 Sergi Casadó-Llombart1
Sergi Casadó-Llombart1 María Velasco-de Andrés1
María Velasco-de Andrés1 Cristina Català1
Cristina Català1 Alejandra Leyton-Pereira1
Alejandra Leyton-Pereira1 Rebeca Gutiérrez-Cózar2Belén Suárez3
Rebeca Gutiérrez-Cózar2Belén Suárez3 Noelia Armiger1
Noelia Armiger1 Esther Carreras1Miriam Esteller4,5,6Elena Ricart4,5,6Ingrid Ordás4,5,6
Esther Carreras1Miriam Esteller4,5,6Elena Ricart4,5,6Ingrid Ordás4,5,6 Javier P. Gisbert6,7
Javier P. Gisbert6,7 María Chaparro6,7María Esteve6,8
María Chaparro6,7María Esteve6,8 Lucía Márquez9David Busquets10Eva Iglesias11,12Esther García-Planella13María Dolores Martín-Arranz14Juliane Lohmann15
Lucía Márquez9David Busquets10Eva Iglesias11,12Esther García-Planella13María Dolores Martín-Arranz14Juliane Lohmann15 C. Korcan Ayata16
C. Korcan Ayata16 Jan Hendrik Niess16,17
Jan Hendrik Niess16,17 Pablo Engel1,2
Pablo Engel1,2 Julián Panés4,5,6
Julián Panés4,5,6 Azucena Salas2,4,6Eugeni Domènech6,18
Azucena Salas2,4,6Eugeni Domènech6,18 Francisco Lozano1,2,3* and ENEIDA Project of GETECCU
Francisco Lozano1,2,3* and ENEIDA Project of GETECCU- 1Immunoreceptors del Sistema Innat i Adaptatiu, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 2Departament de Biomedicina, Facultat de Medicina, Universitat de Barcelona, Barcelona, Spain
- 3Servei d’Immunologia, Centre de Diagnòstic Biomèdic, Hospital Clínic de Barcelona, Barcelona, Spain
- 4Inflammatory Bowel Disease Group, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 5Inflammatory Bowel Disease Unit, Gastroenterology Department, Hospital Clínic de Barcelona, Barcelona, Spain
- 6Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Madrid, Spain
- 7Gastroenterology Unit, Hospital Universitario de La Princesa, Instituto de Investigación Sanitaria Princesa (IIS-IP), Universidad Autónoma de Madrid (UAM), Madrid, Spain
- 8Gastroenterology Department, Hospital Universitari Mútua Terrassa, Terrassa, Spain
- 9Gastroenterology Department, Hospital del Mar and Institut Hospital del Mar Investigacions Mèdiques, Barcelona, Spain
- 10Department of Gastroenterology, Hospital Universitari de Girona Dr Josep Trueta, Girona, Spain
- 11Department of Gastroenterology, Hospital Universitario Reina Sofía, Córdoba, Spain
- 12Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC), Córdoba, Spain
- 13Department of Gastroenterology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
- 14Department of Gastroenterology, and Innate Immunity Group, IdiPAZ Institute for Health Research, La Paz Hospital, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 15Life & Medical Sciences (LIMES) Institute, University of Bonn, Bonn, Germany
- 16Department of Biomedicine, University of Basel, Basel, Switzerland
- 17University Center for Gastrointestinal and Liver Diseases, St. Clara Hospital and University Hospital, Basel, Switzerland
- 18Gastroenterology Department, Hospital Universitari Germans Trias i Pujol, Badalona, Spain
Crohn’s disease (CD) and ulcerative colitis (UC) are inflammatory bowel diseases (IBD) resulting from the interaction of multiple environmental, genetic and immunological factors. CD5 and CD6 are paralogs encoding lymphocyte co-receptors involved in fine-tuning intracellular signals delivered upon antigen-specific recognition, microbial pattern recognition and cell adhesion. While CD5 and CD6 expression and variation is known to influence some immune-mediated inflammatory disorders, their role in IBD remains unclear. To this end, Cd5- and Cd6-deficient mice were subjected to dextran sulfate sodium (DSS)-induced colitis, the most widely used experimental animal model of IBD. The two mouse lines showed opposite results regarding body weight loss and disease activity index (DAI) changes following DSS-induced colitis, thus supporting Cd5 and Cd6 expression involvement in the pathophysiology of this experimental IBD model. Furthermore, DNA samples from IBD patients of the ENEIDA registry were used to test association of CD5 (rs2241002 and rs2229177) and CD6 (rs17824933, rs11230563, and rs12360861) single nucleotide polymorphisms with susceptibility and clinical parameters of CD (n=1352) and UC (n=1013). Generalized linear regression analyses showed association of CD5 variation with CD ileal location (rs2241002CC) and requirement of biological therapies (rs2241002C-rs2229177T haplotype), and with poor UC prognosis (rs2241002T-rs2229177T haplotype). Regarding CD6, association was observed with CD ileal location (rs17824933G) and poor prognosis (rs12360861G), and with left-sided or extensive UC, and absence of ankylosing spondylitis in IBD (rs17824933G). The present experimental and genetic evidence support a role for CD5 and CD6 expression and variation in IBD’s clinical manifestations and therapeutic requirements, providing insight into its pathophysiology and broadening the relevance of both immunomodulatory receptors in immune-mediated disorders.
Introduction
Inflammatory bowel diseases (IBD) are a group of chronic inflammatory conditions of the gastrointestinal tract, including ulcerative colitis (UC) and Crohn’s disease (CD). The precise etiology of IBDs remains unknown, though their relation to multiple and diverse genetic, immunological and environmental factors is accepted. Genome-wide association studies (GWAS) have identified immune-related genes associated to susceptibility and/or clinical manifestations that point to an inappropriate regulation of innate and/or adaptive immune responses in IBD (1). However, these polymorphisms alone do not account for IBD heritability, suggesting that other environmental, epigenetic and genetic factors, including rare variants, must be in place (1).
CD5 and CD6 are paralogs sharing homology in tissue expression patterns, structure and function (2–4). They encode signal-transducing surface co-receptors expressed on all T and B1a cells and involved in the fine tuning of intracellular activation signals delivered upon specific antigen recognition by lymphocyte’s clonotypic receptors (5). Both CD5 and CD6 receptors are composed of an extracellular region encompassing three tandem scavenger receptor cysteine-rich (SRCR) domain repeats, a transmembrane region, and a cytoplasmic region devoid of catalytic activity but well adapted for phosphorylation and association with downstream signaling effectors. Importantly, CD5 and CD6 are physically associated with the T cell receptor complex (TCR) with which co-localize at the center of the immunological synapse (6), providing inhibitory (CD5) and activating/inhibitory (CD6) signals (7). This is likely achieved through interaction with endogenous counter-receptors such as CD166/activated leukocyte cell adhesion molecule (ALCAM) (8), Galectins 1 and 3 (9), and CD318/CUB domain-containing protein 1 (CDCP-1) (10) for CD6, and still ill-defined ligands (CD72, IgVh framework, gp200, gp40-80, gp150, IL-6 and CD5 itself) for CD5 (11–18). Both molecules also act as pattern-recognition receptors (PRRs) for microbial-associated molecular patterns (MAMPs), where CD5 interacts with fungal (β-glucan) (19), viral (hepatitis C virus) (20), and parasitic (E. granulosus teguments) structures (21), while CD6 does it with bacterial (lipopolysaccharide, lipoteichoic acid and peptidoglycan) (22), viral (gp120 HIV-1) (23), and parasitic (E. granulosus tegument) structures (21). This dual role of CD5 and CD6 as both immunomodulatory and microbial PRR receptors is supported by pre-clinical models of infection, autoimmunity and cancer involving Cd5- and Cd6-deficient mouse lines, as well as infusion of wild-type mice with soluble CD5 and CD6 proteins (24–26).
To date, no CD5 or CD6 deficiencies have been reported in humans. However, functionally relevant single nucleotide polymorphisms (SNPs) of CD5 and CD6 have been identified, which act as susceptibility or disease modifier markers in autoimmune and neoplastic processes. Allelic combinations of the CD5 rs2241002 and rs2229177 SNPs resulting in hyper-reactivity to TCR stimulation are associated to more severe systemic lupus erythematosus (SLE) forms, but predict better prognosis in chronic lymphocytic leukemia (CLL) and melanoma (27–29). Moreover, GWAS have involved CD5 (rs2229177) in rheumatoid arthritis susceptibility (30). Regarding CD6, the rs12360861, rs17824933 and rs11230563 SNPs are revealed as disease modifiers in psoriasis, and as susceptibility markers in multiple sclerosis (MS) and Behçet’s disease (31–34). Also, GWAS and meta-analyses have associated the CD6 rs11230563 SNP to IBD susceptibility (35, 36). However, its role as a disease modifier in IBD, and the involvement of other neighboring SNPs from the CD6 and CD5 genes, and from the functionally related CD166/ALCAM gene, are still unknown.
The present work explores the consequence of CD5 and CD6 expression and variation in experimental and clinical IBD. To this end, we first analyzed the impact of Cd5 and Cd6 deficiency on dextran sulphate sodium (DSS)-induced colitis, an experimental model of human IBD (37). Subsequent clinical association studies assessed the impact of CD5 and CD6 variations on different clinically relevant manifestations and therapeutic requirements of CD and UC.
Materials and methods
Mice
Cd5-deficient (Cd5-/-) mice backcrossed to C57BL/6 background were provided by Chander Raman (University of Alabama at Birmingham) (38). Cd6-deficient (Cd6-/-) C57BL/6 mice were obtained through a development agreement with the Knock-Out Mouse Project Repository (KOMP), an international consortium promoted by the National Institutes of Health (NIH; https://www.komp.org) (39). Wild-type C57BL/6 mice from Charles River Laboratories (France) were bred in our animal facility. All mouse procedures were approved by the Animal Experimentation Ethical Committee from University of Barcelona.
DSS-induced mouse colitis model
Colitis was induced by administration of 2% (w/v) 36-50 kDa DSS (MP Biomedicals) in drinking water for 5 days to 11- to 19- week-old wild-type, Cd5-/- and Cd6-/- female mice of C57BL/6 background weighing >20 g. Body weight and disease activity index (DAI) were monitored every day. DAI was scored as follows: rectal bleeding (absent=0, present=1), animal motility (normal=0, reluctant=1, hunched=2), fur appearance (normal=0, ruffled=1, spiky=2) and body weight loss (none=0, 0-5%=1, 5-10%=2, 10-15%=3, >15%=4). At day 8, mice were euthanized by cervical dislocation for collection of blood and organ samples. Colons were measured and weighted, and terminal pieces were collected for histology and RNA extraction. EDTA-anticoagulated blood was centrifuged in heparinized capillaries for 30 min at 1000 xg and hematocrit was calculated as the length of packed red blood cells (RBC) divided by the total blood length (RBC + serum) multiplied by 100. For RBC count, blood was diluted in PBS and RBC were counted with a hemocytometer. For microbiological analyses, mesenteric lymph nodes (mLN) and liver were collected under sterile conditions and disaggregated through a 40 μm nylon mesh for overnight (o/n) seeding at 37 °C on Columbia agar plates with 5% sheep blood (Becton-Dickinson) and colony forming unit (cfu) counting. Pieces of ~2 mm were cut from the terminal colon of mice and submerged in RNA later (Sigma) o/n at 4 °C before being stored dry at -80 °C for further RNA analysis or fixed in PBS containing 4% paraformaldehyde during 48 h for histological studies. RNA was extracted using the TRIzol® Reagent (Life Technologies) and the PureLink™ RNA Mini Kit (Ambion, Life Technologies) following manufacturer’s instructions, with the aid of a QIAGEN TissueLyser. RNA was quantified and retrotranscribed into cDNA by using the High-capacity cDNA Kit (Life Technologies). Cytokine mRNA levels were assessed by real-time quantitative PCR (RT-qPCR) with the TaqMan™ Fast Universal PCR Master Mix No AmpErase™ UNG (Life Technologies) using a 7900HT fast real-time PCR system (Applied Biosystems, Foster City, CA, US) and the following FAM gene expression assays: Mm01179194_m1 (Cd3e), Mm00435532_m1 (Pdcd1), Mm00432423_m1 (Cd79a), Mm01337324_g1 (Ncr1), Mm00447885_m1 (Klrc1), Mm00447885_m1 (Mpo), Mm01324470_m1 (Lcn2), Mm00440502_m1 (Nos2), Mm00801778_m1 (Ifng), Mm00439619_m1 (Il17a), Mm00445259_m1 (Il4), Mm00439614_m1 (Il10), Mm00444241_m1 (Il22), Mm00443260_g1 (Tnf), Mm00434228_m1 (Il1b), Mm00446190_m1 (Il6), Mm01178820_m1 (Tgfb1), Mm00441259_g1 (Ccl3), and Mm04207460_m1 (Cxcl1), Mm00450960_m1 (Tbx21), Mm01261022_m1 (Rorc), Mm00484683_m1 (Gata3) and Mm00475162_m1 (Foxp3), all from Thermo Fisher Scientific. Relative cytokine mRNA expression normalized to Gapdh (Mm99999915_g1) expression was calculated as 2-ΔΔCt, where ΔΔCt = (CTGene of interest sample − CTGAPDH sample) − (CTGene of interest basal − CTGAPDH basal).
For histological analysis, fixed samples were included in paraffin. Three micrometer tissue sections were obtained and stained with hematoxylin-eosin. Histology was scored by two independent evaluators according to the following parameters: degree of inflammation (0-3), goblet cell loss (0-2), abnormal or hyperproliferative crypts (0-3), abscesses (0-1), architectural damage (0-2), transmural damage (0-3). Images were obtained with an Eclipse 50i microscope, using a Pan Fluor 10x/0.30 objective and a Digital Sight DS-5M camera, all from Nikon.
For immunohistochemical analysis, paraffin-embedded 5 μm tissue sections were immersed in xylene and dehydrated in ethanol. After antigen retrieval, tissue sections were blocked with PBS 5% FBS. For myeloperoxidase (MPO) chromogenic immunohistochemistry assay, primary goat anti-mouse MPO polyclonal antibody (R&D Systems) was incubated at 4°C overnight. Then, endogenous peroxidase activity was blocked using PBS 0.3% H2O2 solution for 10 min at room temperature and peroxidase-labelled rabbit anti-goat IgG secondary antibody (SIGMA) was incubated for 1h at room temperature. Tissue sections were stained using 3,3’-diaminobenzidine (DAB; SIGMA) and then hematoxylin staining was performed following standard protocols. Sections were mounted with DPX and visualized at 20x magnifications using a NIKON e600 microscope.
For CD3ϵ and IgM immunofluorescence assay, endogenous biotin was blocked with the Avidin/Biotin blocking kit SP-2001 (VectorLabs) following manufacturer’s indications. Then, primary antibodies rabbit anti-mouse CD3ϵ (Cell signaling, D4V8L; dilution 1/100) and FITC-labelled goat anti-mouse IgM (Southern Biotech; dilution 1/200) were incubated at 4 °C overnight. Biotin-labelled secondary donkey anti-rabbit IgG antibody (Jackson Immunoresearch, dilution 1/200) was incubated for 1 h at room temperature. Finally, A555-labelled streptavidin (Roche, dilution 1/200) was incubated for 20 min at room temperature and samples were mounted with mounting medium (PBS 80% glycerol). Samples were visualized at 10 and 20x magnifications using a NIKON e600 microscope.
DNA samples from patients and controls
Genomic DNA samples from CD (n=1352) and UC patients (n=1013) were retrieved from the ENEIDA biobank upon approval from the Spanish Working Group on CD and UC (GETECCU) (40). Control genomic DNA samples from volunteer donors of the Blood and Tissue Bank (BST) of the Generalitat de Catalunya (n=604) were purified by using the MagNA Pure 96 DNA and Viral NA Large Volume Kit (Roche Diagnostics, Rotkreuz, Switzerland) and the High-throughput robotic workstation MagNa Pure 96 (Roche Diagnostics). The study was approved by the Ethical Committee of Clinical Research of the Hospital Clínic de Barcelona.
SNP genotyping
Genomic DNA samples (20 ng) were subjected to RT-PCR in a LightCycler® 480 Instrument (Roche) using TaqMan Genotyping Master Mix and TaqMan probes for CD5 (rs2229177, rs2241002), CD6 (rs12360861, rs17824933, rs11230563), and CD166/ALCAM (rs6437585) (all from Thermo Fisher), following manufacturer’s instructions. Genotyping failure rate was lower than 0.02 for all SNPs.
Definitions
Location (terminal ileum, colon, ileocolon, and upper gastro-intestinal) and behavior (nonstricturing and nonpenetrating, stricturing, and penetrating) of CD were classified according to the Montreal classification (41). For statistical analysis of location, a value of 1 was assigned to patients with colonic disease, 2 to patients with ileocolonic disease and 3 to patients with ileal disease, independently of upper gastro-intestinal tract involvement. Upper gastro-intestinal tract involvement (presence vs. absence) was assessed independently of distal ileal and colonic involvement. For statistical analysis of extent in UC patients, a value of 0 was assigned to patients with ulcerative proctitis (Montreal classification E1) and a value of 1 was assigned to patients with left-sided UC or extensive UC (Montreal classification E2 and E3). Prognosis was calculated as previously described: patients not requiring any immunomodulatory nor surgical treatment during at least 4 years of follow-up from diagnosis were classified as “good prognosis” while patients requiring two or more immunomodulatory treatments and/or two or more abdominal surgeries were described as “poor prognosis” (42).
Statistical analyses
Statistical analysis in patient/donor cohort studies was performed with R 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria), with the packages ‘SNPassoc’, ‘survival’, and ‘haplo.stats’ available at the Comprehensive R Archive Network (CRAN) repository. The ‘association’ function included in the ‘SNPassoc’ package was used to assess linkage between each SNP and desired clinical variables with generalized linear models. For each analysis, 4 models were generated (codominant, dominant, recessive, log-additive), and the model with lowest Akaike information criterion (AIC) was chosen. Analyzed variables were age of onset (calculated date of diagnosis − date of birth), peripheral arthritis/arthralgia, ankylosing spondylitis, sacroiliitis, sclerosing cholangitis, cutaneous manifestations (pyoderma gangrenosum or erythema nodosum), ocular manifestations (uveitis or iritis), requirement of biological treatments and prognosis. Additionally, location of the disease, presence of stenosis, presence of fistulae and perianal disease were included in the CD cohort and extent of the disease was included in the UC cohort. In all analyses, sex and persistent tobacco consumption were included as co-variants. To generate stenosis-free survival and fistulae-free survival curves, time between enrolment and complication (patients with stenosis or fistulae) or between enrolment and last follow-up (patients without stenosis or fistulae) was calculated. Cox proportional hazards regression was used to assess the linkage between each SNP and stenosis-free survival or fistulae-free survival. Association of SNPs with susceptibility to CD, UC or combined (IBD) was assessed with the ‘association’ function by comparing each cohort with the control cohort. P values were corrected for false discovery rate (FDR) with the ‘p.adjust’ function (q values). The ‘haplo.glm’ function included in the ‘haplo.stats’ package was used to assess linkage between haplotypes and binary clinical variables with generalized linear models, and odds ratio (OR) and confidence intervals (CI) for these associations were obtained with the ‘haplo.cc’ function.
In the study of animal models, normality of data was assessed with the D’Agostino & Pearson normality test. When data was normally distributed, differences were assessed by t-tests, otherwise Mann-Whitney tests were performed. In multiple comparisons, P values were corrected for false discovery rate.
Results
Cd5 and Cd6 deficiency modulate DSS-induced colitis
The putative role of CD5 and CD6 lymphocyte co-receptors in the pathophysiology of IBD was first explored by subjecting Cd5-/- and Cd6-/- mice to the DSS-induced colitis. Cd5-/- mice showed a less aggressive disease than Cd5+/+ controls (Figure 1A), as deduced from lower body weight loss and DAI, in agreement with a previously published result (43). In contrast, Cd6-/- mice showed an exacerbated phenotype with regard to Cd6+/+ controls. Accordingly, Cd6-/- mice presented a higher body weight loss (Figure 1B, left) and a higher DAI (Figure 1B, right), which was seasonally influenced, as observed in spring/summer and autumn/winter variations (Figure 1C).
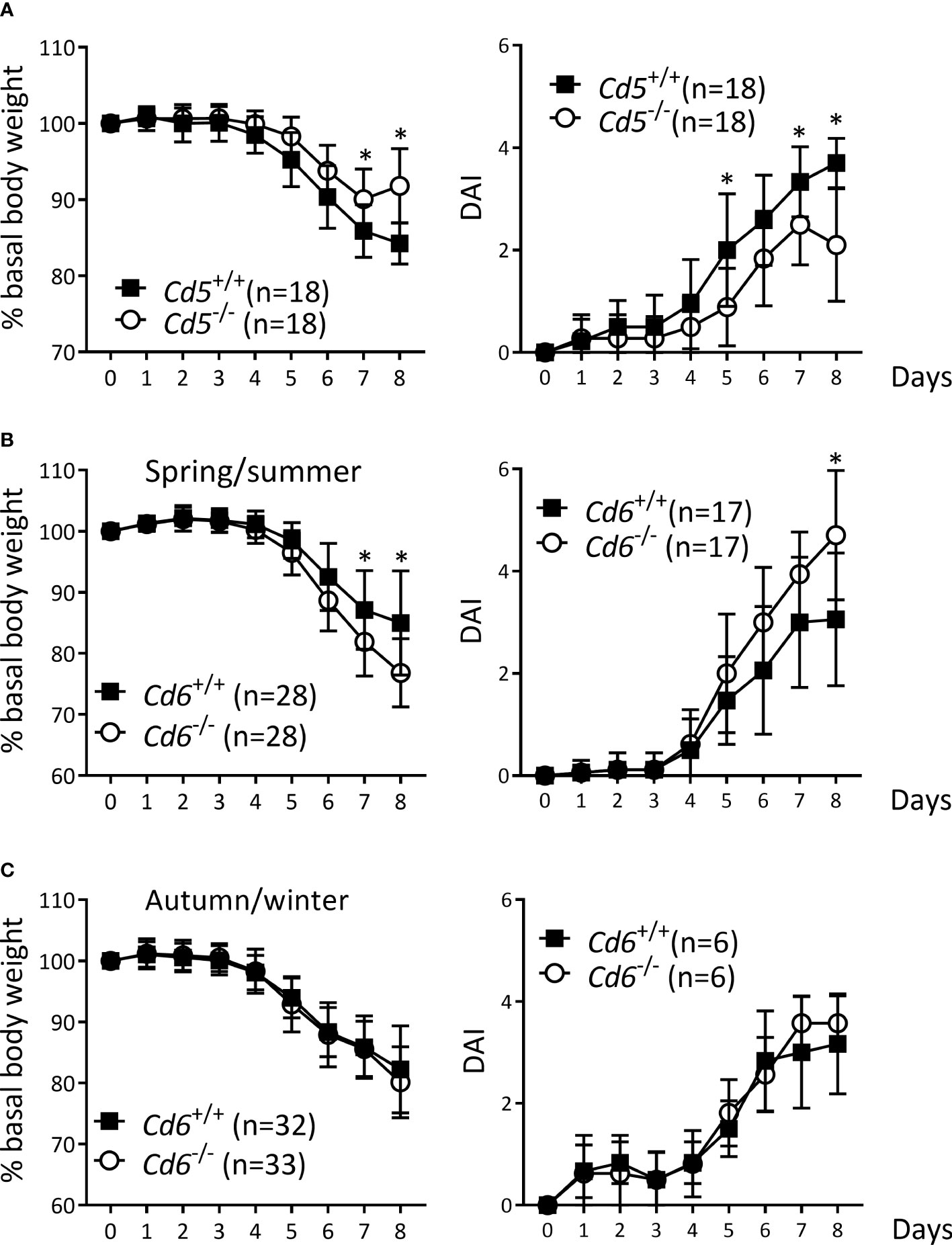
Figure 1 DSS-induced colitis in Cd5-/- and Cd6-/- mice vs. wild-type controls. (A) Percentage of basal body weight (left) and DAI (right) of Cd5-/- mice vs. Cd5+/+ controls. Data combined from two independent experiments is shown. (B) Percentage of basal body weight (left) and DAI (right) of Cd6-/- mice vs. Cd6+/+ controls in spring/summer (between April and September). Basal body weight data are combined from four independent experiments, while DAI data are combined from two independent experiments. (C) Percentage of basal body weight (left) and DAI (right) of Cd6-/- mice vs. Cd6+/+ controls in autumn/winter (between October and February). Basal body weight data are combined from four independent experiments, while DAI data are come from a single experiment. Mean ± SD values are depicted. Statistical differences were assessed by multiple t-tests (one per day) controlled with the FDR approach. *, q<0.01.
Contrary to the Cd5-/- case, the lack of published information of Cd6-/- mice on the DSS-induced colitis model encouraged a deeper evaluation of different experimental parameters at the end of disease follow-up (day 8). No differences were observed in colon length, weight or weight/length ratio relative to Cd6+/+ controls (Figure 2A). As illustrated in Figure 2B, Cd6-/- mice presented increased hematocrit consistent with higher diarrhea-induced fluid loss, and a trend to lower RBC counts together with increased mean corpuscular volume (MCV) consistent with moderate rectal bleeding and erythroblast production, respectively (44). No differences in cfu count were observed in mLN and liver (Figure 2C), arguing against differential bacterial translocation to draining organs as responsible for the differences observed in disease severity. Histological analyses showed noticeable crypt architectural distortion in colon samples from both Cd6+/+ and Cd6-/- mice, with no significant differences between their histology scores (Figure 2D). Immunohistochemical analyses of the colonic mucosa composition revealed no significant differences in terms of granulocyte (MPO+), T cell (CD3ϵ+) and B cell (IgM+) infiltrates (Figure 2E). Gene expression analyses of a wide panel of pro-/anti-inflammatory cytokine and chemokine and transcription factors revealed decreased expression of Ifng, Cd3e, Ncr1 and Gata3 together with increased expression of Il6 and Cxcl1 in Cd6-/- mice with regard to controls (Figure 3A). A trend towards increased expression of Lcn2 was also observed (Figure 3A). No differences were observed regarding expression of Tgfb1, Tnf, Il1b, Il17a, Il10, Il22, Tbx21, Rorc, Foxp3, Ccl3, Klrc1, Pdcd1, Cd79a, Mpo and Nos2 (Supplementary Figure 1). Expression of Il4, a target of GATA3, was also analyzed but it was undetectable in a high proportion of samples. Additional analyses showed significantly increased Il17a/Ifng ratio and a similar trend for Rorc/Gata3 but no differences in the Tbx21/Gata3 ratio in Cd6-/- mice (Figure 3B).
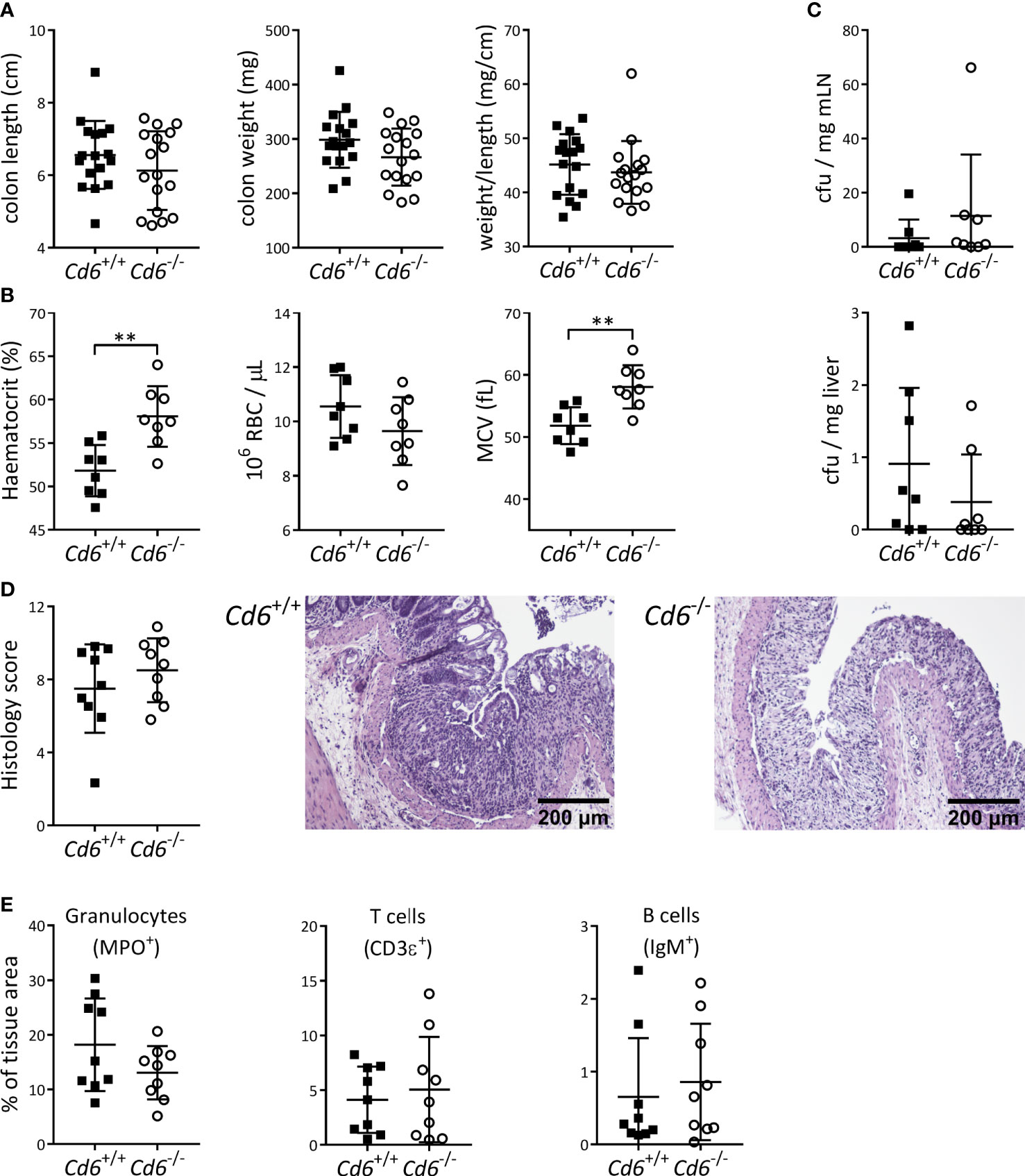
Figure 2 Monitoring of DSS-induced colitis parameters from Cd6-/- mice vs. Cd6+/+ controls at day 8 post-induction. (A) Dot plot showing colon length, weight and weight to length ratio of Cd6-/- (n=17) and Cd6+/+ control (n=17) mice. Mean ± SD values are depicted. Statistical differences were assessed by t-test. (B) Hematocrit, RBC count and mean corpuscular volume (MCV) at day 8 from Cd6-/- (n=8) and Cd6+/+ (n=8). Mean ± SD values are depicted. Statistical differences were assessed by t-test. **, p<0.01. (C) Analysis of microbial translocation into mesenteric lymph nodes (mLN; top) and liver (bottom) from the same mice as in (B) Depicted are mean ± SD of cfu/mg. Statistical differences were assessed by Mann-Whitney tests. (D) Histology score (mean ± SD, left) and representative haematoxylin-eosin stains from DSS-treated Cd6+/+ (center) and Cd6-/- (right) mice. Scale bar: 200 μm. Statistical differences were assessed by t-test. (E) Immunohistochemical analyses of the terminal colon in DSS-treated mice. Percentage of MPO, CD3ϵ and IgM-stained tissue (mean ± SD) from colon sections. Statistical differences were assessed by t-test.
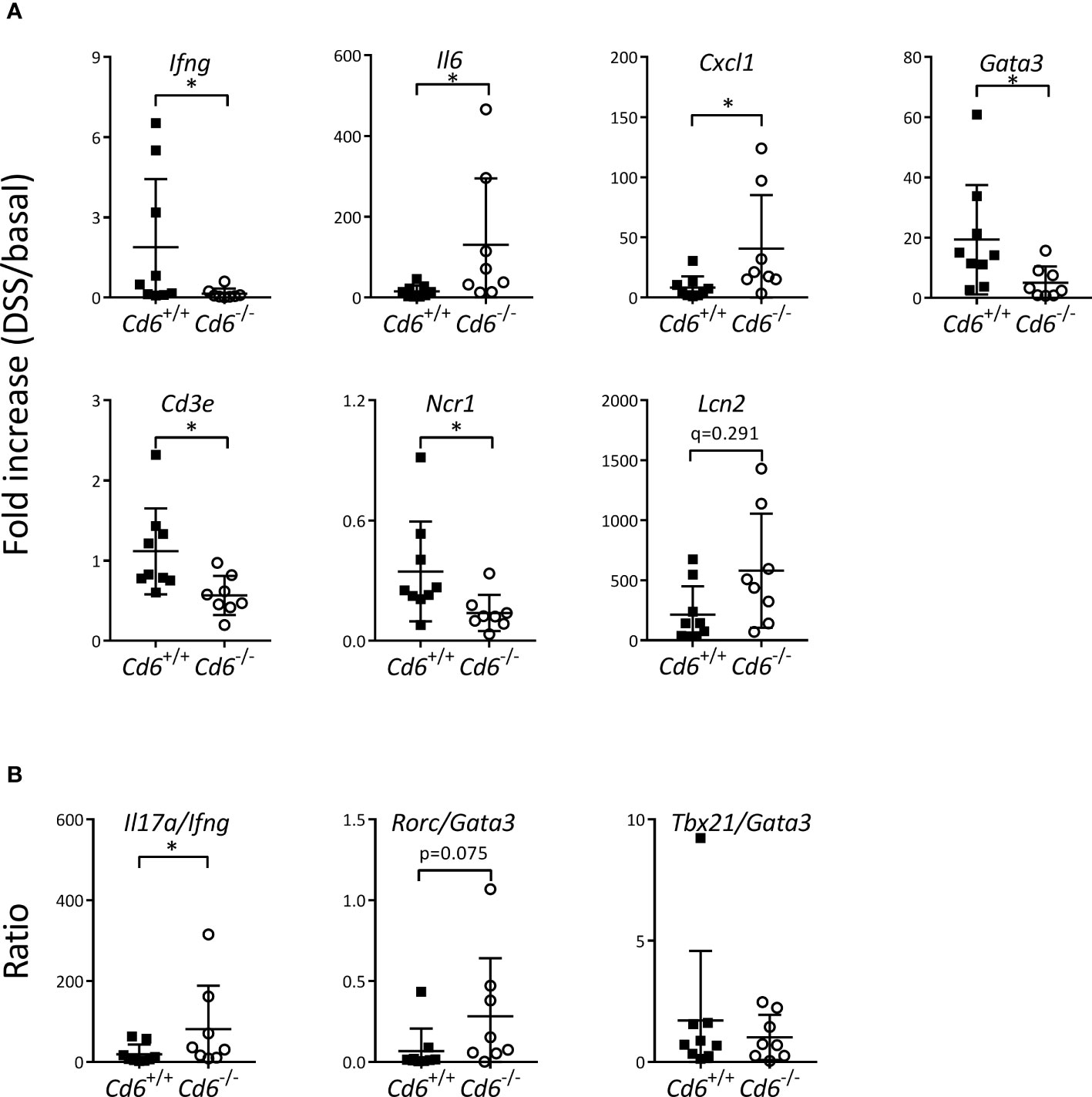
Figure 3 mRNA expression in colons from Cd6-/- vs. Cd6+/+ mice at day 8 post DSS-induced colitis. (A) Relative mRNA expression of different transcripts from colon samples. Depicted are mean ± SD of mRNA fold increase (DSS/basal). (B) Fold increase ratio of indicated mRNA transcripts from colon samples. Ratios were calculated by dividing the fold increase of the following transcripts: Il17a, Ifng, Rorc, Gata3, Tbx21 and Gata3. Statistical differences were assessed by Mann-Whitney tests and corrected for FDR. *, q< 0.1.
CD5 and CD6 variants impact clinical expression of IBD
CD (n=1352) and UC (n=1013) patients from the ENEIDA registry and volunteer blood donor controls (n=604) were genotyped for functionally relevant CD5 (rs2229177, rs2241002), CD6 (rs12360861, rs11230563, rs17824933), and CD166/ALCAM (rs6437585) SNPs. All SNPs were in Hardy-Weinberg equilibrium, except for the rs2241002 in the CD cohort (p=0.0276). Description of the study SNPs and cohorts are shown in Tables 1, 2.

Table 1 Summary of the CD5, CD6 and CD166/ALCAM SNPs analyzed in the present study.
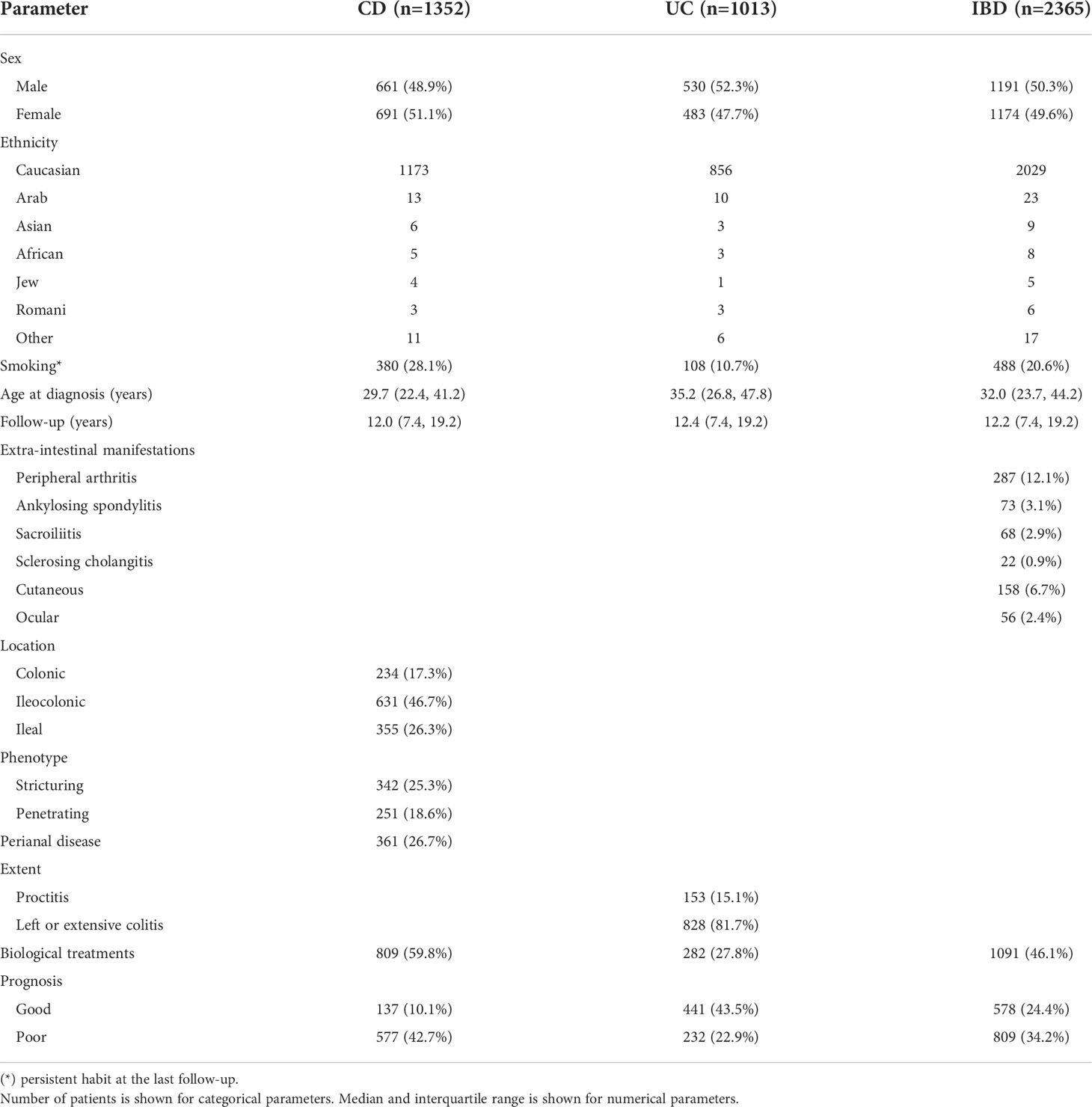
Table 2 Clinical characteristics of the study cohorts.
No association was found for any of the SNPs analyzed with disease susceptibility following comparisons of controls with the CD and UC cohorts, either separately (CD vs. controls, UC vs. controls) or together (IBD vs. controls). Next, the effect of the CD5, CD6 and CD166/ALCAM gene variants on different clinically relevant parameters of CD (age at diagnosis, behavior, location, perianal disease and prognosis) and UC (age at diagnosis, extent and prognosis) was assessed. A significant association was found for the CD5 rs2241002CC genotype with preferential ileal location in the CD cohort (Table 3). Because CD location can influence the risk of developing stenosis and fistulae, association between the SNP and stenosis-free and fistulae-free survival was tested, but no significant results were found. Association between CD5 SNPs and upper-gastrointestinal (GI) tract affectation was also not significant. Haplotypic analyses showed increased need of biologic therapies in CD patients carrying the CD5 rs2241002C rs2229177T haplotype compared with those carrying the most common rs2241002C rs2229177C haplotype (Table 4). Similarly, UC patients carrying the CD5 rs2241002T rs2229177T haplotype had a worse prognosis than those carrying the rs2241002C rs2229177C haplotype (Table 4).
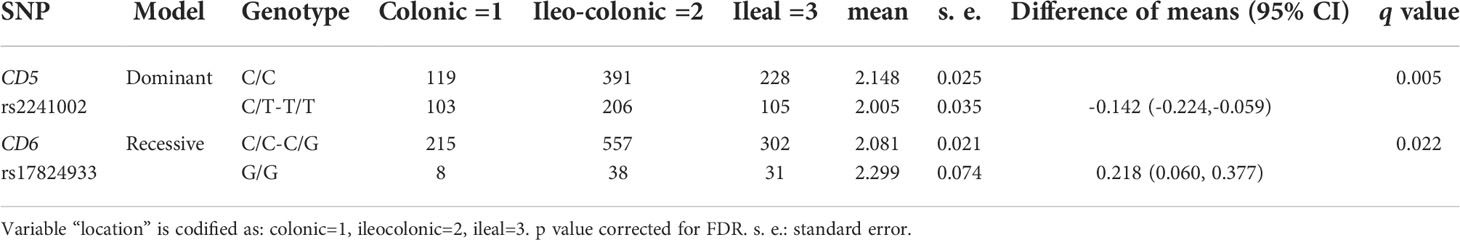
Table 3 Linear regression analysis of CD5 rs2241002 and CD6 rs17824933 SNPs association with CD location. Corrected for sex and smoking.
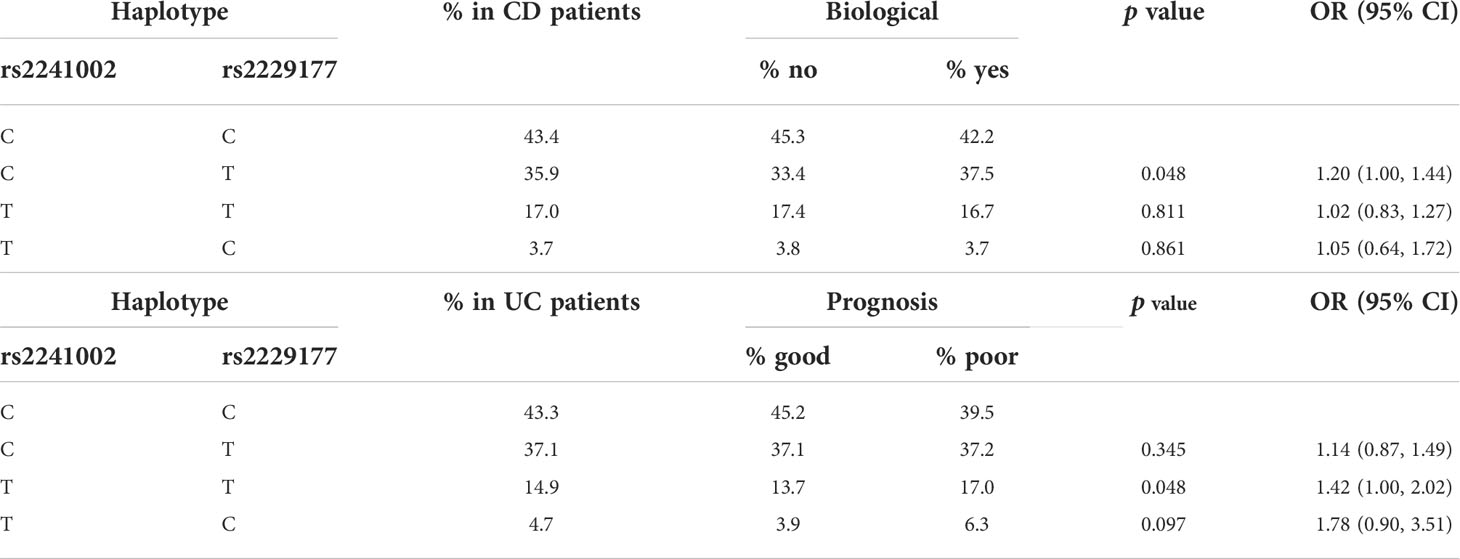
Table 4 Logistic regression analysis of CD5 haplotype association with biological therapy requirement in CD (top half) and to prognosis in UC (bottom half).
Regarding CD6 SNPs associations, the rs17824933GG genotype was associated with preferential ileal location in CD patients (Table 3). This led us to also test association between this SNP and stenosis-free and fistulae-free survival. As seen in Figure 4, the CD6 rs17824933GG genotype was significantly associated with shorter fistula-free survival (HR = 1.56, 95% CI 1.01–12.42, p = 0.046). No significant association was found between CD6 SNPs and upper-GI tract affectation. The CD6 rs17824933GG genotype was also associated with higher extent (left or extensive colitis) in UC patients (Table 5). The CD6 minor rs12360861A allele showed association with better prognosis in CD patients (Table 5). Regarding the appearance of extra-intestinal manifestations, logistic regression analyses showed a significant association of homo- or heterozygous combinations of the CD6 rs17824933G allele with lower risk of ankylosing spondylitis in the whole cohort of IBD patients (Table 5).
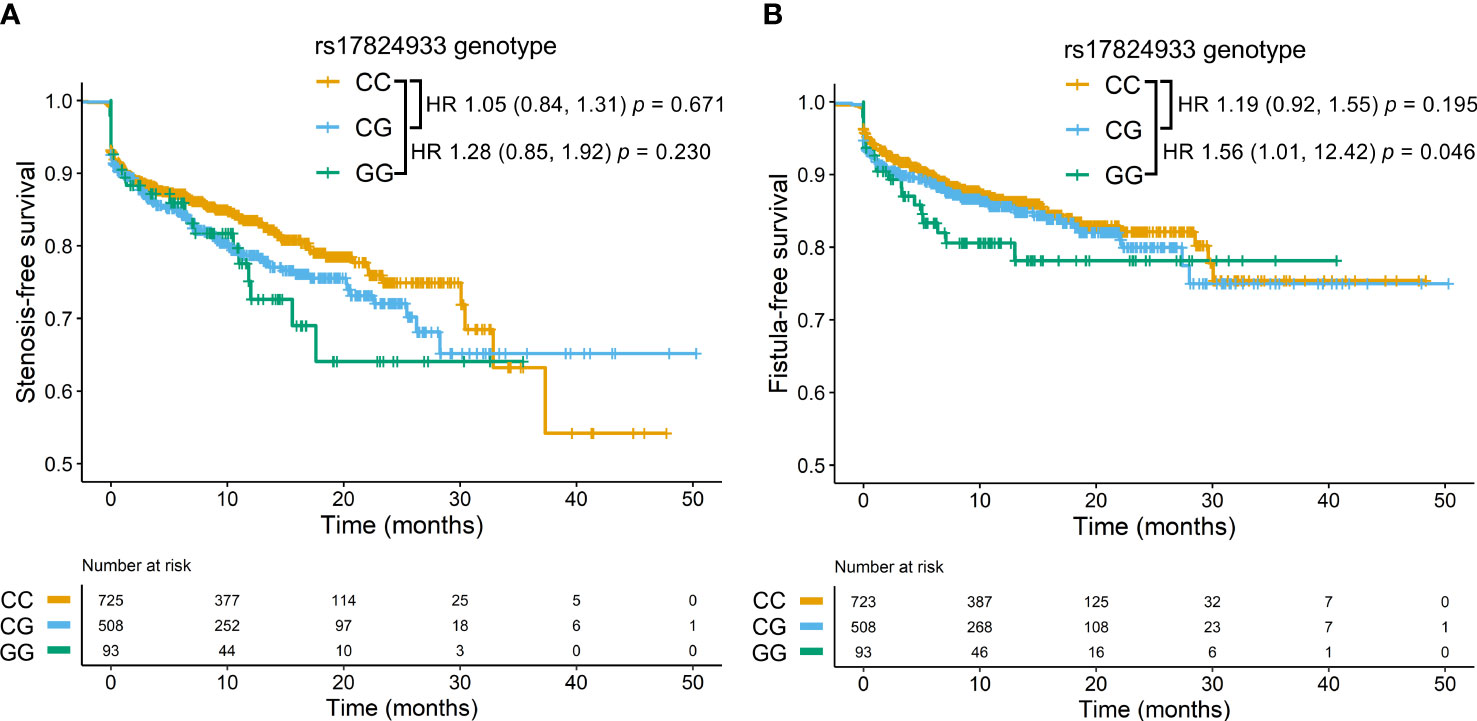
Figure 4 Stenosis and fistulae in CD patients according to rs17824933. Stenosis-free survival (A) and fistulae-free survival (B) of CD patients carrying different CD6 rs17824933 genotypes. Statistical differences were assessed by the Cox proportional hazards model. (A) In the stenosis-free survival analysis hazard ratio (HR) comparing GG and CC genotypes was 1.28, (95% CI 0.85–1.92), p = 0.230, and HR comparing CG and CC genotypes was 1.05, (95% CI 0.84–1.31), p = 0.671. (B) In the fistulae-free survival analysis HR comparing GG and CC genotypes was 1.56, (95% CI 1.01–12.42), p = 0.046, and HR comparing CG and CC genotypes was 1.19, (95% CI 0.92–1.55), p = 0.195.
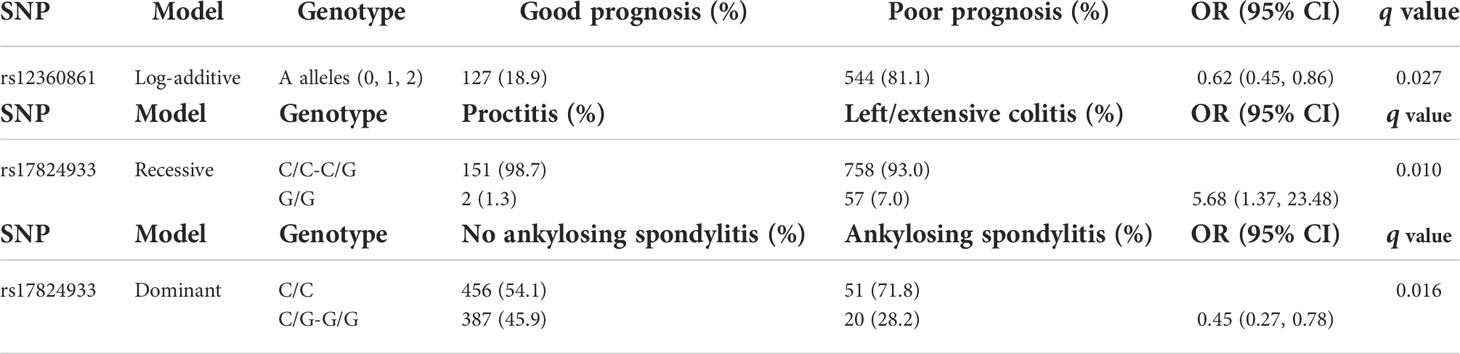
Table 5 Logistic regression analysis of CD6 SNP association with CD prognosis (top), UC extent (middle), and ankylosing spondylitis in IBD patients (bottom), corrected for sex and smoking.
No statistical association was observed with any of the clinical parameters analyzed for CD166/ALCAM rs6437585 SNP, which has been reported to influence CD166/ALCAM transcriptional activity and MS risk (45, 46).
Discussion
We provide experimental and clinical evidence for the involvement of CD5 and CD6 expression and variation in IBD. Previous GWAS and meta-analysis studies have identified the CD6 locus (SNP rs11230563) as a susceptibility marker in CD and UC, thus supporting its contribution to IBD etiopathogenesis (35, 36). Here, we used genetically modified mice and candidate gene-driven association analyses to clinical traits and prognosis with functionally relevant SNPs from the CD5 and CD6 paralogs, as well as from CD166/ALCAM.
Etiopathogenic factors for IBD include host genetic susceptibility, dysregulated immune response, intestinal dysbiosis, and impairment of intestinal epithelial barrier function. Under normal circumstances, there is continuous crosstalk between gut microbiota and the immune system, where gut microbiota modulates the host’s innate and adaptive immunity and vice versa (47). Gut microbiota is in close contact with the intestinal barrier, consisting of an epithelial cell layer and a variety of immune cells of hematopoietic origin. Cells from the intestinal barrier (both epithelial and hematopoietic) sense and signal the presence of microbial components via PRRs, which belong to different structural families such as lectin C-type, leucine-rich repeats (LRR), immunoglobulin (Ig), or scavenger receptor cysteine-rich (SRCR) domains (48). Both CD5 and CD6 are lymphocytic members of the SRCR superfamily, expressed by all T cells and the B1a subset responsible for production of polyreactive natural IgM antibodies (4, 49). CD5 and CD6 are also represented in certain immune cells subsets present in mucosal barriers such as regulatory T (Treg) and B (B1a, Breg) cells, certain macrophage and dendritic cells and innate lymphoid cells (NK, iNKT, ILCs) (50–52). CD166/ALCAM, the best characterized CD6 ligand, is also found in the gastrointestinal epithelial tract (53). Increased expression of both CD6 and CD166/ALCAM has been reported in inflamed mucosa from IBD patients, a fact that is attributed to higher CD6-expressing T cell infiltration rather than surface CD6 expression levels (54). This may relate to quantitative trait loci studies in which the rs11230584 SNP in the intergenic region between CD5 and CD6 modulates expression of both genes in IBD patients but not in healthy controls (55). Taken together, their tissue and cell expression pattern, microbial recognition properties and ability to modulate lymphocyte activation/differentiation and cell adhesion provide the basis for considering both CD5 and CD6 as contributors to IBD pathogenesis.
The observation that both CD5- and CD6-deficient mice differ in their response to DSS-induced colitis further supports their involvement in IBD. In Cd5-/- mice, attenuated DSS-induced colitis was observed in agreement with a previous report (43). The mechanism underlying such attenuated colitis has already been explored and attributed to increased suppressive function of Treg cells from Cd5-/- mice (43), a fact that was not confirmed by others (56). An alternative mechanism could be the increased activation-induced cell death (AICD) in Cd5-/- effector T cells as a result of inhibitory role assigned to the CD5 receptor (7, 57). Further evidence for CD5 expression involvement in IBD comes from recent report showing that inducible Cd5-deficient mice in the autoimmune-prone non-obese diabetic (NOD) background undergo exacerbated DSS-induced colitis by modifying T cell effector function (58).
Regarding Cd6-/- mice, no analysis of DSS-induced colitis has been brought forward, in spite of reports of Cd6-/- mice behavior in several other immune-related inflammatory disease models (i.e., intestinal ischemia-reperfusion, bovine or avian type II collagen-induced arthritis, chronic graft-versus-host disease-induced lupus-like, imiquimod-induced psoriasis-like skin inflammation, experimental autoimmune encephalitis, and autoimmune uveitis) (10, 34, 39, 59–62). CD6 deficiency results in attenuated or exacerbated phenotypes according to mouse background and experimental models responsive to different underlying mechanisms (e.g., increased AICD or defective Treg function). This puzzling situation has been unveiled by CD6 receptor’s multitask signalosome with opposite functions in T cell activation (7). CD6 multifaceted role accounts for past difficulties in classifying it as a co-inhibitory or -stimulatory receptor.
Here we observed that Cd6-/- mice exhibit an increased body weight loss and DAI upon DSS-colitis induction, in conjunction with differential expression of certain mRNA transcripts. This included decreased expression of Ifng —the prototypical Th1 cytokine— and Gata3 —the master regulator of Th2 differentiation—, no differences in Il17a and Il10 expression, and increased expression of Il6 and Cxcl1 —a cytokine and a chemokine involved in the Th17 function, all this pointing to a somehow misbalanced Th1/Th2/Th17 response. Additionally, reduced mRNA expression of Ncr1 —coding for NKp46, one of the NK triggering receptors— and a trend towards increased Lcn2 —coding for lipocalin-2, also named NGAL, a neutrophil secondary granule marker— in colons of Cd6-/- mice undergoing DSS-induced colitis was observed. These findings would fit with the observation that i) decreased NK cell activity would lead to increased granulocyte infiltrate in DSS-induced colitis (63), and ii) increased lipocalin-2 expression would act as marker and a counter reactor of colonic inflammation (64, 65).
Given the multifaceted nature of CD6, the above-mentioned mechanistic findings for exacerbated symptoms during DSS colitis in Cd6-/- mice do not exclude other possibilities such as decreased Treg functionality in Cd6-/- mice (59), a cell subset known for its role in mucosal protection during DSS-induced colitis (66). Another possibility could be the deficient production of natural antibodies, as found in Cd6-/- mice from DBA-1 background (60), and confirmed by us in the C57BL/6 background Cd6-/- mice used here (67). Natural antibodies are an innate component of humoral immunity and have a protective role in IBD (68, 69).
In humans, no CD5 or CD6 deficiencies have been reported. Functionally relevant CD5 or CD6 SNPs previously described act as susceptibility or disease modifier markers for immune-related disorders. Regarding CD5, the rs2241002 and rs2229177 SNPs cause nonsynonymous substitutions at the extracellular (Pro224>Leu) and cytoplasmic (Ala471>Val) regions, respectively, which are relevant to CD5-mediated signal transduction (70). Thus, homozygous carriers of the ancestral Pro224-Ala471 (rs2241002C and rs2229177C) CD5 haplotype are hyper-reactive to TCR/CD3 cross-linking, and present more severe clinical forms of SLE (27) but better CLL and melanoma prognosis (28, 29). Regarding the functionality of CD6 SNPs, the intronic rs17824933G allele identified as a susceptibility marker for MS causes over-expression of a CD6 isoform devoid of the CD166/ALCAM-binding domain (CD6Δd3) concomitant with diminished proliferation and long-term activation of CD4+ T cells (71, 72). Also, the MS-protective haplotype involving the CD6 rs11230563C and rs2074225C SNPs results in higher surface CD6 expression on several lymphocyte subsets (CD4+ and CD8+ naïve T, and NKT cells) (31).
In our genetic analysis the CD5 rs2241002 SNP, which causes a nonsynonymous substitution in the extracellular SRCR domain 2 of CD5 (Pro224>Leu), showed association with CD location. Further analyses showed association of CD5 haplotypes containing the cytoplasmic rs2229177T variant with severity parameters in CD (requirement of biological treatments) and UC (poor prognosis) patients. The rs2229177T variant involves the substitution of ancestral Ala471 for Val, which results in increased CD5 inhibitory capacity (27, 70). This can turn activated lymphocytes less sensitive to AICD and more damaging, thus making more intensive therapies necessary.
Analysis of CD6 SNPs showed association of the rs17824933G allele with preferred ileal CD location and increased UC extent. These results consolidate the damaging effect of the rs17824933G allele in inflammatory diseases, as suggested from its association with more aggressive forms of psoriasis and with MS susceptibility (34, 46). Patients with left-sided or extensive UC also tend to need more aggressive therapies and are at higher risk of developing colorectal cancer (73). A relatively short follow-up (median 12.37 years; Q1 7.43 years; Q3 19.21 years) may underlie the lack of significant differences observed for this SNP regarding prognosis (Supplementary Table 1).
The CD6 rs17824933G allele was further associated with lower risk of ankylosing spondylitis in the whole IBD cohort. This result appears to contradict the above-mentioned deleterious contribution of this variant in UC, as well as in psoriasis and MS. However, this variant also showed association with a more ileal location of CD. Articular extra-intestinal manifestations of IBD are more common in patients with colonic disease than in those with small-bowel disease (74). Thus, preferential ileal location in CD patients may account for the association of rs17824933G with lower ankylosing spondylitis risk.
The study also showed association of the CD6 rs12360861 SNP with prognosis in CD patients but not susceptibility, in agreement with a major genetic contribution to prognosis from loci distinct from those driving disease susceptibility, applicable in this case (42).
As stated above, genetic susceptibility is only one of the known factors in IBD etiopathogenesis. The importance played by other environmental factors is illustrated by seasonal onset and exacerbation patterns in IBD patients (75). We have observed seasonal variations also for Cd6-/- mice regarding susceptibility to DSS-induced colitis. More precisely, the exacerbated DSS-induced colitis phenotype of Cd6-/- mice manifested during the spring/summer but not the autumn/winter season (Figure 1C), reminiscent of other mouse models of human diseases (i.e., EAE) (76). Though incompletely understood, it has been proposed that seasonal variations might be regulated by endogenous circannual rhythms, since they are found even when the animals are subjected to a constant, controlled environment, and genetically regulated (77). Seasonality in Cd5-/- mice could be neither confirmed nor denied, since the two experiments performed were carried out in the summer season (July and September). However, recent unpublished results from a collaborative study show seasonality phenomena in Cd5-/- mice upon mannan-induced psoriatic arthritis induction (Merino R and Merino J, University of Cantabria, Spain).
The main strengths of our study are the use of a large patient cohort, which together with the experimental mouse model highlights a role for CD5 and CD6 in IBD. We also acknowledge some caveats in our study. Separate breeding of Cd5-/- or Cd6-/- mice and their wild-type counterparts can be a source of confusion, which we minimized by periodic colony refresh, the use of high sample sizes, and a large number of repetitions. We measured mRNA expression as a proxy for protein expression, but correlation between mRNA and protein levels is limited. Therefore, further protein expression assays (e.g.: flow cytometry) will be needed to ascertain the molecular mechanisms underlying differences between Cd6-/- and their wild-type counterparts. Similarly, further molecular mechanisms driving clinical SNP associations in IBD patients are to be identified.
In conclusion, our findings support a role for the CD5 and CD6 lymphocyte receptors in the pathophysiology of IBD and hint at their potential in patient stratification and as therapeutic targets. The latter is particularly valid for CD6, where the humanized anti-CD6 mAb Itolizumab currently represents a therapeutic option in several immune-mediated disorders (78), since could modulate the activity of the T cell subsets (i.e., Th1 and Th17) involved in their pathogeny (79).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author/s
Ethics statement
The studies involving human participants were reviewed and approved by Ethical Committee of Clinical Research of the Hospital Clínic de Barcelona. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Comitè Ètic d’Experimentació Animal, Universitat de Barcelona, Spain.
Contributing GETECCU members
Alfredo J. Lucendo, Gastroenterology Department, Hospital General de Tomelloso, IIS-IP, Ciudad Real, Spain, and Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd). Jordi Guardiola, Hospital Universitari de Bellvitge (IDIBELL), l’Hospitalet de Llobregat, Spain. Xavier Calvet, Servei d’Aparell Digestiu, Hospital Universitari Parc Taulí, Departament de Medicina, Universitat Autònoma de Barcelona, Sabadell, Spain, and CIBERehd. Lorenzo Oliván, Hospital San Jorge, Huesca, Spain. Marta Piqueras, Gastroenterology Department, Consorci Sanitari de Terrassa, Barcelona, Spain.
Author contributions
Conceptualization: FL, AS, SC-L, and JP. Mouse studies: SC-L, MV-dA, CC, NA, AL-P, RG-C, EC, MiE, JL, KA, JHN, PE, and AS. Genetic studies: SC-L, MV-dA, CC, NA, AL-P, BS, EC, and JL. Sample and clinical information collection: ER, IO, JPG, MaE, LM, DB, EI, EG-P, MDM-A, AJL, JG, XC, LO, MP, and JP. Statistical analyses and figures: SC-L, EC, and JL. Writing original draft: SC-L and FL. All authors read, critically revised and approved the final version of the manuscript.
Funding
This work was supported by Spanish Ministerio de Economía y Competitividad (MINECO, SAF2016-80535-R) and Ministerio de Ciencia e Innovación (MCIN/AEI/10.13039/501100011033, PID2019-106658RB-I00), co-financed by European Development Regional Fund “A way to achieve Europe” ERDF, and Agència de Gestió d’Ajuts Universitaris i de Recerca from Generalitat de Catalunya (2017/SGR/1582). SC-L, MV-dA, CC, AL-P, and EC are recipients of fellowships from Spanish Ministerio de Educación, Cultura y Deporte (FPU15/02897), Spanish MINECO (BES-2014-069237 and BES-2017-082107), Chilean Agencia Nacional de Investigación y Desarrollo (2018-72190154), and European Community Seventh Framework Program (FP7/2007/2013; 229673), respectively. SC-L and JL are recipients of short-term fellowships from European Federation of Immunological Societies-Immunology Letters (EFIS-IL) and Erasmus+ from the European Union, respectively. The ENEIDA registry of GETECCU is supported by Biogen, Janssen, Takeda and Pfizer. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
We thank Belchin Kostov for statistical analysis support, Marcos Isamat for manuscript editing and critical comments, and Silvia Ariño for technical help with RT-PCR and immunohistochemistry quantification and interpretation. We are indebted to GETECCU and the IDIBAPS Biobank for clinical data and sample procurement.
Conflict of interest
FL is founder and ad-honorem scientific advisor of Sepsia Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.966184/full#supplementary-material
References
1. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474:307–17. doi: 10.1038/nature10209
2. Lecomte O, Bock JB, Birren BW, Vollrath D, Parnes JR. Molecular linkage of the mouse CD5 and CD6 genes. Immunogenetics (1996) 44:385–90. doi: 10.1007/BF02602784
3. Padilla O, Calvo J, Vilà JM, Arman M, Gimferrer I, Places L, et al. Genomic organization of the human CD5 gene. Immunogenetics (2000) 51:993–1001. doi: 10.1007/s002510000235
4. Martínez VG, Moestrup SK, Holmskov U, Mollenhauer J, Lozano F. The conserved scavenger receptor cysteine-rich super family in therapy and diagnosis. Pharmacol Rev (2011) 63:967–1000. doi: 10.1124/pr.111.004523
5. Cho J-H, Sprent J. TCR tuning of T cell subsets. Immunol Rev (2018) 283:129–37. doi: 10.1111/imr.12646
6. Gimferrer I, Calvo M, Mittelbrunn M, Farnós M, Sarrias MR, Enrich C, et al. Relevance of CD6-mediated interactions in T cell activation and proliferation. J Immunol (2004) 173:2262–70. doi: 10.4049/jimmunol.173.4.2262
7. Mori D, Grégoire C, Voisinne G, Celis-Gutierrez J, Aussel R, Girard L, et al. The T cell CD6 receptor operates a multitask signalosome with opposite functions in T cell activation. J Exp Med (2021) 218:e20201011. doi: 10.1084/JEM.20201011
8. Chappell PE, Garner LI, Yan J, Metcalfe C, Hatherley D, Johnson S, et al. Structures of CD6 and its ligand CD166 give insight into their interaction. Structure (2015) 23:1426–36. doi: 10.1016/j.str.2015.05.019
9. Escoda-Ferran C, Carrasco E, Caballero-Baños M, Miró-Julià C, Martínez-Florensa M, Consuegra-Fernández M, et al. Modulation of CD6 function through interaction with galectin-1 and -3. FEBS Lett (2014) 588:2805–13. doi: 10.1016/j.febslet.2014.05.064
10. Enyindah-Asonye G, Li Y, Ruth JH, Spassov DS, Hebron KE, Zijlstra A, et al. CD318 is a ligand for CD6. Proc Natl Acad Sci (2017) 114:E6912–21. doi: 10.1073/pnas.1704008114
11. Van De Velde H, Von Hoegen I, Luo W, Parnes JR, Thielemans K. The b-cell surface protein CD72/Lyb-2 is the ligand for CDS. Nature (1991) 351:662–5. doi: 10.1038/351662a0
12. Biancone L, Bowen MA, Lim A, Aruffo A, Andres G, Stamenkovic I. Identification of a novel inducible cell-surface ligand of CD5 on activated lymphocytes. J Exp Med (1996) 184:811–9. doi: 10.1084/jem.184.3.811
13. Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in b-1 b cells. Sci (80-. ) (1996) 274:1906–9. doi: 10.1126/science.274.5294.1906
14. Calvo J, Places L, Padilla O, Vilà JM, Vives J, Bowen MA, et al. Interaction of recombinant and natural soluble CD5 forms with an alternative cell surface ligand. Eur J Immunol (1999) 29:2119–29. doi: 10.1002/(SICI)1521-4141(199907)29:07<2119::AID-IMMU2119>3.0.CO;2-F
15. Pospisil R, Silverman GJ, Marti GE, Aruffo A, Bowen MA, Mage RG. CD5 is a potential selecting ligand for b-cell surface immunoglobulin: A possible role in maintenance and selective expansion of normal and malignant b cells. Leuk Lymphoma (2000) 36:353–65. doi: 10.3109/10428190009148857
16. Haas KM, Estes DM. The identification and characterization of a ligand for bovine CD5. J Immunol (2001) 166:3158–66. doi: 10.4049/jimmunol.166.5.3158
17. Brown MH, Lacey E. A ligand for CD5 is CD5. J Immunol (2010) 185:6068–74. doi: 10.4049/jimmunol.0903823
18. Zhang C, Xin H, Zhang W, Yazaki PJ, Zhang Z, Le K, et al. CD5 binds to interleukin-6 and induces a feed-forward loop with the transcription factor STAT3 in b cells to promote cancer. Immunity (2016) 44:913–23. doi: 10.1016/j.immuni.2016.04.003
19. Vera J, Fenutria R, Canadas O, Figueras M, Mota R, Sarrias M-R, et al. The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc Natl Acad Sci (2009) 106:1506–11. doi: 10.1073/pnas.0805846106
20. Sarhan MA, Pham TNQ, Chen AY, Michalak TI. Hepatitis c virus infection of human T lymphocytes is mediated by CD5. J Virol (2012) 86:3723–35. doi: 10.1128/JVI.06956-11
21. Mourglia-Ettlin G, Miles S, Velasco-De-Andrés M, Armiger-Borràs N, Cucher M, Dematteis S, et al. The ectodomains of the lymphocyte scavenger receptors CD5 and CD6 interact with tegumental antigens from echinococcus granulosus sensu lato and protect mice against secondary cystic echinococcosis. PloS Negl Trop Dis (2018) 12:e0006891. doi: 10.1371/journal.pntd.0006891
22. Sarrias M-R, Farnós M, Mota R, Sánchez-Barbero F, Ibáñez A, Gimferrer I, et al. CD6 binds to pathogen-associated molecular patterns and protects from LPS-induced septic shock. Proc Natl Acad Sci USA (2007) 104:11724–9. doi: 10.1073/pnas.0702815104
23. Carrasco E, Escoda C, Alvarez-Fenrández C, Sanchez-Palomino S, Carreras E, Gatell JM, et al. A role for scavenger-like lymphocyte receptor CD6 in HIV-1 viral infection. AIDS Res Hum Retroviruses (2014) 30:A49–50. doi: 10.1089/aid.2014.5085.abstract
24. Consuegra-Fernández M, Aranda F, Simões I, Orta M, Sarukhan A, Lozano F. CD5 as a target for immune-based therapies. Crit Rev Immunol (2015) 35:85–115. doi: 10.1615/CritRevImmunol.2015013532
25. Consuegra-Fernández M, Lin F, Fox DA, Lozano F. Clinical and experimental evidence for targeting CD6 in immune-based disorders. Autoimmun Rev (2018) 17:493–503. doi: 10.1016/j.autrev.2017.12.004
26. Velasco-de Andrés M, Casadó-Llombart S, Català C, Leyton-Pereira A, Lozano F, Aranda F. Soluble CD5 and CD6: Lymphocytic class I scavenger receptors as immunotherapeutic agents. Cells (2020) 9:2589. doi: 10.3390/cells9122589
27. Cenit MC, Martínez-Florensa M, Consuegra M, Bonet L, Carnero-Montoro E, Armiger N, et al. Analysis of ancestral and functionally relevant CD5 variants in systemic lupus erythematosus patients. PloS One (2014) 9:e113090. doi: 10.1371/journal.pone.0113090
28. Potrony M, Carreras E, Aranda F, Zimmer L, Puig-Butille J-A, Tell-Martí G, et al. Inherited functional variants of the lymphocyte receptor CD5 influence melanoma survival. Int J Cancer (2016) 139:1297–302. doi: 10.1002/ijc.30184
29. Delgado J, Bielig T, Bonet L, Carnero-Montoro E, Puente XS, Colomer D, et al. Impact of the functional CD5 polymorphism A471V on the response of chronic lymphocytic leukaemia to conventional chemotherapy regimens. Br J Haematol (2017) 177:147–50. doi: 10.1111/bjh.14037
30. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet (2012) 44:1336–40. doi: 10.1038/ng.2462
31. Swaminathan B, Cuapio A, Alloza I, Matesanz F, Alcina A, García-Barcina M, et al. Fine mapping and functional analysis of the multiple sclerosis risk gene CD6. PloS One (2013) 8:e62376. doi: 10.1371/journal.pone.0062376
32. Wagner M, Bilinska M, Pokryszko-Dragan A, Sobczynski M, Cyrul M, Kusnierczyk P, et al. ALCAM and CD6 - multiple sclerosis risk factors. J Neuroimmunol (2014) 276:98–103. doi: 10.1016/j.jneuroim.2014.08.621
33. Zheng M, Zhang L, Yu H, Hu J, Cao Q, Huang G, et al. Genetic polymorphisms of cell adhesion molecules in behçet’s disease in a Chinese han population. Sci Rep (2016) 6:24974. doi: 10.1038/srep24974
34. Consuegra-Fernández M, Julià M, Martínez-Florensa M, Aranda F, Català C, Armiger-Borràs N, et al. Genetic and experimental evidence for the involvement of the CD6 lymphocyte receptor in psoriasis. Cell Mol Immunol (2018) 15:898–906. doi: 10.1038/cmi.2017.119
35. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
36. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet (2016) 48:510–8. doi: 10.1038/ng.3528
37. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol (2014) 104:15.25.1–15.25.14. doi: 10.1002/0471142735.im1525s104
38. Tarakhovsky A, Müller W, Rajewsky K. Lymphocyte populations and immune responses in CD5-deficient mice. Eur J Immunol (1994) 24:1678–84. doi: 10.1002/eji.1830240733
39. Orta-Mascaró M, Consuegra-Fernández M, Carreras E, Roncagalli R, Carreras-Sureda A, Alvarez P, et al. CD6 modulates thymocyte selection and peripheral T cell homeostasis. J Exp Med (2016) 213:1387–97. doi: 10.1084/jem.20151785
40. Zabana Y, Panés J, Nos P, Gomollón F, Esteve M, García-Sánchez V, et al. The ENEIDA registry (Nationwide study on genetic and environmental determinants of inflammatory bowel disease) by GETECCU: Design, monitoring and functions. Gastroenterol Hepatol (2020) 43:551–8. doi: 10.1016/j.gastre.2020.05.006
41. Silverberg MS, Satsangi J, Ahmad T, Arnott IDR, Bernstein CN, Brant SR, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a working party of the 2005 Montreal world congress of gastroenterology. Can J Gastroenterol (2005) 19 Suppl A:5A–36A. doi: 10.1155/2005/269076
42. Lee JC, Biasci D, Roberts R, Gearry RB, Mansfield JC, Ahmad T, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in crohn’s disease. Nat Genet (2017) 49:262–8. doi: 10.1038/ng.3755
43. Dasu T, Qualls JE, Tuna H, Raman C, Cohen DA, Bondada S. CD5 plays an inhibitory role in the suppressive function of murine CD4+ CD25+ treg cells. Immunol Lett (2008) 119:103–13. doi: 10.1016/j.imlet.2008.05.008
44. Kirby C, Baig A, Avlasevich SL, Torous DK, Tian S, Singh P, et al. Dextran sulfate sodium mouse model of inflammatory bowel disease evaluated for systemic genotoxicity via blood micronucleus and pig-a gene mutation assays. Mutagenesis (2020) 35:161–7. doi: 10.1093/mutage/geaa006
45. Zhou P, Du LF, Lv GQ, Yu XM, Gu YL, Li JP, et al. Functional polymorphisms in CD166/ALCAM gene associated with increased risk for breast cancer in a Chinese population. Breast Cancer Res Treat (2011) 128:527–34. doi: 10.1007/s10549-011-1365-x
46. Wagner M, Wiśniewski A, Bilińska M, Pokryszko-Dragan A, Nowak I, Kuśnierczyk P, et al. ALCAM — novel multiple sclerosis locus interfering with HLA-DRB1*1501. J Neuroimmunol (2013) 258:71–6. doi: 10.1016/J.JNEUROIM.2013.02.015
47. Cianci R, Pagliari D, Piccirillo CA, Fritz JH, Gambassi G. The microbiota and immune system crosstalk in health and disease. Mediators Inflamm (2018) 2018:29849485. doi: 10.1155/2018/2912539
48. Gordon S. Pattern recognition receptors: Doubling up for the innate immune response. Cell (2002) 111:927–30. doi: 10.1016/S0092-8674(02)01201-1
49. Zhou ZH, Tzioufas AG, Notkins AL. Properties and function of polyreactive antibodies and polyreactive antigen-binding b cells. J Autoimmun (2007) 29:219–28. doi: 10.1016/j.jaut.2007.07.015
50. Braun M, Müller B, Ter Meer D, Raffegerst S, Simm B, Wilde S, et al. The CD6 scavenger receptor is differentially expressed on a CD56 dim natural killer cell subpopulation and contributes to natural killer-derived cytokine and chemokine secretion. J Innate Immun (2011) 3:420–34. doi: 10.1159/000322720
51. Björklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol (2016) 17:451–60. doi: 10.1038/ni.3368
52. Li H, Burgueño-Bucio E, Xu S, Das S, Olguin-Alor R, Elmets CA, et al. CD5 on dendritic cells regulates CD4+ and CD8+ T cell activation and induction of immune responses. PloS One (2019) 14:e0222301. doi: 10.1371/journal.pone.0222301
53. Levin TG, Powell AE, Davies PS, Silk AD, Dismuke AD, Anderson EC, et al. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology (2010) 139:2072–2082.e5. doi: 10.1053/j.gastro.2010.08.053
54. Ma C, Wu W, Lin R, Ge Y, Zhang C, Sun S, et al. Critical role of CD6highCD4+ T cells in driving Th1/Th17 cell immune responses and mucosal inflammation in IBD. J Crohn’s Colitis (2019) 13:510–24. doi: 10.1093/ecco-jcc/jjy179
55. Peters JE, Lyons PA, Lee JC, Richard AC, Fortune MD, Newcombe PJ, et al. Insight into genotype-phenotype associations through eQTL mapping in multiple cell types in health and immune-mediated disease. PloS Genet (2016) 12:e1005908. doi: 10.1371/journal.pgen.1005908
56. Ordoñez-Rueda D, Lozano F, Sarukhan A, Raman C, Garcia-Zepeda EA, Soldevila G. Increased numbers of thymic and peripheral CD4+ CD25 +Foxp3+ cells in the absence of CD5 signaling. Eur J Immunol (2009) 39:2233–47. doi: 10.1002/eji.200839053
57. Axtell RC, Webb MS, Barnum SR, Raman C. Cutting edge: Critical role for CD5 in experimental autoimmune encephalomyelitis: Inhibition of engagement reverses disease in mice. J Immunol (2004) 173:2928–32. doi: 10.4049/jimmunol.173.5.2928
58. Schuster C, Kiaf B, Hatzihristidis T, Ruckdeschel A, Nieves-Bonilla J, Ishikawa Y, et al. CD5 controls gut immunity by shaping the cytokine profile of intestinal T cells. Front Immunol (2022) 13:906499. doi: 10.3389/fimmu.2022.906499
59. Consuegra-Fernández M, Martínez-Florensa M, Aranda F, de Salort J, Armiger-Borràs N, Lozano T, et al. Relevance of CD6-mediated interactions in the regulation of peripheral T-cell responses and tolerance. Front Immunol (2017) 8:594. doi: 10.3389/fimmu.2017.00594
60. Enyindah-Asonye G, Li Y, Xin W, Singer NG, Gupta N, Fung J, et al. CD6 receptor regulates intestinal Ischemia/Reperfusion-induced injury by modulating natural IgM-producing B1a cell self-renewal. J Biol Chem (2017) 292:661–71. doi: 10.1074/jbc.M116.749804
61. Zhang L, Li Y, Qiu W, Bell BA, Dvorina N, Baldwin WM, et al. Targeting CD6 for the treatment of experimental autoimmune uveitis. J Autoimmun (2018) 90:84–93. doi: 10.1016/J.JAUT.2018.02.004
62. Li Y, Ruth JH, Rasmussen SM, Athukorala KS, Weber DP, Amin MA, et al. Attenuation of murine collagen-induced arthritis by targeting CD6. Arthritis Rheumatol (2020) 72:1505–13. doi: 10.1002/art.41288
63. Hall LJ, Murphy CT, Quinlan A, Hurley G, Shanahan F, Nally K, et al. Natural killer cells protect mice from DSS-induced colitis by regulating neutrophil function via the NKG2A receptor. Mucosal Immunol (2013) 6:1016–26. doi: 10.1038/mi.2012.140
64. Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PloS One (2012) 7:e44328. doi: 10.1371/JOURNAL.PONE.0044328
65. Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, et al. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe (2016) 19:455–69. doi: 10.1016/J.CHOM.2016.03.007
66. Sun X, He S, Lv C, Sun X, Wang J, Zheng W, et al. Analysis of murine and human treg subsets in inflammatory bowel disease. Mol Med Rep (2017) 16:2893–8. doi: 10.3892/MMR.2017.6912/HTML
67. Català C, Velasco-de Andrés M, Leyton-Pereira A, Casadó-Llombart S, Sáez Moya M, Gutiérrez-Cózar R, et al. CD6 deficiency impairs early immune response to bacterial sepsis. iScience (2022) 105078. doi: 10.1016/j.isci.2022.105078
68. Polese L, De Franchis G, Scarpa M, Sturniolo GC, Ruffolo C, Norberto L, et al. B1a lymphocytes in ulcerative colitis. Int J Colorectal Dis (2007) 22:1005–11. doi: 10.1007/S00384-007-0298-7/FIGURES/3
69. Shimomura Y, Mizoguchi E, Sugimoto K, Kibe R, Benno Y, Mizoguchi A, et al. Regulatory role of b-1 b cells in chronic colitis. Int Immunol (2008) 20:729–37. doi: 10.1093/INTIMM/DXN031
70. Carnero-Montoro E, Bonet L, Engelken J, Bielig T, Martínez-Florensa M, Lozano F, et al. Evolutionary and functional evidence for positive selection at the human CD5 immune receptor gene. Mol Biol Evol (2012) 29:811–23. doi: 10.1093/molbev/msr251
71. De Jager PL, Jia X, Wang J, de Bakker PIW, Ottoboni L, Aggarwal NT, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet (2009) 41:776–82. doi: 10.1038/ng.401
72. Kofler DM, Severson CA, Mousissian N, De Jager PL, Hafler DA. The CD6 multiple sclerosis susceptibility allele is associated with alterations in CD4+ T cell proliferation. J Immunol (2011) 187:3286–91. doi: 10.4049/jimmunol.1100626
73. Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural history of adult ulcerative colitis in population-based cohorts: A systematic review. Clin Gastroenterol Hepatol (2018) 16:343–356.e3. doi: 10.1016/j.cgh.2017.06.016
74. Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (N. Y). (2011) 7:235–41.
75. Dharmaraj R, Jaber A, Arora R, Hagglund K, Lyons H. Seasonal variations in onset and exacerbation of inflammatory bowel diseases in children. BMC Res Notes (2015) 8:696. doi: 10.1186/s13104-015-1702-y
76. Teuscher C, Bunn JY, Fillmore PD, Butterfield RJ, Zachary JF, Blankenhorn EP. Gender, age, and season at immunization uniquely influence the genetic control of susceptibility to histopathological lesions and clinical signs of experimental allergic encephalomyelitis: Implications for the genetics of multiple sclerosis. Am J Pathol (2004) 165:1593–602. doi: 10.1016/S0002-9440(10)63416-5
77. Álvarez-Sánchez N, Cruz-Chamorro I, Álvarez-López AI, López-González A, Lacalle Remigio JR, Lardone PJ, et al. Seasonal variations in Macrophages/Microglia underlie changes in the mouse model of multiple sclerosis severity. Mol Neurobiol (2020) 57:4082–9. doi: 10.1007/s12035-020-02017-x
78. Hernández P, Moreno E, Lazaro EA, Rodríguez PC. Therapeutic targeting of CD6 in autoimmune diseases: A review of Cuban clinical studies with the antibodies IOR-T1 and itolizumab. Curr Drug Targets (2016) 17:666–77. doi: 10.2174/1389450117666160201114308
Keywords: Crohn’s disease, inflammatory bowel disease, ulcerative colitis, CD5, CD6
Citation: Casadó-Llombart S, Velasco-de Andrés M, Català C, Leyton-Pereira A, Gutiérrez-Cózar R, Suárez B, Armiger N, Carreras E, Esteller M, Ricart E, Ordás I, Gisbert JP, Chaparro M, Esteve M, Márquez L, Busquets D, Iglesias E, García-Planella E, Martín-Arranz MD, Lohmann J, Ayata CK, Niess JH, Engel P, Panés J, Salas A, Domènech E, Lozano F and ENEIDA Project of GETECCU (2022) Experimental and genetic evidence for the impact of CD5 and CD6 expression and variation in inflammatory bowel disease. Front. Immunol. 13:966184. doi: 10.3389/fimmu.2022.966184
Received: 10 June 2022; Accepted: 31 August 2022;
Published: 21 September 2022.
Edited by:
Peter Hasselblatt, University Hospital Freiburg, GermanyReviewed by:
Lena Sophie Mayer, University of Freiburg Medical Center, GermanyAnna-Maria Globig, Salk Institute for Biological Studies, United States
Wolfgang Reindl, University of Heidelberg, Germany
Copyright © 2022 Casadó-Llombart, Velasco-de Andrés, Català, Leyton-Pereira, Gutiérrez-Cózar, Suárez, Armiger, Carreras, Esteller, Ricart, Ordás, Gisbert, Chaparro, Esteve, Márquez, Busquets, Iglesias, García-Planella, Martín-Arranz, Lohmann, Ayata, Niess, Engel, Panés, Salas, Domènech, Lozano and ENEIDA Project of GETECCU. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Lozano, ZmxvemFub0BjbGluaWMuY2F0