 Shuang-Lan Chen1
Shuang-Lan Chen1 Chun-Meng Li1Wei Li1
Chun-Meng Li1Wei Li1 Qing-Song Liu1
Qing-Song Liu1 Shuang-Yuan Hu1Mao-Yuan Zhao1Dong-Sen Hu2
Shuang-Yuan Hu1Mao-Yuan Zhao1Dong-Sen Hu2 Yan-Wei Hao1Jin-Hao Zeng1,3*Yi Zhang1*
Yan-Wei Hao1Jin-Hao Zeng1,3*Yi Zhang1*- 1Department of Gastroenterology, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Department of Reproductive Medicine, Chengdu Xinan Women’s Hospital, Chengdu, China
- 3TCM Regulating Metabolic Diseases Key Laboratory of Sichuan Province, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
Inflammatory bowel disease (IBD) is a group of disorders that cause chronic inflammation in the intestines, with the primary types including ulcerative colitis and Crohn’s disease. The link between autophagy, a catabolic mechanism in which cells clear protein aggregates and damaged organelles, and intestinal health has been widely studied. Experimental animal studies and human clinical studies have revealed that autophagy is pivotal for intestinal homeostasis maintenance, gut ecology regulation and other aspects. However, few articles have summarized and discussed the pathways by which autophagy improves or exacerbates IBD. Here, we review how autophagy alleviates IBD through the specific genes (e.g., ATG16L1, IRGM, NOD2 and LRRK2), crosstalk of multiple phenotypes with autophagy (e.g., Interaction of autophagy with endoplasmic reticulum stress, intestinal antimicrobial defense and apoptosis) and autophagy-associated signaling pathways. Moreover, we briefly discuss the role of autophagy in colorectal cancer and current status of autophagy-based drug research for IBD. It should be emphasized that autophagy has cell-specific and environment-specific effects on the gut. One of the problems of IBD research is to understand how autophagy plays a role in intestinal tract under specific environmental factors. A better understanding of the mechanism of autophagy in the occurrence and progression of IBD will provide references for the development of therapeutic drugs and disease management for IBD in the future.
1 Introduction
The inflammatory bowel diseases (IBD) are described as complex, recurrent inflammatory conditions which are manifested as Crohn’s disease (CD) and ulcerative colitis (UC). The common clinical symptoms of IBD include severe diarrhea, abdominal pain, and weight loss, among others. If left untreated, the condition can lead to serious complications or the development of colorectal cancer (CRC). The pathogenesis of inflammatory bowel diseases (IBD) is multifactorial and complex, including an unfavorable environment, susceptibility gene variants, abnormal intestinal microbiota, and a mucosal immune and inflammatory response genetically susceptible to the host microbiota (1, 2), but its specific pathogenesis is still not fully elucidated.
Autophagy is a conserved intracellular degradation pathway that helps maintain intracellular homeostasis during stress or malnutrition. Autophagy is prevalent across cells and interacts with other essential cellular homeostatic processes. However, in the intestinal mucosa, cell-type-specific differences in autophagic function may exist (3). A growing number of studies suggest that autophagy may mediate the pathophysiological process of IBD. For example, autophagy regulates the clearance of invading pathogens. When bacteria infect host cells, cytoplasmic vesicles engulf the pathogens to form autophagic vesicles, thereby limiting the pathogens’ access to nutrients. Enhanced autophagy promotes the integration of autophagic vesicles and lysosomes for the degradation of pathogenic microorganisms (4–6). Autophagy maintains cell survival by protecting cells from bacterial toxins, such as intestinal epithelial cells (IECs) and macrophages (7). Autophagy also mediates the functions of innate and adaptive immunity, such as antigen delivery by dendritic cells, inflammatory factor secretion by macrophages, and antimicrobial peptide production by Paneth cells (8, 9). In contrast, autophagy inhibition leads to inflammation and increases the susceptibility to CD (10–12). In summary, impaired autophagy can cause intestinal cell dysfunction, dysbiosis of intestinal microbial and uncontrolled immune response, leading to intestinal inflammation (13–16).
Based on the important impact of autophagy on the gut, in this review, we will discuss how autophagy affects intestinal cells and gut function. We will also discuss how autophagy plays a role in the development of IBD via various pathways, including the genes, cell signaling pathways, ERS, and microorganisms involved. Meanwhile, a brief summary of the current research status of IBD therapeutic agents targeting autophagy is presented. These will further explain the pathogenesis of IBD and provide novel ideas for the development and use of drugs for IBD treatment in the future.
2 Autophagy and the underlying mechanism
In eukaryotic cells, autophagy is an important protein degradation system and a tightly regulated catabolic and organelle renewal process (17). In this process, the cells’ own cytoplasmic proteins and organelles are encapsulated into the vesicles. The vesicles then fuse with lysosomes to form autophagic lysosomes that degrade their encapsulated contents. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (18).
Macroautophagy is a process in which the endoplasmic reticulum, Golgi apparatus, or cytoplasmic membrane surround the material to be degraded into bimodal binding vesicles called autophagosomes. The autophagosome then fuse with lysosomes and degrade the contents. Microphagy implies that the lysosomal membrane directly wraps the long-lived proteins, among other components, and degrades them within the lysosome. CMA is a process of selective protein degradation through a lysosome-dependent mechanism. In this process, intracytoplasmic proteins bind to molecular chaperones and are translocated into the lysosomal compartment and subsequently digested by lysosomal enzymes (19).
Macroautophagy can be roughly divided into four stages (Figure 1): initiation of autophagy, formation of isolation membranes and autophagosomes, fusion of autophagosomes with lysosomes, and cleavage of autophagosomes. Autophagy is initiated by the unc-51-like autophagy-activated kinase 1 (ULK1) complex, which phosphorylates its downstream autophagy proteins FIP200 and autophagy-associated gene 13 (ATG13) (21–23). The transmembrane binding of ATG9 provides the double membrane for the formation of phagosomes, primarily from the endoplasmic reticulum (ER), through the Beclin 1 (BECN1)-phosphatidylinositol-3-kinase (PtdIns3K) complex to produce phosphatidylinositol-3-phosphate (PtdIns3P), which then recruits the WIPI complex (ATG2-ATG18 in yeast). This is followed by phagocytic membrane elongation (24, 25). With the formation of the ATG12 conjugation system (comprising the ATG5-ATG12-ATG16L1 complex, which requires ATG7 and ATG10 activity to form), microtubule-associated protein 1 light chain 3 alpha (MAP1LC3A/LC3) binds to phosphatidylethanolamine (PE) via ATG7 and ATG3, leading to the maturation of autophagosomes (26–28).
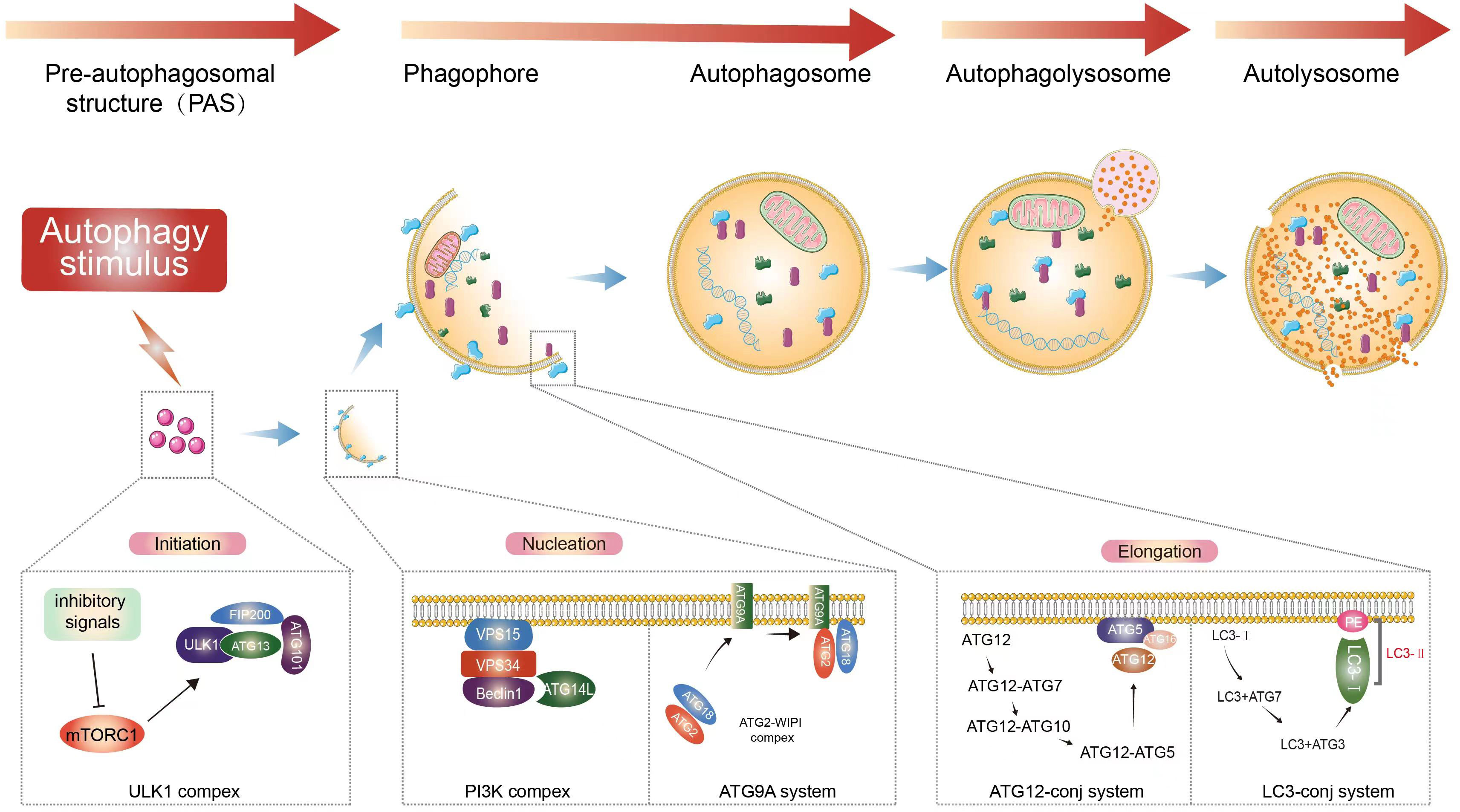
Figure 1 Autophagy process. Extracellular stimuli or recognition of substances that induce the formation of autophagosomes. Macroautophagy starts with inhibitory signals acting on mTOR (20). The ULK1 complex is an important upstream initiator that induces the activation of nucleation complexes such as PtdIns3K and BECN1, which in turn binds to autophagosomes and induces the prolongation of phagocytic membranes (21–23). Binding of LC3 to autophagosomes and control of autophagosome maturation and extension (24, 25). After vesicle formation, autophagosomes fuse with lysosomes to form autophagic lysosomes, releasing the contents (26–28). This is followed by the subsequent degradation of the contents occurs.
3 Autophagy is closely associated with IBD
3.1 Absence or mutations in specific genes of autophagy pathway interfere in IBD
At the genetic level, the autophagy-related 16-like 1 gene (ATG16L1) and immunity-related GTP-ase family M gene (IRGM) as risk factors for CD brought autophagy into focus in IBD (29–32). Subsequent studies have shown that mutations in the autophagy-related genes ULK1, leucine-rich repeat kinase 2 gene (LRRK2), and nucleotide-binding and oligomerisation domain 2 gene (NOD2) are strongly associated with the development of IBD (33–35).
3.1.1 ATG16L1
ATG16L1 is a particularly important autophagy gene. In a German study, a genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1 (29). However, studies in several Asian countries, including Japan, Korea and China, did not show an association between ATG16L1 gene variants and CD (36–38). In the analytical study of correlation with phenotype, ATG16L1 mutations were associated with intestinal luminal stricture and perianal infiltration, and the number of risk alleles was positively associated with the risk and severity of CD development (39).
Cysteine aspartate proteases (caspase) are a group of proteases present in the cytoplasm that are closely related to cell growth, differentiation and especially apoptosis. Thr 300-to-Ala (T300A) polymorphism in ATG16L1 has been found to be a serve as a critical susceptibility factor for CD. It can promote caspase 3-dependent degradation during cellular stress, leading to protein instability (40). In a proapoptotic context, cleaved forms of caspase 3 are retransplanted to degrade BECN1, which hinders the initiation of autophagy (41). ATG16L1T300A expression is associated with an increased risk of CD in response to the reduction of bacterial capture rates and impaired bacterial processing through autophagy. The ATG16L1 coding variant was found to be impaired in the capture of internalized Salmonella in autophagosomes in human IECs (42). Interleukin 22 (IL-22) directly acts on IECs and contributes to the intestinal immune response to pathogen infection (43, 44) and epithelial wound healing (45). However, under certain conditions, IL-22 may cause tissue damage (46). A recent study showed that IL-22 signaling in IEC is regulated by gene ATG16L1 and drives intestinal inflammation in an autophagy-dependent manner (47). ATG16L1 regulation of autophagy is also cell-specific. For example, under inflammatory conditions, ATG16L1 deletion in myeloid cells exerts limited effect on IBD, whereas ATG16L1 deletion in ICE will increases IEC apoptosis, aggravates chronic inflammation. However, IECs can be maintained without autophagy as long as intestinal homeostasis is maintained (48).
3.1.2 IRGM
IRGM is thought to be a shared susceptibility gene for both CD and UC (49). However, it plays a more prominent role in the pathogenesis of CD, and its role in UC seems to be controversial. Because, data from several studies showed a strong association between IRGM variants and CD susceptibility (31, 50, 51), while there was no significant association between susceptibility to UC (50, 52, 53). Further, in a study of the effect of IRGM variants on IBD susceptibility, it was shown that the association signal of IRGM with CD was considerably weaker compared to ATG16L1 (50, 54).
IRGM exerts resistance to intracellular infection through various mechanisms, including regulation of phagosome processing, cell motility, and autophagy. Protein IRGM has no homologs among the ATG genes in yeast, which makes it difficult to assign to it an autophagy-specific function; instead, IRGM has been considered to affect autophagy indirectly (55, 56). IRGM interacts with ULK1 and BECN1 and promotes their co-assembly, thereby regulating the formation of the autophagy initiation complex. Meanwhile, it physically pairs with autophagy protein NOD2 and ATG16L1 to exert antibacterial and anti-inflammatory effects (57). IRGM polymorphisms (rs13361189, rs4958847, and rs11741861) are considered to increase the susceptibility to CD and UC (51, 58). IRGM1 expression deficiency significantly increases the susceptibility to chemically-induced colitis in part by improving the accessibility of commensal bacteria to intestinal tissues (59, 60). For example, altered IRGM expression are predicted to be associated with CD, affecting the autophagic control of Salmonella typhimurium (32). The loss of tight IRGM expression regulation disrupts replication in CD-associated adherent-invasive Escherichia coli controlled through autophagy (61).
3.1.3 NOD2
Genetic polymorphism in NOD2 was the first found to be associated with the risk of CD development and confers the strongest genetic risk (62–64). Around 40–50% of CD patients carry at least one mutation in the NOD2/CARD15 gene (65, 66). However it has heterogeneity and many studies have shown that NOD2 variants play no role in CD in East Asian populations (67–69). In a systematic review and meta-analysis on the association between identified NOD2 mutations and the prognosis of complex CD, it was shown that every NOD2 mutation was associated with a 58% increased risk of surgery (70)
The most common IBD-associated polymorphisms in NOD2 are the amino acid mutations R702W, G908R, and L1007fs. These mutations occur in the C-terminal leucine-rich repeat structural domain responsible for detecting cytoplasmic MDP and have been shown to cause defects in the perception of this bacterial-derived molecule (71, 72). Protein NOD2 directly intersects with autophagy and induces protein ATG16L1 to recruit bacteria to the entry site, where it triggers bacterial autophagy (73, 74). In CD-associated NOD2 shift-mutant pure cells, mutant NOD2 fails to recruit ATG16L1 to the plasma membrane, thereby limiting the autophagic response triggered by intracellular bacterial infection (73). The defective autophagic clearance of such NOD2-associated intracellular bacteria, such as S. typhimurium, adherent invasive E. coli, and Shigella fowleri, can be reversed by the pharmacological induction of autophagy with the autophagy activator rapamycin (73–75). In addition, CD4+ T cell-dependent immunity to bacterial antigens requires effective antigen presentation for the activation of NOD2-mediated autophagy in dendritic cells (74).
3.1.4 LRRK2
Initially, gene LRRK2 mutations were investigated in association with the onset of Parkinson’s. Subsequent studies revealed that functional LRRK2 variants also increase the genetic risk of CD (35, 76). LRRK2 protein kinase is a complex enzyme with kinase and guanosine triphosphatase activities. LRRK2 missense mutations cause overactive LRRK2 protein kinase. The inhibition of LRRK2 protein kinase activity was shown to lead to autophagy stimulation in an atypical manner, a pathway that is independent of the mammalian target of rapamycin (mTOR) and ULK1 but dependent on the activation of BECN1-containing class III PI3 kinases (77). LRRK2-deficient mice exhibit high susceptibility to experimental colitis through modulation of the innate immune response (78). In contrast, recent studies have shown that upregulation of LRRK2 expression promotes intestinal inflammatory responses (79–81). Because LRRK2 protein kinase is able to bind to K48 and induce its ubiquitination, and it also induces degradation of the autophagy initiator Beclin-1 and phosphorylation of Beclin-1 (81), which is a Beclin-1 inactivation mechanism (82). However, this only indicates the susceptibility to intestinal inflammation after exposure to inflammatory stress (81).
3.2 Crosstalk of multiple phenotypes with autophagy is involved in IBD
3.2.1 Endoplasmic reticulum stress interacts with autophagy and is associated with IBD
Endoplasmic reticulum stress (ERS) is the response that activates signaling pathways such as the unfolded protein response (UPR), endoplasmic reticulum overload response, and the caspase-12-mediated apoptotic pathway. These responses can respond to misfolded and unfolded protein aggregation in the lumen of the endoplasmic reticulum and disturbed calcium ion homeostasis. UPR has been found to be initiated by three ER transmembrane sensors, including inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK) and activation of transcription factor 6 (ATF6). Some of the mediators released by ERS can directly induce autophagic vesicle formation and initiate autophagy (83). Findings from recent studies have also confirmed that ERS induces autophagy through multiple pathways, such as the UPR pathway and Akt signaling (84). The induction of autophagy and suppression of inflammation occurs through the inhibition of the PI3K/Akt/mTOR pathway (85, 86).
In the course of IBD, on one hand, ERS regulates autophagy. ER degradation is associated with increased ATF6 activity in the colonic mucosa of patients with UC (87). ERS activates autophagy through the ATF6 -mediated upregulation of death-associated protein kinase 1 (DAPK1) and mediates mAtg9 translocation (88). In IECs this process enhances autophagy for killing bacteria (89). ATF6α acts as an intermediate signaling molecule that regulates its upstream signaling factors and downstream molecules XBP1 and Atg16L1. It is involved in the interaction between ERS and IECs autophagy in IBD (90). In addition, UPR and autophagy also intersect in the PERK-EIF2α-ATF4 pathway (87, 91, 92), where ATF4 induces the expression of the pro-apoptotic cytokine CHOP and increases the transcription of the autophagy gene ATG5 (93). It also induces the expression of DNA damage response 1 (REDD1) during ERS (94), and REDD1 expression in intestinal neutrophils activates autophagy by inhibiting mTOR phosphorylation, which is closely related to the severity of UC (95). Finally, ERS stimulates the IRE1/TRAF2/apoptosis signaling regulatory kinase-1/JNK and IRE1/XBP1 signaling axes to release Beclin1 to increase autophagy (96, 97). On the other hand, autophagy regulates ERS, as evidenced by the fact that the impairment of autophagy promotes endoplasmic reticulum stress. The inhibition of trigger receptor-1 expression on myeloid cells reduces ERS in mice with colitis by restoring macroautophagy and CMA (98). In mice with inactivated IKKα kinase, loss of IKKα function resulted in the reduced stability of ATG16L1, which induced UPR and significantly impaired intestinal epithelial regeneration in mice with acute colitis model (99). Similarly, Atg16L1 knockout mice exhibit more severe colonic inflammation owing to deficient autophagy, resulting in the insufficient degradation of ER to nucleus signaling 1, allowing its excessive accumulation and activation (100).
Appropriate ERS can maintain homeostasis in the intestine, but excess ERS can induce IEC apoptosis and intestinal mucosal barrier dysfunction, and inducing pro-inflammatory cytokine production. Autophagy can also alleviate IBD in several ways; ERS can activate autophagy, but excessive autophagy exacerbates apoptosis and promotes the development of IBD. Therefore, it will be meaningful to explore how the interaction between ERS and autophagy influences the development of IBD, which will help to further explore the pathogenesis of IBD.
3.2.2 Intestinal antimicrobial defense interacts with autophagy and is associated with IBD
Antimicrobial defense is known to be closely related to the expression of autophagy genes (101, 102). Intracellular pathogens attempt to disrupt cell membranes to establish replication niches, cross cell membranes into different compartments, or simply avoid degradation. Autophagy and related pathways can limit such processes to complete the life cycle (103). For extracellular pathogens, autophagy limits infection-induced damage by supporting the activity of specific cell types or inhibiting the production of pro-inflammatory cytokines (103). A network to elucidate the interaction between mucosal bacteria and host autophagic signaling through human intestinal mucosal biopsies showed that patients with CD exhibited greater autophagy and associated signaling cascades than patients with UC. Patients with UC exhibited more severe dysbiosis and a functional phenotype of intestinal mucosal colonizing bacteria (104, 105). Moreover, the populations of the dominant and low-abundance bacteria were positively and negatively correlated with the expression of host autophagy genes, respectively (104). Gene ATG7 conditional knockout mice of IEC exhibited more abundant bacterial invasion and a significant decline in antibacterial or antiparasitic peptide expression in the colonic epithelium (106). Similarly, ATG5-deficient mice showed a decrease in inflammation-controlling bacteria and an increase in the number of pro-inflammatory bacteria (107).
An increased amount of IgA-coated bacteria, which were confirmed as IBD-promoting microbes (108), in the feces of mice with myeloid cell-specific Atg16l1 deficiency and in CD patients carrying the ATG16L1T300A variant (109). The interactive effects of altered ATG16L1 expression on bacteria in IECs and macrophages are widely recognized. Mice with dendritic cell ATG16L1 deletion showed increased susceptibility to infection in DSS-induced colitis (110) but not to S. typhimurium (111). However, ATG16L1 regulates IECs autophagy and is necessary for the clearance and control of S. typhimurium transmission in studies of autophagy gene-specific deletion in IECs (111, 112). Also, CD-associated adherent-invasive E. coli persist in autophagy-deficient IECs. However, it has been shown that autophagy deficiency in ATG16L1 hypomorphic mice alleviates bacterial burden and protects the mouse intestine from severe inflammatory damage (113, 114). Some intestinal bacteria deliver immunomodulatory molecules to immune cells through secretory outer membrane vesicles. Chu and his colleagues (115) showed that outer membrane vesicles require expression of the IBD-related genes ATG16L1 and NOD2 to activate the atypical autophagic pathway. However only in Bacteroides fragilis infection, dendritic cells lacking ATG16L1 fail to induce regulatory T cells to exert a suppressive effect on mucosal inflammation. This also describes genetically how the interaction between autophagy and microbes synergistically promotes beneficial immune responses.
In addition, autophagy can also affect intestinal antimicrobial defense by influencing the formation of secretory granules. The alternative autophagy-based secretion pathway in the atypical secretion of lysozyme is known as secretory autophagy (116). The disruption of secretory autophagy in mouse Paneth cells results in the increased risk of CD in humans when ATG16L1 is mutated (116). In summary, autophagy plays an overall positive role in intestinal bacterial defense, but it has a high degree of cell-type specificity and infection-type specificity in their interactions with the intestinal microbiota
Conversely, host cell autophagy was also influenced by the composition of the intestinal microbiota; e.g., fecal microbiota transplantation increased levels of autophagy-related proteins in the intestinal mucosa of piglets (117). Likewise, bacterial metabolites exert a critical effects on energy homeostasis and autophagy in IECs. For example, colonic cells in germ-free mice are in a state of energy deprivation. In this state, colonic IEC autophagy is enhanced, and when butyrate is added to germ-free colonic cells, it compensates for the deficiency in mitochondrial respiration and limits autophagy (118).
3.2.3 Autophagy-regulated apoptosis is associated with intestinal barrier function in IBD
The strict regulation of autophagy and apoptosis plays a critical role in maintaining tissue homeostasis. Autophagy maintains cell survival in the presence of multiple cellular stressors by degrading long-lived proteins and damaged organelles, while apoptosis eliminates damaged and mutated cells. Autophagy has been shown to exert an inhibitory effect on apoptosis in liver disease (119) and neurological disorders (120). Pott and his colleagues (48) found that mice with IBD selectively deficient in ATG16L1 in IECs showed increased TNF-induced apoptosis, which exacerbated intestinal pathology. However myeloid ATG16L1 deficiency a exerted limited effect on disease. Autophagy protein ATG16L1 in the intestinal epithelium is critical for preventing Paneth cell loss and excessive cell death in an animal model of virus-induced IBD (121). The above findings suggest that the IEC autophagic process can control inflammation-induced apoptosis to maintain intestinal barrier integrity, thereby limiting chronic intestinal inflammation. Alpinetin was shown to enhance intestinal barrier function by driving autophagy to inhibit IEC apoptosis (122). The intracellular protein High Mobility Group Box 1 regulates apoptosis and attenuates inflammation-associated cell injury by protecting the autophagy proteins BECN1 and ATG5 from calpain-mediated cleavage during inflammation (123).
3.3 Autophagy-related signaling pathways are associated with intestinal homeostasis in IBD
3.3.1 mTOR
mTOR is an atypical serine/threonine kinase that interacts with specific junction proteins to form two different macromolecular complexes, mTORC1 and mTORC2. mTORC2 primarily controls the cytoskeleton and motility, whereas mTORC1 primarily controls important cellular processes, including autophagy. Autophagy is negatively regulated by upstream mTOR, and the pharmacological effects of mTOR inhibitors and autophagy stimulators significantly improve experimental colitis and oxidative stress in vivo (124).
3.3.1.1 AMPK/mTOR signaling pathways
Adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK), a key molecule in the regulation of biological energy metabolism, is an AMP-dependent protein kinase. Sodium hydrosulfide (NaHS) activates hepatic autophagy through the AMPK/mTOR pathway and improves non-alcoholic fatty liver disease (125). In colonic tissues, NaHS restored impaired cellular autophagy caused by lipopolysaccharide (LPS) stimulation in rats in vitro and in vivo while improving the signs of UC (126). Xiong and his colleagues (127) found that Sinensetin inhibits apoptosis and alleviates intestinal barrier dysfunction in colitis by promoting autophagy of epithelial cells, but this effect could be reversed by AMPK knockdown. Further, data from Arab (128) showed that dapagliflozin activates the AMPK/mTOR pathway by increasing pAMPK (Thr172)/total AMPK levels and decreasing the p-mTOR (Ser2448)/total mTOR ratio. This is consistent with the action of metformin (an activator of the autophagy machinery) through an AMPK-dependent signaling pathway for limiting dextran sulfate sodium (DSS)-induced damage to the intestinal barrier (129). Similarly, nicotine ameliorates the severity of DSS-induced colitis in a mouse model of UC by modulating AMPK/mTOR pathway-mediated autophagy and regulating apoptosis and proliferation (130).
3.3.1.2 mTOR/NLRP3 signaling pathways
NOD-like receptor thermal protein domain associated protein 3 (NLRP3), a component of inflammatory vesicles, is responsible for the activation of caspase-1 and subsequent pro-IL-1β and pro-IL-18 maturation. It has long been shown that single-nucleotide polymorphisms in the NLRP3 gene are associated with susceptibility to CD (131). There is a reciprocal regulatory relationship between NLRP3 expression and autophagy, with activation of autophagy limiting NLRP3 activity and thereby moderating inflammation (132).
Jesus (133) showed the following: 1) NLRP3 is a novel binding partner for mTOR; 2) NLRP3 plays a key role in the structural blockade of autophagy by promoting mTOR phosphorylation; 3) hypoxia attenuates the colonic inflammatory response by downregulating the binding of mTOR and NLRP3 and activating autophagy. A study by Mai et al. demonstrated that palmatine ameliorated intestinal inflammation by reducing mitochondrial reactive oxygen species (mtROS) production and inhibiting NLRP3 inflammatory vesicles through mitochondrial autophagy (134). Thus, promoting mitochondrial autophagy may be a method to terminate the hyperinflammatory response by inactivating NLRP3 inflammatory vesicles. Andrographolide prevents inflammation-associated colon cancer through the mitochondrial autophagy-mediated inhibition of NLRP3 inflammatory vesicles (135). Similarly, the inhibition of NRLP3 inflammatory vesicles may be an effective mechanism to promote autophagy activation to improve colitis. Ginsenosides, which are anti-inflammatory molecules, were reported to inactivate NLRP3 inflammatory vesicles, thereby inducing mitochondrial autophagy to improve DSS-induced experimental colitis (136).
3.3.1.3 AKT/mTOR signaling pathways
AKT, also known as protein kinase B, is a serine/threonine-specific protein kinase that plays a key role in various cell growth processes. The activation of AKT/mTOR signaling pathway negatively regulates autophagy, and AKT/mTOR signaling participates in pathogenesis of IBD through the regulation of autophagy (137, 138). For example, heat shock transcription factor 2, which promotes autophagy in IECs by inhibiting the PI3K/AKT/mTOR signaling pathway, plays a protective role in UC (139). The overexpression of ring finger protein 8 reduces AKT and mTOR phosphorylation, increases autophagy, and improves intestinal inflammation in UC mice (140). Xianglian pill was also shown to promotes cellular autophagy and attenuates DSS-induced acute colitis in mice by blocking the activation of the PI3K/Akt/mTOR signaling pathway (141).
3.3.2 NF-κB signaling pathways
NF-κB is a transcription factor that activates the transcription of pro-inflammatory factor genes and plays a critical role in the cellular inflammatory response and immune response. The atypical stimulation of NF-κB upregulates of the expression of two activators of selective autophagy, BAG3 and HspB8, thereby regulating the autophagic process (142). Neural-precursor-cell-expressed developmentally down-regulated 4 is a ubiquitin ligase that regulates Beclin-1 stability. NF-κB causes Beclin-1 division to inhibit autophagy through the upregulation of Neural-precursor-cell-expressed developmentally down-regulated 4 (143). Similarly, autophagy inhibits NF-κB activation by inhibiting the NF-κB upstream regulator IκB kinase (IKK) (144) or by recruiting phosphorylated IKK to the autophagic vesicle compartment (145). In addition, ATG16L1 negatively regulates the pro-inflammatory cytokine response through the downregulation of NF-κB expression (146). Autophagy deficiency owing to ATG16L1T300A polymorphism in macrophages promotes the NF-κB-dependent cytokine response and puts CD at an increased risk of disease (147). Ginseng polysaccharides exert beneficial effects, such as regulating intestinal microbiota, protecting the intestinal mucosal barrier, and promoting autophagy. Toll-like receptors (TLR) are key receptors for pathogen recognition and immune activation and are widely present in various immune cells and epithelial cells. Ginseng polysaccharides were demonstrated to improve DSS-induced intestinal inflammation by inhibiting the TLR4-NF-kB pathway and activating mTOR-dependent autophagy (148).
3.3.3 Nrf2/OH-1 signaling pathways
Nuclear transcription factor E2-related factor 2 (Nrf2) is a key transcription factor in the cellular antioxidant response. The pathway involving heme oxygenase-1 (HO-1) is a key signaling pathway that regulates endogenous oxidative stress. Reportedly, p62 SQSTM1 is a target gene of Nrf2 (149), and the Keap1/Nrf2 pathway is strongly associated with autophagy through the autophagic adapter p62 SQSTM1 protein (150). The activation of the Nrf2/HO-1 pathway effectively controlled colonic tissue damage in a trinitro-benzene-sulfonic acid (TNBS)- and DSS-induced colitis model (151, 152). Arab (128) showed that dapagliflozin activates autophagy at least in part by activating the Nrf2/OH-1 pathway to reduce the severity of drug-induced colitis in rats.
3.3.4 Other signaling pathways
The hypoxia-inducible factor-l-dependent induction of Wnt1 disrupts autophagy in epithelial cells, suggesting a role of the Wnt signaling pathway in impaired autophagy in the mucosal epithelial cells of patients with IBD (153). Signal sensors and activator of transcription 3 (STAT3) is a key molecular pathway that can be activated by various pathogens and participates in complex immune disorders (154, 155). Zhang (156) showed that the deletion of High Mobility Group Box 1 protein in the intestine leads to the abnormal regulation of STAT3, which inhibits autophagy. The author also showed that the inhibition of STAT3 restores the autophagic response in bacteria-infected cells, suggesting that STAT3 activation should occur upstream of autophagy regulation. In in vivo and in vitro models of neonatal necrotizing small bowel colitis, TNF-α may induce autophagy through the extracellular signal regulated kinase 1/2 pathway, regulate the apoptosis of neonatal necrotizing small bowel colitis cells IEC-6, and promote disease progression (157). In ATG5-silenced IECs, the MAPK/ERK kinase pathway is activated, which is associated with elevated levels of inflammatory cytokines (158).
4 The role of autophagy in CRC
IBD is at risk of developing into CRC. Based on the key role of autophagy in IBD, the role of autophagy in CRC has also gained attention in recent studies. Autophagy is now emerging as a potent regulator of tumorigenesis by protecting cells from metabolic stress and oxidative damage. However, recent data show that autophagy may promote tumour growth and progression in some conditions (159, 160). Thus, the role of autophagy in tumorigenesis is equally complex.
The role of UVRAG as a tumor suppressor gene has been described among other autophagy-related genes (161). UVRAG regulates BECN1 expression, suggesting that the interaction between UVRAG and BECN1 may be a condition for the tumor suppressive effect. In colon cancer cell lines carrying UVRAG (c.709delA) deletion, reduced UVRAG levels were observed to correlate with impaired autophagy induction (161, 162). Nonsynonymous single-nucleotide polymorphism in ATG16L1T300A was associated with improved overall survival and increased basal production of type I interferon in human CRC (163). In addition, a study analyzing gene ATG5 mutation and expression in gastrointestinal tumors showed that ATG5 protein was well expressed in normal colonic mucosal epithelial cells but was absent in 23% of CRC (164).
In addition to its direct anti-tumor effects, autophagy inhibits CRC by weakening the inflammatory response in the tumor microenvironment and increase the processing and presentation of tumor-associated antigens. This improves anti-tumor immunity. Some chemotherapeutic agents with immunogenic properties have been shown to exhibit immunogenic anti-tumor properties by inducing autophagic cell death (165). Bacillus Calmette - Guerin has been shown to induce autophagic cell death through TLR2 and TLR4 signaling and to radiosensitize colorectal cell lines. In vivo experiments have further demonstrated that Bacillus Calmette - Guerin -mediated radiosensitization is an autophagy-dependent phenomenon (166). In contrast, preoperative hydroxychloroquine treatment improved the therapeutic response to 5-fluorouracil and radiotherapy in patients with advanced CRC. However, chloroquine can inhibit autophagy by blocking the fusion of autophagosomes and lysosomes, sensitizing HT-29 CRC cells to chemotherapy and irradiation (167). In addition, when autophagy genes (e.g., ATG5 or BECN1) were knocked out, cell death and ROS production were enhanced by the oxaliplatin treatment of Caco-2 cells (168, 169). Thus, during CRC, autophagy can both promote tumor survival and lead to tumor cell death, depending on tumor type, CRC stage, and metabolic environment.
5 Pharmacological studies on autophagy in the treatment of IBD
In terms of drug research, the therapeutic targets of IBD are primarily elements related to the maintenance of homeostasis in the intestine, and autophagy has emerged as a new potential therapeutic option for it. Wang and his collaborators (170) then assessed the therapeutic effects of 37 Food And Drug Administration -approved autophagy activators using an embryonic anthracycline-induced cardiotoxicity zebrafish model. They identified spironolactone, pravastatin, and minoxidil as top-ranking drugs for reversing the decline in cardiac function in anthracycline-induced cardiotoxicity and proved that spironolactone and rapamycin activated autophagy in an ATG7-dependent fashion (170). In intestinal diseases, glutamine has been widely used for damage repair in the intestinal mucosa, and reportedly, glutamine can promote IEC autophagy through the mTOR and p38 MAPK signaling pathways, thereby inhibiting stress-induced apoptosis (171). Anakinra, a human IL-1 receptor antagonist produced by genetic recombination technology, has been used in rheumatoid arthritis. Studies have demonstrated that anakinra can restore autophagy levels and reduce il-1-mediated inflammatory responses in patients with chronic granulomatosis, thereby protecting against TNBS-induced colitis, reducing the severity of chronic granulomatosis and promoting recovery from rectal abscesses (15). Sirolimus and everolimus are two rapamycin analogs that have been used clinically. In a retrospective study of refractory IBD that did not respond to conventional treatment in children, 45% of patients with UC and 100% of patients with CD showed clinical remission with sirolimus (172). Meanwhile, treatment with everolimus (an autophagy inducer) also improved CD-like ileitis in IL-10-deficient mice (173). However, whether the rapamycin analogs in the above cases act through autophagy and possibly immunosuppression remains unclear. Furthermore, it is interesting to note that epidemiological studies have shown that smoking is beneficial in UC (174) that the ameliorative effect of nicotine on intestinal inflammation may contribute to this (175–177). Gao and his colleagues (130) confirmed that nicotine ameliorates the severity of DSS-induced colitis in a mouse model of UC by regulating AMPK/mTOR pathway-mediated autophagy and inhibiting apoptosis and proliferation.
However, few of the current abundant of activators or inhibitors have strictly selected one autophagic pathway and one target. Therefore, some of the research has turned towards identifying small m Chu and his colleagues olecules and peptides to precisely modulate autophagy in pathological processes. For example, P140 is a peptide that selectively targets the autophagic process, particularly CMA. P140 effectively affects the processing of endogenous antigens as well as downstream deleterious pro-inflammatory events by interfering with the over-activated autophagic process (178). Importantly, the normal immune system is not affected by this (179) and P140 has been shown to be safe in clinical trials involving patients with systemic lupus erythematosus (180, 181). Preliminary data from current studies also favor P140 as a valuable tool for the treatment of IBDs (182). Spermidine is a natural polyamine that has been shown to improve the weight loss and colonic damage seen in mice with DSS-induced IBD (183). Liu’s study suggests that the improvement of IBD by spermidine is partly due to its activation of macrophage autophagy, allowing it to acquire anti-inflammatory properties (184).
Similarly, some herbal extracts or active ingredients have been shown to ameliorate intestinal inflammation by inducing autophagy; e.g., andrographolide inhibits NLRP3 inflammatory vesicles by enhancing autophagy (135), and tretinoin enhances autophagy by inhibiting the PI3K/AKT/mTOR signaling pathway (138).
Autophagy, owing to its fundamental role in maintaining homeostasis in the intestine, can be an attractive therapeutic target. Although some progress has been made in the study of drugs that induce autophagy through different mechanisms, the treatment of IBD by autophagy modulation is still in its preliminary stages. It is challenging because of the low pharmacological specificity of its targets, the lack of specificity for specific cell classes, and the autophagy-independent effects. Therefore, extensive experimental studies and large-scale clinical trials are necessary to explore and confirm the results.
6 Discussion
Autophagy affects the intestine via multi-pathway and cell-specific and environment-specific mechanisms. The microbial-autophagy-IBD interaction has been described previously, but the diet-autophagy-IBD interaction is poorly understood. Currently, the effects of vitamin D in diet-autophagy-IBD interaction have been studied. The deficiency of this vitamin has been recognized as an important pathological basis for IBD (185). The vitamin D receptor enhances autophagy through Beclin-1 (186), reduces necroptosis (187), and promotes the release of the tight junction protein Claudin-2 to slow intestinal inflammation (188). In addition, anthocyanins belong to a subclass of dietary flavonoids (polyphenolic compounds) that are important in preventing the onset and progression of intestinal inflammation (189) and can accelerate autophagy by regulating the PI3K/AKT/mTOR signaling pathway (190). Haihua and his colleagues further demonstrated that proanthocyanidins may alleviate DSS-induced UC by inducing autophagy (191). Therefore, there are many gaps in our understanding of the diet-autophagy-IBD relationship. These gaps need to be filled with evidence from more experimental and clinical studies.
To date, the number of studies on microautophagy and CMA is insufficient in IBD, and it is yet to be confirmed whether autophagy governs the age and gender in IBD onset. In genetics, the development and application of genetic testing will be particularly important in predicting the occurrence, type, and prognosis of IBD as well as the guidance and utility of drugs, especially for patients with refractory IBD. However, these aspects are still in the preliminary stage of research.
Overall, the current findings indicate that the absence of autophagy favors the exacerbation rather than the induction of intestinal inflammation. However, the inhibition of autophagy can ameliorate intestinal inflammation under certain conditions, such as after appendectomy (192). Therefore, a generalized review of the specific pathways involved in autophagic mechanisms of action in IBD is necessary to further explore the interactions between the environment, infection, and genetics. This will help us to understand how and by what mechanisms autophagy affects the gut and interacts with other cellular processes under specific conditions. These findings may be useful in developing individualized therapeutic regimens.
7 Conclusion
Great strides have been made in our understanding of autophagy within intestinal cells both in a healthy and diseased context. A growing number of autophagic genes and proteins have been shown to be closely associated with intestinal inflammatory processes. Autophagy genes or proteins reduce susceptibility to intestinal injury and maintain intestinal homeostasis by providing endoplasmic reticulum stress protection, phagocytosing pathogen invasion, reducing pathogen attachment, and regulating apoptosis (Figure 2). In the study of autophagy-related signaling pathways and IBD, autophagy protects the intestinal mucosal barrier mainly by regulating cytokines and modulating apoptosis (Figure 2). In addition, as the anti-tumor effects of some chemotherapeutic drugs have been found to be associated with autophagy, the application of autophagy in cancer has received increasing attention. A better understanding of the molecular mechanisms of crosstalk between apoptosis and autophagy may be the key in identifying novel applications of combinatorial treatment to CRC. In pharmacological studies, although some autophagy inducers applied in the clinic have been found to be effective in certain diseases, the studies are still at preclinical stage, and more evidence is needed based on clinical tests.
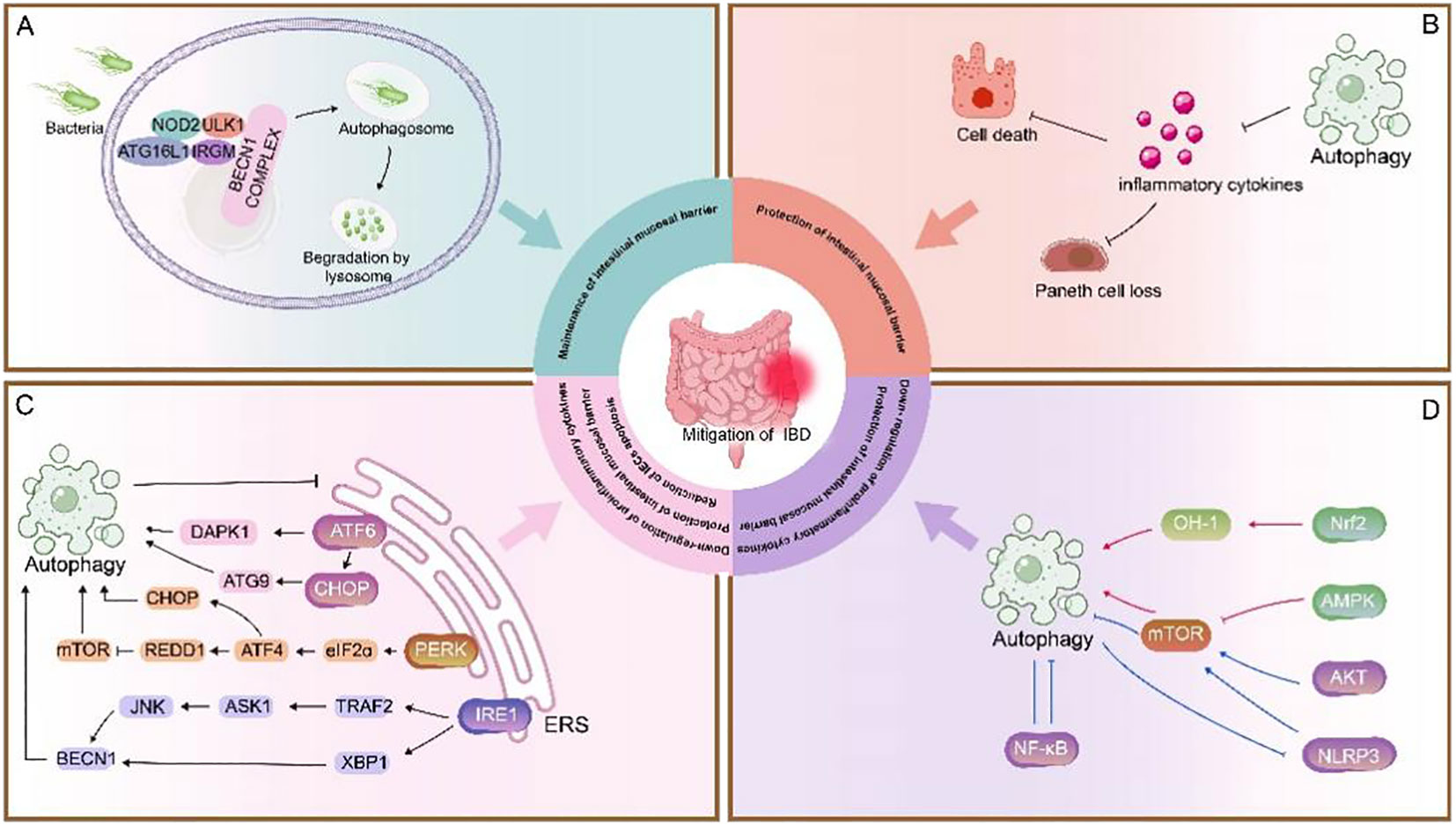
Figure 2 Role of autophagy in IBD. (A) NOD2 recruits ATG16L1 to the plasma membrane at the bacterial entry site, initiating autophagy. Association of IRGM with NOD2 promotes IRGM ubiquitination and the assembly of the core autophagy machinery, promoting xenophagy and intracellular bacterial clearance (135). This process is important for maintenance of intestinal mucosal barrier (48, 121). (B) Autophagy inhibits the secretion of inflammatory cytokines to prevent loss of Paneth cells and exaggerated cell death. (C) The UPR is activated in response to ERS and consists of three pathways: ATF6, PERK, and IRE1. These three stress-related proteins bind to GRP78 when the cell is in a steady state. Under ERS, GRP78 separates from these three receptors and activates the ATF6, PERK, and IRE1 pathways. 1) ATF6 is cleaved in the Golgi apparatus, binds to specific DNA, regulates the pro-apoptotic cytokine CHOP, mediates mAtg9 transport, and activates autophagy; it also activates autophagy by upregulating DAPK1 (88, 89). 2) PERK activates eIF2α through autophosphorylation, thereby activating ATF4, which induces the expression of CHOP and the upregulation of autophagy genes. ATF4 also induces the expression of REDD1 and inhibits mTOR phosphorylation to activate autophagy (87, 91–93). 3) IRE1 splices XBP1 into its active form and can bind to TRAF2 to activate JNK, thereby upregulating Beclin-1 and promoting autophagy. Autophagy and ERS are mutually regulated (96, 97). ERS can promote autophagy, and when ERS is overactive, activated autophagy can inhibit ERS, thereby reduction of IECs apoptosis, down-regulation of proinflammatory cytokines, protection of intestinal mucosal barrier. (D) In this section, the red line represents the Pro-autophagy pathway pathway and the blue line represents the inhibition of autophagy pathway. Autophagy is primarily regulated by the following pathways:1) mTOR is a key negative regulator of autophagy. The AMPK/mTOR pathway positively regulates autophagy (125); The NLRP3/mTOR and AKT/mTOR pathways negatively regulate autophagy (133, 137), whereas the upregulation of autophagy also inhibits NLRP3 inflammatory vesicle activation (132). 2) Nrf2-OH pathway positively regulates autophagy (150).3) The NF-κB pathway negatively regulates autophagy and vice versa (142, 144, 145). Autophagy-related signaling pathways benefit protection of intestinal mucosal barrier and down-regulation of proinflammatory cytokines, protection.
Autophagy is highly cell-specific and environment-specific. Therefore, strict selection of autophagic pathways or targets without affecting other biological processes is the key to autophagy-based therapeutic strategies for IBD. In the future, additional efforts are needed to validate the suitability of autophagy as a therapeutic target for IBD and to determine whether it is effective and feasible. These efforts should perhaps be devoted to the identification of small molecules or peptides for the precise modulation of the autophagic process and to gradually transfer experimental findings to clinical studies.
Author contributions
S-LC, C-ML, WL, Q-SL, D-SH and SY-H retrieved and analyzed concerned literatures. S-LC and Q-SL wrote the manuscript. S-LC, C-ML, WL, M-YZ, J-HZ, S-YH, Y-WH, and YZ revised the manuscript.All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (81973821), Xinglin Scholar Research Promotion Project of Chengdu University of TCM (QJJJ2022010), “Hundred Talents Program” of the Hospital of Chengdu University of Traditional Chinese Medicine(grant no. 22-B09), the Program of Science and Technology Department of Sichuan Province (grant no. 2023NSFSC0039).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviation
ERS, endoplasmic reticulum stress; IBD, inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis; CRC, colorectal cancer; CMA, chaperone mediated autophagy; ULK1, unc-51-like autophagy-activated kinase 1; ATG13, autophagy-associated gene 13; ER, endoplasmic reticulum; BECN1, Beclin 1; MAP1LC3A/LC3, microtubule-associated protein 1 light chain 3 alpha; PE, phosphatidylethanolamine; ATG16L1, autophagy-related 16-like 1 gene; LRRK2, leucine-rich repeat kinase 2; NOD2, nucleotide-binding oligomerization domain-containing protein 2; T300A, Thr 300-to-Ala; caspase 3, Cysteine aspartate protease 3; IL-22, Interleukin22; IECs, intestinal epithelial cells; mTOR, mammalian target of rapamycin; AMPK, Adenosine 5’-monophosphate (AMP)-activated protein kinase; NaHS, Sodium hydrosulfide; LPS, lipopolysaccharide; DSS, dextran sulfate sodium; NLRP3, NOD-like receptor thermal protein domain associated protein 3; mtROS, mitochondrial reactive oxygen species; Nrf2, nuclear transcription factor E2-related factor 2; HO-1, Heme Oxygenase-1; TNBS, trinitro-benzene-sulfonic acid; IKK, IκB kinase; STAT3, Signal sensors and activator of transcription 3; TNF-α, tumor necrosis factor α; UPR, unfolded protein response; IRE1, Inositol requires enzyme 1; PERK, protein kinase R-like ER kinase; ATF6, Activation of transcription factor 6; REDD1, DNA damage response 1; CHOP, C/EBP homologous protein; DAPK1, death-associated protein kinase 1; ASK1, apoptosis signaling regulatory kinase-1.
References
1. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol (2012) 9(10):599–608. doi: 10.1038/nrgastro.2012.152
2. De Souza HS. Fiocchi C.Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol (2016) 13(1):13–27. doi: 10.1038/nrgastro.2015.186
3. Ke P, Shao BZ, Xu ZQ, Chen XW, Liu C. Intestinal autophagy and its pharmacological control in inflammatory bowel disease. Front Immunol (2016) 7:695. doi: 10.3389/fimmu.2016.00695
4. Rich KA, Burkett C. Webster P.Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol (2003) 5(7):455–68. doi: 10.1046/j.1462-5822.2003.00292.x
5. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V, et al. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
6. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group a streptococcus. Science (2004) 306(5698):1037–40. doi: 10.1126/science.1103966
7. Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, et al. Protective role of autophagy against vibrio cholerae cytolysin, a pore-forming toxin from v. cholerae. Proc Natl Acad Sci USA (2007) 104(6):1829–34. doi: 10.1073/pnas.0601437104
8. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol (2015) 8(5):969–78. doi: 10.1038/mi.2015.49
9. Baxt LA, Xavier RJ. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology (2015) 149(3):553–62. doi: 10.1053/j.gastro.2015.06.046
10. Scharl M, Rogler G. Inflammatory bowel disease: dysfunction of autophagy? Dig Dis (2012) 30 Suppl 3:12–9. doi: 10.1159/000342588
11. Strisciuglio C, Duijvestein M, Verhaar AP, Vos AC, van den Brink GR, Hommes DW, et al. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J Crohns Colitis (2013) 7(7):534–41. doi: 10.1016/j.crohns.2012.08.009
12. Kuenzig ME, Yim J, Coward S, Eksteen B, Seow CH, Barnabe C, et al. The NOD2-smoking interaction in crohn's disease is likely specific to the 1007fs mutation and may be explained by age at diagnosis: a meta-analysis and case-only study. EBioMedicine (2017) 21:188–96. doi: 10.1016/j.ebiom.2017.06.012
13. Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol (2014) 20(1):6–21. doi: 10.3748/wjg.v20.i1.6
14. Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol (2013) 4:301. doi: 10.3389/fimmu.2013.00301
15. Van De Veerdonk FL, Dinarello CA. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy (2014) 10(6):1141–2. doi: 10.4161/auto.28638
16. Hosomi S, Kaser A, Blumberg RS. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr Opin Gastroenterol (2015) 31(1):81–8. doi: 10.1097/MOG.0000000000000144
17. Liton PB. The autophagic lysosomal system in outflow pathway physiology and pathophysiology. Exp Eye Res (2016) 144:29–37. doi: 10.1016/j.exer.2015.07.013
18. Wen X, Klionsky DJ. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy (2016) 12(4):617–8. doi: 10.1080/15548627.2016.1158373
19. Sato M, Seki T, Konno A, Hirai H, Kurauchi Y, Hisatsune A, et al. Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells (2016) 21(8):861–73. doi: 10.1111/gtc.12390
20. Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium (2011) 50(3):242–50. doi: 10.1016/j.ceca.2011.04.001
21. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian atg proteins. Autophagy (2010) 6(6):764–76. doi: 10.4161/auto.6.6.12709
22. Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of atg proteins in pre-autophagosomal structure organization. Genes Cells (2007) 12(2):209–18. doi: 10.1111/j.1365-2443.2007.01050.x
23. Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem (2017) 61(6):585–96. doi: 10.1042/EBC20170021
24. Kishi-Itakura C, Koyama-Honda I, Itakura E, Mizushima N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J Cell Sci (2014) 127(Pt 18):4089–102. doi: 10.1242/jcs.156034
25. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell (2019) 176(1-2):11–42. doi: 10.1016/j.cell.2018.09.048
26. Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
27. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature (1998) 395(6700):395–8. doi: 10.1038/26506
28. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature (2000) 408(6811):488–92. doi: 10.1038/35044114
29. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for crohn disease in ATG16L1. Nat Genet (2007) 39(2):207–11. doi: 10.1038/ng1954
30. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi: 10.1038/ng2032
31. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to crohn's disease susceptibility. Nat Genet (2007) 39(7):830–2. doi: 10.1038/ng2061
32. Mccarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and crohn's disease. Nat Genet (2008) 40(9):1107–12. doi: 10.1038/ng.215
33. Retnakumar SV, Muller S. Pharmacological autophagy regulators as therapeutic agents for inflammatory bowel diseases. Trends Mol Med (2019) 25(6):516–37. doi: 10.1016/j.molmed.2019.03.002
34. Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, et al. Genetic variation in the autophagy gene ULK1 and risk of crohn's disease. Inflammation Bowel Dis (2011) 17(6):1392–7. doi: 10.1002/ibd.21486
35. Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, et al. Functional variants in the LRRK2 gene confer shared effects on risk for crohn's disease and parkinson's disease. Sci Transl Med (2018) 10(423). doi: 10.1126/scitranslmed.aai7795
36. Yamazaki K, Onouchi Y, Takazoe M, Kubo M, Nakamura Y, Hata A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with crohn's disease in Japanese patients. J Hum Genet (2007) 52(7):575–83. doi: 10.1007/s10038-007-0156-z
37. Yang SK, Park M, Lim J, Kim H, Ye BD, Lee I, et al. Contribution of IL23R but not ATG16L1 to crohn's disease susceptibility in koreans. Inflammation Bowel Dis (2009) 15(9):1385–90. doi: 10.1002/ibd.20921
38. Zhi J, Zhi FC, Chen ZY, Yao GP, Guan J, Lin Y, et al. Correlation of the autophagosome gene ATG16L1 polymorphism and inflammatory bowel disease. Nan Fang Yi Ke Da Xue Xue Bao (2008) 28(4):649–51.
39. Weersma RK, Stokkers PC, Van Bodegraven AA, van Hogezand RA, Verspaget HW, de Jong DJ, et al. Molecular prediction of disease risk and severity in a large Dutch crohn's disease cohort. Gut (2009) 58(3):388–95. doi: 10.1136/gut.2007.144865
40. Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, et al. A crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature (2014) 506(7489):456–62. doi: 10.1038/nature13044
41. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of beclin-1 inactivates beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis (2010) 1:e18. doi: 10.1038/cddis.2009.16
42. Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a crohn's disease associated ATG16L1 variant. PloS One (2008) 3(10):e3391. doi: 10.1371/journal.pone.0003391
43. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14(3):282–9. doi: 10.1038/nm1720
44. Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol (2015) 16(7):698–707. doi: 10.1038/ni.3180
45. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med (2009) 206(7):1465–72. doi: 10.1084/jem.20082683
46. Wang C, Gong G, Sheh A, Muthupalani S, Bryant EM, Puglisi DA, et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol (2017) 10(6):1504–17. doi: 10.1038/mi.2017.9
47. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med (2018) 215(11):2868–86. doi: 10.1084/jem.20171029
48. Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe (2018) 23(2):191–202.e4. doi: 10.1016/j.chom.2017.12.017
49. Palomino-Morales RJ, Oliver J, Gomez-Garcia M, Lopez-Nevot MA, Rodrigo L, Nieto A, et al. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes Immun (2009) 10(4):356–64. doi: 10.1038/gene.2009.25
50. Glas J, Seiderer J, Bues S, Stallhofer J, Fries C, Olszak T, et al. IRGM variants and susceptibility to inflammatory bowel disease in the German population. PloS One (2013) 8(1):e54338. doi: 10.1371/journal.pone.0054338
51. Rufini S, Ciccacci C, Di Fusco D, Ruffa A, Pallone F, Novelli G, et al. Autophagy and inflammatory bowel disease: association between variants of the autophagy-related IRGM gene and susceptibility to crohn's disease. Dig Liver Dis (2015) 47(9):744–50. doi: 10.1016/j.dld.2015.05.012
52. Moon CM, Shin DJ, Kim SW, Son NH, Park A, Park B, et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflammation Bowel Dis (2013) 19(1):106–14. doi: 10.1002/ibd.22972
53. Lu XC, Tao Y, Wu C, Zhao PL, Li K, Zheng JY, et al. Association between variants of the autophagy related gene–IRGM and susceptibility to crohn's disease and ulcerative colitis: a meta-analysis. PLoS One (2013) 8(11):e80602. doi: 10.1371/journal.pone.0080602
54. Franke A, Mcgovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed crohn's disease susceptibility loci. Nat Genet (2010) 42(12):1118–25. doi: 10.1038/ng.717
55. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science (2006) 313(5792):1438–41. doi: 10.1126/science.1129577
56. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol (2010) 12(12):1154–65. doi: 10.1038/ncb2119
57. Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell (2015) 58(3):507–21. doi: 10.1016/j.molcel.2015.03.020
58. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
59. Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, et al. Irgm1-deficient mice exhibit paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2013) 305(8):G573–84. doi: 10.1152/ajpgi.00071.2013
60. Rogala AR, Schoenborn AA, Fee BE, Cantillana VA, Joyce MJ, Gharaibeh RZ, et al. Environmental factors regulate paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis Model Mech (2018) 11(2). doi: 10.1242/dmm.031070
61. Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in crohn's disease. Nat Genet (2011) 43(3):242–5. doi: 10.1038/ng.762
62. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to crohn's disease. Nature (2001) 411(6837):599–603. doi: 10.1038/35079107
63. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to crohn's disease. Nature (2001) 411(6837):603–6. doi: 10.1038/35079114
64. Vermeire S. NOD2/CARD15: relevance in clinical practice. Best Pract Res Clin Gastroenterol (2004) 18(3):569–75. doi: 10.1016/j.bpg.2003.12.008
65. Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and crohn's disease in German and British populations. Lancet (2001) 357(9272):1925–8. doi: 10.1016/S0140-6736(00)05063-7
66. Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet (2002) 70(4):845–57. doi: 10.1086/339432
67. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol (2015) 12(4):205–17. doi: 10.1038/nrgastro.2015.34
68. Liu JZ, Van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet (2015) 47(9):979–86. doi: 10.1038/ng.3359
69. Park SC, Jeen YT. Genetic studies of inflammatory bowel disease-focusing on Asian patients. Cells (2019) 8(5). doi: 10.3390/cells8050404
70. Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated crohn's disease: a meta-analysis. Am J Gastroenterol (2011) 106(4):699–712. doi: 10.1038/ajg.2011.19
71. Yamamoto S, Ma X. Role of Nod2 in the development of crohn's disease. Microbes Infect (2009) 11(12):912–8. doi: 10.1016/j.micinf.2009.06.005
72. Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology (2003) 124(1):140–6. doi: 10.1053/gast.2003.50019
73. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi: 10.1038/ni.1823
74. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16(1):90–7. doi: 10.1038/nm.2069
75. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive escherichia coli associated with ileal mucosa in crohn's disease. Gastroenterology (2004) 127(2):412–21. doi: 10.1053/j.gastro.2004.04.061
76. Rivas MA, Avila BE, Koskela J, Huang H, Stevens C, Pirinen M, et al. Insights into the genetic epidemiology of crohn's and rare diseases in the ashkenazi Jewish population. PLoS Genet (2018) 14(5):e1007329. doi: 10.1371/journal.pgen.1007329
77. Manzoni C, Mamais A, Roosen DA, Dihanich S, Soutar MP, Plun-Favreau H, et al. mTOR independent regulation of macroautophagy by leucine rich repeat kinase 2 via beclin-1. Sci Rep (2016) 6:35106. doi: 10.1038/srep35106
78. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol (2011) 12(11):1063–70. doi: 10.1038/ni.2113
79. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol (2010) 185(9):5577–85. doi: 10.4049/jimmunol.1000548
80. Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet (2014) 23(16):4201–14. doi: 10.1093/hmg/ddu138
81. Takagawa T, Kitani A, Fuss I, Levine B, Brant SR, Peter I, et al. An increase in LRRK2 suppresses autophagy and enhances dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med (2018) 10(444). doi: 10.1126/scitranslmed.aan8162
82. Levine B, Liu R, Dong X, Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol (2015) 25(9):533–44. doi: 10.1016/j.tcb.2015.05.004
83. Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology (2011) 140(6):1738–47. doi: 10.1053/j.gastro.2011.02.048
84. Qiao D, Zhang Z, Zhang Y, Chen Q, Chen Y, Tang Y, et al. Regulation of endoplasmic reticulum stress-autophagy: a potential therapeutic target for ulcerative colitis. Front Pharmacol (2021) 12:697360. doi: 10.3389/fphar.2021.697360
85. Xue JF, Shi ZM, Zou J, Li XL. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. BioMed Pharmacother (2017) 89:1252–61. doi: 10.1016/j.biopha.2017.01.130
86. Chung YP, Yen CC, Tang FC, Lee KI, Liu SH, Wu CC, et al. Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology (2019) 425:152245. doi: 10.1016/j.tox.2019.152245
87. Treton X, Pedruzzi E, Cazals-Hatem D, Grodet A, Panis Y, Groyer A, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology (2011) 141(3):1024–35. doi: 10.1053/j.gastro.2011.05.033
88. Zhou Y, Zhang S, Dai C, Tang S, Yang X, Li D, et al. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol Toxicol (2016) 32(2):141–52. doi: 10.1007/s10565-016-9323-3
89. Lopes F, Keita AV, Saxena A, Reyes JL, Mancini NL, Al Rajabi A, et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem (2018) 293(9):3073–87. doi: 10.1074/jbc.RA117.000809
90. Stengel ST, Fazio A, Lipinski S, Jahn MT, Aden K, Ito G, et al. Activating transcription factor 6 mediates inflammatory signals in intestinal epithelial cells upon endoplasmic reticulum stress. Gastroenterology (2020) 159(4):1357–74.e10. doi: 10.1053/j.gastro.2020.06.088
91. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ (2007) 14(2):230–9. doi: 10.1038/sj.cdd.4401984
92. Moon HS, Kim B, Gwak H, Suh DH, Song YS. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2alpha) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol Carcinog (2016) 55(4):346–56. doi: 10.1002/mc.22284
93. Rouschop KM, Van Den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest (2010) 120(1):127–41. doi: 10.1172/JCI40027
94. Kimball SR, Jefferson LS. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2alpha phosphorylation, ATF4-dependent cascade. Biochem Biophys Res Commun (2012) 427(3):485–9. doi: 10.1016/j.bbrc.2012.09.074
95. Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, et al. REDD1/Autophagy pathway is associated with neutrophil-driven IL-1beta inflammatory response in active ulcerative colitis. J Immunol (2018) 200(12):3950–61. doi: 10.4049/jimmunol.1701643
96. Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ (2015) 22(6):946–58. doi: 10.1038/cdd.2014.183
97. Rather RA, Bhagat M, Singh SK. Oncogenic BRAF, endoplasmic reticulum stress, and autophagy: crosstalk and therapeutic targets in cutaneous melanoma. Mutat Res Rev Mutat Res (2020) 785:108321. doi: 10.1016/j.mrrev.2020.108321
98. Kokten T, Gibot S, Lepage P, D'Alessio S, Hablot J, Ndiaye NC, et al. TREM-1 inhibition restores impaired autophagy activity and reduces colitis in mice. J Crohns Colitis (2018) 12(2):230–44. doi: 10.1093/ecco-jcc/jjx129
99. Diamanti MA, Gupta J, Bennecke M, De Oliveira T, Ramakrishnan M, Braczynski AK, et al. IKKalpha controls ATG16L1 degradation to prevent ER stress during inflammation. J Exp Med (2017) 214(2):423–37. doi: 10.1084/jem.20161867
100. Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, et al. Defective ATG16L1-mediated removal of IRE1alpha drives crohn's disease-like ileitis. J Exp Med (2017) 214(2):401–22. doi: 10.1084/jem.20160791
101. Fabri M, Realegeno SE, Jo EK, Modlin RL. Role of autophagy in the host response to microbial infection and potential for therapy. Curr Opin Immunol (2011) 23(1):65–70. doi: 10.1016/j.coi.2010.10.010
102. Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol (2012) 24(1):21–31. doi: 10.1016/j.coi.2011.10.006
103. Keller MD, Torres VJ, Cadwell K. Autophagy and microbial pathogenesis. Cell Death Differ (2020) 27(3):872–86. doi: 10.1038/s41418-019-0481-8
104. Wang W, Liu Z, Yue W, Zhu L, Zhong H, Yang C, et al. Mucosa-colonizing microbiota correlate with host autophagy signaling in patients with inflammatory bowel disease. Front Microbiol (2022) 13:875238. doi: 10.3389/fmicb.2022.875238
105. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy (2020) 16(1):38–51. doi: 10.1080/15548627.2019.1635384
106. Tsuboi K, Nishitani M, Takakura A, Imai Y, Komatsu M, Kawashima H. Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus. J Biol Chem (2015) 290(33):20511–26. doi: 10.1074/jbc.M114.632257
107. Yang L, Liu C, Zhao W, He C, Ding J, Dai R, et al. Impaired autophagy in intestinal epithelial cells alters gut microbiota and host immune responses. Appl Environ Microbiol (2018) 84(18). doi: 10.1128/AEM.00880-18
108. Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin a coating identifies colitogenic bacteria in inflammatory bowel disease. Cell (2014) 158(5):1000–10. doi: 10.1016/j.cell.2014.08.006
109. Zhang H, Zheng L, Mcgovern DP, Hamill AM, Ichikawa R, Kanazawa Y, et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. J Immunol (2017) 198(5):2133–46. doi: 10.4049/jimmunol.1601293
110. Zhang H, Wang D, Shihb DQ, Zhang XL. Atg16l1 in dendritic cells is required for antibacterial defense and autophagy in murine colitis. IUBMB Life (2020) 72(12):2686–95. doi: 10.1002/iub.2406
111. Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from salmonella infection. Gastroenterology (2013) 145(6):1347–57. doi: 10.1053/j.gastro.2013.08.035
112. Benjamin JL, Sumpter R Jr., Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe (2013) 13(6):723–34. doi: 10.1016/j.chom.2013.05.004
113. Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, Maurer K, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe (2013) 14(2):216–24. doi: 10.1016/j.chom.2013.07.013
114. Martin PK, Marchiando A, Xu R, Rudensky E, Yeung F, Schuster SL, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol (2018) 3(10):1131–41. doi: 10.1038/s41564-018-0229-0
115. Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science (2016) 352(6289):1116–20. doi: 10.1126/science.aad9948
116. Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science (2017) 357(6355):1047–52. doi: 10.1126/science.aal4677
117. Cheng S, Ma X, Geng S, Jiang X, Li Y, Hu L, et al. Fecal microbiota transplantation beneficially regulates intestinal mucosal autophagy and alleviates gut barrier injury. mSystems (2018) 3(5). doi: 10.1128/mSystems.00137-18
118. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab (2011) 13(5):517–26. doi: 10.1016/j.cmet.2011.02.018
119. Petrovic A, Bogojevic D, Korac A, Golic I, Jovanovic-Stojanov S, Martinovic V, et al. Oxidative stress-dependent contribution of HMGB1 to the interplay between apoptosis and autophagy in diabetic rat liver. J Physiol Biochem (2017) 73(4):511–21. doi: 10.1007/s13105-017-0574-0
120. Fakih W, Mroueh A, Salah H, Eid AH, Obeid M, Kobeissy F, et al. Dysfunctional cerebrovascular tone contributes to cognitive impairment in a non-obese rat model of prediabetic challenge: role of suppression of autophagy and modulation by anti-diabetic drugs. Biochem Pharmacol (2020) 178:114041. doi: 10.1016/j.bcp.2020.114041
121. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med (2017) 214(12):3687–705. doi: 10.1084/jem.20170558
122. Miao Y, Lv Q, Qiao S, Yang L, Tao Y, Yan W, et al. Alpinetin improves intestinal barrier homeostasis via regulating AhR/suv39h1/TSC2/mTORC1/autophagy pathway. Toxicol Appl Pharmacol (2019) 384:114772. doi: 10.1016/j.taap.2019.114772
123. Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest (2015) 125(3):1098–110. doi: 10.1172/JCI76344
124. Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang C, et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine (2018) 35:345–60. doi: 10.1016/j.ebiom.2018.08.035
125. Sun L, Zhang S, Yu C, Pan Z, Liu Y, Zhao J, et al. Hydrogen sulfide reduces serum triglyceride by activating liver autophagy via the AMPK-mTOR pathway. Am J Physiol Endocrinol Metab (2015) 309(11):E925–35. doi: 10.1152/ajpendo.00294.2015
126. Liu Y, Liao R, Qiang Z, Yang W, Cao J, Zeng H. Exogenous H2S protects colon cells in ulcerative colitis by inhibiting NLRP3 and activating autophagy. DNA Cell Biol (2021) 40(6):748–56. doi: 10.1089/dna.2020.6380
127. Xiong YJ, Deng ZB, Liu JN, Qiu JJ, Guo L, Feng PP, et al. Enhancement of epithelial cell autophagy induced by sinensetin alleviates epithelial barrier dysfunction in colitis. Pharmacol Res (2019) 148:104461. doi: 10.1016/j.phrs.2019.104461
128. Arab HH, Al-Shorbagy MY, Saad MA. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem Biol Interact (2021) 335:109368. doi: 10.1016/j.cbi.2021.109368
129. Deng J, Zeng L, Lai X, Li J, Liu L, Lin Q, et al. Metformin protects against intestinal barrier dysfunction via AMPKalpha1-dependent inhibition of JNK signalling activation. J Cell Mol Med (2018) 22(1):546–57. doi: 10.1111/jcmm.13342
130. Gao Q, Bi P, Luo D, Guan Y, Zeng W, Xiang H, et al. Nicotine-induced autophagy via AMPK/mTOR pathway exerts protective effect in colitis mouse model. Chem Biol Interact (2020) 317:108943. doi: 10.1016/j.cbi.2020.108943
131. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to crohn's disease susceptibility. Nat Genet (2009) 41(1):71–6. doi: 10.1038/ng.285
132. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13(3):255–63. doi: 10.1038/ni.2215
133. Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M, et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun (2017) 8(1):98. doi: 10.1038/s41467-017-00213-3
134. Mai CT, Wu MM, Wang CL, Su ZR, Cheng YY, Zhang XJ. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol Immunol (2019) 105:76–85. doi: 10.1016/j.molimm.2018.10.015
135. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy (2014) 10(6):972–85. doi: 10.4161/auto.28374
136. Liu C, Wang J, Yang Y, Liu X, Zhu Y, Zou J, et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem Pharmacol (2018) 155:366–79. doi: 10.1016/j.bcp.2018.07.010
137. Jiang W, Han YP, Hu M, Bao XQ, Yan Y, Chen G. A study on regulatory mechanism of miR-223 in ulcerative colitis through PI3K/Akt-mTOR signaling pathway. Eur Rev Med Pharmacol Sci (2019) 23(11):4865–72. doi: 10.26355/eurrev_201906_18074
138. Zhao J, Sun Y, Shi P, Dong JN, Zuo LG, Wang HG, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol (2015) 26(1):221–8. doi: 10.1016/j.intimp.2015.03.033
139. Zhang F, Wang W, Niu J, Yang G, Luo J, Lan D, et al. Heat-shock transcription factor 2 promotes sodium butyrate-induced autophagy by inhibiting mTOR in ulcerative colitis. Exp Cell Res (2020) 388(1):111820. doi: 10.1016/j.yexcr.2020.111820
140. Zhu Y, Shi Y, Ke X, Xuan L, Ma Z. RNF8 induces autophagy and reduces inflammation by promoting AKT degradation via ubiquitination in ulcerative colitis mice. J Biochem (2020) 168(5):445–53. doi: 10.1093/jb/mvaa068
141. Wang B, Gong Z, Zhan J, Yang L, Zhou Q, Yuan X. Xianglian pill suppresses inflammation and protects intestinal epithelial barrier by promoting autophagy in DSS induced ulcerative colitis mice. Front Pharmacol (2020) 11:594847. doi: 10.3389/fphar.2020.594847
142. Nivon M, Fort L, Muller P, Richet E, Simon S, Guey B, et al. NFkappaB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol Biol Cell (2016) 27(11):1712–27. doi: 10.1091/mbc.e15-12-0835
143. Platta HW, Abrahamsen H, Thoresen SB, Stenmark H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor beclin 1. Biochem J (2012) 441(1):399–406. doi: 10.1042/BJ20111424
144. Shibata Y, Oyama M, Kozuka-Hata H, Han X, Tanaka Y, Gohda J, et al. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat Commun (2012) 3:1061. doi: 10.1038/ncomms2068
145. Gerster R, Eloranta JJ, Hausmann M, Ruiz PA, Cosin-Roger J, Terhalle A, et al. Anti-inflammatory function of high-density lipoproteins via autophagy of IkappaB kinase. Cell Mol Gastroenterol Hepatol (2015) 1(2):171–87.e1. doi: 10.1016/j.jcmgh.2014.12.006
146. Honjo H, Watanabe T, Arai Y, Kamata K, Minaga K, Komeda Y, et al. ATG16L1 negatively regulates RICK/RIP2-mediated innate immune responses. Int Immunol (2021) 33(2):91–105. doi: 10.1093/intimm/dxaa062
147. Gao P, Liu H, Huang H, Sun Y, Jia B, Hou B, et al. The crohn disease-associated ATG16L1(T300A) polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling. Autophagy (2022) 18(11):1–15. doi: 10.1080/15548627.2022.2039991
148. Wang D, Shao S, Zhang Y, Zhao D, Wang M. Insight into polysaccharides from panax ginseng c. a. Meyer in improving intestinal inflammation: modulating intestinal microbiota and autophagy. Front Immunol (2021) 12:683911. doi: 10.3389/fimmu.2021.683911
149. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem (2010) 285(29):22576–91. doi: 10.1074/jbc.M110.118976
150. Kaushal GP, Chandrashekar K, Juncos LA. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int J Mol Sci (2019) 20(15). doi: 10.3390/ijms20153791
151. Li J, Wang H, Zheng Z, Luo L, Wang P, Liu K, et al. Mkp-1 cross-talks with Nrf2/Ho-1 pathway protecting against intestinal inflammation. Free Radic Biol Med (2018) 124:541–9. doi: 10.1016/j.freeradbiomed.2018.07.002
152. Pompili S, Sferra R, Gaudio E, Viscido A, Frieri G, Vetuschi A, et al. Can Nrf2 modulate the development of intestinal fibrosis and cancer in inflammatory bowel disease? Int J Mol Sci (2019) 20(16). doi: 10.3390/ijms20164061
153. Ortiz-Masia D, Cosin-Roger J, Calatayud S, Hernandez C, Alos R, Hinojosa J, et al. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: relevance in IBD. Mucosal Immunol (2014) 7(4):929–38. doi: 10.1038/mi.2013.108
154. Fu XY. STAT3 in immune responses and inflammatory bowel diseases. Cell Res (2006) 16(2):214–9. doi: 10.1038/sj.cr.7310029
155. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
156. Zhang YG, Zhu X, Lu R, Messer JS, Xia Y, Chang EB, et al. Intestinal epithelial HMGB1 inhibits bacterial infection via STAT3 regulation of autophagy. Autophagy (2019) 15(11):1935–53. doi: 10.1080/15548627.2019.1596485
157. Yuan Y, Ding D, Zhang N, Xia Z, Wang J, Yang H, et al. TNF-alpha induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle (2018) 17(11):1390–402. doi: 10.1080/15384101.2018.1482150
158. Kubota M, Kakimoto K, Nakagawa T, Koubayashi E, Nakazawa K, Tawa H, et al. Autophagy deficiency exacerbates colitis through excessive oxidative stress and MAPK signaling pathway activation. PloS One (2019) 14(11):e0225066. doi: 10.1371/journal.pone.0225066
159. Levy J, Cacheux W, Bara MA, L'Hermitte A, Lepage P, Fraudeau M, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat Cell Biol (2015) 17(8):1062–73. doi: 10.1038/ncb3206
160. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev (2011) 25(7):717–29. doi: 10.1101/gad.2016111
161. Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol (2009) 3(4):366–75. doi: 10.1016/j.molonc.2009.05.007
162. Liu W, Meng Y, Zong C, Zhang S, Wei L. Autophagy and tumorigenesis. Adv Exp Med Biol (2020) 1207:275–99. doi: 10.1007/978-981-15-4272-5_20
163. Grimm WA, Messer JS, Murphy SF, Nero T, Lodolce JP, Weber CR, et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut (2016) 65(3):456–64. doi: 10.1136/gutjnl-2014-308735
164. An CH, Kim MS, Yoo NJ, Park SW, Lee SH. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract (2011) 207(7):433–7. doi: 10.1016/j.prp.2011.05.002
165. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell (2016) 166(2):288–98. doi: 10.1016/j.cell.2016.05.051
166. Yuk JM, Shin DM, Song KS, Lim K, Kim KH, Lee SH, et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy (2010) 6(1):46–60. doi: 10.4161/auto.6.1.10325
167. Schonewolf CA, Mehta M, Schiff D, Wu H, Haffty BG, Karantza V, et al. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J Gastrointest Oncol (2014) 6(3):74–82. doi: 10.4251/wjgo.v6.i3.74
168. Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer (2010) 10:370. doi: 10.1186/1471-2407-10-370
169. Shi Y, Tang B, Yu PW, Tang B, Hao YX, Lei X, et al. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in caco-2 cells. PloS One (2012) 7(11):e51076. doi: 10.1371/journal.pone.0051076
170. Wang Y, Lu X, Wang X, Qiu Q, Zhu P, Ma L, et al. atg7-based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circ Res (2021) 129(8):e166–82. doi: 10.1161/CIRCRESAHA.121.319104
171. Sakiyama T, Musch MW, Ropeleski MJ, Tsubouchi H, Chang EB. Glutamine increases autophagy under basal and stressed conditions in intestinal epithelial cells. Gastroenterology (2009) 136(3):924–32. doi: 10.1053/j.gastro.2008.12.002
172. Mutalib M, Borrelli O, Blackstock S, Kiparissi F, Elawad M, Shah N, et al. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J Crohns Colitis (2014) 8(12):1730–4. doi: 10.1016/j.crohns.2014.08.014
173. Matsuda C, Ito T, Song J, Mizushima T, Tamagawa H, Kai Y, et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin Exp Immunol (2007) 148(2):348–59. doi: 10.1111/j.1365-2249.2007.03345.x
174. Bastida G, Beltran B. Ulcerative colitis in smokers, non-smokers and ex-smokers. World J Gastroenterol (2011) 17(22):2740–7. doi: 10.3748/wjg.v17.i22.2740
175. Qin Z, Wan JJ, Sun Y, Wu T, Wang PY, Du P, et al. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J Mol Med (Berl) (2017) 95(2):221–33. doi: 10.1007/s00109-016-1473-5
176. Alsharari SD, Akbarali HI, Abdullah RA, Shahab O, Auttachoat W, Ferreira GA, et al. Novel insights on the effect of nicotine in a murine colitis model. J Pharmacol Exp Ther (2013) 344(1):207–17. doi: 10.1124/jpet.112.198796
177. Hayashi S, Hamada T, Zaidi SF, Oshiro M, Lee J, Yamamoto T, et al. Nicotine suppresses acute colitis and colonic tumorigenesis associated with chronic colitis in mice. Am J Physiol Gastrointest Liver Physiol (2014) 307(10):G968–78. doi: 10.1152/ajpgi.00346.2013
178. Muller S, Monneaux F, Schall N, Rashkov RK, Oparanov BA, Wiesel P, et al. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis Rheum (2008) 58(12):3873–83. doi: 10.1002/art.24027
179. Monneaux F, Parietti V, Briand JP, Muller S. Importance of spliceosomal RNP1 motif for intermolecular T-b cell spreading and tolerance restoration in lupus. Arthritis Res Ther (2007) 9(5):R111. doi: 10.1186/ar2317
180. Zimmer R, Scherbarth HR, Rillo OL, Gomez-Reino JJ, Muller S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann Rheum Dis (2013) 72(11):1830–5. doi: 10.1136/annrheumdis-2012-202460
181. Schall N, Muller S. Resetting the autoreactive immune system with a therapeutic peptide in lupus. Lupus (2015) 24(4-5):412–8. doi: 10.1177/0961203314556138
182. Retnakumar SV, Geesala R, Bretin A, Tourneur-Marsille J, Ogier-Denis E, Maretzky T, et al. Targeting the endo-lysosomal autophagy pathway to treat inflammatory bowel diseases. J Autoimmun (2022) 128:102814. doi: 10.1016/j.jaut.2022.102814
183. Moron B, Spalinger M, Kasper S, Atrott K, Frey-Wagner I, Fried M, et al. Activation of protein tyrosine phosphatase non-receptor type 2 by spermidine exerts anti-inflammatory effects in human THP-1 monocytes and in a mouse model of acute colitis. PloS One (2013) 8(9):e73703. doi: 10.1371/journal.pone.0073703
184. Liu R, Li X, Ma H, Yang Q, Shang Q, Song L, et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, hif-1alpha upregulation and autophagy. Free Radic Biol Med (2020) 161:339–50. doi: 10.1016/j.freeradbiomed.2020.10.029
185. Blaney GP, Albert PJ, Proal AD. Vitamin d metabolites as clinical markers in autoimmune and chronic disease. Ann N Y Acad Sci (2009) 1173:384–90. doi: 10.1111/j.1749-6632.2009.04875.x
186. Lu R, Zhang YG, Xia Y, Sun J. Imbalance of autophagy and apoptosis in intestinal epithelium lacking the vitamin d receptor. FASEB J (2019) 33(11):11845–56. doi: 10.1096/fj.201900727R
187. Shi Y, Cui X, Sun Y, Zhao Q, Liu T. Intestinal vitamin d receptor signaling ameliorates dextran sulfate sodium-induced colitis by suppressing necroptosis of intestinal epithelial cells. FASEB J (2020) 34(10):13494–506. doi: 10.1096/fj.202000143RRR
188. Zhang YG, Lu R, Xia Y, Zhou D, Petrof E, Claud EC, et al. Lack of vitamin d receptor leads to hyperfunction of claudin-2 in intestinal inflammatory responses. Inflammation Bowel Dis (2019) 25(1):97–110. doi: 10.1093/ibd/izy292
189. Farzaei MH, El-Senduny FF, Momtaz S, Parvizi F, Iranpanah A, Tewari D, et al. An update on dietary consideration in inflammatory bowel disease: anthocyanins and more. Expert Rev Gastroenterol Hepatol (2018) 12(10):1007–24. doi: 10.1080/17474124.2018.1513322
190. Zhang H, Luo X, Ke J, Duan Y, He Y, Zhang D, et al. Procyanidins, from castanea mollissima bl. shell, induces autophagy following apoptosis associated with PI3K/AKT/mTOR inhibition in HepG2 cells. BioMed Pharmacother (2016) 81:15–24. doi: 10.1016/j.biopha.2016.04.002
191. Zhang H, Lang W, Liu X, Bai J, Jia Q, Shi Q. Procyanidin A1 alleviates DSS-induced ulcerative colitis via regulating AMPK/mTOR/p70S6K-mediated autophagy. J Physiol Biochem (2022) 78(1):213–27. doi: 10.1007/s13105-021-00854-5
Keywords: signaling pathway, inflammatory bowel disease, autophagy, autophagy-associated gene, endoplasmic reticulum stress, intestinal microflora
Citation: Chen S-L, Li C-M, Li W, Liu Q-S, Hu S-Y, Zhao M-Y, Hu D-S, Hao Y-W, Zeng J-H and Zhang Y (2023) How autophagy, a potential therapeutic target, regulates intestinal inflammation. Front. Immunol. 14:1087677. doi: 10.3389/fimmu.2023.1087677
Received: 02 November 2022; Accepted: 03 April 2023;
Published: 24 April 2023.
Edited by:
Juan Carlos Cutrin, University of Turin, ItalyReviewed by:
Marco Falasca, Curtin University, AustraliaDaniele Souza, Federal University of Minas Gerais, Brazil
Subrata Sabui, University of California, Irvine, United States
Copyright © 2023 Chen, Li, Li, Liu, Hu, Zhao, Hu, Hao, Zeng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Hao Zeng, emVuZ2ppbmhhb0BjZHV0Y20uZWR1LmNu; Yi Zhang, ZHJ6aGFuZ3lpQGNkdXRjbS5lZHUuY24=