 A. Marijke Keestra-Gounder
A. Marijke Keestra-Gounder Prescilla Emy Nagao
Prescilla Emy Nagao- 1Department of Immunology and Microbiology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Laboratory of Molecular Biology and Physiology of Streptococci, Institute of Biology Roberto Alcantara Gomes, Rio de Janeiro State University (UERJ), Rio de Janeiro, Brazil
The inflammasomes are intracellular multimeric protein complexes consisting of an innate immune sensor, the adapter protein ASC and the inflammatory caspases-1 and/or -11 and are important for the host defense against pathogens. Activaton of the receptor leads to formation of the inflammasomes and subsequent processing and activation of caspase-1 that cleaves the proinflammatory cytokines IL-1β and IL-18. Active caspase-1, and in some instances caspase-11, cleaves gasdermin D that translocates to the cell membrane where it forms pores resulting in the cell death program called pyroptosis. Inflammasomes can detect a range of microbial ligands through direct interaction or indirectly through diverse cellular processes including changes in ion fluxes, production of reactive oxygen species and disruption of various host cell functions. In this review, we will focus on the NLRP3, NLRP6, NLRC4 and AIM2 inflammasomes and how they are activated and regulated during infections with Gram-positive bacteria, including Staphylococcus spp., Streptococcus spp. and Listeria monocytogenes.
1. Introduction
Advances in cell microbiology in relation to invasive bacterial infections, signaling molecules, receptors and adhesins on target cells, and cell death mediated by bacterial virulence factors have improved our knowledge of microbial pathogenesis. Pathogen-associated molecular patterns (PAMPs) such as bacterial lipopolysaccharide, flagellin/pili or lipoteichoic acid from pathogenic microorganisms, such as Staphylococcus spp., Streptococcus spp., Listeria monocytogenes are responsible for activating the innate immune system (1–3). However, responses to infection by indirect detection of pathogenic molecules secreted by microorganisms and the action of proteins from the superfamily of leucine-rich repeats of the nucleotide-binding domain (NLR), in order to interrupt the signaling pathways of the host immunity, demonstrated that effector-triggered immunity promoted disruption of host cell signaling. As innocuous microorganisms do not provide pathogenic molecules to host, immunity triggered by effectors is particular for pathogenic organisms (4, 5).
Inflammasomes are signaling protein complexes that induce immune responses with the activation of inflammatory caspases, and consequent pro-inflammatory cell death called pyroptosis (6). Inflammasome activation plays a crucial role in protecting against bacterial infections organizing an effective innate immune response by controlling bacterial load and modulating the nature and magnitude of the adaptive immune response (7). Dysregulated inflammasome activation can result in an exaggerated innate immune response with tissue damage in the host. The recognition of PAMPs or microbe-associated molecular patterns (MAMPs) by phagocytes promotes the activation of inflammasomes - NLRP1, NLRP3, NLRP6, NLRC4 and AIM2 - with recruitment of adapter ASC for activation of caspase-1, secretion of interleukins pro-inflammatory such as IL-1β and IL-18, and consequent induction of pyroptosis (8). Pyroptosis, a form of cell death in immune cells, can also be activated in non-immune cells, such as hepatocytes, epithelial cells, endothelial cells, and keratinocytes (9, 10). Therefore, studies on inflammasome functions and biological responses remain of interest in improving treatments and/or prevention of inflammatory and infectious diseases. Currently, important virulence mechanisms used to avoid detection of inflammasomes and the functional consequences in host cells to evade pyroptosis induction have been studied in different Gram-positive pathogens. Therefore, this overview will focus on inflammasome activation and regulation by Gram-positive bacteria.
2. Inflammasomes
The innate immune system is the first barrier pathogens encounter and that will protect the host from infections. Pattern recognition receptors (PRRs) are a class of innate immune receptors that detect PAMPs (11, 12). One family of the PRRs are the nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) (13). To date, 22 NLRs have been identified in humans and they are categorized into subfamilies based on the type of N-terminal signaling domain (13). The NLRP receptors contain a pyrin domain (PYD) and NLRC receptors contain a caspase activating recruitment domain (CARD) (14). Both NLRPs and NLRCs can form a multiprotein complex called the inflammasome that consists of the adaptor apoptosis associated speck-like protein containing a CARD (ASC) and caspase-1 (15). ASC contains a PYD and a CARD domain and binds the NLRs via either PYD-PYD or CARD-CARD interaction, respectively. The interaction of the NLR with ASC leads to the recruitment of procaspase-1 that subsequently is cleaved into the active caspase-1. Caspase-1 cleaves the proforms of interleukin (IL)-1β and IL-18 and gasdermin D (16–18). Cleaved gasdermin D inserts itself into the plasma membrane to form pores that mediate the release of IL-1β and IL-18 and drive the cell death program pyroptosis (19, 20). Inflammasome activation requires two steps of stimulation; signal 1 is known as ‘priming’, and signal 2 as ‘activation’ (21). Priming can be achieved by activation of Toll-like receptors (TLRs) to promote the activation of NF-κB leading to the transcriptional expression of key components of the inflammasome, such as pro-Il-1β, pro-IL-18 and the NLR itself. Signal 2 is the stimulus that activates the inflammasome (Figure 1). In addition to PAMPs, Damage/Danger Associated Molecular Patterns (DAMPs) which are endogenous danger signals released from damaged tissues can also trigger inflammasome activation (22). In this review, we will focus on the NLRP3, NLRP6, NLRC4 and AIM2 inflammasomes and their activation by Gram-positive bacterial pathogens.
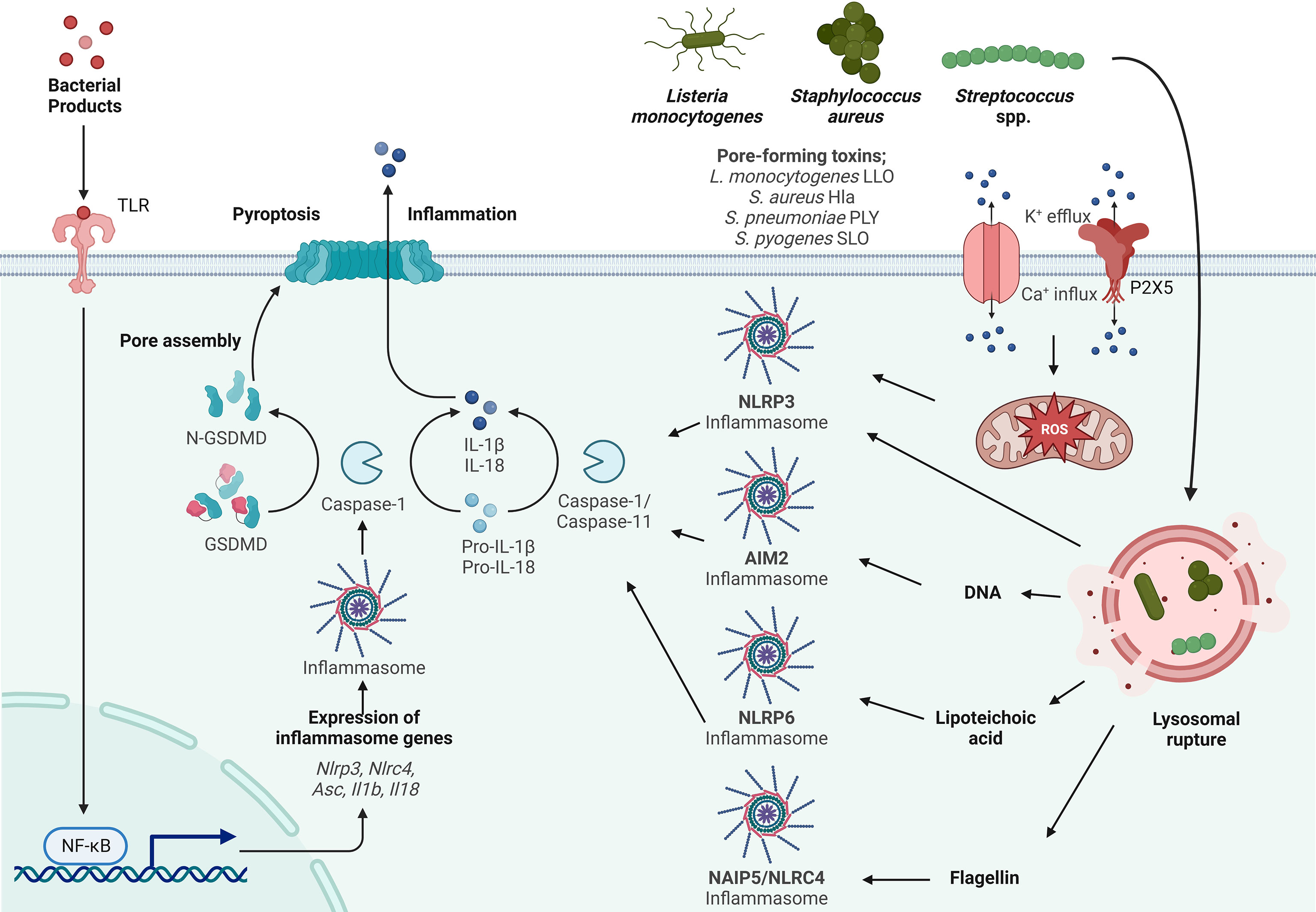
Figure 1 Inflammasome activation by Gram-positive bacteria. Activation of the inflammasome typically starts by priming (signal 1) through TLR activation resulting in the transcriptional upregulation of inflammasome components such as Asc, Nlrp3, Nlrc4, IL-1β and IL-18. Activation (signal 2) is initiated by a variety of PAMPs from Listeria monocytogenes, Streptococcus spp. and Staphylococcus spp., such as lipoteichoic acid (LTA), DNA and flagellin. LTA activates the NLRP6 inflammasome, DNA activates the AIM2 inflammasome and flagellin activates the NLRC4 inflammasome. Listeria monocytogenes, Streptococcus spp. and Staphylococcus spp. express pore forming toxins (PFTs) that are inserted into the plasma membrane to facilitate access to the host cytosol. K+ efflux and Ca+ influx through these pores and the P2X5 receptor (a member of the P2X purinergic receptor family of ligand-gated cation channels) results in activation of the NLRP3 inflammasome. In addition to PAMP activation and ion fluxes, the inflammasome is also activated by damage-associated molecular patterns (DAMPs) such as lysosomal rupture and production of mitochondrial reactive oxygen species (mtROS) that activates the NLRP3 inflammasome. Sensing of PAMPs and DAMPs by the NLR initiates the formation of the inflammasome consisting of ASC and Caspase-1, which in turn cleaves pro-IL-1β and pro-IL-18. Gasdermin D (GSDMD) is also cleaved and inserts into the membrane, forming pores leading to the release of the proinflammatory cytokines IL-1β and IL-18 and the induction of pyroptosis.
2.1. The NLRP3 inflammasome
NLRP3 is expressed in innate immune cells, mainly macrophages, and NLRP3 is one of the most widely studied inflammasome-forming NLRs. The NLRP3 inflammasome is activated by an increasing number of stimulators, including viruses, fungi, bacteria, pore-forming toxins, reactive oxygen species (ROS), extracellular ATP, potassium efflux and calcium influx, mitochondrial dysfunction, nigericin, urate crystals, amyloid-B, and various environmental insults such as asbestos and silica. Direct binding of a ligand to NLRP3, however, remains unclear since all stimuli are structurally and chemically very different (23). Instead, it is believed that NLRP3 can sense a common cellular event that is induced by the wide variety of stimuli, such as potassium efflux or production of ROS, however the underlying mechanism remains to be determined. The NLRP3 inflammasome is important for maintaining a balanced in the gastrointestinal tract since Nlrp3-/- mice displayed a unique intestinal microbiota and are more susceptible to experimental colitis (24–26). Nlrp3-/- mice were also found to have reduced expression of AMPs, which could potentially influence the microbiota composition (25).
NLRP3 is also involved in the non-canonical inflammasome which is activated by the binding of cytosolic lipopolysaccharides (LPS) of Gram-negative bacteria to the murine inflammatory caspase-11 (27, 28). In humans, the orthologs of murine caspase-11 are caspase-4 and -5 (29). Cytosolic LPS induces self-cleavage and oligomerization of caspase-11 that subsequently processes gasdermin D to induce pore formation in the plasma membrane. The pore formation leads to potassium efflux, which in turn activates the NLRP3inflammasome resulting in the processing of pro-IL-1β and pro-IL-18 and secretion of IL-1β and IL-18 (30, 31). Caspase-11 is widely expressed in a variety of cells of both the hematopoietic- and non-hematopoietic lineage, including epithelial cells and macrophages (32). It was suggested that since caspase-11 directly binds to LPS it only contributes to defense against gram-negative bacteria, however, a recent study reported that caspase-11 plays a crucial role in generating immune responses against Listeria monocytogenes and Staphylococcus aureus infection (see paragraph 2.3. The NLRP6 inflammasome) (33).
2.2. The NLRC4 inflammasome
The NLRC4 inflammasome is activated in response to flagellin and the needle and inner rod protein of Type III and Type IV secretion systems (T3SS, T4SS) of Gram-negative bacteria, such as Salmonella Typhimurium, Shigella flexneri, Legionella pneumophila and Pseudomonas aeruginosa. These ligands do not directly bind to NLRC4, but instead, NLRC4 requires the NLR family apoptosis inhibitory proteins (NAIPs) to sense and bind to the bacterial ligands. Murine NAIP1 binds the needle protein, NAIP2 the inner rod protein and NAIP5/6 binds flagellin (34–36). Humans have two isoforms, NAIP and the extended NAIP*. NAIP* binds to flagellin, whereas NAIP binds the needle and rod proteins (37–39). Direct binding of flagellin to NAIP5 changes the conformation of NAIP5 allowing recruitment and binding of NLRC4, which promotes further recruitment and activation of the next incoming NLRC4 protein, resulting in a wheel-shaped structure consisting of a single flagellin-NAIP5 and NLRC4 protomers (40–44). Upon completion of the formation of the wheel-shaped structure, the CARDs of NLRC4 interact and recruit ASC molecules to initiate ASC polymerization into long filaments, known as the ASC specks. The CARD of ASC is exposed on the outside as serves as a docking station for pro-caspase-1 (45). The NLRC4 inflammasome can also function without ASC, since macrophages deficient of ASC display similar levels of pyroptosis in response to NLRC4 ligands (46, 47). The gram-positive bacteria Staphylococcus aureus and Listeria monocytogenes have also been demonstrated to activate the NLRC4 inflammasome. The specific ligand and the exact underlying mechanism how S. aureus (non-flagellated, no T3SS or T4SS) can activate the NLRC4 inflammasome is currently unknown (48). L. monocytogenes flagellin activates the NLRC4 inflammasome (49, 50).
2.3. The NLRP6 inflammasome
NLPR6 is a lesser studied NLR and is predominantly expressed in the intestine. Similar to the NLRP3 and NLRC4 inflammasome, NLRP6 assembles into a multiprotein complex consisting of ASC and caspase-1, promoting cleavage and secretion of inflammatory cytokines and cleavage of gasdermin D to initiate pyroptosis (51). One of the first reports on NLRP6 demonstrated that it is important in maintaining homeostasis of the microbiome (52). Changes in the composition of the microbiome results in an imbalance in the microbial community that is associated with a wide range of human diseases, such as inflammatory bowel disease, cancer, obesity, and diabetes. Dysbiosis can be established in multiple different ways, for example during infections, caused by mutation in host factors or changes to the diet. Additionally, the host immune system has also been demonstrated to be important in maintaining homeostasis of the microbiome. NLRP6 activation was one of the first host factors of the innate immune system suggested to regulate the microbiome composition (52). It was demonstrated that Asc-/-, Caspase-1-/-, Nlrp6-/- and Il18-/- mice, but not Il1b-/- or Il1R-/- mice, developed severe colitis after dextran sodium sulphate (DSS) treatment and that co-housed wild type mice were also more susceptible to DSS treatment, indicating that the microbiota is responsible for the exacerbation of DSS-induced colitis (51, 52). These findings, however, were later challenged as it was demonstrated that Nlrp6-/- and Asc-/- mice and littermate control mice did not reveal any differences in the microbiome composition, nor were they more susceptible to DSS treatment (53). They argued that the differences in the microbiome could be associated with the facilities where the mice are housed. In the report by Levy and co-workers, it was demonstrated that the microbiota-modulated metabolite taurine can activate the NLRP6 inflammasome resulting in pro-IL-18 processing and IL-18 secretion. IL-18 mediates production of antimicrobial peptides (AMPs) and a balanced AMP landscape is required for the maintenance of intestinal homeostasis (51). In contrast, the levels of the metabolites histamine and spermine were increased in Asc-/- mice, and both histamine and spermine suppressed IL-18 production in colon explants. These data indicate that microbiota-modulated metabolites can either positively or negatively regulate the NLRP6 inflammasome and their absence/presence in the gastrointestinal tract can have major effects on gut health and disease (51). NLRP6 expression in intestinal epithelial cells protects against colorectal cancer and colitis via a mechanism dependent on caspase-1 mediated IL-18 production (52–55). NLRP6 has also been demonstrated to be expressed in goblet cells where it regulates mucus production (56). Interestingly, Nlrp6-/- mice are more resistant to infection with Listeria monocytogenes, Salmonella Typhimurium and Escherichia coli, demonstrating NLRP6 can act as a negative regulator in innate immune signaling and host defense (57). Similarly, NLRP6 serves as a negative regulator during infection with Staphylococcus aureus (58). In a later report it was demonstrated that in response to Listeria monocytogenes and Staphylococcus aureus infection, NLRP6 directly binds to lipoteichoic acid (LTA) in macrophages leading to the recruitment of both caspase-1 and caspase-11 resulting in IL-18 secretion (33). In addition to its role in the host defense against bacterial species, NLRP6 can also bind to RNA from enteric viruses, and it can sense microbiota-associated metabolites (51, 59). In response to viral RNA, however, the NLRP6 does not form an inflammasome with ASC and caspase-1, but instead binds the RNA helicase DHX15 and interacts with mitochondrial antiviral signaling protein (MAVS) to induce a type I interferon (T1IFN) response (59). Although NLRP6 can form the classical inflammasome, many aspects of NLRP6 are still poorly understood; NLRP6 plays a major role in maintenance of microbiota homeostasis, can bind distinct ligands, is anti-bacterial and anti-viral, but is also a negative regulator of innate immune signaling. Future studies will have to determine the underlying mechanisms of NLRP6 activation and immune signaling during infections.
2.4. The AIM2 inflammasome
Absent in melanoma 2 (AIM2) is a member of the HIN200 family and is a cytosolic sensor of double-stranded DNA (dsDNA). It is the first non-NLR receptor identified that can form an inflammasome with ASC and caspase-1 (60–63). Upon binding of bacterial, viral and mitochondrial dsDNA to the HIN200 domain of AIM2, multiple sensors cluster together resulting in binding of ASC via PYD-PYD interaction. The CARD domain of ASC subsequently recruits procaspase-1 to the complex allowing for autocleavage of caspase-1, processing of pro-IL-1β, pro-IL18 and gasdermin D, resulting in cytokine secretion and pyroptosis. Gram-positive bacteria, including but not limited to Staphylococcus aureus and Listeria monocytogenes release dsDNA in the cytosol after escaping the phagolysosome that activates the AIM2 inflammasome (64, 65).
3. Gram-positive bacteria trigger inflammasome activation
Host immune programs, including inflammasomes, play a crucial role in limiting the spread of pathogens and maintaining tissue function and integrity. Thus, pro-inflammatory transcription factors orchestrate the magnitude and duration of immune responses. Dysregulation of immune response can lead to uncontrolled inflammation promoting serious disease (66–68). In this way, several pathogenic microorganisms have evolved mechanisms to prevent inflammasome activation.
3.1. Listeria monocytogenes - Advantage of inflammasome activation
L. monocytogenes is a Gram-positive bacterium responsible for listeriosis acquired by contaminated food. Murine or human macrophages infected with L. monocytogenes demonstrated activation of multiple inflammasomes, including AIM2-ASC, NLRP3-caspase-1, NAIP5-NLRC4, NLRP6-caspase-11, caspase-1, caspase-11, and NLRP1B (69, 70). In addition, cytosolic listeriolysin O (LLO) and lipoteichoic acid (LTA) have also been implicated in inflammasome activation (Table 1).
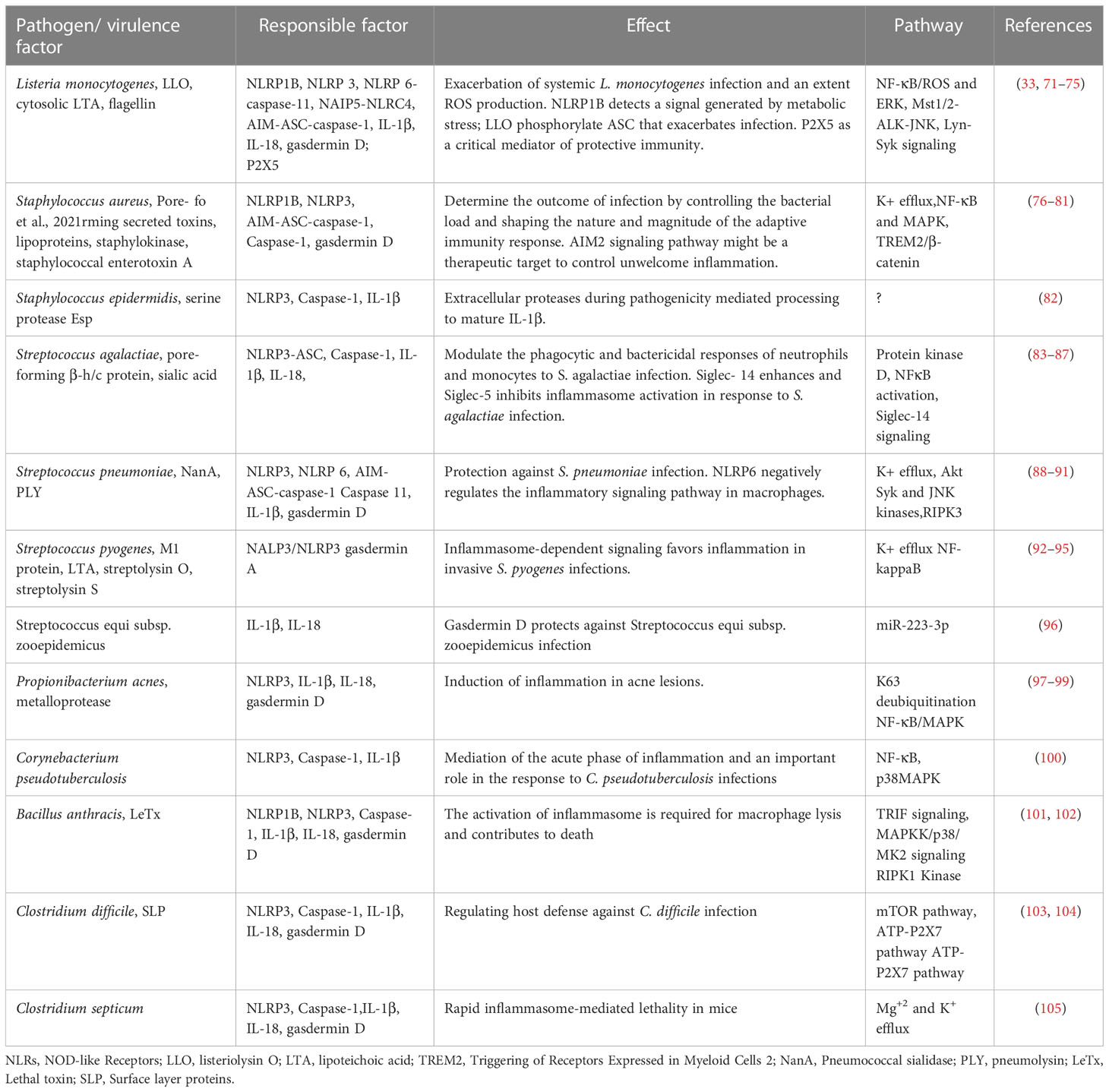
Table 1 Inflammasome activation by Gram positive bacteria.
After endocytosis, L. monocytogenes promotes lysosomal disruption, leading to the release of lysosomal contents, translocation of Listeria to the cytoplasm and inflammasome activation. NLRP3 is activated when L. monocytogenes escapes the vacuole in conditions with limited cytosolic replication and lysosomal damage, and NLRC4 is activated by the bacterial flagellum. However, AIM2 recognition may predominate in conditions with high cytosolic replication. AIM2-ASC-caspase-1 is the inflammasome sensor of L. monocytogenes DNA and its expression requires type I IFN signaling (106). AIM2 connects DNA directly, resulting in atypical inflammasome with the activation of caspase-1, IL-1β and IL-18 and consequent induction of pyroptosis (71, 107).
Gao and co-workers (72) showed that infection by L. monocytogenes induced the activation of the NLRP3 inflammasome through the Mst1/2-ALK pathways; as well as the participation of the interaction between Nek7 and NLRP3 via JNK that promoted the intensification of the host defense against Listeria infection by increasing the maturation and release of the pro-inflammatory cytokine IL-1β. Furthermore, pore-forming toxin and LLO played a critical role in the process. Recently, an increase in transcriptional levels of inflammatory factors was demonstrated, where levels of proteins associated with NLRP3, NF-κB and ERK were increased after Listeria infection and consequent inflammation of the central nervous system (73). Therefore, blocking inflammasome activation could decrease inflammation and improve patient survival and prognosis as a potential therapeutic intervention target for L. monocytogenes infection (108).
Interestingly, mouse macrophages infected with L. monocytogenes activated NLRP6 and caspase-11. NLRP6, activated by Gram-positive bacteria or lipoteichoic acid (LTA) in the host cytosol, has a function to limit inflammation and to regulate systemic infection by pathogens (109, 110). Activation of NLRP6 in macrophages infected by L. monocytogenes or LTA induced caspase-11 processing which promoted IL-1β/IL-18 maturation, caspase-1 activation and regulation of IL-18 secretion, revealing a novel signaling pathway by which the NLRP6 inflammasome was activated by cytosolic LTA, recruiting pro-inflammatory caspases in systemic infection by Gram-positive bacterial pathogens (33).
NLRP1B is activated under conditions that lead to a decrease in cytosolic ATP (76), suggesting that energy reduction may allow NLRP1B to detect intracellular pathogens, including Gram-positive bacteria. The pore-forming toxin and LLO were required for the rupture of the vacuolar membrane, reduction of ATP levels and energy stress through disruption of mitochondrial function by L. monocytogenes. In addition, Listeria invasion into the cytosol may contribute to ATP depletion. Listeria-infected fibroblasts expressed NLRP1B, pro-caspase-1 and IL-1β, suggesting that NLRP1B detected a signal generated by metabolic stress (74). Another study showed that LLO was recruited and oligomerized leading to activation of Lyn and Syk kinases in lipid membrane rafts. Upregulation of ASC phosphorylation was verified in a secondary step, showing that ASC phosphorylation can utilize another signaling pathway in addition to LLO-mediated Lyn-Syk kinases. These data demonstrated that pathogens could activate different inflammasomes in order to control immune responses and benefit the spread of the pathogen in the host (75).
During inflammasome activation, the increased influx of macrophages may be detrimental to the defense against L. monocytogenes in vivo. The Mint3-mediated pathway, which regulates ATP production in macrophages, has contributed to the regulation of severe listeriosis in mice, suggesting a potential therapeutic target (111). In this way, the regulation of gasdermin D by caspase-1 was enhanced in Mint3–/– mice infected with Listeria, accompanied by a strong induction of pyroptosis and bacterial shedding (112). Other molecules have also been described as activating inflammasomes; among them the P2X7 receptor, a potent activator of NLRP3 inflammasome and the release of the pro-inflammatory cytokines IL-1β and IL-18 (113). However, very little is known about P2X5, belong to the P2X family, in the immune system. Recently, P2X5 has been shown to play an important role in protective immune responses associated with inflammasome activity, including in vivo infection by L. monocytogenes (114).
The protein kinase, p38-regulated/activated protein kinase (PRAK), contributes to the regulation of cell proliferation, stress responses and cell migration. Furthermore, PRAK dysfunction impaired the antibacterial activity of neutrophils and the formation of neutrophil extracellular traps (NETs) (115). Assays with PRAK-deficient mice showed high mortality when compared to wild-type mice after infection by L. monocytogenes. In addition, Listeria-induced autophagic activities were reduced in the absence of PRAK. In this way, PRAK potentiated bactericidal activity with increased ROS production, inflammasome activation and autophagy induction, providing a new perspective for innate immunity against listeriosis (116).
3.2. Staphylococcus spp
Staphylococcus aureus is responsible for human infections such as pneumonia, endocarditis and sepsis with high morbidity/mortality and consequent increase in hospital costs (117). S. aureus can manipulate cell death by apoptosis, necrosis or pyroptosis to colonize and establish infection in different hosts (118). The inflammasome plays a crucial role in rising an effective immune response against S. aureus infection, controlling bacterial load and the magnitude of the adaptive immune response (7). S. aureus pore-forming toxins (α-toxin, LukAB and PVL) induced NLRP3-ASC inflammasome activation in human and murine phagocytes in vitro. In addition, methicillin-resistant S. aureus strain activated NLRP3-ASC inflammasomes through K+ efflux, caspase-1 activation, and release of IL-1β and IL-18 cytokines with induction of pyroptosis (77, 78). Peptidoglycan resistant to lysozyme strongly inhibited IL-1β production in response to infection in vitro and in vivo, suggesting that the S. aureus subverts IL-1β secretion by modifying its peptidoglycan from cell wall (119). Thus, new therapeutic approaches targeting peptidoglycan degradation in host immune cells could control bacterial infections.
ASC assembly and AIM2 activation by intracellular S. aureus after suppression of perforin-2 were described for the first time by Pastar and co-workers (79). In this work, S. aureus-infected diabetic foot ulcers triggered the AIM2 inflammasome activation resulting in activation of gasdermin D, and increased levels of IL-1β in tissue samples from chronic ulcers in diabetic patients, contributing to prolonged inflammation. Additionally, phagocytosis and bacterial degradation were related to the activation of NLRP3 inflammasomes and IL-1β secretion in response to both live S. aureus and S. aureus peptidoglycan. Interestingly, IL-1β production was mediated by the NLRP3 activation, and the secretion of IL-6, KC and CCL2 was AIM2-dependent (120). Together, the NLRP3/AIM2 inflammasomes played a critical role in S. aureus infection and could be a therapeutic target to control unwanted inflammation.
The involvement of extracellular vesicles serving as a secretory pathway for the transport of protected bacterial cargo (virulence factors, surface glycoproteins, proteases, etc) to host cells have been reported in several Gram-positive bacteria. Activation of NLRP3 by S. aureus extracellular vesicles have been shown to be dependent on the load of pore-forming toxins and K+ efflux. The critical role of pore-forming toxins was associated with S. aureus extracellular vesicles in triggering inflammasome activation, modulating of innate host immune response during S. aureus infection. In this way, extracellular vesicles serve as an important secretion system that allows the transport of molecules to host cells during infections to modulate cellular functions (121). Similarly, after endocytosis of S. aureus by bovine mammary epithelial cells, the bacteria escaped from the endosomal vesicles, causing inflammation and cell death through the K+ efflux and NLRP3 activation, and consequently pyroptosis of MAC-T cells (80).
Staphylococcal enterotoxin A promotes intestinal barrier dysfunction, triggering an inflammatory response with activation of the NLRP3 inflammasome and the NF-κB/MAPK signaling pathways. Selective inhibitors of NF-κB/MAPK pathways in THP-1 cells attenuated the expression of inflammasome-associated proteins that were upregulated by staphylococcal enterotoxin A. Thus, the development of compounds acting on enterotoxin A may be a promising strategy for the prevention and treatment of diseases caused by the toxin (81). Moreover, the triggering of receptors expressed in myeloid cells 2 (TREM2) is considered a protective factor for the host against bacterial infection; however, their role is unclear. TREM2 or its adapter molecule DAP12 activates β-catenin, the molecule responsible for maintaining cell viability and preventing cell death, in myeloid cells stimulated by M-CSF. Study showed that TREM2 inhibited macrophage pyroptosis induced by S. aureus and Streptococcus pneumoniae, providing a new host defense mechanism against pathogenic microorganisms (122–124). In addition, TREM2 promoted the stabilization and phosphorylation of β-catenin in the nucleus and cytoplasm and down-regulation of NLRP3 inflammasome activation. TREM2/β-catenin also inhibited ASC oligomerization and ASC-NLRP3 association with suppressed pyroptosis in macrophages infected by pyogenic bacteria (80).
Potential role of staphylokinase in the virulence of community associated S. aureus remains unknown. Furthermore, a significant role of staphylokinase in the acute pneumonia model involved the activation of innate immune signals, especially pathways related to the NLRP3 inflammasome. The investigations revealed that staphylokinase contributed to NLRP3 inflammasome activation, increasing K+ efflux, ROS production and NF-κB signaling, exacerbating pulmonary infection. These data improve our understanding of the mechanism of action of staphylokinase in bacterial pathogenesis (80).
Recently, researchers showed that Staphylococcus epidermidis strains induced caspase-1-independent IL-1β release and NLRP3 inflammasome activation in keratinocytes, providing important information during the S. epidermidis-keratinocyte interaction to activate the innate host defense (82).
3.3. Streptococcus spp
3.3.1. Streptococcus agalactiae
Streptococcus agalactiae is an opportunistic β-hemolytic bacterium of the female genitourinary tract that can ascend during pregnancy, causing premature births and/or abortions. The high vaginal colonization of S. agalactiae is the main cause of vertical transmission, resulting in invasive infections such as pneumonia and neonatal meningitis (125). Understanding the pathogenesis of S. agalactiae will allow the development of effective vaccines and therapies against the pathogen (126). The secretion of IL-1β and IL-18 in human macrophages treated with β-hemolysin from S. agalactiae was associated with NLRP3 inflammasome activation during bacterial infection-mediated fetal death (127).
Several bacterial pathogens have evolved mechanisms to presentation sialic acid-binding immunoglobulin-like lectin ligands (Siglecs) on their cell surface to inhibit immune responses by molecular mimicry (83). S. agalactiae manipulates host immune cells through sialic acid mimicry, involving CD33-related inhibitory Siglecs to promote evasion of the host immune system. Siglecs are inhibitory receptors containing immunoreceptor tyrosine-based inhibition motifs in their intracellular signaling domains. Siglec-14 and -5 modulate the phagocytic and bactericidal responses of neutrophils and monocytes to S. agalactiae. The innate spectrum can also be modulated by the opposing mechanisms performed by Siglec-5, which inhibits inflammasome activation, and Siglec-14, which potentiates inflammasome activation in response to S. agalactiae infection, and may represent a therapeutic target against excessive inflammatory responses promoted by S. agalactiae (84). Additionally, vimentin has been described as a ligand of Siglec-14 after its secretion by activated macrophages, which may increase inflammasome activation (128). In addition, β protein induced binding to the Siglec-7 receptor, decreasing pro-IL-1β cleavage and IL-1β secretion, which may represent a mechanism to prevent innate immunity (129).
The induction of proinflammatory responses by protein kinase D (PKD) in S. agalactiae-mediated immune activation has been reported in murine and human macrophages (130). Protein kinase D is involved in cellular activities such as modulating cell signaling and vesicular traffic, transcription factor regulation, cellular detoxification, and inflammasome activation (85, 86). Study using placental macrophages infected by S. agalactiae showed the essential role of PKD during NLRP3 inflammasome formation/activation, inflammatory cytokine release, and NF-κB signaling processes (87). To date, the NLRP3 inflammasome has contributed significantly to host defenses, with β-hemolysin being the main stimulus of S. agalactiae to trigger a highly effective immune response through the NLRP3 inflammasome activation.
3.3.2. Streptococcus pneumoniae
Streptococcus pneumoniae is a microorganism responsible for invasive diseases such as community-acquired pneumonia, sepsis, meningitis and otitis media, leading to high morbidity and mortality worldwide. However, the mechanisms used to activate the immune response remains to be clarified. Studies have shown NLRP3-dependent variation in IL-1β secretion by human cells infected with S. pneumoniae. Macrophages infected with serotypes that expressed low/non-hemolytic pneumolysin (PLY) release smaller amounts of IL-1β compared to macrophages infected with serotypes expressing active PLY (131). Additionally, the NLRP3 inflammasome was differentially activated by serotypes expressing PLY variants that triggered the production of IL-1 β and IL-18 cytokines (132). Hoegen and co-workers (133) showed the participation of PLY as an inducer of NLRP3 inflammasome and IL-1β expression in human THP-1 cells. In human neutrophils, PLY-mediated NLRP3 activation resulted in K+ efflux, caspase-1 activation, and pro-IL-1β cleavage (134). Furthermore, studies with murine neutrophils indicated that IL-1β activation was NLRP3 inflammasome-dependent, but AIM2 and NLRC4 inflammasomes-independent (135). In addition to NLRP3, PLY can also activate AIM2 inflammasomes (136).
Inactivation of NLRP3 or AIM2 inflammasomes increased mortality and bacterial colonization in the host, demonstrating the protective role of inflammasomes against S. pneumoniae infection (88). Moreover, IL-1β secretion was partially dependent on NLRP6 inflammasome activation during macrophage infection. Nlrp6−/− mice showed reduced mortality and bacterial colonization, as well as lower neutrophil and macrophage recruitment compared to wild-type mice, showing that NLRP6 plays a detrimental role in host defense against S. pneumoniae (89). Another study also demonstrated that Syk and JNK kinases were phosphorylated in a PLY-dependent manner during inflammasome activation by S. pneumoniae (90).
Receptor-interacting protein kinase 3 (RIPK3), a serine/threonine kinase, regulates both pro-inflammatory signaling for microbial elimination and excessive inflammation (137). Activation of the pulmonary NLRP3 inflammasome by S. pneumoniae was RIPK3-dependent. RIPK3 deficiency induced a decrease in NLRP3 expression, reduced IL-1β production, reduced bacterial clearance, with consequent severe lung damage and high mortality. RIPK3 also induced mitochondrial Ca2+ uptake, mitochondrial ROS production, and NLRP3 inflammasome activation via the AKT pathway against S. pneumoniae infection (138). In the lungs of infected mice, macrophage ATF3 activates the NLRP3 inflammasome to induce IL-1β secretion, which subsequently stimulates IL-17 secretion by γδ T cells. Interestingly, ATF3 (activating transcription factor-3) an important factor in the oxidative stress pathway of the endoplasmic reticulum promoted the activation and assembly of the NLRP3 inflammasome by ROS regulation during early S. pneumoniae infection. In addition, intracellular Ca2+ and ATP homeostasis, production of IL-1β and IL-23, and IL-17 secretion were also modulated by ATF3, demonstrating an important role in the survival of the host and in the elimination of S. pneumoniae (139).
Another important virulence factor, pneumococcal NanA, promoted extensive surface desialylation of infected cells and exaggerated inflammatory responses after infection. NanA induced the activation of multiple inflammatory pathways and cell death through the unregulated transfer of signals between TLR2 and siglec-5. NanA-mediated desialylation increased ASC oligomerization, caspase-1 activation and proteolytic cleavage of gasdermin D in infected THP-1 cells, suggesting that NanA inactivation may be a target to ameliorate inflammation and cytotoxicity caused by S. pneumonia (91).
Interestingly, biological aging favors a decrease in inflammasome activation during the inflammatory process caused by S. pneumoniae, showing that the elderly are more susceptible to S. pneumoniae. Elderly murine models demonstrated that misfolded proteins became increased with age, contributing to decreased assembly and activation of the NLRP3 inflammasome during S. pneumoniae infection (140).
3.3.3. Streptococcus pyogenes
Streptococcus pyogenes is a pathogen responsible for injury and tissue destruction, bacteremia, multiple organ failure and death worldwide. Infections caused by S. pyogenes are highly contagious and can occur through hand contact with nasal secretions, airborne droplets, or surfaces contaminated with bacteria. Ineffective treatment of S. pyogenes infections can result in post-infection sequelae such as acute rheumatic fever and glomerulonephritis (141).
NLRP3 inflammasome signaling during S. pyogenes infection occurs through multiple mechanisms, depending on the cell line used, S. pyogenes M serotypes and in vitro or in vivo models. S. pyogenes LTA is a potent priming agent for the NLRP3 inflammasome in mouse bone marrow-derived macrophages, but not in differentiated human THP-1 cells (92). Isogenic mutant S. pyogenes revealed that streptolysin O (SLO) and streptolysin S were the main factors for inflammasome signaling in macrophages (142). SLO forms pores in host cell membranes, sufficient to release intracellular contents such as K+ and IL-1β (143). SLO also stimulates the activation of NLRP3 and caspase-1 in THP-1 macrophage-like cells (93). SLO-induced IL-1β-mediated inflammasome secretion required TLR and NF-κB-mediated cell signaling (94). In addition, a study demonstrated that partially active SLO, and therefore less cytotoxic, induced IL-1β secretion and delayed pyroptotic cell death. In this way, SLO-induced K+ efflux was able to activate the NLRP3 inflammasome (144). Moreover, macrophages were able to release IL-1β through P2X7 in response to S. pyogenes infection (145). Additionally, activation of caspase-1 by S. pyogenes required NF-κB, but was independent of P2X7R and TLR signaling (94).
Cysteine protease (SpeB) is able to cleave IL-1β contributing to alternative inflammasome-independent pathways. IL-1β signaling may protect the host but also lead to severe S. pyogenes infection (92). Cleavage of gasdermin A by the virulence factor SpeB was recently reported. This mechanism promoted a rapid burst of inflammation that limited invasive infection. Interestingly, SpeB directly cleaves gasdermin A, a family member expressed in keratinocytes, to release a pore-forming lytic subunit that can intercalate into membranes and lyse eukaryotic cells (95). These results demonstrated the important function of gasdermin A in defense of keratinocytes against the invasion of S. pyogenes, acting as a sensor of proteases of pathogenic bacteria.
3.3.4. Streptococcus equi subsp. zooepidemicus
Streptococcus equi subsp. zooepidemicus (Sez) is an important pathogen responsible for infections such as meningitis, endocarditis and pneumonia in humans, acute and chronic endometritis in horses, and zoonosis (146). Studies have shown that microRNAs (miRNAs) play a crucial regulatory role in the human body. Regulation of the NLRP3 Inflammasome by Sez strains involved the downregulation of miR-223-3p in murine macrophages. Computational analyzes showed that miR-223-3p targets NLRP3 mRNA, whose overexpression of miR-223-3p suppressed NLRP3 inflammasome activation during Sez infection. Furthermore, miRNA-223-3p through regulation of the NLRP3/caspase-1 pathway caused inhibition of IL-1β and IL-18 secretion in response to the pathogen. Additionally, inhibition of miR-223-3p caused NLRP3 inflammasome hyperactivation. Consequently, miR-223-3p contributes to suppression of NLRP3 inflammasome activation and may be a therapeutic target for the treatment of Sez (96). Moreover, Gasdermin D-deficient mice were susceptible to intraperitoneal bacterial infection, with serious damage to the spleen and reduced secretion of cytokines IL-1β and IL-18 during in vivo infection (147). Furthermore, pore formation and pyroptosis in macrophages was gasdermin D-dependent, suggesting a defensive role of gasdermin D against Sez, providing a potential therapeutic target.
3.4. Cutibacterium acnes
Cutibacterium acnes (formerly called Propionibacterium acnes) plays a significant role in the development of acne, triggering immunologic defense that are vital for the pathogenesis and clinical appearance of acne vulgaris (148). In addition, C. acnes can also induce intervertebral disc degeneration (149). C. acnes produces proteases that participate in the inflammatory response of acne causing degradation of the extracellular matrix and proteolytic detachment of follicular keratinocytes. Activation of the NLRP3 inflammasome was dependent on metalloproteinase expression by C. acnes and ROS generation in sebocytes (97). C. acnes also induced ROS production, NLRP3 inflammasome activation and IL-8 release in HaCaT keratinocytes. Furthermore, the authors described the anti-inflammatory potential of the medicinal herb Paris polyphylla in reducing the inflammatory responses, proliferation and migration of keratinocytes caused by C. acnes, suggesting a new therapeutic for the treatment of acne (150). In addition, celastrol isolated from the Celastraceae family also exhibited anti-inflammatory activities by suppressing NLRP3 inflammasome activation promoted by C. acnes through inhibition of K63 deubiquitination of NLRP3, and providing evidence for its application in disease therapy (151).
Previously published data demonstrated increased activation of the NLRP3 inflammasome, caspase-1, caspase-5, IL-1β, IL-18 and gasdermin D after infection of C. acnes-infected nucleus pulposus tissue. In this study, it was also shown that the addition of the inflammasome inhibitor NLRP3 MCC950 and the thioredoxin binding protein (TXNIP) reduced the secretions of IL-1β and IL-18, implying an inflammatory response activated by C. acnes via the TXNIP-NLRP3 pathway. In this way, MCC950 can alleviate the inflammatory lesion and pyroptosis of C. acnes-infected cells, slowing the progression of disc degeneration, providing a new direction for the treatment of intervertebral disc degeneration (98).
The NLRP3 inflammasome plays a key role in acne lesions, raising interest in the suppression of this mechanism in the treatment of acne. Activation of NF-κB induced an increase in the expression of inflammatory factors during NLRP3 inflammasome activation. Thus, the suppression of the NF-κB pathway and the activation of the ERK1/2 MAPK signal transduction pathways caused inhibition of caspase-1 and NLRP3 expression, which could be considered therapeutic targets in the treatment of acne (99).
3.5. Corynebacterium pseudotuberculosis
Corynebacterium pseudotuberculosis, a facultative intracellular microorganism, belongs to the Corynebacterium-Mycobacterium-Nocardia-Rhodococcus group responsible for caseous lymphadenitis in goats and sheep. C. pseudotuberculosis is reported worldwide as the cause of significant economic losses affecting meat, wool and milk production, being difficult to detect in subclinically infected animals, to control and eradicate (152). C. pseudotuberculosis infections cause pyogranulomas and abscesses in organs and lymph nodes, with an increase in inflammatory cytokines. IL-1β is an important cytokine during the inflammatory phase in response to pathogens. Zhou and colleagues first described the activation of the NLRP3 inflammasome in IL-1β secretion in macrophages after infection by C. pseudotuberculosis. Activation of NLRP3 was independent of AIM2, but dependent on NF-κB, p38MAPK and partially dependent on TLR4 pathways. These results contribute to the knowledge of the mechanism of IL-1β secretion and of the host’s pro-inflammatory immune response in macrophages infected with C. pseudotuberculosis (100).
3.6. Bacillus anthracis
Bacillus anthracis, spore-forming Gram-positive bacteria, efficiently kills infected hosts through the systemic action of secreted edema toxin (EdTx) and lethal toxin (LeTx) (153). LeTx targets polymorphonuclear and mononuclear cells during the early stages of infection, paralyzing the host’s innate immune defenses and allowing bacteria to spread and cause systemic infection (154). Furthermore, direct or direct activation of caspase-1 by the NLRP1B inflammasome in response to LeTx was related to macrophage killing (101). Recent publication has shown the involvement of TNFR1, TNFR2 receptors, and the MAPKK/p38/MK2 signaling pathway in LeTx-intoxicated macrophages during NLRP3 inflammasome activation dependent on RIPK1 activity (155).
In contrast to activation of the NLRP3 sensor, little is known about the requirements for NLRP1B activation. Cell death was induced by LeTx toxin and mediated by the NLRP1B inflammasome and proteasome activity. However, inhibition of components such as caspase-1, K+ channel, cathepsin B and heat shock can block this activity. In this way, proteasome inhibition could delay the time of death caused by the LeTx toxin (102). When IL-1β and IL-18 are present, the LeTx toxin activates NLRP1B in toxin-responsive rodent macrophages, while resistant macrophages only suffer the consequences of cleavage of LeTx’s mitogen-activated protein kinase and others subtracts. Importantly, LeTx was not able to regulate or activate IL-1β release or induce the formation of murine neutrophil NETs. LeTx also did not induce neutrophil pyroptosis, despite cleaving cytosolic mitogen-activated protein kinase substrates, suggesting a complex mechanism of interaction between different cell types and mediators in vivo that differs from in vitro or ex vivo models (156). Therefore, it is important to know the inflammasome activation in a multicellular context of inflammatory response to a bacterial toxin.
3.7. Clostridioides difficile
Clostridioides difficile is the leading cause of hospital-acquired infection, resulting in pseudomembranous colitis, toxic megacolon, and even death. The diagnosis of high inflammatory level is a predictor of severe disease with high morbidity and mortality. Toxins A and B produced by C. difficile can stimulate a potent pro-inflammatory response with activation of IL-1β, IL-6, IL-8 and TNFα with inflammasome activation. C. difficile toxins A/B also synergize with MyD88 in the production of IL-23. Furthermore, inhibition of Rho protein GTPase activity by toxins A/B induced the cleavage of pro-IL-1β and IL-8, resulting in the recruitment of innate immune cells (157).
The pyrine inflammasome does not directly recognize pathogens or host-derived molecules, but responds to disturbances in cytoplasmic homeostasis caused by infections leading to inactivation of RhoA GTPase. Activation of these signaling pathways increases IL-1β production and promotes IL-1 receptor expression with uncontrolled activation of the pyrin inflammasome in neutrophils (158). Furthermore, pyrin inflammasome activation can be affected by RIPK3, which modulates the rapamycin (mTOR) pathway, regulating the expression of the gene encoding pyrin and pyrin inflammasome activation (103).
Interactions between bacterial virulence factors and cell receptors are fundamental as possible targets for treating bacterial infectious diseases. Surface layer proteins of C. difficile (SLPs) bind to the lipid rafts, induced caspase-1, and IL-1β production in dose-dependent manners (159). The production of IL-1β induced by C. difficile in macrophages was dependent on caspase-1, MyD88 and TLR2, considered critical in the production of pro-IL-1β. Additionally, the toxigenic C. difficile in the cytosol was necessary for inflammasome activation through the ATP-P2X7 pathway with consequent loss of membrane integrity, release of intracellular content and caspase-1-dependent cell death. In addition, SLPs were released from pyroptotic cells. Consequently, MyD88 and TLR2 were essential components in pro-IL-1β cleavage and inflammasome activation via ATP-P2X7, responsible for inflammation-mediated bacterial clearance during C. difficile infection (104). Together, these results contribute to the identification and understanding of the essential factors of infection caused by C. difficile, allowing new therapeutic strategies to prevent the disease.
3.8. Clostridium spp
Recently, evidence has demonstrated the involvement of mixed-lineage protein kinase (MLKL) in mediating the immune response against severe gas gangrene and enterocolitis caused by Clostridium perfringens. MLKL plays an important role in innate immunity during the process of necroptosis. Regarding C. perfringens, MLKL promoted bacterial eradication, increased host survival and resolution of inflammation, demonstrating that MLKL-NLRP3 inflammasome confers protection to invading pathogens (160).
Another species, Clostridium septicum, a pathogen that causes sepsis and gas gangrene, was able to activate the inflammasome complex in mice and humans. C. septicum secretes a α-toxin responsible for binding to glycosylphosphatidylinositol proteins anchored in the plasma membrane of host cells, forming pores that allow the efflux of Mg2+ and K+ ions. The efflux of these cytosolic ions triggers NLRP3 inflammasome activation, caspase-1 and gasdermin D activation, IL-1β and IL-18 secretion. The innate detection of the α-toxin is essential for the recognition of C. septicum infection, whose therapeutic blockade may be able to prevent sepsis and death caused by microbial toxins (105).
4. Cross-regulation of inflammasome and autophagy activation by Gram-positive bacteria
The induction of autophagy acts as the first line of defense during infection by intracellular pathogens, including Gram-positive bacteria (161). Autophagy is a mechanism that acts on cellular homeostasis, allowing the generation of amino acids, recycling and elimination of non-functional organelles (162, 163). Thus, autophagy is upregulated in response to the need for amino acids and damaged organelles, including mitochondria (mitophagy).
The connection between autophagy and inflammasome activation was first reported in 2008. Autophagy dysfunction can lead to hyperinflammation and excessive activation of the NLRP3 inflammasome, acting as an inflammasome regulator. Elimination by autophagy of NLRP3 inflammasome components reduced inflammasome activation and inflammatory response. On the other hand, the inflammasome can also regulate the autophagic process necessary for the balance of the inflammatory response, preventing excessive and harmful inflammation (164). Autophagy has a protective role in some NLRP3 inflammasome-associated inflammatory diseases, including sepsis caused by many Gram-positive bacteria (165). CD46 is a glycoprotein expressed by human cells that binds to pathogens such as S. pyogenes. Mechanisms employed for intracellular S. pyogenes autophagy include the binding of the CD46 receptor with bacterial M Protein. After CD46 activation, S. pyogenes is targeted to autophagosomes. CD46 can control early infection of the pathogen by autophagy, as shown that the CD46-Cyt-1/GOPC pathway participated in directing S. pyogenes to autophagic degradation. This pathway induces the formation of autophagosomes that efficiently sequester and degrade pathogens, optimizing the innate cellular defense against pathogenic microorganisms (166).
Pathogens in the cytosol are ubiquitinated and captured by LC3-positive double membranes via autophagy receptors (167). Interestingly, SLO S. pyogenes promoted escape from phagosomes, ubiquitination and recognition by the p62, NDP52 and NBR1 autophagic adapters. Furthermore, studies have shown that the secreted protein, NAD-glycohydrolase (Nga), is translocated to the host cytosol via SLO, promoting inhibition of LC3 formation and suppression of phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3)-dependent autophagosome formation during early infection, facilitating intracellular proliferation of S. pyogenes (168). The p62, NDP52 and NBR1 adapters are critical for the recognition of ubiquitinated S. pyogenes and recruitment of LC3 before autophagic degradation. Moreover, expression of SpeB S. pyogenes protected the bacterium from autophagy through p62, NDP52 and NBR1 degradation (169). Interestingly, L. monocytogenes suppressed LC3-associated phagocytosis through modulation of mitochondrial Ca2+ signaling to survive in the intracellular environment. Macrophage invasion by L. monocytogenes induced mitochondrial Ca2+ uptake through the mitochondrial Ca2+ uniporter, promoting increased production of acetyl-coenzyme A by pyruvate dehydrogenase, decreasing the formation of LC3-associated phagocytosis. Consequently, modulation of mitochondrial Ca2+ signaling increased Listeria survival in the cytosol, demonstrating the important role of mitochondrial metabolism in Listeria-host cell interactions (170). Furthermore, LLO forms pores in the phagosomal membrane immediately after uptake by bacteria, and L. monocytogenes is able to escape autophagic degradation (171). The virulence factor, ActA, from L. monocytogenes is implicated in intracellular motility and evasion of autophagy. Despite contradictory results with ΔactA mutants, results with HeLa cells showed a time-dependent increase in colocalization with LC3, which may represent an important escape mechanism from ActA-dependent autophagy (172). Moreover, the synergistic effects of ActA and L. monocytogenes phospholipases (PlcA and B) for escape from autophagy was reported. PlcA and plcB mutants were rapidly targeted to autophagosomes compared to wild-type bacteria (173).
Previous studies demonstrated that blocking autophagy potentiated inflammasome activity, while stimulation of autophagy promoted inflammasome ubiquitination with the recruitment of the p62 autophagic adapter, which aided in autophagosome targeting. In this way, autophagy accompanied inflammasome activation to moderate inflammation (174). Studies have also shown that impaired mitophagy increases activation of NLRP3, while induction of mitophagy reduces activation of NLRP3 (175). Supporting this notion, a study by Shi and co-workers (174) showed colocalization of the NLRP3 inflammasome with autophagosomes and that inhibition of the autophagic pathway by post-translational modifications of NLRP3 caused increased activation of the NLRP3 inflammasome, suggesting a negative feedback to prevent the excessive inflammation. Furthermore, induction of autophagy increased IL-1β secretion in response to NLRP3 inflammasome activation in macrophages (176). The regulation of IL-1β release by the autophagic machinery is complex and requires further investigation, and may be influenced by different inflammasome activators, autophagy inducer/inhibitor and cell lineage types.
Interestingly, experiments performing silencing of NLRP3 or ASC in microglia suppressed caspase-1 activation and increased autophagy. In addition, inhibition of autophagy by the NLRP3 inflammasome occurred through cleavage of the TRIF signaling molecule by caspase-1 (177). Inflammasome activation promoted a rapid blockade of caspase-1 dependent mitophagy, resulting in the accumulation of mitochondrial DNA and dysfunctional mitochondria. The increase in damaged mitochondria produced the increase in mitochondrial ROS allowing greater activation of the inflammasome, amplification of the inflammatory response and pyroptosis in macrophages (178).
NLRP3 autophagy-inflammasome regulation is very complex and the influence is bidirectional (Figure 2). However, the relationship between autophagy and the NLRP3 inflammasome should be further explored. For example, an in-depth analysis to understand the physiological, pathological and inflammatory processes of autophagy relevant to the future development of drugs to treat inflammatory diseases.
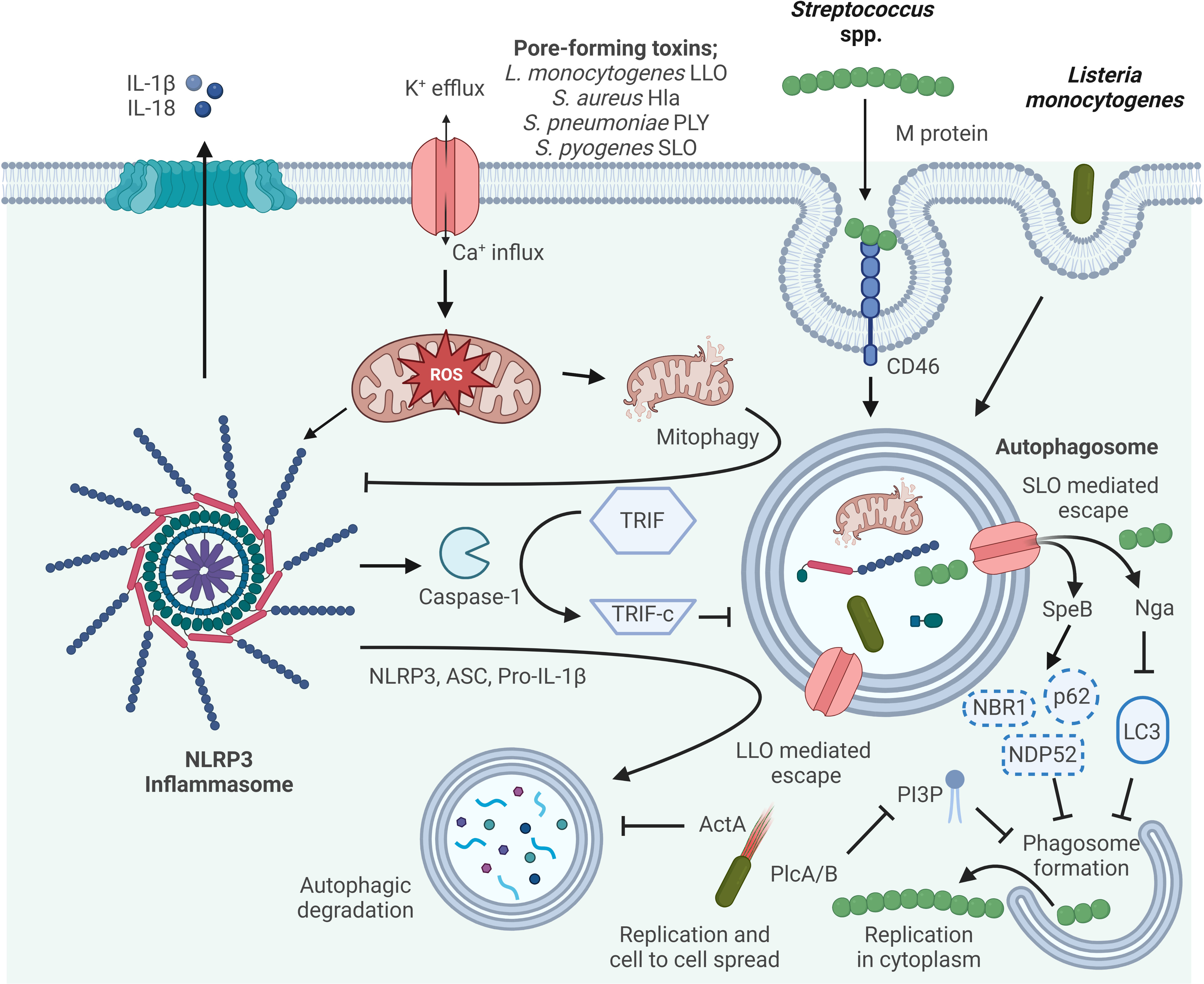
Figure 2 Mechanisms of autophagy/NLRP3 inflammasome by Gram-positive bacteria interaction. Autophagy can inhibit NLRP3 inflammasome activation by reducing ASC, increasing NLRP3 phosphorylation, and eliminating ROS. Activated Caspase-1 directly cleaves TRIF, thereby diminishing TRIF-mediated autophagy. The M protein of S. pyogenes binds the CD46 receptor to facilitate internalization, and subsequently S. pyogenes escapes from the phagosome via streptolysin O (SLO)-dependent pore formation. In the host cytosol, streptococcal pyrogenic exotoxin B (SpeB) degrades the autophagy adapter proteins p62, NDP52 and NBR1, and NAD-glycohydrolase (Nga) disrupts LC3 formation thereby inhibiting phagosome formation and escape from autophagy allowing for S. pyogenes cytosolic replication. LLO, listeriolysin O, pore-forming acts to damage and disrupt the vacuole membrane. Surface protein A (ActA) recruits the host Arp2/3 complex and Ena/VASP to the bacterial surface, which disguises the bacteria from autophagic recognition. Phosphatidylinositol-specific phospholipase C (Plc) A and B expressed by L. monocytogenes interfere with the production of phosphatidylinositol 3-phosphate (PI3P), a phospholipid required for phagosome formation. Escape from autophagy allows for L. monocytogenes replicating within the host cytosol and cell-to-cell spread.
5. Concluding remarks and future perspectives
Recent work has expanded our understanding of inflammasome activation and how Gram-positive bacteria can activate, evade, and manipulate the inflammasome and the downstream mediated immune responses. The success or failure of a pathogen is dependent on its ability to adapt to a new host environment. The host immune system on the other hand has evolved to effectively defend against the pathogens attack. This attack-defense strategy has been the driving force for both the host and the pathogen to develop new defenses to cope with new attack mechanisms. For example, Gram-positive bacteria insert pore-forming toxins (PFTs) into the cell membrane to facilitate the infection cycle. Some PFTs are also involved in disruption of the phagosome, thereby gaining cytosolic access, which is critical for their virulence strategy and lifestyle. However, changes in cytosolic potassium concentrations as a consequence of pore formation, or release of PAMPs from a ruptured phagosome, leads to activation of inflammasomes. This continuous host-pathogen interaction has put selective pressure on the pathogen to make changes to their virulence factors to try to inhibit immune responses or to avoid immune recognition. Similarly, inflammasomes have evolved to respond not only directly by binding to microbial ligands, but also indirectly by sensing common infection strategies employed by a variety of pathogens. Understanding this constant battle and the underlying mechanisms by which Gram-positive bacteria modulate host innate immune responses is essential for future therapeutic development.
Author contributions
AMK-G and PN wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors and their work were supported by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) [E-26/010.001628/2019], Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Sub-Reitoria de Pós-Graduação e Pesquisa da Universidade do Estado do Rio de Janeiro (SR-2/UERJ) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001 and CAPES-PrInt 88881.311598/2018-01. Work in A.M.K-G.’s laboratory is supported by grants from the NIAID of the NIH under Award Numbers AI154043, AI164154 and AI173121.
Acknowledgments
Figures 1 and 2 were created with BioRender.com. Adapted from “Suppression of Inflammasome by IRF4 and IRF8 is Critical for T Cell Priming”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bülow S, Zeller L, Werner M, Toelge M, Holzinger J, Entzian C, et al. Bactericidal/Permeability-increasing protein is an enhancer of bacterial lipoprotein recognition. Front Immunol (2018) 9:2768. doi: 10.3389/fimmu.2018.02768
2. Johansson J, Freitag NE. Regulation of listeria monocytogenes virulence. Microbiol Spectr (2019) 7(4). doi: 10.1128/microbiolspec.GPP3-0064-2019
3. Moriyama K, Nishida O. Targeting cytokines, pathogen-associated molecular patterns, and damage-associated molecular patterns in sepsis via blood purification. Int J Mol Sci (2021) 22(16):8882. doi: 10.3390/ijms22168882
4. Jones JD, Dangl JL. The plant immune system. Nature (2006) 444(7117):323–9. doi: 10.1038/nature05286
5. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
6. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362(6417):956–60. doi: 10.1126/science.aar7607
7. Evavold CL, Kagan JC. How inflammasomes inform adaptive immunity. J Mol Biol (2018) 430(2):217–37. doi: 10.1016/j.jmb.2017.09.019
8. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
9. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe (2014) 16(2):249–56. doi: 10.1016/j.chom.2014.07.002
10. Liu J, Du S, Kong Q, Zhang X, Jiang S, Cao X, et al. HSPA12A attenuates lipopolysaccharide-induced liver injury through inhibiting caspase-11-mediated hepatocyte pyroptosis via PGC-1α-dependent acyloxyacyl hydrolase expression. Cell Death Differ (2020) 27(9):2651–67. doi: 10.1038/s41418-020-0536-x
11. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
12. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449(7164):819–26. doi: 10.1038/nature06246
13. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
14. Bryant C, Fitzgerald KA. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol (2009) 19(9):455–64. doi: 10.1016/j.tcb.2009.06.002
15. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
16. Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J, et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. J Immunol (1991) 147(9):2964–9. doi: 10.4049/jimmunol.147.9.2964
17. Fantuzzi G, Puren AJ, Harding MW, Livingston DJ, Dinarello CA. Interleukin-18 regulation of interferon gamma production and cell proliferation as shown in interleukin-1beta-converting enzyme (caspase-1)-deficient mice. Blood (1998) 91(6):2118–25. doi: 10.1182/blood.V91.6.2118
18. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
19. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
20. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
21. Patel MN, Carroll RG, Galván-Peña S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med (2017) 23(2):165–80. doi: 10.1016/j.molmed.2016.12.007
22. Bryant C, Fitzgerald KA. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol (2009) 19(9):455–64. doi: 10.1016/j.tcb.2009.06.002
23. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
24. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi: 10.1016/j.immuni.2010.03.003
25. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflammation Bowel Dis (2011) 17(6):1359–72. doi: 10.1002/ibd.21478
26. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut (2010) 59(9):1192–9. doi: 10.1136/gut.2009.197822
27. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
28. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
29. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
30. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
31. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D'Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45(10):2918–26. doi: 10.1002/eji.201545655
32. Viganò E, Mortellaro A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol (2013) 43(9):2240–5. doi: 10.1002/eji.201343800
33. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram-positive pathogen infection. Cell (2018) 175(6):1651–64.e14. doi: 10.1016/j.cell.2018.09.047
34. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi: 10.1038/nature10394
35. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
36. Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J Immunol (2013) 191(8):3986–9. doi: 10.4049/jimmunol.1301549
37. Wen J, Xuan B, Liu Y, Wang L, He L, Meng X, et al. Updating the NLRC4 inflammasome: From bacterial infections to autoimmunity and cancer. Front Immunol (2021) 12:702527. doi: 10.3389/fimmu.2021.702527
38. Romagnoli KM, Handler SM, Hochheiser H. Home care: More than just a visiting nurse. BMJ Qual Saf (2013) 22(12):972–4. doi: 10.1136/bmjqs-2013-002339
39. Kortmann J, Brubaker SW, Monack DM. Cutting edge: Inflammasome activation in primary human macrophages is dependent on flagellin. J Immunol (2015) 195(3):815–9. doi: 10.4049/jimmunol.1403100
40. Tenthorey JL, Haloupek N, López-Blanco JR, Grob P, Adamson E, Hartenian E, et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science (2017) 358(6365):888–93. doi: 10.1126/science.aao1140
41. Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk TH, Huizinga EG. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved n- and c-terminal regions of flagellin. J Biol Chem (2012) 287(46):38460–72. doi: 10.1074/jbc.M112.393512
42. Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science (2015) 350(6259):399–404. doi: 10.1126/science.aac5489
43. Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science (2015) 350(6259):404–9. doi: 10.1126/science.aac5789
44. Diebolder CA, Halff EF, Koster AJ, Huizinga EG, Koning RI. Cryoelectron tomography of the NAIP5/NLRC4 inflammasome: Implications for NLR activation. Structure (2015) 23(12):2349–57. doi: 10.1016/j.str.2015.10.001
45. Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S, et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci USA (2015) 112(43):13237–42. doi: 10.1073/pnas.1507579112
46. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
47. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8(6):471–83. doi: 10.1016/j.chom.2010.11.007
48. Paudel S, Ghimire L, Jin L, Baral P, Cai S, Jeyaseelan S. NLRC4 suppresses IL-17A-mediated neutrophil-dependent host defense through upregulation of IL-18 and induction of necroptosis during gram-positive pneumonia. Mucosal Immunol (2019) 12(1):247–57. doi: 10.1038/s41385-018-0088-2
49. Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by listeria monocytogenes. J Clin Immunol (2010) 30(5):693–702. doi: 10.1007/s10875-010-9425-2
50. Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, et al. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci USA (2011) 108(30):12419–24. doi: 10.1073/pnas.1019041108
51. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163(6):1428–43. doi: 10.1016/j.cell.2015.10.048
52. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi: 10.1016/j.cell.2011.04.022
53. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, Martens L, et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity (2017) 47(2):339–48.e4. doi: 10.1016/j.immuni.2017.07.011
54. Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186(12):7187–94. doi: 10.4049/jimmunol.1100412
55. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA (2011) 108(23):9601–6. doi: 10.1073/pnas.1100981108
56. Birchenough GM, Nyström EE, Johansson ME, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science (2016) 352(6293):1535–42. doi: 10.1126/science.aaf7419
57. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488(7411):389–93. doi: 10.1038/nature11250
58. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog (2018) 14(9):e1007308. doi: 10.1371/journal.ppat.1007308
59. Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science (2015) 350(6262):826–30. doi: 10.1126/science.aab3145
60. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10(3):266–72. doi: 10.1038/ni.1702
61. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
62. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
63. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323(5917):1057–60. doi: 10.1126/science.1169841
64. Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol Cell Host Microbe (2010) 7(5):412–9. doi: 10.1016/j.chom.2010.04.004
65. Feng S, Yang Y, Liu Z, Chen W, Du C, Hu G., et al Intracellular bacteriolysis contributes to pathogenicity of staphylococcus aureus by exacerbating AIM2-mediated inflammation and necroptosis Virulence (2022) 13(1):1684–96. doi: 10.1080/21505594.2022.2127209
66. Ratsimandresy RA, Indramohan M, Dorfleutner A, Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol (2017) 14(1):127–42. doi: 10.1038/cmi.2016.35
67. Lugrin J, Martinon F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol Rev (2018) 281(1):99–114. doi: 10.1111/imr.12618
68. Ta A, Vanaja SK. Inflammasome activation and evasion by bacterial pathogens. Curr Opin Immunol (2021) 68:125–33. doi: 10.1016/j.coi.2020.11.006
69. Man SM, Karki R, Kanneganti TD. DNA-Sensing inflammasomes: regulation of bacterial host defense and the gut microbiota. Pathog Dis (2016) 74(4):ftw028. doi: 10.1093/femspd/ftw028
70. Sanchez-Garrido J, Slater SL, Clements A, Shenoy AR, Frankel G. Vying for the control of inflammasomes: The cytosolic frontier of enteric bacterial pathogen-host interactions. Cell Microbiol (2020) 22(4):e13184. doi: 10.1111/cmi.13184
71. Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA, et al. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol (2010) 185(2):818–21. doi: 10.4049/jimmunol.1000724
72. Gao A, Tang H, Zhang Q, Liu R, Wang L, Liu Y, et al. Mst1/2-ALK promotes NLRP3 inflammasome activation and cell apoptosis during listeria monocytogenes infection. J Microbiol (2021) 59(7):681–92. doi: 10.1007/s12275-021-0638-2
73. Yuan L, Zhu Y, Huang S, Lin L, Jiang X, Chen S. NF-κB/ROS and ERK pathways regulate NLRP3 inflammasome activation in listeria monocytogenes infected BV2 microglia cells. J Microbiol (2021) 59(8):771–81. doi: 10.1007/s12275-021-0692-9
74. Neiman-Zenevich J, Stuart S, Abdel-Nour M, Girardin SE, Mogridge J. Listeria monocytogenes and shigella flexneri activate the NLRP1B inflammasome. Infect Immun (2017) 85(11):e00338–17. doi: 10.1128/IAI.00338-17
75. Tanishita Y, Sekiya H, Inohara N, Tsuchiya K, Mitsuyama M, Núñez G, et al. Listeria toxin promotes phosphorylation of the inflammasome adaptor ASC through Lyn and syk to exacerbate pathogen expansion. Cell Rep (2022) 38(8):110414. doi: 10.1016/j.celrep.2022.110414
76. Chavarría-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog (2013) 9(6):e1003452. doi: 10.1371/journal.ppat.1003452
77. Melehani JH, James DB, DuMont AL, Torres VJ, Duncan JA. Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog (2015) 11:e1004970. doi: 10.1371/journal.ppat.1004970
78. Ezekwe EA Jr., Weng C, Duncan JA. ADAM10 cell surface expression but not activity is critical for staphylococcus aureus α-hemolysin-mediated activation of the NLRP3 inflammasome in human monocytes. Toxins (2016) 8:95. doi: 10.3390/toxins8040095
79. Pastar I, Sawaya AP, Marjanovic J, Burgess JL, Strbo N, Rivas KE, et al. Intracellular staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest (2021) 131(24):e133727. doi: 10.1172/JCI133727
80. Wang X, Liu M, Geng N, Du Y, Li Z, Gao X, et al. Staphylococcus aureus mediates pyroptosis in bovine mammary epithelial cell via activation of NLRP3 inflammasome. Vet Res (2022) 53(1):10. doi: 10.1186/s13567-022-01027-y
81. Liu C, Chi K, Yang M, Guo N. Staphylococcal enterotoxin a induces intestinal barrier dysfunction and activates NLRP3 inflammasome via NF-κB/MAPK signaling pathways in mice. Toxins (Basel) (2022) 14(1):29. doi: 10.3390/toxins14010029
82. Rademacher F, Bartels J, Gläser R, Rodewald M, Schubert S, Drücke D, et al. Staphylococcus epidermidis-derived protease esp mediates proteolytic activation of Pro−IL-1β in human keratinocytes. J Invest Dermatol (2022) 142(10):2756–2765.e8. doi: 10.1016/j.jid.2022.04.010
83. Carlin AF, Lewis AL, Varki A, Nizet V. Group b streptococcal capsular sialic acids interact with siglecs (immunoglobulin-like lectins) on human leukocytes. J Bacteriol (2007) 189(4):1231–7. doi: 10.1128/JB.01155-06
84. Tsai CM, Riestra AM, Ali SR, Fong JJ, Liu JZ, Hughes G, et al. Siglec-14 enhances NLRP3-inflammasome activation in macrophages. J Innate Immun (2020) 12(4):333–43. doi: 10.1159/000504323
85. Rozengurt E. Protein kinase d signaling: multiple biological functions in health and disease. Physiol (Bethesda) (2011) 26(1):23–33. doi: 10.1152/physiol.00037.2010
86. Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, et al. Protein kinase d at the golgi controls NLRP3 inflammasome activation. J Exp Med (2017) 214(9):2671–93. doi: 10.1084/jem.20162040
87. Sutton JA, Rogers LM, Dixon BREA, Kirk L, Doster R, Algood HM, et al. Protein kinase d mediates inflammatory responses of human placental macrophages to group b streptococcus. Am J Reprod Immunol (2019) 81(3):e13075. doi: 10.1111/aji.13075
88. Feng S, Chen T, Lei G, Hou F, Jiang J, Huang Q, et al. Absent in melanoma 2 inflammasome is required for host defence against streptococcus pneumoniae infection. Innate Immun (2019) 25(7):412–9. doi: 10.1177/1753425919860252
89. Xu D, Wu X, Peng L, Chen T, Huang Q, Wang Y, et al. The critical role of NLRP6 inflammasome in streptococcus pneumoniae infection In vitro and In vivo. Int J Mol Sci (2021) 22(8):3876. doi: 10.3390/ijms22083876
90. Feng S, Huang Q, Ye C, Wu R, Lei G, Jiang J, et al. Syk and JNK signaling pathways are involved in inflammasome activation in macrophages infected with streptococcus pneumoniae. Biochem Biophys Res Commun (2018) 507(1-4):217–22. doi: 10.1016/j.bbrc.2018.11.011
91. Tseng YW, Chang CC, Chang YC. Novel virulence role of pneumococcal NanA in host inflammation and cell death through the activation of inflammasome and the caspase pathway. Front Cell Infect Microbiol (2021) 11:613195. doi: 10.3389/fcimb.2021.613195
92. Richter J, Brouwer S, Schroder K, Walker MJ. Inflammasome activation and IL-1β signalling in group a streptococcus disease. Cell Microbiol (2021) 23(9):e13373. doi: 10.1111/cmi.13373
93. Valderrama JA, Riestra AM, Gao NJ, LaRock CN, Gupta N, Ali SR, et al. Group a streptococcal m protein activates the NLRP3 inflammasome. Nat Microbiol (2017) 2(10):1425–34. doi: 10.1038/s41564-017-0005-6
94. Harder J, Franchi L, Muñoz-Planillo R, Park JH, Reimer T, Núñez G. Activation of the Nlrp3 inflammasome by streptococcus pyogenes requires streptolysin O and NF-kappa b activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol (2009) 183(9):5823–9. doi: 10.4049/jimmunol.0900444
95. LaRock DL, Johnson AF, Wilde S, Sands JS, Monteiro MP, LaRock CN. Group a streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature (2022) 605(7910):527–31. doi: 10.1038/s41586-022-04717-x
96. Li G, Zong X, Cheng Y, Xu J, Deng J, Huang Y, et al. miR-223-3p contributes to suppressing NLRP3 inflammasome activation in streptococcus equi ssp. zooepidemicus infection. Vet Microbiol (2022) 269:109430. doi: 10.1016/j.vetmic.2022.109430
97. Li ZJ, Choi DK, Sohn KC, Seo MS, Lee HE, Lee Y, et al. Propionibacterium acnes activates the NLRP3 inflammasome in human sebocytes. J Invest Dermatol (2014) 134(11):2747–56. doi: 10.1038/jid.2014.221
98. He D, Zhou M, Bai Z, Wen Y, Shen J, Hu Z. Propionibacterium acnes induces intervertebral disc degeneration by promoting nucleus pulposus cell pyroptosis via NLRP3-dependent pathway. Biochem Biophys Res Commun (2020) 526(3):772–9. doi: 10.1016/j.bbrc.2020.03.161
99. Fang F, Xie Z, Quan J, Wei X, Wang L, Yang L. Baicalin suppresses propionibacterium acnes-induced skin inflammation by downregulating the NF-κB/MAPK signaling pathway and inhibiting activation of NLRP3 inflammasome. Braz J Med Biol Res (2020) 53(12):e9949. doi: 10.1590/1414-431X20209949
100. Zhou Z, Li H, Tian S, Yi W, Zhou Y, Yang H, et al. Critical roles of NLRP3 inflammasome in IL-1β secretion induced by corynebacterium pseudotuberculosis in vitro. Mol Immunol (2019) 116:11–7. doi: 10.1016/j.molimm.2019.09.016
101. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38(2):240–4. doi: 10.1038/ng1724
102. Newman ZL, Crown D, Leppla SH, Moayeri M. Anthrax lethal toxin activates the inflammasome in sensitive rat macrophages. Biochem Biophys Res Commun (2010) 398(4):785–9. doi: 10.1016/j.bbrc.2010.07.039
103. Sharma D, Malik A, Balakrishnan A, Malireddi RKS, Kanneganti TD. RIPK3 promotes mefv expression and pyrin inflammasome activation via modulation of mTOR signaling. J Immunol (2020) 205(10):2778–85. doi: 10.4049/jimmunol.2000244
104. Liu YH, Chang YC, Chen LK, Su PA, Ko WC, Tsai YS, et al. The ATP-P2X7 signaling axis is an essential sentinel for intracellular clostridium difficile pathogen-induced inflammasome activation. Front Cell Infect Microbiol (2018) 8:84. doi: 10.3389/fcimb.2018.00084
105. Jing W, Pilato JL, Kay C, Feng S, Tuipulotu DE, Mathur A, et al. Clostridium septicum α-toxin activates the NLRP3 inflammasome by engaging GPI-anchored proteins. Sci Immunol (2022) 7(71):eabm1803. doi: 10.1126/sciimmunol.abm1803
106. Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe (2010) 7(5):412–9. doi: 10.1016/j.chom.2010.04.004
107. Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, et al. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol (2010) 40(6):1545–51. doi: 10.1002/eji.201040425
108. Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci (2017) 11:63. doi: 10.3389/fncel.2017.00063
109. Bottai D, Gröschel MI, Brosch R. Type VII secretion systems in gram-positive bacteria. Curr Top Microbiol Immunol (2017) 404:235–65. doi: 10.1007/82-2015-5015
110. Seregin SS, Golovchenko N, Schaf B, Chen J, Eaton KA, Chen GY. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol (2017) 10(2):434–45. doi: 10.1038/mi.2016.55
111. Sakamoto T, Seiki M. Mint3 enhances the activity of hypoxia-inducible factor-1 (HIF-1) in macrophages by suppressing the activity of factor inhibiting HIF-1. J Biol Chem (2009) 284(44):30350–9. doi: 10.1074/jbc.M109.019216
112. Uematsu T, Tsuchiya K, Kobayashi N, Seiki M, Inoue JI, Kaneko S, et al. Mint3 depletion-mediated glycolytic and oxidative alterations promote pyroptosis and prevent the spread of listeria monocytogenes infection in macrophages. Cell Death Dis (2021) 12(4):404. doi: 10.1038/s41419-021-03691-y
113. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol (2006) 176(7):3877–83. doi: 10.4049/jimmunol.176.7.3877
114. Jeong YH, Walsh MC, Yu J, Shen H, Wherry EJ, Choi Y. Mice lacking the purinergic receptor P2X5 exhibit defective inflammasome activation and early susceptibility to listeria monocytogenes. J Immunol (2020) 205(3):760–6. doi: 10.4049/jimmunol.1901423
115. Wang Y, Wang Y, Wu J, Liu C, Zhou Y, Mi L, et al. PRAK is required for the formation of neutrophil extracellular traps. Front Immunol (2019) 10:1252. doi: 10.3389/fimmu.2019.01252
116. Mi L, Wang Y, Xu H, Wang Y, Wu J, Dai H, et al. PRAK promotes the pathogen clearance by macrophage through regulating autophagy and inflammasome activation. Front Immunol (2021) 12:618561. doi: 10.3389/fimmu.2021.618561
117. Missiakas D, Winstel V. Selective host cell death by staphylococcus aureus: A strategy for bacterial persistence. Front Immunol (2021) 11:621733. doi: 10.3389/fimmu.2020.621733
118. Alphonse MP, Rubens JH, Ortines RV, Orlando NA, Patel AM, Dikeman D, et al. Pan-caspase inhibition as a potential host-directed immunotherapy against MRSA and other bacterial skin infections. Sci Transl Med (2021) 13(601):eabe9887. doi: 10.1126/scitranslmed.abe9887
119. Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe (2010) 7(1):38–49. doi: 10.1016/j.chom.2009.12.008
120. Feng S, Yang Y, Liu Z, Chen W, Du C, Hu G., et al Intracellular bacteriolysis contributes to pathogenicity of staphylococcus aureus by exacerbating AIM2-mediated inflammation and necroptosis. Virulence (2022) 13(1):1684–96. doi: 10.1080/21505594.2022.2127209
121. Wang X, Eagen WJ, Lee JC. Orchestration of human macrophage NLRP3 inflammasome activation by staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci USA (2020) 117(6):3174–84. doi: 10.1073/pnas.1915829117
122. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. aureus evades macrophage killing through NLRP3-dependent effects on mitochondrial trafficking. Cell Rep (2018) 22:2431–41. doi: 10.1016/j.celrep.2018.02.027
123. Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol (2009) 10:734–43. doi: 10.1038/ni.1744
124. Guo Y, Mishra A, Weng T, Chintagari NR, Wang Y, Zhao C, et al. Wnt3a mitigates acute lung injury by reducing P2X7 receptor-mediated alveolar epithelial type I cell death. Cell Death Dis (2014) 5:e1286. doi: 10.1038/cddis.2014.254
125. Raabe VN, Shane AL. Group b Streptococcus (Streptococcus agalactiae). Microbiol Spectr (2019) 7(2). doi: 10.1128/microbiolspec.GPP3-0007-2018
126. Kurian NK, Modi D. Mechanisms of group b streptococcus-mediated preterm birth: lessons learnt from animal models. Reprod Fertil (2022) 3(3):R109–20. doi: 10.1530/RAF-21-0105
127. Boldenow E, Gendrin C, Ngo L, Bierle C, Vornhagen J, Coleman M, et al. Group b streptococcus circumvents neutrophils and neutrophil extracellular traps during amniotic cavity invasion and preterm labor. Sci Immunol (2016) 1(4):eaah4576. doi: 10.1126/sciimmunol.aah4576
128. dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun (2015) 6:6574. doi: 10.1038/ncomms7574
129. Fong JJ, Tsai CM, Saha S, Nizet V, Varki A, Bui JD. Siglec-7 engagement by GBS β-protein suppresses pyroptotic cell death of natural killer cells. Proc Natl Acad Sci USA (2018) 115(41):10410–5. doi: 10.1073/pnas.1804108115
130. Upadhyay K, Park JE, Yoon TW, Halder P, Kim YI, Metcalfe V, et al. Group b streptococci induce proinflammatory responses via a protein kinase D1-dependent pathway. J Immunol (2017) 198(11):4448–57. doi: 10.4049/jimmunol.1601089
131. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog (2010) 6(11):e1001191. doi: 10.1371/journal.ppat.1001191
132. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol (2011) 187(1):434–40. doi: 10.4049/jimmunol.1003143
133. Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, et al. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin b release. J Immunol (2011) 187(10):5440–51. doi: 10.4049/jimmunol.1100790
134. Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, et al. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on k+ efflux. J Immunol (2015) 194(4):1763–75. doi: 10.4049/jimmunol.1401624
135. Zhang T, Du H, Feng S, Wu R, Chen T, Jiang J, et al. NLRP3/ASC/Caspase-1 axis and serine protease activity are involved in neutrophil IL-1β processing during streptococcus pneumoniae infection. Biochem Biophys Res Commun (2019) 513(3):675–80. doi: 10.1016/j.bbrc.2019.04.004
136. Surabhi S, Cuypers F, Hammerschmidt S, Siemens N. The role of NLRP3 inflammasome in pneumococcal infections. Front Immunol (2020) 11:614801. doi: 10.3389/fimmu.2020.614801
137. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
138. Huang HR, Cho SJ, Harris RM, Yang J, Bermejo S, Sharma L, et al. RIPK3 activates MLKL-mediated necroptosis and inflammasome signaling during streptococcus infection. Am J Respir Cell Mol Biol (2021) 64(5):579–91. doi: 10.1165/rcmb.2020-0312OC
139. Lee S, Kim GL, Kim NY, Kim SJ, Ghosh P, Rhee DK. ATF3 stimulates IL-17A by regulating intracellular Ca2+/ROS-dependent IL-1β activation during streptococcus pneumoniae infection. Front Immunol (2018) 9:1954. doi: 10.3389/fimmu.2018.01954
140. Cho SJ, Rooney K, Choi AMK, Stout-Delgado HW. NLRP3 inflammasome activation in aged macrophages is diminished during streptococcus pneumoniae infection. Am J Physiol Lung Cell Mol Physiol (2018) 314(3):L372–87. doi: 10.1152/ajplung.00393.2017
141. Kanwal S, Vaitla P. Streptococcus pyogenes. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2022).
142. Hancz D, Westerlund E, Valfridsson C, Aemero GM, Bastiat-Sempe B, Orning P, et al. Streptolysin O induces the ubiquitination and degradation of pro-IL-1β. J Innate Immun (2019) 11(6):457–68. doi: 10.1159/000496403
143. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin d pore structure reveals preferential release of mature interleukin-1. Nature (2021) 593(7860):607–11. doi: 10.1038/s41586-021-03478-3
144. Keyel PA, Roth R, Yokoyama WM, Heuser JE, Salter RD. Reduction of streptolysin O (SLO) pore-forming activity enhances inflammasome activation. Toxins (Basel) (2013) 5(6):1105–18. doi: 10.3390/toxins5061105
145. Westerlund E, Valfridsson C, Yi DX, Persson JJ. The secreted virulence factor NADase of group a streptococcus inhibits P2X7 receptor-mediated release of IL-1β. Front Immunol (2019) 10:1385. doi: 10.3389/fimmu.2019.01385
146. Skive B, Rohde M, Molinari G, Braunstein TH, Bojesen AM. Streptococcus equi subsp. zooepidemicus invades and survives in epithelial cells. Front Cell Infect Microbiol (2017) 7:465. doi: 10.3389/fcimb.2017.00465
147. Xu G, Guo Z, Liu Y, Yang Y, Lin Y, Li C, et al. Gasdermin d protects against streptococcus equi subsp. zooepidemicus infection through macrophage pyroptosis. Front Immunol (2022). doi: 10.3389/fimmu.2022.1005925
148. Qin M, Pirouz A, Kim MH, Krutzik SR, Garbán HJ, Kim J. Propionibacterium acnes induces IL-1β secretion via the NLRP3 inflammasome in human monocytes. J Invest Dermatol (2014) 134(2):381–8. doi: 10.1038/jid.2013.309
149. Tang G, Han X, Lin Z, Qian H, Chen B, Zhou C, et al. Propionibacterium acnes accelerates intervertebral disc degeneration by inducing pyroptosis of nucleus pulposus cells via the ROS-NLRP3 pathway. Oxid Med Cell Longev (2021) 2021:4657014. doi: 10.1155/2021/4657014
150. Yang S, Jiang Y, Yu X, Zhu L, Wang L, Mao J, et al. Polyphyllin I inhibits propionibacterium acnes-induced IL-8 secretion in HaCaT cells by downregulating the CD36/NOX1/ROS/NLRP3/IL-1β pathway. Evid Based Complement Alternat Med (2021) 2021:1821220. doi: 10.1155/2021/1821220
151. Yan CY, Ouyang SH, Wang X, Wu YP, Sun WY, Duan WJ, et al. Celastrol ameliorates propionibacterium acnes/LPS-induced liver damage and MSU-induced gouty arthritis via inhibiting K63 deubiquitination of NLRP3. Phytomedicine (2021) 80:153398. doi: 10.1016/j.phymed.2020.153398
152. Haas DJ, Dorneles EM, Spier SJ, Carroll SP, Edman J, Azevedo VA, et al. Molecular epidemiology of corynebacterium pseudotuberculosis isolated from horses in California. Infect Genet Evol (2017) 49:186–94. doi: 10.1016/j.meegid.2016.12.011
153. Liu S, Zhang Y, Moayeri M, Liu J, Crown D, Fattah RJ, et al. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature (2013) 501(7465):63–8. doi: 10.1038/nature12510
154. Liu S, Miller-Randolph S, Crown D, Moayeri M, Sastalla I, Okugawa S, et al. Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of bacillus anthracis infections in mice. Cell Host Microbe (2010) 8(5):455–62. doi: 10.1016/j.chom.2010.10.004
155. Van Hauwermeiren F, Van Opdenbosch N, Van Gorp H, de Vasconcelos N, van Loo G, Vandenabeele P, et al. Bacillus anthracis induces NLRP3 inflammasome activation and caspase-8-mediated apoptosis of macrophages to promote lethal anthrax. Proc Natl Acad Sci USA (2022) 119(2):e2116415119. doi: 10.1073/pnas.2116415119
156. Greaney AJ, Portley MK, O'Mard D, Crown D, Maier NK, Mendenhall MA, et al. Frontline science: Anthrax lethal toxin-induced, NLRP1-mediated IL-1β release is a neutrophil and PAD4-dependent event. J Leukoc Biol (2020) 108(3):773–86. doi: 10.1002/JLB.4HI0320-028R
157. Donlan A, Petri WA Jr. The inflammasome and type-2 immunity in clostridium difficile infection. Clin Colon Rectal Surg (2020) 33(2):67–72. doi: 10.1055/s-0040-1701231
158. Cowardin CA, Kuehne SA, Buonomo EL, Marie CS, Minton NP, Petri WA Jr. Inflammasome activation contributes to interleukin-23 production in response to clostridium difficile. mBio (2015) 6(1):e02386–14. doi: 10.1128/mBio.02386-14
159. Chen Y, Huang K, Chen LK, Wu HY, Hsu CY, Tsai YS, et al. Membrane cholesterol is crucial for clostridium difficile surface layer protein binding and triggering inflammasome activation. Front Immunol (2020) 11:1675. doi: 10.3389/fimmu.2020.01675
160. Liu Y, Xing LH, Li FX, Wang N, Ma YZ, Li JW, et al. Mixed lineage kinase-like protein protects against Clostridium perfringens infection by enhancing NLRP3 inflammasome-extracellular traps axis. iScience (2022) 25(10):105121. doi: 10.1016/j.isci.2022.105121
161. Siqueira MDS, Ribeiro RM, Travassos LH. Autophagy and its interaction with intracellular bacterial pathogens. Front Immunol (2018) 9:935. doi: 10.3389/fimmu.2018.00935
162. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med (2017) 6(1):24. doi: 10.1186/s40169-017-0154-5
163. Krakauer T. Inflammasomes, autophagy, and cell death: The trinity of innate host defense against intracellular bacteria. Mediators Inflammation (2019) 2019:2471215. doi: 10.1155/2019/2471215
164. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. doi: 10.1038/nature07383
165. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
166. Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe (2009) 6(4):354–66. doi: 10.1016/j.chom.2009.09.006
167. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol (2014) 12(2):101–14. doi: 10.1038/nrmicro3160
168. Toh H, Nozawa T, Minowa-Nozawa A, Hikichi M, Nakajima S, Aikawa C, et al. Group a Streptococcus modulates RAB1- and PIK3C3 complex-dependent autophagy. Autophagy (2020) 16(2):334–46. doi: 10.1080/15548627.2019.1628539
169. Barnett TC, Liebl D, Seymour LM, Gillen CM, Lim JY, Larock CN, et al. The globally disseminated M1T1 clone of group a streptococcus evades autophagy for intracellular replication. Cell Host Microbe (2013) 14(6):675–82. doi: 10.1016/j.chom.2013.11.003
170. Li T, Kong L, Li X, Wu S, Attri KS, Li Y, et al. Listeria monocytogenes upregulates mitochondrial calcium signalling to inhibit LC3-associated phagocytosis as a survival strategy. Nat Microbiol (2021) 6(3):366–79. doi: 10.1038/s41564-020-00843-2
171. Shaughnessy LM, Hoppe AD, Christensen KA, Swanson JA. Membrane perforations inhibit lysosome fusion by altering pH and calcium in listeria monocytogenes vacuoles. Cell Microbiol (2006) 8(5):781–92. doi: 10.1111/j.1462-5822.2005.00665.x
172. Mostowy S, Sancho-Shimizu V, Hamon M, Simeone R, Brosch R, Johansen T, et al. p62 and NDP52 proteins target intracytosolic shigella and listeria to different autophagy pathways. J Biol Chem (2011) 286:26987–95. doi: 10.1074/jbc.M111.223610
173. Tattoli I, Sorbara MT, Philpott DJ, Girardin SE. Stalling autophagy: a new function for listeria phospholipases. Microb Cell (2014) 1:48–50. doi: 10.15698/mic2014.01.124
174. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13(3):255–63. doi: 10.1038/ni.2215
175. Wu J, Li X, Zhu G, Zhang Y, He M, Zhang J. The role of resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp Cell Res (2016) 341(1):42–53. doi: 10.1016/j.yexcr.2016.01.014
176. Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife (2015) 4:e11205. doi: 10.7554/eLife.11205
177. Lai M, Yao H, Shah SZA, Wu W, Wang D, Zhao Y, et al. The NLRP3-caspase 1 inflammasome negatively regulates autophagy via TLR4-TRIF in prion peptide-infected microglia. Front Aging Neurosci (2018) 10:116. doi: 10.3389/fnagi.2018.00116
Keywords: inflammasome, Gram-positive bacteria, mechanisms of activation, autophagy, regulation
Citation: Keestra-Gounder AM and Nagao PE (2023) Inflammasome activation by Gram-positive bacteria: Mechanisms of activation and regulation. Front. Immunol. 14:1075834. doi: 10.3389/fimmu.2023.1075834
Received: 21 October 2022; Accepted: 06 January 2023;
Published: 24 January 2023.
Edited by:
Hideki Hara, Asahikawa Medical University, JapanReviewed by:
Yasuyuki Matsuda, Department of Microbiology and Immunochemistry, JapanKei Sakamoto, Nagasaki University, Japan
Copyright © 2023 Keestra-Gounder and Nagao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prescilla Emy Nagao, cG5hZ2FvQHVvbC5jb20uYnI7, cG5hZ2FvQHVlcmouYnI=
†ORCID: A. Marijke Keestra-Gounder, orcid.org/0000-0002-2231-652X
Prescilla Emy Nagao, orcid.org/0000-0001-6007-0033