 Yuxi Zhang
Yuxi Zhang Xiaoqing Xu
Xiaoqing Xu Hui Cheng1,2,3,4*
Hui Cheng1,2,3,4* Fusheng Zhou
Fusheng Zhou- 1Department of Dermatology, The First Affiliated Hospital, Anhui Medical University, Hefei, Anhui, China
- 2Institute of Dermatology, Anhui Medical University, Hefei, Anhui, China
- 3Key Laboratory of Dermatology (Anhui Medical University), Ministry of Education, Hefei, Anhui, China
- 4Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Hefei, Anhui, China
Psoriasis is a chronic inflammatory skin disease occurring worldwide, with multiple systemic complications, which seriously affect the quality of life and physical and mental health of patients. The pathogenesis of psoriasis is related to the environment, genetics, epigenetics, and dysregulation of immune cells such as T cells, dendritic cells (DCs), and nonimmune cells such as keratinocytes. Absent in melanoma 2 (AIM2), a susceptibility gene locus for psoriasis, has been strongly linked to the genetic and epigenetic aspects of psoriasis and increased in expression in psoriatic keratinocytes. AIM2 was found to be activated in an inflammasome-dependent way to release IL-1β and IL-18 to mediate inflammation, and to participate in immune regulation in psoriasis, or in an inflammasome-independent way by regulating the function of regulatory T(Treg) cells or programming cell death in keratinocytes as well as controlling the proliferative state of different cells. AIM2 may also play a role in the recurrence of psoriasis by trained immunity. In this review, we will elaborate on the characteristics of AIM2 and how AIM2 mediates the development of psoriasis.
Introduction
Psoriasis is a chronic systemic inflammatory skin disease, as well as a polygenic genetic disorder, the etiology of which is not completely clear (1, 2). Psoriasis invades most regions and races worldwide, affecting up to 125 million individuals (3). People with psoriasis are more likely to suffer from Crohn’s disease (4), cardiovascular disorders (5), psoriatic arthritis (6), depression (7), cancer (8) and obesity (9). The most common type of psoriasis is psoriasis vulgaris, which clinically features pink or red plaques distinct from normal skin and covered with white scales, often symmetrically distributed on the elbows, calves, scalp, abdomen, and back (10). Psoriasis is susceptible to recurrence and requires long-term treatment; many patients suffer from a huge physical and mental burden, as well as an enormous financial burden.
Psoriasis is considered to be associated with environmental factors, genetic factors, and disturbances between immune and nonimmune cells, and in recent years, epigenetic regulatory mechanisms have also been found to play a significant role in development of psoriasis (11, 12). Psoriasis is often easily irritated by factors such as injury, drugs, infections, stress, or other stimuli (13). Unlike Mendelian diseases, psoriasis is a complex polygenic genetic disease that often presents the characteristics of family aggregation. Through several studies researchers identified the importance of the psoriasis susceptibility gene 1 (PSORS1), which was the main susceptibility locus of psoriasis, accounts for 35-50% of disease heritability (14, 15). More than 40 susceptibility loci for psoriasis have been identified via genome-wide association studies (GWASs) and immunochip studies (16). The IL-23/IL-17 axis is the most important part of the immunomodulatory mechanism in psoriasis, especially IL-17, which plays a central role in disease progression (17). Besides, other cytokines are also actively involved in the immune regulation of psoriasis. Among them, IL-18 expression is significantly correlated with the severity of psoriasis (18). IL-23 binding to IL-1β or IL-18 promotes IL-17 production (19, 20). IL-1β and IL-23 can also activate γδ T cells and amplify T helper cell 17 (Th17) responses, while IL-18 reacts synergistically with IL-23 to similarly activate γδ T cells and induce Th1 immune responses (20, 21).
In addition to the above influencing factors, the inflammasome also participates in the pathogenesis of psoriasis (22). AIM2 polymerizes with other proteins to form the AIM2 inflammasome, which is one of the inflammasomes closely associated with psoriasis (22). The AIM2 inflammasome was found to be expressed increasingly in psoriatic skin, especially in keratinocytes (23). In addition to the role of the AIM2 inflammasome, AIM2 inflammasome-independent way may also play a significant role in the pathogenesis of psoriasis. For example, a recent study showed that AIM2 expression in Treg cells contributes to restraining the autoimmunity response (24).
In this review, we focus on the characterization of AIM2 and its relationship with psoriasis, how AIM2 plays an inflammasome-dependent and possibly inflammasome-independent role in the development of psoriasis, and how AIM2 may potentially modulate psoriasis relapse by training immunity.
AIM2 inflammasome
Since Martinon et al. (25) came up with the concept of the inflammasome in 2002, it has been described in many immune-related diseases, acting as an important part of innate immunity. Inflammasomes are intracellular multiprotein complexes (26). When encountering dangers such as microbes or self-DNA, the inflammasome assembles under the guidance of its specific pattern-recognition receptors (PRRs) to respond to pathogen-associated molecular patterns (PAMPs) (27). PAMPs include cell wall components of microorganisms, microorganisms, or nucleic acids. PRRs consist of PRRs localized on the plasma membrane and PRRs localized in the cytoplasm, while absent in melanoma 2-like receptors (ALRs) are intracytoplasmic PRRs that specifically recognize PAMPs in the cytoplasm (28).
AIM2 is a cytoplasmic dsDNA sensor, and an initial member of the ALRs family for inflammasome sensors (29, 30). AIM2 belongs to the PYHIN family and has one or two hematopoietic, interferon-inducible and nuclear (HIN) domains at the C-terminus that recognize and bind to cytoplasmic DNA, and a Pyrin domain (PYD) at the N-terminus that interacts with the PYD domain of the adapter protein, which called apoptosis-associated speck-like protein containing a CARD(ASC) (31, 32). ASC binds to procaspase-1 through CARD-CARD homologous protein interaction and completes the assembly of the AIM2 inflammasome complex (31). The caspase-1 generated by cleavage after procaspase-1 activation cleaves gasdermin-D (GSDMD) and inserts its N-terminal into the cell membrane to form a hole that cytokines can be released through, and the hole made of GSDMD insertion could result in a special cell death named pyroptosis and further exacerbate inflammation (33). Activated caspase-1 cleaves proIL-1 and proIL-18, and leading to the activation of downstream IL‐1β and IL‐18 and an inflammatory response (Figure 1A) (34–36). IL-18 is a member of the IL-1 family, takes part in immune cells such as Th1, Th2, natural killer (NK) cells, IL‐17‐producing γδ T cells, and macrophage activation, and acts as an IFN γ‐inducing factor to activate NF-κB to trigger inflammation (37). In human skin, the accumulated DNA in the cytoplasm of keratinocytes activates the AIM2 inflammasome and leads to the release of IL-1β, and keratinocytes are recognized as the main source of IL-1β (38). IL-1β is increased in keratinocytes of psoriatic skin and promotes the development of T-cell-dependent skin inflammation (39).
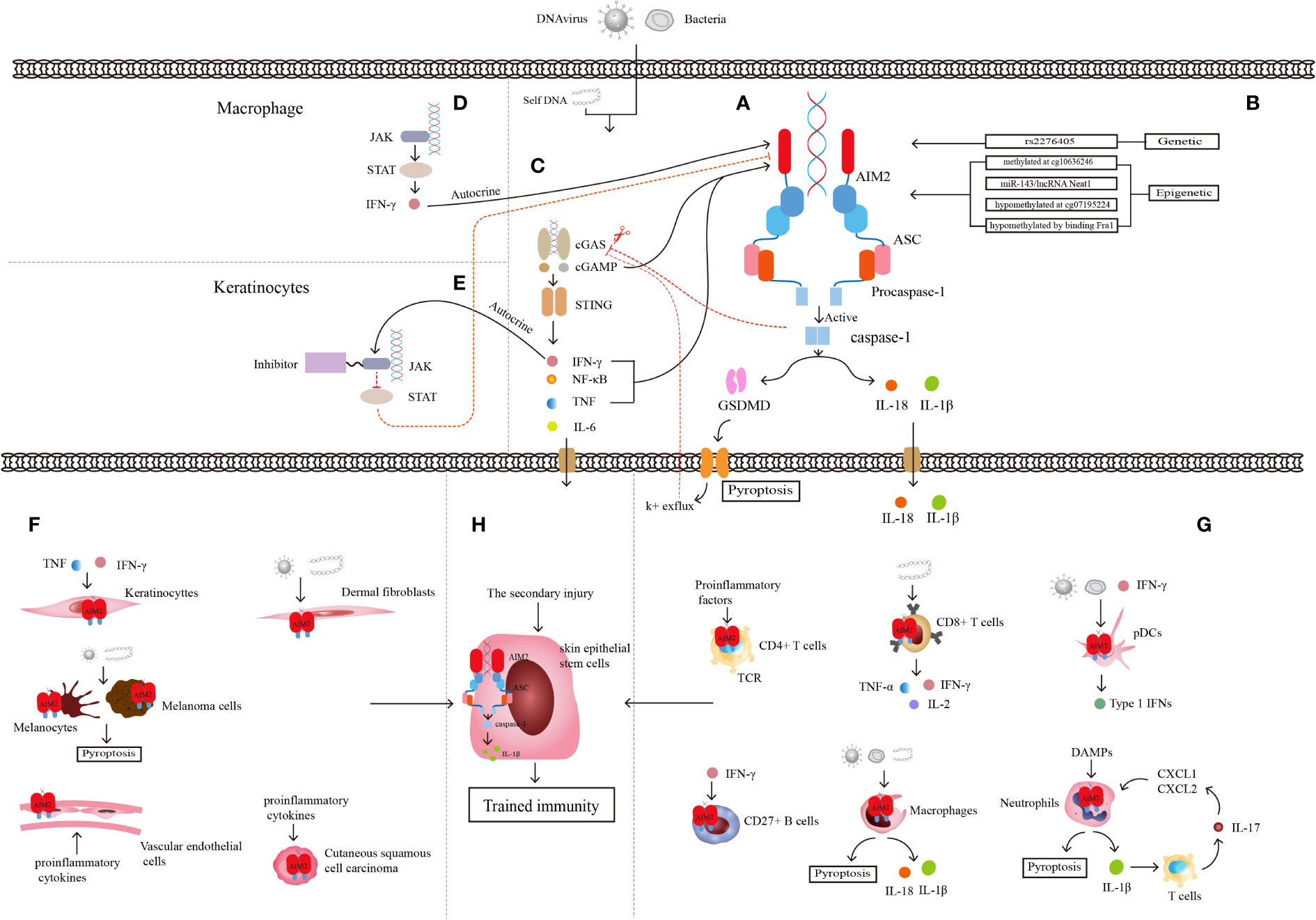
Figure 1 Characteristics of AIM2 inflammasome. (A) Structure of AIM2 inflammasome. (B) Genetic regulation and epigenetic regulation of AIM2. (C) Relationship between cGAS-STING pathway and AIM2 inflammasome. (D) Relationship between JAK-STAT pathway and AIM2 inflammasome in macrophages linked by autocrine secretion of IFN-γ. (E) Relationship between JAK-STAT pathway and AIM2 inflammasome in keratinocytes after JAK inhibitor application. JAK-STAT and cGAS-STING pathway are linked through the autocrine of IFN-γ. (F) Some non-immune cells mediating inflammation through AIM2 inflammasome. (G) Some immune cells mediating inflammation through AIM2 inflammasome. (H) Skin epithelial stem cells are involved in trained immunity through AIM2 inflammasome and the downstream inflammatory cytokine IL-1β. AIM2 integrates the trained immune memory properties of immune cells with non-immune cells.
AIM2 can only be bound by dsDNA of a suitable length. If the length of dsDNA is less than 80bp, AIM2 cannot effectively oligomerize around it, so the inflammasome cannot be activated by the short dsDNA, and AIM2 may only play a regulatory role under this circumstance (40, 41). The activated AIM2 inflammasome plays an important role in the occurrence and development of tumors, the innate immune response, proliferation and apoptosis of cells, and neurodegenerative disorders (30, 42).
Genetic and epigenetic control of the Aim2 inflammasome
Mutation of the AIM2 gene often drives the development of different diseases. The stop-gain variant in the AIM2 gene, whose definite site is rs2276405 (Figure 1B), is significantly enriched in psoriasis (43). Methylation of the AIM2 gene at the site cg10636246 drives the relationship between trauma exposure, posttraumatic stress disorder (PTSD), the level of C-reactive protein (CRP) and other inflammatory mediators, such as IL-6, IL-10, and TNF-α, in the body (44, 45). MicroRNA (miRNA), a non-coding RNA (ncRNA) of approximately 22 nucleotides in length, binds to messenger RNA (mRNA) targets of inflammasome genes for post-transcriptional regulation of inflammasome expression (46). Researchers transfected microRNA-143 (miR-143) (Figure 1B), which has been shown to target several inflammation-related genes, into the Jurkat cell line and found significant increases in mRNA expression of both AIM2 and ASC (47). Another type of ncRNA, long noncoding RNA (lncRNA), which is defined as untranslated RNA more than 200 nucleotides in length, can regulate several specific cis- or trans-acting elements (48). Researchers have identified nuclear enriched abundant transcript 1 (Neat1), a kind of lncRNA that can be transcribed from multiple endocrine tumor loci. The lncRNA Neat1 (Figure 1B) can reduce the inflammatory response by promoting the assembly of AIM2, activating the oligomerization of caspase-1, and producing cytokines (49).
In addition to AIM2, researchers have explored a 600-bp-long CpG island exists in the transcription initiation site of the ASC gene named PYCARD/TMSI, the methylation state of this CpG island is associated with the expression level of ASC. The unmethylated CpG site signifies the active transcription of ASC, the hypermethylated site indicates the inactive state, while the aberrant methylation of that is associated with gene silence (50, 51). Some experiments also proved that ASC methylation affects inflammasome activation (52).
Aim2 inflammasome-related signals
AIM2 can also interact with multiple signaling pathways. The Janus kinase/signal transduction and activator of transcription (JAK/STAT) signaling pathway is strongly linked to immune-mediated diseases, and the mutation and polymorphisms of JAK and STAT genes result in several inflammatory and autoimmune diseases, include psoriasis (53). The mouse experiments with the atherosclerotic cardiovascular disease model show that JAK/STAT mutation led to a series of changes within cells, such as metabolic disorder, abnormal proliferation, DNA oxidative damage, and replication error. The final result is activation of the AIM2 inflammasome and the production of mature IL-1β and IL-18 (Figure 1D) (54). JAK-STAT signaling expression is upregulated in lupus erythematosus skin lesions, and the application of JAK1 inhibitors significantly reduces the expression of proinflammatory mediators such as AIM2 and improves skin lesions in lupus mouse models (55). Quercetin was found to reduce caspase-1 expression and aggregation of the AIM2 inflammasome and the production of IL-18 and IL-1β by inhibiting IFN-induced JAK-STAT expression in human keratinocytes, thus reducing skin inflammation (Figure 1E) (56).
Activation of the AIM2 inflammasome in nonimmune cells
Keratinocytes are among the components that form the physical barrier of the skin and regulate the immune response of the skin microenvironment through the secretion of proinflammatory cytokines, chemokines, antimicrobial peptides (AMPs) and growth factors (57). Keratinocytes directly or indirectly crosstalk with immune cells through these proinflammatory cytokines and chemokines and can also express related receptors to recruit immune cells to secrete more proinflammatory ingredients, leading to immune disease (Figure 1F) (58). Keratinocytes detect a wide range of PAMPs by expressing abundant PRRs (23, 59), including AIM2 (60). Although researchers found that AIM2 is expressed only in the Langerhans cells and melanocytes of healthy skin, AIM2 can be upregulated significantly in keratinocytes when skin suffers from harmful conditions such as psoriasis or wounds. AIM2 and its downstream cytokine IL-1β always play an important role in the immune defense response of keratinocytes, and keratinocytes are considered to be the main source of IL-1β in skin cells (59). Although the cytosolic DNA receptor cyclic GMP-AMP synthase and the stimulator of interferon genes (cGAS-STING) is also a DNA recognition receptor, only AIM2 can trigger the secretion of IL-1β in keratinocytes (61). When keratinocytes are infiltrated with proinflammatory cytokines such as IFN-γ and TNF-α, AIM2 inflammasome activation and downstream IL-1β release have been proven to result in psoriasis lesions (62).
Arboviruses induce the expression of the AIM2 inflammasome in human dermal fibroblasts, and the inhibition of caspase-1 restrains the effect of AIM2 but enhances viral replication, showing that the function of dermal fibroblasts is positively associated with early infection through AIM2 (63). Researchers used the EP1 pulse protocol to observe the expression of cytokine mRNA in mouse skin after pDNA electrotransfer for four hours, and as they expected, the expression of AIM2 in both fibroblasts and keratinocytes was upregulated, describing the role of the DNA recognition receptor of AIM2 in fibroblasts (64).
Skin epithelial stem cells, which contribute significantly to skin wound healing, were first discovered to have the function of inflammatory memory as nonimmune cells. This trained immunity is mediated by the AIM2 inflammasome. Researchers believe that recurring immune skin diseases such as psoriasis may be associated with AIM2-mediated inflammatory memory in skin epithelial stem cells (65).
Although scientists have found that AIM2 activation in keratinocytes drives the development of many immune diseases, some other skin cells have the opposite expression; for example, AIM2 is restrictedly expressed in melanocytes in normal skin (23). Wang and his colleagues also proved that the viral dsDNA analog could activate AIM2 and then induce apoptosis of primary cultured normal human melanocytes. Knockdown of AIM2 can partially reduce apoptosis of melanocytes, and the reason may be related to the low expression of AIM2 in melanocytes (66). However, AIM2 plays an immunosuppressive role in the melanoma microenvironment in mouse model based on that AIM2-dependent IL-1β and IL-18 promote tumor growth and that AIM2 acts as a negative regulator of the STING pathway, inhibiting STING-mediated type I IFN secretion and migration of CD8+ T cells to tumors (67).
Cutaneous squamous cell carcinoma originates from skin keratinocytes, and chronic inflammation is one of the important pathogenic risk factors. The protein and mRNA expression of AIM2 is increased in cutaneous squamous cell carcinoma cells, and AIM2 was discovered to possibly be involved in the regulation of the cell cycle as well as cell survival and apoptosis (68).
Vascular endothelial cells also express AIM2 and respond to inflammation by upregulating AIM2 expression. For mice of acute or chronic vascular injury, activation of AIM2 leads to limited endothelial cell regeneration (69). Inhibition of AIM2 also reduced IL-1β and IL-18 expression levels in vascular endothelial cells in atherosclerosis-prone mouse model (70).
Activation of the AIM2 inflammasome in immune cells
Overactivation or suppression of the innate and adaptive immune systems are considered one of the pathogeneses in immune-mediated diseases, and psoriasis is one of the diseases in which immune cells play an important role. Psoriasis-associated immune cells, including DCs, monocytes, macrophages, neutrophils, mast cells, and T cells, play a central role in the development and progression of the disease (71). The comprehensive analysis of core genes and innate immune cells in psoriasis shows that AIM2 has a positive correlation with activated dendritic cells and M1 macrophages, as well as a negative correlation with resting mast cells (72), suggesting a close association of AIM2 inflammasomes with immune cells (Figure 1G) (Table 1).
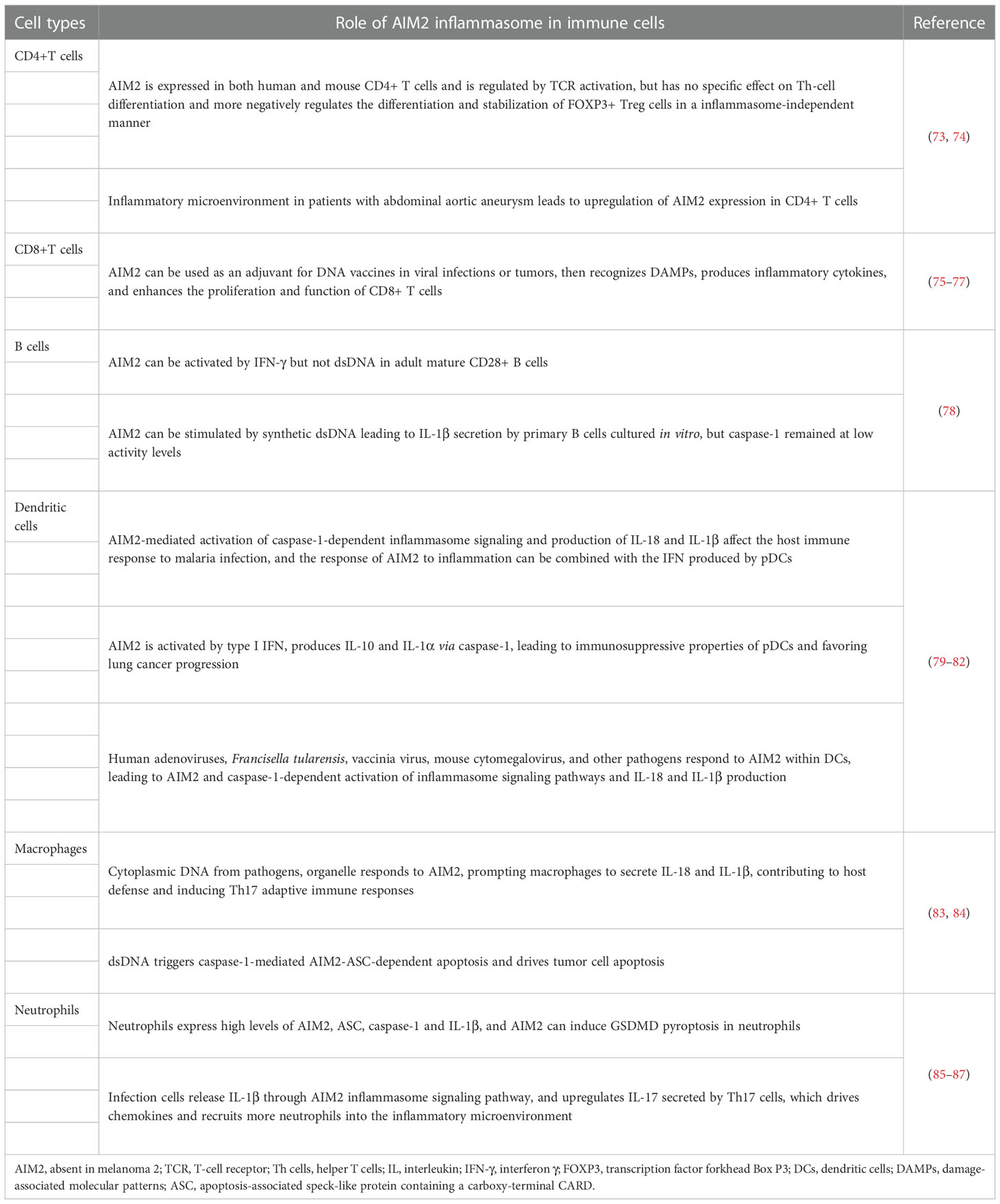
Table 1 The activation of AIM2-inflammasome in immune cells.
AIM2 was found to be expressed in CD4+ T cells in both humans and mice and regulated by T-cell receptor (TCR) activation, but there were no significant changes in the levels of differentiated Th1, Th2, and Th17 cells from CD4+ T cells in AIM2-knockout mice, and the expression of specific cytokines secreted by these cells was also not altered significantly. In vitro cellular experiments demonstrated that AIM2 is not involved in the differentiation of these T-cell subpopulations. CD4+ T cells can differentiate into Th-cell subpopulations and Treg cells, but since ASC deficiency cannot affect transcription factor forkhead Box P3 (FOXP3) production or differentiation of FOXP3+ Treg cells, AIM2 cannot regulate Treg cells in an inflammasome-dependent way (73). Researchers also found that various proinflammatory factors in patients with abdominal aortic aneurysms cause an imbalance of CD4+ T cells and upregulate the expression of AIM2 in them (74). AIM2 can be added as an adjuvant to DNA vaccines to mediate caspase-1 activation and IL-1β and IL-18 production in mouse models, as well as pyroptosis, to release damage-associated molecular patterns (DAMPs) and transmit inflammatory signals to surrounding cells (75). This kind of vaccine would enhance the induction of multifunctional CD8+ T cells, which express IL-2, TNF-α, and IFN-γ, and the vaccines have been proven to be effective in preventing virus-induced myocarditis in a mouse model (76). Moreover, this special vaccine with AIM2 as the adjuvant can also activate the proliferation of mouse CD8+ T cells and enhance their antitumor activity (77).
Analysis of adult blood and cord blood revealed that AIM2 is preferentially expressed in adult mature CD27+ B cells and that AIM2 activation is dependent on IFN-γ rather than IFN-α and synthetic dsDNA. Human primary B cells cultured in vitro can be stimulated by synthetic dsDNA to release IL-1β, but caspase-1 remains at low activity levels. The regulatory expression of AIM2 in human B cells cultured in vitro, human in vivo B cells, and mouse B cells is still not fully understood, since part of the regulatory effects is the opposite (78).
Monocytes can differentiate into macrophages that remain in tissue and dendritic cells that roam around the tissue as prototypical antigen-presenting cells, and both can discriminate, phagocytose and present microbial and foreign matter (88–90). There are three specific cutaneous DC populations, pDCs, dermal myeloid DCs (mDCs), and epidermal Langerhans cells (LCs), each of which appears to have a significant impact on inflammatory skin disease. These cells can act as antigen-presenting cells or an important cellular source of pathogenic cytokines, but only pDCs can activate AIM2, which mediates disease progression in an inflammasome-dependent manner (91, 92). In malaria, deficiency of AIM2 or caspase-1 significantly reduces IL-1β production in pDCs, conventional DCs (cDCs), and macrophages, and type I IFN regulates inflammasome activation and IL-1β production, as we all know that pDCs are a major source of type I IFN so that the AIM2 inflammasome response to infection can then be cross-linked to type I IFN signaling (79). In lung cancer tissues, pDCs have been found to activate the AIM2 inflammasome through type I IFN signaling, conducive to caspase-1 aggregation and the production of IL-1α and IL-10 but not IL-1β and IL-18 indirectly, leading to the immunosuppressive properties of pDCs and thus benefiting tumor progression (80). Previous studies confirmed that many pathogens could respond to AIM2 activation in DCs and macrophages of human and mouse and induce AIM2 inflammasome cleavage of mature proIL-1β and proIL-18 in succession (81, 82).
Macrophages can form the first line of defense to protect the body from harmful substances via antigen presentation, phagocytosis, and inflammasome activation (93, 94). During the inflammatory response, many cytokines, such as IL-6, IL-1β, TNFα (83), and excessive reactive oxygen species (ROS) (95), are released from activated cultured macrophages, which can activate the AIM2 inflammasome (96). In contrast, when the AIM2 inflammasomes from human or mouse immune cells encounter dsDNA from microorganisms, other organelles, and extracellular DNA, macrophages are activated to secrete IL-1β and IL-18, conducive to host defense and inducing a Th17 adaptive immune response (83). In addition, dsDNA has been found to trigger AIM2-ASC-dependent apoptosis dominated by caspase-1-induced pyroptosis in mouse macrophages (84). Disruption of Parkinson’s disease-associated mitochondrial serine protease HtrA2 activity leads to an increased response of the AIM2 inflammasome in macrophages (97). The above evidence also shows that AIM2 inflammasome activation in macrophages mediates immune responses in various infections.
Neutrophils are the first and most plentiful cells that permeate through the inflammatory region, where they can receive DAMPs (98) and then release many secretory vesicles containing proinflammatory factors and phagocytose pathogens to antagonize inflammation (99). Researchers have proven that neutrophils are an important source of IL-1β in both mice and human and that higher mRNA and protein levels of AIM2, ASC, and caspase-1 and AIM2 can induce GSDMD pyroptosis in neutrophils (85, 86). AIM2-mediated secretion of IL-1β upregulates IL-17 release by T cells of a murine model of atherosclerosis, which then drives chemokines such as CXC motif chemokine ligand 1 (CXCL1) and CXCL2 to recruit more neutrophils during inflammation (87).
AIM2-inflammasome as a target for disease treatment
In recent years, targeted treatment of AIM2 has been proven successful in autoinflammatory diseases. Scientists have discovered that double-stranded RNA-dependent protein kinase (PKR) plays an important role in the activation of multiple inflammasomes, including the AIM2 inflammasome. Knockout of PKR or medicinal inhibitor application neutralizes inflammasome activation in mouse peritoneal macrophages under microbial infection and dsDNA trigger (100). Ahn et al. found that fructose-arginine (FA), a main component of Korean red ginseng, reduced the secretion of caspase-1, IL-1β, and IL-18 from mouse macrophages or human monocytes triggered AIM2 activation. They also demonstrated that FA could restrain the formation of ASC and the incision of GSDMD on the plasma membrane (101). Chung et al. reported that EFLA945, a constituent of red grape vine leaf extracts, prevents dsDNA from entering macrophages. Thus, EFLA945 reduces the oligomerization of ASC and inhibits the activation of caspase-1 and the release of the downstream cytokines IL-1β and IL-18. It is vital that EFLA945 also suppresses the mouse model immune response caused by IMQ (102). Liu et al. revealed that tripartite motif 11 (TRIM11) degrades AIM2 when in vitro cultured human macrophages or mice are infected with the virus and regulates the degradation of AIM2 through the autophagic cargo receptor p62 to mediate selective autophagy in macrophages (103). Dang et al. detected an interesting metabolic circuit between inflammasome activation and cellular cholesterol content regulation in mouse macrophages. Excess overabundance of cholesterol triggers the activation of AIM2. Therefore, macrophages activated by cholesterol could make use of cholesterol-25-hydroxylase (Ch25 h), an oxysterol that inhibits the synthesis of cholesterol, to restrain the AIM2 inflammasome from being activated (104).
Whole-exome SNP array revealed AIM2 in psoriasis
Our team has proven that AIM2 is one of 15 new risk genes/loci of psoriasis through the whole-exome single nucleotide polymorphism (SNP) array for the first time. We used a large-scale whole-exome array and a three-stage case-control design to analyze the susceptibility gene of psoriasis, including 17,614 cases and 25,216 controls. Our meta-analysis of 76 psoriasis-related SNPs in the Exome Asian array and Exome Fine array and replication phase identified 11 variants in 11 different new susceptibility genes located in nonhuman leukocyte antigen (HLA) regions, including AIM2 (rs2276405, P=3.22 × 10−9, odds ratio (OR) =0.83), which is a stop-gain gene variant locus (Figure 1B) (43). Even though GWAS have revealed more than 40 psoriasis susceptibility genes or loci, the majority of these genetic variants are located in noncoding genomic regions, and low frequency and rare genetic variants cannot be well explained; due to the small sample size, this method cannot detect some small-effect loci, the whole-exome SNP array can be a good complement to this deficiency (105–107).
Epigenetic control of the Aim2 in psoriasis
Our team reported that cg07195224 (Figure 1B), a CPG site of AIM2, located in the promoter region of the gene, is significantly hypomethylated in psoriatic lesions. The intensity of the promoter-associated peak is negatively correlated with the level of methylation at this CPG site and positively correlated with the level of AIM2 mRNA expression. Meanwhile, our team identified Fra1 transcription factor (Figure 1B) target binds upstream of AIM2 transcriptional start site, whose upregulation leads to AIM2 inflammasome activation. Binding of transcription factors may lead to DNA hypomethylation, thereby maintaining the open structural domains of chromatin, and increased chromatin accessibility is closely linked to psoriasis pathogenesis (108).
Evidence of Aim2 inflammasome-related signals in psoriasis
The cGAS-STING is a DNA recognition signaling pathway that can induce programmed cell death, regulate the cell life cycle, prevent pathogen infections, and be involved in autoimmune or inflammatory diseases (109). When dsDNA in macrophages or keratinocytes is recognized by cGAS, it can activate the synthesis of cyclic GMP-AMP (cGAMP), and STING is activated by cGAMP induction in turn and triggers the downstream Interferon regulatory Factor 3 (IRF3)/IFN and NF-κB/IL-6 signaling cascades (110). The cGAS-STING signaling pathway activates AIM2 inflammasomes in two ways. Firstly, cGAMP is directly involved in the initiation and activation of cGAS-DNA induced inflammasomes, but STING not participates in this step and plays only a partial role in AIM2-dependent IL-1β secretion. Secondly, binding of cGAMP and STING upregulate the cytokines such as IFN-β and TNF, which are important signals in inflammasome components activation, then initiate and activate AIM2 inflammasome (111). Researchers found increased expression of STING and its downstream cytokines in skin lesions of psoriasis patients and the imiquimod (IMQ)-induced psoriasiform mouse model. Knockdown of STING reduced skin symptoms in a murine psoriasiform inflammation model, which might be related to the reduced IL-1β produced by keratinocytes (112). AIM2 inflammasome also inhibit cGAS-STING pathway by two methods. One is activated caspase-1 binds and cleaves cGAS directly, leads to the reduced cGAMP and downstream cytokine production (113). The other is caspase-1-activated GSDMD leads to potassium ion efflux from the membrane pore, which inhibits the activation of cGAS and cGAMP, then conduces to the reduced IFN-β production in turn (114). AIM2-deficient cells showed enhanced expression of cGAS-STING because the inhibition of AIM2 inflammasome pathway will reduce the pyroptosis, which leads to cGAMP and STING aggregation, as well as increased STING downstream components (115). IFN-β produced by cGAS-STING pathway also can activate the JAK-STAT pathway, cross-link AIM2 inflammasome and these signaling pathways (Figure 1C), mediate the development of psoriasis together.
AIM2-mediated trained immunity in psoriasis
Former outlooks insisted that only adaptive immunity could built immunological memory, but recent studies have changed that point of view (116, 117). The immune systems of plants and invertebrates, as well as vertebrates, have the function of securing immune memory after the first stimulation (118) and reinforcing responses to protect from a secondary infection, a phenomenon called trained immunity or innate immune memory (119–121). Immune memory is triggered by the interaction of PRRs and PAMPs (120). Trained immunity, different from classical immunological memory, driven by signals of transcription factors and epigenetic reprogramming, does not induce permanent genetic changes (122, 123). Naik et al. described trained immunity in inflammation-experienced epithelial stem cells, and AIM2 and the downstream IL-1β played a key role in this immune response process (65), the first demonstration that trained immunity can be established not only by immune cells but also in nonimmune cells (Figure 1H).
Trained immunity of cells may be associated with psoriasis
Stromal cells are defined as the heterogeneous cell group of mesenchymal origin, which includes fibroblasts, reticular stromal cells, and tissue-specific connective tissue cells such as osteoblasts and adipocytes (124, 125). The trait of secretory, immunomodulatory, and homing properties of these cells contributes to the function of supporting immune cells, building immune memory and immunoregulatory properties as well as antimicrobial activity (126–128). Mesenchymal stromal cells can regulate innate and adaptive immune responses via inflammatory cytokines released into the microenvironment (129). Lin et al. reported for the first time that when mesenchymal stromal cells were repeatedly exposed to lipopolysaccharide (LPS) in mice, the activation of NF-κB was increased significantly, enhancing the anti-inflammatory effect of macrophages, and this process was associated with histone methylation (130). Gingival fibroblasts have been found to maintain a low-grade inflammatory response until the second encounter with LPS and then respond rapidly to reduce inflammation after engagement (131). Synovial cells from patients with rheumatoid arthritis can display an inflammatory memory function similar to the inflammatory memory function of T cells. Endothelial cells also show an altered (rising or falling) inflammatory response after secondary stimulation (132).
Vascular endothelial cells continuously detect relevant danger signals such as pathogens, metabolic substances or cytokines in the blood, leading to an inflammatory response upon activation (133). The concept that vascular endothelial cells are conditioned innate immune cells was proposed by Mai et al. (134). Because vascular endothelial cells can experience epigenetic changes upon stimulation, leading to the generation of immune memory (135).
The group of 2 innate lymphoid cells (ILC2s) are regarded as tissue-resident lymphocytes. The ILC2s have been found to lack antigen-specific receptors, and thus memory ILC2s have no antigen specificity, which was considered to be consistent with the characteristics of innate immune memory (136). Skin ILC2s can participate in atopic dermatitis (AD), another chronic recurrent inflammatory disease, through activation by bacterial or other stimuli and then acquiring immune memory, leading to skin damage relapse (137, 138).
Monocytes are reputed to be the prophase of macrophages. When tissues suffer from infections, adult monocytes remove inflamed tissues and differentiate into macrophages that clear up the pathogen (139). Research has shown that when monocytes encounter Candida albicans (140) and Bacille Calmette-Guérin (BCG) vaccination (141), intensified protection against infections is induced, which is known as trained immunity. Trained immunity of monocytes/macrophages may be associated with the inflammasomes (139).
NK cells are innate lymphocytes that have shown the characteristic of adaptive immunity, in addition to the main feature of responding rapidly against pathogens and tumors in an antigen-independent manner (142). NK cells have been found to stimulate increased protective mechanisms against viral infections in mice after infection with influenza virus or cowpox virus (143), and there is also extensive evidence in primate disease models of viruses to demonstrate this stimulation (144). When NK cells are in a noninfected environment, they can be nonspecifically activated by proinflammatory cytokines and possess a stronger inflammatory response than resting NK cells when tested after a period of time (118, 145).
Does the AIM2 inflammasome-induced trained immunity link to psoriasis recurrence?
Several studies in recent years that obtained startling results from a series of psoriasis modeling experiments in mice showed that skin resident memory T (TRM) cells (146), memory-like γδ T cells (147), keratinocytes and skin epithelial stem cells (EpSCs) (65) drive the relapse of psoriasis. When mouse skin exposed to psoriatic-like stimulation, EPSCs may remember the stimuli and ensures that the epidermis responds more rapidly and intensively to subsequent diverse challenges (65, 148). EpSCs are basal keratinocytes that obtain inflammatory memory in the background of psoriatic skin inflammation, and chromatin domains remain accessible for rapid activation and opening in response to secondary inflammatory stimulation (65, 149). The rapid and enhanced acquired inflammatory memory is particularly dependent on the AIM2 inflammasome and its downstream component IL-1β in EpSCs, and the induction of epidermal AIM2 expression to promote inflammatory repair could occur regardless of the activity of IMQ or other stimuli (65). Several studies have proven that inflammasomes and inflammasome-dependent IL-1β expression play an important role in immune memory (150, 151).
Numerous studies have shown that there is a strong correlation between trained immunity and epigenetic reprogramming, such as DNA methylation (152, 153). Previous work by our team has demonstrated increased chromatin accessibility in psoriatic lesions in patients with psoriasis. Most of the differential peaks in chromatin accessibility detected in psoriatic lesions overlapped with the peaks of DNase I hypersensitivity in keratinocytes, and more than 95% of psoriasis-associated CPGs found in psoriatic lesions were hypomethylated (108). Our team has demonstrated that cg07195224 is included in the promoter region of the AIM2 gene, which is the CPG site of significant hypomethylation in psoriasis (154).
With all that in mind, we can speculate that AIM2 might be the key marker modulating trained immunity in epidermal keratinocytes, as well as an important factor leading to the relapse of psoriasis, which needs more evidence for verification.
Inflammasome-independent roles of AIM2 may related to psoriasis
In addition to its critical role in the inflammasome and trained immunity, AIM2 has also been implicated in several other ways to modulate skin homeostasis, which might potentially be involved in psoriasis occurrence, development, or recurrence (Figure 2). For example, AIM2 might promote the proliferation of epithelial cells (155) and regulate the function of Treg cells (24); both of these cells are instrumental cell types that execute important functions in psoriasis development.
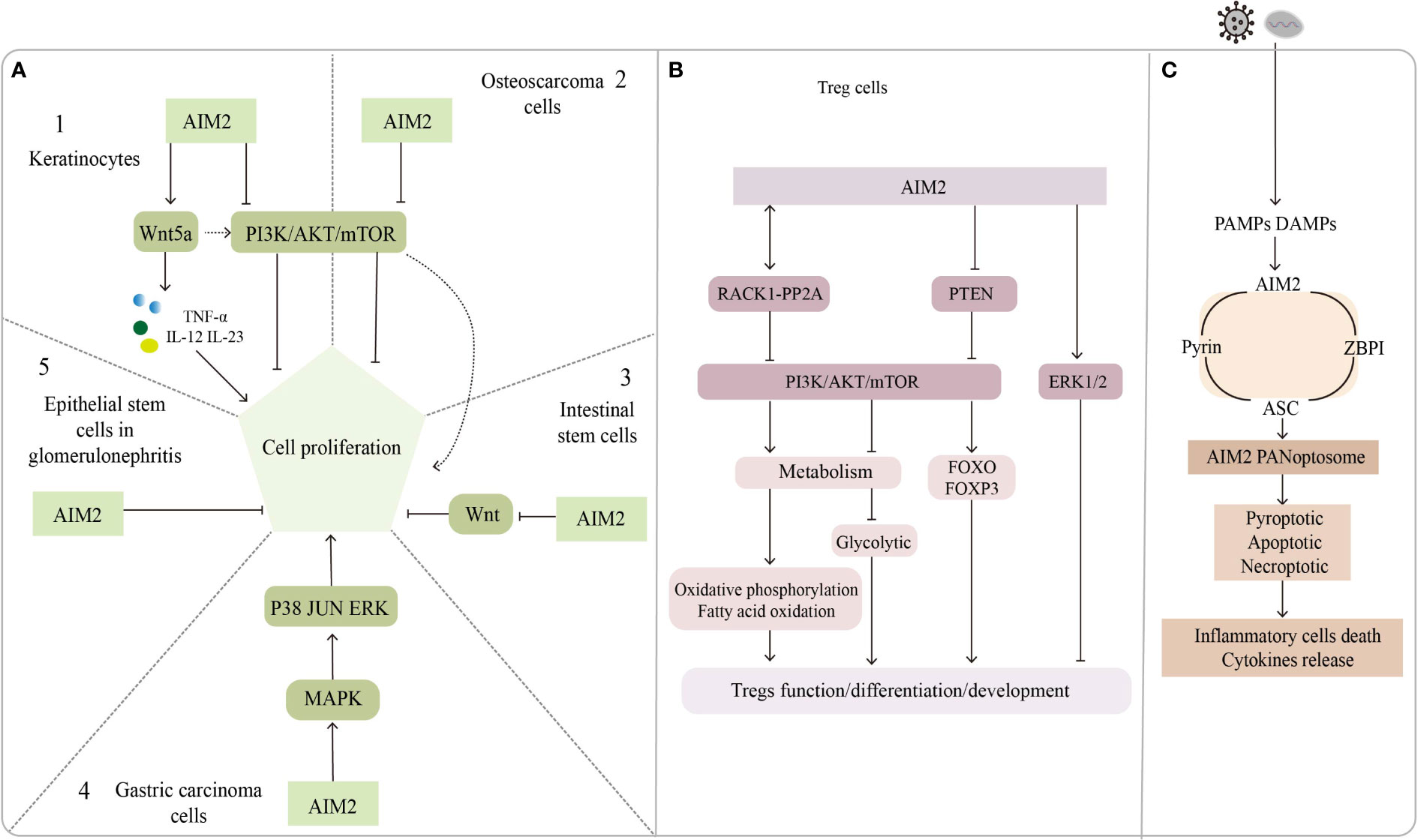
Figure 2 The inflammasome-independent way of AIM2. (A) Regulatory role of AIM2 in cell proliferation. A1, A2: AIM2 promotes keratinocytes proliferation through the Wnt5a pathway; PI3K/AKT pathway promotes keratinocytes and osteosarcoma cells proliferation, but AIM2 inhibits it. A3: AIM2 inhibits the proliferation of intestinal stem cells by suppressing the Wnt pathway, AIM2 activates PI3K/AKT pathway via Wnt5a and promotes intestinal stem cell proliferation. A4: AIM2 promotes gastric carcinoma cell proliferation through MAPK pathway. A5: AIM2 put a curb on epithelial stem cells proliferation in glomerulonephritis. (B) Role of AIM2 in Treg cells function/development/differentiation. AIM2 interacts with RACK1-PP2A protein to inhibit AKT/mTOR pathway, leading to metabolite changes that regulate Treg function and stability; AIM2 inhibits Treg proliferation through ERK1/2 pathway or by inhibiting PI3K/AKT pathway. (C) AIM2 forms a complex named AIM2 PANoptosome with pyrin, ASC and ZBPI, which mediates inflammatory cell death and cytokine release.
AIM2 might disturb the proliferation of epithelial cells
Keratinocytes are a type of epithelial cell that might modulate psoriasis development by both directedly triggering an immune cascade and passively responding to cytokines when the skin microenvironment is altered. Several lines of evidence suggest that genetic modification of some genes might lead to psoriasis-like phenotypes but does not engage the systemic inflammatory response. For example, mice constitutively activating STAT3 in keratinocytes develop a psoriasis phenotype (156). Specific deletion of the NF-κB-responsive gene IκBa in keratinocytes leads to skin inflammation, including epithelial compartment and immune cell infiltration (157).
Recently, GWAS identified dozens of susceptible genes that might contribute to keratinocyte hyperproliferation and aberrant immune response (158). Some of these genes increased or decreased the risk of affecting the disease phenotypes when they were specifically deleted in keratinocytes. Keratinocyte-specific loss of function of target genes such as TNIP1, TNFAIP3, or CD109 in mice does not spontaneously develop visible skin inflammation, but when treated with IMQ, these mice were more likely to develop or show severe psoriasis symptoms than control mice (159–161). Meanwhile, JunB/c-Jun double knockout and N-WASP knockout in mice keratinocytes directly lead to hyperplasia and chronic inflammation (162, 163). In contrast, IL17RA and TRAF6 deletion in keratinocytes might protect mice from skin inflammation (164, 165). In summary, keratinocytes are key for psoriasis development, and their proliferation can be controlled by a single molecular or signaling pathway.
We noticed that most of these genes modulate psoriasis through the secretion of IL-23, and that downstream signaling molecules of the AIM2 inflammasome may act in conjunction with IL-23 to regulate psoriasis development, besides AIM2 might be directly involved in keratinocyte proliferation in an inflammasome-independent manner. AIM2 mutations have been found in 56% of small bowel cancers (166) but in only 2% of melanomas. Man and his colleagues found that AIM2-deficient mice presented uncontrolled proliferation of intestinal stem cells through an abnormal wingless (Wnt) signaling pathway, while inflammasome function was found to be undisrupted by assessing IL-1β, IL-18, and caspase-1 (155). Feng et al. reported that AIM2 might promote cell proliferation in gastric cancer via the mitogen-activated protein kinase (MAPK) pathway (167), leading us to suspect that AIM2 might also control keratinocyte proliferation by augmenting the Wnt or MAPK pathways. Indeed, Wnt5a increases keratinocyte proliferation by secreting TNF-α, IL-12, and IL-23 but repressing the Notch1 pathways which could activate the AIM2 inflammasome (168, 169). In contrast, AIM2 might repress proliferation by inactivating the phosphatidylinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway in osteosarcoma cells (170). Wilson et al. shown that AIM2 inhibits tumor cell proliferation by suppressing AKT phosphorylation in colonic epithelial cells and colon cancer cells, does not affect body inflammation and inflammasomes activation (171). PI3K/AKT signaling pathway also play an important role in the regulation of keratinocyte hyperproliferation in psoriasis (172), leading us to assume that AIM2 might regulate the proliferative state of keratinocytes in psoriasis by interacting with the PI3K/AKT signaling pathway, although the inhibitory effect of AIM2 is not very strong. The ability of AIM2 to inhibit cell proliferation was also found in glomerulonephritis (173). In summary, AIM2 can activate or inhibit cell proliferation via different signaling pathways. The exact role of AIM2 in modulating keratinocytes might be context-dependent.
AIM2 might regulate the function of Treg cells in psoriasis
Treg cells are a kind of suppressive helper T cells that express high levels of IL-10 and the transcription factor FOXP3. By releasing TGF-β and IL-10, Treg cells can inhibit effector immune T cells, thus playing important roles in maintaining immunological self-tolerance and executing host defense immune responses in a healthy state (174, 175). Even though the percentage or frequency of circulating and infiltrating Tregs in psoriasis is controversial among studies (176, 177), impaired function has been confirmed by several groups; for example, the suppressive ability of Tregs was decreased, showing reduced CD73 expression and an inactive CD73/AMPK/mTOR pathway (178).
In psoriasis, Treg cell dysfunction can be lead to an imbalance in Th17/Treg cells, but the detailed mechanism underlying this phenomenon remains elusive (179). Recently, several epigenetic markers that cause imbalanced Th17/Treg cells have been identified in inflammatory psoriasis. Wu et al. found that miRNA-210 and miRNA-126 can promote the differentiation of Th17 cells, thus leading to an increased Th17/Treg ratio and psoriatic-like phenotypes in genetically engineered mice (180, 181). Ma and his colleagues found that the mis-regulated Notch1 signaling pathway increased the Th17/Treg ratio in psoriasis (182). In summary, impaired suppressive function of Tregs or an increased Th17/Treg ratio would cause psoriasis, mainly because these conditions can lead to an abnormal proinflammatory cytokine environment, such as upregulated IL-17A and IFN-γ and downregulated IL-10 and TGF-β.
AIM2 can regulate the development and function of Treg cells by regulating signaling pathways. The PI3K/AKT signaling pathway and its upstream or downstream components play critical roles in regulating Treg cell differentiation, development and function (183), and AIM2 was proved to suppress PI3K/AKT activation in an inflammasome-independent way (171). AIM2 in non-small cell lung cancer cells can promote extracellular signal-regulated kinase 1/2(ERK1/2) phosphorylation, the function has nothing with inflammasomes, while ERK1/2 was found to inhibit the development of FOXP3+ Treg cells (73).
Accumulating evidence suggests that metabolic processes are involved in the regulation of growth, differentiation, localization, and function of Treg cells (184). The regulatory effects of metabolic substrates such as lipids, amino acids, and glucose are dependent mainly on the cell state and the external environment and are largely flexible and dynamic. These metabolites have been found to contribute to the activation and maintenance function of Treg cells (185). FOXP3 is not only essential for Treg differentiation but also for regulating the expression of critical genes encoding metabolic molecules in Treg cells (186). For instance, in vitro analysis showed that FOXP3 activated the proliferation of Treg cells, which was inhibited by lactate (187). More recently, Chou et al. found that AIM2 promoted oxidative phosphorylation of lipids while suppressing aerobic glycolysis in Treg cells, an effect works in part by attenuating AKT phosphorylation and the mTOR and MYC signaling pathways, rather than by the activation of the AIM2 inflammasome, Treg stability is essential for regulating autoimmune diseases (24). These findings support the notion that regulation of metabolism is important for the suppressive capabilities of Treg cells and contributes to the maintenance of homeostasis at the skin barrier.
Chou’s finding attracted us to believe that regulation of Treg cell function through AIM2 can mediate the pathogenesis of psoriasis or other inflammatory diseases.
AIM2 might control keratinocyte cell death
The AIM2 inflammasome could trigger the innate immune response by producing mature proinflammatory cytokines such as IL-18 and IL-1β, and it might also activate cell necroptosis when facing infections. Additionally, dead cells release more cytokines and inflammatory mediators, initiating the signaling cascade to eliminate infected cells or triggering chronic inflammation (188).
Recent findings suggested that necroptosis mediated by receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like protein (MLKL) has been implicated in psoriasis development (189–191). The expression of RIPK1 is elevated in psoriatic lesions compared with no lesion skin and atopic dermatitis, but downregulated RIPK1 enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling in psoriasis (192). Furthermore, RIPK1 potentially mediates the interactions between RIPK3 and Z-DNA binding protein 1 (ZBP1), thus preventing the activation of RIPK3 and the upstream kinase protein MLKL (193). Conditional knockout of RIPK1 in the epidermis could induce ZBP1-dependent necroptosis and skin inflammation (194, 195). Recently, Lee and colleagues described a new multiprotein complex, named PANoptosis, which could detect and respond to herpes simplex virus 1 infection. In this study, the author found that AIM2 regulates the innate sensors pyrin and ZBP1 to drive inflammatory signaling and trigger cell death, which is characterized by the activation of critical pyroptotic, apoptotic and necroptotic molecules. Since AIM2 interacts with pyrin, ZBP1 and ASC to form the complex, they called it the AIM2 PANoptosome (196). Even though whether AIM2 PANoptosome plays roles in psoriasis has not been determined, we still think AIM2 might be a good target for drug development in the future.
Conclusions and perspectives
Psoriasis is a chronic inflammatory skin disease that is closely related to many complications and seriously affects the appearance and mental health of patients. Treatment modalities for psoriasis include traditional oral drugs, topical medication, and phototherapy. Many oral or topical medications can have some negative effects on the body after long-term use (197). In recent years, biological agents have gradually become the first-line treatment for moderate to severe psoriasis (198). Although biologics anticytokines and antiproinflammatory pathways are highly targeted and effective, they also have disadvantages such as high price, leading to reduced immunity of patients, easy infection, and easy loss of efficacy, so there is still a need to develop new effective biological agents for personalized treatment of psoriasis (3). In recent years, AIM2 has been known as an important component in the pathogenesis of psoriasis, but whether AIM2 could be the next effective drug target for psoriasis is not yet clear. Inhibitors of AIM2 or molecules such as ASC and caspase-1 in the AIM2 inflammasome pathway have also been shown to be effective in cancers and some other diseases (199, 200). The downstream proinflammatory cytokines of the AIM2 inflammasome, IL-1β, and IL-18, can activate inflammatory cascade responses in psoriatic skin, such as the IL-23/Th17/IL-17 signaling axis, there are several corresponding targeted drugs for which there are currently a large number of successful cases in the treatment of psoriasis (201). However, the mechanisms by which AIM2 causes psoriasis involve the activation of various cytokines, proinflammatory pathways, and several signaling pathways in cells that are still not fully understood. There is also no direct evidence to show that AIM2 can regulate the progression of psoriasis by regulating the proliferation, metabolism, and apoptosis of related cells or by mediating trained immunity to regulate the recurrence of psoriasis. Moreover, the function of AIM2 in adaptive immune cells is also not fully understood. Therefore, we can only expect more research on these interesting mechanisms in the future.
Author contributions
FZ proposed the idea of the paper and contributed to the review of this work with HC, YZ conceived and wrote the article, and XX made constructive revisions to the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Fund Project of National Natural Science Foundation of China (4601020204).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Rendon A, Schäkel K. Psoriasis pathogenesis and treatment. Int J Mol Sci (2019) 20:1475. doi: 10.3390/ijms20061475
3. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. JAMA (2020) 323:1945–60. doi: 10.1001/jama.2020.4006
4. De Francesco MA, Caruso A. The gut microbiome in psoriasis and crohn’s disease: Is its perturbation a common denominator for their pathogenesis? Vaccines (Basel) (2022) 10:244. doi: 10.3390/vaccines10020244
5. Hu SC-S, Lan C-CE. Psoriasis and cardiovascular comorbidities: Focusing on severe vascular events, cardiovascular risk factors and implications for treatment. Int J Mol Sci (2017) 18:E2211. doi: 10.3390/ijms18102211
6. Boehncke W-H. Psoriasis and psoriatic arthritis: Flip sides of the coin? Acta Derm Venereol (2016) 96:436–41. doi: 10.2340/00015555-2385
7. Koo J, Marangell LB, Nakamura M, Armstrong A, Jeon C, Bhutani T, et al. Depression and suicidality in psoriasis: review of the literature including the cytokine theory of depression. J Eur Acad Dermatol Venereol (2017) 31:1999–2009. doi: 10.1111/jdv.14460
8. Loft ND, Vaengebjerg S, Skov L. Cancer risk in patients with psoriasis: should we be paying more attention? Expert Rev Clin Immunol (2020) 16:479–92. doi: 10.1080/1744666X.2020.1754194
9. Coimbra S, Catarino C, Santos-Silva A. The triad psoriasis-obesity-adipokine profile. J Eur Acad Dermatol Venereol (2016) 30:1876–85. doi: 10.1111/jdv.13701
10. Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet (2007) 370:263–71. doi: 10.1016/S0140-6736(07)61128-3
11. Deng Y, Chang C, Lu Q. The inflammatory response in psoriasis: a comprehensive review. Clin Rev Allergy Immunol (2016) 50:377–89. doi: 10.1007/s12016-016-8535-x
12. Srivastava AK, Chand Yadav T, Khera HK, Mishra P, Raghuwanshi N, Pruthi V, et al. Insights into interplay of immunopathophysiological events and molecular mechanistic cascades in psoriasis and its associated comorbidities. J Autoimmun (2021) 118:102614. doi: 10.1016/j.jaut.2021.102614
13. Kamiya K, Kishimoto M, Sugai J, Komine M, Ohtsuki M. Risk factors for the development of psoriasis. Int J Mol Sci (2019) 20:4347. doi: 10.3390/ijms20184347
14. Fan X, Yang S, Huang W, Wang Z-M, Sun L-D, Liang Y-H, et al. Fine mapping of the psoriasis susceptibility locus PSORS1 supports HLA-c as the susceptibility gene in the han Chinese population. PloS Genet (2008) 4:e1000038. doi: 10.1371/journal.pgen.1000038
15. Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and haplotype analysis supports HLA-c as the psoriasis susceptibility 1 gene. Am J Hum Genet (2006) 78:827–51. doi: 10.1086/503821
16. Mahil SK, Capon F, Barker JN. Genetics of psoriasis. Dermatologic Clinics (2015) 33:1–11. doi: 10.1016/j.det.2014.09.001
17. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol (2018) 201:1605–13. doi: 10.4049/jimmunol.1800013
18. Rasmy H, Mikhael N, Ismail S. Interleukin-18 expression and the response to treatment in patients with psoriasis. Arch Med Sci (2011) 7:713–9. doi: 10.5114/aoms.2011.24144
19. Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature (2010) 467:967–71. doi: 10.1038/nature09447
20. Shimoura N, Nagai H, Fujiwara S, Jimbo H, Yoshimoto T, Nishigori C. Interleukin (IL)-18, cooperatively with IL-23, induces prominent inflammation and enhances psoriasis-like epidermal hyperplasia. Arch Dermatol Res (2017) 309:315–21. doi: 10.1007/s00403-017-1735-2
21. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
22. Ciążyńska M, Olejniczak-Staruch I, Sobolewska-Sztychny D, Narbutt J, Skibińska M, Lesiak A. The role of NLRP1, NLRP3, and AIM2 inflammasomes in psoriasis: Review. Int J Mol Sci (2021) 22:5898. doi: 10.3390/ijms22115898
23. de Koning HD, Bergboer JGM, van den Bogaard EH, van Vlijmen-Willems IMJJ, Rodijk-Olthuis D, Simon A, et al. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp Dermatol (2012) 21:961–4. doi: 10.1111/exd.12037
24. Chou W-C, Guo Z, Guo H, Chen L, Zhang G, Liang K, et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature (2021) 591:300–5. doi: 10.1038/s41586-021-03231-w
25. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
26. Awad F, Assrawi E, Louvrier C, Jumeau C, Georgin-Lavialle S, Grateau G, et al. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol Ther (2018) 187:133–49. doi: 10.1016/j.pharmthera.2018.02.011
27. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
28. Kesavardhana S, Kanneganti T-D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol (2017) 29:201–10. doi: 10.1093/intimm/dxx018
29. Kawasaki T, Kawai T. Discrimination between self and non-Self-Nucleic acids by the innate immune system. Int Rev Cell Mol Biol (2019) 344:1–30. doi: 10.1016/bs.ircmb.2018.08.004
30. Wang B, Bhattacharya M, Roy S, Tian Y, Yin Q. Immunobiology and structural biology of AIM2 inflammasome. Mol Aspects Med (2020) 76:100869. doi: 10.1016/j.mam.2020.100869
31. Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu T-M, et al. Molecular basis of caspase-1 polymerization and its inhibition by a novel capping mechanism. Nat Struct Mol Biol (2016) 23:416. doi: 10.1038/nsmb.3199
32. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
33. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
34. Fan X, Jiao L, Jin T. Activation and immune regulation mechanisms of PYHIN family during microbial infection. Front Microbiol (2022) 12:809412. doi: 10.3389/fmicb.2021.809412
35. Forouzandeh M, Besen J, Keane RW, de Rivero Vaccari JP. The inflammasome signaling proteins ASC and IL-18 as biomarkers of psoriasis. Front Pharmacol (2020) 11:1238. doi: 10.3389/fphar.2020.01238
36. Lindberg E, Baumer Y, Stempinski ES, Rodante JA, Powell-Wiley TM, Dey AK, et al. Nanotomography of lesional skin using electron microscopy reveals cytosolic release of nuclear DNA in psoriasis. JAAD Case Rep (2021) 9:9–14. doi: 10.1016/j.jdcr.2020.12.024
37. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev (2018) 281:138–53. doi: 10.1111/imr.12616
38. Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev (2015) 265:6–21. doi: 10.1111/imr.12296
39. Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y, et al. A critical role of the IL-1β-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol (2019) 139:146–56. doi: 10.1016/j.jid.2018.07.025
40. Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, Sohn J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun (2015) 6:7827. doi: 10.1038/ncomms8827
41. Sharma M, de Alba E. Structure, activation and regulation of NLRP3 and AIM2 inflammasomes. Int J Mol Sci (2021) 22:872. doi: 10.3390/ijms22020872
42. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6:291. doi: 10.1038/s41392-021-00687-0
43. Zuo X, Sun L, Yin X, Gao J, Sheng Y, Xu J, et al. Whole-exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat Commun (2015) 6:6793. doi: 10.1038/ncomms7793
44. Miller MW, Maniates H, Wolf EJ, Logue MW, Schichman SA, Stone A, et al. CRP polymorphisms and DNA methylation of the AIM2 gene influence associations between trauma exposure, PTSD, and c-reactive protein. Brain Behav Immun (2018) 67:194–202. doi: 10.1016/j.bbi.2017.08.022
45. Hawn SE, Neale Z, Wolf EJ, Zhao X, Pierce M, Fein-Schaffer D, et al. Methylation of the AIM2 gene: An epigenetic mediator of PTSD-related inflammation and neuropathology plasma biomarkers. Depress Anxiety (2022) 39:323–33. doi: 10.1002/da.23247
46. Poli G, Fabi C, Bellet MM, Costantini C, Nunziangeli L, Romani L, et al. Epigenetic mechanisms of inflammasome regulation. Int J Mol Sci (2020) 21:E5758. doi: 10.3390/ijms21165758
47. Momeni M, Reza Mirzaei M, Zainodini N, Hassanshahi G, Arababadi MK. MiR-143 induces expression of AIM2 and ASC in jurkat cell line. Iran J Immunol (2013) 10:103–9.
48. Wang W, Yang N, Yang Y-H, Wen R, Liu C-F, Zhang T-N. Non-coding RNAs: Master regulators of inflammasomes in inflammatory diseases. J Inflamm Res (2021) 14:5023–50. doi: 10.2147/JIR.S332840
49. Zhang P, Cao L, Zhou R, Yang X, Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun (2019) 10:1495. doi: 10.1038/s41467-019-09482-6
50. Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Epigenetic regulation of ASC/TMS1 expression: potential role in apoptosis and inflammasome function. Cell Mol Life Sci (2014) 71:1855–64. doi: 10.1007/s00018-013-1524-9
51. Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res (2000) 60:6236–42.
52. Meier K, Drexler SK, Eberle FC, Lefort K, Yazdi AS. Silencing of ASC in cutaneous squamous cell carcinoma. PloS One (2016) 11:e0164742. doi: 10.1371/journal.pone.0164742
53. Tzeng H-T, Chyuan I-T, Lai J-H. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: A focus on molecular mechanisms and therapeutic potential. Biochem Pharmacol (2021) 193:114760. doi: 10.1016/j.bcp.2021.114760
54. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
55. Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective janus kinase 1 inhibition is a promising therapeutic approach for lupus erythematosus skin lesions. Front Immunol (2020) 11:344. doi: 10.3389/fimmu.2020.00344
56. Lee K-M, Kang JH, Yun M, Lee S-B. Quercetin inhibits the poly(dA:dT)-induced secretion of IL-18 via down-regulation of the expressions of AIM2 and pro-caspase-1 by inhibiting the JAK2/STAT1 pathway in IFN-γ-primed human keratinocytes. Biochem Biophys Res Commun (2018) 503:116–22. doi: 10.1016/j.bbrc.2018.05.191
57. Ortiz-Lopez LI, Choudhary V, Bollag WB. Updated perspectives on keratinocytes and psoriasis: Keratinocytes are more than innocent bystanders. Psoriasis (Auckl) (2022) 12:73–87. doi: 10.2147/PTT.S327310
58. Tüzün Y, Antonov M, Dolar N, Wolf R. Keratinocyte cytokine and chemokine receptors. Dermatologic Clinics (2007) 25:467–76. doi: 10.1016/j.det.2007.06.003
59. Kopfnagel V, Wittmann M, Werfel T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1β secretion. Exp Dermatol (2011) 20:1027–9. doi: 10.1111/j.1600-0625.2011.01382.x
60. Jiang Y, Tsoi LC, Billi AC, Ward NL, Harms PW, Zeng C, et al. Cytokinocytes: the diverse contribution of keratinocytes to immune responses in skin. JCI Insight (2020) 5:142067. doi: 10.1172/jci.insight.142067
61. Piipponen M, Li D, Landén NX. The immune functions of keratinocytes in skin wound healing. Int J Mol Sci (2020) 21:8790. doi: 10.3390/ijms21228790
62. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Göss C, Anz D, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med (2011) 3:82ra38. doi: 10.1126/scitranslmed.3002001
63. Ekchariyawat P, Hamel R, Bernard E, Wichit S, Surasombatpattana P, Talignani L, et al. Inflammasome signaling pathways exert antiviral effect against chikungunya virus in human dermal fibroblasts. Infect Genet Evol (2015) 32:401–8. doi: 10.1016/j.meegid.2015.03.025
64. Bosnjak M, Znidar K, Sales Conniff A, Jesenko T, Markelc B, Semenova N, et al. In vitro and in vivo correlation of skin and cellular responses to nucleic acid delivery. Biomed Pharmacotherapy (2022) 150:113088. doi: 10.1016/j.biopha.2022.113088
65. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature (2017) 550:475–80. doi: 10.1038/nature24271
66. Wang S, Liu D, Ning W, Xu A. Cytosolic dsDNA triggers apoptosis and pro-inflammatory cytokine production in normal human melanocytes. Exp Dermatol (2015) 24:298–300. doi: 10.1111/exd.12621
67. Fukuda K, Okamura K, Riding RL, Fan X, Afshari K, Haddadi N-S, et al. AIM2 regulates anti-tumor immunity and is a viable therapeutic target for melanoma. J Exp Med (2021) 218:e20200962. doi: 10.1084/jem.20200962
68. Farshchian M, Nissinen L, Siljamäki E, Riihilä P, Piipponen M, Kivisaari A, et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget (2017) 8:45825–36. doi: 10.18632/oncotarget.17573
69. Lüsebrink E, Goody PR, Lahrmann C, Flender A, Niepmann ST, Zietzer A, et al. AIM2 stimulation impairs reendothelialization and promotes the development of atherosclerosis in mice. Front Cardiovasc Med (2020) 7:582482. doi: 10.3389/fcvm.2020.582482
70. Paulin N, Viola JR, Maas SL, de Jong R, Fernandes-Alnemri T, Weber C, et al. Double-strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation (2018) 138:321–3. doi: 10.1161/CIRCULATIONAHA.117.033098
71. Schön MP. Adaptive and innate immunity in psoriasis and other inflammatory disorders. Front Immunol (2019) 10:1764. doi: 10.3389/fimmu.2019.01764
72. Gong X, Wang W. Profiles of innate immune cell infiltration and related core genes in psoriasis. BioMed Res Int (2021) 2021:6656622. doi: 10.1155/2021/6656622
73. Lozano-Ruiz B, Tzoumpa A, Martínez-Cardona C, Moreno D, Aransay AM, Cortazar AR, et al. Absent in melanoma 2 (AIM2) regulates the stability of regulatory T cells. Int J Mol Sci (2022) 23:2230. doi: 10.3390/ijms23042230
74. Wang H, Wei G, Cheng S, Wang D, Ma J, Xin S. Circulatory CD4-positive T-lymphocyte imbalance mediated by homocysteine-induced AIM2 and NLRP1 inflammasome upregulation and activation is associated with human abdominal aortic aneurysm. J Vasc Res (2020) 57:276–90. doi: 10.1159/000508077
75. Suschak JJ, Wang S, Fitzgerald KA, Lu S. Identification of Aim2 as a sensor for DNA vaccines. J Immunol (2015) 194:630–6. doi: 10.4049/jimmunol.1402530
76. Yin L, Chai D, Yue Y, Dong C, Xiong S. AIM2 Co-immunization with VP1 is associated with increased memory CD8 T cells and mounts long lasting protection against coxsackievirus B3 challenge. Front Cell Infect Microbiol (2017) 7:247. doi: 10.3389/fcimb.2017.00247
77. Chai D, Shan H, Wang G, Zhang Q, Li H, Fang L, et al. Combining DNA vaccine and AIM2 in H1 nanoparticles exert anti-renal carcinoma effects via enhancing tumor-specific multi-functional CD8+ T-cell responses. Mol Cancer Ther (2019) 18:323–34. doi: 10.1158/1535-7163.MCT-18-0832
78. Svensson A, Patzi Churqui M, Schlüter K, Lind L, Eriksson K. Maturation-dependent expression of AIM2 in human b-cells. PloS One (2017) 12:e0183268. doi: 10.1371/journal.pone.0183268
79. Yu X, Du Y, Cai C, Cai B, Zhu M, Xing C, et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat Commun (2018) 9:4964. doi: 10.1038/s41467-018-07384-7
80. Sorrentino R, Terlizzi M, Crescenzo VGD, Popolo A, Pecoraro M, Perillo G, et al. Human lung cancer–derived immunosuppressive plasmacytoid dendritic cells release IL-1α in an AIM2 inflammasome-dependent manner. Am J Pathol (2015) 185:3115–24. doi: 10.1016/j.ajpath.2015.07.009
81. Eichholz K, Bru T, Tran TTP, Fernandes P, Welles H, Mennechet FJD, et al. Immune-complexed adenovirus induce AIM2-mediated pyroptosis in human dendritic cells. PloS Pathog (2016) 12:e1005871. doi: 10.1371/journal.ppat.1005871
82. Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol (2010) 11:395–402. doi: 10.1038/ni.1864
83. Li H, Li Y, Song C, Hu Y, Dai M, Liu B, et al. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis via AIM2 inflammasome activation in LPS-induced ALI/ARDS. J Inflammation Res (2021) 14:4839–58. doi: 10.2147/JIR.S321513
84. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ (2013) 20:1149–60. doi: 10.1038/cdd.2013.37
85. Bakele M, Joos M, Burdi S, Allgaier N, Pöschel S, Fehrenbacher B, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem (2014) 289:5320–9. doi: 10.1074/jbc.M113.505636
86. Chauhan D, Demon D, Vande Walle L, Paerewijck O, Zecchin A, Bosseler L, et al. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep (2022) 23:e54277. doi: 10.15252/embr.202154277
87. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages and Th17 cells for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
88. Germic N, Frangez Z, Yousefi S, Simon H-U. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ (2019) 26:715–27. doi: 10.1038/s41418-019-0297-6
89. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6Chi monocytes controls their differentiation into iNOS+ macrophages or monocyte-derived dendritic cells. Immunity (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
90. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity (2013) 39:925–38. doi: 10.1016/j.immuni.2013.10.004
91. Verna G, Liso M, Cavalcanti E, Bianco G, Di Sarno V, Santino A, et al. Quercetin administration suppresses the cytokine storm in myeloid and plasmacytoid dendritic cells. Int J Mol Sci (2021) 22:8349. doi: 10.3390/ijms22158349
92. Wang A, Bai Y. Dendritic cells: The driver of psoriasis. J Dermatol (2020) 47:104–13. doi: 10.1111/1346-8138.15184
93. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
94. Oishi Y, Manabe I. Macrophages in inflammation, repair and regeneration. Int Immunol (2018) 30:511–28. doi: 10.1093/intimm/dxy054
95. Trachalaki A, Tsitoura E, Mastrodimou S, Invernizzi R, Vasarmidi E, Bibaki E, et al. Enhanced IL-1β release following NLRP3 and AIM2 inflammasome stimulation is linked to mtROS in airway macrophages in pulmonary fibrosis. Front Immunol (2021) 12:661811. doi: 10.3389/fimmu.2021.661811
96. Ngoungoure FP, Owona BA. Withaferin a modulates AIM2 inflammasome and caspase-1 expression in THP-1 polarized macrophages. Exp Cell Res (2019) 383:111564. doi: 10.1016/j.yexcr.2019.111564
97. Rodrigue-Gervais IG, Doiron K, Champagne C, Mayes L, Leiva-Torres GA, Vanié P, et al. The mitochondrial protease HtrA2 restricts the NLRP3 and AIM2 inflammasomes. Sci Rep (2018) 8:8446. doi: 10.1038/s41598-018-26603-1
98. Son S, Yoon S-H, Chae BJ, Hwang I, Shim D-W, Choe YH, et al. Neutrophils facilitate prolonged inflammasome response in the DAMP-rich inflammatory milieu. Front Immunol (2021) 12:746032. doi: 10.3389/fimmu.2021.746032
99. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe (2014) 15:526–36. doi: 10.1016/j.chom.2014.04.011
100. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature (2012) 488:670–4. doi: 10.1038/nature11290
101. Ahn H, Han B-C, Lee S-H, Lee G-S. Fructose-arginine, a non-saponin molecule of Korean red ginseng, attenuates AIM2 inflammasome activation. J Ginseng Res (2020) 44:808–14. doi: 10.1016/j.jgr.2020.06.002
102. Chung I-C, Yuan S-N, OuYang C-N, Hu S-I, Lin H-C, Huang K-Y, et al. EFLA 945 restricts AIM2 inflammasome activation by preventing DNA entry for psoriasis treatment. Cytokine (2020) 127:154951. doi: 10.1016/j.cyto.2019.154951
103. Liu T, Tang Q, Liu K, Xie W, Liu X, Wang H, et al. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep (2016) 16:1988–2002. doi: 10.1016/j.celrep.2016.07.019
104. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell (2017) 171:1057–1071.e11. doi: 10.1016/j.cell.2017.09.029
105. Tang H, Jin X, Li Y, Jiang H, Tang X, Yang X, et al. A large-scale screen for coding variants predisposing to psoriasis. Nat Genet (2014) 46:45–50. doi: 10.1038/ng.2827
106. Sun L-D, Cheng H, Wang Z-X, Zhang A-P, Wang P-G, Xu J-H, et al. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat Genet (2010) 42:1005–9. doi: 10.1038/ng.690
107. Zhang X-J, Huang W, Yang S, Sun L-D, Zhang F-Y, Zhu Q-X, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet (2009) 41:205–10. doi: 10.1038/ng.310
108. Tang L, Wang M, Shen C, Wen L, Li M, Wang D, et al. Assay for transposase-accessible chromatin using sequencing analysis reveals a widespread increase in chromatin accessibility in psoriasis. J Invest Dermatol (2021) 141:1745–53. doi: 10.1016/j.jid.2020.12.031
109. Wan D, Jiang W, Hao J. Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front Immunol (2020) 11:615. doi: 10.3389/fimmu.2020.00615
110. Pan Y, You Y, Sun L, Sui Q, Liu L, Yuan H, et al. The STING antagonist h-151 ameliorates psoriasis via suppression of STING/NF-κB-mediated inflammation. Br J Pharmacol (2021) 178:4907–22. doi: 10.1111/bph.15673
111. Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med (2017) 214:3611–26. doi: 10.1084/jem.20171749
112. Yu Y, Xue X, Tang W, Su L, Zhang L, Zhang Y. Cytosolic DNA−Mediated STING-dependent inflammation contributes to the progression of psoriasis. J Invest Dermatol (2022) 142:898–906.e4. doi: 10.1016/j.jid.2021.08.430
113. Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, et al. Inflammasome activation triggers caspase-1-Mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity (2017) 46:393–404. doi: 10.1016/j.immuni.2017.02.011
114. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin d restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity (2018) 49:413–426.e5. doi: 10.1016/j.immuni.2018.07.006
115. Corrales L, Woo S-R, Williams J, McWhirter SM, Dubensky TW, Gajewski TF. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J Immunol (2016) 196:3191–8. doi: 10.4049/jimmunol.1502538
116. Netea MG, Quintin J, van der Meer JWM. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
117. Netea MG, van der Meer JWM. Trained immunity: An ancient way of remembering. Cell Host Microbe (2017) 21:297–300. doi: 10.1016/j.chom.2017.02.003
118. Sun JC, Ugolini S, Vivier E. Immunological memory within the innate immune system. EMBO J (2014) 33:1295–303. doi: 10.1002/embj.201387651
119. Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, Netea MG. Trained immunity: Reprogramming innate immunity in health and disease. Annu Rev Immunol (2021) 39:667–93. doi: 10.1146/annurev-immunol-102119-073855
120. Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: Opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol (2014) 21:534–45. doi: 10.1128/CVI.00688-13
121. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
122. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
123. Rusek P, Wala M, Druszczyńska M, Fol M. Infectious agents as stimuli of trained innate immunity. Int J Mol Sci (2018) 19:E456. doi: 10.3390/ijms19020456
124. Schildberg FA, Donnenberg VS. Stromal cells in health and disease. Cytometry Part A (2018) 93:871–5. doi: 10.1002/cyto.a.23600
125. Viswanathan S, Shi Y, Galipeau J, Krampera M, Leblanc K, Martin I, et al. Mesenchymal stem versus stromal cells: International society for cell & gene therapy (ISCT®) mesenchymal stromal cell committee position statement on nomenclature. Cytotherapy (2019) 21:1019–24. doi: 10.1016/j.jcyt.2019.08.002
126. Tokoyoda K, Hauser AE, Nakayama T, Radbruch A. Organization of immunological memory by bone marrow stroma. Nat Rev Immunol (2010) 10:193–200. doi: 10.1038/nri2727
127. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol (2012) 12:383–96. doi: 10.1038/nri3209
128. Soliman H, Theret M, Scott W, Hill L, Underhill TM, Hinz B, et al. Multipotent stromal cells: One name, multiple identities. Cell Stem Cell (2021) 28:1690–707. doi: 10.1016/j.stem.2021.09.001
129. Saccu G, Menchise V, Gai C, Bertolin M, Ferrari S, Giordano C, et al. Bone marrow mesenchymal Stromal/Stem cell-derived extracellular vesicles promote corneal wound repair by regulating inflammation and angiogenesis. Cells (2022) 11:3892. doi: 10.3390/cells11233892
130. Lin T, Pajarinen J, Kohno Y, Huang J-F, Maruyama M, Romero-Lopez M, et al. Trained murine mesenchymal stem cells have anti-inflammatory effect on macrophages, but defective regulation on T-cell proliferation. FASEB J (2019) 33:4203–11. doi: 10.1096/fj.201801845R
131. Zaric SS, Coulter WA, Shelburne CE, Fulton CR, Zaric MS, Scott A, et al. Altered toll-like receptor 2-mediated endotoxin tolerance is related to diminished interferon beta production. J Biol Chem (2011) 286:29492–500. doi: 10.1074/jbc.M111.252791
132. Klein K, Frank-Bertoncelj M, Karouzakis E, Gay RE, Kolling C, Ciurea A, et al. The epigenetic architecture at gene promoters determines cell type-specific LPS tolerance. J Autoimmun (2017) 83:122–33. doi: 10.1016/j.jaut.2017.07.001
133. Li X, Fang P, Sun Y, Shao Y, Yang WY, Jiang X, et al. Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine-induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol (2020) 28:101373. doi: 10.1016/j.redox.2019.101373
134. Mai J, Virtue A, Shen J, Wang H, Yang X-F. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol (2013) 6:61. doi: 10.1186/1756-8722-6-61
135. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med (2008) 205:2409–17. doi: 10.1084/jem.20081188
136. Martinez-Gonzalez I, Mathä L, Steer CA, Takei F. Immunological memory of group 2 innate lymphoid cells. Trends Immunol (2017) 38:423–31. doi: 10.1016/j.it.2017.03.005
137. Martinez-Gonzalez I, Ghaedi M, Steer CA, Mathä L, Vivier E, Takei F. ILC2 memory: Recollection of previous activation. Immunol Rev (2018) 283:41–53. doi: 10.1111/imr.12643
138. Imai Y, Yasuda K, Nagai M, Kusakabe M, Kubo M, Nakanishi K, et al. IL-33-Induced atopic dermatitis-like inflammation in mice is mediated by group 2 innate lymphoid cells in concert with basophils. J Invest Dermatol (2019) 139:2185–2194.e3. doi: 10.1016/j.jid.2019.04.016
139. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science (2014) 345:1251086. doi: 10.1126/science.1251086
140. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
141. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LAB, Jacobs C, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun (2014) 6:152–8. doi: 10.1159/000355628
142. Smith SL, Kennedy PR, Stacey KB, Worboys JD, Yarwood A, Seo S, et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv (2020) 4:1388–406. doi: 10.1182/bloodadvances.2019000699
143. Gillard GO, Bivas-Benita M, Hovav A-H, Grandpre LE, Panas MW, Seaman MS, et al. Thy1+ NK [corrected] cells from vaccinia virus-primed mice confer protection against vaccinia virus challenge in the absence of adaptive lymphocytes. PloS Pathog (2011) 7:e1002141. doi: 10.1371/journal.ppat.1002141
144. Reeves RK, Li H, Jost S, Blass E, Li H, Schafer JL, et al. Antigen-specific NK cell memory in rhesus macaques. Nat Immunol (2015) 16:927–32. doi: 10.1038/ni.3227
145. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16:112–23. doi: 10.1038/nri.2015.9
146. Fenix K, Wijesundara DK, Cowin AJ, Grubor-Bauk B, Kopecki Z. Immunological memory in imiquimod-induced murine model of psoriasiform dermatitis. Int J Mol Sci (2020) 21:7228. doi: 10.3390/ijms21197228
147. Hartwig T, Pantelyushin S, Croxford AL, Kulig P, Becher B. Dermal IL-17-producing γδ T cells establish long-lived memory in the skin. Eur J Immunol (2015) 45:3022–33. doi: 10.1002/eji.201545883
148. Niec R, Rudensky AY, Fuchs E. Inflammatory adaptation in barrier tissues. Cell (2021) 184:3361–75. doi: 10.1016/j.cell.2021.05.036
149. Larsen SB, Cowley CJ, Sajjath SM, Barrows D, Yang Y, Carroll TS, et al. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell (2021) 28:1758–74.e8. doi: 10.1016/j.stem.2021.07.001
150. Teufel LU, Arts RJW, Netea MG, Dinarello CA, Joosten LAB. IL-1 family cytokines as drivers and inhibitors of trained immunity. Cytokine (2022) 150:155773. doi: 10.1016/j.cyto.2021.155773
151. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell (2018) 172:147–161.e12. doi: 10.1016/j.cell.2017.11.034
152. Tercan H, Riksen NP, Joosten LAB, Netea MG, Bekkering S. Trained immunity: Long-term adaptation in innate immune responses. Arterioscler Thromb Vasc Biol (2021) 41:55–61. doi: 10.1161/ATVBAHA.120.314212
153. van der Heijden CDCC, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and trained immunity. Antioxid Redox Signal (2018) 29:1023–40. doi: 10.1089/ars.2017.7310
154. Zhou F, Wang W, Shen C, Li H, Zuo X, Zheng X, et al. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol (2016) 136:779–87. doi: 10.1016/j.jid.2015.12.029
155. Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell (2015) 162:45–58. doi: 10.1016/j.cell.2015.06.001
156. Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med (2005) 11:43–9. doi: 10.1038/nm1162
157. Rebholz B, Haase I, Eckelt B, Paxian S, Flaig MJ, Ghoreschi K, et al. Crosstalk between keratinocytes and adaptive immune cells in an IkappaBalpha protein-mediated inflammatory disease of the skin. Immunity (2007) 27:296–307. doi: 10.1016/j.immuni.2007.05.024
158. Ogawa K, Okada Y. The current landscape of psoriasis genetics in 2020. J Dermatol Sci (2020) 99:2–8. doi: 10.1016/j.jdermsci.2020.05.008
159. Devos M, Mogilenko DA, Fleury S, Gilbert B, Becquart C, Quemener S, et al. Keratinocyte expression of A20/TNFAIP3 controls skin inflammation associated with atopic dermatitis and psoriasis. J Invest Dermatol (2019) 139:135–45. doi: 10.1016/j.jid.2018.06.191
160. Ippagunta SK, Gangwar R, Finkelstein D, Vogel P, Pelletier S, Gingras S, et al. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc Natl Acad Sci USA (2016) 113:E6162–71. doi: 10.1073/pnas.1606996113
161. Zhang H, Carnevale G, Polese B, Simard M, Thurairajah B, Khan N, et al. CD109 restrains activation of cutaneous IL-17-Producing γδ T cells by commensal microbiota. Cell Rep (2019) 29:391–405.e5. doi: 10.1016/j.celrep.2019.09.003
162. Li H, Yao Q, Mariscal AG, Wu X, Hülse J, Pedersen E, et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun (2018) 9:1420. doi: 10.1038/s41467-018-03704-z
163. Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of jun proteins. Nature (2005) 437:369–75. doi: 10.1038/nature03963
164. Matsumoto R, Dainichi T, Tsuchiya S, Nomura T, Kitoh A, Hayden MS, et al. Epithelial TRAF6 drives IL-17-mediated psoriatic inflammation. JCI Insight (2018) 3:121175. doi: 10.1172/jci.insight.121175
165. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity (2011) 35:596–610. doi: 10.1016/j.immuni.2011.08.001
166. Schulmann K, Brasch FE, Kunstmann E, Engel C, Pagenstecher C, Vogelsang H, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology (2005) 128:590–9. doi: 10.1053/j.gastro.2004.12.051
167. Feng X, Song Z, Huang Q, Jia J, Zhang L, Zhu M, et al. AIM2 promotes gastric cancer cell proliferation via the MAPK signaling pathway. J Healthc Eng (2022) 2022:8756844. doi: 10.1155/2022/8756844
168. Kim JE, Bang SH, Choi JH, Kim CD, Won CH, Lee MW, et al. Interaction of Wnt5a with Notch1 is critical for the pathogenesis of psoriasis. Ann Dermatol (2016) 28:45–54. doi: 10.5021/ad.2016.28.1.45
169. Zheng Q, Lei Y, Hui S, Tong M, Liang L. HDAC3 promotes pulmonary fibrosis by activating NOTCH1 and STAT1 signaling and up-regulating inflammasome components AIM2 and ASC. Cytokine (2022) 153:155842. doi: 10.1016/j.cyto.2022.155842
170. Zheng J, Liu C, Shi J, Wen K, Wang X. AIM2 inhibits the proliferation, invasion and migration, and promotes the apoptosis of osteosarcoma cells by inactivating the PI3K/AKT/mTOR signaling pathway. Mol Med Rep (2022) 25:53. doi: 10.3892/mmr.2021.12569
171. Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis by interfering with DNA-PK–dependent akt activation. Nat Med (2015) 21:906–13. doi: 10.1038/nm.3908
172. Gao J, Guo J, Nong Y, Mo W, Fang H, Mi J, et al. 18β-glycyrrhetinic acid induces human HaCaT keratinocytes apoptosis through ROS-mediated PI3K-akt signaling pathway and ameliorates IMQ-induced psoriasis-like skin lesions in mice. BMC Pharmacol Toxicol (2020) 21:41. doi: 10.1186/s40360-020-00419-0
173. Chung H, Komada T, Lau A, Chappellaz M, Platnich JM, de Koning HD, et al. AIM2 suppresses inflammation and epithelial cell proliferation during glomerulonephritis. J Immunol (2021) 207:2799–812. doi: 10.4049/jimmunol.2100483
174. Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
175. Sabat R, Wolk K, Loyal L, Döcke W-D, Ghoreschi K. T Cell pathology in skin inflammation. Semin Immunopathol (2019) 41:359–77. doi: 10.1007/s00281-019-00742-7
176. Zhang L, Li Y, Yang X, Wei J, Zhou S, Zhao Z, et al. Characterization of Th17 and FoxP3(+) treg cells in paediatric psoriasis patients. Scand J Immunol (2016) 83:174–80. doi: 10.1111/sji.12404
177. Zhang L, Yang X-Q, Cheng J, Hui R-S, Gao T-W. Increased Th17 cells are accompanied by FoxP3(+) treg cell accumulation and correlated with psoriasis disease severity. Clin Immunol (2010) 135:108–17. doi: 10.1016/j.clim.2009.11.008
178. Yan K, Xu W, Huang Y, Zhang Z, Huang Q, Xin KZ, et al. Methotrexate restores the function of peripheral blood regulatory T cells in psoriasis vulgaris via the CD73/AMPK/mTOR pathway. Br J Dermatol (2018) 179:896–905. doi: 10.1111/bjd.16560
179. Nussbaum L, Chen Y l., Ogg G s. Role of regulatory T cells in psoriasis pathogenesis and treatment. Br J Dermatol (2021) 184:14–24. doi: 10.1111/bjd.19380
180. Wu R, Li X, Li S, Tang G, Zhang S, Zhu Y, et al. Decreased microRNA-126 expression in psoriatic CD4+ T cells promotes T-helper 17 cell differentiation and the formation of dermatitis in imiquimod-induced psoriasis-like mice. J Dermatol (2022) 49:432–40. doi: 10.1111/1346-8138.16272
181. Wu R, Zeng J, Yuan J, Deng X, Huang Y, Chen L, et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest (2018) 128:2551–68. doi: 10.1172/JCI97426
182. Ma L, Xue H, Gao T, Gao M, Zhang Y. Notch1 signaling regulates the Th17/Treg immune imbalance in patients with psoriasis vulgaris. Mediators Inflamm (2018) 2018:3069521. doi: 10.1155/2018/3069521
183. Nadya NA, Tezuka H, Ohteki T, Matsuda S, Azuma M, Nagai S. PI3K-akt pathway enhances the differentiation of interleukin-27-induced type 1 regulatory T cells. Immunol (2017) 152:507–16. doi: 10.1111/imm.12789
184. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17:618–25. doi: 10.1038/ni.3466
185. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish t(reg)-cell function. Nature (2013) 499:485–90. doi: 10.1038/nature12297
186. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight (2017) 2:e89160. doi: 10.1172/jci.insight.89160
187. Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, et al. GARP dampens cancer immunity by sustaining function and accumulation of regulatory T cells in the colon. Cancer Res (2019) 79:1178–90. doi: 10.1158/0008-5472.CAN-18-2623
188. Lu F, Lan Z, Xin Z, He C, Guo Z, Xia X, et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J Cell Physiol (2020) 235:3207–21. doi: 10.1002/jcp.29268
189. Duan X, Liu X, Liu N, Huang Y, Jin Z, Zhang S, et al. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis (2020) 11:134. doi: 10.1038/s41419-020-2328-0
190. Honda T, Yamamoto O, Sawada Y, Egawa G, Kitoh A, Otsuka A, et al. Receptor-interacting protein kinase 3 controls keratinocyte activation in a necroptosis-independent manner and promotes psoriatic dermatitis in mice. J Allergy Clin Immunol (2017) 140:619–22.e6. doi: 10.1016/j.jaci.2017.02.027
191. Speir M, Nowell CJ, Chen AA, O’Donnell JA, Shamie IS, Lakin PR, et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat Immunol (2020) 21:54–64. doi: 10.1038/s41590-019-0550-7
192. Saito N, Honma M, Shibuya T, Iinuma S, Igawa S, Kishibe M, et al. RIPK1 downregulation in keratinocyte enhances TRAIL signaling in psoriasis. J Dermatol Sci (2018) 91:79–86. doi: 10.1016/j.jdermsci.2018.04.007
193. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature (2020) 580:391–5. doi: 10.1038/s41586-020-2129-8
194. Devos M, Tanghe G, Gilbert B, Dierick E, Verheirstraeten M, Nemegeer J, et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J Exp Med (2020) 217:e20191913. doi: 10.1084/jem.20191913
195. Lin J, Kumari S, Kim C, Van T-M, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature (2016) 540:124–8. doi: 10.1038/nature20558
196. Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti T-D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature (2021) 597:415–9. doi: 10.1038/s41586-021-03875-8
197. Kim J, Krueger JG. Highly effective new treatments for psoriasis target the IL-23/Type 17 T cell autoimmune axis. Annu Rev Med (2017) 68:255–69. doi: 10.1146/annurev-med-042915-103905
198. Rizvi S, Chaudhari K, Syed BA. The psoriasis drugs market. Nat Rev Drug Discov (2015) 14:745–6. doi: 10.1038/nrd4763
199. Soriano-Teruel PM, García-Laínez G, Marco-Salvador M, Pardo J, Arias M, DeFord C, et al. Identification of an ASC oligomerization inhibitor for the treatment of inflammatory diseases. Cell Death Dis (2021) 12:1155. doi: 10.1038/s41419-021-04420-1
200. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci USA (2018) 115:E6065–74. doi: 10.1073/pnas.1722041115
Keywords: psoriasis, AIM2, inflammasome, keratinocytes, epigenetic, trained immunity
Citation: Zhang Y, Xu X, Cheng H and Zhou F (2023) AIM2 and Psoriasis. Front. Immunol. 14:1085448. doi: 10.3389/fimmu.2023.1085448
Received: 31 October 2022; Accepted: 04 January 2023;
Published: 18 January 2023.
Edited by:
Rajendranath Ramasawmy, Universidade Nilton Lins, BrazilReviewed by:
Yue Xing, New York University, United StatesQian Yin, Florida State University, United States
Copyright © 2023 Zhang, Xu, Cheng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Cheng, YWhtdS5jaDgxM0AxNjMuY29t; Fusheng Zhou, YmlvemhvdWZzQDE2My5jb20=