 Sara Signa1
Sara Signa1 Arinna Bertoni1,2
Arinna Bertoni1,2 Federica Penco1
Federica Penco1 Roberta Caorsi1
Roberta Caorsi1 Alessia Cafaro3
Alessia Cafaro3 Giuliana Cangemi3Stefano Volpi1,2
Giuliana Cangemi3Stefano Volpi1,2 Marco Gattorno1*†
Marco Gattorno1*† Francesca Schena1†
Francesca Schena1†- 1Center for Autoinflammatory Diseases and Immunodeficiencies, Istituto di Ricovero e cura a carattere scientifico (IRCCS) Istituto Giannina Gaslini, Genoa, Italy
- 2Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal-Child Sciences (DINOGMI), University of Genoa, Genoa, Italy
- 3Chromatography and Mass Spectrometry Section, Central Laboratory of Analysis, Istituto di Ricovero e cura a carattere scientifico (IRCCS) Istituto Giannina Gaslini, Genoa, Italy
Deficiency of Adenosine deaminase 2 (DADA2) is a monogenic autoinflammatory disorder presenting with a broad spectrum of clinical manifestations, including immunodeficiency, vasculopathy and hematologic disease. Biallelic mutations in ADA2 gene have been associated with a decreased ADA2 activity, leading to reduction in deamination of adenosine and deoxyadenosine into inosine and deoxyinosine and subsequent accumulation of extracellular adenosine. In the early reports, the pivotal role of innate immunity in DADA2 pathogenic mechanism has been underlined, showing a skewed polarization from the M2 macrophage subtype to the proinflammatory M1 subtype, with an increased production of inflammatory cytokines such as TNF-α. Subsequently, a dysregulation of NETosis, triggered by the excess of extracellular Adenosine, has been implicated in the pathogenesis of DADA2. In the last few years, evidence is piling up that adaptive immunity is profoundly altered in DADA2 patients, encompassing both T and B branches, with a disrupted homeostasis in T-cell subsets and a B-cell skewing defect. Type I/type II IFN pathway upregulation has been proposed as a possible core signature in DADA2 T cells and monocytes but also an increased IFN-β secretion directly from endothelial cells has been described. So far, a unifying clear pathophysiological explanation for the coexistence of systemic inflammation, immunedysregulation and hematological defects is lacking. In this review, we will explore thoroughly the latest understanding regarding DADA2 pathophysiological process, with a particular focus on dysregulation of both innate and adaptive immunity and their interacting role in the development of the disease.
Introduction
Since the first description of Adenosine Deaminase 2 deficiency (DADA2) in 2014 (1, 2), we have witnessed an exponential expansion of its possible clinical phenotypes. The clinical manifestations range from the initial descriptions of systemic inflammation with fever, early onset strokes and vasculopathy (3–10) to immune dysregulation (hypogammaglobulinemia, lymphoproliferation, increased infection rate) (5, 11–13) and hematological abnormalities (PRCA, bone marrow (BM) failure, cytopenias) (5, 6, 13–17), with several reports of different features overlapping (12, 18).
The polymorphic DADA2 clinical presentation reflects the ADA2 defect’s deep impact on different immune response branches (Figure 1), even if the possible presence of different clinical manifestations in patients with the same mutation (3, 8, 19, 20) advocates for further factors influencing the clinical phenotype. Recently a pivotal work unraveled in DADA2 an association of partial loss of ADA2 activity with the vasculitic phenotype and predominant missense mutations, whereas complete absent activity was observed in hematologic disease (13).
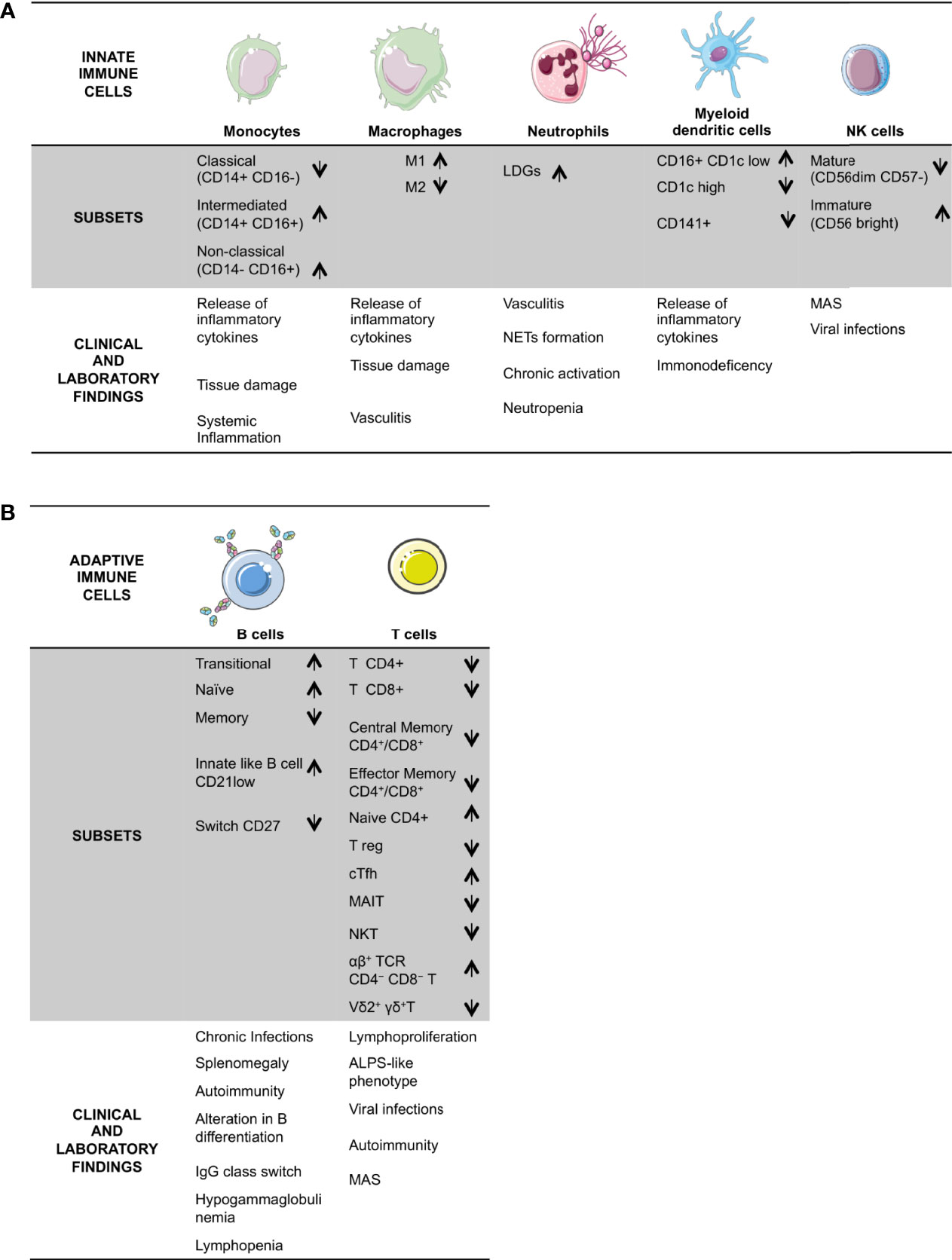
Figure 1 Schematic overview of DADA2 alterations in immune cells and the described related clinical signs and laboratory findings. DADA2 is associated with a wide variety of clinical manifestations. Here is a summary of clinical and immunological features, grouped in the two branches of the immune system; innate (A) and adaptive (B). LDG, Low Density Granulocytes; Treg, regulatory T cells; cTfh, circulating Tfh; MAIT, mucosal-associated invariant T; MAS, macrophage activation syndrome; ALPS; Autoimmune Lymphoproliferative Syndrome.
ADA2 is a dimeric enzyme that, similarly to its ADA1 isoform, catalyzes the deamination of adenosine and 2’-deoxyadenosine into inosine and deoxyinosine respectively (21–23). ADA2 exhibits not only an enzymatic activity, but also a growth factor role (22, 23) and it is required for a proper monocytes-T cells interaction (23, 24).
Notably, the determination of ADA2 activity is essential in the current diagnosis of DADA2, since pathogenic variants may not be detected by conventional genetic testing and may require the incorporation of additional diagnostic methods (25, 26). Few methods are currently available for a reliable ADA2 enzyme testing (1, 13, 27–29), some of them based on chromatographyc assays for quantification of adenosine and inosine (1, 7, 9, 30).
Spectrophotometric methods, with less specificity, have been reported (31–34). Recently, a novel enzymatic assay based on liquid chromatography-tandem mass spectrometry, that allows the accurate determination of the ADA2 activity starting from very small amounts of plasma spotted on DBS has been described (35).
The Impact of ADA2 Deficiency on the Cross Talk Between Innate and Adaptive Immunity
ADA2 protein is highly expressed in immune cells, particularly in myeloid ones (23, 24, 36), from which is actively secreted (23) and binds several immune cell lineages, such as monocytes but also B cells, neutrophils and NK cell (24). Therefore, the impact of ADA2 deficiency on different branches of the immune system is not unexpected.
The present review aims to clarify innate and adaptive immunity’s main peculiarities identified in DADA2 patients, and their possible interconnections leading to DADA2 polymorphic manifestations (Figure 2).
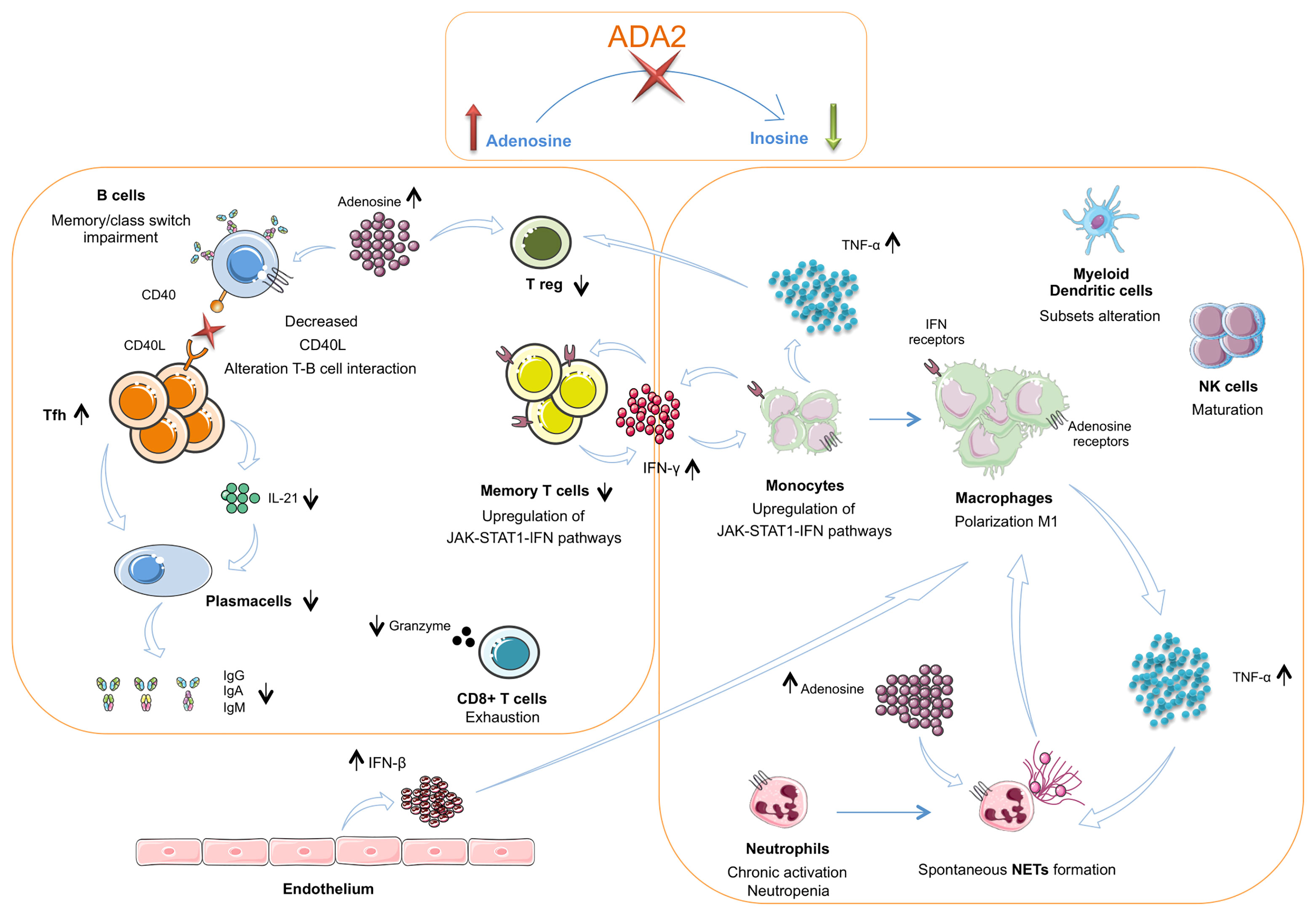
Figure 2 The interplay between innate and adaptive immune cells in DADA2. The insufficient or absent ADA2 enzymatic activity leads to a reduction of adenosine conversion into inosine and to an accumulation of adenosine in the extracellular space. In the innate immune system this leads to a chronic neutrophils activation and NETosis dysregulation with spontaneous NETs formation. Macrophages show a polarization towards the M1 subtype with consequent hypersecretion of inflammatory cytokines, especially TNF-α Dysregulation of NETosis and IFN-β secreted in large amount by endothelium also contribute to the M1 polarization. TNF-α acts on neutrophils contributing to maintaining their chronic activation state. CD56bright immature NK cells are increased. Dendritic cells present an altered subsets distribution. TNF-α, together with adenosine, acts also on Treg cells, that result diminished. In the T cell compartment, Tfh cells show an increased frequency but an impaired secretion of IL-21, crucial for B-cell function. Furthermore, T cells present a reduced proportion of the memory subsets and an alteration of CD40L expression. This defect is associated with an impaired B cell proliferation, differentiation and switching with consequent lower levels of immunoglobulins. CD8+ T cells show an exhausted phenotype and produce low amounts of granzyme A. Both T cells and monocytes show an upregulation of JAK – STAT1 – IFN pathways. In monocytes this activation contributes to the M1 macrophage polarization. TNF-α, Tumor necrosis factor α; IFN β, Interferon β; NETs, Neutrophils extracellular traps.
Monocytes and Macrophages, the Drivers of Inflammatory Cytokines
Since the original reports, the pivotal role of monocytes and macrophages inducing the inflammatory response in DADA2 has been outlined. Zhou and colleagues (1) showed a skewed in vitro monocyte differentiation, with increased proinflammatory M1 macrophages and relatively decreased anti-inflammatory M2 macrophages subset in DADA2 patients. This monocyte/macrophage polarization is supposed to induce a release of proinflammatory cytokines, leading to downstream endothelial disruption (1, 37). Notably, the lentiviral correction of defective ADA2 enzymatic activity with ADA2 reconstitution in patients macrophages is able to restrain inflammation (36, 38).
Despite initial reports not being univocal, a preferential binding of ADA2 to CD16+ monocytes instead to the “classical” CD16- monocytes is reported (24). An in-depth immune-phenotyping analysis of prevalently untreated DADA2 patients showed significantly reduced proportions of classical monocytes (CD14+ CD16-) with an increased proportions of intermediated (CD14+ CD16+) and non-classical (CD14- CD16+) monocytes (12). This finding has been confirmed by Watanabe et al (39), who analyzed CD14+ monocytes subpopulations through single cell RNA sequencing, identifying a higher frequency of nonclassical monocytes and an up-regulation of M1 macrophage markers compared with healthy donors. Indeed, non-classical monocytes are a prominent source of TNF-α compared with classical monocytes (40–42), and these findings are in line with the pathogenic role of TNF-α. Interestingly, no differences were found comparing monocyte transcriptomes of patients with a hematologic phenotype versus a nonhematologic phenotype, suggesting that the explanation for the BM failure observed in DADA2 is to be sought elsewhere (39, 42).
A possible pivotal role in DADA2 pathogenesis has been recently attributed to a dysregulation of the IFN type I and II axis. An upregulation of several pathways involved in the immune response, including IFNα/β and IFNγ signaling, was evident in all monocyte subsets (39). This observation was in line with the identification of elevated plasma levels of IFNγ. Similar findings were reported in other studies profiling transcriptome and proteome analysis on whole blood samples or gene expression on DADA2 PBMCs (37, 43). The authors suggested that elevated IFNγ may activate cell signaling, leading to monocytes differentiation into M1 macrophages and release of TNFα (39). Interestingly, Yap and colleagues (12) identified a twofold higher level of expression of CD169/SIGLEC1 on all subsets of ADA2-deficient monocytes compared to controls. CD169/SIGLEC1 is a cell adhesion protein induced by type I IFN-signaling and a potential myeloid cell-specific biomarker for an overt type I IFN response on monocytes (44). This finding is in line with reported evidence of upregulated type I IFN signaling in DADA2 with elevation of IFN score (12, 37, 43–48).
Finally, an upregulation of STAT1 phosphorylation in DADA2 monocytes after stimulation with IFNα and IFNγ was recently shown (37) confirming similar findings regarding STAT1 central role in altered DADA2 immune response (43, 49).
Together with IFN type I, the latter study also identified a clear NFKB signature in the whole blood of untreated patients, with normalization of both findings after anti-TNF treatment (37).
Natural Killer (NK) Cells and Dendritic Cells (DCs): The Producers of Interferons in DADA2?
NK cells are mainly involved in antiviral and antitumor response (50–52). ADA2 can bind NK cells, so NK is a target cell to ADA2 (23). After the initial reports showing a reduction in NK numbers (24) in DADA2 patients a consensual reduction of mature CD56 dim CD57- NK cells, displaying high cytotoxicity and low cytokine production, was found (3, 12). An increased proportion of immature CD56 bright NK cells, known as NK regulatory (53), was also identified (12). These later cells are characterized by low cytotoxicity and high cytokine production, including TNF-α. It would be fascinating to investigate the functional role of this subset, particularly to understand the possible involvement in some peculiar clinical features described in DADA2, such as recurrent viral infections (12, 27, 54, 55) and macrophage activation syndrome (MAS) (1, 5, 56) (Figure 1).
The role of DCs in DADA2 patients has not yet been investigated thoroughly. ADA2 is highly expressed in DCs1, which secrete ADA2 continuously (23). The in-depth immune phenotyping of DCs (12) showed that ADA2-deficiency does not impact on overall numbers and proportions of DCs. However, the distribution of myeloid DCs (mDCs) subsets was altered, with significant reduction in the proportion of CD1c-hi and CD141+ subsets. Conversely, a significantly increased proportion of CD16+CD1c lo/− mDCs was found in DADA2 patients compared with healthy controls. These DCs, or non-classical like monocytes, have potent T cell stimulatory capacity and produce proinflammatory cytokines upon TLR stimulation by TNF-α, IL6, IL12 (12). Thus, these cells may play a role in inducing and maintaining the inflammatory state observed in DADA2. In addition, reduced proportions of mDCs may contribute to the overall defect in T and B cell immune responses observed in vivo in DADA2 patients. It could be speculated that the DCs activated by exogenous triggers and the plasmocytoid DCs (pDCs) stimulated by self-antigens may secrete huge amounts of type I IFN. In turn, this process can act as an autocrine and paracrine stimulus on pDCs, monocytes and other cells extending the inflammatory cascade. So far, functional studies supporting this hypothesis are missing.
Neutrophils and Interplay With Innate Immune Cells
Neutrophils generate and release adenosine at sites of inflammation (57–59) and express receptors for adenosine (24), which may play several effects on their function (57). The zebrafish DADA2 model presented neutropenia (1), which may be part of the clinical phenotype (7, 60, 61). In DADA2 patients a marked upregulation of neutrophil expressed gene transcripts, a chronic activation of neutrophils and an increased expression of myeloperoxidase in PBMCs, possibly leading to endothelial damage (46), were demonstrated. Carmona-Rivera and colleagues (62) described the pathogenic role of neutrophils and the possible involvement of NETosis in the induction of tumor necrosis factor (TNF)-α release from macrophages (Figure 2).
DADA2 patients present a higher proportion of low-density granulocytes (LDGs), which have been partly implicated in the pathogenesis of vasculitis through the formation of neutrophil extracellular traps (NETs) (63–65).
The increase of circulating LDGs, prone to spontaneous NETs formation, was observed in DADA2 patients in active disease, with a significant reduction in disease remission after anti–TNF therapy. Adenosine was demonstrated to trigger NETs formation, by engaging A1 and A3 adenosine receptors (ARs) and the induction of reactive oxygen species (ROS)– and peptidylarginine deaminase (PAD)–dependent pathways. M1 macrophages incubated with NETs derived from DADA2 patients released significantly greater amounts of TNF-α (66).
Further works are necessary to evaluate neutrophils’ role in DADA2 pathogenesis, possibly examining immune pathways activation with single cells technology, as already performed for other immune cells lines (39, 49) and their interactions with other cellular types.
T Cells, Unconventional T Cells (NKT, γδT, MAIT) and B Cells: Only Supporting Actors or Deserve a Main Role?
ADA2 increases the rate of proliferation of monocyte-activated CD4+ T cells, acting as growth factor and induces T cell-dependent differentiation of monocytes into macrophages, stimulating macrophage proliferation (22, 23). Therefore, ADA2 deficiency impact on T cell compartment was not unexpected. Despite an initial report of normal T cells subsets (1), subsequent studies showed a reduction in helper and cytotoxic T subpopulations (12, 18, 55, 67) and a significant increase of cTfh cell frequency, likely secondary to the ongoing chronic inflammation (67). DADA2 cTfh cells present an impaired IL-21 production, a crucial cytokine for B-cell help function. A further alteration of T cells is linked to a reduced expression of CD40 ligand (CD40L), associated with impaired helper activity toward B-cells, indicating also an alteration in the T–B cell interaction process (67).
More recently, an impaired differentiation of CD4+ and CD8+ memory T cells in vivo has been highlighted (12, 67). CD4+ and CD8+ T cells in DADA2 patients display an exacerbated senescent/exhausted phenotype, as shown by cellular markers, such as increased CD95 and CD57, decreased CD28 and increased CX3CR1 (12). ADA2-deficient CD8+ TEM/TEMRA cells secrete lower levels of granzyme A (12), whereas cytokines secretion and effector function of CD4+ T cells seem unaffected. Furthermore, a significantly reduced proportion of Treg cells was recently identified in DADA2 patients (12) and lower proportions of unconventional NKT (12, 24) and mucosal-associated invariant T (MAIT) (12) were also evident.
These data, together with a remarkably lower proportion of Vγδ2+T cells (12), suggest that DADA2 CD8+ T cells may be involved in the development of refractory/recurrent viral infections observed in some patients (54, 68–70). The defect in ADA2-deficient cytotoxic lymphocytes may be also linked to rare DADA2-associated manifestations, such as hemophagocytic lymphohistiocytosis (HLH) (1, 5, 56) or CD3+ CD8+ large granular lymphocytes (11, 18).
An increased proportion of CD4-CD8- αß+ TCR+ cells is also reported in several patients (18–20, 71), consistently with an ALPS-like phenotype (3, 11, 71, 72).
Profiling of the TCR repertoires at the single-cell levels showed no significant clonal dominance of T cells, most TCRs were individual-private, and there were no disease-specific TCRs indicating as unlikely the possibility of a common pathogenic background (49, 73).
T-cells transcriptomic analysis in DADA2 revealed an activation of IFN pathways (both IFNα/β and IFNγ signaling) as a T cells signature and STAT1 as a hub gene in the gene network of T cell activation and cytotoxicity (49). Notably, many functions of IFNγ have been ascribed to direct STAT1- mediated induction of immune effector genes. STAT1 activation could also be mediated by cross regulation of cellular responses to other cytokines and inflammatory factors (74).
As mentioned above, defective B cell function is linked to defective cTfh help towards B cells (67), but also an intrinsic defect of B cell lineage itself has been shown (12, 67).
ADA2 binds B cells (23), which have also the ability to produce and secrete ADA2 (67). The involvement of the B-cell compartment in DADA2 was evident since first reports. Zhou et al. (1) described a B cells defect in terminal maturation and a tendency towards apoptosis, consistent with a clinical phenotype characterized by hypogammaglobulinemia and recurrent infections together with the typical inflammatory features (18) (Figure 1).
The most striking effect of ADA2 deficiency on B cells is the impairment of their differentiation, which seems compromised at several levels. A reduction of memory B cells is confirmed by different studies (1, 2, 18, 27, 55, 67). Recent findings (12, 18, 67) show that ADA2-deficiency alters not only the generation and/or maintenance of the memory B cell pool, but also the ability of B cells to undergo Ig class switching, mainly to IgA secreting cells (12). In the study by Yap et al, the absolute levels of CD38 and IgM expressed were significantly higher on transitional, naïve and memory B cells from DADA2 patients than healthy donors (12). An increase in naive (12, 27, 55) and transitional B cells subsets (12, 18) has been also found. In conclusion, DADA2 B cells populations seem to be less mature than those of healthy controls (12), with an impairment in their ability to proliferate and differentiate. DADA2 naıve B-cells display also a reduced capacity to respond to TLR 9 agonist, CD40L, and anti-human Ig stimulation, suggesting an intrinsic proliferation defect induced by mutations of ADA2 (67). Furthermore, an expansion of CD21low B cells has been shown (67), possibly being a potential predisposing factor to autoimmunity (18, 75).
It has to be noted that, compared with HDs, BM from DADA2 patients may exhibit a reduced number of mature and immature populations belonging to different hematopoietic lineages, B lineage included (36). Notably, the aberrant distribution and phenotype of peripheral B cells subsets in DADA2 patients could be the consequence of a primary block of maturation of B precursors in the BM, as recently shown in one untreated patient, with an accumulation of pre-B cells and immature B cells (12).
Discussion
Since the first studies on DADA2 (1, 2) it was clear that DADA2 pathogenesis and clinical heterogeneity could not be completely explained by macrophage involvement alone, possibly implicating different immune cell lineages participation (Figure 2). In fact, DADA2 is an atypical autoinflammatory disease, in which the causative gene is not involved in the canonical inflammatory pathways, but rather affects purines metabolism and signaling (24, 76, 77). Monocytes/macrophages remain central actors in the pathogenesis of vasculitis-related phenotype (37), but other mechanisms have been recently shown to contribute to endothelial disruption, such as increased NETosis and NETs stimulatory effect on macrophages itself (62).
Additional works unraveled alterations across all the immune system, possibly linked to the pleiotropic clinical features in DADA2 (Figure 1): an intrinsic defect of B cell compartment may be related not only to recurrent and chronic infections (12, 18, 27), but also to autoimmunity development (18, 48, 75), which can be additionally influenced by a T reg defect (12) and an hyperactivation of IFN type axis (12, 37, 44–48, 78).
Predisposition to viral infections may be justified by CD8+ alterations and Vγδ2+T cells defect (12) together with the increased proportion of NK regulatory cells (12). These alterations could be also implicated in the reported HLH episodes (1, 7, 56, 79).
Hematological manifestations have been described (7, 14, 17, 18, 38, 39, 43, 55, 60, 80, 81), but their pathophysiology is still mostly obscure. In addition to the maturation block of patients hematopoietic progenitors (HPC) (12, 36), patients’ marrow plasma (19, 71) showed an inhibitory effect on growth and differentiation of healthy BM precursors, possibly suggesting a humoral inhibitory effect. ADA2 may have a role in hematopoietic processes besides its inflammation-associated effects. Recently a genotype-phenotype correlation (13) demonstrated that mutations leading to complete (or almost) loss of ADA2 activity are associated with a prevalent hematological phenotype. We could hypothesize that the coexistence of inflammatory, immunodeficiency and hematologic phenotypes could be independent of ADA2 enzyme activity per se, because it is known that ADA2 protein binds to multiple immune cells (22, 23) and may exert different effects on different target cells, possibly acting as growth factor also for HPC.
Specific studies investigating DADA2 patients BM microenvironment and hematopoiesis regulation are mandatory to elucidate the possibility of an intrinsic defect in DADA2 HPC or extrinsic factors in BM niche (19, 71).
Definitely, so far few cytokines have shown a central role in DADA2: TNF-α and, lately, type I and type II IFN (82).
TNF-α is one of the main mediators of vascular inflammation (83) and several studies showed detectable levels of TNF-α in the peripheral blood and affected tissues from DADA2 patients with active disease (1, 9, 37, 39). Its biological effects include activation of other immune cells such as macrophages, DCs, T-cells and B-cells (84) and inhibition of Treg-cells, consistently with reduced Tregs number in DADA2 patients.
Notably, reduced ADA2 activity has been associated with intracellular accumulation of deoxyinosine, interfering with the cellular methionine cycle through the inhibition of SAM synthetase activity. This mechanism drives epigenomic hypomethylation and overexpression of immune-stimulatory endogenous retroviral elements, inducing IFN-β production (77).
How the activation of type I IFN in DADA2 patients contributes to the pathophysiology is still not completely clarified. It is known that IFNα/β enhance NK cytotoxicity, effector CD4+ and CD8+ T cell responses and B cell differentiation into plasma cells and antibody production, essential mechanisms leading to autoimmune response (78).
Recent studies show an activation of the IFN–JAK–STAT1 pathway in DADA2 patients (37, 43), mainly in monocytes (39) and T cells (49). The IFN-STAT1 axis could be an efficient inducer of M1 macrophages polarization, with a possible reciprocal cross-regulation of TNF and IFN production (73).
Interestingly, IFNγ and TNF-α have been described as possibly implicated in the pathophysiology of aplastic anemia (85) and abnormal STAT1 activation was demonstrated in BM samples of few aplastic anemia patients (86). Given the recent observation of STAT1 axis activation in DADA2 patients, its possible role in DADA2 hematological compartment defect should be explored.
At the moment, DADA2 treatment may differ based on clinical phenotype: most inflammatory features can be effectively controlled using TNF inhibitors, with reduction of stroke recurrence (18, 87, 88), improvement of blood vessels endothelial integrity and resolution of inflammatory myeloid cell infiltrates (37). Nevertheless, TNFi are not so effective at controlling hematological manifestations (13, 60, 61, 87, 89, 90). For these patients, hematopoietic stem cell transplantation has been reported reverting both hematological and inflammatory features and restoring ADA2 activity (15, 16, 18). Recent gene therapy preclinical studies demonstrate the correction of proinflammatory profile in patients’ macrophages after lentiviral-mediated ADA2 gene transfer (36, 38) and recovery of stem cell proliferation and colony forming units capacity in CD34+ HSPC (38). Further exploration of IFNs role in DADA2 pathophysiological process may open new therapeutic options for these patients, such as JAK-inhibitors or anti-IFNγ antibodies, as well as NETs inhibitors if NETosis involvement in DADA2 pathogenesis will be reinforced.
In conclusion, ADA2 deficiency has a complex impact on both innate and adaptive immunity cell lineages. Recent studies helped to start clarifying the pathophysiological mechanism underlying several clinical manifestations, but wide gaps still need to be filled. Future therapeutic approaches, such as gene therapy, will need to take into consideration the spread impact of ADA2 deficiency on the immune system.
Author Contributions
FS and SS conceived the idea, reviewed the literature, and wrote the first draft of the manuscript. AB and FP created the figures. RC, MG, SV, AC, and GC critically reviewed the manuscript and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by the Italian Ministry of Health—Ricerca Finalizzata (grant RF-2019-12370600) to MG, and partially funded by the Italian Ministry of Health, RC2021.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors would like to thank patients and family members for their continuous support to their research projects. The authors are also grateful to Associazione per la Lotta all’Ictus Cerebrale (ALICe) and to www.servier.com for available images. We gratefully thank all Italian citizens allocated the 5 × 1000 share of their tax payment in support of health Research.
Footnotes
- ^ https://www.proteinatlas.org/ENSG00000093072-ADA2/immune+cell, https://www.proteinatlas.org/ENSG00000093072-ADA2/single+cell+type.
References
1. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset Stroke and Vasculopathy Associated With Mutations in ADA2. N Engl J Med (2014) 370(10):911–20. doi: 10.1056/NEJMoa1307361
2. Elkan PN, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant Adenosine Deaminase 2 in a Polyarteritis Nodosa Vasculopathy. N Engl J Med (2014) 370(10):921–31. doi: 10.1056/NEJMoa1307362
3. Van Montfrans JM, Hartman EAR, Braun KPJ, Hennekam EAM, Hak EA, Nederkoorn PJ, et al. Phenotypic Variability in Patients With ADA2 Deficiency Due to Identical Homozygous R169Q Mutations. Rheumatol (United Kingdom) (2016) 55(5):902–10. doi: 10.1093/rheumatology/kev439
4. Pinto B, Deo P, Sharma S, Syal A, Sharma A. Expanding Spectrum of DADA2: A Review of Phenotypes, Genetics, Pathogenesis and Treatment. Clin Rheumatol (2021) 40 (10): 3883–96. doi: 10.1007/s10067-021-05711-w
5. Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A Case Series of Adenosine Deaminase 2-Deficient Patients Emphasizing Treatment and Genotype-Phenotype Correlations. J Rheumatol (2015) 42(8):1532–4. doi: 10.3899/jrheum.150024
6. Özen S, Batu ED, Taşkiran EZ, Özkara HA, Ünal Ş, Güleray N, et al. A Monogenic Disease With a Variety of Phenotypes: Deficiency of Adenosine Deaminase 2. J Rheumatol (2020) 47(1):117–25. doi: 10.3899/jrheum.181384
7. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the Clinical Phenotype of Adenosine Deaminase 2 Deficiency. J Pediatr (2016) 7:316–20. doi: 10.1016/j.jpeds.2016.06.058
8. Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol (2016) 68(9):2314–22. doi: 10.1002/art.39699
9. Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 Deficiency (DADA2) as an Unrecognised Cause of Early Onset Polyarteritis Nodosa and Stroke: A Multicentre National Study. Ann Rheum Dis (2017) 76(10):1648–56. doi: 10.1136/annrheumdis-2016-210802
10. Sharma A, Naidu GSRSNK, Sharma V, Jha S, Dhooria A, Dhir V, et al. Deficiency of Adenosine Deaminase 2 in Adults and Children: Experience From India. Arthritis Rheumatol (2021) 73(2):276–85. doi: 10.1002/art.41500
11. Trotta L, Martelius T, Siitonen T, Hautala T, Hämäläinen S, Juntti H, et al. ADA2 Deficiency: Clonal Lymphoproliferation in a Subset of Patients. J Allergy Clin Immunol (2018) 141(4):1534–1537.e8. doi: 10.1016/j.jaci.2018.01.012
12. Yap JY, Moens L, Lin M-W, Kane A, Kelleher A, Toong C, et al. Intrinsic Defects in B Cell Development and Differentiation, T Cell Exhaustion and Altered Unconventional T Cell Generation Characterize Human Adenosine Deaminase Type 2 Deficiency. J Clin Immunol (2021) 41(8):1915–35. doi: 10.1007/s10875-021-01141-0
13. Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and Functional Correlates of Disease Phenotype in Deficiency of Adenosine Deaminase 2 (DADA2). J Allergy Clin Immunol (2020) 145(6):1664–72.e10. doi: 10.1016/j.jaci.2019.12.908
14. Hashem H, Egler R, Dalal J. Refractory Pure Red Cell Aplasia Manifesting as Deficiency of Adenosine Deaminase 2. J Pediatr Hematol Oncol (2017) 39(5):e293–6. doi: 10.1097/MPH.0000000000000805
15. Hashem H, Kumar AR, Müller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic Stem Cell Transplantation Rescues the Hematological, Immunological, and Vascular Phenotype in DADA2. Blood (2017) 130(24):2682–8. doi: 10.1182/blood-2017-07-798660
16. Hashem H, Bucciol G, Ozen S, Unal S, Bozkaya IO, Akarsu N, et al. Hematopoietic Cell Transplantation Cures Adenosine Deaminase 2 Deficiency: Report on 30 Patients. J Clin Immunol (2021) 41(7):1663–47. doi: 10.1007/s10875-021-01098-0
17. Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet (2018) 103(6):930–47. doi: 10.1016/j.ajhg.2018.10.027
18. Barron KS, Aksentijevich I, Deuitch NT, Stone DL, Hoffmann P, Videgar-Laird R, et al. The Spectrum of the Deficiency of Adenosine Deaminase 2: An Observational Analysis of a 60 Patient Cohort. Front Immunol (2022) 12. doi: 10.3389/fimmu.2021.811473
19. Dell’Orso G, Grossi A, Penco F, Caorsi R, Palmisani E, Terranova P, et al. Case Report: Deficiency of Adenosine Deaminase 2 Presenting With Overlapping Features of Autoimmune Lymphoproliferative Syndrome and Bone Marrow Failure. Front Immunol (2021) 12(October):1–8. doi: 10.3389/fimmu.2021.754029
20. Saettini F, Fazio G, Corti P, Quadri M, Bugarin C, Gaipa G, et al. Two Siblings Presenting With Novel ADA2 Variants, Lymphoproliferation, Persistence of Large Granular Lymphocytes, and T-Cell Perturbations. Clin Immunol (2020) 218(April):2–4. doi: 10.1016/j.clim.2020.108525
21. Zavialov AV, Engström Å. Human ADA2 Belongs to a New Family of Growth Factors With Adenosine Deaminase Activity. Biochem J (2005) 391(1):51–7. doi: 10.1042/BJ20050683
22. Zavialov AV, Yu X, Spillmann D, Lauvau G, Zavialo AV. Structural Basis for the Growth Factor Activity of Human Adenosine Deaminase ADA2. J Biol Chem (2010) 285(16):12367–77. doi: 10.1074/jbc.M109.083527
23. Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human Adenosine Deaminase 2 Induces Differentiation of Monocytes Into Macrophages and Stimulates Proliferation of T Helper Cells and Macrophages. J Leukoc Biol (2010) 88(2):279–90. doi: 10.1189/jlb.1109764
24. Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human Adenosine Deaminases ADA1 and ADA2 Bind to Different Subsets of Immune Cells. Cell Mol Life Sci (2017) 74(3):555–70. doi: 10.1007/s00018-016-2357-0
25. Rama M, Duflos C, Melki I, Bessis D, Bonhomme A, Martin H, et al. A Decision Tree for the Genetic Diagnosis of Deficiency of Adenosine Deaminase 2 (DADA2): A French Reference Centres Experience. Eur J Hum Genet (2018) 26(7):960–71. doi: 10.1038/s41431-018-0130-6
26. Shinar Y, Ceccherini I, Rowczenio D, Aksentijevich I, Arostegui J. Special Report ISSAID/EMQN Best Practice Guidelines for the Genetic Diagnosis of Monogenic Autoinflammatory Diseases in the Next-Generation Sequencing Era. Clin Chem (2020) 166(4):525–36. doi: 10.1093/clinchem/hvaa024
27. Schepp J, Proietti M, Frede N, Buchta M, Hübscher K, Rojas Restrepo J, et al. Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol (2017) 69(8):1689–700. doi: 10.1002/art.40147
28. Bowers SM, Gibson KM, Cabral DA, Brown KL. Adenosine Deaminase 2 Activity Negatively Correlates With Age During Childhood. Pediatr Rheumatol (2020) 18(1):54. doi: 10.1186/s12969-020-00446-5
29. Muraoka T, Katsuramaki T, Shiraishi H, Yokoyama MM. Automated Enzymatic Measurement of Adenosine Deaminase Isoenzyme Activities in Serum. Anal Biochem (1990) 187(2):268–72. doi: 10.1016/0003-2697(90)90455-I
30. Van Der Weyden MB, Bailey L. A Micromethod for Determining Adenosine Deaminase and Purine Nucleoside Phosphorylase Activity in Cells From Human Peripheral Blood. Clin Chim Acta (1978) 82(1–2):179–84. doi: 10.1016/0009-8981(78)90041-4
31. Schnappauf O, Sampaio Moura N, Aksentijevich I, Stoffels M, Ombrello AK, Hoffmann P, et al. Sequence-Based Screening of Patients With Idiopathic Polyarteritis Nodosa, Granulomatosis With Polyangiitis, and Microscopic Polyangiitis for Deleterious Genetic Variants in ADA2. Arthritis Rheumatol (2021) 73(3):512–9. doi: 10.1002/art.41549
32. Ito K, Sakura N, Usui T, Uchino H. Screening for Primary Immunodeficiencies Associated With Purine Nucleoside Phosphorylase Deficiency or Adenosine Deaminase Deficiency. J Lab Clin Med (1977) 90(5):844–8.
33. Ito M, Nihira H, Izawa K, Yasumi T, Nishikomori R, Iwaki-Egawa S. Enzyme Activity in Dried Blood Spot as a Diagnostic Tool for Adenosine Deaminase 2 Deficiency. Anal Biochem (2021) 628(June):114292. doi: 10.1016/j.ab.2021.114292
34. Iwaki-Egawa S, Watanabe Y, Matsuno H. Correlations Between Matrix Metalloproteinase-9 and Adenosine Deaminase Isozymes in Synovial Fluid From Patients With Rheumatoid Arthritis. J Rheumatol (2001) 28(3):485–9.
35. Cafaro A, Pigliasco F, Barco S, Penco F, Schena F, Caorsi R, et al. A Novel LC – MS/MS-Based Method for the Diagnosis of ADA2 Deficiency From Dried Plasma Spot. Molecules (2021) 26(8): 5707. doi: 10.3390/molecules26185707
36. Zoccolillo M, Brigida I, Barzaghi F, Scala S, Hernández RJ, Basso-Ricci L, et al. Lentiviral Correction of Enzymatic Activity Restrains Macrophage Inflammation in Adenosine Deaminase 2 Deficiency. Blood Adv (2021) 5(16):3174–87. doi: 10.1182/bloodadvances.2020003811
37. Deuitch NT, Yang D, Lee PY, Yu X, Moura NS, Schnappauf O, et al. TNF-Inhibition in Vasculitis Management in Adenosine Deaminase 2 Deficiency (DADA2). J Allergy Clin Immunol (2022) 149(5):1812–16.e6. doi: 10.1016/j.jaci.2021.10.030
38. Hong Y, Casimir M, Houghton BC, Zhang F, Jensen B, Omoyinmi E, et al. Lentiviral Mediated ADA2 Gene Transfer Corrects the Defects Associated With De Fi Ciency of Adenosine Deaminase Type 2. Front Immunol (2022) 13:852830. doi: 10.3389/fimmu.2022.852830
39. Watanabe N, Gao S, Wu Z, Batchu S, Kajigaya S, Diamond C, et al. Analysis of Deficiency of Adenosine Deaminase 2 Pathogenesis Based on Single-Cell RNA Sequencing of Monocytes. J Leukoc Biol (2021) 110(3):409–24. doi: 10.1002/JLB.3HI0220-119RR
40. Belge K-U, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, et al. The Proinflammatory CD14 + CD16 + DR ++ Monocytes Are a Major Source of TNF. J Immunol (2002) 168(7):3536–42. doi: 10.4049/jimmunol.168.7.3536
41. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of Monocytes and Dendritic Cells in Blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
42. Ehlers L, Meyts I. What a Difference ADA2 Makes: Insights Into the Pathophysiology of ADA2 Deficiency From Single-Cell RNA Sequencing of Monocytes. J Leukoc Biol (2021) 110(3):405–7. doi: 10.1002/JLB.5CE0421-186R
43. Nihira H, Izawa K, Ito M, Umebayashi H, Okano T, Kajikawa S, et al. Detailed Analysis of Japanese Patients With Adenosine Deaminase 2 Deficiency Reveals Characteristic Elevation of Type II Interferon Signature and STAT1 Hyperactivation. J Allergy Clin Immunol (2021) 148(2):550–62. doi: 10.1016/j.jaci.2021.01.018
44. Uettwiller F, Sarrabay G, Rodero MP, Rice GI, Lagrue E, Marot Y, et al. ADA2 Deficiency: Case Report of a New Phenotype and Novel Mutation in Two Sisters. RMD Open (2016) 2(1):1–5. doi: 10.1136/rmdopen-2015-000236
45. Insalaco A, Moneta GM, Pardeo M, Caiello I, Messia V, Bracaglia C, et al. Variable Clinical Phenotypes and Relation of Interferon Signature With Disease Activity in ADA2 Deficiency. J Rheumatol (2019) 46(5):523–6. doi: 10.3899/jrheum.180045
46. Belot A, Wassmer E, Twilt M, Lega JC, Zeef LAH, Oojageer A, et al. Mutations in CECR1 Associated With a Neutrophil Signature in Peripheral Blood. Pediatr Rheumatol (2014) 12(1):1–7. doi: 10.1186/1546-0096-12-44
47. Rice GI, Melki I, Frémond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J Clin Immunol (2017) 37(2):123–32. doi: 10.1007/s10875-016-0359-1
48. Skrabl-Baumgartner A, Plecko B, Schmidt WM, König N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune Phenotype With Type I Interferon Signature in Two Brothers With ADA2 Deficiency Carrying a Novel CECR1 Mutation. Pediatr Rheumatol (2017) 15(1):1–5. doi: 10.1186/s12969-017-0193-x
49. Wu Z, Gao S, Watanabe N, Batchu S, Kajigaya S, Diamond C, et al. Single-Cell Profiling of T Lymphocytes in Deficiency of Adenosine Deaminase 2. J Leukoc Biol (2022) 111(2):301–12. doi: 10.1002/JLB.5A0621-314R
50. Brauning A, Rae M, Zhu G, Fulton E, Admasu TD, Stolzing A, et al. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Cells (2022) 11(6):1–25. doi: 10.3390/cells11061017
51. Cronk JM, Fafoutis E, Brown MG. Licensing Natural Killers for Antiviral Immunity. Pathogens (2021) 10(7):1–14. doi: 10.3390/pathogens10070908
52. Vaněk O, Kalousková B, Abreu C, Nejadebrahim S, Skořepa O. Natural Killer Cell-Based Strategies for Immunotherapy of Cancer. Adv Protein Chem Struct Biol (2022) 129:91–133. doi: 10.1016/bs.apcsb.2022.02.001
53. Laroni A, Armentani E, Kerlero de Rosbo N, Ivaldi F, Marcenaro E, Sivori S, et al. Dysregulation of Regulatory CD56bright NK Cells/T Cells Interactions in Multiple Sclerosis. J Autoimmun (2016) 72:8–18. doi: 10.1016/j.jaut.2016.04.003
54. Arts K, Bergerson JRE, Ombrello AK, Similuk M, Oler AJ, Agharahimi A, et al. Warts and DADA2: A Mere Coincidence? J Clin Immunol (2018) 38(8):836–43. doi: 10.1007/s10875-018-0565-0
55. Van Eyck L, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic Stem Cell Transplantation Rescues the Immunologic Phenotype and Prevents Vasculopathy in Patients With Adenosine Deaminase 2 Deficiency. J Allergy Clin Immunol (2015) 135(1):283–287.e5. doi: 10.1016/j.jaci.2014.10.010
56. Iyengar VV, Chougule A, Gowri V, Taur P, Prabhu S, Bodhanwala M, et al. DADA2 Presenting as Nonimmune Hemolytic Anemia With Recurrent Macrophage Activation Syndrome. Pediatr Blood Cancer (2021) 5(October):5–6. doi: 10.1002/pbc.29461
57. Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The Adenosine/Neutrophil Paradox Resolved: Human Neutrophils Possess Both A1 and A2 Receptors That Promote Chemotaxis and Inhibit O-2 Generation, Respectively. J Clin Invest (1990) 5(4):1150–7. doi: 10.1172/JCI114547
58. Barletta KE, Ley K, Mehrad B. Regulation of Neutrophil Function by Adenosine. Arterioscler Thromb Vasc Biol (2012) 32(4):856–64. doi: 10.1161/ATVBAHA.111.226845
59. Junger WG. Immune Cell Regulation by Autocrine Purinergic Signalling. Nat Rev Immunol (2011) 11(3):201–12. doi: 10.1038/nri2938
60. Michniacki TF, Hannibal M, Ross CW, Frame DG, DuVall AS, Khoriaty R, et al. Hematologic Manifestations of Deficiency of Adenosine Deaminase 2 (DADA2) and Response to Tumor Necrosis Factor Inhibition in DADA2-Associated Bone Marrow Failure. J Clin Immunol (2018) 38(2):166–73. doi: 10.1007/s10875-018-0480-4
61. Cipe FE, Aydogmus C, Serwas NK, Keskindemirci G, Boztuğ K. Novel Mutation in CECR1 Leads to Deficiency of ADA2 With Associated Neutropenia. J Clin Immunol (2018) 38(3):273–7. doi: 10.1007/s10875-018-0487-x
62. Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, et al. Deficiency of Adenosine Deaminase 2 Triggers Adenosine-Mediated NETosis and TNF Production in Patients With DADA2. Blood (2019) 134(4):395–406. doi: 10.1182/blood.2018892752
63. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil Extracellular Traps Induce Endothelial Dysfunction in Systemic Lupus Erythematosus Through the Activation of Matrix Metalloproteinase-2. Ann Rheum Dis (2015) 74(7):1417–24. doi: 10.1136/annrheumdis-2013-204837
64. O’Neil LJ, Kaplan MJ, Carmona-Rivera C. The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus. Vol 8 J Clin Med (2019) p:1325. doi: 10.3390/jcm8091325
65. van Dam LS, Kraaij T, Kamerling SWA, Bakker JA, Scherer UH, Rabelink TJ, et al. Intrinsically Distinct Role of Neutrophil Extracellular Trap Formation in Antineutrophil Cytoplasmic Antibody–Associated Vasculitis Compared to Systemic Lupus Erythematosus. Arthritis Rheumatol (2019) 71:2047–58. doi: 10.1002/art.41047
66. Lee PY. NETing the Mechanism of Inflammation in DADA2. Blood (2019) 134(4):338–9. doi: 10.1182/blood.2019001251
67. Schena F, Penco F, Volpi S, Pastorino C, Caorsi R, Kalli F, et al. Dysregulation in B-Cell Responses and T Follicular Helper Cell Function in ADA2 Deficiency Patients. Eur J Immunol (2021) 51(1):206–19. doi: 10.1002/eji.202048549
68. Le Voyer T, Boutboul D, Ledoux-Pilon A, de Fontbrune FS, Boursier G, Latour S, et al. Late-Onset EBV Susceptibility and Refractory Pure Red Cell Aplasia Revealing Dada2. J Clin Immunol (2020) 40(6):948–53. doi: 10.1007/s10875-020-00812-8
69. Staples E, Simeoni I, Stephens JC, Allen HL, Wright P, Davies EG, et al. ADA2 Deficiency Complicated by EBV-Driven Lymphoproliferative Disease. Clin Immunol (2020) 215(January):22–5. doi: 10.1016/j.clim.2020.108443
70. Brooks JP, Rice AJ, Ji W, Lanahan SM, Konstantino M, Dara J, et al. Uncontrolled Epstein-Barr Virus as an Atypical Presentation of Deficiency in ADA2 (Dada2). J Clin Immunol (2021) 41(3):680–3. doi: 10.1007/s10875-020-00940-1
71. Barzaghi F, Minniti F, Mauro M, De Bortoli M, Balter R, Bonetti E, et al. ALPS-Like Phenotype Caused by ADA2 Deficiency Rescued by Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol (2019) 10(JAN). doi: 10.3389/fimmu.2018.02767
72. Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 Mimicking Autoimmune Lymphoproliferative Syndrome in the Absence of Livedo Reticularis and Vasculitis. Pediatr Blood Cancer (2018) 65(4):10–3. doi: 10.1002/pbc.26912
73. Mesa-Nuñez C, Mortellaro A. T Cells and Monocytes: A Dangerous Liaison in Adenosine Deaminase 2 Deficiency. J Leukoc Biol (2022) 111(2):297–9. doi: 10.1002/JLB.3CE1021-561R
74. Hu X, Ivashkiv LB. Cross-Regulation of Signaling Pathways by Interferon-Gamma: Implications for Immune Responses and Autoimmune Diseases. Immunity (2009) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
75. Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol (2018) 38(5):569–78. doi: 10.1007/s10875-018-0525-8
76. Papa R, Picco P, Gattorno M. The Expanding Pathways of Autoinflammation: A Lesson From the First 100 Genes Related to Autoinflammatory Manifestations. Adv Protein Chem Struct Biol (2020) 120:1–44. doi: 10.1016/bs.apcsb.2019.11.001
77. Dhanwani R, Takahashi M, Mathews IT, Lenzi C, Romanov A, Watrous JD, et al. Cellular Sensing of Extracellular Purine Nucleosides Triggers an Innate IFN-β Response. Sci Adv (2020) 6(30):eaba3688. doi: 10.1126/sciadv.aba3688
78. Crow MK, Olferiev M, Kirou KA. Type I Interferons in Autoimmune Disease. Annu Rev Pathol: Mech Dis (2019) 14:369–93. doi: 10.1146/annurev-pathol-020117-043952
79. Crayne C, Cron RQ. Pediatric Macrophage Activation Syndrome, Recognizing the Tip of the Iceberg. Eur J Rheumatol (2020) 7(1):13–20. doi: 10.5152/eurjrheum.2019.19150
80. Süleyman M, Tan Ç, Uner A, İnkaya Ç, Aytaç S, Büyükaşık Y, et al. Adenosine Deaminase Type II Deficiency: Severe Chronic Neutropenia, Lymphoid Infiltration in Bone Marrow, and Inflammatory Features. Immunol Invest (2020) 51(3):558–66. doi: 10.1080/08820139.2020.1853153
81. Pilania RK, Banday AZ, Sharma S, Kumrah R, Joshi V, Loganathan S, et al. Deficiency of Human Adenosine Deaminase Type 2 – A Diagnostic Conundrum for the Hematologist. Front Immunol (2022) 13(May):1–10. doi: 10.3389/fimmu.2022.869570
82. Lee PY, Aksentijevich I, Zhou Q. Mechanisms of Vascular Inflammation in Deficiency of Adenosine Deaminase 2 (DADA2). Semin Immunopathol (2022) 44(3):269–80. doi: 10.1007/s00281-022-00918-8
83. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. The Role of Tumor Necrosis Factor Alpha (Tnf-α) in Autoimmune Disease and Current Tnf-α Inhibitors in Therapeutics. Int J Mol Sci (2021) 22(5):1–16. doi: 10.3390/ijms22052719
84. Cella M, Engering A, Pinet V, Pieters J LA. Inflammatory Stimuli Induce Accumulation of MHC Class. Nature (1997) 388(6644):782–7. doi: 10.1038/42030
85. Giudice V, Selleri C. Aplastic Anemia: Pathophysiology. Semin Hematol (2022) 59(1):13–20. doi: 10.1053/j.seminhematol.2021.12.002
86. Rosenberg JM, Peters JM, Hughes T, Lareau CA, Ludwig LS, Massoth LR, et al. JAK Inhibition in a Patient With a STAT1 Gain-of-Function Variant Reveals STAT1 Dysregulation as a Common Feature of Aplastic Anemia. Med [NY] (2022) 3(1):42–57.e5. doi: 10.1016/j.medj.2021.12.003
87. Cooray S, Omyinmi E, Hong Y, Papadopoulou C, Harper L, Al-Abadi E, et al. Anti-Tumour Necrosis Factor Treatment for the Prevention of Ischaemic Events in Patients With Deficiency of Adenosine Deaminase 2 (DADA2). Rheumatology (2021) 60(9):4373–8. doi: 10.1093/rheumatology/keaa837
88. Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med (2019) 380(16):1582–4. doi: 10.1056/NEJMc1801927
89. Göschl L, Winkler S, Dmytrus J, Heredia RJ, Lagler H, Ramharter M, et al. Unreported Missense Mutation in the Dimerization Domain of ADA2 Leads to ADA2 Deficiency Associated With Severe Oral Ulcers and Neutropenia in a Female Somalian Patient—Addendum to the Genotype-Phenotype Puzzle. J Clin Immunol (2019) 40(1)223–6. doi: 10.1007/s10875-019-00700-w
Keywords: adenosine deaminase 2 (ADA 2), DADA2, innate immunity, adaptive immunity, TNF-α, interferon
Citation: Signa S, Bertoni A, Penco F, Caorsi R, Cafaro A, Cangemi G, Volpi S, Gattorno M and Schena F (2022) Adenosine Deaminase 2 Deficiency (DADA2): A Crosstalk Between Innate and Adaptive Immunity. Front. Immunol. 13:935957. doi: 10.3389/fimmu.2022.935957
Received: 04 May 2022; Accepted: 10 June 2022;
Published: 11 July 2022.
Edited by:
Pui Y. Lee, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Durga Prasanna Misra, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), IndiaBenzeeta Pinto, Manipal Hospital, India
Copyright © 2022 Signa, Bertoni, Penco, Caorsi, Cafaro, Cangemi, Volpi, Gattorno and Schena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Gattorno, bWFyY29nYXR0b3Jub0BnYXNsaW5pLm9yZw==
†These authors share senior authorship