 Gabriel Orozco
Gabriel Orozco Meera Gupta
Meera Gupta Roberto Gedaly
Roberto Gedaly Francesc Marti
Francesc Marti- 1Department of Surgery - Transplant Division, College of Medicine, University of Kentucky, Lexington, KY, United States
- 2Alliance Research Initiative [Treg cells to Induce Liver Tolerance (TILT) Alliance], University of Kentucky College of Medicine, Lexington, KY, United States
- 3Lucille Parker Markey Cancer Center, University of Kentucky, College of Medicine, Lexington, KY, United States
Numerous preclinical studies have provided solid evidence supporting adoptive transfer of regulatory T cells (Tregs) to induce organ tolerance. As a result, there are 7 currently active Treg cell-based clinical trials in solid organ transplantation worldwide, all of which are early phase I or phase I/II trials. Although the results of these trials are optimistic and support both safety and feasibility, many experimental and clinical unanswered questions are slowing the progression of this new therapeutic alternative. In this review, we bring to the forefront the major challenges that Treg cell transplant investigators are currently facing, including the phenotypic and functional diversity of Treg cells, lineage stability, non-standardized ex vivo Treg cell manufacturing process, adequacy of administration route, inability of monitoring and tracking infused cells, and lack of biomarkers or validated surrogate endpoints of efficacy in clinical trials. With this plethora of interrogation marks, we are at a challenging and exciting crossroad where properly addressing these questions will determine the successful implementation of Treg cell-based immunotherapy in clinical transplantation.
Introduction
Since the inception of transplant programs, the discovery and use of immunosuppressive drugs have played a critical role in preserving allograft function. After several decades of implementation, these immunosuppressive regimens have efficiently decreased the incidence of acute graft loss. However, long-term and chronic allograft rejection rates remain pervasive and, together with the severity of side effects in the allograft recipient population, makes the pursuit of therapeutic alternatives a medical necessity. A better understanding of self-tolerance mechanisms has facilitated different approaches aiming at rebalancing alloantigen-reactive conventional T-cells (Tconv) and immunosuppressive regulatory T cells (Tregs). This is a clear conceptual shift from the current standard multidrug-based protocols focused on halting effector immune responses.
CD25hiFoxP3+ Treg cells represent 1-5% of circulating CD4+ T lymphocytes and are essential in maintaining peripheral immune tolerance and homeostasis. After transplantation, the frequency of circulating Tregs in tolerant recipients is higher compared to patients with acute allograft rejection (1, 2). Increasing evidence also suggests that the balance between graft-reactive effector cells and graft-protective suppressor Tregs plays a role in organ engraftment and long-term allograft survival (3, 4).
Despite a decade of major progress in Treg research, technical limitations and significant gaps in our knowledge of Treg cell biology continue to hinder our ability to harness the therapeutic potential of these cells to induce allograft tolerance. This review summarizes achievements, current status and future challenges in the clinical implementation of Treg cell-based immunotherapy in solid organ transplant (SOT) recipients.
Achievements and Current Status of Clinical Trials
The ability to isolate and expand Treg cells under good manufacturing practice (GMP)-compliant conditions paved the way for the clinical use of adoptive Treg cell transfer to induce allospecific tolerance in SOT patients. The first pilot study in SOT was reported by Todo et al. (5) in 10 liver transplant recipients using donor-specific Treg-enriched cell product in combination with standard immunosuppressive drugs that were gradually discontinued over a period of 18 months. All 10 recipients maintained stable graft function. Seven patients successfully achieved weaning of drugs between 16 and 33 months. All three patients who developed mild rejection during the immunosuppression weaning process underwent transplantation for autoimmune liver disease, which original autoimmune effector-regulatory unbalance may account for the difficult long-term control of effector responses. Since Todo’s report, five more original manuscripts have been published to date in SOT, four of them in kidney transplant patients and another in liver recipients (summarized in Table 1). Across all studies with at least one-year follow-up, fifty-four SOT recipients who received a single infusion of autologous Tregs had 100% survival, no episodes of graft loss, no increased risk of infection, and no report of de novo cancer (6–10). Only two patients suffered mild adverse events: one experienced mild and transient cytokine release syndrome (9), and another developed donor-specific antibodies one-year-post transplant and primary disease recurrence after a two-year follow-up (7). Furthermore, among 28 kidney transplant recipients receiving autologous transfer therapy with Tregs, the ONE study reported a significant decrease in the incidence of viral infections after transplant (12). Like the Todo et al. study, the stability of transplant function in Harden et al. study (10) also permitted minimization of immunosuppression, revealing a significant reduction of inflammatory cell populations in the transplanted organ as a result of Treg transfer. Overall, the published results support feasibility and safety of Treg infusion procedures in SOT patients and disclosed promising early data on feasibility of drug immunosuppressive minimization/discontinuation (Table 1). They are also uncovering multiple challenges that may harness the progression of immunotherapies in the clinic, including phenotypic and functional diversity of Treg cells, lineage stability, optimization of ex vivo Treg cell manufacturing process, adequacy of administration route, inability of monitoring and tracking infused cells, absence of organ specificity/trafficking markers in Treg cells, and lack of biomarkers or validated surrogate endpoints of efficacy in clinical trials. Importantly, measurements and report outcomes are often not comparable among different trials or centers, which makes it difficult to standardize methodologies and verify and validate data for consistency.
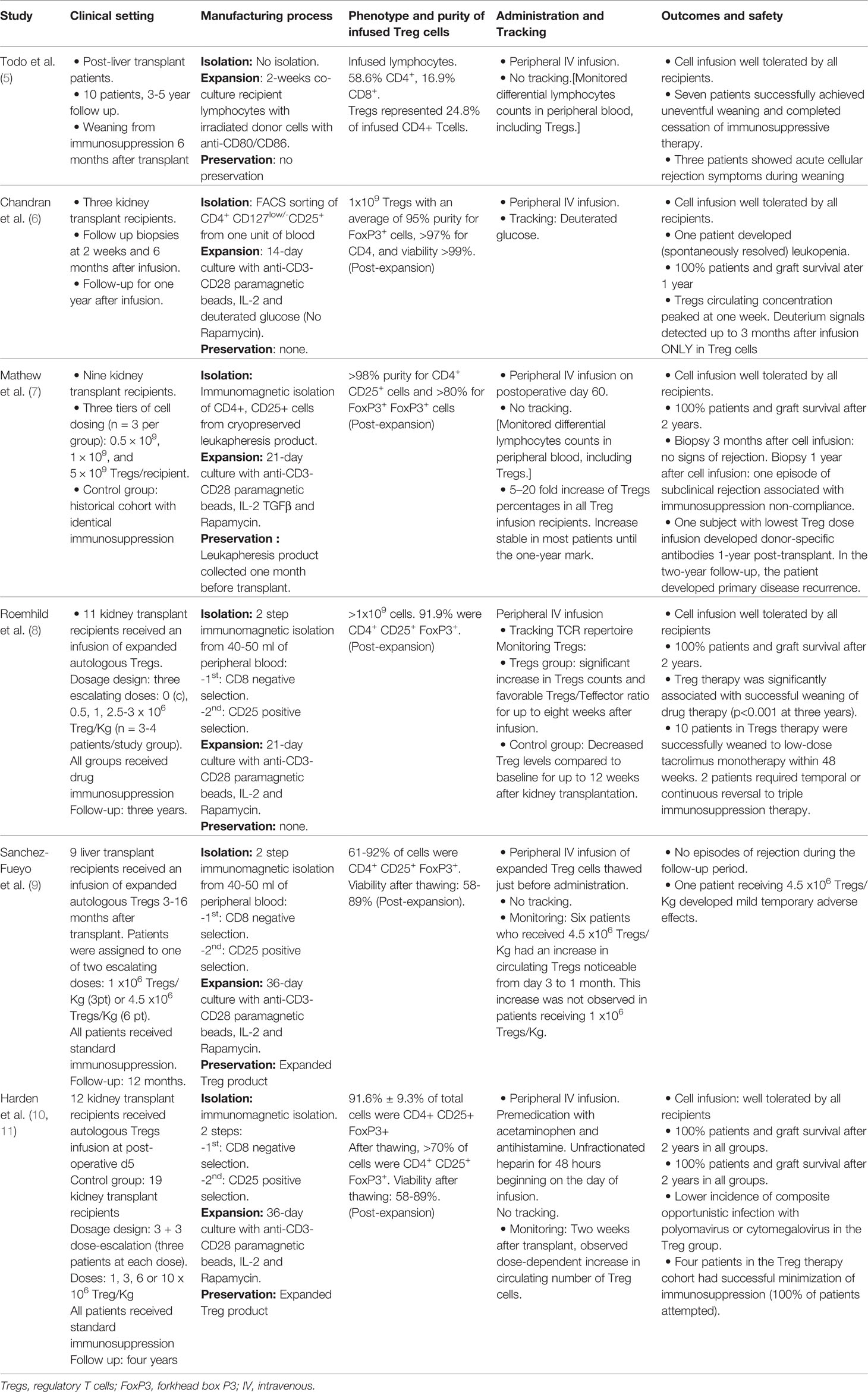
Table 1 Published studies evaluating Treg transfer therapy after solid organ transplantation.
Phenotypic Diversity
The efforts to characterize Tregs have revealed a broad spectrum of phenotypes in cells capable of engaging different suppressive mechanisms to control particular immune effector cell responses. The initial identification of these suppressor cells as CD4+ CD25+ T cells was substantiated by mouse experiments where their removal led to severe autoimmunity, which could be prevented after reconstituting these cells back to circulation (13, 14). In 2003, the forkhead box transcription factor FoxP3 was identified as an essential molecular marker of Treg cell development, differentiation and function. Since then, FoxP3 been considered as the defining Treg cell lineage “master-regulator” (15) and CD4+ CD25+ FoxP3+ as the distinctive core Treg phenotype. Expression of the interleukin-7 receptor (IL-7R) α chain (CD127) on the surface of Treg cells inversely correlates with FoxP3 expression and is another convenient marker for Treg cells as it provides an additional distinction between CD127high (FoxP3-/low) and CD127-/low (FoxP3high) subpopulations. In combination with CD25 during flow cytometry analysis, CD127 can be used as biomarker for analysis and, because of the expression on the cell membrane, for isolation of Tregs (16, 17).
Treg cells can be also categorized by the expression of another membrane marker, CD45RA. Consequently, functionally suppressive Treg population can be distinguished between naïve resting Tregs, with high proliferative potential (FoxP3low CD25low CD45RA+), and terminally differentiated, short-living Treg cells with low proliferative potential (FoxP3high CD25high CD45RA-) (18, 19). Accordingly, as proposed by Arroyo-Hornero et al. (20) and supported by Canavan et al. in Crohn’s disease patients (21), the segregation of the initial population of Treg cells based on the expression of CD45RA should be taken into consideration as CD45RA+ Tregs, but not CD45RA-, maintain a stable Treg signature after expansion. In a similar context, the expression of the Ikaros transcription factor family member, Helios, has been associated with lineage-committed, thymus-originated FoxP3+ Treg cell and, therefore, regarded as potential biomarker for therapeutic competent Treg cells. In mice, Helios+ and Helios− Treg subpopulations are phenotypically and functionally distinct and express different TCR repertoires (22, 23). However, similar studies in human Tregs have not generated consistent results (24–27). A recent report by Lam et al. (28) suggests that Helios expression in Treg cells may be an important marker of lineage stability, although it does not have a direct role in the maintenance of the lineage-committed state. Co-expression of the surface markers T cell immunoreceptor with Ig and ITIM domains (TIGIT) and Fc Receptor-like 3 (FCRL3, CD307c) in Helios+ FoxP3 Treg cells (29) could facilitate reliable identification and selection of Helios+ Treg cells.
A shortcoming in the published literature of Treg cell-based clinical trials is the inconsistency and poor definition of the initial population of isolated Treg cells. Further phenotypic characterization is needed to identify the most appropriate population from which to expand FoxP3+ Tregs for use in adoptive transfer approaches, which will also help to determine whether a unique initial phenotypic Treg cell profile is well-suited to fulfill the specific demands of different target tissues.
Lineage Stability
Lineage stability refers to the capability of a Treg cell to sustain immunosuppressive function in different environments and generate a progeny with similar characteristics after replication. The stable expression of signature genes for Treg cell lineage commitment should be considered as a critical parameter in the clinical competent population of Treg cells. Epigenetic changes such as DNA methylation, histone modification, and non-coding RNA synthesis regulate gene expression and cellular differentiation (30). Despite the phenotypic variability of Tregs, the epigenetic pattern can be used to identify a cell lineage with a stable immunosuppressive function. Indeed, the type of Treg-specific CpG hypomethylation pattern (TrHMP) is regarded as a more specific biomarker of functionally stable Treg than mere FoxP3 expression (31). The TrHMP includes hypomethylation of signature genes such as FoxP3, CTLA, GITR, and Helios (32), is heritable, independent of FoxP3 expression, and persists after TCR stimulation and in different culture conditions (30, 32). TrHMP is also linked to the suppressive strength of Treg cells as observed with in vitro induced (iTregs) cells. Despite the expression of FoxP3, iTregs show a TrHMP similar to activated Tconv, are less suppressive and demonstrate less lineage commitment than natural, thymus-originated Tregs (nTregs) (32). The strong association between Treg lineage stability and specific epigenetic imprinting supports the use of TrHMP as biomarker for Treg lineage determination/stability and the inclusion among the most reliable parameters currently available as criterion for the identification of functional Treg cells in experimental settings. However, the methylation status pattern is not included in any reported clinical study as a release criterion for clinically competent Treg cells (Table 1). In zaddition, the capacity to track lineage stability of infused Tregs in vivo in the clinic is limited (7). In Chandran et al. study, the authors transferred deuterium-labeled Tregs into kidney transplant recipients. The fact that deuterium signals were only detected in the Treg population within three months post-infusion suggests the lineage stability of infused cells (6).
Manufacturing Process
While the Treg ability to inhibit the effector immune reactions that trigger graft rejection has been demonstrated in numerous pre-clinical studies, their low concentration in peripheral blood has become a major obstacle to their clinical application (33). However, refinements in the manufacturing process under Good Manufacturing Practices (GMP)-compatible conditions now facilitate escalating the cellular yield up to 2,000-fold (8). This process includes three main steps: isolation, expansion, and preservation.
Isolation
The two most common methods for Treg isolation are Fluorescence-Activated Cell Sorting (FACS) and immuno-magnetic cell separation. FACS has been primarily used for research and analytical purposes, but recent adaptations to comply with GMP legislation have allowed its clinical use. FACS can distinguish very specific cellular subpopulations, sort cells based on the degree of expression of particular markers and discriminate several subpopulations simultaneously. However, FACS-based isolation of Treg cells for human therapy relies only on extracellular markers to identify the target population (34). Another technical limitation of this method is that the sorting efficiency is reduced when the population of interest is rare, requiring lengthy processing times from a large initial cell population (35). Still, some groups have successfully isolated Treg cells for clinical interventions with FACS (6, 36, 37), and the progress towards using more complex membrane marker combinations to define the initial Treg population may help a broader use of this technology as cell isolation procedure.
Immunomagnetic cell separation is the current method of choice for Treg isolation in clinical trials. Biotechnology companies have developed closed, automatic systems to comply with GMP regulations. In this method, magnetized particles are conjugated with antibodies, and consecutive steps of negative and positive selection allow the isolation of a specific Treg cell population. The purity of the isolation can increase by selecting multiple markers during a single pass of negative selection (e.g., CD8 and CD19) (11, 38). Most published Treg-based clinical trials in SOT reported the use of magnetic immunoselection isolation technique as a two-step procedure with initial CD8 depletion and subsequent CD25 enrichment (7–10). A significant loss of targeted cells after each selection step, the necessity of fine-tune optimization to find the optimal balance between cell yield and purity, the lack of discriminatory capacity between low or high expression of cellular markers, and the elevated cost of the procedure (specific equipment and supplies) are some of the shortcomings associated with Treg isolation by magnetic immunoselection. As such, more versatile GMP technologies are needed to improve yield and purity of clinical-grade quality Treg cell isolates and facilitate the standard implementation of this technology in clinical practice.
Expansion
Tregs constitute 1-5% of the total circulating CD4+ T lymphocyte population (39). These low numbers and favoring cell purity over yield in the isolation process make ex vivo expansion a critical step towards successful cell therapy implementation. The main strategy for ex vivo expansion is establishing cell culture conditions to preferentially activate and expand Treg cells while preventing the replication of other potential contaminant cell types. Expansion protocols can produce up to 2,000-fold amplification of Treg cell numbers (36) and are based on the concomitant engagement of the T cell receptor (TCR) and the costimulatory receptor CD28, and high doses of the T cell growth and survival factor IL-2. Addition of mTOR inhibitors (e.g., rapamycin, everolimus) promotes the selective expansion and suppressive activity of Tregs (40, 41) while preventing Tconv activation and growth. Mechanistic evidence supports that mTOR signaling pathway is a critical regulator of effector Tconv homeostasis and function but not of Tregs (42–44). In fact, PI3K/Akt/mTOR activation represses Treg differentiation, and the inhibition of the Akt pathway is crucial to promote the activation of FoxP3 (45–47). Metabolically, Tconv depends on the mTOR-driven glycolytic pathway for a rapid supply of energy and molecular precursors (48); in contrast, the energy demand of Treg cells is fulfilled by the constant crosstalk between glycolytic and oxidative mitochondrial metabolic arms (49–51).
Prolonged stimulation of Tregs triggers epigenetic changes leading to suboptimal TCR signaling and progressive hypermethylation of Treg-specific demethylated regions (52). These epigenetic alterations can change the quality of the final cell product by promoting Treg conversion to Tconv or reducing their suppressive function (52, 53). Upon activation, Tregs may undergo a progressive shift from CD45RA+ to CD45RA- phenotype (54). Upon further expansion, the CD45RA- fraction experiences a decline in both FoxP3 expression and suppressive activity (54). As mentioned, adding an mTOR inhibitor such as rapamycin sustains the expansion and suppressive activity of Tregs (40), but also induces the conversion of conventional CD4+ T cells into iTregs. However, these iTregs do not possess the TrHMP hypomethylated signature of Treg genes and can revert into non-suppressive cells in the absence of rapamycin. Therefore, as suggested by Battaglia et al. (40), careful attention and appropriate quality controls must be in place when mTOR inhibitors are included in the expansion protocol for Treg cell therapy. For clinical application, the initiation of the expansion phase with highly purified and well-defined population of Treg cells seems the appropriate strategy. Overall, these studies highlight the importance of optimizing cell culture conditions (composition and duration) and quality control assessments in the expansion protocols for Treg manufacturing (52, 54). The progress of Treg immunotherapy demands establishing relevant mechanistic links between pre- and post-expansion phenotype, suppressor function and epigenetic profile of Treg cell populations with corresponding clinically relevant outcomes of operational tolerance or reduced rejection.
Preservation
For far-reaching applications of Treg cell-based therapy, it is essential to ensure the stability of the cell product during storage, including optimal cell viability, recovery and functionality. Widening the window between the collection and application of Tregs for adoptive therapy would increase the flexibility of their clinical use. Because preservation techniques can potentially change the yield, viability, and activity of Tregs, they are considered a new therapeutic biological product from a regulatory point of view (55).
Tregs can be cryopreserved before isolation (as peripheral blood mononuclear cells, PBMCs), just after isolation, or after the expansion phase. Treg cell recovery rates from cryopreserved PBMCs fluctuate between 35 to 63% (55–61). Using isolated Tregs cryopreserved in liquid nitrogen for up to one year, Peters et al. reported a viability of 70-80%, with a suppressive capacity that was significantly impaired after thawing but recovered after activation (38). Kaiser et al. found better recovery rates and cellular viability by using cryogenic solution of 5% DMSO instead of 10% DMSO (55). Cryopreserved Tregs after three or four cycles of re-stimulation did not alter their original phenotype or suppressive function (56). Different groups have reported using cryopreserved Treg cells in SOT patients. Mathew et al. cryopreserved the leukapheresis product approximately one month before kidney transplant, their expansion protocol lasted 21 days, and the infusion of Tregs was given 60 days after surgery (7). Harden et al. and Sanchez-Fueyo et al. cryopreserved the Treg product after isolation and expansion and thawing was performed at the bedside of the patient prior to administration (9, 10). Fraser et al. reported the feasibility of infusing pre-expanded cryopreserved Tregs, showing a reported cell recovery >90%, viability >75% and suppressive function of >80% (11).
Using fresh starting material may have recognized advantages, but the ability to cryopreserve also allows for a more flexible, convenient, efficient -and less expensive- manufacturing process that can be easily managed and scheduled in cellular therapy laboratories (9, 10). However, the effect of cryopreservation on Treg phenotypic and functional parameters and on subsequent clinical outcomes has to be properly established. Such assessments would have a profound logistical impact on clinical trial design, infusion timelines and testing requirements for future studies.
Administration Route
Too often, the cell delivery method is an overlooked factor that may have a direct effect on treatment bioavailability to the target organ and, as such, a determining factor for assessments of feasibility, safety and efficacy outcomes of treatment (62, 63). There are two principal methods to introduce cells into the body: systemic delivery and local delivery into the organ. The most common method for Treg cell infusion is systemic intravenous (IV) injection. IV injection allows for wide distribution of cells throughout the body, and it has the advantage of being minimally invasive with low/minimal safety risks in early phase clinical studies. With this methodology, there are several hurdles to overcome in order to deliver cells to the target organ and have them engrafted. IV delivered cells have to pass through the lungs before they can distribute throughout the body. This pulmonary “first-pass” effect results in significant entrapment of cells (64) caused by the estimated size of Tregs (10–15 μm diameter) (65–68), as observed with microsphere particles of this size (64, 69). Similarly, clinical studies with IV-delivered stem cell infusion showed that the majority of cells get trapped in the lungs after intravenous administration (64, 69, 70). Likewise, systemic infusion of expanded tumor infiltrating lymphocytes (TILs) resulted in higher concentrations of cells in lung, liver, and spleen (71).
The optimal method of therapeutic cell delivery will always depend on the mechanism of action of the cell product. Since Tregs cannot exert their organ-protective effect distally, the delivery system must reach the target organ or allow Treg cells to migrate toward it. The alternative to systemic infusion is the direct local delivery into the organ. This approach can provide a high concentration of Tregs in a first passage where all injected cells have opportunity to interact with post-capillary endothelia of the target organ (72). Direct intra-arterial infusion of stem cells into the brain has proven to significantly enhance cellular engraftment and concentration in animal models of brain ischemia when compared to systemic IV administration (73–75). Also reported, infusion of radiolabeled TILs in the hepatic artery is followed by a rapid increase and slow decline in the intensity signal of the liver (76). However, a disadvantage of local injection is that it may cause further local damage in tissue that, such as a SOT, is already particularly sensitive. It has also been shown that, although direct injection increased localization, it did not necessarily increase engraftment or survival (77). Animal models using direct intra-arterial delivery of mesenchymal stromal cells to the kidney have shown retention of cells in the renal cortex (78) and induction of a favorable tolerogenic milieu after transplantation (79–85). To the best of our knowledge, all currently active clinical protocols using adoptive transferred Treg cells in transplantation are using systemic IV delivery of cells. As safety is the necessary focus of these phase I/(II) studies, alternative routes of cell administration have become an understudied area that remains to be properly addressed. Developing efficient cell delivery protocols could significantly improve the effective implementation and outcomes of Treg-based cell therapy in SOT.
Monitoring and Tracking Infused Cells
Regardless of the infusion route, the success of any cell-based immunotherapy relies on efficacy of cell trafficking and recruitment to the targeted area where they must remain functional. Tracking these adoptive cells in vivo becomes critical to evaluate their delivery, biodistribution and therapeutic response. However, our ability to longitudinally interrogate the migration and fate of infused Treg cells throughout the body remains elusive. In fact, it has become one of the most challenging limitations in current Treg cell immunotherapy studies.
There are only a few studies reporting the in vivo assessment of the distribution and fate of infused Treg cells in humans. Oo et al. used single-photon emission computed tomography (SPECT) to track the distribution of autologous Tregs marked with 111Indium tropolonate (111In) in four patients with autoimmune hepatitis. At 24 hours, they detected a predominant distribution within the liver (22-44%), spleen (11-24%), and bone marrow (9-13%). Tregs persisted in the liver for 72 hours until the 111In was no longer detectable (86). Bluestone et al. used non-radioactive labeling of deoxyribose with deuterium for tracking Treg cells after infusion in type-1 diabetes patients (37). They observed a peak concentration of circulating Tregs between days 7 and 14 with a subsequent decline. Ninety days later, the concentration was 25% of the maximum, and one year after, labeled Tregs were still detected. They reported an initial fast decay phase of infused cells with a half-life of 19.6 days, followed by a slower decay phase. Chandran et al. demonstrated similar kinetic and stability pattern: Tregs peaked in the first week, had a bi-phasic decay, and were still detectable circulating one month after infusion, but were undetectable at the 3-month mark (6). However, a study in non-human primates reported strikingly different results: using carboxyfluorescein succinimidyl ester (CFSE)-labeled cells, Singh et al. observed a rapid decrease of Tregs in peripheral blood during the first three days after infusion, and were barely detectable after 16 days. The uptake and clearance of infused Tregs in bone marrow and lymph nodes followed a similar pattern as with concentrations in blood. They also reported a significant change in phenotype, with less than 30% of CFSE-labeled cells holding the CD25+FoxP3+ phenotype by day 16 (87). Although cell manufacturing and labeling protocols differed among studies, and accounting for possible inter-species variability, the inconsistent results among available studies underscore current limitations to assess in vivo trafficking, homing and fate of infused Treg cells to the transplanted organ.
Novel approaches for monitoring the biodistribution and organ trafficking efficacy of adoptive Treg cells after infusion are in dire need. In the absence of standard non-invasive modalities to assess treatment responses, allograft biopsy analyses of FoxP3 mRNA expression in the transplanted organ, either alone or as a ratio with GranzymeB, are used as surrogate markers for infiltrated Treg and Teff cells, respectively (88–101). New non-invasive imaging technologies such as SPECT, Positron Emission Tomography (PET), Magnetic Resonance Imaging (MRI) or hybrid modalities such as MRI-SPECT in combination with computational biology (102–104) still require validation and standardization. However, they are among emerging technologies that, once implemented into clinical practice, will significantly help improve the efficacy of current cell-based therapy protocols.
Antigen-Specific Treg Cells
The generation of antigen-specific Treg cells is a valuable new approach to provide local, more restricted, immune tolerance (105–109) with cells that are efficiently trafficking to tissues that express cognate antigens (110, 111). Efforts to generate antigen-specific Tregs are currently focused on two different strategies: ex-vivo induction of Tregs by stimulation of antigen-directed CD4+ effector Tconv cells (112), and engineering synthetic T-cell receptors (TCRs) or chimeric antigen receptors (CARs) with target-tissue specificity (113). The reported lineage instability of iTregs cells under inflammatory conditions precludes the clinical use of these cells. To potentially overcome the limitations to generate clinically efficient antigen-directed iTreg cells, gene-editing or transgenic approaches are being applied to induce stable expression of FoxP3 or other Treg signature proteins, as well as to identify key gene targets and pathways involved in the regulation of Treg function and stability (112, 114–118).
On the other hand, the genetic introduction of engineered TCRs and CARs can provide antigen-specificity to polyclonal Tregs (106, 111, 113, 119–121). The ectopic expression of TCRs in Treg cells allows the targeting of processed intracellular antigens presented by HLA molecules. Several pre-clinical studies have demonstrated translational potential of this approach (120, 122–125). However, the HLA-restricted physiological activation limits the application of engineered TCRs and may acquire harmful specificities when mispaired with endogenous TCRs. Interestingly, enforcing the expression of MHC-I-restricted TCRs or not functional low affinity Tconv TCRs (126), enable human Treg cells to bypass the MHC requirement for antigen recognition. Also, instead of using exogenous TCRs isolated from Tconv cells, there is the option of using specific Treg TCRs, which have shown some structural differences (127–129). Another strategy may entail the creation of universal Treg donor cell lines by sequential genetic modifications of MHC molecules (130).
CARs are modular artificial receptors that combine an extracellular antigen-recognition domain and intracellular signaling and costimulatory domains. CAR-engineered effector T cells are being used to reprogram effector Tconv to target tumor cells in patients with blood cancers (131–134). The major advantage of CARs is their ability to recognize whole proteins expressed in target tissues unrestricted to MHC class I or II presentation. Therefore, unlike TCR-modified Treg, CAR-Tregs cells could be applicable to a larger number of patients. However, the design of the CAR should consider specific traits of the host Treg cell, such as the determination of optimal specificity and affinity/avidity of antigen recognition and identification of co-stimulatory signaling domains and accessory molecules that enhance suppressive activity without jeopardizing Treg lineage stability. Recent in-depth reviews comprehensively discuss current status and future prospects of engineered TCR- and CAR-Treg cells in different clinical settings (135, 136).
Currently, a multi-center clinical trial is investigating safety and tolerability of CAR-Treg therapy in HLA-A2 mismatched kidney transplant recipients (NCT04817774) (Table 2). These advanced genetic technologies in cell therapy should be implemented in clinical settings with restricted safety precautions and quality control assessments. Among the latter, the complex nature of Treg functional fitness needs careful attention to any TCR genetic manipulation as the maintenance of Treg identity depends on a fine-tuned strength of antigen-specific stimulation (113, 137, 138).
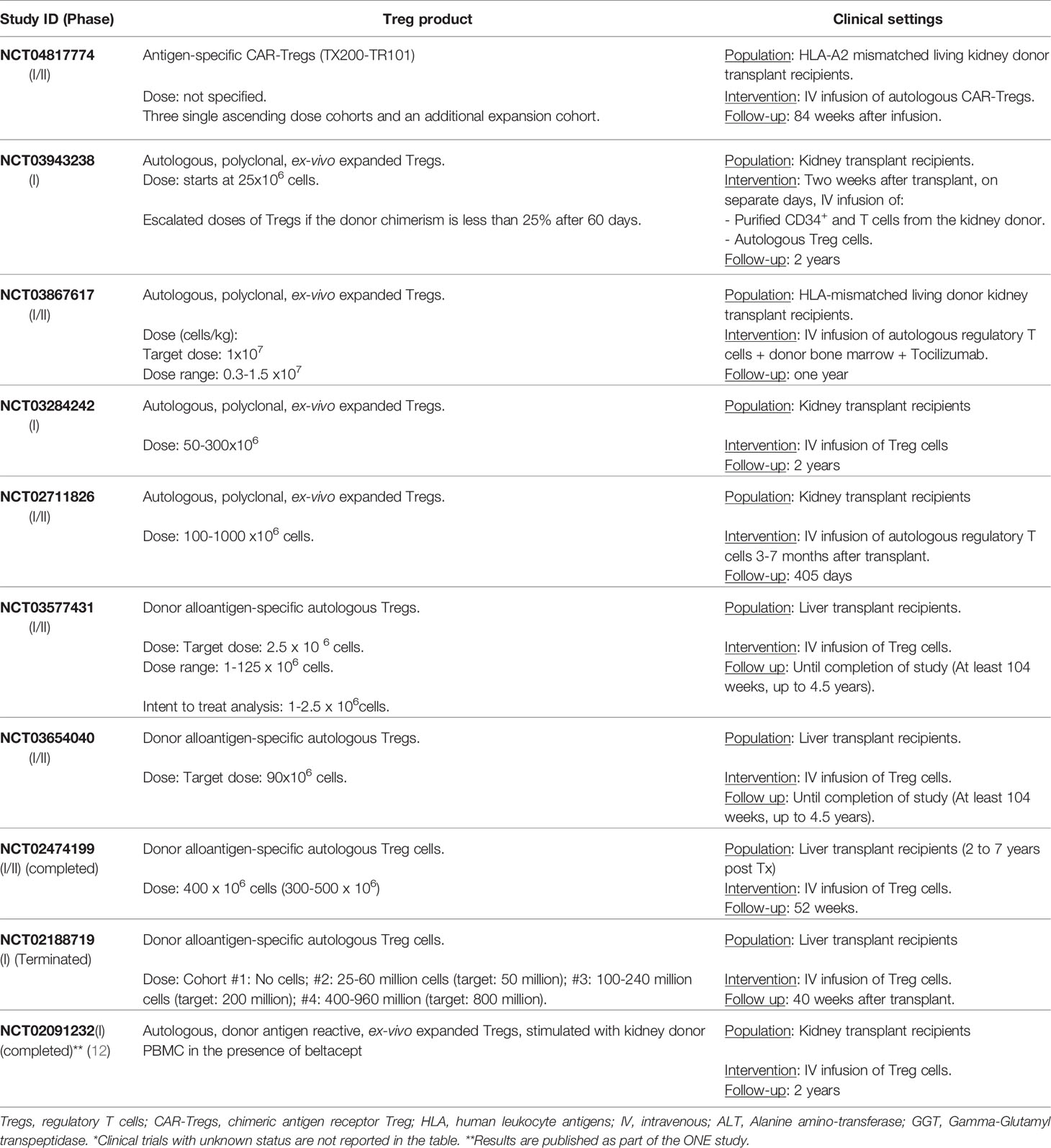
Table 2 Clinical trials evaluating Treg transfer therapy after solid organ transplantation*.
Efficacy: Endpoints and Biomarkers
Traditional primary endpoints for treatment efficacy in transplant clinical trials include graft survival, death with a functioning graft, and quality of life (QoL). These ‘patient-centered’ endpoints are commonly evaluated by surrogate endpoints which, by definition, require adequate validation and should demonstrate robust ability to predict meaningful benefits. Effective use of surrogate endpoints offers the promise of more efficient assessment by providing earlier answers to questions of therapeutic efficacy. Common surrogate endpoints to assess transplant allograft survival include: subclinical, acute cellular, antibody-mediated and steroid-resistant rejection episodes.
The use of biomarkers as surrogates has facilitated the assessment of treatment efficacy in numerous clinical studies. Currently, there is no validated biomarker for treatment efficacy in organ transplantation, which is a fundamental limitation in clinical studies. QoL is a complex endpoint difficult to evaluate because it includes multiple physical, emotional and intellectual parameters that are subjective in nature (139). Although Treg transfer therapy studies are early phase I/II safety and feasibility trials, the evaluation of clinical efficacy is still a critical unresolved issue. To aggravate this limitation, current good short-term outcomes of standard drug immunosuppression regimens demand long-term evaluation of large cohorts to assess the efficacy of any novel therapeutic intervention (140, 141). Implementation of standardized measurable outcomes of direct relevance to patients (including graft function and QoL) is an obvious shortcoming of current SOT clinical trials. Multicentric trials such as Treg Adoptive Therapy for Subclinical Inflammation in Kidney Transplantation (TASK), TReg Adoptive Cell Therapy (TRACT), or the ONE Study are positive initial attempts to unify criteria of cell manufacturing and evaluation of SOT Treg-based clinical studies. However, current protocols still vary in essential criteria and further efforts are required to develop common designs in future clinical trials.
Conclusion
The indispensable role of Treg cells to immune homeostasis and sustaining self-tolerance has awakened an exciting field of research in SOT. Aiming at improving the QoL and long-term outcomes of transplant recipients, Treg cell therapy appears as an attractive alternative to current standard immunosuppressive treatments. Recent bench-to-bedside progress is paving the way towards the successful application of cellular therapies to achieve transplant tolerance. We hope this review will help the reader appreciate the enormous therapeutic potential and also the challenges of Treg cell-based immunotherapy in transplantation.
Author Contributions
Conception and design of the study, RG and FM. GO, RG, and FM wrote the review. MG provided significant revisions to the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Treg Cells to Induce Liver Tolerance (TILT) Alliance Research Initiative of the College of Medicine at the University of Kentucky.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li Y, Koshiba T, Yoshizawa A, Yonekawa Y, Masuda K, Ito A, et al. Analyses of Peripheral Blood Mononuclear Cells in Operational Tolerance After Pediatric Living Donor Liver Transplantation. Am J Transplant (2004) 4(12):2118–25. doi: 10.1111/j.1600-6143.2004.00611.x
2. Demirkiran A, Kok A, Kwekkeboom J, Kusters JG, Metselaar HJ, Tilanus HW, et al. Low Circulating Regulatory T-Cell Levels After Acute Rejection in Liver Transplantation. Liver Transpl (2006) 12(2):277–84. doi: 10.1002/lt.20612
3. Hanidziar D, Koulmanda M. Inflammation and the Balance of Treg and Th17 Cells in Transplant Rejection and Tolerance. Curr Opin Organ Transplant (2010) 15(4):411–5. doi: 10.1097/MOT.0b013e32833b7929
4. Teshima T, Maeda Y, Ozaki K. Regulatory T Cells and Il-17-Producing Cells in Graft-Versus-Host Disease. Immunotherapy (2011) 3(7):833–52. doi: 10.2217/imt.11.51
5. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A Pilot Study of Operational Tolerance With a Regulatory T-Cell-Based Cell Therapy in Living Donor Liver Transplantation. Hepatology (2016) 64(2):632–43. doi: 10.1002/hep.28459
6. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am J Transplant (2017) 17(11):2945–54. doi: 10.1111/ajt.14415
7. Mathew JM, HV J, LeFever A, Konieczna I, Stratton C, He J, et al. A Phase I Clinical Trial With Ex Vivo Expanded Recipient Regulatory T Cells in Living Donor Kidney Transplants. Sci Rep (2018) 8(1):7428. doi: 10.1038/s41598-018-25574-7
8. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T Cells for Minimising Immune Suppression in Kidney Transplantation: Phase I/IIa Clinical Trial. BMJ (2020) 371:m3734. doi: 10.1136/bmj.m3734
9. Sanchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, Safety, and Biological Activity of Regulatory T Cell Therapy in Liver Transplantation. Am J Transplant (2020) 20(4):1125–36. doi: 10.1111/ajt.15700
10. Harden PN, Game DS, Sawitzki B, van der Net JB, Hester J, Bushell A, et al. Feasibility, Long-Term Safety, and Immune Monitoring of Regulatory T Cell Therapy in Living Donor Kidney Transplant Recipients. Am J Transplant (2021) 21(4):1603–11. doi: 10.1111/ajt.16395
11. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A Rapamycin-Based Gmp-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Mol Ther Methods Clin Dev (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
12. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory Cell Therapy in Kidney Transplantation (the One Study): A Harmonised Design and Analysis of Seven Non-Randomised, Single-Arm, Phase 1/2a Trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
13. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
14. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing Il-2 Receptor Alpha-Chains (Cd25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
15. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
16. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. Cd127 Expression Inversely Correlates With Foxp3 and Suppressive Function of Human Cd4+ T Reg Cells. J Exp Med (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
17. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of Interleukin (Il)-2 and Il-7 Receptors Discriminates Between Human Regulatory and Activated T Cells. J Exp Med (2006) 203(7):1693–700. doi: 10.1084/jem.20060468
18. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the Cd45ra+ Subpopulation of Cd4+Cd25high T Cells Gives Rise to Homogeneous Regulatory T-Cell Lines Upon In Vitro Expansion. Blood (2006) 108(13):4260–7. doi: 10.1182/blood-2006-06-027409
19. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human Cd4+ T Cells Expressing the Foxp3 Transcription Factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
20. Arroyo Hornero R, Betts GJ, Sawitzki B, Vogt K, Harden PN, Wood KJ. Cd45ra Distinguishes Cd4+Cd25+Cd127-/Low Tsdr Demethylated Regulatory T Cell Subpopulations With Differential Stability and Susceptibility to Tacrolimus-Mediated Inhibition of Suppression. Transplantation (2017) 101(2):302–9. doi: 10.1097/TP.0000000000001278
21. Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, et al. Developing In Vitro Expanded Cd45ra+ Regulatory T Cells as an Adoptive Cell Therapy for Crohn’s Disease. Gut (2016) 65(4):584–94. doi: 10.1136/gutjnl-2014-306919
22. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros Transcription Factor Family Member, Differentiates Thymic-Derived From Peripherally Induced Foxp3+ T Regulatory Cells. J Immunol (2010) 184(7):3433–41. doi: 10.4049/jimmunol.0904028
23. Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, et al. Helios(+) and Helios(-) Treg Subpopulations Are Phenotypically and Functionally Distinct and Express Dissimilar Tcr Repertoires. Eur J Immunol (2019) 49(3):398–412. doi: 10.1002/eji.201847935
24. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in Expression Level of Helios and Neuropilin-1 Do Not Distinguish Thymus-Derived From Extrathymically-Induced Cd4+Foxp3+ Regulatory T Cells. PloS One (2015) 10(10):e0141161. doi: 10.1371/journal.pone.0141161
25. Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- Cells Coexist Within the Natural Foxp3+ T Regulatory Cell Subset in Humans. J Immunol (2013) 190(5):2001–8. doi: 10.4049/jimmunol.1201379
26. Ayyoub M, Raffin C, Valmori D. Comment on "Helios+ and Helios- Cells Coexist Within the Natural Foxp3+ T Regulatory Cell Subset in Humans". J Immunol (2013) 190(9):4439–40. doi: 10.4049/jimmunol.1390018
27. MacDonald KG, Han JM, Himmel ME, Huang Q, Kan B, Campbell AI, et al. Response to Comment on "Helios+ and Helios- Cells Coexist Within the Natural Foxp3+ T Regulatory Cell Subset in Humans". J Immunol (2013) 190(9):4440–1. doi: 10.4049/jimmunol.1390019
28. Lam AJ, Uday P, Gillies JK, Levings MK. Helios Is a Marker, Not a Driver, of Human Treg Stability. Eur J Immunol (2022) 52(1):75–84. doi: 10.1002/eji.202149318
29. Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, et al. Coexpression of Tigit and Fcrl3 Identifies Helios+ Human Memory Regulatory T Cells. J Immunol (2015) 194(8):3687–96. doi: 10.4049/jimmunol.1401803
30. Delcuve GP, Rastegar M, Davie JR. Epigenetic Control. J Cell Physiol (2009) 219(2):243–50. doi: 10.1002/jcp.21678
31. Ohkura N, Sakaguchi S. Transcriptional and Epigenetic Basis of Treg Cell Development and Function: Its Genetic Anomalies or Variations in Autoimmune Diseases. Cell Res (2020) 30(6):465–74. doi: 10.1038/s41422-020-0324-7
32. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell Receptor Stimulation-Induced Epigenetic Changes and Foxp3 Expression Are Independent and Complementary Events Required for Treg Cell Development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
33. Lam AJ, Hoeppli RE, Levings MK. Harnessing Advances in T Regulatory Cell Biology for Cellular Therapy in Transplantation. Transplantation (2017) 101(10):2277–87. doi: 10.1097/TP.0000000000001757
34. McIntyre CA, Flyg BT, Fong TC. Fluorescence-Activated Cell Sorting for Cgmp Processing of Therapeutic Cells. BioProcess Int (2010) 8(6):44–53. Available at: https://bioprocessintl.com/manufacturing/cell-therapies/fluorescence-activated-cell-sorting-for-cgmp-processing-of-therapeutic-cells-297340/
35. Iyer RK, Bowles PA, Kim H, Dulgar-Tulloch A. Industrializing Autologous Adoptive Immunotherapies: Manufacturing Advances and Challenges. Front Med (Lausanne) (2018) 5:150. doi: 10.3389/fmed.2018.00150
36. Trzonkowski P, Szarynska M, Mysliwska J, Mysliwski A. Ex Vivo Expansion of Cd4(+)Cd25(+) T Regulatory Cells for Immunosuppressive Therapy. Cytometry A (2009) 75(3):175–88. doi: 10.1002/cyto.a.20659
37. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 Diabetes Immunotheraphy Using Polyclonal Regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
38. Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJ, Joosten I. Clinical Grade Treg: Gmp Isolation, Improvement of Purity by Cd127 Depletion, Treg Expansion, and Treg Cryopreservation. PloS One (2008) 3(9):e3161. doi: 10.1371/journal.pone.0003161
39. Tresoldi E, Dell’Albani I, Stabilini A, Jofra T, Valle A, Gagliani N, et al. Stability of Human Rapamycin-Expanded Cd4+Cd25+ T Regulatory Cells. Haematologica (2011) 96(9):1357–65. doi: 10.3324/haematol.2011.041483
40. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin Selectively Expands Cd4+Cd25+Foxp3+ Regulatory T Cells. Blood (2005) 105(12):4743–8. doi: 10.1182/blood-2004-10-3932
41. Gedaly R, De Stefano F, Turcios L, Hill M, Hidalgo G, Mitov MI, et al. Mtor Inhibitor Everolimus in Regulatory T Cell Expansion for Clinical Application in Transplantation. Transplantation (2019) 103(4):705–15. doi: 10.1097/TP.0000000000002495
42. Ono M. Control of Regulatory T-Cell Differentiation and Function by T-Cell Receptor Signalling and Foxp3 Transcription Factor Complexes. Immunology (2020) 160(1):24–37. doi: 10.1111/imm.13178
43. Richards DM, Delacher M, Goldfarb Y, Kagebein D, Hofer AC, Abramson J, et al. Treg Cell Differentiation: From Thymus to Peripheral Tissue. Prog Mol Biol Transl Sci (2015) 136:175–205. doi: 10.1016/bs.pmbts.2015.07.014
44. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The Kinase Tak1 Integrates Antigen and Cytokine Receptor Signaling for T Cell Development, Survival and Function. Nat Immunol (2006) 7(8):851–8. doi: 10.1038/ni1355
45. Haxhinasto S, Mathis D, Benoist C. The Akt-Mtor Axis Regulates De Novo Differentiation of Cd4+Foxp3+ Cells. J Exp Med (2008) 205(3):565–74. doi: 10.1084/jem.20071477
46. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T Cell Receptor Signaling Controls Foxp3 ExpressionVia Pi3k, Akt, and Mtor. Proc Natl Acad Sci USA (2008) 105(22):7797–802. doi: 10.1073/pnas.0800928105
47. Wang P, Zhang Q, Tan L, Xu Y, Xie X, Zhao Y. The Regulatory Effects of Mtor Complexes in the Differentiation and Function of Cd4(+) T Cell Subsets. J Immunol Res (2020) 2020:3406032. doi: 10.1155/2020/3406032
48. Salmond RJ. Mtor Regulation of Glycolytic Metabolism in T Cells. Front Cell Dev Biol (2018) 6:122. doi: 10.3389/fcell.2018.00122
49. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory Cd4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
50. Hashimoto H, McCallion O, Kempkes RWM, Hester J, Issa F. Distinct Metabolic Pathways Mediate Regulatory T Cell Differentiation and Function. Immunol Lett (2020) 223:53–61. doi: 10.1016/j.imlet.2020.04.011
51. Galgani M, De Rosa V, La Cava A, Matarese G. Role of Metabolism in the Immunobiology of Regulatory T Cells. J Immunol (2016) 197(7):2567–75. doi: 10.4049/jimmunol.1600242
52. Ou K, Hamo D, Schulze A, Roemhild A, Kaiser D, Gasparoni G, et al. Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function. Front Cell Dev Biol (2021) 9:751590. doi: 10.3389/fcell.2021.751590
53. Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Helios-Deficient Tregs Is Associated With Conversion to a T-Effector Phenotype and Enhanced Antitumor Immunity. Proc Natl Acad Sci USA (2016) 113(22):6248–53. doi: 10.1073/pnas.1604765113
54. Marek N, Bieniaszewska M, Krzystyniak A, Juscinska J, Mysliwska J, Witkowski P, et al. The Time Is Crucial for Ex Vivo Expansion of T Regulatory Cells for Therapy. Cell Transplant (2011) 20(11-12):1747–58. doi: 10.3727/096368911X566217
55. Kaiser D, Otto NM, McCallion O, Hoffmann H, Zarrinrad G, Stein M, et al. Freezing Medium Containing 5% Dmso Enhances the Cell Viability and Recovery Rate After Cryopreservation of Regulatory T Cell Products Ex Vivo and In Vivo. Front Cell Dev Biol (2021) 9:750286. doi: 10.3389/fcell.2021.750286
56. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive Ex Vivo Expansion of Human Natural Regulatory T Cells (T(Regs)) With Minimal Loss of In Vivo Functional Activity. Sci Transl Med (2011) 3(83):83ra41. doi: 10.1126/scitranslmed.3001809
57. Golab K, Leveson-Gower D, Wang XJ, Grzanka J, Marek-Trzonkowska N, Krzystyniak A, et al. Challenges in Cryopreservation of Regulatory T Cells (Tregs) for Clinical Therapeutic Applications. Int Immunopharmacol (2013) 16(3):371–5. doi: 10.1016/j.intimp.2013.02.001
58. MacDonald KN, Ivison S, Hippen KL, Hoeppli RE, Hall M, Zheng G, et al. Cryopreservation Timing Is a Critical Process Parameter in a Thymic Regulatory T-Cell Therapy Manufacturing Protocol. Cytotherapy (2019) 21(12):1216–33. doi: 10.1016/j.jcyt.2019.10.011
59. Sattui S, de la Flor C, Sanchez C, Lewis D, Lopez G, Rizo-Patron E, et al. Cryopreservation Modulates the Detection of Regulatory T Cell Markers. Cytometry B Clin Cytom (2012) 82(1):54–8. doi: 10.1002/cyto.b.20621
60. Elkord E. Frequency of Human T Regulatory Cells in Peripheral Blood Is Significantly Reduced by Cryopreservation. J Immunol Methods (2009) 347(1-2):87–90. doi: 10.1016/j.jim.2009.06.001
61. Van Hemelen D, Oude Elberink JN, Heimweg J, van Oosterhout AJ, Nawijn MC. Cryopreservation Does Not Alter the Frequency of Regulatory T Cells in Peripheral Blood Mononuclear Cells. J Immunol Methods (2010) 353(1-2):138–40. doi: 10.1016/j.jim.2009.11.012
62. Aijaz A, Vaninov N, Allen A, Barcia RN, Parekkadan B. Convergence of Cell Pharmacology and Drug Delivery. Stem Cells Transl Med (2019) 8(9):874–9. doi: 10.1002/sctm.19-0019
63. Milone MC, Bhoj VG. The Pharmacology of T Cell Therapies. Mol Ther Methods Clin Dev (2018) 8:210–21. doi: 10.1016/j.omtm.2018.01.010
64. Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary Passage Is a Major Obstacle for Intravenous Stem Cell Delivery: The Pulmonary First-Pass Effect. Stem Cells Dev (2009) 18(5):683–92. doi: 10.1089/scd.2008.0253
65. Du M, Kalia N, Frumento G, Chen F, Zhang Z. Biomechanical Properties of Human T Cells in the Process of Activation Based on Diametric Compression by Micromanipulation. Med Eng Phys (2017) 40:20–7. doi: 10.1016/j.medengphy.2016.11.011
66. Teague TK, Munn L, Zygourakis K, McIntyre BW. Analysis of Lymphocyte Activation and Proliferation by Video Microscopy and Digital Imaging. Cytometry (1993) 14(7):772–82. doi: 10.1002/cyto.990140710
67. Pollizzi KN, Waickman AT, Patel CH, Sun IH, Powell JD. Cellular Size as a Means of Tracking Mtor Activity and Cell Fate of Cd4+ T Cells Upon Antigen Recognition. PloS One (2015) 10(4):e0121710. doi: 10.1371/journal.pone.0121710
68. Rathmell JC, Farkash EA, Gao W, Thompson CB. Il-7 Enhances the Survival and Maintains the Size of Naive T Cells. J Immunol (2001) 167(12):6869–76. doi: 10.4049/jimmunol.167.12.6869
69. Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. Stem Cell Transplantation: The Lung Barrier. Transplant Proc (2007) 39(2):573–6. doi: 10.1016/j.transproceed.2006.12.019
70. Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The Dynamic in Vivo Distribution of Bone Marrow-Derived Mesenchymal Stem Cells After Infusion. Cells Tissues Organs (2001) 169(1):12–20. doi: 10.1159/000047856
71. Fisher B, Packard BS, Read EJ, Carrasquillo JA, Carter CS, Topalian SL, et al. Tumor Localization of Adoptively Transferred Indium-111 Labeled Tumor Infiltrating Lymphocytes in Patients With Metastatic Melanoma. J Clin Oncol (1989) 7(2):250–61. doi: 10.1200/JCO.1989.7.2.250
72. Visioni A, Kim M, Wilfong C, Blum A, Powers C, Fisher D, et al. Intra-Arterial Versus Intravenous Adoptive Cell Therapy in a Mouse Tumor Model. J Immunother (2018) 41(7):313–8. doi: 10.1097/CJI.0000000000000235
73. Misra V, Ritchie MM, Stone LL, Low WC, Janardhan V. Stem Cell Therapy in Ischemic Stroke: Role of IV and Intra-Arterial Therapy. Neurology (2012) 79(13 Suppl 1):S207–12. doi: 10.1212/WNL.0b013e31826959d2
74. Li L, Jiang Q, Ding G, Zhang L, Zhang ZG, Li Q, et al. Effects of Administration Route on Migration and Distribution of Neural Progenitor Cells Transplanted Into Rats With Focal Cerebral Ischemia, an Mri Study. J Cereb Blood Flow Metab (2010) 30(3):653–62. doi: 10.1038/jcbfm.2009.238
75. Lundberg J, Jussing E, Liu Z, Meng Q, Rao M, Samen E, et al. Safety of Intra-Arterial Injection With Tumor-Activated T Cells to the Rabbit Brain Evaluated by Mri and Spect/Ct. Cell Transplant (2017) 26(2):283–92. doi: 10.3727/096368916X693347
76. Takayama T, Makuuchi M, Sekine T, Terui S, Shiraiwa H, Kosuge T, et al. Distribution and Therapeutic Effect of Intraarterially Transferred Tumor-Infiltrating Lymphocytes in Hepatic Malignancies. A Preliminary Report. Cancer (1991) 68(11):2391–6. doi: 10.1002/1097-0142(19911201)68:11<2391::aid-cncr2820681110>3.0.co;2-7
77. Chabner KT, Adams GB, Qiu J, Moskowitz M, Marsters ES, Topulos GP, et al. Direct Vascular Delivery of Primitive Hematopoietic Cells to Bone Marrow Improves Localization But Not Engraftment. Blood (2004) 103(12):4685–6. doi: 10.1182/blood-2003-12-4145
78. Sierra-Parraga JM, Munk A, Andersen C, Lohmann S, Moers C, Baan CC, et al. Mesenchymal Stromal Cells Are Retained in the Porcine Renal Cortex Independently of Their Metabolic State After Renal Intra-Arterial Infusion. Stem Cells Dev (2019) 28(18):1224–35. doi: 10.1089/scd.2019.0105
79. Gregorini M, Bosio F, Rocca C, Corradetti V, Valsania T, Pattonieri EF, et al. Mesenchymal Stromal Cells Reset the Scatter Factor System and Cytokine Network in Experimental Kidney Transplantation. BMC Immunol (2014) 15:44. doi: 10.1186/s12865-014-0044-1
80. Zulpaite R, Miknevicius P, Leber B, Strupas K, Stiegler P, Schemmer P. Ex-Vivo Kidney Machine Perfusion: Therapeutic Potential. Front Med (Lausanne) (2021) 8:808719. doi: 10.3389/fmed.2021.808719
81. Pool M, Eertman T, Sierra Parraga J, t Hart N, Roemeling-van Rhijn M, Eijken M, et al. Infusing Mesenchymal Stromal Cells Into Porcine Kidneys During Normothermic Machine Perfusion: Intact Mscs Can Be Traced and Localised to Glomeruli. Int J Mol Sci (2019) 20(14). doi: 10.3390/ijms20143607
82. Pool MBF, Vos J, Eijken M, van Pel M, Reinders MEJ, Ploeg RJ, et al. Treating Ischemically Damaged Porcine Kidneys With Human Bone Marrow- And Adipose Tissue-Derived Mesenchymal Stromal Cells During Ex Vivo Normothermic Machine Perfusion. Stem Cells Dev (2020) 29(20):1320–30. doi: 10.1089/scd.2020.0024
83. Brasile L, Henry N, Orlando G, Stubenitsky B. Potentiating Renal Regeneration Using Mesenchymal Stem Cells. Transplantation (2019) 103(2):307–13. doi: 10.1097/TP.0000000000002455
84. Lohmann S, Pool MBF, Rozenberg KM, Keller AK, Moers C, Moldrup U, et al. Mesenchymal Stromal Cell Treatment of Donor Kidneys During Ex Vivo Normothermic Machine Perfusion: A Porcine Renal Autotransplantation Study. Am J Transplant (2021) 21(7):2348–59. doi: 10.1111/ajt.16473
85. Thompson ER, Bates L, Ibrahim IK, Sewpaul A, Stenberg B, McNeill A, et al. Novel Delivery of Cellular Therapy to Reduce Ischemia Reperfusion Injury in Kidney Transplantation. Am J Transplant (2021) 21(4):1402–14. doi: 10.1111/ajt.16100
86. Oo YH, Ackrill S, Cole R, Jenkins L, Anderson P, Jeffery HC, et al. Liver Homing of Clinical Grade Tregs After Therapeutic Infusion in Patients With Autoimmune Hepatitis. JHEP Rep (2019) 1(4):286–96. doi: 10.1016/j.jhepr.2019.08.001
87. Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of Rapamycin Over Tacrolimus in Preserving Nonhuman Primate Treg Half-Life and Phenotype After Adoptive Transfer. Am J Transplant (2014) 14(12):2691–703. doi: 10.1111/ajt.12934
88. Speletas M, Argentou N, Germanidis G, Vasiliadis T, Mantzoukis K, Patsiaoura K, et al. Foxp3 Expression in Liver Correlates With the Degree But Not the Cause of Inflammation. Mediators Inflamm (2011) 2011:827565. doi: 10.1155/2011/827565
89. Amoras Eda S, Gomes ST, Freitas FB, Santana BB, Ishak G, Ferreira de Araujo MT, et al. Intrahepatic Mrna Expression of Fas, Fasl, and Foxp3 Genes Is Associated With the Pathophysiology of Chronic Hcv Infection. PloS One (2016) 11(5):e0156604. doi: 10.1371/journal.pone.0156604
90. Sakamoto R, Asonuma K, Zeledon Ramirez ME, Yoshimoto K, Nishimori A, Inomata Y. Forkhead Box P3 (Foxp3) Mrna Expression Immediately After Living-Donor Liver Transplant. Exp Clin Transplant (2009) 7(1):8–12.
91. Grimbert P, Mansour H, Desvaux D, Roudot-Thoraval F, Audard V, Dahan K, et al. The Regulatory/Cytotoxic Graft-Infiltrating T Cells Differentiate Renal Allograft Borderline Change From Acute Rejection. Transplantation (2007) 83(3):341–6. doi: 10.1097/01.tp.0000248884.71946.19
92. Mansour H, Homs S, Desvaux D, Badoual C, Dahan K, Matignon M, et al. Intragraft Levels of Foxp3 Mrna Predict Progression in Renal Transplants With Borderline Change. J Am Soc Nephrol (2008) 19(12):2277–81. doi: 10.1681/ASN.2008030254
93. Bestard O, Cruzado JM, Mestre M, Caldes A, Bas J, Carrera M, et al. Achieving Donor-Specific Hyporesponsiveness Is Associated With Foxp3+ Regulatory T Cell Recruitment in Human Renal Allograft Infiltrates. J Immunol (2007) 179(7):4901–9. doi: 10.4049/jimmunol.179.7.4901
94. Bestard O, Cruzado JM, Rama I, Torras J, Goma M, Seron D, et al. Presence of Foxp3+ Regulatory T Cells Predicts Outcome of Subclinical Rejection of Renal Allografts. J Am Soc Nephrol (2008) 19(10):2020–6. doi: 10.1681/ASN.2007111174
95. Xu Y, Jin J, Wang H, Shou Z, Wu J, Han F, et al. The Regulatory/Cytotoxic Infiltrating T Cells in Early Renal Surveillance Biopsies Predicts Acute Rejection and Survival. Nephrol Dial Transplant (2012) 27(7):2958–65. doi: 10.1093/ndt/gfr752
96. Zuber J, Brodin-Sartorius A, Lapidus N, Patey N, Tosolini M, Candon S, et al. Foxp3-Enriched Infiltrates Associated With Better Outcome in Renal Allografts With Inflamed Fibrosis. Nephrol Dial Transplant (2009) 24(12):3847–54. doi: 10.1093/ndt/gfp435
97. Graca L, Cobbold SP, Waldmann H. Identification of Regulatory T Cells in Tolerated Allografts. J Exp Med (2002) 195(12):1641–6. doi: 10.1084/jem.20012097
98. Bunnag S, Allanach K, Jhangri GS, Sis B, Einecke G, Mengel M, et al. Foxp3 Expression in Human Kidney Transplant Biopsies Is Associated With Rejection and Time Post Transplant But Not With Favorable Outcomes. Am J Transplant (2008) 8(7):1423–33. doi: 10.1111/j.1600-6143.2008.02268.x
99. Veronese F, Rotman S, Smith RN, Pelle TD, Farrell ML, Kawai T, et al. Pathological and Clinical Correlates of Foxp3+ Cells in Renal Allografts During Acute Rejection. Am J Transplant (2007) 7(4):914–22. doi: 10.1111/j.1600-6143.2006.01704.x
100. Batsford S, Dickenmann M, Durmuller U, Hopfer H, Gudat F, Mihatsch M. Is Monitoring of Foxp3 Treg Cells in Renal Transplants During Acute Cellular Rejection Episodes Useful? Clin Nephrol (2011) 75(2):101–6. doi: 10.5414/NHX01378
101. Kollins D, Stoelcker B, Hoffmann U, Bergler T, Reinhold S, Banas MC, et al. Foxp3+ Regulatory T-Cells in Renal Allografts: Correlation With Long-Term Graft Function and Acute Rejection. Clin Nephrol (2011) 75(2):91–100.
102. Salcido-Ochoa F, Allen JC Jr. Biomarkers and a Tailored Approach for Immune Monitoring in Kidney Transplantation. World J Transplant (2017) 7(6):276–84. doi: 10.5500/wjt.v7.i6.276
103. Madill-Thomsen K, Perkowska-Ptasinska A, Bohmig GA, Eskandary F, Einecke G, Gupta G, et al. Discrepancy Analysis Comparing Molecular and Histology Diagnoses in Kidney Transplant Biopsies. Am J Transplant (2020) 20(5):1341–50. doi: 10.1111/ajt.15752
104. Bloom RD, Bromberg JS, Poggio ED, Bunnapradist S, Langone AJ, Sood P, et al. Cell-Free DNA and Active Rejection in Kidney Allografts. J Am Soc Nephrol (2017) 28(7):2221–32. doi: 10.1681/ASN.2016091034
105. Tran GT, Hodgkinson SJ, Carter N, Verma ND, Robinson CM, Plain KM, et al. Autoantigen Specific Il-2 Activated Cd4(+)Cd25(+)T Regulatory Cells Inhibit Induction of Experimental Autoimmune Neuritis. J Neuroimmunol (2020) 341:577186. doi: 10.1016/j.jneuroim.2020.577186
106. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a Chimeric Antigen Receptor Specific for Donor Hla Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am J Transplant (2017) 17(4):931–43. doi: 10.1111/ajt.14185
107. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
108. Stephens LA, Malpass KH, Anderton SM. Curing Cns Autoimmune Disease With Myelin-Reactive Foxp3+ Treg. Eur J Immunol (2009) 39(4):1108–17. doi: 10.1002/eji.200839073
109. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human Regulatory T Cells With Alloantigen Specificity Are More Potent Inhibitors of Alloimmune Skin Graft Damage Than Polyclonal Regulatory T Cells. Sci Transl Med (2011) 3(83):83ra42. doi: 10.1126/scitranslmed.3002076
110. Elinav E, Waks T, Eshhar Z. Redirection of Regulatory T Cells With Predetermined Specificity for the Treatment of Experimental Colitis in Mice. Gastroenterology (2008) 134(7):2014–24. doi: 10.1053/j.gastro.2008.02.060
111. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of Murine Colitis and Its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol Ther (2014) 22(5):1018–28. doi: 10.1038/mt.2014.41
112. Freudenberg K, Lindner N, Dohnke S, Garbe AI, Schallenberg S, Kretschmer K. Critical Role of Tgf-Beta and Il-2 Receptor Signaling in Foxp3 Induction by an Inhibitor of DNA Methylation. Front Immunol (2018) 9:125. doi: 10.3389/fimmu.2018.00125
113. Selck C, Dominguez-Villar M. Antigen-Specific Regulatory T Cell Therapy in Autoimmune Diseases and Transplantation. Front Immunol (2021) 12:661875. doi: 10.3389/fimmu.2021.661875
114. Aarts-Riemens T, Emmelot ME, Verdonck LF, Mutis T. Forced Overexpression of Either of the Two Common Human Foxp3 Isoforms Can Induce Regulatory T Cells From Cd4(+)Cd25(-) Cells. Eur J Immunol (2008) 38(5):1381–90. doi: 10.1002/eji.200737590
115. Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. Cd4(+) T Cells From Ipex Patients Convert Into Functional and Stable Regulatory T Cells by Foxp3 Gene Transfer. Sci Transl Med (2013) 5(215): 215ra174. doi: 10.1126/scitranslmed.3007320
116. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. Crispr-Based Gene Editing Enables Foxp3 Gene Repair in Ipex Patient Cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
117. Yamaguchi T, Kishi A, Osaki M, Morikawa H, Prieto-Martin P, Wing K, et al. Construction of Self-Recognizing Regulatory T Cells From Conventional T Cells by Controlling Ctla-4 and Il-2 Expression. Proc Natl Acad Sci USA (2013) 110(23):E2116–25. doi: 10.1073/pnas.1307185110
118. Akamatsu M, Mikami N, Ohkura N, Kawakami R, Kitagawa Y, Sugimoto A, et al. Conversion of Antigen-Specific Effector/Memory T Cells Into Foxp3-Expressing Treg Cells by Inhibition of Cdk8/19. Sci Immunol (2019) 4(40):eaaw2707. doi: 10.1126/sciimmunol.aaw2707
119. Sadelain M, Brentjens R, Riviere I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
120. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of Human Islet-Specific Regulatory T Cells by Tcr Gene Transfer. J Autoimmun (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
121. Bezie S, Charreau B, Vimond N, Lasselin J, Gerard N, Nerriere-Daguin V, et al. Human Cd8+ Tregs Expressing a Mhc-Specific Car Display Enhanced Suppression of Human Skin Rejection and Gvhd in Nsg Mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
122. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered Antigen-Specific Human Regulatory T Cells: Immunosuppression of FVIII-Specific T- and B-Cell Responses. Blood (2015) 125(7):1107–15. doi: 10.1182/blood-2014-04-566786
123. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered Mbp-Specific Human Tregs Ameliorate Mog-Induced Eae Through Il-2-Triggered Inhibition of Effector T Cells. J Autoimmun (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
124. Fujio K, Okamoto A, Araki Y, Shoda H, Tahara H, Tsuno NH, et al. Gene Therapy of Arthritis With Tcr Isolated From the Inflamed Paw. J Immunol (2006) 177(11):8140–7. doi: 10.4049/jimmunol.177.11.8140
125. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front Immunol (2017) 8:1313. doi: 10.3389/fimmu.2017.01313
126. Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, et al. Tcr Affinity and Specificity Requirements for Human Regulatory T-Cell Function. Blood (2012) 119(15):3420–30. doi: 10.1182/blood-2011-09-377051
127. Picarda E, Bezie S, Usero L, Ossart J, Besnard M, Halim H, et al. Cross-Reactive Donor-Specific Cd8(+) Tregs Efficiently Prevent Transplant Rejection. Cell Rep (2019) 29(13):4245–55.e6. doi: 10.1016/j.celrep.2019.11.106
128. Picarda E, Bezie S, Venturi V, Echasserieau K, Merieau E, Delhumeau A, et al. Mhc-Derived Allopeptide Activates Tcr-Biased Cd8+ Tregs and Suppresses Organ Rejection. J Clin Invest (2014) 124(6):2497–512. doi: 10.1172/JCI71533
129. Beringer DX, Kleijwegt FS, Wiede F, van der Slik AR, Loh KL, Petersen J, et al. T Cell Receptor Reversed Polarity Recognition of a Self-Antigen Major Histocompatibility Complex. Nat Immunol (2015) 16(11):1153–61. doi: 10.1038/ni.3271
130. Lanza R, Russell DW, Nagy A. Engineering Universal Cells That Evade Immune Detection. Nat Rev Immunol (2019) 19(12):723–33. doi: 10.1038/s41577-019-0200-1
131. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-Lineage Cells and Regression of Lymphoma in a Patient Treated With Autologous T Cells Genetically Engineered to Recognize Cd19. Blood (2010) 116(20):4099–102. doi: 10.1182/blood-2010-04-281931
132. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
133. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
134. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. Cd19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults With Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci Transl Med (2013) 5(177):177ra38. doi: 10.1126/scitranslmed.3005930
135. Raffin C, Vo LT, Bluestone JA. Treg Cell-Based Therapies: Challenges and Perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
136. Amini L, Greig J, Schmueck-Henneresse M, Volk HD, Bezie S, Reinke P, et al. Super-Treg: Toward a New Era of Adoptive Treg Therapy Enabled by Genetic Modifications. Front Immunol (2020) 11:611638. doi: 10.3389/fimmu.2020.611638
137. Zhang AH, Yoon J, Kim YC, Scott DW. Targeting Antigen-Specific B Cells Using Antigen-Expressing Transduced Regulatory T Cells. J Immunol (2018) 201(5):1434–41. doi: 10.4049/jimmunol.1701800
138. Lamarthee B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E, et al. Transient Mtor Inhibition Rescues 4-1bb Car-Tregs From Tonic Signal-Induced Dysfunction. Nat Commun (2021) 12(1):6446. doi: 10.1038/s41467-021-26844-1
139. Burckhardt CS, Anderson KL. The Quality of Life Scale (Qols): Reliability, Validity, and Utilization. Health Qual Life Outcomes (2003) 1:60. doi: 10.1186/1477-7525-1-60
140. Tanriover B, Jaikaransingh V, MacConmara MP, Parekh JR, Levea SL, Ariyamuthu VK, et al. Acute Rejection Rates and Graft Outcomes According to Induction Regimen Among Recipients of Kidneys From Deceased Donors Treated With Tacrolimus and Mycophenolate. Clin J Am Soc Nephrol (2016) 11(9):1650–61. doi: 10.2215/CJN.13171215
141. Willoughby LM, Schnitzler MA, Brennan DC, Pinsky BW, Dzebisashvili N, Buchanan PM, et al. Early Outcomes of Thymoglobulin and Basiliximab Induction in Kidney Transplantation: Application of Statistical Approaches to Reduce Bias in Observational Comparisons. Transplantation (2009) 87(10):1520–9. doi: 10.1097/TP.0b013e3181a484d7
Keywords: regulatory T-cells, tolerance induction, transplantation, cellular therapy, adoptive therapies
Citation: Orozco G, Gupta M, Gedaly R and Marti F (2022) Untangling the Knots of Regulatory T Cell Therapy in Solid Organ Transplantation. Front. Immunol. 13:883855. doi: 10.3389/fimmu.2022.883855
Received: 25 February 2022; Accepted: 07 April 2022;
Published: 01 June 2022.
Edited by:
Marco Romano, King’s College London, United KingdomReviewed by:
Ethan Menahem Shevach, National Institutes of Health (NIH), United StatesNina Pilat, Medical University of Vienna, Austria
Copyright © 2022 Orozco, Gupta, Gedaly and Marti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesc Marti, Zm1hcnQzQHVreS5lZHU=