MM, Bizzarro B, Sá-Nunes A, Rios FJ, Jancar S, et al. Activation of PAF-Receptor Induces Regulatory Dendritic Cells Through PGE2 and IL-10. Prostaglandins Leukot Essent Fatty Acids (2013) 89(5): 319–26. doi: 10.1016/j.plefa.2013.09.003
123. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but Not Transforming Growth Factor-Beta Are Essential for the Differentiation of Interleukin 17-Producing Human T Helper Cells. Nat Immunol (2007) 8(9):9427–9. doi: 10.1038/ni1496
124. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, Cytokine Profile and Function of Human Interleukin 17-Producing Helper T Cells. Nat Immunol (2007) 8(9):950–7. doi: 10.1038/ni1497
125. Drolet A-M, Thivierge M, Turcotte S, Hanna D, Maynard B, Stankovà J, et al. Platelet-Activating Factor Induces Th17 Cell Differentiation. Mediators Inflammation (2011) 913802. doi: 10.1155/2011/913802
126. Travers JB, Murphy RC, Johnson CA, Pei Y, Morin SM, Clay KL, et al Identification and Pharmacological Characterization of Platelet-Activating Factor and Related 1-Palmitoyl Species in Human Inflammatory Blistering Diseases. Prostaglandins Other Lipid Mediat (1998) 56(5–6):305–24. doi: 10.1016/S0090-6980(98)00060-4
127. Grandel KE, Farr RS, Wanderer AA, Eisenstadt TC, Wasserman SI. Association of Platelet-Activating Factor With Primary Acquired Cold Urticaria. N Engl J Med (1985) 313(7):405–9. doi: 10.1056/NEJM198508153130702
128. Harrison KA, Romer E, Weyerbacher J, Ocana JA, Sahu RP, Murphy RC, et al. Enhanced Platelet-Activating Factor Synthesis Facilitates Acute and Delayed Effects of Ethanol-Intoxicated Thermal Burn Injury. J Invest Dermatol (2018) 138(11):2461–9. doi: 10.1016/j.jid.2018.04.039
129. Sahu RP, Ocana JA, Harrison KA, Ferracini M, Touloukian CE, Al-Hassani M, et al. Chemotherapeutic Agents Subvert Tumor Immunity by Generating Agonists of Platelet-Activating Factor. Cancer Res (2014) 74(23):7069–78. doi: 10.1158/0008-5472.CAN-14-2043
130. Sahu RP, Turner MJ, DaSilva SC, Rashid BM, Ocana JA, Perkins SM, et al. The Environmental Stressor Ultraviolet B Radiation Inhibits Murine Antitumor Immunity Through Its Ability to Generate Platelet-Activating Factor Agonists. Carcinogenesis (2012) 33(7):1360–7. doi: 10.1093/carcin/bgs152
131. Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-Derived Microvesicles Modulate the Establishment of Metastatic Melanoma in a Phosphatidylserine-Dependent Manner. Cancer Lett (2009) 283(2):168–75. doi: 10.1016/j.canlet.2009.03.041
132. Sato S, Kume K, Ito C, Ishii S, Shimizu T. Accelerated Proliferation of Epidermal Keratinocytes by the Transgenic Expression of the Platelet-Activating Factor Receptor. Arch Dermatol Res (1999) 291(11):614–21. doi: 10.1007/s004030050463
133. Biancone L, Cantaluppi V, Del Sorbo L, Russo S, Tjoelker LW, Camussi G, et al. Platelet-activating Factor Inactivation by Local Expression of Platelet-activating Factor Acetyl-Hydrolase Modifies Tumor Vascularization and Growth. Clin Cancer Res (2003) 9(11):4214–20.
134. Im SY, Ko HM, Kim JW, Lee HK, Ha TY, Lee HB, et al. Augmentation of Tumor Metastasis by Platelet-Activating Factor. Cancer Res (1996) 56(11):2662–5.
135. Shimada A, Ota Y, Sugiyama Y, Inoue S, Sato S, Kume K, et al. In Situ Expression of Platelet-Activating Factor (PAF)-Receptor Gene in Rat Skin and Effects of PAF on Proliferation and Differentiation of Cultured Human Keratinocytes. J Invest Dermatol (1998) 110(6):889–93. doi: 10.1046/j.1523-1747.1998.00202.x
136. Tudor DV, Bâldea I, Lupu M, Kacso T, Kutasi E, Hopârtean A, et al. COX-2 as a Potential Biomarker and Therapeutic Target in Melanoma. Cancer Biol Med (2020) 17(1):20–31. doi: 10.20892/j.issn.2095-3941.2019.0339
137. Melnikova VO, Villares GJ, Bar-Eli M. Emerging Roles of PAR-1 and PAFR in Melanoma Metastasis. Cancer Microenviron (2008) 1(1):103–11. doi: 10.1007/s12307-008-0002-7
138. Braeuer RR, Zigler M, Villares GJ, Dobroff AS, Bar-Eli M. Transcriptional Control of Melanoma Metastasis: The Importance of the Tumor Microenvironment. Semin Cancer Biol (2011) 21(2):83–8. doi: 10.1016/j.semcancer.2010.12.007
139. Xu X, Wang Y, Chen Z, Sternlicht MD, Hidalgo M, Steffensen B. Matrix Metalloproteinase-2 Contributes to Cancer Cell Migration on Collagen. Cancer Res (2005) 65(1):130–6.
140. Sahu RP, Harrison KA, Weyerbacher J, Murphy RC, Konger RL, Garrett JE, et al. Radiation Therapy Generates Platelet-Activating Factor Agonists. Oncotarget (2016) 7(15):20788–800. doi: 10.18632/oncotarget.7878
141. Darst M, Al-Hassani M, Li T, Yi Q, Travers JM, Lewis DA, et al. Augmentation of Chemotherapy-Induced Cytokine Production by Expression of the Platelet-Activating Factor Receptor in a Human Epithelial Carcinoma Cell Line. J Immunol (2004) 172(10):6330–5. doi: 10.4049/jimmunol.172.10.6330
142. da Silva Junior IA, de SousaAndrade LN, Jancar S, Chammas R, et al. Platelet Activating Factor Receptor Antagonists Improve the Efficacy of Experimental Chemo- and Radiotherapy. Clinics (Sao Paulo Brazil) (2018) 73(suppl 1):e792s. doi: 10.6061/clinics/2018/e792s
143. Onuchic AC, Machado CML, Saito RF, Rios FJ, Jancar S, Chammas R. Expression of PAFR as Part of a Prosurvival Response to Chemotherapy: A Novel Target for Combination Therapy in Melanoma. Mediators Inflamm (2012) 2012:175408. doi: 10.1155/2012/175408
144. Sreevidya CS, Fukunaga A, Khaskhely NM, Masaki T, Ono R, Nishigori C, et al. Agents That Reverse UV-Induced Immune Suppression and Photocarcinogenesis Affect DNA Repair. J Invest Dermatol (2010) 130(5):1428–37. doi: 10.1038/jid.2009.329
145. Sreevidya CS, Khaskhely NM, Fukunaga A, Khaskina P, Ullrich SE. Inhibition of Photocarcinogenesis by Platelet-Activating Factor or Serotonin Receptor Antagonists. Cancer Res (2008) 68(10):3978–84. doi: 10.1158/0008-5472.CAN-07-6132
146. de Oliveira SI, Andrade LNS, Onuchic AC, Nonogaki S, Fernandes PD, Pinheiro MC, et al. Platelet-Activating Factor Receptor (PAF-R)-Dependent Pathways Control Tumour Growth and Tumour Response to Chemotherapy. BMC Cancer (2010) 10(1):200. doi: 10.1186/1471-2407-10-200
147. Heon Seo K, Ko H-M, Kim H-A, Choi J-H, Jun Park S, Kim K-J, et al. Platelet-Activating Factor Induces Up-Regulation of Antiapoptotic Factors in a Melanoma Cell Line Through Nuclear Factor-κb Activation. Cancer Res (2006) 66(9):4681–6. doi: 10.1158/0008-5472.CAN-05-3186
148. Baj-Krzyworzeka M, Szatanek R, Węglarczyk K, Baran J, Urbanowicz B, Brański P, et al. Tumour-Derived Microvesicles Carry Several Surface Determinants and mRNA of Tumour Cells and Transfer Some of These Determinants to Monocytes. Cancer Immunol Immunother (2006) 55(7):808–18. doi: 10.1007/s00262-005-0075-9
149. Li T, Southall MD, Yi Q, Pei Y, Lewis D, Al-Hassani M, et al. The Epidermal Platelet-Activating Factor Receptor Augments Chemotherapy-Induced Apoptosis in Human Carcinoma Cell Lines*. J Biol Chem (2003) 278(19):16614–21. doi: 10.1074/jbc.M211287200
150. McVey MJ, Weidenfeld S, Maishan M, Spring C, Kim M, Tabuchi A, et al. Platelet Extracellular Vesicles Mediate Transfusion-Related Acute Lung Injury by Imbalancing the Sphingolipid Rheostat. Blood (2021) 137(5):690–701. doi: 10.1182/blood.2020005985
151. Liangpunsakul S, Rahmini Y, Ross RA, Zhao Z, Xu Y, Crabb DW. Imipramine Blocks Ethanol-Induced ASMase Activation, Ceramide Generation, and PP2A Activation, and Ameliorates Hepatic Steatosis in Ethanol-Fed Mice. Am J Physiol Gastrointest Liver Physiol (2012) 302(5):G515–23. doi: 10.1152/ajpgi.00455.2011
152. Yang Y, Yin J, Baumgartner W, Samapati R, Solymosi EA, Reppien E, et al. Platelet-Activating Factor Reduces Endothelial Nitric Oxide Production: Role of Acid Sphingomyelinase. Eur Respir J (2010) 36(2):417–27. doi: 10.1183/09031936.00095609
153. Chauhan SJ, Thyagarajan A, Chen Y, Travers JB, Sahu RP. Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells. Int J Mol Sci (2020) 21(22):8517. doi: 10.3390/ijms21228517
154. Thyagarajan A, Kadam SM, Liu L, Kelly LE, Rapp CM, Chen Y, et al. Gemcitabine Induces Microvesicle Particle Release in a Platelet-Activating Factor-Receptor-Dependent Manner via Modulation of the MAPK Pathway in Pancreatic Cancer Cells. Int J Mol Sci (2018) 20(1):32. doi: 10.3390/ijms20010032
155. Nakatsuji T, Tang DC, Zhang L, Gallo RL, Huang CM. Propionibacterium Acnes CAMP Factor and Host Acid Sphingomyelinase Contribute to Bacterial Virulence: Potential Targets for Inflammatory Acne Treatment. PloS One (2011) 6(4):e14797. doi: 10.1371/journal.pone.0014797
156. Ma J, Gulbins E, Edwards MJ, Caldwell CC, Fraunholz M, Becker KA, et al. Staphylococcus Aureus α-Toxin Induces Inflammatory Cytokines via Lysosomal Acid Sphingomyelinase and Ceramides. Cell Physiol Biochem (2017) 43(6):2170–84. doi: 10.1159/000484296
157. Appelqvist H, Wäster P, Eriksson I, Rosdahl I, Ollinger K. Lysosomal Exocytosis and Caspase-8-Mediated Apoptosis in UVA-Irradiated Keratinocytes. J Cell Sci (2013) 126(Pt 24):5578–84. doi: 10.1242/jcs.130633
158. Kim S, Kim Y, Lee Y, Chung JH. Ceramide Accelerates Ultraviolet-Induced MMP-1 Expression Through JAK1/STAT-1 Pathway in Cultured Human Dermal Fibroblasts. J Lipid Res (2008) 49(12):2571–81. doi: 10.1194/jlr.M800112-JLR200
159. Calignano A, Cirino G, Meli R, Persico P. Isolation and Identification of Platelet-Activating Factor in UV-Irradiated Guinea Pig Skin. J Pharmacol Methods (1988) 19(1):89–91. doi: 10.1016/0160-5402(88)90049-6
160. Sheng Y, Birkle DL. Release of Platelet Activating Factor (PAF) and Eicosanoids in UVC-Irradiated Corneal Stromal Cells. Curr Eye Res (1995) 14(5):341–7. doi: 10.3109/02713689508999931
161. Zhang Q, Seltmann H, Zouboulis CC, Travers JB. Activation of Platelet-Activating Factor Receptor in SZ95 Sebocytes Results in Inflammatory Cytokine and Prostaglandin E2 Production. Exp Dermatol (2006) 15(10):769–74. doi: 10.1111/j.1600-0625.2006.00458.x
162. Wolverton JE, Al-Hassani M, Yao Y, Zhang Q, Travers JB. Epidermal Platelet-Activating Factor Receptor Activation and Ultraviolet B Radiation Result in Synergistic Tumor Necrosis Factor-Alpha Production. Photochem Photobiol (2010) 86(1):231–5. doi: 10.1111/j.1751-1097.2009.00618.x
163. Koga MM, Bizzarro B, Sá-Nunes A, Rios FJ, Jancar S. Boosting Adaptive Immunity: A New Role for PAFR Antagonists. Sci Rep (2016) 6(1):39146. doi: 10.1038/srep39146
164. Sahu RP. Expression of the Platelet-Activating Factor Receptor Enhances Benzyl Isothiocyanate-Induced Apoptosis in Murine and Human Melanoma Cells. Mol Med Rep (2015) 12(1):394–400. doi: 10.3892/mmr.2015.3371
165. Pei Y, Barber LA, Murphy RC, Johnson CA, Kelley SW, Dy LC, et al. Activation of the Epidermal Platelet-Activating Factor Receptor Results in Cytokine and Cyclooxygenase-2 Biosynthesis. J Immunol (1998) 161(4):1954–61.
166. Elmets CA, Viner JL, Pentland AP, Cantrell W, Lin H-Y, Bailey H, et al. Chemoprevention of Nonmelanoma Skin Cancer With Celecoxib: A Randomized, Double-Blind, Placebo-Controlled Trial. J Natl Cancer Institute (2010) 102(24):1835–44. doi: 10.1093/jnci/djq442
167. Tang JY, Aszterbaum M, Athar M, Barsanti F, Cappola C, Estevez N. Basal Cell Carcinoma Chemoprevention With Nonsteroidal Anti-Inflammatory Drugs in Genetically Predisposed PTCH1+/- Humans and Mice. Cancer Prev Res (Philadelphia Pa) (2010) 3(1):25–34. doi: 10.1158/1940-6207.CAPR-09-0200
Keywords: microvesicle particles, platelet-activating factor, ultraviolet light, UVB, platelet-activating factor receptor, immunosuppression, local inflammation, aSMase inhibitors
Citation: Frommeyer TC, Gilbert MM, Brittain GV, Wu T, Nguyen TQ, Rohan CA and Travers JB (2022) UVB-Induced Microvesicle Particle Release and Its Effects on the Cutaneous Microenvironment. Front. Immunol. 13:880850. doi: 10.3389/fimmu.2022.880850
Received: 21 February 2022; Accepted: 04 April 2022;
Published: 06 May 2022.
Edited by:
Alicia R. Mathers, University of Pittsburgh, United StatesReviewed by:
Andrzej T. Slominski, University of Alabama at Birmingham, United StatesStephen Balmert, University of Pittsburgh, United States
Nabiha Yusuf, University of Alabama at Birmingham, United States
Copyright © 2022 Frommeyer, Gilbert, Brittain, Wu, Nguyen, Rohan and Travers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey B. Travers, amVmZnJleS50cmF2ZXJzQHdyaWdodC5lZHU=
†These authors have contributed equally to this work and share first authorship