 Ann E. Sluder1*
Ann E. Sluder1* Susan Raju Paul1
Susan Raju Paul1 Leonard Moise2
Leonard Moise2 Christina Dold3
Christina Dold3 Guilhem Richard2†Laura Silva-Reyes3Laurie A. Baeten4Anja Scholzen5†Patrick M. Reeves1
Guilhem Richard2†Laura Silva-Reyes3Laurie A. Baeten4Anja Scholzen5†Patrick M. Reeves1 Andrew J. Pollard3Anja Garritsen5
Andrew J. Pollard3Anja Garritsen5 Richard A. Bowen4
Richard A. Bowen4 Anne S. De Groot2
Anne S. De Groot2 Christine Rollier3†Mark C. Poznansky1*
Christine Rollier3†Mark C. Poznansky1*- 1Vaccine and Immunotherapy Center, Massachusetts General Hospital, Boston, MA, United States
- 2EpiVax, Inc., Providence, RI, United States
- 3Oxford Vaccine Group, Department of Paediatrics, The National Institute for Health Research (NIHR) Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
- 4Department of Biomedical Sciences, Colorado State University, Fort Collins, CO, United States
- 5Innatoss Laboratories B.V., Oss, Netherlands
T cell-mediated immunity plays a central role in the control and clearance of intracellular Coxiella burnetii infection, which can cause Q fever. Therefore, we aimed to develop a novel T cell-targeted vaccine that induces pathogen-specific cell-mediated immunity to protect against Q fever in humans while avoiding the reactogenicity of the current inactivated whole cell vaccine. Human HLA class II T cell epitopes from C. burnetii were previously identified and selected by immunoinformatic predictions of HLA binding, conservation in multiple C. burnetii isolates, and low potential for cross-reactivity with the human proteome or microbiome. Epitopes were selected for vaccine inclusion based on long-lived human T cell recall responses to corresponding peptides in individuals that had been naturally exposed to the bacterium during a 2007-2010 Q fever outbreak in the Netherlands. Multiple viral vector-based candidate vaccines were generated that express concatemers of selected epitope sequences arranged to minimize potential junctional neo-epitopes. The vaccine candidates caused no antigen-specific reactogenicity in a sensitized guinea pig model. A subset of the vaccine epitope peptides elicited antigenic recall responses in splenocytes from C57BL/6 mice previously infected with C. burnetii. However, immunogenicity of the vaccine candidates in C57BL/6 mice was dominated by a single epitope and this was insufficient to confer protection against an infection challenge, highlighting the limitations of assessing human-targeted vaccine candidates in murine models. The viral vector-based vaccine candidates induced antigen-specific T cell responses to a broader array of epitopes in cynomolgus macaques, establishing a foundation for future vaccine efficacy studies in this large animal model of C. burnetii infection.
1 Introduction
Q fever, a disease caused by the Gram-negative bacterium Coxiella burnetii, is highly infectious; a single inhaled organism can result in acute disease in humans, leading in ~5% of cases to chronic disease that can be severe (1–3). Zoonotic Q fever outbreaks have been documented in several countries including Australia and the Netherlands, and has been of concern to the United States (US) Department of Defense because of high seroconversion rates detected among military personnel serving in the Middle East (4–6). Doxycycline and other antibiotics can be used to treat acute Q fever, following clinical and serological confirmation of C. burnetii infection (2). However, a mild or asymptomatic infection may evade diagnosis, and can in some individuals subsequently progress to a persistent chronic infection, especially in those with specific risk factors such as advanced age or a history of cardiac valve surgery, or more commonly to post-infection Q fever fatigue syndrome (2, 7). A vaccine is thus considered critical to the prevention and control of Q fever in occupational and biodefense settings. Regulatory approval of Q-VAX®, an inactivated vaccine used in Australia, has been hindered in the US and Europe by reactogenicity in previously exposed individuals, and Q-VAX® vaccination is contraindicated for individuals with a positive skin test for cell-mediated immune reactions to C. burnetii antigen or with serologically detected circulating anti-Coxiella antibodies (8–10). There is thus a need for an efficacious but less reactogenic vaccine for occupational and biodefense purposes.
Both humoral and cellular immune responses contribute to the control of C. burnetii infection and to the responses to whole cell vaccines (11–13). Results from studies in murine infection models suggest that T cell responses, particularly Th1 responses, are necessary for effective infection control (14–18), while antibodies alone are insufficient to clear infection (14, 17, 19). Vaccines comprising epitopes selected based on T cell responses observed in mice previously exposed to live or inactivated whole cell C. burnetii can reduce disease following infection challenge in mice (20, 21).
The objective of the Q-VaxCelerate program is to develop a non-reactogenic T cell-targeted vaccine that will protect against C. burnetii infection and disease in humans (22). A systematic process of computational epitope prediction and experimental validation, including screening for human T cell recall responses in individuals naturally exposed to the bacterium during the 2007-2010 Dutch Q fever outbreak, identified promiscuous human leukocyte antigen (HLA) class II epitope clusters from C. burnetii that are associated with long-lived T cell memory in humans (23). The corresponding peptide sequences represent candidates for inclusion in a vaccine that aims to elicit sustained T cell memory that can subsequently be recalled and boosted by natural exposure in an immunogenetically diverse human population.
Based on these previous epitope profiling data, we generated multiple viral vector-based vaccine candidates expressing epitope concatemers designed to establish anti-C. burnetii cellular immunity in humans. Vaccine development requires establishing immune response correlates in animal models that can inform selection of safe and effective vaccine doses for human testing (24, 25). To this end, we tested the vaccine candidates in three established animal models of immune responses to C. burnetii. The epitope concatemer vaccines did not elicit antigen-specific hypersensitivity responses in a guinea pig model of Q fever vaccine reactogenicity. While several of the vaccine epitopes stimulated antigenic recall responses in a mouse model of Q fever, vaccine immunogenicity was dominated by a single epitope in this model and this was insufficient to confer protection against C. burnetii challenge. However, broader immunogenic responses to the multi-epitope antigens were observed in cynomolgus macaques, suggesting that this nonhuman primate may be a more appropriate model for the evaluation of human-targeted T cell vaccines.
2 Materials And Methods
2.1 Bacteria
Axenic stock C. burnetii Nine Mile strain (Cb9M) was prepared using acidified citrate cysteine medium (ACCM-2) pH 4.75 (Sunrise Science Products, San Diego, CA, USA). Propagation was as described (26, 27), seeding cell free culture media with 1 x 106 genome equivalents (GE) of Cb9M phase 1 RSA 411 (BEI Resources, Manassas, VA, USA). Flasks were incubated for nine days on a shaker (75 rpm) at 37°C in a 2.5% O2, 5% CO2 environment. Bacteria were recovered by centrifugation at 14,000 x g for 30 min, resuspended in sucrose phosphate buffer (28) and stored at -80°C. The stock aliquots were found to contain 2.17 x 107 GE per µL as determined by qPCR (see below). Infectivity of stock was confirmed previously by passage through mice (A/JCr) (27).
2.1.1 Quantitative PCR (qPCR)
Genomic DNA was extracted from samples using standard techniques (QIAamp DNA mini blood kit, Qiagen, Valencia, CA, USA) and qPCR was performed to detect C. burnetii targets using LSI VetMax™ Coxiella burnetii Absolute Quantification kit (Life Technologies, Lissieu, France).
2.2 Reference Vaccine
The veterinary vaccine, Coxevac® was obtained from CEVA Sante Animale (Lot 0101Eg1A, Libourne, France). This formalin inactivated phase I C. burnetii corpuscular antigen formulation is preserved with thiomersal and marketed for annual use in ruminants. Dosing in this study was based upon the assumption that Coxevac® was standardized at 100 µg of antigen per mL (29). The vaccine was determined to contain 86 µg of protein per mL (BCA protein assay, Pierce Biotechnology, Rockford, IL, USA) and qPCR indicated that the vaccine contained 1.3x108 GE per mL.
2.3 Experimental Animals and Ethics Statement
2.3.1 Ethics Statement
Mice for immunogenicity studies were used in accordance with the UK Animals (Scientific Procedures) Act 1986 under project license number 30/3385 granted by the UK Home Office; the experimental protocols were approved by the Oxford University Animal Welfare and Ethical Review Committee (AWERB). Animal research protocols for murine vaccine challenge studies were reviewed and approved by the Colorado State University (CSU) Institutional Animal Care and Use Committee (IACUC) (16–6,844). Animal research protocols for guinea pig experiments were also reviewed and approved by the CSU IACUC (14-5305A, 16-6844A). The animal research protocol for vaccine immunogenicity assessment in cynomolgus macaques was reviewed and approved by the Massachusetts General Hospital IACUC (2019N000076). All animal experimental activities were conducted in full compliance with the applicable institutional, federal and international regulations, and protocols were further reviewed and approved by the US Defense Threat Reduction Agency’s Animal Care and Use Review Office.
2.3.2 Mice
For vaccine immunogenicity and dosing studies, 6- to 8-week old female C57BL/6 mice were obtained from Harlan (UK). Mice were housed in specific pathogen-free conditions.
For studies involving C. burnetii-infected mice, female wild-type C57BL/6 mice (6–8 weeks old) were obtained from Charles River Laboratories (Wilmington, MA, USA). Mice were maintained under BSL3 conditions in microisolator cages (Smart Flow, Tecniplast, Westchester, PA, USA) at the Regional Biocontainment Laboratory, Colorado State University, Fort Collins, CO, USA. Animals were provided water and rodent chow ad libitum and evaluated daily to detect changes in body weight, body condition, behavior, and activity level.
2.3.3 Guinea Pigs
Female Dunkin-Hartley Crl:HA guinea pigs (300 g/35-42 days of age) were purchased from Charles River Laboratories (Wilmington, MA, USA). All guinea pigs were maintained under biosafety level (BSL) 3 conditions in isolator cages (Smart Flow, Techniplast, Westchester, PA, USA) at the Regional Biocontainment Laboratory at Colorado State University (Fort Collins, Colorado, USA). Animals were provided water and guinea pig chow ad libitum. Dried fruit and hay pellets were offered for enrichment. Clinical evaluations were made daily on each animal throughout the study period to detect changes in body weight, body condition, behavior and activity level. Body temperature was documented at four hour intervals by temperature recorders (Thermochron iButton®, Maxim Integrated Products, Inc., Sunnyvale, CA, USA) implanted in the abdominal cavity via flank incision.
2.3.4 Cynomolgus Macaques
Male cynomolgus macaques (4-7 kg) were purchased from Charles River Laboratories (Wilmington, MA, USA). All macaques were housed in social pairs in modulated stainless steel caging equipped with a perch or resting surface and providing sufficient space for physical activity. Animals were provided water ad libitum, with standard Purina Lab Monkey Diet and ¼ piece of fresh produce provided daily. Manipulanda supplied included standard rubber, plastic, and stainless steel enrichments as well as novel toys and enrichment creations distributed weekly. Animals were assessed for abnormal or undesirable behaviors in twice-weekly behavioral rounds. Following vaccine administration, animals were monitored daily for signs of discomfort, and for signs of irritation or inflammation at the site of vaccine injection.
2.4 Assessment of Class II Epitope Antigenicity in C57BL/6 Mice
C57BL/6 mice (n=3 per group) were inoculated intranasally with 1x105 GE of C. burnetii Nine Mile or with saline control. The mice were evaluated clinically from day 7 to termination with body weight and clinical score recorded daily. Mice were euthanized at day 51 post-infection and splenocytes recovered for ELISpot analysis.
2.4.1 Ex Vivo ELISpot Assay of Splenocytes from C. burnetii-Infected Mice
Isolation and assay of splenocytes from C. burnetii-infected mice were carried out under BSL3 containment. The frequency of epitope-specific splenocytes was determined by IFNγ ELISpot assay using the colorimetric Mabtech IFNγ ELISpot Kit with pre-coated plates according to the manufacturer’s protocol. Washed splenocytes in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum (FCS, Atlanta Biologicals) were added at 2.5x105 cells per well. Assays included 4 μg/mL anti-CD28 (BD Pharmingen, USA) and 4 μg/ml anti-CD137 (BioCell, USA) as co-stimulants. Individual peptides were added at 2 µg/mL in triplicate wells. Triplicate wells were stimulated with 2 μg/mL Concanavalin A (ConA; Sigma Aldrich) as a positive control for cell viability, and six replicate wells with medium containing 1% DMSO were used for background determination. Triplicate wells stimulated with 9 μg/mL purified recombinant Com1 protein (gift from Dr. Wei-Mei Ching, Viral and Rickettsial Diseases Department, US Naval Medical Research Center, Silver Spring, MD, USA) or with 2 μg/mL of Coxevac® as positive controls for C. burnetii-specific antigenic recall responses (the amount of Coxevac® included in the assays was limited by the concentration of the commercial formulation). Raw spot counts were recorded by photographic imaging of ELISpot plates under an OMAX microscope within the BSL3 facility. ELISpot well images were transferred electronically and raw spot counts were analyzed using the AID ELISpot software v7.0 (AID Diagnostika GmbH). Results were calculated as the average number of spots in the peptide wells minus the average number of spots in the background determination wells, adjusted to spots per one million cells. High spot density in the ConA-stimulated wells resulted in lack of separation between individual spots that precluded accurate counting by either the analysis software or manual counting; based on approximate manual counts, these wells averaged >800 spot-forming units per one million cells.
2.5 Vaccine Computational Design
String-of-epitopes constructs were designed by concatenating select epitopes head-to-tail in random order. To avoid production of neo-epitopes at epitope junctions, the VaxCAD algorithm was applied to arrange epitopes in an order that diminishes potential junctional immunogenicity (30). The final epitope arrangements did not require insertion of spacers between epitopes to disrupt potential junctional immunogenicity unresolved by VaxCAD. The potential for transmembrane insertion was assessed using TMHMM v2.0 [https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 (31)]. No transmembrane stretches necessitating epitope rearrangement were predicted. Concatemer-encoding nucleic acid sequences, including an N-terminal tissue plasminogen activator (TPA) signal sequence and C-terminal V5 expression tag and codon-optimization for expression in human cells, presented no obvious major concerns when analyzed for RNA secondary structure using the RNAfold WebServer [http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (32)] and the RNAstructure Fold Web Server [http://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Fold/Fold.html (33)].
Epitope concatemer-protein fusion designs utilized protein sequences for C. burnetii Com1 (UniProt ID: H7C7D7) or Mycobacterium tuberculosis HSP70 (UniProt ID: P9WMJ9).
2.6 Vaccine Construct Generation and Production
An E1- and E3-deficient human adenovirus serotype 5, the ChAdOx2 derivative of the chimpanzee adenovirus (ChAd) 68 (34), and Modified vaccinia virus-Ankara (MVA (35) were used as vaccine vectors. DNA sequences corresponding to vaccine antigen designs were synthesized and cloned into plasmids containing attR1 and attR2 recombination sites (Gateway® Life technologies, CA, USA) under control of a CMV promotor. Antigen-encoding inserts were then transferred into the vaccine vectors by recombination as previously described (36–38). The adenoviral vaccine candidates were produced as previously described using the HEK293A cell line (Invitrogen), which contains a stably integrated copy of the E1 gene that supplies the E1 proteins (E1a and E1b) required to generate recombinant adenovirus (39), by the Viral Vector Core Facility of the Jenner Institute, University of Oxford, UK. MVA vaccines were produced using the DF-1 cell line and purified using sucrose cushion centrifugation (40). Virally vectored vaccines were formulated in endotoxin-free PBS. All constructs were verified by sequencing. Vaccine constructs generated are summarized in Table 1 and described further in the Results.
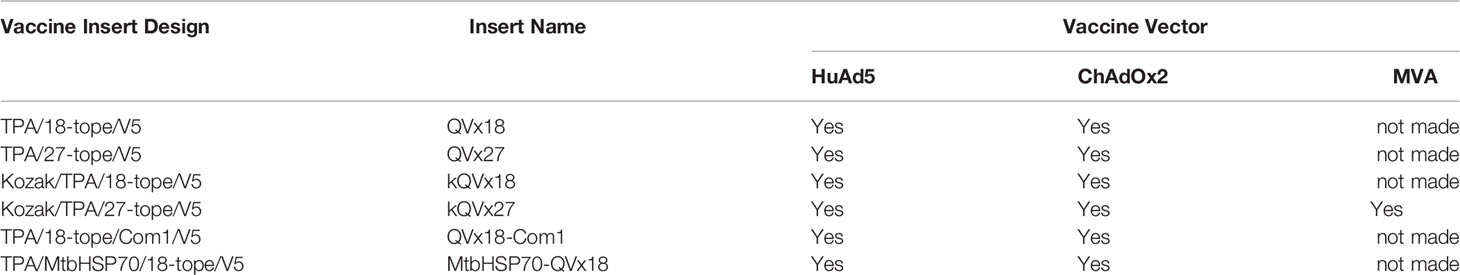
Table 1 Summary of epitope concatemer vaccine constructs.
2.7 Assessment of Vaccine Insert Expression
2.7.1 Western Blot Analysis of Epitope Concatemer Expression
Plasmid clones carrying verified antigen-encoding inserts were transfected into HEK293 cells (2.5 µg of plasmid DNA). Controls underwent a mock transfection. Cells were harvested at 24, 48, or 72 hr after transfection, and proteins in cell lysates were separated by polyacrylamide gel electrophoresis (NU-Page pre-cast BIS-TRIS 4-12% gradient gels). Separated proteins were transferred to PVDF membranes. Blots were probed with anti-V5 antibody (rabbit IgG Poly29038, Biolegend catalog #903802). Bound anti-V5 antibody was detected using an anti-rabbit secondary antibody (mouse IgG anti-rabbit-HRP, Rockland catalog #18-8816-31) visualized using the Pierce ECL system (catalog #32106).
2.7.2 Flow Cytometry Analysis
HeLa cells were infected with adenoviruses expressing transgenes at a MOI of 100. Transfected and infected HeLa cells were left overnight at 37°C with 5% CO2. Vaccine antigen expression was assessed by flow cytometry using FITC-conjugated rabbit anti-V5 Tag antibody (#A190-120A, Bethyl Laboratories).
2.8 Assessment of Vaccine Immunogenicity in C57BL/6 Mice
2.8.1 Vaccinations
Mice received HuAd5 vaccines (109 infectious units, IU) and MVA-kQVx27 boost vaccinations (107 IU) as indicated; all vaccines were delivered intramuscularly (IM) in saline. Each dose was delivered as 50 µL in each hind leg (100 µL total). Splenocytes were harvested 21 days (HuAd5 vaccines) or 7 days (MVA-kQVx27 boost) after the final vaccination. Briefly, spleens were mashed and filtered into a single cell suspension and red blood cells lysed in ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 M Na2EDTA, pH ∼ 7.3) before resuspension in complete α-MEM containing 10% FCS, 2 mM l-glutamine, 0.1% β-mercaptoethanol, 100 U penicillin and 100 μg streptomycin.
2.8.2 Ex Vivo IFNγ ELISpot Assay of Epitope-Specific Responses to Vaccine Constructs
A Mouse IFN-γ FluoroSpot kit (Mabtech) was used to measure antigen-specific T cell responses in mouse spleen tissue. Anti-IFN-γ monoclonal capture antibodies were diluted in PBS to a concentration of 15 μg/ml. The IPFL plate membrane was washed with 15 μL of 35% ethanol per well for no more than 60 s and then washed five times with 200 μL of sterile H2O per well. 100 μL of capture antibody was added to each well and the plate was sealed for overnight incubation at 4°C. The plate was washed five times with 200 μL of sterile PBS per well the following day and the wells were then blocked with DMEM containing 10% FBS for 30 mins at room temperature. Stimuli, either DMSO (1:100) as a negative control, ConA (12 μg/mL) as a positive control, individual peptides corresponding to vaccine epitopes (2 µg/mL), or a Com1 peptide pool (6 µg/mL) composed of a series of overlapping peptides covering the full-length Com1 protein sequence (15mers overlapping by 11 amino acids; manufactured by thinkpeptides, ProImmune), were added to the appropriate wells, followed by 2 x 105 splenocytes from the appropriate sample to each corresponding well. The plate was sealed and incubated overnight at 37°C in a 5% CO2 incubator. Cells were removed from the wells the following day and the plate was washed five times with 200 μL of PBS per well. Anti-IFN-γ-R4-6A2-BAM detection antibody was diluted in PBS containing 0.1% BSA to a concentration of 1:200. 100 μL of detection antibody mixture was added to each well and incubated for two hours at room temperature. Anti-BAM-490 fluorophore conjugate was diluted to a concentration of 1:200 with PBS containing 0.1% BSA and, after washing the plate five times with PBS, 100 μL of this dilution were added to each well. The plate was wrapped in aluminium foil and incubated for one hour in the dark at room temperature. The plate was washed five times with PBS before adding 50 μL of fluorescence enhancer to each well and incubating in the same manner for 15 mins. The plate was emptied of all liquid and the underdrain was removed. Plates were completely dried in the dark at room temperature prior to spot counting with AID ELISpot software 8.0 (Autoimmune Diagnostika). Excitation 490 nm/emission 510 nm (FITC) wavelengths were used to measure IFN-γ.
2.9 Vaccine Efficacy Study in C57BL/6 Mice
Sixty C57BL/6 mice (6-8 weeks old at study initiation) were randomly assigned to 6 groups of 10 animals each, and vaccinated as indicated in Table 2. At study day 140 (16 days after the second boost), mice were chemically restrained (intraperitoneal ketamine, 100 mg/kg, and xylazine, 10 mg/kg) for intranasal inoculation with 2x103 GE (as determined by qPCR) of C. burnetii in 20 µL. Mice were euthanized at day 154 (two weeks after challenge). Spleens were harvested and weighed, and spleen-to-body weight ratio determined. Splenocytes were collected and the spleen bacterial burden was determined from homogenates by qPCR.

Table 2 Vaccination groups and schedule for C57BL/6 vaccine efficacy study.
2.10 Assessment of Vaccine Reactogenicity in Guinea Pigs
Female guinea pigs were sensitized by intranasal inoculation with 106 GE of C. burnetii Nine Mile strain or saline in 100 µL volume, as described previously (41). Blood samples were collected from chemically restrained animals by venipuncture at day 42 post-sensitization for serum collection and evaluation of serological status. Reactogenicity of vaccine candidates relative to positive and negative controls was evaluated by intradermal (i.d.) challenge at day 42 post-infection (p.i) (Table 3). Challenge with 1 µg Coxevac® whole cell vaccine (Ceva Sante Animale, Libourne, France) was used as a positive control; negative controls consisted of saline injections. Gross reactions were monitored at 3 hr and 8 hr post-challenge and daily thereafter. On day 7 post-challenge, animals were anesthetized (ketamine 40 mg/kg and xylazine 5 mg/kg, i.p.) and euthanized with Beuthanasia (i.p.). Inoculation sites were excised from euthanized animals by punch biopsy (12mm AcuPunch, Acuderm Inc., Ft. Lauderdale, FL, USA) and collected in 10% formalin for paraffin embedding and stained with hematoxylin and eosin for histopathological review. Histological reactions at injection sites were scored by an experimenter blinded to the treatment group, using the criteria previously described (41). Briefly, a score of 0 indicates no inflammation, 1 indicates localized macrophage dominated inflammation, 2 macrophage dominated inflammation with limited tissue infiltrations, 3 lymphocytic inflammatory infiltrates extending into the deep dermis, 4 edema and increased pyogranulomatous inflammation extending deep into the subcutis, and 5 widespread pyogranulomatous inflammation including necrosis.

Table 3 Challenge inoculations for reactogenicity testing in guinea pigs.
2.10.1 Serology
Serological status of sensitized guinea pigs was determined by ELISA using the Q Fever antibody test kit (IDEXX Laboratories Inc., Westbrook, ME, USA) with the secondary antibody replaced with peroxidase conjugated protein A/G at 1:10,000 dilution (EMD Millipore Corp., Billerica, MA, USA); the kit substrate is 3,3’,5,5’-tetramethylbenzidine (TMB). Results are presented as optical density at 450 nm (OD450). Day 0 samples were tested at the kit recommended dilution of 1:400 and D42 samples were tested at 1:1000.
2.11 Assessment of Vaccine Immunogenicity in Cynomolgus Macaques
2.11.1 Vaccinations and Sample Collection
Four male Mauritian cynomolgus macaques (4-7 kg) were assigned to two vaccine groups (n=2 per group). On the day of vaccination, the animals were sedated (0.015 mg/kg dexmedetomidine hydrochloride plus 2-4 mg/kg ketamine) and vaccinated intramuscularly on the arm with 500 μl of vaccine in isotonic phosphate-buffered saline (PBS). Animals received either ChAdOx2-QVx18-Com1 or ChAdOx2-QVx27 (each formulated at 2x106 IU/μl) as prime vaccination. Heterologous boost vaccinations, administered 8 and 11 weeks after the prime vaccination, were performed using MVA-kQVx27 (formulated at 2x105 IU/μl). The timing of blood draws and spacing of vaccinations were based on established University of Oxford protocols for these vaccine platforms (42). After each vaccination, the animals were monitored for 15 minutes for any local or systemic reaction to the vaccine, then released from sedation by administration of 0.3 mg/kg atipamezole hydrochloride.
Whole blood (up to 50 mL) was drawn into K2EDTA vacutainer tubes from peripheral veins prior to vaccination and at 2, 4, 8, 9, 11, 12 and 14 weeks after the prime vaccination. Peripheral blood mononuclear cells (PBMC) were isolated from these samples using a 60% percoll gradient. In brief, whole blood was diluted with PBS at a ratio of 1:2. The diluted whole blood was layered on top of 60% percoll at a ratio of 1:1. The sample was centrifuged at 800 g for 30 min at 15°C with no brakes applied during deceleration. The buffy coat that was collected after centrifugation was washed in PBS, and thereafter erythrocytes lysed with red blood cell lysis buffer. Post-lysis the cells were washed and resuspended in RPMI medium supplemented with 10% fetal bovine serum, 1% L-glutamine, 100 international units/mL penicillin, and 100 µg mL streptomycin. For each stimulation condition, one million cells were plated per well in a 96-well round bottom plate, and rested for 2 hours at 37°C with 5% CO2.
2.11.2 Ex Vivo Measurement of Immunogenic T Cell Responses
Plated PBMCs from individual animals were cultured at 37°C with 5% CO2 for 25 hr. in the presence of one of 7 vaccine epitope peptide pools, a pool of peptides representing the full Com1 protein sequence, a peptide concatemer of non-Coxiella sequences, or no peptide stimulation. Vaccine epitope peptide pools comprised the following sets (epitopes are as defined in Table 4): Pool A (p14, p15, p20, p21); Pool B (p18, p22, p37, p38); Pool C (p04, p12, p17, p26); Pool D (p27, p30, p42, p43); Pool E (p02; p45; p48); Pool F (p19, p23, p46, p47); Pool G (p06, p24, p31, p50); epitopes in italics are included in QVx27 but not QVx18 constructs, though p02 is derived from Com1 and this epitope is thus present in the QVx18-Com1 fusion antigen. The final peptide concentration in the stimulation cultures with the Coxiella peptide pools was 2 µg/mL of each vaccine epitope peptide. The final concentration of the Com1 peptide pool was 6 µg/mL. The non-Coxiella peptide concatemer (Biotin-[PEG4]-LEQLERVKR–VSGLEQLESIINFEKLTEWTS-RVKR-EKFDPLGQLSIFYHKTY-amide) contained two control sequences: a sequence from ovalbumin (bold) encompassing the immunodominant epitope SIINFEKL and a scrambled peptide sequence (italics) with no significant homology to mammalian proteins. The concentration of the non-Coxiella peptide concatemer in the stimulation cultures was 1 µg/mL. The Com1 peptide pool was included as a stimulation condition from week 4 post-prime vaccination, and the non-Coxiella epitope concatemer from week 8 after the prime vaccination. Brefeldin A (5 µg/mL) was added for the final 5 hours. Cell viability in all PBMC preparations was confirmed by responses to a 5 hr. stimulation with phorbol-12-myristate 13-acetate (40.5 nM) and ionomycin (669.3 nM) in the presence of Brefeldin A (5 µg.mL).
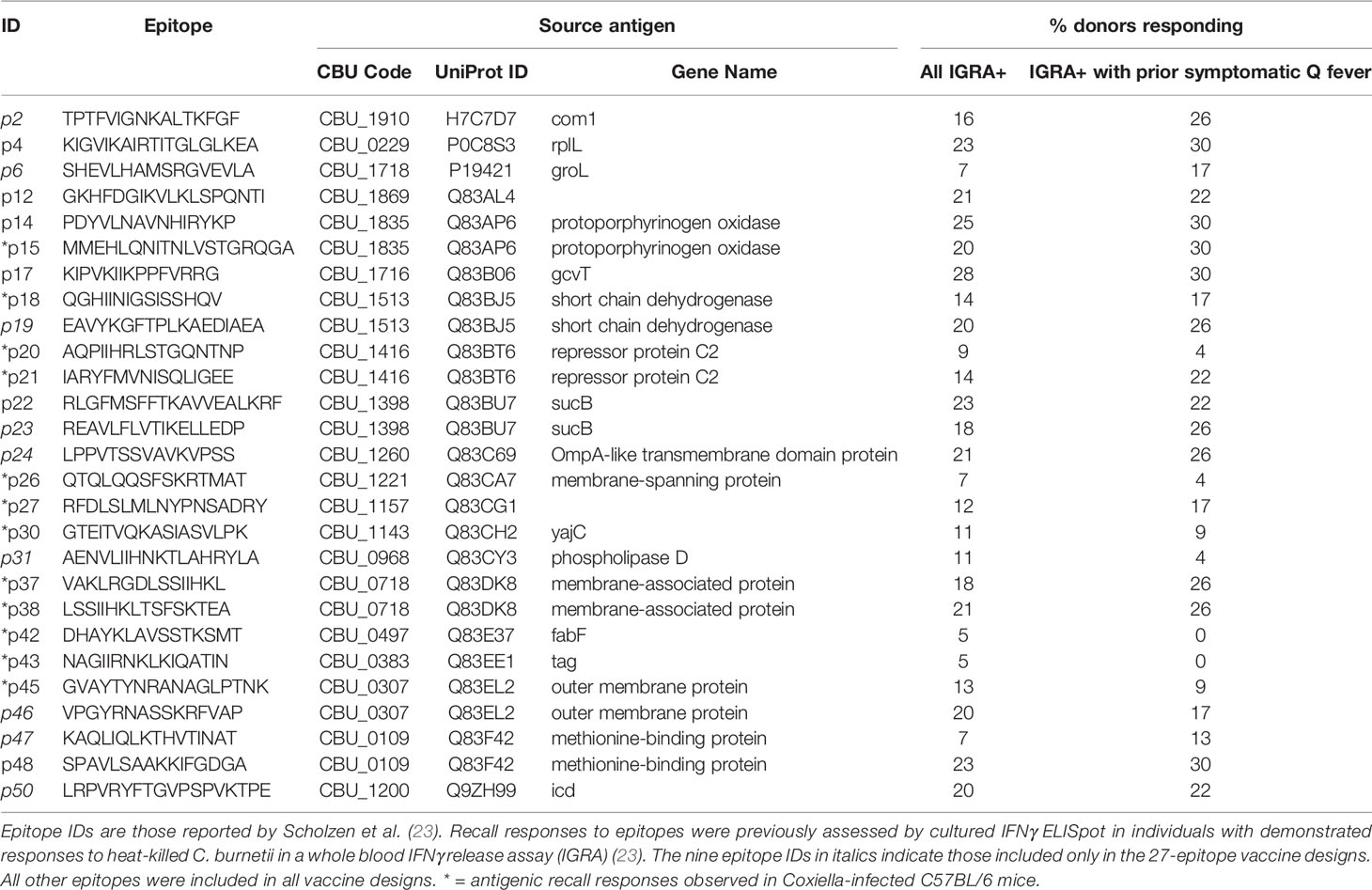
Table 4 HLA class II epitopes selected for vaccine design.
T cell activation status following peptide stimulation was evaluated by flow cytometry. Stimulated cells were washed in PBS, and thereafter were incubated with 1:250 dilution of Zombie UV fixable viability dye (Biolegend) for 30 minutes at room temperature. Cells were washed and resuspended with cell staining buffer (PBS containing 1 mg/mL bovine serum albumin and 2 mM Na2EDTA), then incubated for 30 minutes with antibodies against cell surface antigens: CD3 (SP34-2, BUV395, BD Biosciences), CD4 (OKT4, BV510, Biolegend), CD8 (SK1, APC Fire750, Biolegend), CD11c (3.9, BV785, Biolegend), CD20 (2H7, PerCP-Cy5.5, Biolegend), CD16 (3G8, PerCP-Cy5.5, Biolegend), CD14 (M5E2, PerCP-Cy5.5, Biolegend) and CD66b (TET2, PerCP-Vio770, Miltenyi Biotec). Surface-stained cells were then washed, fixed and permeabilized using isotonic fixation/permeabilization buffer (eBioscience). After this, cells were incubated for 30 minutes with antibodies against intracellular antigens: CD154 (24-31, APC, Biolegend), IFNγ (B27, PE, Biolegend), IL-2 (MQ1-17H12, BV421, Biolegend), Perforin (Pf-344, FITC, Mabtech), TNFα (Mab11, BV711, Biolegend) and T-bet (4B10, PE-Cy7, Biolegend). Fully stained cells were washed and treated with 1.6% paraformaldehyde for 10 minutes. The fully fixed cells were washed and resuspended in cell staining buffer, then stored overnight at 4°C. Data were acquired the next day on a BD LSRFortessa X-20 and analysed using FlowJo™ Software (BD Life Sciences).
2.12 Statistical Analysis
Statistical analyses were performed using GraphPad Prism v9 (San Diego, CA, US).
3 Results
3.1 Generation of Multi-Epitope T Cell-Targeted Vaccines
3.1.1 Epitope Selection
To support rational selection of epitopes for inclusion in a vaccine for prevention of Q fever disease in humans, previous work identified 44 promiscuous HLA class II epitope clusters from C. burnetii that were associated with long-lived human T cell recall responses in individuals naturally exposed to the bacterium during a 2007-2010 Q fever outbreak in the Netherlands (23). The identified epitope clusters are derived from multiple antigens, are conserved in seven publicly available C. burnetii genome sequences, and cover eight human HLA-DR supertype alleles. A subset of these epitope clusters, including 23 epitopes that elicited recall responses in at least 10% of individuals with known prior exposure to C. burnetii or previous symptomatic Q fever, were selected for inclusion in multi-epitope concatemers as vaccine candidates (Table 4).
In anticipation of testing vaccines comprising these human antigenic HLA class II epitopes in a C57BL/6 mouse C. burnetii infection model, we assessed C57BL/6 post-infection antigenic recall responses to 26 epitopes associated with observed recall responses in humans and predicted binding potential for the MHC class II I-Ab allele of C57BL/6 mice [assessed using the EpiMatrix algorithm (30)]. Two of the epitopes (p43, p45) as well as recombinant Com1 protein consistently stimulated IFNγ T-cell recall responses in splenocytes from C. burnetii-infected mice, with additional peptides inducing elevated recall responses in some individual mice (Figure 1). The veterinary vaccine Coxevac induced variable ex vivo recall responses despite containing whole cell antigens, perhaps due to interference with ex vivo cellular antigen processing and presentation by the formalin inactivation used in Coxevac production (43) or to constraints on the volume of commercial formulation that could be included in the assay. To expand the potential for vaccine responses in the C57BL/6 model, four additional epitopes (p20, p26, p42, and p43) were selected based on these murine antigenic recall responses for inclusion in vaccine designs, despite eliciting recall responses in <10% of the C. burnetii-exposed humans evaluated in the previous human antigenicity screening (Table 4).
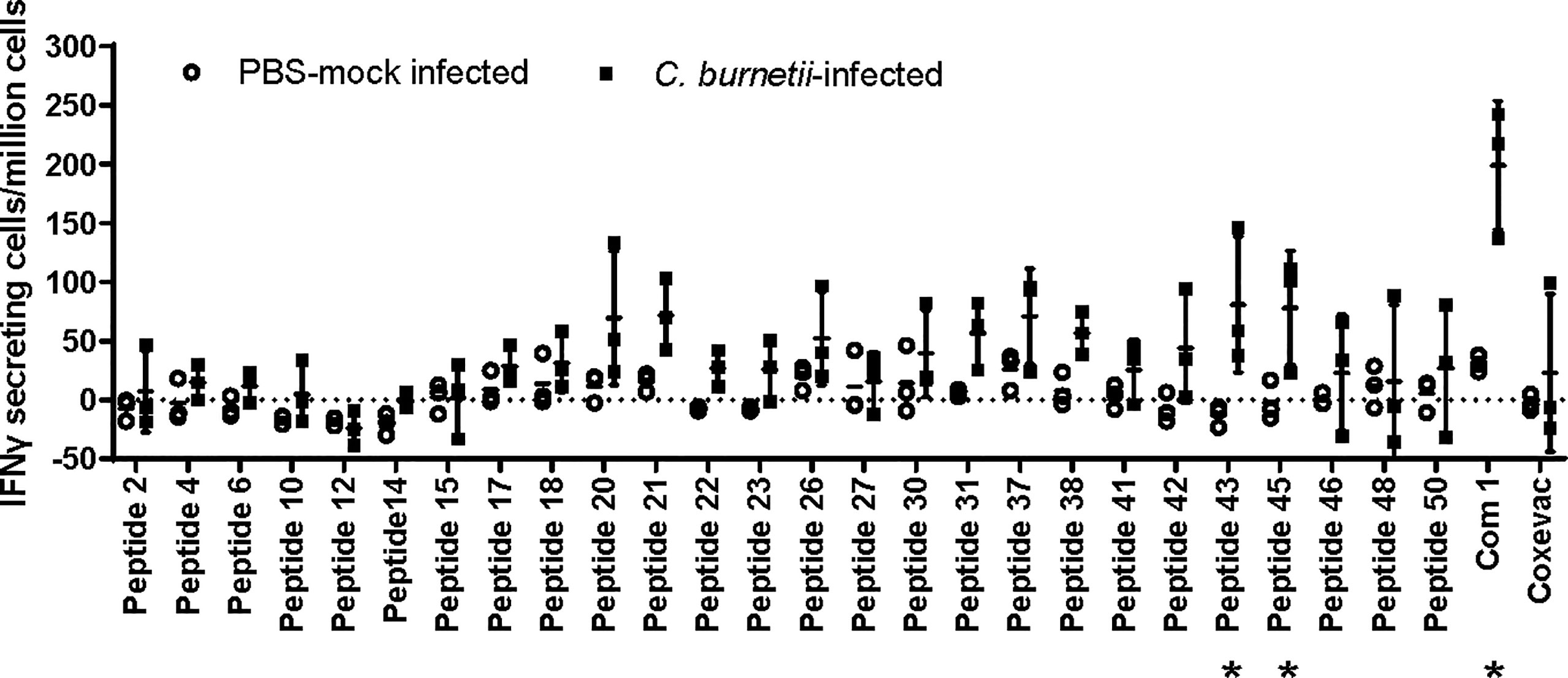
Figure 1 Antigenic recall responses in Coxiella-infected C57BL/6 mice. C57BL/6 mice (n=3 per group) were infected intranasally with 1x105 GE of C. burnetii Nine Mile strain, or mock-infected with PBS. Mice were terminated at day 51 post-infection and splenocytes recovered for ELISpot analysis. Splenocytes were stimulated with individual peptides, recombinant Com1, or the veterinary vaccine Coxevac in the presence of costimulants anti-CD28 and anti-CD173. Data are presented as IFNγ-secreting cells (spot-forming units)/million splenocytes; bars indicate group mean and standard deviation. * = p <0.05 for differences between mock-infected and C. burnetii-infected animals by two-way ANOVA with Sidak’s multiple comparison test.
3.1.2 Multi-Epitope Antigen Design
Four multi-epitope antigen designs were developed that incorporate the selected human immunoreactive peptide sequences (Figure 2A). Epitope concatemers were designed from two epitope sets: (a) all 27 selected epitopes (concatemer QVx27) and (b) an 18-epitope subset (QVx18) comprising 14 epitopes associated with particularly strong recall responses (stimulated:unstimulated response ratio >3) in at least 10% of C. burnetii-exposed humans (23) plus the four additional epitopes selected based on antigenic recall responses in C. burnetii-infected mice. Each epitope set was randomly concatenated and analyzed for non-specific potential immunogenicity at epitope junctions as well as the potential for transmembrane insertion, and rearrangements were made to minimize the potential of both. Immunogenicity at epitope junctions was assessed for mouse (C57BL/6) and human supertype HLA class II alleles to generate antigen designs with pre-clinical to clinical translation potential. The final epitope arrangements have no predicted immunogenicity at the epitope junctions nor potential for transmembrane insertion (Figures S1, S2). Final antigen designs for each concatemer incorporated a C-terminal V5 epitope tag for expression monitoring and N-terminal tissue plasminogen activator (TPA) signal sequence for concatemer secretion and uptake by the exogenous antigen processing pathway (Table 5).
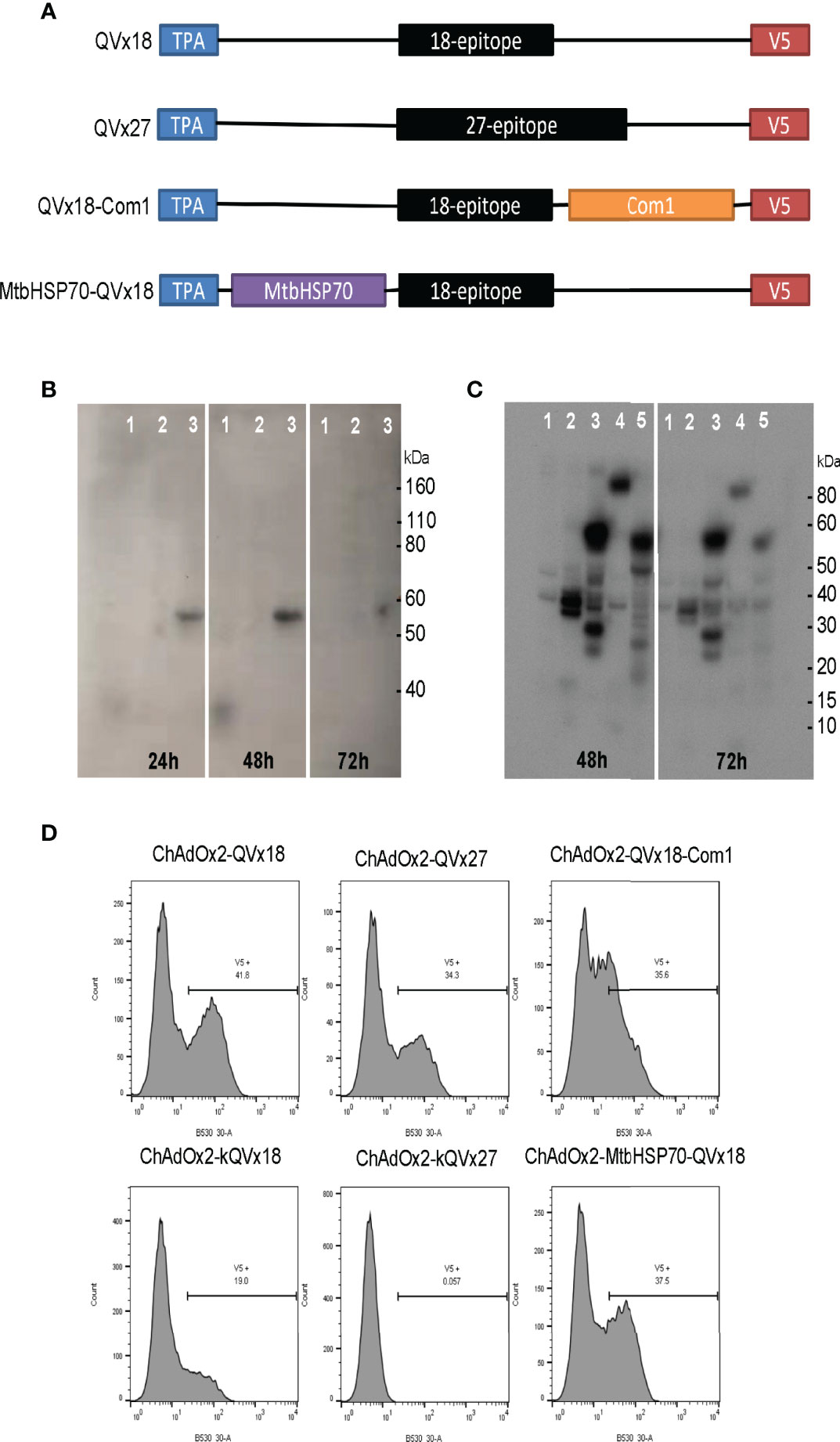
Figure 2 Design and expression of multi-epitope vaccine antigens. (A) Vaccine antigen designs. Sequences of 18-epitope and 27-epitope concatemers are given in Table 5. (B, C) Western blot analysis of epitope concatemer expression in HEK293 cells transfected with plasmids encoding the vaccine antigen inserts. Expected molecular weights: QVx18 concatemer, 36 kDa; QVx27 concatemer; 54 kDa; QVx18-Com1, 59 kDa; MtbHSP70-QVx18, 102 kDa. Vaccine antigen designs evaluated in (B) 1: QVx18; 2: empty plasmid; 3: QVx27. Vaccine antigen designs evaluated in (C) 1: empty plasmid; 2: kQVx18; 3: QVx18-Com1; 4: MtbHSP70-QVx18; 5: kQVx27. (D) Expression of vaccine antigens in HeLa cells infected with ChAdOx2 vaccine constructs detected by anti-V5 flow cytometry.

Table 5 Epitope concatemer vaccine designs.
Two additional antigen designs were informed by literature reports describing the activity of protein-concatemer fusion vaccines (44, 45): (a) fusion of the 18-epitope concatemer to full-length Com1, a known target of anti-Coxiella antibodies (46), to generate a combination vaccine targeting both T cells and B cells; and (b) fusion of the 18-epitope concatemer to Mycobacterium tuberculosis HSP70 (MtbHSP70), which has multiple immune-adjuvanting activities that might promote responses to the concatemer epitopes (47, 48). Fusions of the 18-epitope concatemer to the N-terminus or C-terminus of Com1 and to the C-terminus of MtbHSP70, in conjunction with inclusion of the N-terminal TPA signal sequence and the C-terminal V5 expression tag, were evaluated for junctional neoepitopes. Based on the results, construct designs fusing the 18-epitope concatemer to the N-terminus of Com1 and to the C-terminus of MtbHSP70 were selected for production.
3.1.3 Vaccine Constructs
The four vaccine antigen designs were each produced in two different adenoviral vectors (Table 1). Constructs in a human Ad5-derived (HuAd5) vaccine vector were produced for murine studies, although these will not be candidates for clinical use due to pre-existing adenovirus immunity in humans. To circumvent such pre-existing immunity, ChAdOx2, a derivative of the chimpanzee adenovirus (ChAd) 68, was utilized as a clinically-relevant vaccine delivery vector (34, 39). In addition, vaccine insert variants that incorporated a Kozak consensus translation initiation sequence were generated in both adenoviral vectors for the QVx18 and QVx27 antigen designs (Table 1). Vaccination regimens combining an adenoviral prime vaccine with a heterologous boost using a Modified Vaccinia Ankara (MVA)-vectored vaccine can increase the breadth and robustness of immune responses to a vaccine antigen (42, 49, 50). Therefore, an MVA-vectored vaccine expressing the TPA/27-epitope/V5 concatemer (MVA-kQVx27), which could be used as a heterologous boost for any of the adenovirus vaccines, was also produced.
Western blot detection of the C-terminal V5 expression tag confirmed expression of proteins of the expected sizes from plasmid clones bearing each of the vaccine inserts (Figures 2B, C). Vaccine insert expression from the adenoviral constructs was evaluated by flow cytometry, confirming antigen expression from the ChAdOx2 constructs (Figure 2D). Notably, the ChAdOx2 variants incorporating the Kozak sequence (kQVx18 and kQVx27) exhibited lower levels of detected antigen expression (Figure 2D), as did the HuAd5 constructs (Figure S3).
3.2 Immunogenicity of Epitope Concatemer Vaccines in Mice
To determine if adenoviral delivery of the epitope concatemer vaccines induced T cell responses, the vaccine constructs were tested for immunogenicity in C57BL/6 mice. Mice were vaccinated with HuAd5 vaccines expressing either the QVx18 or QVx27 concatemer (Table 1). Splenocytes were harvested at 21 days post-vaccination and tested in ELIspot assays for IFNγ recall responses to twelve peptides for which antigenic recall responses had previously been observed in Coxiella-infected C57BL/6 (Table 4) (23). Specific responses were detected to 8 of these peptides in at least a subset of vaccinated animals (Figures 3A, B). Epitope-specific responses were also detected in C57BL/6 mice immunized with the MVA-kQVx27 vaccine (Figure 3C). Responses were strongest to epitope p45. Of note, p45 largely overlaps with one of the class II epitopes included in a peptide vaccine previously reported to confer partial protection against C. burnetii infection in C57BL/6 mice (21).
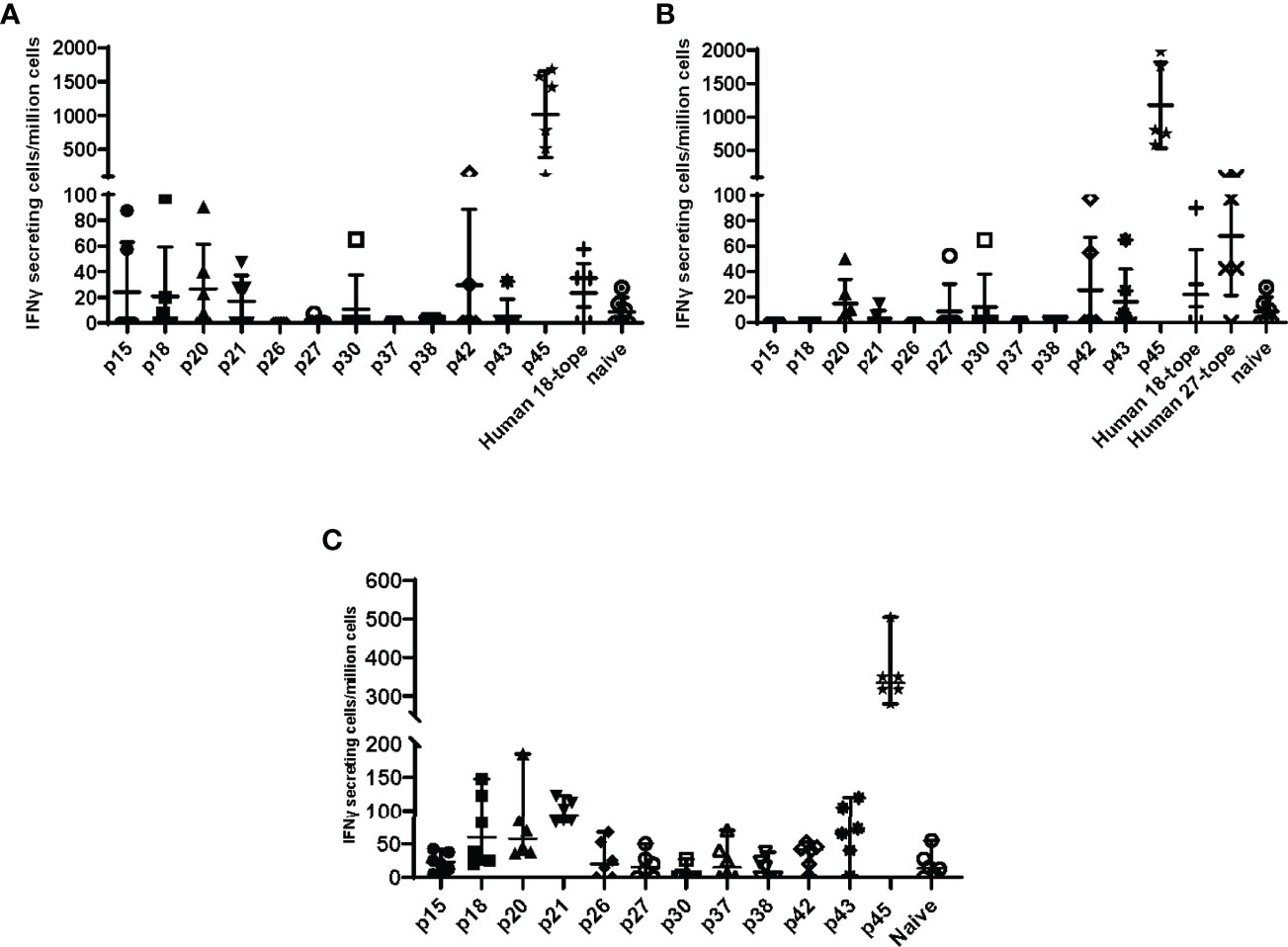
Figure 3 Epitope concatemer vaccine immunogenicity in C57BL/6 mice. Mice received a single dose of (A) HuAd5-QVx18 (n=5/group), (B) HuAd5-QVx27 (n=5/group), or (C) MVA-kQvX27 (n=6/group), administered IM in saline; naïve mice were unvaccinated. Splenocytes were harvested 21 days post-HuAD5 vaccination or 7 days post-MVA vaccination, and tested for peptide-specific T cell responses based on IFNγ production as measured by direct ELISpot assays. Peptides used for stimulation of splenocytes of infected mice are indicated along the X axis. Negative control splenocytes from naïve mice were stimulated with a pool of all assayed peptides. Responses to individual peptides were determined for those epitopes for which antigenic recall responses were observed in Coxiella-infected mice (see Table 4). Responses to the remaining epitopes were assessed by stimulation with pools of peptides representing epitopes common to both concatemer designs (Human 18-tope) or those unique to the 27-epitope design (Human 27-tope). Data are presented as IFNγ-secreting cells (spot-forming units)/million splenocytes for each individual mouse per group; bars indicate group mean and standard deviation.
Heterologous prime-boost and prime-boost-boost vaccination schedules were compared using the HuAd5-QVx27 and MVA-kQVx27 vaccine constructs to determine if responses to epitopes other than p45 were increased following boost vaccinations. MVA boost vaccinations were delivered 9 and 12 weeks after the HuAd5 prime vaccination, and animals were sacrificed 7 days after the final MVA boost. The response to p45 remained the dominant response under all vaccination schedules (Figure S4). The remaining four adenoviral vaccine constructs (fusion proteins, Kozak sequence addition) were tested to determine whether these specific design variations might support an immunogenic response to a broader range of epitopes. However, results for these four vaccine constructs were similar, with the epitope p45 again eliciting the strongest responses (Figure S5).
Based on the combined results of the expression and immunogenicity studies, the QVx18 and QVx27 adenoviral vaccines were selected for additional characterization and testing. The QVx18-Com1 adenoviral vaccines were also evaluated in further studies, to assess the impact of including a known target of anti-Coxiella antibodies.
3.3 Evaluation of Vaccine Efficacy in a Murine Vaccine-Challenge Model
The QVx18, QVx18-Com1, and QVx27 vaccines were tested in a C57BL/6 mouse vaccine-challenge study, using a HuAd5 prime-MVA boost-boost vaccination schedule (Table 2). Satellite immunogenicity groups (3 mice/group; Table S1) received a prime HuAd5 vaccination and either two MVA boosts (2 animals) or no boost (1 animal). Epitope-specific responses were confirmed by ELISpot assays, with the strongest responses again being observed against p45 (Figure S6). The challenge study was powered to detect an effect size relative to unvaccinated animals approximately 50% of the protective effect of the veterinary vaccine Coxevac® (27). Compared to control animals, Coxevac®-immunized animals showed significantly (p<0.001) reduced spleen-to-body weight ratios and bacterial burden upon challenge infection. In contrast, no definitive differences in either of these two disease endpoints were observed between epitope vaccine groups and the unvaccinated control animals (Figure 4). These results indicate that a robust T cell response to the p45 epitope alone is not sufficient to confer protection against a subsequent C. burnetii infection challenge in this murine infection model. This is consistent with results from other murine vaccine-challenge studies in which multi-epitope T cell responses but not responses to single epitopes conferred protection against C. burnetii challenge (21).
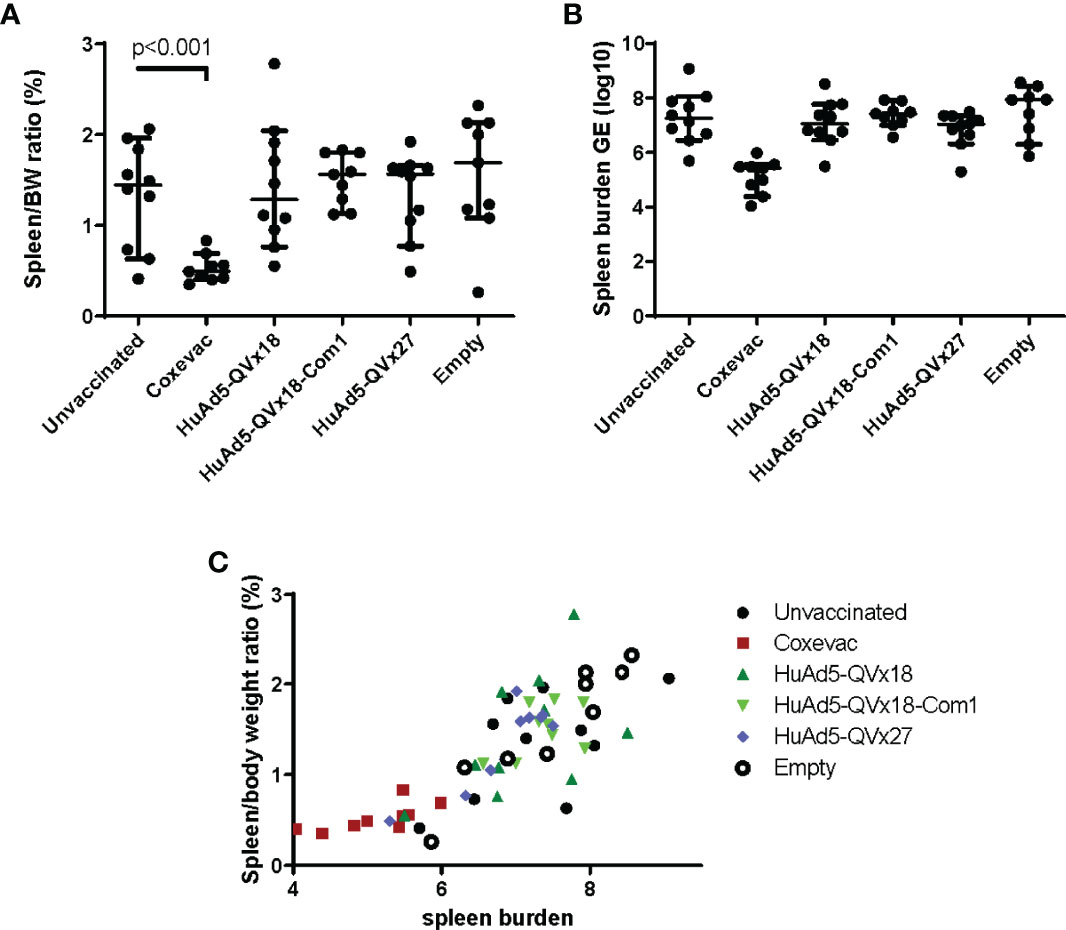
Figure 4 Murine vaccine efficacy study. C57BL/6 mice were vaccinated using a heterologous prime-boost-boost schedule as indicated in Table 2. Group labels in the graphs indicate the prime vaccine administered. Sixteen days after the second MVA-kQVx27 boost, mice were intranasally challenged with C. burnetii (Nine Mile strain). Mice were euthanized at two weeks after challenge. (A) Spleens were harvested and weighed, and spleen-to-body weight ratio determined. Only the Coxevac®-immunized group was significantly different from the unvaccinated control group (p<0.001; two-tailed t test). (B) Splenocytes were collected and the spleen bacterial burden was determined from homogenates by qPCR. No treatment groups were significantly different from the unvaccinated controls. (C) The relationship between spleen-to-body weight ratio and bacterial burden is plotted for each individual mouse.
3.4 Comparison of Vaccine Candidates to a Whole-Cell Control Vaccine in a Guinea Pig Model of Q Fever Vaccine Reactogenicity
The selected ChAdOx2 vaccine candidates and MVA-kQVx27 were evaluated for reactogenicity relative to positive and negative controls (Table 3) in a C. burnetii-sensitized guinea pig model (41). Induction of anti-C. burnetii immune responses were confirmed in sensitized animals prior to vaccine antigen challenge by detection of circulating C. burnetii-specific antibodies (Figure S7). Sensitization of animals to Coxiella antigens was confirmed using the formalin-inactivated phase I whole cell vaccine Coxevac® as a standardized positive control challenge antigen. The Coxevac® challenge sites in C. burnetii-sensitized animals developed detectable erythema and induration, with reactions reaching full extent within 24-72 hours post-challenge and persisting through termination on day 7 post-challenge (Table S2), consistent with previous observations in this model (41). No gross reactions were noted at Coxevac® challenge sites in saline control animals. No reactions were observed to challenge with a control saline solution in either saline- or C. burnetii-sensitized animals.
Reactions to ChAdOx2-vectored vaccines were noted within 8 hours post-challenge in both saline- and C. burnetii-sensitized animals; these reactions subsided by day 7 post-challenge (Table S2). Minimal reactions were noted to the corresponding empty ChAdOx2 vector. Reactions to the MVA-vectored vaccine and to the empty MVA vector were observed in both sensitized and unsensitized animals, and these reactions persisted through day 7 post-challenge (Table S2).
Tissues were collected for histological assessment at termination on day 7 post-challenge. No histological reactions were observed at negative control (saline) inoculation sites in either sensitized or unsensitized animals. Significant histological reactions consistent with pyogranulomatous changes were noted at positive control (Coxevac®) inoculation sites in sensitized animals (Figure 5). Mild to moderate histological changes were observed at inoculation sites for ChAdOx2 or MVA vectored-vaccine candidates in both sensitized and unsensitized animals (Figure 5). Similar reactions were observed to empty ChAdOx2 and MVA vectors.
Figure 5 Histological assessment of reactions to vaccine challenge in guinea pigs. Female guinea pigs (n=3/group) were sensitized by intranasal inoculation with 106 GE of C. burnetii Nine Mile strain or saline, as described previously (41). Intradermal vaccine challenges (summarized in Table 3) are indicated on the X axes. Histological lesions of skin biopsies were scored on a graded scale (41). Data are presented as the mean score from two skin sections per inoculation site for each animal; bars indicate group mean and standard deviation.
Altogether, these results indicate that reactions occurred to the viral vaccine vectors independent of prior C. burnetii-sensitization, and that there was no reactogenicity specific to the presence of the epitope concatemer vaccine antigens in this guinea pig model.
3.5 Immunogenicity of Epitope Concatemer Vaccines in Nonhuman Primates
Based on the results of the mouse immunogenicity studies of the epitope concatemer vaccines, two vaccine candidates (ChAdOx2-QVx18-Com1 and ChAdOx2-QVx27) were advanced to initial immunogenicity studies in Mauritian cynomolgus macaques (n=2 per group), using a ChAdOx2 prime-MVA boost-boost vaccination schedule (Figure 6A) to maximize the possibility of observing immunogenic responses. Three of the animals shared a single major histocompatibility complex (MHC) haplotype, and one animal (9219) was homozygous for this haplotype (Table S3). The MVA-kQVx27 vaccine, which included all the epitopes in the concatemers expressed by both prime vaccines (see Tables 1, 5), was used for all heterologous boost vaccinations. Animals were visually examined twice per week for any signs of injection site reactions. No reactions were observed following the prime vaccinations. After the first MVA boost all animals exhibited a minor transient weight loss (Figure S8).
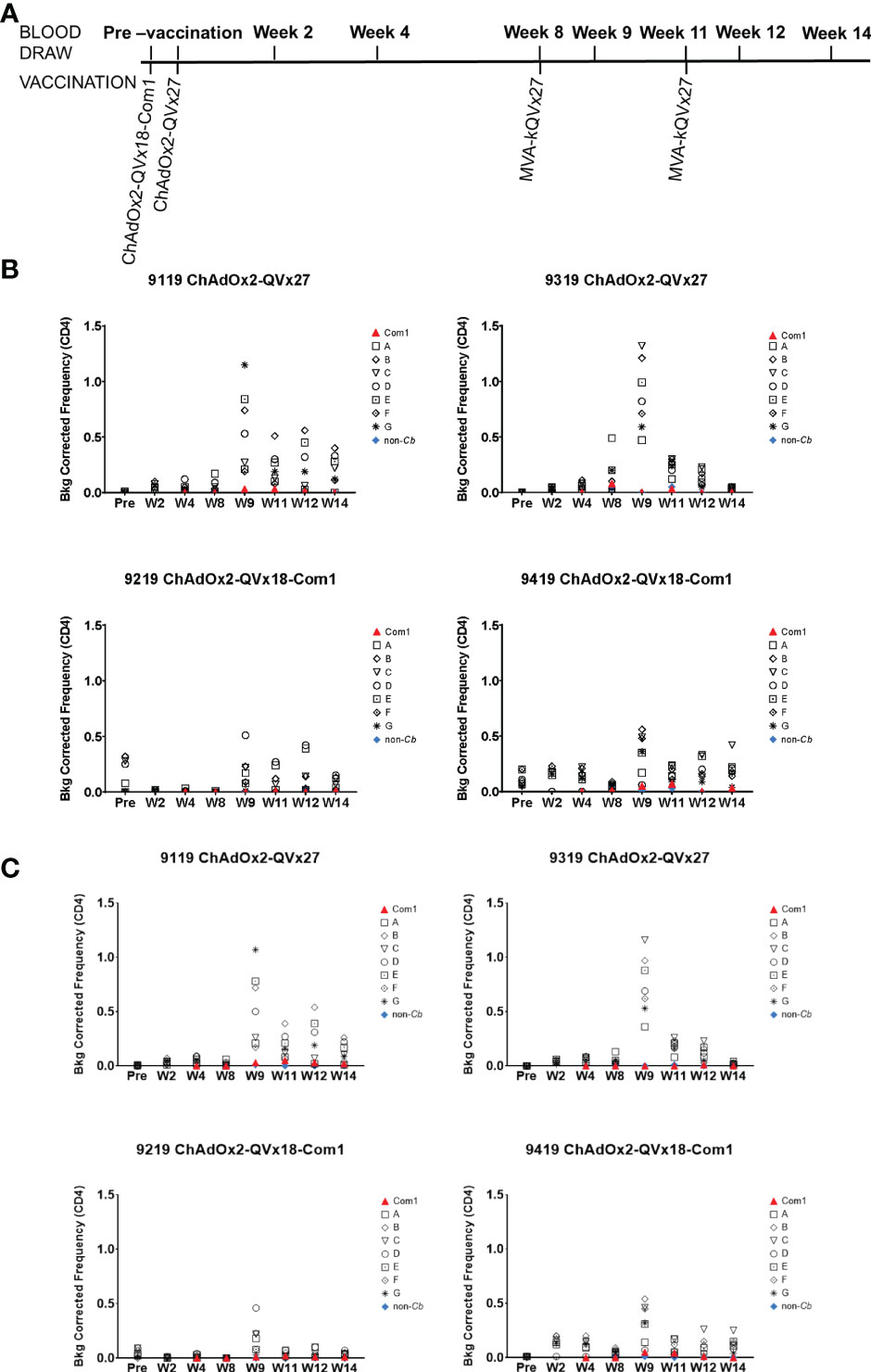
Figure 6 Ex vivo CD4+ T cell recall responses to vaccine epitopes in vaccinated cynomolgus macaques. (A) Schedule of activities for nonhuman primate vaccine immunogenicity study. Cynomolgus macaques received prime vaccinations in Week 0 using the indicated ChAdOx2 vaccine constructs (1x109 IU/vaccination, delivered IM) (n = 2 for each vaccine construct). All animals received MVA-kQVx27 boost vaccinations (1x108 IU/vaccination, delivered IM) at Weeks 8 and 11. Animals 9119 and 9319 received ChAdOx2-QVx27 prime vaccination. Animals 9219 and 9419 received ChAdOx2-QVx18-Com1 prime vaccination. (B, C) PBMCs from individual animals were stimulated ex vivo by the indicated epitope peptide pools (A-G, see Methods), a pool of peptides representing the full Com1 protein sequence, a short concatemer of non-Coxiella peptide sequences (non-Cb), or no peptide. T cell activation status following peptide stimulation was evaluated by flow cytometry. Study time points are indicated on the X axis (Pre, pre-vaccination; W, Week). The frequencies of (B) CD4+ IFNγ+ or (C) CD4+ CD154+ IFNγ+ T cells are shown as a percentage of total CD4+ T cells (Y axis). Data shown are background corrected (peptide stimulated minus unstimulated). Individual macaque identification numbers and prime vaccine are noted in each graph title.
Antigen-specific CD4+ T cell responses were observed in all animals one week after the first boost, as indicated by the increased frequency of CD4+ T cells expressing IFNγ and the early activation marker CD154 in response to peptide stimulation, (see week 9 in Figures 6B, C). CD4+ T cells expressing the transcription factor T-bet were also increased following immunization (Figure S9), paralleling the contribution of T-bet to responses to inactivated whole cell C. burnetii vaccines (11, 18). Antigen-specific CD8+ T cell responses were also observed, reflected by increased frequencies of both CD8+ IFNγ+ (Figure 7A) and CD8+ T-bet+ (Figure 7B). The two animals receiving the ChAdOx2-QVx27 prime vaccination showed greater responses than did the two primed with the ChAdOx2-QVx18-Com1 vaccine, though determining whether this reflects true differences in prime vaccine function or simple variations in responses by individual macaques would require a larger study.
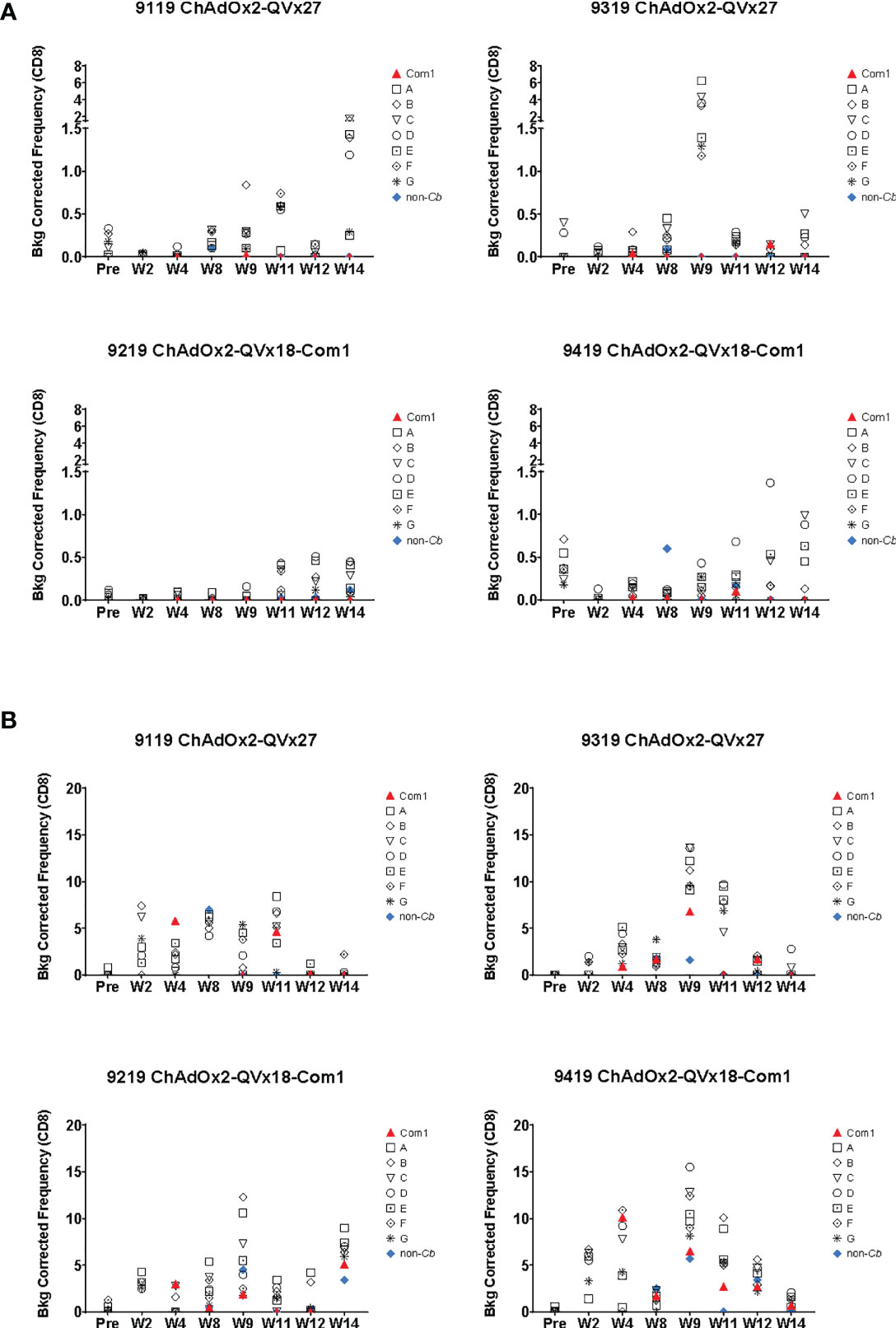
Figure 7 Ex vivo CD8+ T cell recall responses to vaccine epitopes in vaccinated cynomolgus macaques. Study time points are indicated on the X axis (Pre = pre-vaccination; W = Week); see Figure 6A for the corresponding vaccination schedule. Individual macaque identification numbers and prime vaccine are noted in each graph title. MVA-kQVx27 boost vaccinations were administered at Week 8 and Week 11. PBMCs from individual animals were stimulated ex vivo by indicated epitope peptide pools (A-G, see Methods), a pool of peptides representing the full Com1 protein sequence, a short concatemer of non-Coxiella peptide sequences (non-Cb), or no peptide. T cell activation status following peptide stimulation was evaluated by flow cytometry. The frequencies of (A) CD8+ IFNγ+ or (B) CD8+ T-bet+ T cells are shown as a percentage of total CD8+ T cells (Y axis). Data shown are background corrected (peptide stimulated-unstimulated).
Responses were observed to multiple peptide pools in all animals, indicating that cynomolgus macaques exhibit responses to a broader range of the vaccine-encoded human-targeted HLA class II epitopes than were observed in C57BL/6 mice. These results demonstrate that delivery of human T cell epitope concatemers via viral vaccine vectors can elicit T cell responses to multiple epitopes in non-human primates, although the most effective vaccination schedule, the duration of the induced T cell responses, and the magnitude of any recall response upon infection challenge remain to be determined.
4 Discussion
We describe here the design and initial preclinical testing of multi-epitope T cell-targeted vaccine candidates for the prevention of Q fever disease in humans. Promiscuous HLA class II epitope clusters included in these vaccines were selected from C. burnetii antigens to which the host immune system is expected to be exposed during course of infection (23). Although the specific roles in the pathophysiology of infection are not known for many of these antigens, the durable T cell memory responses detected 7-10 years after infection associate the selected epitopes with presumed post-infection immunity. Whether exposure to these epitopes separately from an active infection can confer protection against subsequent Q fever disease remains to be determined, and a key objective of the studies reported here was assessment of potential correlates of immune response, safety and efficacy in established experimental animal models of C. burnetii infection (51).
C57BL/6 mice were selected for use in initial vaccine immunogenicity and efficacy tests, as they exhibit less severe disease and lower mortality in response to C. burnetii infection compared to other mouse strains (52), and are arguably more reflective of the acute non-lethal disease observed in humans. Twelve of the twenty-seven human epitopes included in these vaccines stimulated antigenic recall responses in splenocytes from C. burnetii-infected C57BL/6 mice (Table 4 and Figure 1). However, vaccine immunogenicity in the C57BL/6 mouse model was dominated by a single epitope (p45), and this was insufficient to confer protection against infection challenge. Whether the narrow immunogenicity of these vaccines in C57BL/6 is due to suboptimal recognition of the human-targeted epitopes in the C57BL/6 MHC background, to poor processing and presentation of the epitope concatemers, or to other factors remains unknown. In this context, three observations suggest that the limited immunogenicity is not due simply to truncated translation of the vaccine antigens: Expression of full-length concatemers in cultured cells was confirmed by immunological detection of the C-terminal V5 expression tag (Figure 2). The dominant p45 epitope is in the center of the epitope concatemers (see Table 5 and Figures S1, S2), and there is no apparent bias toward higher immunogenicity of N-terminal epitopes. In addition, mice vaccinated with the HuAd5-QVx18-Com1 vaccine, in which Com1 is C-terminal to the epitope concatemer, exhibit robust T cell responses to Com1 (Figure S5).
Guinea pigs played a central role in the identification of the causative infectious agent for Q fever (53), and they more fully replicate aspects of human acute Q fever than do mice (51, 54). Unfortunately, reagents and protocols for analyzing cellular immunology in this species are limited, and little information is available regarding recognition of human T cell epitopes by the guinea pig immune system. However, the guinea pig is an established model for assessing the reactogenicity of candidate Q fever vaccines (41, 55). We therefore tested the T cell vaccine candidates in a sensitized guinea pig reactogenicity model. The observation of transient reactions to the viral vectors (ChAdOx2 and MVA) in both sensitized and unsensitized animals is consistent with the transient reactions reported in human tests of ChAd- and MVA-vectored vaccines expressing non-Coxiella antigens (56, 57). While the results indicate that the antigens encoded in the vaccine candidates do not elicit specific hypersensitivity responses, we note that immunogenicity of these antigens in guinea pigs was not directly demonstrated. As the recognition of the encoded epitopes by guinea pig MHCs is unknown, we cannot definitively rule out the potential for specific reactogenic responses in a different species.
Aerosol exposure models of acute Q fever have been established in both rhesus and cynomolgus macaques, and the latter have been suggested as a preferred non-human primate model on the basis of higher similarity of pathological responses to those in humans (58, 59). Macaque MHC classes are homologous to those of humans, although they do not exhibit simple one-to-one correlation with human HLA subtypes due to the more extensive expansion of MHC genes in macaques (60). Notably, the repertoire of MHC class II epitopes from Mycobacterium tuberculosis recognized in both cynomolgus and rhesus macaques largely overlaps that of human T cells (61). Thus, macaque cellular responses to T cell vaccines are expected to be more predictive of human responses than are those of the small animal models. We advanced two of our vaccine candidates to an initial immunogenicity study in cynomolgus macaques. Specific CD4+ responses following vaccination were observed to multiple peptide pools in all animals, which had distinct MHC genotypes, indicating that cynomolgus macaques exhibit responses to a broader range of the human-targeted HLA class II epitopes encoded in these vaccines than was observed in C57BL/6 mice. Intriguingly, specific CD8+ responses were also observed in vaccinated macaques. Whether these responses arise from cryptic MHC class I epitopes within the vaccine-encoded class II sequences or from unconventional MHC class II restricted CD8+ T cells (62) remains to be investigated.
Defining an appropriate development path for vaccines expected to demonstrate efficacy only in the context of the human immune system has been recognized as a limitation in emerging disease response readiness, requiring consideration of regulatory requirements on a case-by-case basis (63, 64). In particular, species differences in MHC binding preferences present challenges in the preclinical development of human-targeted T cell vaccines, especially for a select agent such as C. burnetii for which intentional human exposure would not be considered safe or ethical (65, 66), as vaccine responses in animal models may not fully reflect the potential responses to such vaccines in humans. These challenges are reflected in the studies reported here for human-targeted multi-epitope Q fever vaccines. The lack of established tools for cellular immunology limits the utility of the guinea pig in defining correlates of immunity or protection for development of such T cell-targeted vaccines. The greater availability of immunological reagents and relatively low cost have made the mouse a favored research model for Q fever as for other diseases. However, the combined results of our murine immunogenicity and vaccine-challenge studies indicate that the C57BL/6 mouse is not an effective model for preclinical testing of these specific human-targeted multi-epitope vaccines.
The broad immunogenic responses to the vaccine candidates in cynomolgus macaques provide a foundation for further preclinical and clinical evaluation of a T cell-targeted vaccine for Q fever. Reagents and protocols are established for assessment of both humoral and cellular immune responses in macaques, enabling animal efficacy studies capable of defining correlates of protection bridging animal model results with human clinical responses. In this context, some consideration of the design of animal efficacy studies is warranted. Sterilizing immunity is a common efficacy objective in vaccine studies and could be reasonably considered a correlate of protection from disease. However, in the aerosol-challenge macaque model, vaccination with the efficacious commercial vaccine Q-VAX reduced but did not prevent bacteremia, though disease symptoms and pathology were reduced or abrogated in vaccinated animals compared to unvaccinated animals (59). Thus, prevention of acute Q fever disease may not require fully sterilizing immunity, and requiring an endpoint in the macaque model that is not reached by a vaccine with proven efficacy in humans may represent an unachievably stringent objective for development of a new Q fever vaccine. The suitability of sterilizing immunity as an efficacy endpoint is particularly questionable for T cell-targeted vaccines intended to induce infection-clearing, and thereby disease-limiting, cellular immunity rather than infection-blocking humoral immunity. For novel vaccine development programs anticipating such challenges, the US Food and Drug Administration recommends early discussions with agency staff regarding the design of studies that would support regulatory review (66). The immunogenic responses in macaques to the vaccine candidates described here provide a basis for such a discussion to define an expanded preclinical testing plan that could inform and justify first-in-human testing of a T cell-targeted vaccine for Q fever.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Colorado State University Institutional Animal Care and Use Committee; Massachusetts General Hospital Institutional Animal Care and Use Committee; Oxford University Animal Care and Ethical Review Committee; US Defense Threat Reduction Agency Animal Care and Use Review Office.
Author Contributions
AES, SRP, LM, LB, AS, PR, RB, AP, AG, CR, and MP conceptualized and designed the study and experiments. Experiments were performed by SRP, CD, GR, LS-R, and LB. Data were analyzed and interpreted by AES, SRP, LM, CD, GR, LB, AS, PR, CR, and MCP. GR, LM, PR, RB, ADG, CR, and MP contributed critical reagents, materials and analytic tools. AES, LM, AG, RB, ADG, CR, and MP acquired funding and supervised research activities. AES, SRP, LM, LB and CR wrote the manuscript and GR, PR, AS, AP, AG, RB, and MP critically reviewed and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by contract HDTRA1-15-C-0020 from the US Defense Threat Reduction Agency (www.dtra.mil), awarded to Massachusetts General Hospital (MCP Lead Principal Investigator); work by authors at other institutions was supported by subcontracts under the prime contract award to MGH.
Conflict of Interest
MP, AES, ADG, LM, AG, and AS are named inventors on patent application WO 2019/183627 A1, “Coxiella burnetii epitopes for T cell-targeted Q fever vaccines”.
AG is a senior officer and shareholder and AS was an employee of Innatoss Laboratories B.V., which provides diagnostic screening for Q fever. ADG is a senior officer and shareholder, and LM and GR are employees of EpiVax, Inc., a company specializing in immunoinformatic analysis. Innatoss Laboratories B.V and EpiVax, Inc., own patents to technologies utilized by associated authors in the research reported here.
AP is Chair of UK Dept. Health and Social Care’s (DHSC) Joint Committee on Vaccination & Immunisation (JCVI), and is a member of the WHO’s SAGE. AP is an NIHR Senior Investigator. The views expressed in this article do not necessarily represent the views of DHSC, JCVI, NIHR or WHO. CR, CD, and AP are named inventors on a patent application in the field of meningococcal vaccines. AP waives all his rights to any patent.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Nicole Marlenee, Stephanie Porter, Danielle Adney, Paul Gordy, Airn Tolnay, Angelo Izzo, and the CSU Experimental Pathology Facility for technical assistance, Ghazel Mukhtar for assisting with preliminary data analysis, Tyler Roady for assisting in manuscript preparation, and Timothy Brauns for helpful discussions. The authors thank the Jenner Institute Viral Vector Core Facility and the staff at the Oxford University Functional Genomics Facility. The following individuals are acknowledged for dedicated and flexible support of the macaque immunogenicity study during the early months of the SARS-CoV-2 pandemic and the associated local public health restrictions: Dr. Joanne Morris and staff of the MGH Center for Comparative Medicine for vaccinating and collecting samples from the macaques; Dr. Ruxandra Sirbulescu, Laura Valine, Michael Chapin, Dr. Huabiao Chen and Dr. Sen Han for assisting with the preparation of macaque samples; and Heather Cahill from the MGH Flow Cytometry Core for assisting with data collection. Coxiella burnetii (Strain Nine Mile Phase I RSA 411, NR-134) was obtained through BEI Resources, National Institute of Allergy and Infectious Disease of the US National Institutes of Health. We thank Ceva Biomune for providing the Coxevac® vaccine.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.901372/full#supplementary-material
References
1. Akamine CM, Perez ML, Lee JH, Ing MB. Q Fever in Southern California: A Case Series of 20 Patients From a VA Medical Center. Am J Trop Med Hyg (2019) 101(1):33–9. doi: 10.4269/ajtmh.18-0283
2. Eldin C, Melenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, et al. From Q Fever to Coxiella burnetii Infection: A Paradigm Change. Clin Microbiol Rev (2017) 30(1):115–90. doi: 10.1128/CMR.00045-16
3. Hopper B, Cameron B, Li H, Graves S, Stenos J, Hickie I, et al. The Natural History of Acute Q Fever: A Prospective Australian Cohort. QJM (2016) 109(10):661–8. doi: 10.1093/qjmed/hcw041
4. Anderson AD, Baker TR, Littrell AC, Mott RL, Niebuhr DW, Smoak BL. Seroepidemiologic Survey for Coxiella burnetii Among Hospitalized US Troops Deployed to Iraq. Zoonoses Public Health (2011) 58(4):276–83. doi: 10.1111/j.1863-2378.2010.01347.x
5. Dahlgren FS, McQuiston JH, Massung RF, Anderson AD. Q Fever in the United States: Summary of Case Reports From Two National Surveillance Systems, 2000-2012. Am J Trop Med Hyg (2015) 92(2):247–55. doi: 10.4269/ajtmh.14-0503
6. Royal J, Riddle MS, Mohareb E, Monteville MR, Porter CK, Faix DJ. Seroepidemiologic Survey for Coxiella burnetii Among US Military Personnel Deployed to Southwest and Central Asia in 2005. Am J Trop Med Hyg (2013) 89(5):991–5. doi: 10.4269/ajtmh.12-0174
7. Kampschreur LM, Oosterheert JJ, Hoepelman AI, Lestrade PJ, Renders NH, Elsman P, et al. Prevalence of Chronic Q Fever in Patients With a History of Cardiac Valve Surgery in an Area Where Coxiella burnetii Is Epidemic. Clin Vaccine Immunol (2012) 19(8):1165–9. doi: 10.1128/CVI.00185-12
8. Marmion BP. A Guide to Q Fever and Q Fever Vaccination. Parkville, Victoria, Australia: CSL Biotherapies (2009).
9. Schoffelen T, Wong A, Rumke HC, Netea MG, Timen A, van Deuren M, et al. Adverse Events and Association With Age, Sex and Immunological Parameters of Q Fever Vaccination in Patients at Risk for Chronic Q Fever in the Netherlands 2011. Vaccine (2014) 32(49):6622–30. doi: 10.1016/j.vaccine.2014.09.061
10. Sellens E, Bosward KL, Willis S, Heller J, Cobbold R, Comeau JL, et al. Frequency of Adverse Events Following Q Fever Immunisation in Young Adults. Vaccines (Basel) (2018) 6(4):83. doi: 10.3390/vaccines6040083
11. Reeves PM, Raju Paul S, Baeten L, Korek SE, Yi Y, Hess J, et al. Novel Multiparameter Correlates of Coxiella burnetii Infection and Vaccination Identified by Longitudinal Deep Immune Profiling. Sci Rep (2020) 10(1):13311. doi: 10.1038/s41598-020-69327-x
12. Shannon JG, Heinzen RA. Adaptive Immunity to the Obligate Intracellular Pathogen Coxiella burnetii. Immunol Res (2009) 43(1-3):138–48. doi: 10.1007/s12026-008-8059-4
13. Zhang G, Zhang Y, Samuel JE. Components of Protective Immunity. Adv Exp Med Biol (2012) 984:91–104. doi: 10.1007/978-94-007-4315-1_5
14. Zhang G, Russell-Lodrigue KE, Andoh M, Zhang Y, Hendrix LR, Samuel JE. Mechanisms of Vaccine-Induced Protective Immunity Against Coxiella burnetii Infection in BALB/c Mice. J Immunol (2007) 179(12):8372–80. doi: 10.4049/jimmunol.179.12.8372
15. Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. T Cells Are Essential for Bacterial Clearance, and Gamma Interferon, Tumor Necrosis Factor Alpha, and B Cells Are Crucial for Disease Development in Coxiella burnetii Infection in Mice. Infect Immun (2007) 75(7):3245–55. doi: 10.1128/IAI.01767-06
16. Read AJ, Erickson S, Harmsen AG. Role of CD4+ and CD8+ T Cells in Clearance of Primary Pulmonary Infection With Coxiella burnetii. Infect Immun (2010) 78(7):3019–26. doi: 10.1128/IAI.00101-10
17. Gregory AE, van Schaik EJ, Fratzke AP, Russell-Lodrigue KE, Farris CM, Samuel JE. Soluble Antigens Derived From Coxiella burnetii Elicit Protective Immunity in Three Animal Models Without Inducing Hypersensitivity. Cell Rep Med (2021) 2(12):100461. doi: 10.1016/j.xcrm.2021.100461
18. Ledbetter L, Cherla R, Chambers C, Zhang Y, Mitchell WJ, Zhang G. Major Histocompatibility Complex Class II-Restricted, CD4(+) T Cell-Dependent and -Independent Mechanisms Are Required for Vaccine-Induced Protective Immunity Against Coxiella burnetii. Infect Immun (2020) 88(3):e00824–19. doi: 10.1128/IAI.00824-19
19. Humphres RC, Hinrichs DJ. Role of Antibody in Coxiella burnetii Infection. Infect Immun (1981) 31(2):641–5. doi: 10.1128/iai.31.2.641-645.1981
20. Xiong X, Jiao J, Gregory AE, Wang P, Bi Y, Wang X, et al. Identification of Coxiella burnetii CD8+ T-Cell Epitopes and Delivery by Attenuated Listeria Monocytogenes as a Vaccine Vector in a C57BL/6 Mouse Model. J Infect Dis (2017) 215(10):1580–9. doi: 10.1093/infdis/jiw470
21. Xiong X, Qi Y, Jiao J, Gong W, Duan C, Wen B. Exploratory Study on Th1 Epitope-Induced Protective Immunity Against Coxiella burnetii Infection. PloS One (2014) 9(1):e87206. doi: 10.1371/journal.pone.0087206
22. Reeves PM, Paul SR, Sluder AE, Brauns TA, Poznansky MC. Q-Vaxcelerate: A Distributed Development Approach for a New Coxiella burnetii Vaccine. Hum Vaccin Immunother (2017) 13(12):2977–81. doi: 10.1080/21645515.2017.1371377
23. Scholzen A, Richard G, Moise L, Baeten LA, Reeves PM, Martin WD, et al. Promiscuous Coxiella burnetii CD4 Epitope Clusters Associated With Human Recall Responses Are Candidates for a Novel T-Cell Targeted Multi-Epitope Q Fever Vaccine. Front Immunol (2019) 10:207. doi: 10.3389/fimmu.2019.00207
24. Long CM. Q Fever Vaccine Development: Current Strategies and Future Considerations. Pathogens (2021) 10(10):1223. doi: 10.3390/pathogens10101223
25. Plotkin SA. Complex Correlates of Protection After Vaccination. Clin Infect Dis (2013) 56(10):1458–65. doi: 10.1093/cid/cit048
26. Omsland A, Heinzen RA. Life on the Outside: The Rescue of Coxiella burnetii From Its Host Cell. Annu Rev Microbiol (2011) 65:111–28. doi: 10.1146/annurev-micro-090110-102927
27. Reeves PM, Raju Paul S, Baeten LA, Korek SE, Yi Y, Hess J, et al. Novel Multiparameter Correlates of Coxiella burnetii Infection and Vaccination Identified by Longitudinal Deep Immune Profiling. Sci Rep (2019) 10:13311. doi: 10.1038/s41598-020-69327-x
28. Kersh GJ, Oliver LD, Self JS, Fitzpatrick KA, Massung RF. Virulence of Pathogenic Coxiella burnetii Strains After Growth in the Absence of Host Cells. Vector-Borne Zoonot (2011) 11(11):1433–8. doi: 10.1089/vbz.2011.0670
29. Arricau-Bouvery N, Souriau A, Bodier C, Dufour P, Rousset E, Rodolakis A. Effect of Vaccination With Phase I and Phase II Coxiella burnetii Vaccines in Pregnant Goats. Vaccine (2005) 23(35):4392–402. doi: 10.1016/j.vaccine.2005.04.010
30. Moise L, Gutierrez A, Kibria F, Martin R, Tassone R, Liu R, et al. iVAX: An Integrated Toolkit for the Selection and Optimization of Antigens and the Design of Epitope-Driven Vaccines. Hum Vaccin Immunother (2015) 11(9):2312–21. doi: 10.1080/21645515.2015.1061159
31. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting Transmembrane Protein Topology With a Hidden Markov Model: Application to Complete Genomes. J Mol Biol (2001) 305(3):567–80. doi: 10.1006/jmbi.2000.4315
32. Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, et al. Vienna RNA Package 2.0. Algorithms Mol Biol (2011) 6:26. doi: 10.1186/1748-7188-6-26
33. Reuter JS, Mathews DH. RNAstructure: Software for RNA Secondary Structure Prediction and Analysis. BMC Bioinf (2010) 11:129. doi: 10.1186/1471-2105-11-129
34. Morris SJ, Sebastian S, Spencer AJ, Gilbert SC. Simian Adenoviruses as Vaccine Vectors. Future Virol (2016) 11(9):649–59. doi: 10.2217/fvl-2016-0070
35. Sebastian S, Gilbert SC. Recombinant Modified Vaccinia Virus Ankara-Based Malaria Vaccines. Expert Rev Vaccines (2016) 15(1):91–103. doi: 10.1586/14760584.2016.1106319
36. Wang C, Dulal P, Zhou X, Xiang Z, Goharriz H, Banyard A, et al. A Simian-Adenovirus-Vectored Rabies Vaccine Suitable for Thermostabilisation and Clinical Development for Low-Cost Single-Dose Pre-Exposure Prophylaxis. PloS Negl Trop Dis (2018) 12(10):e0006870. doi: 10.1371/journal.pntd.0006870
37. Rollier CS, Spencer AJ, Sogaard KC, Honeycutt J, Furze J, Bregu M, et al. Modification of Adenovirus Vaccine Vector-Induced Immune Responses by Expression of a Signalling Molecule. Sci Rep (2020) 10(1):5716. doi: 10.1038/s41598-020-61730-8
38. Marsay L, Dold C, Paterson GK, Yamaguchi Y, Derrick JP, Chan H, et al. Viral Vectors Expressing Group B Meningococcal Outer Membrane Proteins Induce Strong Antibody Responses But Fail to Induce Functional Bactericidal Activity in Mice. J Infect (2022) 84:658–67. doi: 10.1016/j.jinf.2022.02.032
39. Dicks MD, Spencer AJ, Edwards NJ, Wadell G, Bojang K, Gilbert SC, et al. A Novel Chimpanzee Adenovirus Vector With Low Human Seroprevalence: Improved Systems for Vector Derivation and Comparative Immunogenicity. PloS One (2012) 7(7):e40385. doi: 10.1371/journal.pone.0040385
40. McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, et al. Enhanced T-Cell Immunogenicity of Plasmid DNA Vaccines Boosted by Recombinant Modified Vaccinia Virus Ankara in Humans. Nat Med (2003) 9(6):729–35. doi: 10.1038/nm881
41. Baeten LA, Podell BK, Sluder AE, Garritsen A, Bowen RA, Poznansky MC. Standardized Guinea Pig Model for Q Fever Vaccine Reactogenicity. PloS One (2018) 13(10):e0205882. doi: 10.1371/journal.pone.0205882
42. Capone S, Reyes-Sandoval A, Naddeo M, Siani L, Ammendola V, Rollier CS, et al. Immune Responses Against a Liver-Stage Malaria Antigen Induced by Simian Adenoviral Vector AdCh63 and MVA Prime-Boost Immunisation in non-Human Primates. Vaccine (2010) 29(2):256–65. doi: 10.1016/j.vaccine.2010.10.041
43. di Tommaso A, de Magistris MT, Bugnoli M, Marsili I, Rappuoli R, Abrignani S. Formaldehyde Treatment of Proteins can Constrain Presentation to T Cells by Limiting Antigen Processing. Infect Immun (1994) 62(5):1830–4. doi: 10.1128/iai.62.5.1830-1834.1994
44. Tobias J, Jasinska J, Baier K, Kundi M, Ede N, Zielinski C, et al. Enhanced and Long Term Immunogenicity of a Her-2/Neu Multi-Epitope Vaccine Conjugated to the Carrier CRM197 in Conjunction With the Adjuvant Montanide. BMC Cancer (2017) 17(1):118. doi: 10.1186/s12885-017-3098-7
45. Webster DP, Dunachie S, Vuola JM, Berthoud T, Keating S, Laidlaw SM, et al. Enhanced T Cell-Mediated Protection Against Malaria in Human Challenges by Using the Recombinant Poxviruses FP9 and Modified Vaccinia Virus Ankara. Proc Natl Acad Sci USA (2005) 102(13):4836–41. doi: 10.1073/pnas.0406381102
46. Vigil A, Ortega R, Nakajima-Sasaki R, Pablo J, Molina DM, Chao CC, et al. Genome-Wide Profiling of Humoral Immune Response to Coxiella burnetii Infection by Protein Microarray. Proteomics (2010) 10(12):2259–69. doi: 10.1002/pmic.201000064
47. Brauns T, Leblanc P, Gelfand JA, Poznanski M. Could Mycobacterial Hsp70-Containing Fusion Protein Lead the Way to an Affordable Therapeutic Cancer Vaccine? Expert Rev Vaccines (2015) 14(3):435–46. doi: 10.1586/14760584.2015.979797
48. Leblanc P, Moise L, Luza C, Chantaralawan K, Lezeau L, Yuan J, et al. VaxCelerate II: Rapid Development of a Self-Assembling Vaccine for Lassa Fever. Hum Vaccin Immunother (2014) 10(10):3022–38. doi: 10.4161/hv.34413
49. Rollier CS, Hill AVS, Reyes-Sandoval A. Influence of Adenovirus and MVA Vaccines on the Breadth and Hierarchy of T Cell Responses. Vaccine (2016) 34(38):4470–4. doi: 10.1016/j.vaccine.2016.07.050
50. Rollier CS, Verschoor EJ, Verstrepen BE, Drexhage JA, Paranhos-Baccala G, Liljestrom P, et al. T- and B-Cell Responses to Multivalent Prime-Boost DNA and Viral Vectored Vaccine Combinations Against Hepatitis C Virus in Non-Human Primates. Gene Ther (2016) 23(10):753–9. doi: 10.1038/gt.2016.55
52. Scott GH, Williams JC, Stephenson EH. Animal Models in Q Fever: Pathological Responses of Inbred Mice to Phase I Coxiella burnetii. J Gen Microbiol (1987) 133(3):691–700. doi: 10.1099/00221287-133-3-691
53. Hirschmann JV. The Discovery of Q Fever and Its Cause. Am J Med Sci (2019) 358(1):3–10. doi: 10.1016/j.amjms.2019.04.006
54. Russell-Lodrigue KE, Zhang GQ, McMurray DN, Samuel JE. Clinical and Pathologic Changes in a Guinea Pig Aerosol Challenge Model of Acute Q Fever. Infect Immun (2006) 74(11):6085–91. doi: 10.1128/IAI.00763-06
55. Wilhelmsen CL, Waag DM. Guinea Pig Abscess/Hypersensitivity Model for Study of Adverse Vaccination Reactions Induced by Use of Q Fever Vaccines. Comp Med (2000) 50(4):374–8.
56. de Barra E, Hodgson SH, Ewer KJ, Bliss CM, Hennigan K, Collins A, et al. A Phase Ia Study to Assess the Safety and Immunogenicity of New Malaria Vaccine Candidates ChAd63 CS Administered Alone and With MVA Cs. PloS One (2014) 9(12):e115161. doi: 10.1371/journal.pone.0115161
57. de Graaf H, Payne RO, Taylor I, Miura K, Long CA, Elias SC, et al. Safety and Immunogenicity of ChAd63/MVA Pfs25-IMX313 in a Phase I First-In-Human Trial. Front Immunol (2021) 12:694759. doi: 10.3389/fimmu.2021.694759
58. Waag DM, Byrne WR, Estep J, Gibbs P, Pitt ML, Banfield CM. Evaluation of Cynomolgus (Macaca Fascicularis) and Rhesus (Macaca Mulatta) Monkeys as Experimental Models of Acute Q Fever After Aerosol Exposure to Phase-I Coxiella burnetii. Lab Anim Sci (1999) 49(6):634–8.
59. Waag DM, England MJ, Tammariello RF, Byrne WR, Gibbs P, Banfield CM, et al. Comparative Efficacy and Immunogenicity of Q Fever Chloroform:Methanol Residue (CMR) and Phase I Cellular (Q-Vax) Vaccines in Cynomolgus Monkeys Challenged by Aerosol. Vaccine (2002) 20(19-20):2623–34. doi: 10.1016/S0264-410X(02)00176-7
60. Wiseman RW, Karl JA, Bohn PS, Nimityongskul FA, Starrett GJ, O'Connor DH. Haplessly Hoping: Macaque Major Histocompatibility Complex Made Easy. ILAR J (2013) 54(2):196–210. doi: 10.1093/ilar/ilt036
61. Mothe BR, Lindestam Arlehamn CS, Dow C, Dillon MBC, Wiseman RW, Bohn P, et al. The TB-Specific CD4(+) T Cell Immune Repertoire in Both Cynomolgus and Rhesus Macaques Largely Overlap With Humans. Tuberculosis (Edinb) (2015) 95(6):722–35. doi: 10.1016/j.tube.2015.07.005
62. Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, et al. Cytomegalovirus Vectors Violate CD8+ T Cell Epitope Recognition Paradigms. Science (2013) 340(6135):1237874. doi: 10.1126/science.1237874
63. De Groot AS, Moise L, Olive D, Einck L, Martin W. Agility in Adversity: Vaccines on Demand. Expert Rev Vaccines (2016) 15(9):1087–91. doi: 10.1080/14760584.2016.1205951
64. Blue Ribbon Study Panel on Biodefense. A National Blueprint for Biodefense: Leadership and Major Reform Needed to Optimize Efforts – Bipartisan Report of the Blue Ribbon Study Panel on Biodefense. Washington, D.C: Hudson Institute (2015).
65. Aebersold P. FDA Experience With Medical Countermeasures Under the Animal Rule. Adv Prev Med (2012) 2012:507571. doi: 10.1155/2012/507571
Keywords: Q fever, Coxiella burnetii, ChAdOx2, multi-epitope vaccine, modified vaccinia Ankara (MVA), interferon gamma (IFNγ), T cell vaccine, non-human primate (NHP)
Citation: Sluder AE, Raju Paul S, Moise L, Dold C, Richard G, Silva-Reyes L, Baeten LA, Scholzen A, Reeves PM, Pollard AJ, Garritsen A, Bowen RA, De Groot AS, Rollier C and Poznansky MC (2022) Evaluation of a Human T Cell-Targeted Multi-Epitope Vaccine for Q Fever in Animal Models of Coxiella burnetii Immunity. Front. Immunol. 13:901372. doi: 10.3389/fimmu.2022.901372
Received: 21 March 2022; Accepted: 20 April 2022;
Published: 16 May 2022.
Edited by:
Anthony E. Gregory, University of California, Irvine, United StatesReviewed by:
Erin J. Van Schaik, Texas A&M Health Science Center, United StatesDavid H. Walker, University of Texas Medical Branch at Galveston, United States
Copyright © 2022 Sluder, Raju Paul, Moise, Dold, Richard, Silva-Reyes, Baeten, Scholzen, Reeves, Pollard, Garritsen, Bowen, De Groot, Rollier and Poznansky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann E. Sluder, YXNsdWRlckBtZ2guaGFydmFyZC5lZHU=; Mark C. Poznansky, bXBvem5hbnNreUBtZ2guaGFydmFyZC5lZHU=
†Present Address: Guilhem Richard, EpiVax Therapeutics, Inc., Providence, RI, United States
Anja Scholzen, Byondis B.V., Nijmegen, The Netherlands
Christine Rollier, School of Biosciences and Medicine, University of Surrey, Guildford, United Kingdom