 Tewodros Shibabaw
Tewodros Shibabaw Banchamlak Teferi2
Banchamlak Teferi2 Birhanu Ayelign
Birhanu Ayelign- 1Department of Biochemistry, School of Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
- 2Department of Clinical Pharmacy, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
- 3Department of Immunology and Molecular Biology, School of Biomedical and Laboratory Science, College of Medicine and Health Science, University of Gondar, Gondar, Ethiopia
- 4Research School of Biology, College of Science, Australian National University, Canberra, ACT, Australia
Metastatic breast cancer is one of the most common and well-known causes of death for women worldwide. The inflammatory tumor cell and other cancer hallmarks dictate the metastatic form and dissemination of breast cancer. Taking these into account, from various components of the tumor microenvironment, a pro-inflammatory infiltrative cell known as Th-17 plays an immense role in breast cancer proliferation, invasiveness, and metastasis. It has been demonstrated that IL-17, a pleiotropic pro-inflammatory cytokine generated by Th-17, is upregulated in a metastatic form of breast cancer. Recent research updates stated that chronic inflammation and mediators like cytokines and chemokines are causative hallmarks in many human cancers, including breast cancer. Therefore, IL-17 and its multiple downward signaling molecules are the centers of research attention to develop potent treatment options for cancer. They provide information on the role of IL-17-activated MAPK, which results in tumor cell proliferation and metastasis via NF-kB-mediated expression of MMP signaling. Overall, this review article emphasizes IL-17A and its intermediate signaling molecules, such as ERK1/2, NF-kB, MMPs, and VEGF, as potential molecular targets for the prevention and treatment of breast cancer.
Introduction
Breast cancer (BC) is the most frequently diagnosed metastatic cancer among women (1). It is a molecularly diverse disease that involves complex processes that result in initiation, progression, and metastasis (2). The tumors of breast origin can be classified either by the gene expression pattern of three receptors, such as estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), or through its nodal metastasis (3). Like other solid tumors, breast cancer starts locally and spreads into distant organs—metastatic breast cancer (2). Breast cancer metastasis is a usual hallmark of cancer and leads to treatment failure, leading to the death of many patients. Around 10%–15% of breast cancer patients experience metastasis, leading to death (2). The bone is the most common site of breast cancer metastasis for about 75% of patients with late-stage BC (4). Usually, metastatic breast cancer has a poor prognosis, with 73% of the patients having less-than-5-year survival (5, 6). According to studies, several factors may affect the pathogenesis and prognosis of breast cancer (6). The genetic mutation of tumor cells is responsible for the proliferation, uncontrolled growth, and spreading ability of the primary tumor cell malignancy invasiveness and distant migration (7–9). Inflammatory condition is also a prognostic factor in metastatic breast cancer and contributes to cancer development and progression. Particularly, immune cells—including Th-17, tumor-associated macrophage, neutrophils, natural killer (NK) cells, and γδT cells and mediators in the microenvironment—facilitate angiogenesis and proliferation (10–12). Th-17 is one of the inflammatory CD4+ cells that play an essential role in cancer pathogenesis and anti-tumor immune response (13). Notably, in breast cancer, Th-17 cells are positively related to IL-6, IL-1β, and IL-17 expression and negatively correlated with increased metastatic lymph nodes and tumor cell angiogenesis. IL-17-induced inflammatory mediators such as G-CSF, IL-6, and CXCL1 stimulate the expansion and recruitment of dysfunctional myeloid cells to establish a proangiogenic and immune-suppressive tumor environment that enhances tumor growth and metastasis (14). This results in the formation of a metastatic secondary tumor. However, how the cytokine of the microenvironment promotes tumor metastasis remains a research question. For the effective migration and metastasis of breast cancer cells in the vascular or lymphatic drainage system, chemical mediators such as calcium-dependent zinc-containing endopeptidases like MMPs must be required for the degradation of the ECM as well as VEGF and IL-8 for vascularization during intravasation and extravasation processes, respectively, and reach to the bone (15). A study showed that the expression and activation of MMPs are mediated through TNF-α and IL-1 secreted by tumor cells, and IL-17A secreted from the microenvironment plays a role on the regulation of different MMPs (16, 17). There are five major classes of MMPs depending on their function and the substrates that they digest, including matrilysins (MMP-7 and MMP-26), collagenases (MMP-1, MMP-8, and MMP-13), stromelysins (MMP-3, MMP-10, and MMP-11), gelatinase (MMP-2 and MMP-9), and membrane-associated metalloproteinases (MMP-14, MMP-15, MMP-16, MMP-17, MMP-23A/B, MMP-24, and MMP-25) (18, 19). A retrospective SEER study on 25,323 women presenting with stage IV BC explored that 26.8% and 12.8% had overall survival of 5 and 10 years, respectively (20, 21). It explained that there is a strong association between elevated rates of IL-17 and Th-17 cell infiltration and estrogen receptor (ER)-negative and triple-negative molecular subtypes of BC (22).
In the mammary gland tumor microenvironment, excessive infiltration of Th-17 cells, NK, and γδT cells are associated with poor prognostic factors for staging, overall, and disease-free survival (23, 24). Furthermore, the functional contribution of human Th-17 cells to tumor immunity remains unclear since both pro- and anti-tumor effects have been observed. According to the data, in both the 4T1 and E0771 tumor models, increased Th-17 was seen at the early stage of tumor progression, peaked at the middle cancer stage, and then markedly declined at the late stage (25). This review further elaborates on the interaction of IL-17A with its heterodimer single-pass transmembrane receptor (IL17RA/IL17RC). In turn, TRAF-6/TAK-1 joins the MAPK pathway and upregulates the subsequent phosphorylation of extracellular signal-regulated kinases (ERK1/2) in all human breast cancer, thus leading to uncontrolled growth, proliferation, and resistance to traditional chemotherapeutic agents such as docetaxel (26). In addition, these reviews will discuss the IL-17/NF-kB-associated incidence of bone metastatic breast cancer. The progression and metastasis of BC thought to be controlled through locally infiltrated Th-17 cells produce inflammatory cytokines (IL-17A). This results from activating the IL-17A-IL-6-STAT-3 pathways, NF-kB-mediated production of MMPs, and vascular endothelial cell growth factor (VEGF) (27). In supporting the abovementioned scenario0, IL-17A also triggers the growth and proliferation of tumor cells through the IL-17A/MAPK pathways (14, 28). Overall, following metastasized breast cancer diagnosis, biological signaling pathways are the foundation of current anti-cancer therapies. Therefore, it is crucial to thoroughly understand the molecular and immunological mechanisms to classify and design appropriate treatment for breast cancer (29). This review article aimed to illustrate the dysregulated MAPK and NF-kB pathways in response to IL-17A/IL-17AR/CR interaction in bone metastatic breast cancer and its therapeutic options.
Mechanism of Th-17 cell polarization and the diversity of IL-17 and IL-17R
The third independent lineage of the CD4+ T cell subset, designated as “Th-17 cell”, produces IL-17A, and a related family of IL-17 cytokines was discovered in 2005 (30–33). Currently, there are six related IL-17 family members such as IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F (30, 34, 35). Both IL-17F and IL-17A share the same structural similarities and are secreted in homodimeric (two IL-17A or two IL-17F) or heterodimeric (IL-17A/IL-17F) forms. These forms are biologically active and connected by disulfide bonds formed by its cysteine residues (36). Further studies have shown that “Th-17” cells are also capable of secreting IL-21, IL-22, and GM-CSF (37). IL-21 creates an amplification loop for the further generation of Th-17 cells (38), whereas IL-17A is mostly secreted as a heterodimeric form with IL-17F, suggesting that the activity of IL-17A is partially attributed to the most potent form of IL-17A/IL-17F heterodimeric cytokine (36, 39)., Apart from Th-17, IL-17 can also be secreted by γδT cells, natural killer (NK) cells, NK T cells, mast cells, granulocytes, a subset of CD8+ T cells, known as Tc17 cells, and “innate lymphoid cells” (35, 40). The tumor cells, breast cancer-associated macrophage (41), and cancer-associated fibroblasts (CAF) secrete chemokines such as MCP-1 or CCL2, CCL20 (MIP-3A) (42, 43), and CXCL12 (SDF-1) as chemo-attraction of CD4+ T cell during differentiation of Th-17 cell and even for the selective attraction of Th-17 cell infiltration and/or its migration into the site of the tumor microenvironment (28). The ability of naive CD4+ T cells to undergo lineage polarization into distinct effector subsets is mediated by master transcription factors (44). These master transcription factors play opposing roles in Th-1/Th-2/Th-17/Treg cell fate decisions; retinoic acid receptor-related orphan receptor-γt (ROR-γt) is induced during Th-17 cell differentiation and strongly suppresses other Th cell polarization of the gene expression (45). Downstream of STAT3 signaling is the Th-17 master regulator ROR-γt. This transcription factor directly regulates the expression of IL-17A and IL-17F, along with other Th-17-specific genes (44, 46). Therefore, the polarization of naïve CD4+ T cell to Th-17 cells takes two significant steps in the reaction process. Activation of naïve CD4+ T cell is the first step of the reaction, mediated by cytokines (IL-6, IL-23, and TGF-β) secreted by a professional APC such as dendritic cells and macrophages (Figure 1) (30, 47). Taken together, polarizing cytokines produced by APC of the tumor microenvironment and cancer stem cells are the driving force for differentiation, maturation, and survival (IL-23 mediated) of the Th-17 phenotype (48, 49). In the context of inflammation driven by cytokines such as tumor necrosis factor (TNF), there is a clear synergy with IL-17F, reaching a pro-inflammatory gene signature very far from that induced by the combination of TNF-α and IL-17A (50). Depending on amino acid homology with IL-17A, the remaining related families, such as IL-17C, IL-17D, and IL-17E, have been identified and are significantly divergent from IL-17A (51). Research scholars stated that the inflammatory potency of IL-17F becomes more amplified when expressed and works together with tumor necrosis factor (TNF-α) (52). The signal transduction of each member of the IL-17 family is through its binding to specific interleukin-17 receptors (IL-17R) (34). The tissue distribution of IL-17RAs is almost in every cell type, whereas IL-17RC is predominantly expressed in epithelial cells, endothelial cells, fibroblasts, osteoblasts, and limited expression in myeloid cells (53–55). Based on sequence homology to IL−17RA, additional receptors have been identified in the IL−17R family such as IL−17RB, IL−17RC, IL−17RD, and IL−17RE. Indeed not only IL−17RA but also IL−17RC is required for the action of both IL−17A and IL−17F. Therefore, it showed that IL-17A or IL-17F binds the receptor complex called IL-17RA–IL-17RC to drive the expression of a gene involved in the inflammation, proliferation, angiogenesis, and metastasis of primary tumor cells through NF-kB and MAPK activation (56–58).
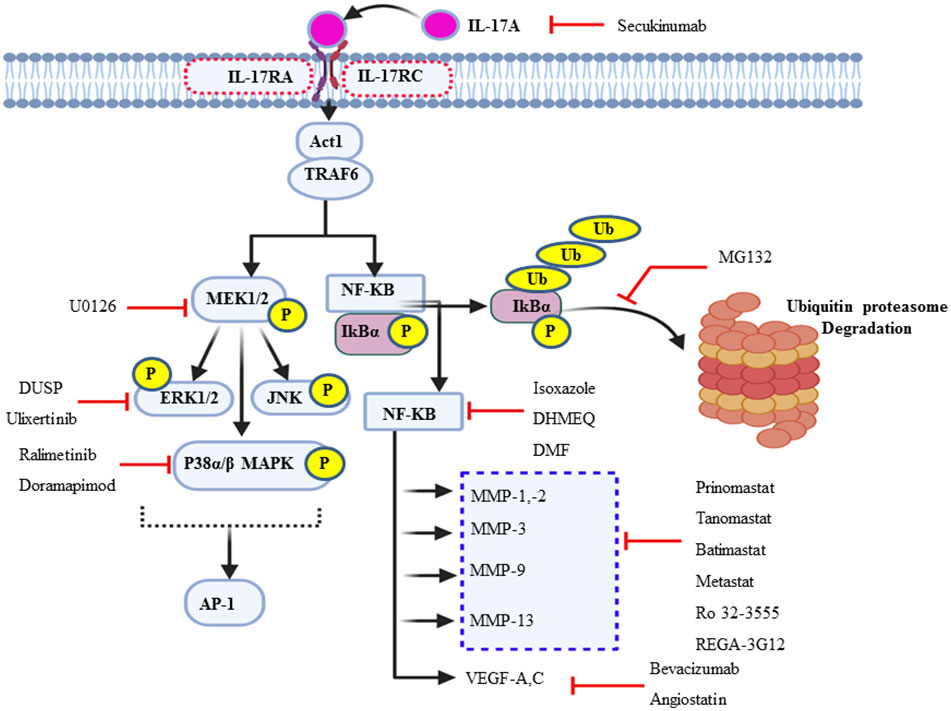
Figure 1 Schematic illustration of Th-17 polarization and its cytokine signature. Chemokines, including CCL2 and MCP-1, are secreted by cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAM), and antigen-presenting cells (APC) that promote CD4+ cell recruitment. Breast cancer cells, CAFs, TAMs, and dendritic cells are part of the stem cell and its microenvironment that produces different polarizing cytokines, such as IL-6, IL-23, and TGF-β. In turn, such polarizing cytokines activate distinct transcription factor cascades within naïve CD4+ T cells and influence T cell differentiation into distinct effector T cell subtypes, mainly Th-17 effector cell subset that produces all IL-17 signature cytokines (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F) and IL-21 and IL-22.
IL-17A/MAPK signaling promotes the proliferation of breast cancer cell
IL-17 plays a pivotal role in the tumor microenvironment, from the initial stages of tumorigenesis to its invasiveness, proliferation, and distant migration (14). Therefore, after the dysregulated interaction of IL-17 with its receptor, there will be a feed-forward expression of other inflammatory molecules, such as IL-6 through NF-kB. In turn, IL-6 joins the vicious cycle or loop via the additional activation of NF-kB through the IL-6/STAT-3/NF-kB axis (14). A study on a variety of BC cell line in a mouse model explored that IL-17 has a pro-tumoral effect and contributes to chemotherapeutic resistance (for example, paclitaxel). IL-17A/E induces c-RAF and ERK1/2 phosphorylation by p70S6 kinase; in turn, it activates the MAPK signaling pathway and contributes to BC taxane resistance. All IL-17R receptors contain an extracellular domain (ligand binding site), a transmembrane domain, and a cytoplasmic domain “SEFIR” (56). The SEFIR domain of IL-17RA is a conserved cytoplasmic motif that depends on an adapter molecule ACT1 (also known as TRAF3IP2) and TNF receptor−associated factor 6 (TRAF6) to recruit the rest of its downstream signalings such as MAPK (p38, JNK, and ERK1/2) (59, 60) and the NF-kB pathway (Figure 2) (34) (61–63). Taking this step further, ACT1 recruitment relays on the SEFIR domain, then TARF-6 binds to ACT-1 and form the IL-17R complex (64). In addition, ACT-1 also plays a non-degradative ubiquitination of TRAF-6 through its U-box, like lysine-63 (K63) E3 ligase domain (32). In turn, the ubiquitination of TRAF6 provides a scaffold for the recruitment and activation of TAK-1 (MAPKKK)-mediated activation of MEK-1/2 (65). Elevated IL-17A or IL-17B expression is strongly associated with poor prognosis outcomes for patients who present with BC (22, 66). To stimulate cells to proliferate or differentiate, these short-lived signaling events need to be converted into longer-lasting ones that can sustain the signal and relay it downstream to the nucleus (67). ERK (ERK1 and ERK2) is activated upon phosphorylation by MEK (MEK1 and MEK2), which is itself activated when phosphorylated by TAK-1 (Figure 2). In support of this, studies showed that the type of IL-17 response is context dependent, i.e., different Il-17 families affect the cell with different effector molecules differently concerning cell lines like MCF7, T47D, BT20, MDA-MB468, MD-MB157, and MDA-MB231 (Table 1) (68, 69). Few preclinical studies support the antitumorigenic properties, particularly of IL-17-E. In contrast, many more clinical as well as preclinical studies explained pro-tumorigenesis with the exposures of IL-17A, IL-17F, and IL-17B (75). Moreover, similar to that of the overexpression or mutation of receptor tyrosine kinase, most cancers, including breast cancer-associated lesions, that lead to constitutive or uncontrolled activation of ERK signaling (55) were due to either the overexpression of IL-17A/IL-17RA and ACT1-TRAF-6 or the activating mutation of TAK-1 molecule (63, 76, 77). However, there is also amplification or deregulation of its nuclear transcription factor targets, such as AP-1 (dimeric transcription factor of c-Fos and c-Jun) (78). AP-1 does not always promote cell proliferation but has anti-proliferative activities (79). In turn, activated c-Jun-containing AP-1 allows positive G1-to-S-phase progression, proliferation, and differentiation regulators such as cyclin D1 to be turned on and transcribed (80) or represses or turns off tumor suppressor p53 (blocker of CD1 and cyclin A/E via P21) and p16 expression (81–83). Additionally, the study showed that IL-17A is produced by BC TILs and responsible for docetaxel chemoresistance, angiogenesis, and its proliferation potential through the ERK1/2 pathway and induction of phosphorylation of EGFR in collaboration with IL-17ER (69). It proposed that multi-target inhibition, i.e., not only targeting IL-17A/IL-17AR/CR but also inhibiting its co-worker IL17E/IL-17ER, maximizes the clinical efficiency or potency of anti-EGFR such as panitumumab or rrastuzumab for treatments of BC (84, 85). Among MAPK signaling, p38 and ERK1/2 are the most commonly activated in tumorigenesis and migration of BC (86–88). During BC cell proliferation and invasion, IL-17 is suggested to be critical for p38 MAPK activation. The activated p38 MAPK, in turn, promotes the production of cytokines (TGFβ and TNFα) and interleukins (IL-6, IL-8, and IL-1β) within the tumor microenvironment, all of which are known to play a role in promoting tumor growth, angiogenesis, invasion, and metastasis (89).
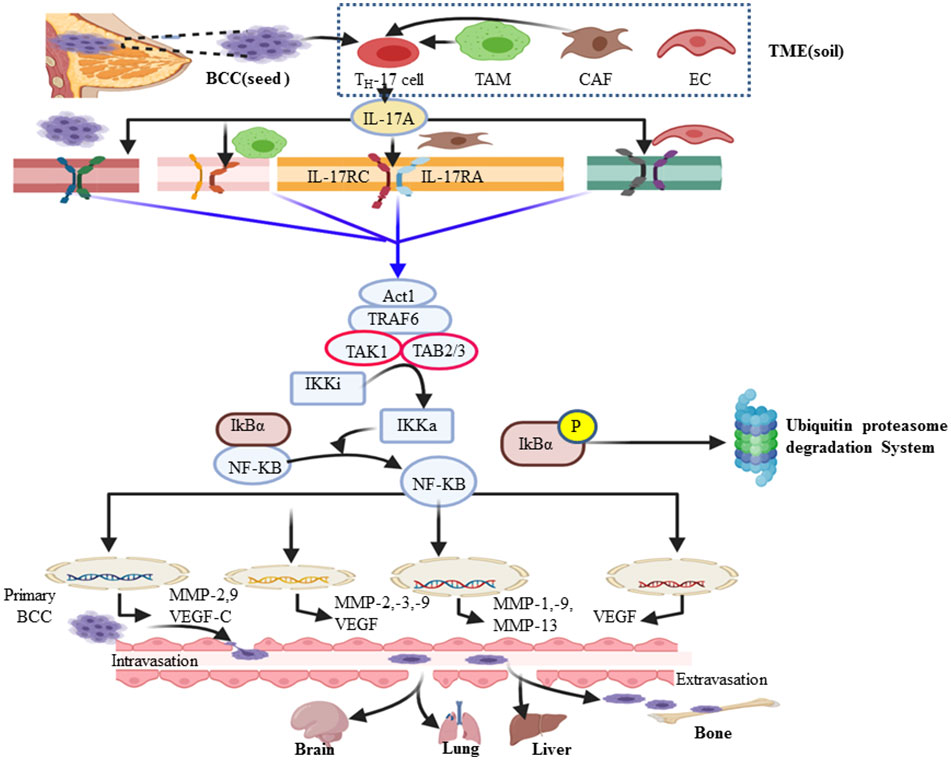
Figure 2 An overview of the IL-17A/MAPK signaling pathway in the proliferation of breast cancer. ERK1/2, JNK, and p38 MAPK are the major effector molecules of this signal cascade and lead to phosphorylate multiple transcription factors, particularly activator protein one (AP-1), which is a hetero-dimeric composition of c-Jun and c-Fos proteins. Once active, it translocates to the nucleus and orchestrates the expression and function of many proliferative genes or cell cycle regulators such as cyclin D1 (G1-to-S phase) and cyclin A/E (S-to-G2 phase). In turn, it increases cell growth or proliferation and survival.
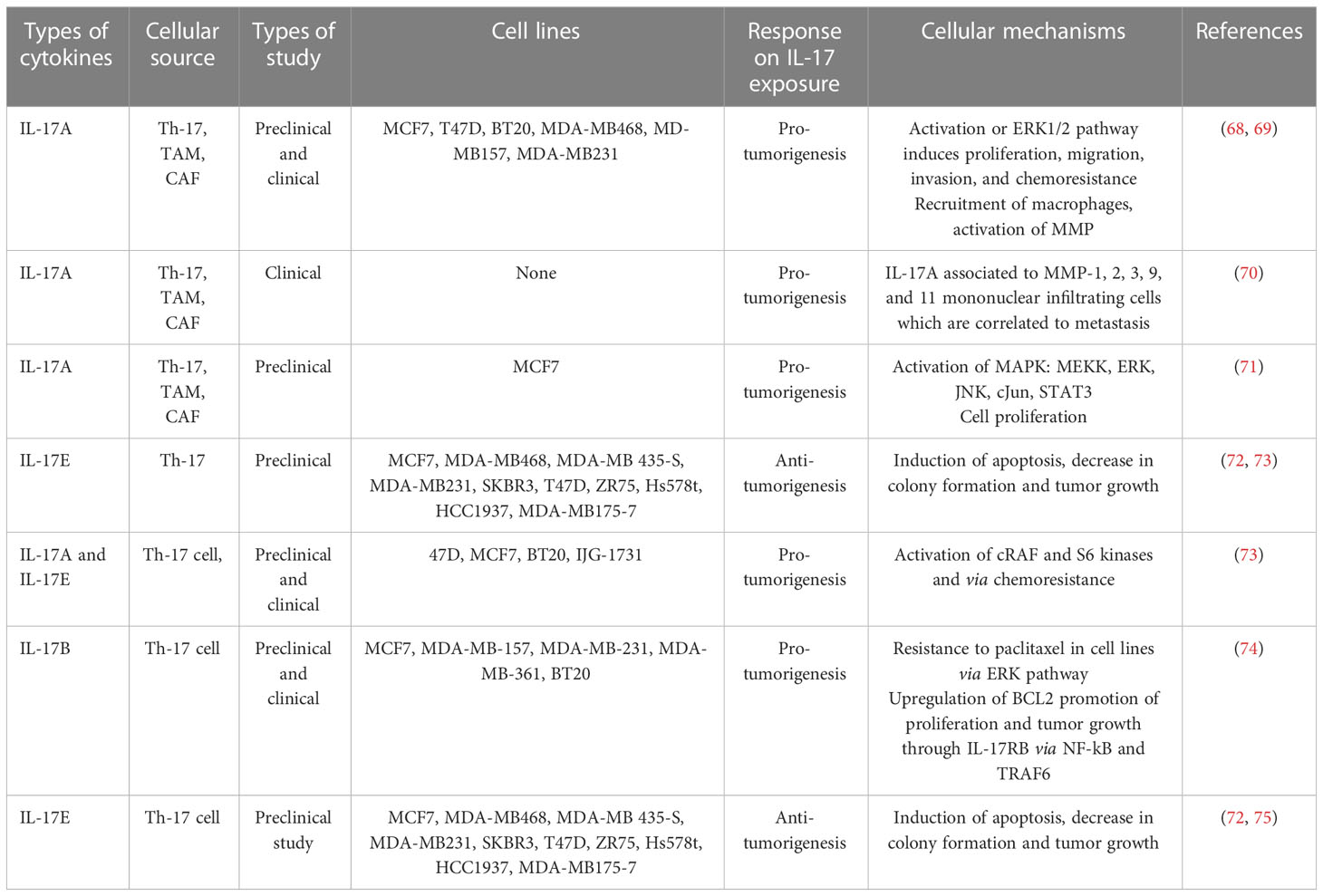
Table 1 In vivo and in vitro roles of IL-17 cytokines in breast cancer.
IL-17A//NF-kB/MMPs axis promotes bone metastatic breast cancer
Upon the interaction of IL-17A with its corresponding receptor, the u-box domain of Act1 is essential for IL-17-induced NF-kB activation (90). ACT-1-mediated ubiquitination of TRAF-6 acts as a scaffolding intermediate of the IL-17A signaling pathway (40). TRAF6 is also a signaling adaptor molecule that plays a key role as an E3 ubiquitin ligase and ubiquitin-conjugating enzyme (E2) complex composed of Ubc13 and Uev1A (91). Subsequently ubiquitinated TRAF-6 recruits a protein kinase complex involving TGF-β-activated kinase 1 (TAK1) and TAK1-binding proteins (TAB2–TAB3) (60). TAK1, a member of the MAP kinase kinase (MAP3K) family, then activates the inactive IkB kinase (IKKi) complex (IKKα/β/γ) into its activated form (IKKa) via phosphorylation (92). In turn, IKKa then phosphorylates the IκB subunit of the NF-κB/IκB complex, marking IκB for E3 ubiquitin ligase–proteasomal proteolysis (42, 43). Ubiquitin (Ub) itself can be further ubiquitinated and form a polyubiquitin (poly-Ub) chain on IκB. Then, IκB becomes recognizable by the proteasome. Ub–proteasome-based degradation of IκB makes NF-kB free of it, translocates to the nucleus, and acts on a wide spectrum of the NF-kB gene response element involved in the inflammation and metastasis of cancer (Figure 2) (93). The major cause of cancer-associated morbidity and mortality is its metastasis and colonization of other organs like bone, lung, liver, and brain in the case of BC (94, 95). Thus, cancer develops after migration to other anatomic sites, which are called secondary tumors (96). NF-kB key transcription factor plays a role in the expression and activity of MMPs (16, 17, 97). This, in turn, defines as many of the effects of IL-17A that are correlated with the TRAF-6-mediated activation of NF-kB. Therefore, the NF-kB-mediated expression of MMP-2 and -9, respectively, are the most important driving force in the invasiveness and metastasis of various human cancers such as colorectal cancer (98), hepatocellular cancer (99), nasopharyngeal carcinoma (100), and non-small cell lung cancer (101). Similarly, the researchers explored that the NF-kB-mediated invasiveness, migration, and metastasis of BC also rely on the increased expression of MMP-2, MMP-13, MMP-9, and MMP-1 (Figure 3) (102–104). MMP-13, known as collagenase-3, plays in ECM physical barrier degradation and increases the invasive capacities of the malignant cells (70). In support of this, MMP-13 mRNA and its protein expression in BC serve as independent biomarkers of poor prognosis or shorter overall survival (95,) (105). In addition, tumor-associated macrophage (M2Ф) secretes MMP-13 and MMP-3 which are involved in the promotion of metastasis via the IL-17/IL-8 axis (105–107). Similar to M2Ф, the CAF cells of the microenvironment also secretes MMP-1, MMP-11, MMP-9, and MMP-13 (Figure 3). In the clinical diagnosis of high-grade (grade-3) breast cancer, the study showed exuberantly increases MMP-2 and MMP-9 mRNA and protein expression (19, 108). Furthermore, several other studies support a pro-tumorigenesis role of IL-17 in BC, and the details are presented in Table 1. Those studies also elaborate that the level of IL-17 was increased and correlated with the expansion of the disease. Moreover, p38/NF-kB-mediated transcription products such as TNF-α, MMPs (MMP-2 and MMP-9), VEGF (also called VEGF-A and located at chromosome 6p12), VEGF-C, and PGE1/2 facilitate the invasion and metastasis of cancer (34, 41,) (109). Judah Folkman (father of angiogenesis) stated in 1974 that no tumor could grow beyond 2 mm3 unless they are vascularized, and tumors could be restricted to tiny sizes (110). New blood vessel formation from the existing vasculature establishment of a tumor blood supply plays a central role in distant metastasis in breast cancer (111, 112). A tumor cell secretes VEGF that interacts with VEGF receptors on the endothelial cell membrane and stimulates migration, proliferation, and neo-vessel formation from the adjacent established blood vessel (113). The VEGF family includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (114). VEGF-A and its receptors VEGFR-1 and VEGFR-2 play major roles in pathological angiogenesis, including tumor angiogenesis, whereas VEGF-C/D and their receptor VEGFR-3 primarily function as critical regulators of lymphangiogenesis (113). A solid tumor microenvironment responds to low oxygen tension by enhancing the hypoxia-inducible factor (HIF) response (112). As a result, evidence showed that HIF-1α and HIF-2α activate several hypoxia-inducible gene pathways involved in angiogenesis and glycolysis (115, 116). On the other hand, activated NF-kB also promotes anti-apoptotic gene transcription (Bcl-2, Bcl-XL, and BCL-W) and proliferative gene expression (cyclin D) (14, 117, 118). In turn, Bcl-2 (sometimes called master regulators of anti-apoptosis) not only promotes cancer cell proliferation and invasion but also allows the chemo- and immunotherapeutic resistance of cancer cells (119). In this regard, apoptosis evasion via the over-expression of Bcl-2 or Bcl-XL has recently been proposed as a hallmark of cancer (120). A research conducted by Cochaud et al. explored that ER-negative BC is rich in increased infiltration of IL-17A-producing cells and PDL1 levels (28, 121). IL-17 promotes the expression of CCL17 and CCL22 and facilitates Treg cell migration to suppress antitumor immunity (122, 123). Thus, IL-17A also enhances proliferation and metastasis via inhibiting tumor apoptosis and suppressing antitumor immunity (53, 55, 121, 124, 125) [i.e., through decreasing CD4 T helper 1 (TH-1) cells and increasing Treg cell] (126).
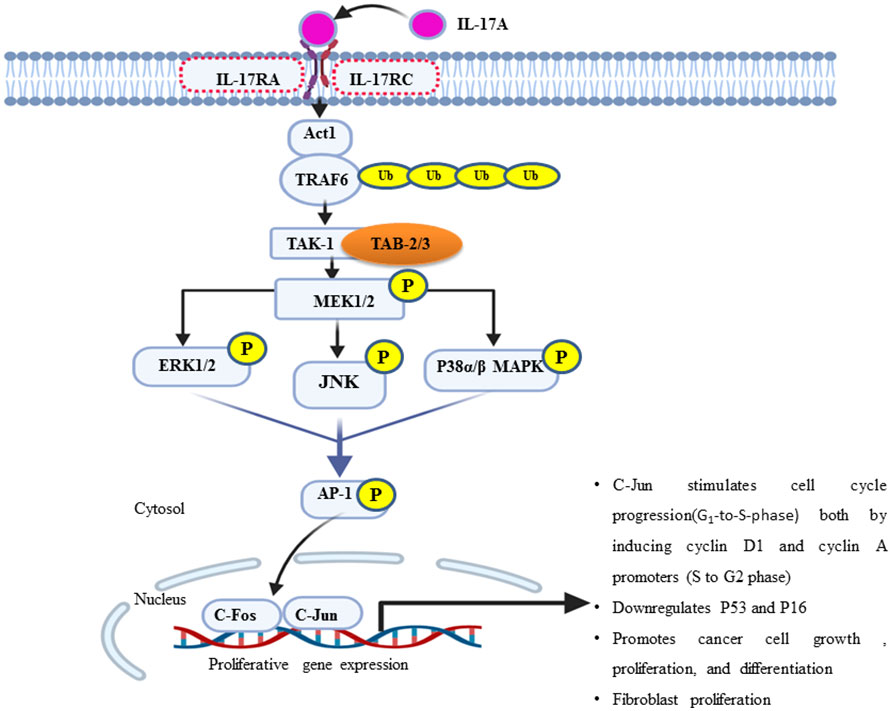
Figure 3 Diagram summarizing the mechanism of the IL-17A/NF-kB pathway in breast cancer metastasis and other organs like the brain. IL-17A can be secreted by breast cancer (BC) cells and many cells in the breast cancer microenvironment such as tumor-associated macrophages, cancer-associated fibroblasts, Th-17 cell, γδT cells, and endothelial cells. In turn, IL-17 and other Th-17 derived cytokines influence the tumor microenvironment by directly promoting transformed cell properties and the nearby stromal cell activity. IL-17A binds with IL-17RA–IL-17RC receptor and transduces signaling via the adaptor protein nuclear factor (NF)-k activator (Act1). Many IL-17 target genes contain the promoter’s region that binds with NF-kB. In turn, the NF-kB signal pathway tend to be activated and promote the expression of genes encoded for angiogenesis and metastasis. The major steps of bone metastatic BC include extravasation, circulatory journey, extravasation in distant sites, and ultimately metastatic colonization of bone or other target organs (brain, lung, and liver).
IL-17 signaling cascade as a therapeutic target of breast cancer metastasis
As elaborated well above, IL-17A is potentially significant in the growth, proliferation, and progression of human cancer, including breast cancer (28). Thus, in the animal model experiment, IL-17A is considerably a therapeutic target during the chemotherapeutic management of breast cancer since its inhibition decreases cancer progression, migration, and distant metastasis. As discussed earlier, IL-17A mediates cancer cell invasiveness and metastasis via MMP-2, MMP-9, and MMP-13. Furthermore, IL-17A stimulates MMP-9 mRNA expression, and MMP-9 inhibitors can inhibit the IL-17A-dependent invasion and metastasis of BCCs (17). The relation between IL-17A and its downstream MMP activity and breast cancer metastasis through MAPK and NF-Kb suggests the possibility of various strategies connected with blocking these checkpoints and kinase enzyme activity. Therefore, strategies target IL-17A by blocking downstream signaling molecules like MAPK or inhibiting specific gene products like MMPs. On the other hand, MMP can be inhibited simply by targeting the master transcription factor known as NF-kB.
IL-17A as a potential therapeutic target
The role of IL-17 still has controversy and needs advanced research. IL-17-producing cells of both lymphocytic and myeloid origins or the microenvironment of the cancer cell as well as their suggested pro- and antitumorigenic functions in an organ-dependent context all contribute to make it purely a challenge to bring science to clinical practice (75). In support of this, a study showed the anti-tumorigenesis effects of IL-17E exposure to the breast cancer cell lines of MCF7, MDA-MB468, MDA-MB435-S,MDA-MB231, SKBR3,T47D, ZR75, Hs578t, HCC1937, and MDA-MB175-7 (Table 1) (34, 75). Th-17 cell infiltration with a common pro-inflammatory signature cytokine, IL-17A, is a crucial player in the proliferation, growth, migration, and dissemination of many cancer cells, including BC and many more types of cancer. Table 1 shows a summary of the current correlation findings between different IL-17 subtypes producing T cells and their overall mechanism of pro-tumorigenesis in various breast cancer cell lines (75). As a result, research scholars understand the molecular mechanism of IL-17 in the development and progress of breast cancer and try to target the development of potential therapeutic options to tackle this life-threatening non-communicable disease. The Food and Drug Administration approved the fully-humanized anti-IL-17A monoclonal antibody secukinumab (AIN457) as an acceptable therapeutic choice for psoriasis, rheumatoid arthritis, ankylosing spondylitis, and other inflammatory diseases (Figure 4) (47, 127–129). Treating mice with ER- or triple-negative breast cancer by secukinumab boosts the antitumor immunity such as CD4+ and CD8+ T cells and decreases both the expression of PDL-1 and Treg cell infiltration (121). Interestingly, a combination treatment approach [anti-IL-17A (secukinumab) and anti-PDL1 (pembrolizumab)] improved antitumor immunity in support of its eradication (130).
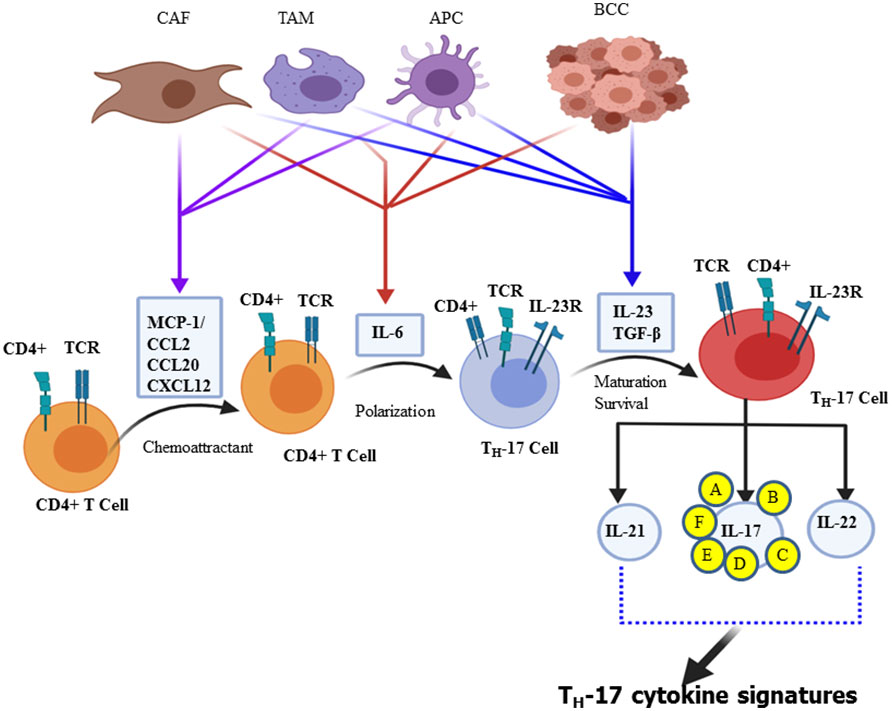
Figure 4 Schematic summary of the actions of various inhibitors of IL-17A and its multiple downstream signal transduction pathways. In addition to the synthetic and natural inhibitors of these signaling target molecules, extracellular signal-regulated kinase can be endogenously regulated by short negative feedback loop via dephosphorylation, dual-specificity phosphatase.
MAPK as a potential therapeutic target
Cochaud et al. reported that the IL-17A/IL-17AR interaction not only stimulates the proliferation and growth of the human BC cell line but also is responsible for chemoresistance (docetaxel). This pathologic mechanism of IL-17A was due to the upregulation of the ERK1/2 JNK and p38 MAPK pathway via MEK1/2 (69). Because p38 MAPK, ERK1/2, and MEK1/2 are the “Achilles heel” of tumor growth and cancer cell survival, targeting them helps negatively interrupt the typical proliferation environment of the cancer cell. The MEK1/2 inhibitor U0126 (IC50 = 0.5 uM) chemically inhibits MAPK signaling (131, 132). The ERK1/2 expression in de novo is negatively regulated by MAP kinase phosphatases (MKPs) or dual-specificity protein phosphatase through a negative feedback loop (133). In addition, ulixertinib, a reversible ATP-competitive small-molecule ERK1/2 kinase inhibitor, has shown promising results in phase 1 clinical trials (134). Furthermore, there is also a selective p38 MAPK α and β isoform inhibitor, ralimetinib (LY2228820 dimesylate), not only for BC patients but also for some other human cancer, including glioblastoma, multiple myeloma, ovarian, and lung cancer (Figure 4) (135, 136). The IC50 of LY2228820 for p38 alpha = 5.3 nM and for p38 beta = 3.2 nM. BIRB-796 (doramapimod) is also an inhibitor of p38 with IC50 for p38 alpha = 38 nM, for p38 beta = 65 nM, and for p38 delta = 520 nM (136).
NF-kB as a potential therapeutic target
In mammals, there are five prominent member of the NF-κB family of transcription factors such as p50, p52, c-Rel, RelA (p65), and RelB (137). TRAF6 is the first signal transducer in NF-kB activation via IκB kinase (IKK) complex-mediated phosphorylation of the inhibitor of NF-κB proteins (IκBs) followed by ubiquitin–proteasomal degradation in response to IL-17. The IKK complex maintains NF-kB in the inactive state (NF-kB- IκB dimer) in the cytosol of unstimulated cells. Gene profiling analysis revealed that the constitutive activation of the NF-kB may be a key regulator (138) and a driving force for the pathogenesis of a variety of solid tumors, including BC and TNBCs (139). NF-κB is a set of transcription factors that play in various inflammation and immunity-associated diseases. It is also involved in different cancer progression and survival (140). The natural compound extracted from Curcuma and its isoxazole analog has many properties, such as anti-inflammatory and antitumor properties, especially TNBC cell lines and HL-60 in human leukemia by counteracting NF-kB activation (141).
Dehydroxymethylepoxyquinomicin (DHMEQ) is a synthetic and selective inhibitor of NF-kB at the site of its translocation (142, 143). Studies have shown that a substantial reduction of the activation of NF-kB is observed in TNBC patients presently treated with DHMEQ (142). Furthermore, dimethyl fumarate also effectively blocks NF-kB activity in multiple BCC lines (144). MG132 is another synthetic compound that targets and prevents the ubiquitin–proteasome degradation of NF-kB inhibitor, IkBα, or β (Figure 4) (145, 146).
VEGF as a therapeutic target
Angiogenesis is essential for breast cancer progression and metastasis (147). The uncontrolled expression and activity of VEGF are very common in different cancer types, including BC, as reliable biomarkers for angiogenesis and vascularization (148). IL-17 induces the expression of specific chemokines like chemokine ligand 2 (CCL-2) and vascular endothelial growth factor (VEGF), which promotes IL-17-producing angiogenic macrophage, which can contribute to the microenvironment and angiogenesis (14). In support of this, a study conducted on the 4T1 BC cell line in the murine model explored that, with the administration of IL-17, VEGF mediated vasculogenesis and increased microvascular density (149, 150). The exuberant expression and circulatory detection of VEGF mRNA are predictors of poor prognosis factors (151). Thus, increased VEGF expression has been associated with poor response to tamoxifen or chemotherapy in patients with advanced breast cancer (111). Inhibiting or targeting VEGF is the most promising mode of chemotherapy for different types of solid tumors, including BC, and it also interrupts its metastatic ability. The monoclonal antibody drug bevacizumab targets and inhibits the activity of the soluble form of VEGF-A ligand due to the loss of its structural conformation. This results in inhibiting VEGF-mediated angiogenesis, metastasis, and tumor survival (148, 152). Therefore, bevacizumab led to a prominent prolongation in mean progression-free survival from 15.6 to 20.2 months (Figure 4) (153). Clinical and preclinical studies showed that exposure to trastuzumab significantly decreased VEGF in HER-2-overexpressing cells (154). Moreover, angiostatin is an endogenous inhibitor of angiogenesis or suppressor of neovascularization through negative inhibition of endothelial cell migration and proliferation. In turn, it augments tumor inhibition. Angiostatin is found naturally in humans and several other animals (152).
MMP as a therapeutic target
MMPs are potential pharmacological therapeutic targets for treating invasive breast cancer (155). Endogenously, TIMP has natural and clinically significant therapeutic effects via inhibition of many MMPs. Rebimastat, an inhibitor of MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-13, and MMP-14, significantly abolishes tumor growth and abrogates BC metastasis (156). Similarly, broad-spectrum batimastat has a broad-spectrum inhibition of virtually all MMP members (157, 158). In contrast, considering specificity and selectivity, anti-MMP-2 and anti-MMP-1—such as AG-3340, BAY 12-9566, and BMS-257291—and Ro 32-3555, respectively, are used as specific therapeutic options (155, 159). Furthermore, small molecules such as tanomastat, prinomastat, and rebimastat inhibit MMP-2, MMP-3, MMP-8, MMP-9, and MMP-13; MMP-2, MMP-3, MMP-9, MMP-13, and MMP-14; and MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-13, and MMP-14, respectively (156, 158). In addition, a murine monoclonal antibody called REGA-3G12 inhibits MMP-9 without influencing the function of MMP-2 (Figure 4) (160).
Conclusions and future direction
Breast cancer is still one of the leading threats to women’s life. In the stages of the disease, cancer can spread to distant organs, including the brain and bone, where chemotherapy is not easily accessible. Achieving effective cancer therapy is significantly hampered by inflammatory cancer microenvironments. Thus, targeting IL-17A signaling pathways provides a promising future approach to developing novel treatment options. Considering contradictory results observed in other research regarding the pro- and anti-cancer nature of the Th-17 cell, an individualized adjustment may be required with different cell lines and even in various stages of cancer to tackle or target the IL-17A downstream signaling axis. Targeting the IL-17/IL-17R axis in breast cancer as relayed in clinical and preclinical models surprisingly produces excellent outcomes due to the types of cell line stage of the disease and the exposure status of the cell with IL-17. The evidence suggested that targeting and reprogramming multiple downstream signaling pathways of IL-17A may be an essential complementary option to promote the efficacy of conventional chemotherapy to treat breast cancer metastasis. Therefore, further research is needed in the future to develop anti-cancer strategies that target IL-17 signatures and their signaling pathways.
Author contributions
TS was involved in the conception, study design, execution, acquisition of data, analysis, and interpretation, and drafting and critical review of the paper. BT and BA were involved in literature search and drafting and critical review of the paper. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to forward our deepest gratitude to the authors of the articles that we used in this review report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IL-17, interleukin-17; ACT1, NF-kappa B activator 1; MAPK, mitogen-activated protein kinase; BC, breast cancer; BCC, breast cancer cell; MMP, matrix metalloproteinases; NF-kB, nuclear factor kB; STAT-3, signal transducer and the activator of transcription 3; EGFR, epidermal growth factor receptor; ECM, extracellular matrix; APC, antigen-presenting cells; GM-CSF, granulocyte–macrophage colony-stimulating factor; VEGF, vascular endothelial growth factor; TRAF6, TNF receptor−associated factor 6; FDA, Food and Drug Administration.
References
2. Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis (2018) 5(2):77–106. doi: 10.1016/j.gendis.2018.05.001
3. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
4. Yousefi M, Nosrati R, Salmaninejad A, Dehghani S, Shahryari A, Saberi A, et al. Organ-specific metastasis of breast cancer: Molecular and cellular mechanisms underlying lung metastasis. Cell Oncol (2018) 41:123–40. doi: 10.1007/s13402-018-0376-6
5. Valastyan S, Weinberg RA. Tumor metastasis: Molecular insights and evolving paradigms. Cell (2011) 147(2):275–92. doi: 10.1016/j.cell.2011.09.024
6. Rabbani SA, Mazar AP. Evaluating distant metastases in breast cancer: From biology to outcomes. Cancer Metastasis Rev (2007) 26:663–74. doi: 10.1007/s10555-007-9085-8
7. Paget S. The distribution of secondary growths in cancer of the breast. Lancet (1889) 1889:571–3. doi: 10.1016/S0140-6736(00)49915-0
8. Wang M, Xia F, Wei Y, Wei X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res (2020) 8(1):1–20. doi: 10.1038/s41413-020-00105-1
9. Chiang AC, Massagué J. Molecular basis of metastasis. New Engl J Med (2008) 359(26):2814–23. doi: 10.1056/NEJMra0805239
10. Xu L-L, Li Z-J, Niu X-L, Deng WM. The mechanisms of IL-17A on promoting tumor metastasis. Int Rev Immunol (2017) 36(6):360–9. doi: 10.1080/08830185.2017.1356296
11. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity (2005) 22(3):285–94. doi: 10.1016/j.immuni.2005.01.011
12. O'Brien RL, Roark CL, Born WK. IL-17-producing γδ T cells. Eur J Immunol (2009) 39(3):662–6. doi: 10.1002/eji.200839120
13. Karpisheh V, Ahmadi M, Abbaszadeh-Goudarzi K, Mohammadpour Saray M, Barshidi A, Mohammadi H, et al. The role of Th17 cells in the pathogenesis and treatment of breast cancer. Cancer Cell Int (2022) 22(1):108. doi: 10.1186/s12935-022-02528-8
14. Yang B, Kang H, Fung A, Zhao H, Wang T, Ma D, et al. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflammation (2014) 2014:12. doi: 10.1155/2014/623759
15. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct targeted Ther (2020) 5(1):1–17. doi: 10.1038/s41392-020-0134-x
16. Feng M, Wang Y, Chen K, Bian Z, Wu J, Gao Q, et al. IL-17A promotes the migration and invasiveness of cervical cancer cells by coordinately activating MMPs expression via the p38/NF-κB signal pathway. PloS One (2014) 9(9):e108502. doi: 10.1371/journal.pone.0108502
17. Koslawsky D, Zaretsky M, Alcalay R, Mazor O, Aharoni A, Papo N, et al. A bi-specific inhibitor targeting IL-17A and MMP-9 reduces invasion and motility in MDA-MB-231 cells. Oncotarget (2018) 9(47):28500. doi: 10.18632/oncotarget.25526
18. Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Trans Sci (2017) 147:1–73. doi: 10.1016/bs.pmbts.2017.02.005
19. Balduyck M, Zerimech F, Gouyer V, Lemaire R, Hemon B, Grard G, et al. Specific expression of matrix metalloproteinases 1, 3, 9 and 13 associated with invasiveness of breast cancer cells in vitro. Clin Exp metastasis (2000) 18(2):171–8. doi: 10.1023/A:1006762425323
20. Eng LG, Dawood S, Sopik V, Haaland B, Tan PS, Bhoo-Pathy N, et al. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Res Treat (2016) 160(1):145–52. doi: 10.1007/s10549-016-3974-x
21. Westphal T, Gampenrieder SP, Rinnerthaler G, Yuan C, Zeng Z, Zhang L, et al. Cure in metastatic breast cancer. memo-Magaz Eur Med Oncol (2018) 11(3):172–9. doi: 10.1007/s12254-018-0426-9
22. Chen WC, Lai YH, Chen HY, Guo HR, Su IJ, Chen HH, et al. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology (2013) 63(2):225–33. doi: 10.1111/his.12156
23. Fasoulakis Z, Kolios G, Papamanolis V, Kontomanolis EN. Interleukins associated with breast cancer. Cureus (2018) 10(11). doi: 10.7759/cureus.3549
24. Sun J, Cui H, Gao Y, Xu H, Deng L, Shi Y, et al. TGF-α overexpression in breast cancer bone metastasis and primary lesions and TGF-α enhancement of expression of procancer metastasis cytokines in bone marrow mesenchymal stem cells. BioMed Res Int (2018) 2018. doi: 10.1155/2018/6565393
25. Huang Y, Ma C, Zhang Q, Ye J, Wang F, Zhang Y, et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget (2015) 6(19):17462. doi: 10.18632/oncotarget.3958
26. Abotaleb M, Kubatka P, Caprnda M, Varghese E, Zolakova B, Zubor P, et al. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed Pharmacother (2018) 101:458–77. doi: 10.1016/j.biopha.2018.02.108
27. Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol (2013) 182(1):10–20. doi: 10.1016/j.ajpath.2012.08.041
28. Cochaud S, Giustiniani J, Thomas C, Laprevotte E, Garbar C, Savoye AM, et al. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci Rep (2013) 3(1):1–10. doi: 10.1038/srep03456
29. Ferraro MG, Piccolo M, Misso G, Maione F, Montesarchio D, Caraglia M, et al. Breast cancer chemotherapeutic options: A general overview on the preclinical validation of a multi-target ruthenium (III) complex lodged in nucleolipid nanosystems. Cells (2020) 9(6):1412. doi: 10.3390/cells9061412
30. Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol (2006) 6(4):329–34. doi: 10.1038/nri1807
31. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
32. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends Immunol (2017) 38(5):310–22. doi: 10.1016/j.it.2017.01.006
33. Mojsilović S, Jauković A, Santibañez JF, Bugarski D. Interleukin-17 and its implication in the regulation of differentiation and function of hematopoietic and mesenchymal stem cells. Mediators Inflammation (2015) 2015. doi: 10.1155/2015/470458
34. Fabre JAS, Giustiniani J, Garbar C, Merrouche Y, Antonicelli F, Bensussan A. The interleukin-17 family of cytokines in breast cancer. Int J Mol Sci (2018) 19(12):3880. doi: 10.3390/ijms19123880
35. Chen X-W, Zhou S-F. Inflammation, cytokines, the IL-17/IL-6/STAT3/NF-κB axis, and tumorigenesis. Drug Design Dev Ther (2015) 9:2941. doi: 10.2147/DDDT.S86396
36. Potteaux S, Lehmann-Che J, Bensussan A, Le Naour R, Merrouche Y. Multiple roles of the interleukin IL-17 members in breast cancer and beyond. J Cell Immunol (2020) 2(2):55–64.
37. Muls N, Nasr Z, Dang HA, Sindic C, Van Pesch V. IL-22, GM-CSF and IL-17 in peripheral CD4+ T cell subpopulations during multiple sclerosis relapses and remission. impact of corticosteroid therapy. PloS One (2017) 12(3):e0173780. doi: 10.1371/journal.pone.0173780
38. Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine (2008) 41(2):84–91. doi: 10.1016/j.cyto.2007.09.014
39. Brembilla NC, Senra L, Boehncke W-H. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
40. Bulek K, Li X. TRAF regulation of IL-17 cytokine signaling. Front Immunol (2019) 10:1293. doi: 10.3389/fimmu.2019.01293
41. Zhu X, Mulcahy LA, Mohammed RA, Lee AH, Franks HA, Kilpatrick L, et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res (2008) 10(6):1–11. doi: 10.1186/bcr2195
42. Shen F, Gaffen SL. Structure–function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine (2008) 41(2):92–104. doi: 10.1016/j.cyto.2007.11.013
43. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17–dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol (2007) 8(3):247–56. doi: 10.1038/ni1439
44. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol (2019) 16(7):634–43. doi: 10.1038/s41423-019-0220-6
45. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
46. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell (2012) 151(2):289–303. doi: 10.1016/j.cell.2012.09.016
47. Shibabaw T. Inflammatory cytokine: IL-17A signaling pathway in patients present with COVID-19 and current treatment strategy. J Inflammation Res (2020) 13:673–80. doi: 10.2147/JIR.S278335
48. Kuwabara T, Ishikawa F, Kondo M, Antonicelli F, Merrouche Y, Bensussan A, et al. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflammation (2017) 2017. doi: 10.1155/2017/3908061
49. Fabre J, Giustiniani J, Garbar C, Nakae S. Targeting the tumor microenvironment: The protumor effects of IL-17 related to cancer type. Int J Mol Sci (2016) 17(9):1433. doi: 10.3390/ijms17091433
50. Miossec P. Update on interleukin-17: A role in the pathogenesis of inflammatory arthritis and implication for clinical practice. RMD Open (2017) 3(1):e000284. doi: 10.1136/rmdopen-2016-000284
51. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34(2):149–62. doi: 10.1016/j.immuni.2011.02.012
52. Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology (2011) 134(1):8–16. doi: 10.1111/j.1365-2567.2011.03465.x
53. Welte T, Zhang XH-F. Interleukin-17 could promote breast cancer progression at several stages of the disease. Mediators Inflammation (2015) 2015:804347. doi: 10.1155/2015/804347
54. Miossec P, Kolls JK. Targeting IL-17 and TH 17 cells in chronic inflammation. Nat Rev Drug discovery. (2012) 11(10):763–76. doi: 10.1038/nrd3794
55. Méndez-García LA, Nava-Castro KE, Ochoa-Mercado T, Palacios-Arreola MI, Ruiz-Manzano RA, Segovia-Mendoza M, et al. Breast cancer metastasis: Are cytokines important players during its development and progression? J Interferon Cytokine Res (2019) 39(1):39–55. doi: 10.1089/jir.2018.0024
56. Goepfert A, Lehmann S, Wirth E, Rondeau JM. The human IL-17A/F heterodimer: a two-faced cytokine with unique receptor recognition properties. Sci Rep (2017) 7(1):1–13. doi: 10.1038/s41598-017-08360-9
57. Romano M, De Francesco F, Zarantonello L, et al. From inflammation to cancer in inflammatory bowel disease: molecular perspectives. Anticancer Res (2016) 36(4):1447–60.
58. Neill DR, McKenzie AN. TH9: The latest addition to the expanding repertoire of IL-25 targets. Immunol Cell Biol (2010) 88(5):502–4. doi: 10.1038/icb.2010.43
59. Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Cytokine (2013) 62(2):175–82. doi: 10.1016/j.cyto.2013.03.014
60. Kawai T, Akira S. Signaling to NF-κB by toll-like receptors. Trends Mol Med (2007) 13(11):460–9. doi: 10.1016/j.molmed.2007.09.002
61. Bie Q, Jin C, Zhang B, Dong H. IL-17B: A new area of study in the IL-17 family. Mol Immunol (2017) 90:50–6. doi: 10.1016/j.molimm.2017.07.004
62. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harbor Perspect Biol (2018) 10(4):a028522. doi: 10.1101/cshperspect.a028522
63. Huang F, Kao C-Y, Wachi S, Thai P, Ryu J, Wu R, et al. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-κB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol (2007) 179(10):6504–13. doi: 10.4049/jimmunol.179.10.6504
64. Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine (2008) 41(2):105–13. doi: 10.1016/j.cyto.2007.09.015
65. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64(2):477–85. doi: 10.1016/j.cyto.2013.07.022
66. Furuta S, Jeng Y-M, Zhou L, Huang L, Kuhn I, Bissell MJ, et al. IL-25 causes apoptosis of IL-25R–expressing breast cancer cells without toxicity to nonmalignant cells. Sci Trans Med (2011) 3(78):78ra31–1. doi: 10.1126/scitranslmed.3001374
67. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter, et al. Signaling through enzyme-linked cell-surface receptors. In: Molecular biology of the cell 4th edition. New York: Garland Science (2002). Available at: https://www.ncbi.nlm.nih.gov/books/NBK26822/
68. Song X, Wei C, Li X. The potential role and status of IL-17 family cytokines in breast cancer. Int Immunopharmacol (2021) 95:107544. doi: 10.1016/j.intimp.2021.107544
69. Cochaud S, Giustiniani J, Thomas C, Laprevotte E, Garbar C, Savoye AM, et al. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci Rep (2013) 3:3456. doi: 10.1038/srep03456
70. Culhaci N, Metin K, Copcu E, Dikicioglu E. Elevated expression of MMP-13 and TIMP-1 in head and neck squamous cell carcinomas may reflect increased tumor invasiveness. BMC Canc (2004) 4(1):1–8. doi: 10.1186/1471-2407-4-42
71. Kim G, Khanal P, Lim SC, Yun HJ, Ahn SG, Ki SH, et al. Interleukin-17 induces AP-1 activity and cellular transformation via upregulation of tumor progression locus 2 activity. Carcinogenesis (2013) 34(2):341–50. doi: 10.1093/carcin/bgs342
72. Furuta S, Jeng YM, Zhou L, Huang L, Kuhn I, Bissell MJ, et al. IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells. Sci Transl Med (2011) 3(78):78ra31. doi: 10.1126/scitranslmed.3001374
73. Mombelli S, Cochaud S, Merrouche Y, Garbar C, Antonicelli F, Laprevotte E, et al. IL-17A and its homologs IL-25/IL-17E recruit the c-RAF/S6 kinase pathway and the generation of pro-oncogenic LMW-e in breast cancer cells. Sci Rep (2015) 5:11874. doi: 10.1038/srep11874
74. Huang CK, Yang CY, Jeng YM, et al. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene (2014) 33(23):2968–77. doi: 10.1038/onc.2013.268
75. Kuen DS, Kim BS, Chung Y. IL-17-Producing cells in tumor immunity: Friends or foes? Immune net (2020) 20(1):e6. doi: 10.4110/in.2020.20.e6
76. Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cell Mol Immunol (2010) 7(3):164–74. doi: 10.1038/cmi.2010.21
77. Li X, Bechara R, Zhao J, et al. IL-17 receptor–based signaling and implications for disease. Nat Immunol (2019) 20(12):1594–602. doi: 10.1038/s41590-019-0514-y
78. Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Canc (2003) 3(11):859–68. doi: 10.1038/nrc1209
79. Lopez-Bergami P, Lau E, Ronai Ze. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Canc (2010) 10(1):65–76. doi: 10.1038/nrc2681
80. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (2016) 94(12):1313–26. doi: 10.1007/s00109-016-1475-3
81. Schreiber M, Kolbus A, Piu F, Szabowski A, Möhle-Steinlein U, Tian J, et al. Control of cell cycle progression by c-jun is p53 dependent. Genes Dev (1999) 13(5):607–19. doi: 10.1101/gad.13.5.607
82. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci (2004) 117(25):5965–73. doi: 10.1242/jcs.01589
83. Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol (2009) 220(2):292–6. doi: 10.1002/jcp.21791
84. Mombelli S, Cochaud S, Merrouche Y, Garbar C, Antonicelli F, Laprevotte E, et al. IL-17A and its homologs IL-25/IL-17E recruit the c-RAF/S6 kinase pathway and the generation of pro-oncogenic LMW-e in breast cancer cells. Sci Rep (2015) 5(1):1–10. doi: 10.1038/srep11874
85. Flynn JF, Wong C, Wu JM. Anti-EGFR therapy: mechanism and advances in clinical efficacy in breast cancer. J Oncol (2009) 2009:526963. doi: 10.1155/2009/526963
86. Zhu C, Qi X, Chen Y, Sun B, Dai Y, Gu Y, et al. PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in IGF-1-induced VEGF-c upregulation in breast cancer. J Cancer Res Clin Oncol (2011) 137(11):1587. doi: 10.1007/s00432-011-1049-2
87. Rosenthal DT, Iyer H, Escudero S, Bao L, Wu Z, Ventura AC, et al. p38γ promotes breast cancer cell motility and metastasis through regulation of RhoC GTPase, cytoskeletal architecture, and a novel leading edge behavior. Cancer Res (2011) 71(20):6338–49. doi: 10.1158/0008-5472.CAN-11-1291
88. Meng F, Zhang H, Liu G, et al. p38γ mitogen-activated protein kinase contributes to oncogenic properties maintenance and resistance to poly (ADP-ribose)-polymerase-1 inhibition in breast cancer. Neoplasia (2011) 13(5):472–82. doi: 10.1593/neo.101748
89. Patnaik A, Haluska P, Tolcher AW, Erlichman C, Papadopoulos KP, Lensing JL, et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 dimesylate), in patients with advanced cancer. Clin Cancer Res (2016) 22(5):1095–102. doi: 10.1158/1078-0432.CCR-15-1718
90. Wu L, Zepp J, Li X. Function of Act1 in IL-17 family signaling and autoimmunity. In: Current topics in innate immunity II. Springer (2012). 946:223–35. doi: 10.1007/978-1-4614-0106-3_13
91. Dainichi T, Matsumoto R, Mostafa A, Kabashima K. Immune control by TRAF6-mediated pathways of epithelial cells in the EIME (epithelial immune microenvironment). Front Immunol (2019) 10:1107. doi: 10.3389/fimmu.2019.01107
92. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell (2000) 103(2):351–61. doi: 10.1016/S0092-8674(00)00126-4
93. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
94. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev oncogenesis (2013) 18(1-2):43. doi: 10.1615/CritRevOncog.v18.i1-2.40
95. Zhang B, Cao X, Liu Y, Cao W, Zhang F, Zhang S, et al. Tumor-derived matrix metalloproteinase-13 (MMP-13) correlates with poor prognosis of invasive breast cancer. BMC Canc (2008) 8(1):83. doi: 10.1186/1471-2407-8-83
96. Hanahan D, Weinberg RA. The hallmarks of cancer. cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
97. Weng C-J, Chau C-F, Hsieh Y-S, Yang SF, Yen GC. Lucidenic acid inhibits PMA-induced invasion of human hepatoma cells through inactivating MAPK/ERK signal transduction pathway and reducing binding activities of NF-κB and AP-1. Carcinogenesis (2008) 29(1):147–56. doi: 10.1093/carcin/bgm261
98. Ren H, Wang Z, Zhang S, Ma H, Wang Y, Jia L, et al. IL-17A promotes the migration and invasiveness of colorectal cancer cells through NF-κB-Mediated MMP expression. Oncol Res Featuring Preclinical Clin Cancer Ther (2016) 23(5):249–56. doi: 10.3727/096504016X14562725373716
99. Li J, Lau GK-K, Chen L, Dong SS, Lan HY, Huang XR, et al. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PloS One (2011) 6(7):e21816. doi: 10.1371/journal.pone.0021816
100. Wang L, Ma R, Kang Z, Zhang Y, Ding H, Guo W, et al. Effect of IL-17A on the migration and invasion of NPC cells and related mechanisms. PloS One (2014) 9(9):e108060. doi: 10.1371/journal.pone.0108060
101. Wu Z, He D, Zhao S, Wang H. IL-17A/IL-17RA promotes invasion and activates MMP-2 and MMP-9 expression via p38 MAPK signaling pathway in non-small cell lung cancer. Mol Cell Biochem (2019) 455(1-2):195–206. doi: 10.1007/s11010-018-3483-9
102. Roy LD, Sahraei M, Schettini JL, Gruber HE, Besmer DM, Mukherjee P, et al. Systemic neutralization of IL-17A significantly reduces breast cancer associated metastasis in arthritic mice by reducing CXCL12/SDF-1 expression in the metastatic niches. BMC Canc (2014) 14(1):225. doi: 10.1186/1471-2407-14-225
103. Bartsch JE, Staren ED, Appert HE. Matrix metalloproteinase expression in breast cancer. J Surg Res (2003) 110(2):383–92. doi: 10.1016/S0022-4804(03)00007-6
104. Kotepui M, Punsawad C, Chupeerach C, Songsri A, Charoenkijkajorn L, Petmitr S, et al. Differential expression of matrix metalloproteinase-13 in association with invasion of breast cancer. Contemp Oncol (2016) 20(3):225. doi: 10.5114/wo.2016.61565
105. Pivetta E, Scapolan M, Pecolo M, Wassermann B, Abu-Rumeileh I, Balestreri L, et al. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res (2011) 13(5):R105. doi: 10.1186/bcr3047
106. Park S-H, Kim J-H, Lee D-H, Kang JW, Song HH, Oh SR, et al. Luteolin 8-c-β-fucopyranoside inhibits invasion and suppresses TPA-induced MMP-9 and IL-8 via ERK/AP-1 and ERK/NF-κB signaling in MCF-7 breast cancer cells. Biochimie (2013) 95(11):2082–90. doi: 10.1016/j.biochi.2013.07.021
107. Han J, Bae SY, Oh SJ, Lee J, Lee JH, Lee HC, et al. Zerumbone suppresses IL-1β-induced cell migration and invasion by inhibiting IL-8 and MMP-3 expression in human triple-negative breast cancer cells. Phytother Res (2014) 28(11):1654–60. doi: 10.1002/ptr.5178
108. Sullu Y, Demirag GG, Yildirim A, Karagoz F, Kandemir B. Matrix metalloproteinase-2 (MMP-2) and MMP-9 expression in invasive ductal carcinoma of the breast. Pathology-Res Practice. (2011) 207(12):747–53. doi: 10.1016/j.prp.2011.09.010
109. Numasaki M, Fukushi J-i, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood (2003) 101(7):2620–7. doi: 10.1182/blood-2002-05-1461
110. Folkman J. Tumor angiogenesis. Adv Cancer Res (1974) 19:331–58. doi: 10.1016/S0065-230X(08)60058-5
111. Schneider BP, Miller KD. Angiogenesis of breast cancer. J Clin Oncol (2005) 23(8):1782–90. doi: 10.1200/JCO.2005.12.017
112. Fox SB, Generali DG, Harris AL. Breast tumour angiogenesis. Breast Cancer Res (2007) 9(6):1–11. doi: 10.1186/bcr1796
113. Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. Genes Canc (2011) 2(12):1097–105. doi: 10.1177/1947601911423031
114. Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol (2005) 6(2):1–10. doi: 10.1186/gb-2005-6-2-209
115. Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von hippel-lindau-associated renal cell carcinoma. Mol Cell Biol (2005) 25(13):5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005
116. Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J Cell Mol Med (2005) 9(4):777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x
117. Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-κ b transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene (2003) 22(1):90–7. doi: 10.1038/sj.onc.1206056
118. Carrington EM, Zhan Y, Brady JL, Zhang JG, Sutherland RM, Anstee NS, et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Different (2017) 24(5):878–88. doi: 10.1038/cdd.2017.30
119. Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis (2018) 9(2):1–14. doi: 10.1038/s41419-017-0035-2
120. García-Aranda M, Pérez-Ruiz E, Redondo M. Bcl-2 inhibition to overcome resistance to chemo-and immunotherapy. Int J Mol Sci (2018) 19(12):3950. doi: 10.3390/ijms19123950
121. Ma Y-F, Chen C, Li D, Liu M, Lv ZW, Ji Y, et al. Targeting of interleukin (IL)-17A inhibits PDL1 expression in tumor cells and induces anticancer immunity in an estrogen receptor-negative murine model of breast cancer. Oncotarget (2017) 8(5):7614. doi: 10.18632/oncotarget.13819
122. Yang Z, Zhang B, Li D, et al. Mast cells mobilize myeloid-derived suppressor cells and treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PloS One (2010) 5(1):e8922. doi: 10.1371/journal.pone.0008922
123. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV. Regulatory T cells exhibit distinct features in human breast cancer. Immunity (2016) 45(5):1122–34. doi: 10.1016/j.immuni.2016.10.032
124. Dawod B, Liu J, Gebremeskel S, Yan C, Sappong A, Johnston B, et al. Myeloid-derived suppressor cell depletion therapy targets IL-17A-expressing mammary carcinomas. Sci Rep (2020) 10(1):1–17. doi: 10.1038/s41598-020-70231-7
125. Qian X, Chen H, Wu X, Hu L, Huang Q, Jin Y, et al. Interleukin-17 acts as double-edged sword in anti-tumor immunity and tumorigenesis. Cytokine (2017) 89:34–44. doi: 10.1016/j.cyto.2015.09.011
126. Egen JG, Ouyang W, Wu LC. Human anti-tumor immunity: Insights from immunotherapy clinical trials. Immunity (2020) 52(1):36–54. doi: 10.1016/j.immuni.2019.12.010
127. Cafarotti S. Severe acute respiratory syndrome–Coronavirus-2 infection and patients with lung cancer: The potential role of interleukin-17 target therapy. J Thorac Oncol (2020) 15(7):e101–3. doi: 10.1016/j.jtho.2020.04.015
128. Ritchlin CT, Krueger JG. New therapies for psoriasis and psoriatic arthritis. Curr Opin Rheumatol (2016) 28(3):204. doi: 10.1097/BOR.0000000000000274
129. Fletcher JM, Moran B, Petrasca A, et al. IL-17 in inflammatory skin diseases psoriasis and hidradenitis suppurativa. Clin Exp Immunol (2020) 1(2):121–34. doi: 10.1111/cei.13449
130. Planes-Laine G, Rochigneux P, Bertucci F, Chrétien AS, Viens P, Sabatier R, et al. PD-1/PD-L1 targeting in breast cancer: The first clinical evidences are emerging–a literature review. Cancers (2019) 11(7):1033. doi: 10.3390/cancers11071033
131. Newton R, Cambridge L, Hart LA, Stevens DA, Lindsay MA, Barnes PJ, et al. The MAP kinase inhibitors, PD098059, UO126 and SB203580, inhibit IL-1β-dependent PGE2 release via mechanistically distinct processes. Br J Pharmacol (2000) 130(6):1353–61. doi: 10.1038/sj.bjp.0703431
132. Zlobina M, Steblyanko YY, Shklyaeva M, Kharchenko VV, Salova AV, Kornilova ES. MAP-kinase pathway inhibitors U0126 and PD98059 differentially affect organization of the tubulin cytoskeleton after stimulation of EGF receptor endocytosis. Cell Tissue Biol (2015) 9(4):290–8. doi: 10.1134/S1990519X15040124
133. Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther (2018) 187:45–60. doi: 10.1016/j.pharmthera.2018.02.007
134. Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer discovery. (2018) 8(2):184–95. doi: 10.1158/2159-8290.CD-17-1119
135. Cicenas J, Zalyte E, Rimkus A, et al. JNK, p38, ERK, and SGK1 inhibitors in cancer. Multidiscip Digital Publishing Institute (2018) 10(1):1. doi: 10.3390/cancers10010001
136. Campbell RM, Anderson BD, Brooks NA, Brooks HB, Chan EM, De Dios A, et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol Cancer Ther (2014) 13(2):364–74. doi: 10.1158/1535-7163.MCT-13-0513
137. Eluard B, Thieblemont C, Baud V. NF-κB in the new era of cancer therapy. Trends Cancer (2020) 6(8):677–87. doi: 10.1016/j.trecan.2020.04.003
138. Ossovskaya V, Wang Y, Budoff A, Xu Q, Lituev A, Potapova O, et al. Exploring molecular pathways of triple-negative breast cancer. Genes Canc (2011) 2(9):870–9. doi: 10.1177/1947601911432496
139. Barbie TU, Alexe G, Aref AR, Li S, Zhu Z, Zhang X, et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J Clin Invest (2014) 124(12):5411–23. doi: 10.1172/JCI75661
140. Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harbor Perspect Biol (2009) 1(4):a000034. doi: 10.1101/cshperspect.a000034
141. Labbozzetta M, Notarbartolo M, Poma P. Can NF-κB be considered a valid drug target in neoplastic diseases? our point of view. Int J Mol Sci (2020) 21(9):3070. doi: 10.3390/ijms21093070
142. Poma P, Labbozzetta M, D'Alessandro N, Notarbartolo M. NF-κB is a potential molecular drug target in triple-negative breast cancers. OMICS: A J Integr Biol (2017) 21(4):225–31. doi: 10.1089/omi.2017.0020
143. Laprevotte E, Cochaud S, Du Manoir S, Lapierre M, Dejou C, Philippe M, et al. The IL-17B-IL-17 receptor b pathway promotes resistance to paclitaxel in breast tumors through activation of the ERK1/2 pathway. Oncotarget (2017) 8(69):113360. doi: 10.18632/oncotarget.23008
144. Kastrati I, Siklos MI, Calderon-Gierszal EL, El-Shennawy L, Georgieva G, Thayer EN, et al. Dimethyl fumarate inhibits the nuclear factor κB pathway in breast cancer cells by covalent modification of p65 protein. J Biol Chem (2016) 291(7):3639–47. doi: 10.1074/jbc.M115.679704
145. Minn AJ, Bevilacqua E, Yun J, Rosner MR, et al. Identification of novel metastasis suppressor signaling pathways for breast cancer. Cell Cycle (2012) 11(13):2452–7. doi: 10.4161/cc.20624
146. Narayanan S, Cai C-Y, Assaraf YG, Guo HQ, Cui Q, Wei L, et al. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Res Updates. (2020) 48:100663. doi: 10.1016/j.drup.2019.100663
147. Longatto Filho A, Lopes JM, Schmitt FC. Angiogenesis and breast cancer. J Oncol (2010) 2010:576384. doi: 10.1155/2010/576384
148. Kristensen TB, Knutsson ML, Wehland M, Laursen BE, Grimm D, Warnke E, et al. Anti-vascular endothelial growth factor therapy in breast cancer. Int J Mol Sci (2014) 15(12):23024–41. doi: 10.3390/ijms151223024
149. Du J-W, Xu K-Y, Fang L-Y, et al. Interleukin-17, produced by lymphocytes, promotes tumor growth and angiogenesis in a mouse model of breast cancer. Mol Med Rep (2012) 6(5):1099–102. doi: 10.3892/mmr.2012.1036
150. Bian G, Zhao WY. To the Editor IL-17, an important prognostic factor and potential therapeutic target for breast cancer? Eur J Immunol (2014) 44(2):604–5. doi: 10.1002/eji.201343875
151. Bando H. Vascular endothelial growth factor and bevacitumab in breast cancer. Breast Canc (2007) 14(2):163–73. doi: 10.2325/jbcs.968
152. Madu CO, Wang S, Madu CO, et al. Angiogenesis in breast cancer progression, diagnosis, and treatment. J Canc (2020) 11(15):4474. doi: 10.7150/jca.44313
153. Dickler MN, Barry WT, Cirrincione CT, et al. Phase III trial evaluating letrozole as first-line endocrine therapy with or without bevacizumab for the treatment of postmenopausal women with hormone receptor–positive advanced-stage breast cancer: CALGB 40503 (Alliance). J Clin Oncol (2016) 34(22):2602. doi: 10.1200/JCO.2015.66.1595
154. Epstein M, Ayala R, Tchekmedyian N, et al. HER2-overexpressing human breast cancer xenografts exhibit increased angiogenic potential mediated by vascular endothelial growth factor (VEGF). Breast Cancer Res Treat (2002) (233 SPRING ST, NEW YORK, NY 10013 USA: SPRINGER) 76:S143–S143.
155. Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Institute. (2001) 93(3):178–93. doi: 10.1093/jnci/93.3.178
156. Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther (2018) 17(6):1147–55. doi: 10.1158/1535-7163.MCT-17-0646
157. Kumar A, Bhatnagar S, Kumar A. Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am J Pathol (2010) 177(1):248–60. doi: 10.2353/ajpath.2010.091176
158. Mohan V, Talmi-Frank D, Arkadash V, et al. Matrix metalloproteinase protein inhibitors: highlighting a new beginning for metalloproteinases in medicine. Metalloproteinases Med (2016) 3:31–47. doi: 10.2147/MNM.S65143
159. Fields GB. Mechanisms of action of novel drugs targeting angiogenesis-promoting matrix metalloproteinases. Front Immunol (2019) 10:1278. doi: 10.3389/fimmu.2019.01278
Keywords: bone metastasis, breast cancer, inflammation, antitumor effects, MAPK, NF-kB, MMPs, interleukin-17A
Citation: Shibabaw T, Teferi B and Ayelign B (2023) The role of Th-17 cells and IL-17 in the metastatic spread of breast cancer: As a means of prognosis and therapeutic target. Front. Immunol. 14:1094823. doi: 10.3389/fimmu.2023.1094823
Received: 10 November 2022; Accepted: 21 February 2023;
Published: 13 March 2023.
Edited by:
Yusuf Omosun, Morehouse School of Medicine, United StatesReviewed by:
Pooya Farhangnia, Iran University of Medical Sciences, IranZhen Wang, Zhejiang University, China
Chanitra Thuwajit, Mahidol University, Thailand
Copyright © 2023 Shibabaw, Teferi and Ayelign. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Birhanu Ayelign, YmlyaGFudWF5ZWxpZ240MkBnbWFpbC5jb20=