 Eleni Maneta1*†
Eleni Maneta1*† Evmorfia Aivalioti1†
Evmorfia Aivalioti1† Simon Tual-Chalot2
Simon Tual-Chalot2 Besa Emini Veseli3
Besa Emini Veseli3 Aikaterini Gatsiou2
Aikaterini Gatsiou2 Kimon Stamatelopoulos1,4
Kimon Stamatelopoulos1,4 Konstantinos Stellos2,3,5,6*
Konstantinos Stellos2,3,5,6*- 1Department of Clinical Therapeutics, National and Kapodistrian University of Athens Medical School, Athens, Greece
- 2Biosciences Institute, Vascular Biology and Medicine Theme, Faculty of Medical Sciences, Newcastle University, Newcastle Upon Tyne, United Kingdom
- 3Department of Cardiovascular Research, European Center for Angioscience (ECAS), Heidelberg University, Mannheim, Germany
- 4Translational and Clinical Research Institute, Vascular Biology and Medicine Theme, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom
- 5German Centre for Cardiovascular Research (DZHK), Partner Site Heidelberg/Mannheim, Mannheim, Germany
- 6Department of Cardiology, University Hospital Mannheim, University of Heidelberg, Mannheim, Germany
Sepsis is a life-threatening clinical syndrome characterized by multiorgan dysfunction caused by a dysregulated or over-reactive host response to infection. During sepsis, the coagulation cascade is triggered by activated cells of the innate immune system, such as neutrophils and monocytes, resulting in clot formation mainly in the microcirculation, a process known as immunothrombosis. Although this process aims to protect the host through inhibition of the pathogen’s dissemination and survival, endothelial dysfunction and microthrombotic complications can rapidly lead to multiple organ dysfunction. The development of treatments targeting endothelial innate immune responses and immunothrombosis could be of great significance for reducing morbidity and mortality in patients with sepsis. Medications modifying cell-specific immune responses or inhibiting platelet–endothelial interaction or platelet activation have been proposed. Herein, we discuss the underlying mechanisms of organ-specific endothelial dysfunction and immunothrombosis in sepsis and its complications, while highlighting the recent advances in the development of new therapeutic approaches aiming at improving the short- or long-term prognosis in sepsis.
Introduction
The latest definition by the Third International Consensus Definition Task Force defines sepsis as a life-threatening organ dysfunction resulting from a dysregulated host response to infection (1). Despite a better understanding of the underlying pathophysiological mechanisms and improvements to existing therapeutic algorithms, morbidity and mortality in patients with sepsis still remain high. It is estimated that 2.8 million deaths per year are attributed to sepsis, with one in three hospital deaths related to it (2, 3). Sepsis can originate from any infecting organism, but the most frequently identified ones are Staphylococcus aureus (S. aureus), Pseudomonas species, and Escherichia coli (E. coli). Among different sites susceptible to infection, the main ones leading to sepsis are the lungs (64%), the abdomen (20%), the bloodstream (15%), and the renal and genitourinary tract (14%) (2).
Activated components of innate immunity act as the first line of host immune defense against invading pathogens (4). Apart from their direct role in clearing the microorganism, neutrophils, monocytes, and the complement can interact with platelets, and the coagulation cascade leading to the local formation of thrombi in microvessels. This process, known as “immunothrombosis,” contributes to the recognition and restriction of a pathogen’s spread, tissue invasion, and survival, thus consisting an essential component of intravascular immunity (5). Although this distinct innate immune mechanism seems initially beneficial for the host, uncontrolled activation of this process in sepsis can trigger aberrant thrombosis in the microcirculation. During immunothrombosis, immune cells interact also with endothelial cells, which acquire a proadhesive and prothrombotic phenotype (5). Indeed, coagulation abnormalities are common in patients with sepsis, with severity ranging from subclinical coagulation activation to devastating thrombotic and hemorrhagic complications (6). In sepsis, both the activation of the coagulation cascade and the dysregulation of the endogenous anticoagulant and fibrinolytic mechanisms lead to microvascular thrombosis and tissue ischemia, which in turn contribute to tissue injury and multiorgan failure (7). Specifically, high thrombin and d-dimer plasma levels, both markers of coagulation activation, are considered indicative of severe sepsis (8, 9). Accordingly, decreased plasma levels of natural anticoagulants, such as protein C, protein S, and antithrombin (AT), have been found in sepsis and are associated with worse disease prognosis (9–11). Disseminated intravascular coagulation (DIC) is estimated to occur in up to 30%–50% of patients with severe sepsis, and it is associated with a twofold higher mortality risk (12). DIC is characterized by endothelial dysfunction and dysregulation of procoagulant, anticoagulant, and fibrinolytic factors (8). In sepsis-associated DIC, immunothrombosis is the underlying pathophysiological mechanism that triggers the initiation of DIC (5). Most of our knowledge regarding immunothrombosis suggests that it is a process mainly taking place in the microvasculature, affecting both arterioles and venules.
Despite the high prevalence of coagulation abnormalities and their potential deleterious effect on the outcome of sepsis patients, the current treatment approach is generally focused on antibiotics, fluid resuscitation, vasopressors, as well as oxygen and ventilatory support (13). Apart from heparin’s use for venous thromboembolism (VTE) prophylaxis in patients with sepsis, effective treatments specifically targeting the process of immunothrombosis, aiming to abrogate the devastating complications while maintaining its beneficial effects for the host, do not yet exist (13). A better understanding of the underlying mechanisms and cellular interactions in sepsis-related immunothrombosis is crucial to identifying new therapeutic targets that would open the field for the development of new treatments in sepsis. Our study summarizes the current literature regarding the role of endothelial cells and innate immune cells in immunothrombosis in sepsis and discusses clinical and preclinical evidence on drugs targeting immunothrombosis in sepsis.
Overview of hemostasis and thrombosis
The process of thrombus formation inside a blood vessel, which can lead to partial or total vessel occlusion, requires platelets’ accumulation and activation as well as activation of the coagulation cascade (14). The binding of platelet glycoprotein Ib (GP Ib) receptor to von Willebrand factor (vWF) and the interaction of platelet glycoprotein IIb/IIIa (GP IIb/IIIa) receptor with fibrinogen are essential steps for platelet adhesion and aggregation, respectively (14). The coagulation cascade involves the activation of a series of clotting factors and is classically divided into three pathways: the extrinsic, the intrinsic, and the common pathway. The extrinsic pathway is initiated when tissue factor (TF) from the damaged vessel wall interacts with activated clotting factor VII (aFVII), while the intrinsic pathway begins when factor XII (FXII) interacts with the exposed collagen and becomes activated (aFXII). Both pathways end up in factor X (FX) activation, which in turn, along with activated factor V (aFV), cleaves prothrombin into thrombin. Afterward, thrombin activates fibrinogen into fibrin and also cleaves factor XIII (FXIII) into activated factor XIII (aFXIII), resulting in clot stabilization (common pathway) (15, 16). In normal hemostasis, along with the coagulation cascade activation, endogenous anticoagulant mechanisms are being recruited in order to prevent uncontrolled clot expansion and thrombosis. Tissue factor pathway inhibitor (TFPI), AT, activated protein C (APC), and protein S, as well as thrombomodulin (TM), which along with endothelial protein C receptor (EPCR) catalyzes the thrombin-mediated protein C activation, are some of the main physiologic anticoagulant mechanisms (17–20). Finally, under normal conditions, plasminogen is activated by tissue plasminogen activator (tPA) into plasmin, which catalyzes clot dissolution and fibrinolysis, while plasminogen activator inhibitor (PAI) serves as a tPA inhibitor (21, 22).
Endothelial dysfunction in sepsis
Signals leading to endothelial activation
Many of the clinical manifestations related to sepsis can be attributed to endothelial dysfunction which constitutes one of the most prominent underlying pathophysiological mechanisms of sepsis and sepsis-related complications. Although a variety of inflammatory diseases are characterized by endothelial dysfunction, sepsis is linked to a distinctive procoagulant, proadhesive and apoptotic profile of endothelial cells (23). Normal endothelium contributes to vascular health through the expression of antithrombotic and anti-inflammatory molecules (23, 24). In sepsis, the endothelium is activated directly by pathogen-associated molecular patterns (PAMPs) of bacteria, viruses, and fungi or indirectly via neutrophil extracellular traps (NETs) and proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 (IL-1) (Figure 1A) (24–27). NETs are extracellular structures that are composed of cell-free DNA (cfDNA), histones, and antimicrobial proteins and are primarily released through a cell death process called NETosis or alternatively, through a process that does not require cell death, named non-lytic NETosis (28). PAMPs activate endothelial cells via pattern recognition receptors (PRRs), among which toll-like receptors (TLRs) exert the most profound inflammatory response in sepsis (29). Interestingly, endothelial activation during sepsis, as indicated by high circulating levels of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), E-selectin, P-selectin, and vWF, is correlated with disease severity and mortality (30–32).
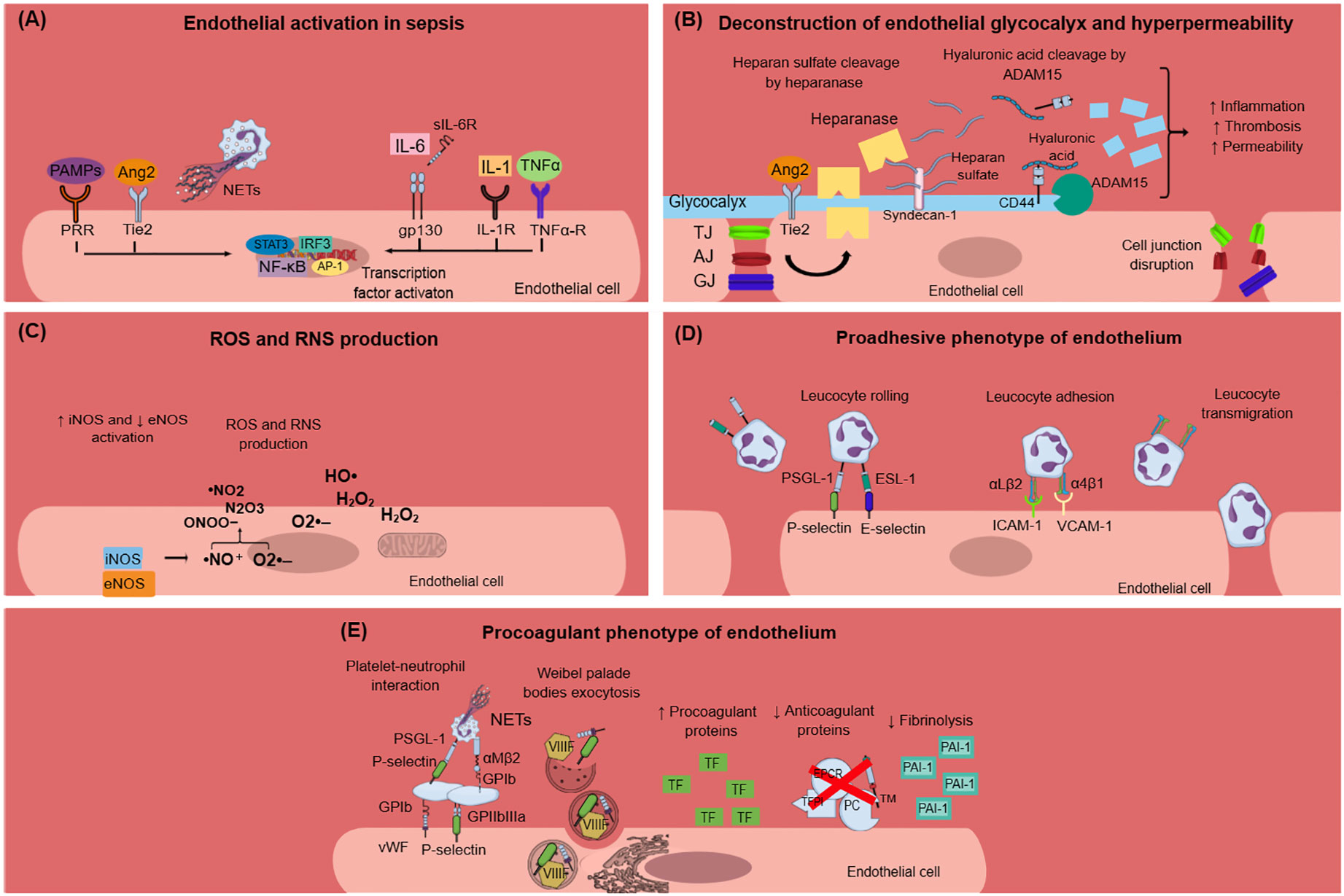
Figure 1 Endothelial dysfunction in sepsis. (A) PAMPs, TNF-α, IL-1, IL-6, Ang2, and NETs activate endothelial cells during sepsis. This results in intracellular signaling activation, leading to transcription factors activation, and mainly NF-κB. (B) Endothelial glycocalyx damage during sepsis is mediated through heparanase and ADAM15, which cleave heparan sulfate and CD44, respectively. Glycocalyx deconstruction results in increased inflammation, thrombosis, and hyperpermeability in sepsis. Furthermore, sepsis leads to endothelial cell junctions’ disruption, which subsequently leads to increased endothelial permeability. (C) Sepsis is characterized by dysregulated nitric oxide synthase activation, leading to the production of reactive nitrogen species. Moreover, infection triggers endothelial cells to produce ROS, which in turn destroy the endothelium. (D) Endothelium activation during sepsis results in the expression of E-selectin, P-selectin, VCAM-1, and ICAM-1 on the endothelial cell surface and subsequent leukocyte rolling, adhesion, and transmigration through endothelial cells. Leukocytes express specific ligands for binding with endothelial receptors such as PSGL-1 and ESL-1 for leukocyte rolling and αLβ2 and α4β1 integrins for leucocyte adhesion. (E) Activated endothelial cells during sepsis express molecules such as vWF and P-selectin on their surface, which mediate platelet aggregation. This leads to the formation of platelet-neutrophil aggregates and subsequent neutrophil activation. Weibel–Palade bodies contain procoagulant factors such as VIIIF, vWF, and P-selectin that can undergo exocytosis under sepsis conditions. The procoagulant phenotype of the endothelium is further characterized by increased production of TF and subsequent extrinsic coagulation pathway activation, decreased production of anticoagulant proteins (TFPI, EPCR, TM, PC), and increased production of PAI-1, which inhibits fibrinolysis. Ang2, angiopoietin 2; ADAM-15; disintegrin and metalloproteinase 15; AJ, adherens junctions; AP-1, activation of activator protein-1; eNOS, endothelial nitric oxide synthase; ESL-1, E-selectin ligand; GJ, gap junctions; gp130, glycoprotein 130; H2O2, hydrogen peroxide; HO·, hydroxyl radical; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; IL-1R, interleukin-1 receptor; iNOS, inducible nitric oxide synthase; IRF3, interferon regulatory transcription factor-3; NETs, neutrophil extracellular traps; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; ·NO, nitric oxide; ·NO2, nitrogen dioxide; N2O3, dinitrogen trioxide; O2·–, superoxide anion; ONOO−, peroxynitrite; PAI-1, plasminogen activator inhibitor-1; PAMPs, pathogen associated molecular patterns; PC, protein C; PRR, pattern recognition receptor; PSGL-1, P-selectin glycoprotein ligand-1; RNS, reactive nitrogen species; ROS, reactive oxygen species; sIL-6R: soluble interleukin-6 receptor; STAT3, signal transducer and activator of transcription 3; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TJ, tight junctions; TNF-α, tumor necrosis factor alpha; TNF-α-R, TNF-α receptor; VIIIF, VIII factor; VCAM-1, vascular cell adhesion molecule 1; vWF, von Willebrand factor; WPBs, Weibel–Palade bodies.
Endothelial barrier dysfunction
One of the first signs of endothelial dysfunction in sepsis is endothelial glycocalyx (eGC) deconstruction (33) (Figure 1B). The endothelial glycocalyx is a carbohydrate-rich layer that covers the luminal surface of endothelial cells and serves numerous protective roles for the vascular system (34, 35). Specifically, eGC regulates vascular permeability and endothelial response to shear stress and inhibits leucocyte and platelet adhesion as well as coagulation cascade activation and hence microthrombus formation (36). During sepsis, distinctive mechanisms have been identified that promote eGC damage, which subsequently enhances endothelial dysfunction and sepsis complications. In vivo sepsis models suggest that sepsis-induced reduction in eGC thickness leads to increased albumin extravasation and circulating glycocalyx products such as hyaluronic acid and CD44 (37). The underlying mechanism can be explained by a cascade of reactions where disintegrin and metalloproteinase 15 (ADAM15) cleave CD44 of eGC, leading to an increase in plasma levels of soluble CD44 and hyaluronic acid. This is followed by a decrease in endothelial barrier integrity and a disruption of vascular endothelial (VE)-cadherin and β-catenin, which are essential components of adherens cell junctions (37). Similarly, endothelial tight and gap junctions were found to be disrupted in sepsis (38). A variety of eGC products are emerging as biomarkers in sepsis as they add important prognostic value. For example, increased syndecan-1 (SYN-1) plasma levels during sepsis are correlated with the requirement for renal replacement therapy, respiratory failure, multiple organ dysfunction syndrome (MODS), and predict coagulation failure and mortality (39–41). Sphingosine-1-phosphate (S1P) is another eGC component that predicts sepsis coagulation failure and mortality (40), while increased plasma endocan levels, another marker of eGC destruction, are also associated with the need for mechanical ventilation and with higher mortality in sepsis patients (42, 43). Angiopoietin-2 (Ang-2) is a growth factor produced by the endothelium under inflammatory conditions. It has been linked to increased vascular permeability in sepsis through enhancing endothelial heparanase secretion, which in turn cleaves heparan sulfate, the main component of eGC (34). Increased heparanase production by Ang-2 was shown to be mediated by the inhibition of endothelium-stabilizing receptor Tie2 signaling (34). Additionally, Tie2 tyrosine kinase inhibition by Ang2 in sepsis resulted in a procoagulant phenotype of endothelial cells (44). Interestingly, increased Ang-2 has been associated with higher mortality, respiratory, liver, and kidney failure, and predicted DIC severity in patients with sepsis (44–47).
Dysregulation of nitric oxide pathway
Nitric oxide (NO) is a soluble gas produced by endothelial cells and studied in depth for its protective role on the vascular endothelium. NO promotes vasodilation and is capable of inhibiting platelet activation and adherence to the endothelium (24). NO has another crucial role in impeding leucocyte adhesion on the vascular wall (24). This is possibly mediated through the downregulation of P-selectin, chemokine, E-selectin, ICAM-1, and VCAM-1 expression in endothelial cells (24). In preclinical sepsis models, an imbalance between endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) activity has been demonstrated. Specifically, eNOS activity was found to be decreased compared to the upregulated iNOS activity. Overproduced NO by iNOS reacts with superoxide anion resulting in reactive nitrogen species (RNS) production, which contributes to endothelial dysfunction (38) (Figure 1C). Experimental and clinical studies are inconclusive regarding NO levels and their effects on the endothelium during sepsis. Some studies suggest that NO production increases during sepsis and this is associated with poor prognosis, while others support that NO levels are lower in sepsis patients compared to healthy controls (48–51). New evidence suggests that increased NO bioavailability in sepsis may be beneficial for the host without deteriorating vasodilation and subsequent hypotension, as has been proposed in previous studies (52). Overall, the dysregulation of the NO pathway during sepsis merits further investigation.
Proadhesive endothelial cell phenotype
Expression of proadhesive molecules in activated endothelial cells poses a hallmark process for the progression of sepsis-induced endothelial dysfunction (Figure 1D). Activated endothelium interacts both with leukocytes and platelets, while platelet–leucocyte aggregates deposit on endothelium and further enhance local inflammation (23). The infection triggers immune and endothelial cells to produce reactive oxygen species (ROS) and inflammatory and anti-inflammatory cytokines. Reversely, cytokines stimulate endothelial cells to express adhesion molecules such as selectins, integrins, ICAMs, and VCAM-1 (38). These molecules interact with leukocytes, which roll, bind to the endothelium, and transmigrate through it. Furthermore, ROS released by immune cells during sepsis destroy the endothelium and result in increased permeability (38).
Prothrombotic endothelial cell phenotype
Accumulating evidence suggests that endothelial cells can interact with immune cells, triggering thrombus formation, a process implicated in the pathogenesis of numerous diseases, such as antiphospholipid syndrome, coronary artery disease, and sepsis (53–58). Both experimental and clinical studies, presented in Table 1, explain how endothelial cells can lose their anticoagulant properties and lead to thrombosis when stimulated directly or indirectly by pathogens (Figure 1E). A large body of evidence suggests that upon sepsis-induced endothelium activation, there is increased TF expression and secretion accompanied by enhanced TF activity (26, 60, 62–64, 72). Complement, especially C5a, and the inactive terminal C complex (iTCC) formed by mixing C5b6, C7, C8, and C9, were also found to stimulate endothelial cells to produce active TF (77, 78) (Table 2). Beyond TF, other important mechanisms contributing to immunothrombosis are the dysfunctional fibrinolytic and anticoagulant properties of the endothelium. Accumulating experimental data confirm that pathogen invasion is associated with increased endothelial expression of PAI-1 and decreased expression of EPCR, TM, and TFPI, as well as decreased protein C activation (25, 63, 68, 69, 72). Complement C3 activation stimulated by Shiga toxin was also shown to result in endothelial TM reduction and enhanced thrombus formation (82) (Table 2). Despite decreased endothelial TM expression during sepsis, soluble TM is increased in patients with sepsis and predicts acute kidney injury, multiple organ failure, and mortality (96–99). A possible illustrative mechanism is that apoptotic endothelial cells release their intracellular proteins into circulation. Similarly, elevated TFPI plasma levels are associated with overt DIC in patients with sepsis, in contrast to the reduced TFPI expression by endothelium (100). Accordingly, increased plasma levels of PAI-1 have been correlated with DIC in patients with sepsis and predicted mortality (101). Circulating levels of proteins C and S have also been evaluated, and clinical data support their deficiency in sepsis (102). Importantly, in patients with severe sepsis, decreased plasma levels of protein C and increased syndecan-1, which characterize endothelial damage, were associated with whole blood hypocoaguability in the context of DIC, as measured by thromboelastography (103).
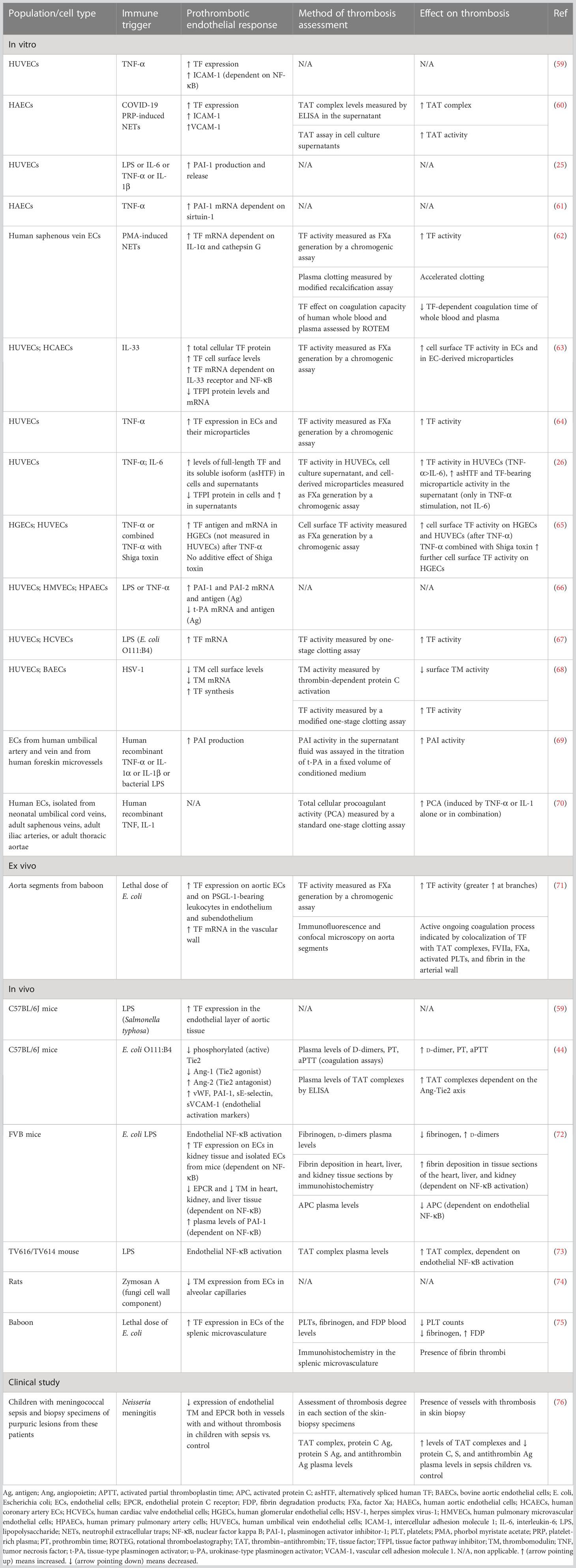
Table 1 Role of endothelial cells in immunothrombosis.
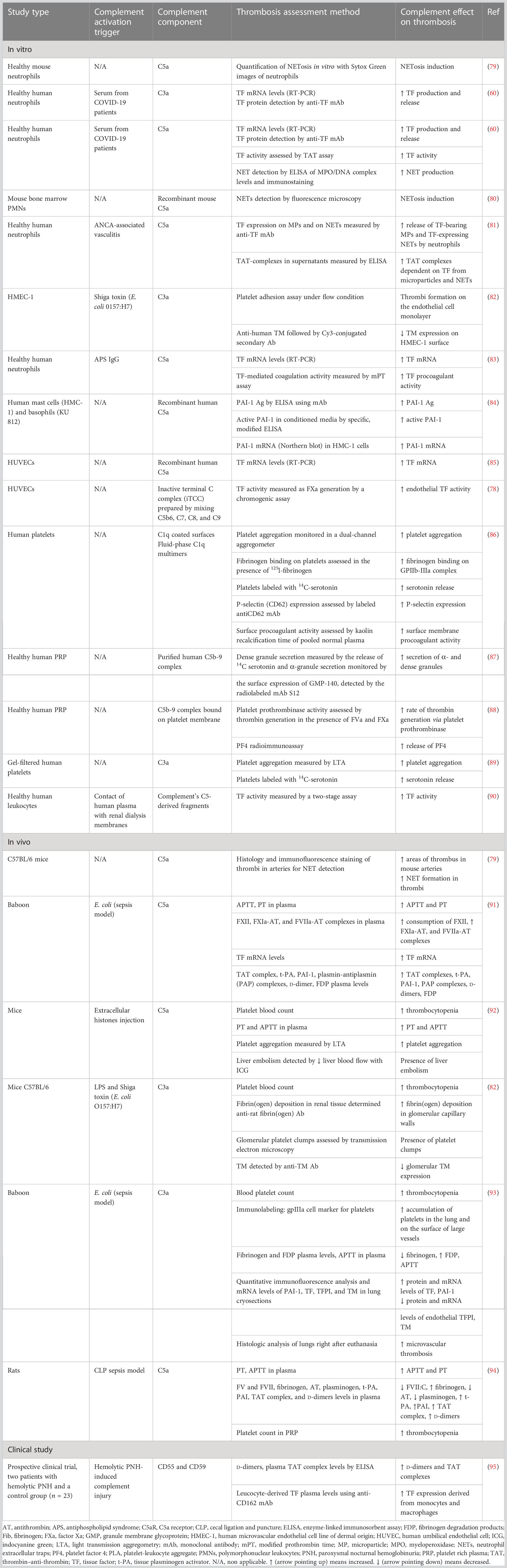
Table 2 Complement role in immunothrombosis.
Transcription factor activation
Endothelial cell activation, following pathogen invasion, is mediated by intracellular signaling, which includes transcription factor activation such as nuclear factor kappa B (NFκB), activator protein-1 (AP-1), interferon regulatory transcription factor-3 (IRF3), and signal transducer and activator of transcription 3 (STAT3) (38, 104) (Figure 1A). Specifically, activation of endothelial NFκB by pathogens has been found to mediate increased expression of procoagulant molecules such as PAI-1 and TF, while fibrinolytic and anticoagulant factors such as TM, APC, and EPCR diminished in a way that was also dependent on NFκB (72, 73). Indeed, NFκB has been associated with increased circulating d-dimers and tissue fibrin deposition in an endotoxemia mouse model (72).
Role of Weibel–Palade bodies
Weibel–Palade bodies (WPBs) are organelles inside endothelial cells containing procoagulant proteins such as vWF, P-selectin, and factor VIII (FVIII), which can rapidly undergo exocytosis under specific circumstances (33). Lipopolysaccharide (LPS) has been shown to induce WPBs’ content secretion both in vitro and in vivo (33) (Figure 1E). Ultra-long vWF strings can be produced in vitro after endothelial cell activation by Shiga toxin. Furthermore, the Shiga toxin prevented cleavage of these vWF strings by inactivating ADAMTS13 (105). It has been found that in preclinical sepsis models, increased markers of endothelial activation are associated with abnormal coagulation markers, while some studies have additionally demonstrated the subsequent thrombus formation in the microcirculation. Dysregulated coagulation was indicated by markers such as high levels of circulating d-dimers, fibrin, fibrin degradation products (FDPs), thrombin-antithrombin (TAT) complexes prolonged prothrombin time (PT), activated partial thromboplastin time (aPTT), and/or decreased levels of plasma fibrinogen and platelets (44, 71, 72).
Neutrophils in immunothrombosis
Apart from endothelial dysfunction, which facilitates immunothrombosis in sepsis, activated cells of the innate immune system, in particular neutrophils and monocytes, also play a significant role (Figure 2). Neutrophils are an essential part of the host’s innate immune response and are typically the first responders to acute inflammation (28). In sepsis, PAMPs and damage-associated molecular patterns (DAMPs) can activate neutrophils by binding to PRRs. Activated neutrophils exert their antimicrobial activity mainly through three processes: phagocytosis, degranulation, and the release of NETs (28).
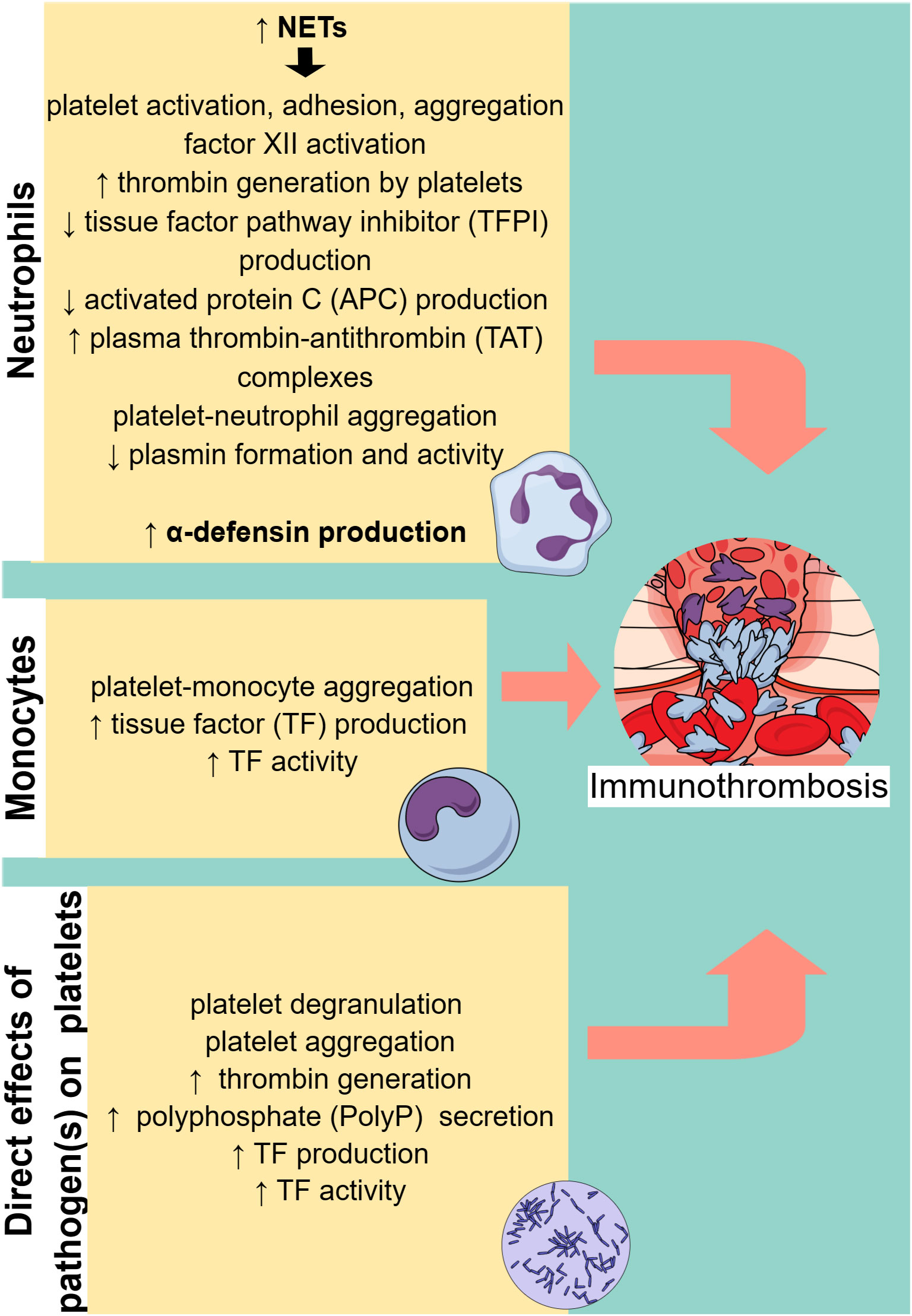
Figure 2 Cell-specific immunothrombosis mechanisms. Neutrophils and monocytes can induce thrombosis through specific mechanisms, while pathogens can directly activate platelets, leading to thrombosis. NETs, neutrophil extracellular traps. ↑ (arrow pointing up) means increased. ↓ (arrow pointing down) means decreased.
Induction of NETosis in sepsis
A variety of different pathogens, bacterial toxins, and cytokines can induce NETosis (28, 106). NETosis can also be enhanced by activated platelets (28, 60, 107–109). More specifically, in a sepsis mouse model, it was found that LPS binding to platelet TLR4 stimulated platelets to interact with neutrophils and induce NET production in the liver sinusoids and capillaries (107). The binding of platelet P-selectin to P-selectin glycoprotein ligand 1 (PGSL1) on neutrophils was found to be a key mechanism for NETosis induction in mice (110). However, this finding was not reproduced in studies with human neutrophils, and different agonists, where an antibody against P-selectin did not affect platelet-mediated NETosis (111, 112). The interaction of platelet-derived high mobility group protein B1 (HMGB1) with the receptor for advanced glycation end products (RAGE) on neutrophils was also found to be a potential mechanism for NETosis stimulation by platelets, possibly through induction of autophagy in neutrophils (112, 113). Moreover, it was shown that LPS and Pam3CSK4, a synthetic lipopeptide, through binding to platelet TLR4 and TLR2, respectively, activated platelets to interact with neutrophils and induce NETosis through platelet GP1b-neutrophil β2-integrin (CD18) interaction. Platelet-derived vWF, platelet factor 4 (PF4), and thromboxane A2 (TXA2) were essential mediators of this process (111). Furthermore, experimental studies have shown that platelet-derived CXCL4/CCL5 heterodimers and β-defensin 1 enhanced NET production (114). Besides platelet–neutrophil interaction and subsequent NETosis induction, it was found that also complement cleavage products, in particular, C3a and C5a anaphylatoxins could stimulate NETosis in sepsis animal models or in neutrophils stimulated by LPS or platelet-rich plasma from COVID-19 patients (60, 80, 115–117) (Table 2). Moreover, complement may play a role in the inhibition of NET degradation (118).
Role of NETs in endothelial dysfunction
It has been shown that NETs, apart from their beneficial role in clearing the invading microorganism, are implicated in the pathogenesis of many diseases in which endothelial dysfunction is present, such as sepsis and cardiovascular diseases. Therefore, there has been a growing interest in studying the role of NETs in endothelial dysfunction in sepsis (119). In vitro studies suggest that NETs can promote endothelial dysfunction, as indicated by increased endothelial expression of ICAM-1, VCAM-1, E-selectin, TF, and vWF (60, 62, 109, 120–123). Cathepsin G is a component of NETs, capable of processing pro-IL-1α into IL-1α, which has been found to enhance VCAM-1 and ICAM-1 expression on the endothelial surface (62). Moreover, NET-induced ICAM-1 and E-selectin expression on endothelial cells was found to be dependent on the triggering receptor expressed on myeloid cell-1 (TREM-1). TREM-1 is an orphan immune receptor expressed on various types of cells, such as immune cells, platelets, and endothelial cells. Apart from mediating the NET-induced proadhesive phenotype of endothelial cells, TREM-1 potentiated NETosis triggered by LPS (121). NETs have also been associated with increased platelet adhesion on endothelial cells through enhancement of vWF expression on the endothelium (120). Both NETs and histones have been found to increase TF mRNA and TF activity in human aortic endothelial cells (HAECs) and human saphenous vein endothelial cells (HSVECs), resulting in plasma clotting acceleration (60, 62, 123). Specifically for histones, their effect on TF was mediated by TLR2 or TLR4, which in turn activated AP-1 and NFκB transcription factors (123). As one of the major components of NETs, histones have been further studied for their impact on endothelium, with results suggesting cytotoxicity in a concentration-dependent manner (124). Citrullinated histone H3 (CitH3) was found to increase endothelium permeability in vitro (125). Furthermore, images using intravital microscopy showed that CitH3 resulted in microvascular endothelial barrier dysfunction, by disrupting the adherens junctions and reorganizing the actin cytoskeleton without causing cell death (126). Accordingly, NETs have been found to decrease endothelial expression of VE-cadherin and Zonula occludens (ZO)-1 proteins, which are major components of adherens and tight junctions, respectively (109).
Prothrombotic role of NETs
NETs not only contribute to endothelial dysfunction but have also been found to exert procoagulant and prothrombotic activity through various mechanisms, as shown in Table 3. NETs could serve as a scaffold and inducer of platelet adhesion, activation, and aggregation, and this effect was largely mediated by specific NET components like histones H3 and H4 (134). Nets not only served as a scaffold for platelet adhesion but also for red blood cells (RBCs), promoting the generation of red (RBC-rich) thrombus. Plasma proteins, implicated in thrombosis, such as vWF, fibronectin, and fibrinogen could also bind to NETs, while NETs contributed to thrombin-mediated fibrin generation (134). Histones, in particular H3 and H4, could activate platelets to promote plasma thrombin generation, through binding to platelet TLR2 and TLR4 (131). Notably, this effect was enhanced when histones were in complex with DNA (131). In contrast, another study showed that the addition of deoxyribonuclease (DNase), which destroys the DNA network of NETs, enhanced histone-mediated, platelet-dependent thrombin generation, presumably due to increased exposure to histones (128). Besides NETs-induced platelet-dependent mechanisms of thrombin formation, NETs were capable of stimulating thrombin generation also in platelet-poor plasma via activation of the intrinsic coagulation pathway (128). This effect was largely attenuated with DNase administration, suggesting the crucial role of cell-free DNA (cfDNA) in that process. Accordingly, DNA–histone complexes measured in platelet poor plasma from sepsis patients were increased compared to plasma from healthy subjects, and cfDNA was positively correlated with thrombin generation (128). NETs, possibly due to their negatively charged extracellular DNA surface, could bind and promote activation of factor XII, thus triggering the contact pathway of coagulation (146). Interaction of NETs with membrane-derived microparticles released by activated neutrophils enhanced NET-mediated intrinsic coagulation pathway activation and subsequent thrombin formation (139). In support, it was demonstrated that NETs were capable of promoting pulmonary clot formation in mice with septicemia caused by LPS and heat-killed E. coli (140). However, despite robust evidence on the effect of NET components on thrombosis, the role of intact NETs may merit further investigation. Specifically, an in vitro study showed that although cfDNA and histones, separately, were capable of coagulation activation, intact NETs could not (147). One possible explanation for this is that the complex interactions between DNA and histones within the NET structure abrogate the thrombogenic negative charge of DNA and cover the histones’ binding sites. Moreover, the absence of other blood cells, endothelium, and blood flow in an in vitro model compared to an in vivo one may have contributed to the absence of the procoagulant effect of intact NETs (147).
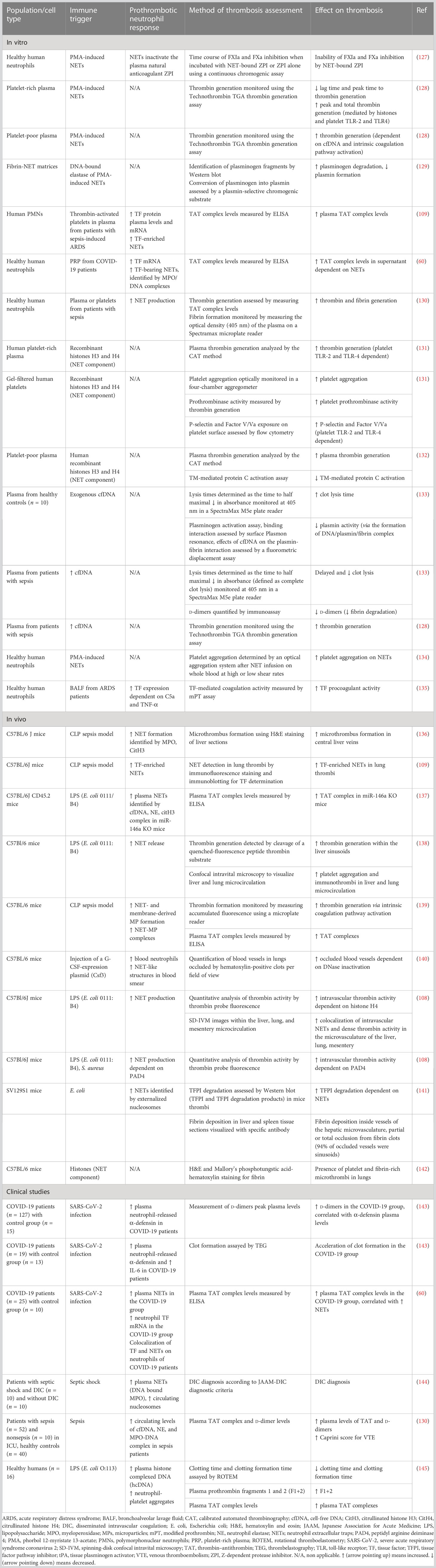
Table 3 Role of neutrophils in immunothrombosis.
Disruption of endogenous anticoagulant and fibrinolytic mechanisms by NETs
NETs were also shown to interfere with the endogenous anticoagulant mechanisms. More specifically, extracellular nucleosomes within NETs facilitated TFPI degradation by neutrophil elastase on the surface of activated neutrophils, thus abrogating TFPI-mediated inhibition of of the extrinsic coagulation pathway (141). Neutrophil elastase bound to DNA complexes was also shown to cleave plasminogen into fragments, resulting in decreased plasmin production and impaired fibrinolysis (129). Apart from decreased plasmin generation, cfDNA was capable of binding to plasmin and fibrin at the same time, and the formed complex was shown to be responsible for decreased plasmin-mediated fibrin clot dissolution (133). Besides cfDNA, histones, specifically H3 and H4, could also interact with TM and protein C, leading to the inhibition of APC generation (132).
Prognostic utility of NETs in sepsis
Therefore, experimental data support the crucial role of NETs in triggering thrombus formation. From a clinical point of view, it was shown that increased NET-forming capacity was significantly associated with thrombocytopenia, increased PT, aPTT, d-dimers, and low fibrinogen in patients with sepsis at intensive care unit (ICU) admission, while it could also predict DIC development and mortality after ICU admission (106). Noteworthy, specific components of NETs, such as circulating levels of cfDNA, neutrophil elastase, and the myeloperoxidase–DNA complex in patients with sepsis, are positively correlated with the risk of VTE (130). Increased MPO-DNA levels on days 3 and 7 during sepsis hospitalization were associated with decreased mean arterial pressure, a lower PaO2/FIO2 ratio, an increased sepsis-related organ failure assessment (SOFA) score, and 28-day mortality (148).
TF production and release by neutrophils
Regarding the production and release of TF by neutrophils, there is conflicting evidence. It was reported that neutrophils in the setting of appropriate interaction with injured endothelial cells or after stimulation with P-selectin or the peptide formyl-MetLeuPhe (Fmlp) were able to produce functional TF, initiating the extrinsic coagulation pathway (149, 150). Moreover, C3a and C5a anaphylatoxins were found to stimulate neutrophils to produce and release active TF (Table 2) (60, 83, 90). However, these findings have been questioned in other studies, and it was proposed that granulocytes may acquire TF produced by monocytes (151, 152). With respect to the presence of TF in NETs, it was found that neutrophils from patients with sepsis could release large amounts of functional TF-bearing NETs, and autophagy was implicated in that process (151).
Role of neutrophil-derived α-defensins in immunothrombosis
Another potential mechanism through which neutrophils may contribute to immunothrombosis is by secreting antimicrobial peptides. Specifically, α-defensins, which are secreted by neutrophils as part of the host innate immune response, have been found elevated in the plasma of septic patients (153). Recently, an in vivo study showed that α-defensins accelerate and stabilize fibrin clot formation, while also disrupting fibrinolysis (154). Consistently, circulating α-defensins have been correlated with the acceleration of clot formation and d-dimers plasma levels in COVID-19 patients (143, 155).
Monocytes in immunothrombosis
Monocytes and macrophages have been found to play a vital role in sepsis-induced immunothrombosis (Figure 2). TF expression by monocytes seems to be the most potent pathway leading to coagulation cascade activation in sepsis (156–160). While both hematopoietic and non-hematopoietic cells produce TF and enhance coagulation in an endotoxemia mouse model, monocytes are the main source of circulating TF (161). Additionally, other blood cells have been found to acquire TF released by monocytes through microparticles (161). Numerous studies depicted in Table 4 suggest that pathogens induce monocyte-derived TF expression and release in the circulation, which in turn activates the extrinsic coagulation pathway. The binding of LPS to transmembrane receptors such as TLR4 in monocytes induces TF mRNA expression via NF-κB activation (166). Moreover, the interaction of pathogen components either with TLRs or directly with intracellular pathways in monocytes can result in inflammasome activation and subsequent TF release via pyroptosis (163, 166). Pyroptosis is a type of proinflammatory cell death induced by caspase-1 (canonical inflammasome) or caspase-11 (noncanonical inflammasome) and is characterized by gasdermin D-dependent pore formation in the cell membrane, subsequent osmotic cell lysis, and finally the release of cytosolic content. Indeed, LPS or type III secretion system (T3SS) rod proteins have been found to trigger in vivo caspase-11 and caspase-1 activation, respectively, leading to TF release (163). Gasdermin D-mediated pore formation induced by caspase-11 has also been proposed to activate TF on macrophages (162). Specifically, pore formation on the cell membrane can induce calcium influx, which triggers phosphatidylserine exposure on the membrane, followed by TF activation (162). Furthermore, sphingomyelin, another membrane lipid, is involved in the activation of TF to its procoagulant form. This process occurs on the macrophage cell membrane and is dependent on sphingomyelin hydrolysis by acid sphingomyelinase (ASMase) (167).
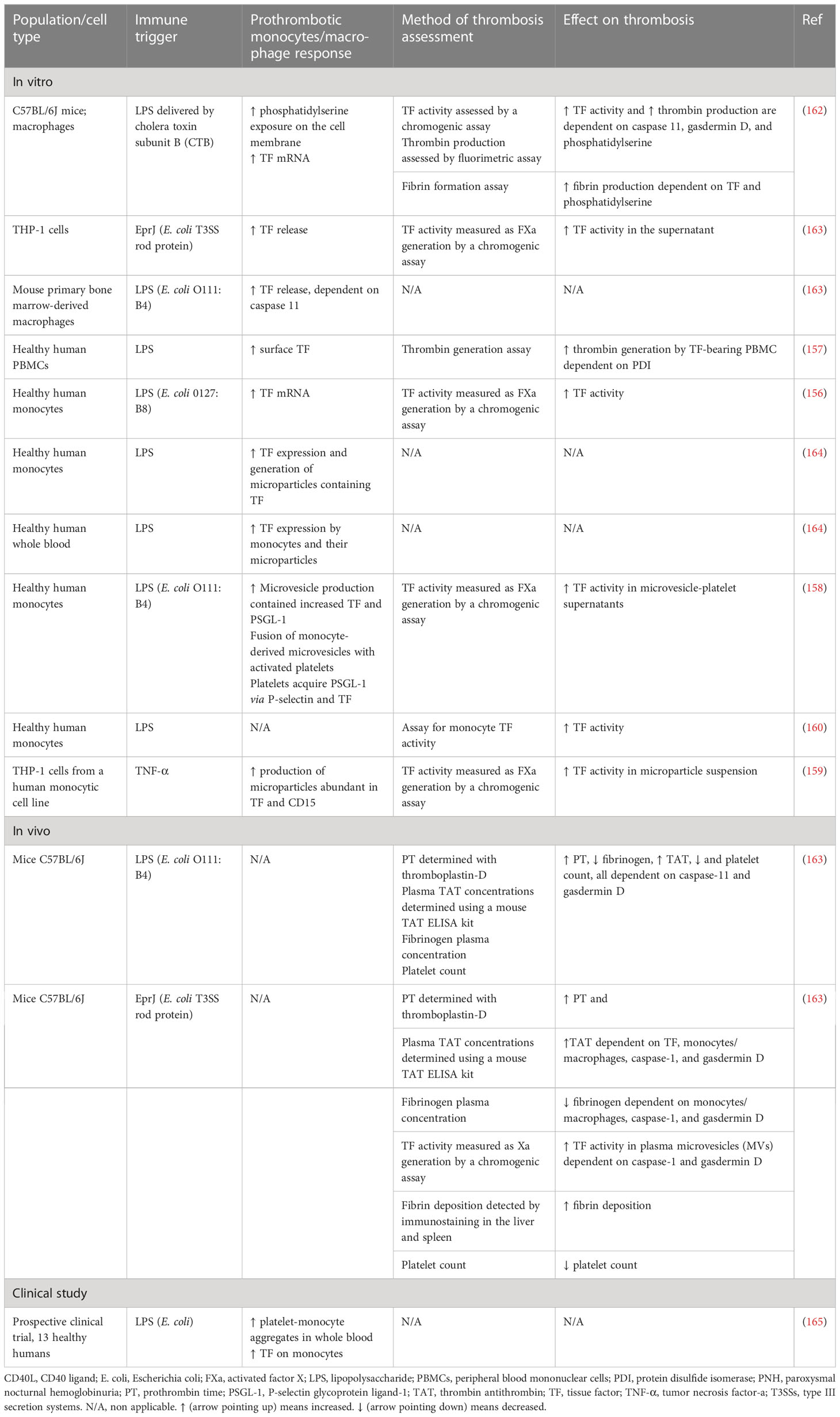
Table 4 Role of monocytes/macrophages in immunothrombosis.
Tissue factor production is additionally triggered by HMGB1, which is a DAMP released by myeloid cells, platelets, and hepatocytes. HMGB1 binds LPS, enhances endocytosis of the LPS-HMGB1 complex in monocytes, and is associated with increased TF release via caspase-11-induced pyroptosis (168). Furthermore, TF expression and activity are also induced by the interaction of monocytes or their microparticles with activated platelets (158, 164). Monocyte activation by pathogens is followed by increased PSGL-1 expression and the release of TF- and PSGL-1-bearing microparticles. These microparticles can fuse in vitro with platelets, leading to increased TF activity (158). Additionally, a prospective clinical study confirms that LPS endotoxemia enhances platelet–monocyte aggregation, TF expression, and platelet activation (165). On the other hand, thrombin generated by coagulation cascade activation binds to monocytes via protease-activated receptor-1 (PAR-1), leading to increased TF expression. Accordingly, inhibition of PAR-1 by vorapaxar (a PAR-1 antagonist) has been found to attenuate sepsis-induced coagulation, as indicated by decreased plasma prothrombin fragments and TAT complexes (169).
Platelets in immunothrombosis
Platelet activation in sepsis
Accumulating evidence suggests a complex interaction between platelets and different immune cells. Increased platelet activation and platelet–leukocyte aggregates have been found in a variety of thrombotic conditions, such as unstable angina, myocardial infarction, stroke, and sepsis (170–173). Multiple roles of platelets have been demonstrated in experimental and clinical studies. Platelets were suggested to contribute to tissue regeneration and myocardial recovery following acute coronary syndrome (ACS) (174–178). On the contrary, increased platelet activation and reactivity have been linked with a poor prognosis in several diseases, including ACS (179), dementia (180), diabetes (181, 182), and sepsis (183). Specifically, in sepsis, thrombocytopenia is associated with increased disease severity and an ominous prognosis (184). Decreased platelet count is attributed to excessive peripheral consumption of platelets resulting from many causes, among which are the interaction of platelets with immune cells, the microcirculation thrombosis, and the direct binding of pathogens to platelets (184). Beyond innate immune cells, which play a crucial role in immunothrombosis, pathogens can directly activate platelets, which in turn promote thrombosis (Figure 2). Recently, it has been found that during sepsis there is accelerated megakaryocyte maturation, triggered by PAMPs, DAMPs, TNF-α, or other cytokines. This subpopulation of megakaryocytes presents a stronger immunological profile and produces fewer platelets compared to megakaryocytes undergoing normal maturation induced by thrombopoietin (185). A variety of receptors on platelets interact with PAMPs, or pathogens coated by antibodies. Numerous pathogens have been proven to activate platelets and increase their aggregation, as presented in Table 5. Pathogens coated by immunoglobulins or pentraxins bind to low-affinity receptors for the constant fragment (Fc) of immunoglobulin (Ig) G (FcγRIIa) on platelets, leading to dense and α-granules secretion. FcγRIIa triggers an intracellular pathway for platelet activation by phosphorylating the tyrosine kinases Src, Syk, and phospholipase c gamma 2 (PLCγ2) (188). Glycoproteins IIb/IIIa (GP IIb/IIIa) and Iba (GPIba), expressed on the surface of activated platelets (202), constitute another receptor category that promotes platelet degranulation and aggregation after binding pathogen components (180, 188, 189, 202). Bacteria have also been found to activate platelets through TLR-2 and TLR-4 (192, 194, 203). Specifically, intracellular signaling of TLR-2 is mediated by PI3 kinase, which activates the Rap1 guanine nucleotide (192). TLR-4 activation in platelets is followed by a complicated intracellular signaling pathway that implicates NFκB. The majority of NFκB signaling proteins are present in platelets, presenting nongenomic effects and resulting in degranulation (203, 204). Moreover, LPS-induced platelet aggregation in vitro is dependent on both TLR-4 and TLR-2 platelet receptors as well as on myeloid differentiation factor 88 (MyD88) and cGMP-dependent protein kinase intracellular pathways. Accordingly, LPS accelerated in vivo thrombus formation and occlusion in FeCl3-injured carotid artery of mice, and this effect was dependent on the MyD88 signaling pathway (194).
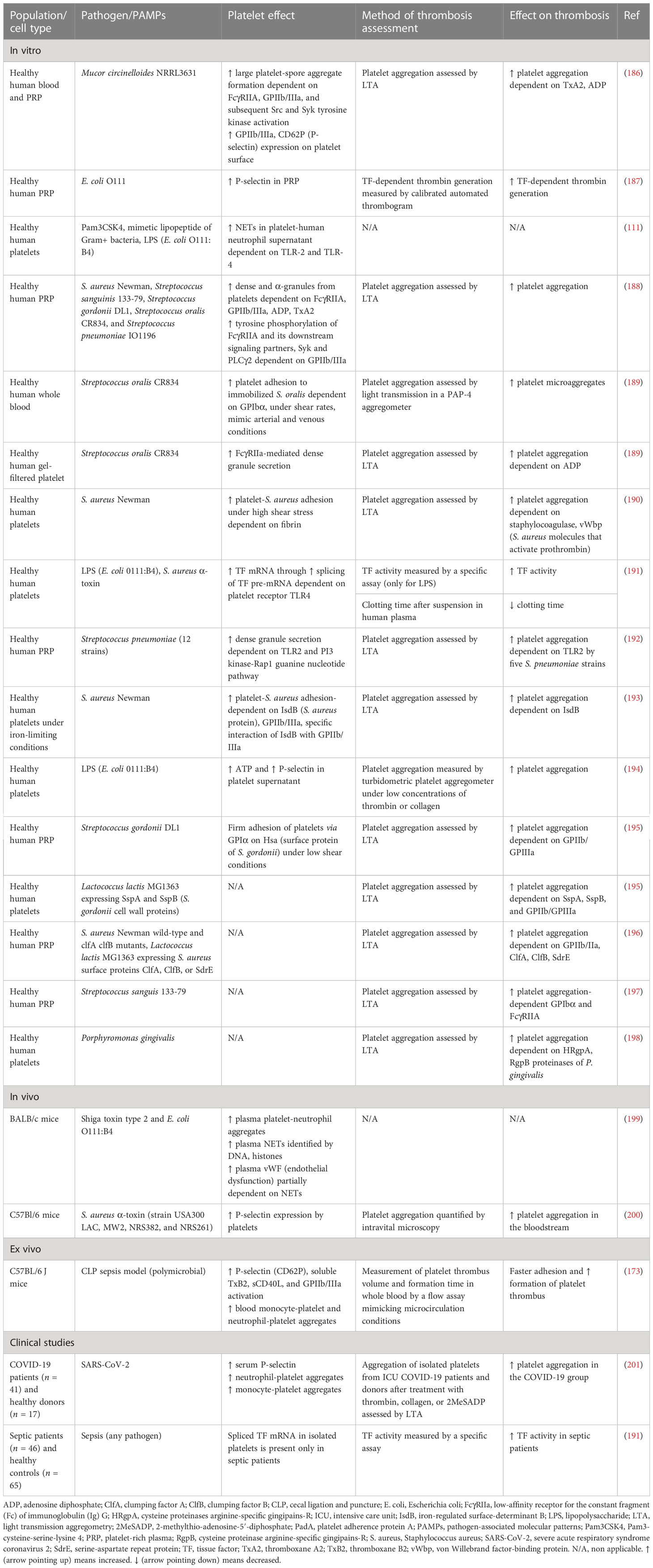
Table 5 Role of pathogen-activated platelets in immunothrombosis.
Platelet aggregation and thrombus formation in sepsis
All the aforementioned platelet receptors induce immunothrombosis through different pathways after being triggered by PAMPs. The surface of activated platelets promotes thrombin generation and, subsequently, fibrin and clot formation (205). Both dense and α-granules are secreted by pathogen-activated platelets and enhance their aggregation. Specific proteins secreted by platelets are involved in the process of immunothrombosis, such as P-selectin, PF4, vWF from α-granules, and ADP/ATP from dense granules (188). Activated platelets have also been found to release polyphosphate (PolyP), an inorganic polymer that exerts procoagulant activity. In vitro, PolyP initiates the contact pathway by FXII activation, resulting in enhanced thrombin generation and clot formation (206). The procoagulant activity of PolyP has also been confirmed in a sepsis mouse model where LPS-induced Polyp is released by platelets, resulting in increased intravascular thrombin activity (108). Protein disulfide isomerase (PDI) is found in the platelet endoplasmic reticulum and is released into circulation by endothelial cells or platelets (157, 207). The pathophysiological mechanism explaining PDI thrombotic effect in vivo is not fully elucidated. Activated platelets increase the secretion of PDI, which binds to the GPIIb/IIIa integrin and results in enhanced GPIIb/IIIa-binding affinity. An in vivo study suggests that TNF-α activated endothelium induces PDI secretion leading to platelet-neutrophil aggregation through GPIIb/IIIa and αΜβ2 receptors (207). Another possible mechanism has been proposed by an in vitro study finding that PDI enhances TF-mediated thrombin generation in peripheral blood mononuclear cells (PBMCs) triggered by LPS (157). Platelet-derived TF levels and activity have also been found to increase in vitro after activation by LPS, E. coli, or S. aureus α-toxin. TLR-4 is one of the platelet receptors mediating TF production by splicing of TF pre-mRNA (187, 191). Indeed, a clinical study confirms that platelets from septic patients present increased TF activity in contrast to healthy ones (191). Furthermore, increased expression of P-selectin and GPIIb/IIIa has been found in a sepsis mouse model and is associated with neutrophil-platelet aggregates and thrombus formation (173). Similarly, a prospective clinical trial found that COVID-19 infection amplifies platelet activation, as indicated by P-selectin, and promotes platelet aggregation (201).
Targeting immunothrombosis in sepsis
Clinical evidence on drugs targeting immunothrombosis in sepsis
The efficacy of FDA-approved drugs commonly used in clinical practice, such as heparin, P2Y12, and GPIIb/IIIa inhibitors, has been tested in patients with sepsis. AT, another FDA-approved drug indicated for the prevention of peri-operative and peripartum thromboembolic events in patients with hereditary AT deficiency, has been also assessed (Table 6; Figure 3) (239). In addition to FDA-approved drugs, a variety of currently non-FDA-approved agents, including recombinant TM, APC, and TFPI, have been evaluated in sepsis (Table 7; Figure 3). According to the international guidelines for sepsis and septic shock, neither anticoagulant nor antiplatelet medications are recommended for sepsis or DIC treatment, except for low molecular weight heparin (LMWH) or unfractionated heparin (UFH) for VTE prevention (13). On the contrary, AT replacement therapy and recombinant TM are recommended for sepsis-associated DIC in Japan (274). The efficacy of AT in sepsis is controversial, as derived from studies depicted in Table 6 and meta-analyses (275). Decreased AT plasma levels and DIC presence have been correlated with higher efficacy of AT treatment (221, 234, 275). Furthermore, bleeding risk in patients receiving AT is increased, especially in co-administration with heparin (275). Studies assessing the efficacy of TM have shown more promising results, as TM administration resulted in decreased mortality and was not associated with increased bleeding events in sepsis-induced coagulopathy (276, 277). APC (or drotrecogin alfa activated), despite its antithrombotic effects, is not used in clinical practice as it has not improved survival and has been associated with increased bleeding risk in sepsis (271, 278). Similarly, heparin is not recommended in sepsis management, although some studies have found a beneficial effect on 28-day mortality (279–281). Bleeding events following heparin treatment in sepsis are an important disadvantage of its use (279). Antiplatelet agents, beyond aspirin, have been tested to a lesser extent than anticoagulants, resulting in scarce data regarding their effects on the survival of sepsis patients (215–217). As for aspirin, a recent study showed no effect on mortality in septic patients (282). However, there are multiple underlying mechanisms through which aspirin could affect sepsis, specifically through its known anti-inflammatory properties. As a result, aspirin’s effects on sepsis should not be attributed exclusively to its antithrombotic properties (282). Therefore, drugs targeting immunothrombosis are not approved for clinical use in sepsis due to both inconclusive results for their efficacy and an increased risk for side effects.
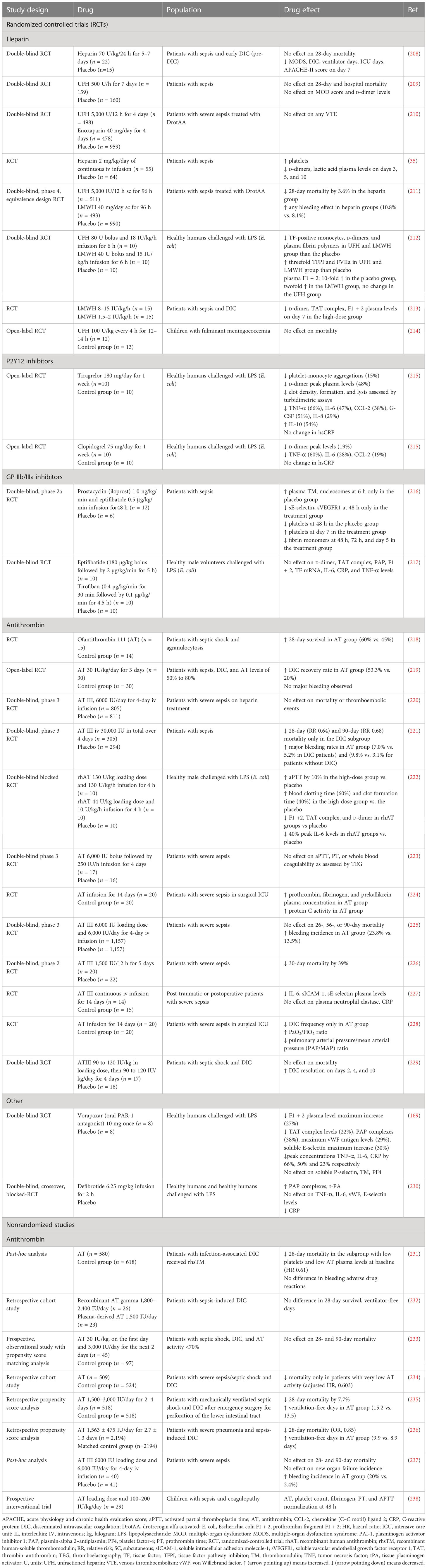
Table 6 Clinical studies assessing current anticoagulant and antithrombotic drugs in sepsis outcomes.
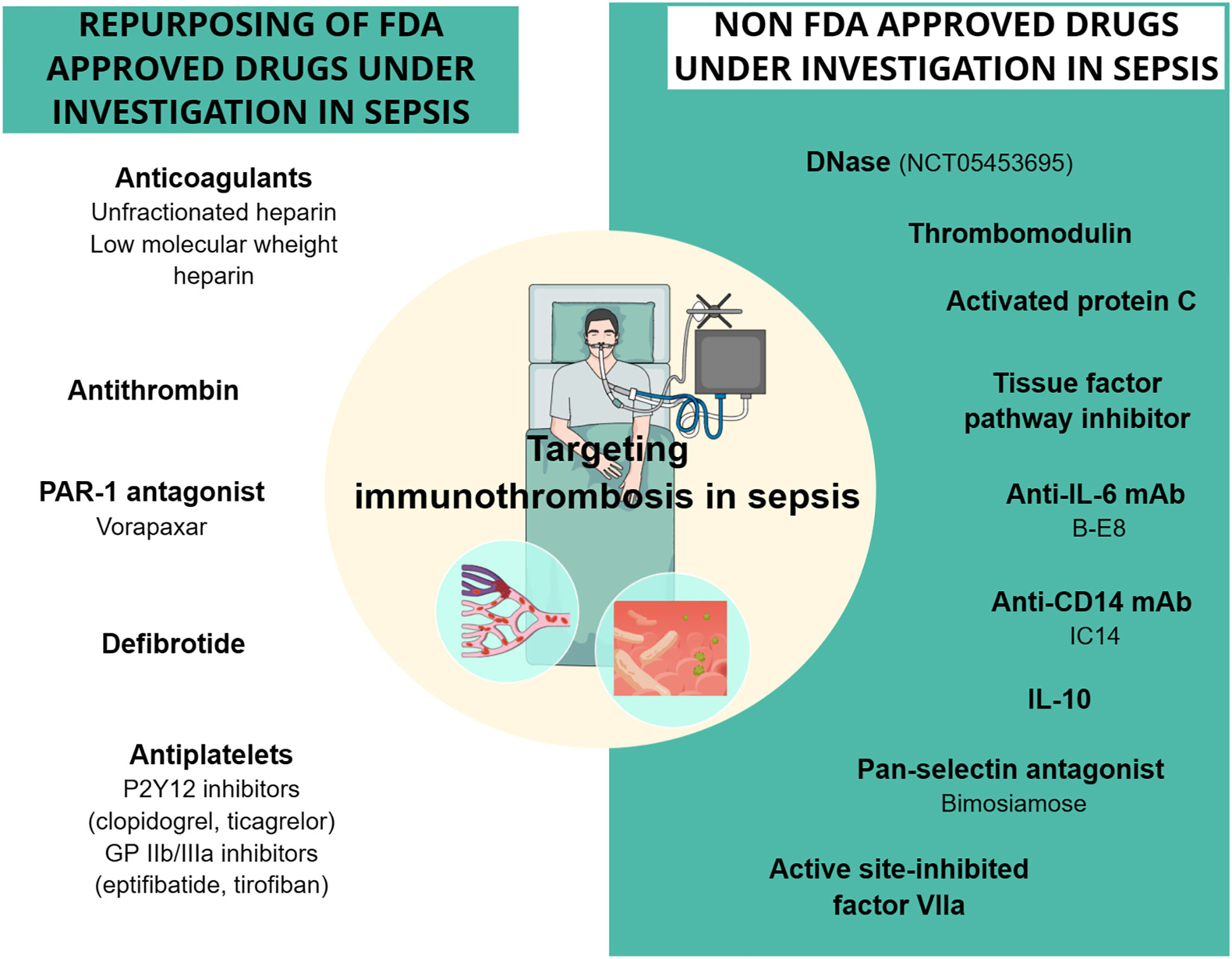
Figure 3 Drugs targeting sepsis-related immunothrombosis in clinical trials. Both FDA-approved and unapproved drugs have been studied in clinical trials for their effect on sepsis-related immunothrombosis. The drugs depicted in the figure showed conflicting results regarding their effect on mortality rates and coagulation parameters in sepsis patients. DNase is the only one with no clinical results yet, as the phase 1 study (NCT05453695) using DNase intervention has recently started. CD14, cluster of differentiation 14; DNase, deoxyribonuclease; GPIIb/IIIa, glycoprotein IIb/IIIa; IL, interleukin; mAb, monoclonal antibody; PAR-1, protease-activated receptor-1.
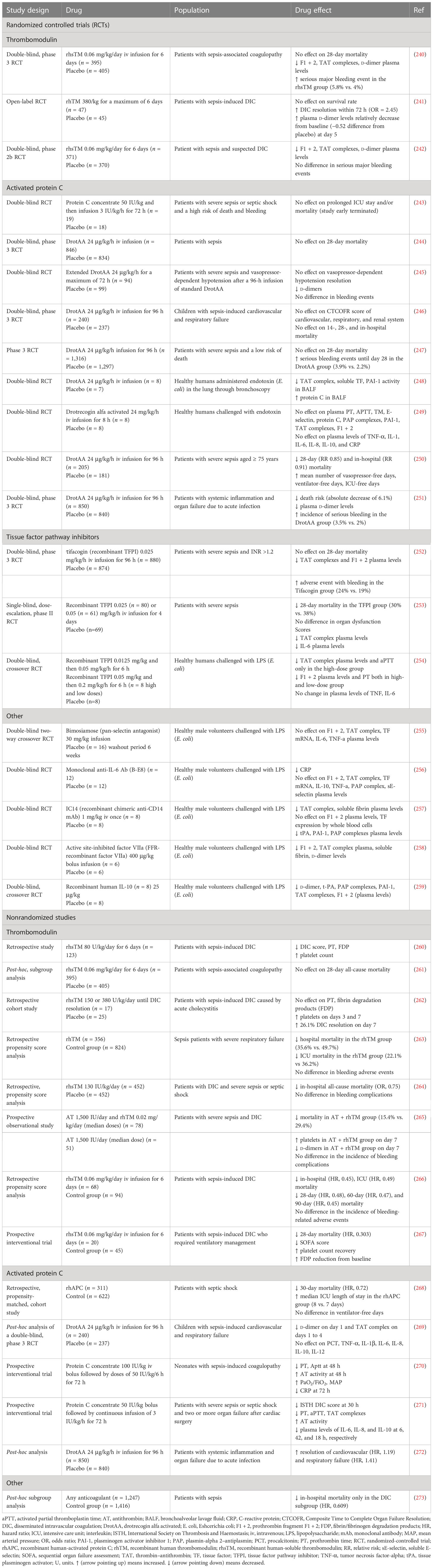
Table 7 Clinical evidence on drugs targeting immunothrombosis in sepsis.
Preclinical evidence on potential molecules targeting immunothrombosis in sepsis
The general failure of clinical trials to identify pharmaceutical agents with an acceptable risk-to-benefit ratio poses the need for discovering new molecules that could mitigate the deleterious effects of immunothrombosis in sepsis. A variety of such molecules have been examined in sepsis animal models, showing promising results, as depicted in Table 8 and Figure 4. Administration of histidine-rich glycoprotein (HRG), a plasma protein found to be significantly depleted in septic conditions, attenuated immunothrombi formation in the lungs of septic mice (294). Reduced neutrophil adherence to endothelium due to shape changes and decreased NET generation, as well as reduced endothelial cell activation, all mediated by HRG, were suggested to be among the potential mechanisms responsible for this effect (294). Another study investigated the beneficial role of Annexin A1, an anti-inflammatory protein found in low levels in patients with sepsis, on LPS-induced thrombosis (287, 299). Interestingly, Annexin A1 mimetic peptide attenuated platelet aggregation and resulted in delayed and reduced microthrombi formation in the cerebral microcirculation (287).
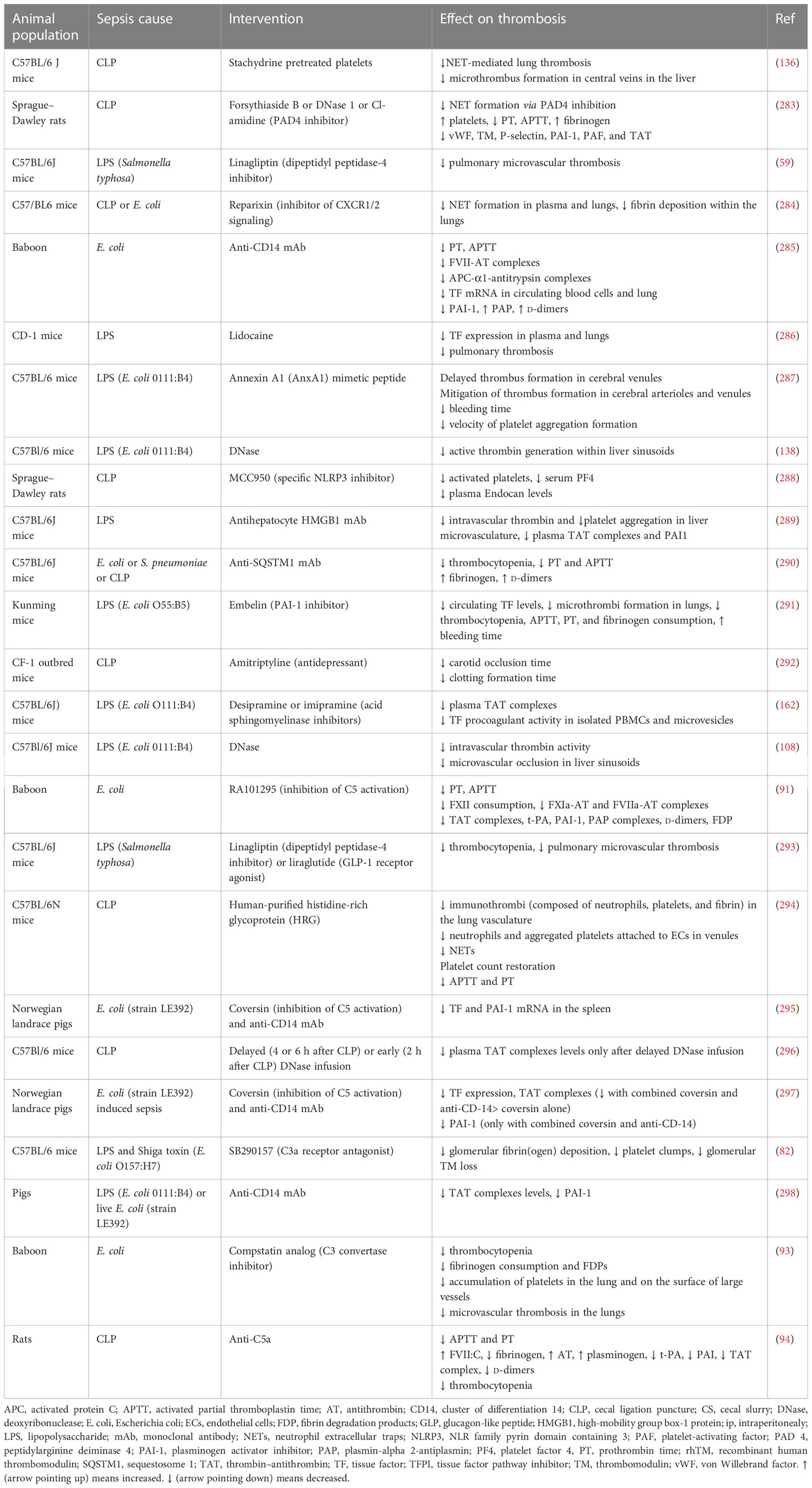
Table 8 Preclinical evidence of novel drugs targeting immunothrombosis in sepsis.
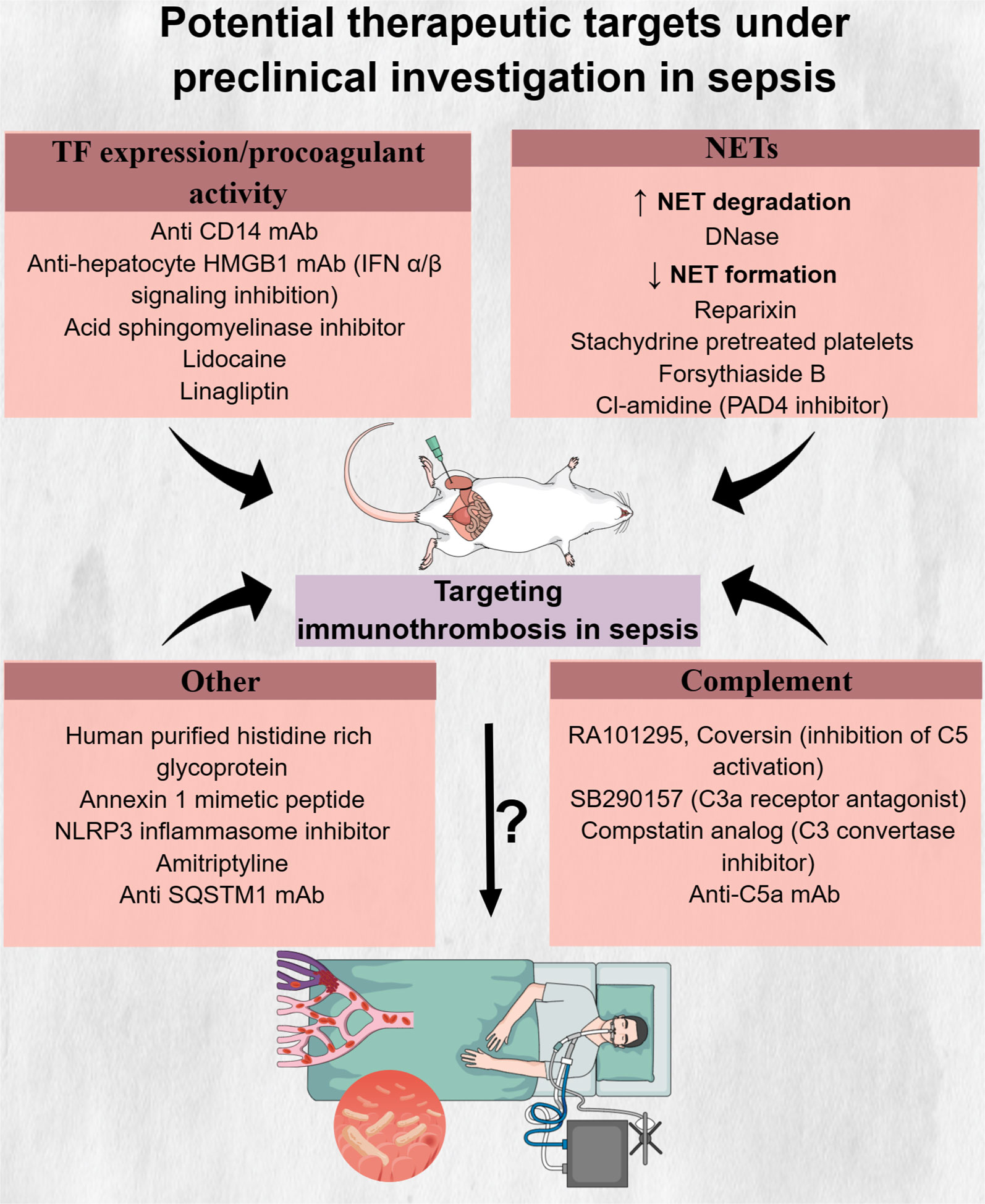
Figure 4 Potential therapeutic targets under preclinical investigation in sepsis. Inhibitors of innate immune components, like neutrophil extracellular traps and complement components C5 and C3, drugs interfering with tissue factor formation and procoagulant activity as well as molecules with multiple actions, have shown promising results in reducing immunothrombosis in preclinical sepsis models. However, the safety, tolerability, and effectiveness of these drugs in abrogating the deleterious effects of immunothrombosis in patients with sepsis are still unknown. C5, complement 5; C3, complement 3; CD14, cluster of differentiation 14; DNase, deoxyribonuclease; HMGB1, high mobility group box 1; IFN, interferon; mAb, monocloncal antibody; NETs, neutrophil extracellular traps; NLRP3, NLR family pyrin domain containing 3; PAD4, protein arginine deiminase; SQSTM1, sequestosome 1; TF, tissue factor.
A variety of inhibitory molecules targeting pathways involved in TF expression in immune or endothelial cells or in TF procoagulant activity have also been proposed. Disrupting TLR4-mediated LPS signaling through cluster of differentiation 14 (CD14) inhibition in immune cells was one potential pathway to target. Specifically, CD14 is a protein responsible for LPS transfer to its cell surface receptor complex, TLR4/myeloid differentiation factor 2 (MD-2) (a co-receptor of TLR4) (285). Inhibitory monoclonal antibody (mAb) against CD14 led to decreased activation of the extrinsic coagulation pathway through suppression of TF expression in leukocytes while also enhancing fibrinolysis through decreasing plasma PAI-1 (285). Apart from anti-CD14 mAb, lidocaine could also inhibit LPS-induced TF expression in monocytes through upregulation of suppressor of cytokine signaling 3 (SOCS3), which in turn downregulated the TLR4-TF pathway (286). Another potential pathway that could serve as a therapeutic target for mitigating immunothrombosis was shown to be type 1 interferon (IFN-1)-induced upregulation of TF procoagulant activity. Type 1 IFNs are cytokines that increase in response to bacterial infection and are characterized by antimicrobial properties. Inhibition of the expression of IFN-1 receptors or downstream effectors (e.g., HMGB1) in macrophages reduced gram-negative bacteria-induced DIC (289). TF procoagulant activity was also found to be decreased by acid sphingomyelinase inhibitors. In response to LPS, acid sphingomyelinase translocates to the cell membrane and hydrolyzes sphingomyelin, which, in resting conditions, maintains TF in an encrypted form with minimal procoagulant activity. Therefore, inhibition of acid sphingomyelinase inhibitors could maintain sphingomyelin levels on cell membranes despite LPS stimulation, resulting in decreased TF activity and reduced TAT levels in a sepsis mouse model (167). Linagliptin, a dipeptidyl peptidase-4 inhibitor used for diabetes treatment, was also found to ameliorate LPS-induced pulmonary thrombosis in mice via decreased endothelial TF expression. This effect was shown to be mediated through linagliptin-induced activation of the Akt/eNOS pathway and subsequent NO production (59). Noteworthy, linagliptin or the glucagon-like peptide-1 (GLP-1) analog, liraglutide, were shown to have further inhibitory effects on platelet thrombin generation and platelet aggregation via the cyclic AMP (cAMP)-protein kinase A (PKA) signaling pathway (293). Among other clinically approved drugs, amitriptyline, a tricyclic antidepressant, was also capable of reducing thrombosis through TNF-α level reduction (292).
The NLRP3 inflammasome, a complex responsible for IL-1β and IL-18 release and a crucial mediator of pyroptosis, became activated in platelets in a sepsis rat model and correlated with lung and renal injury (300, 301). Blockage of NLRP3, using a selective inhibitor, led to decreased platelet activation and offered protection from organ dysfunction in rats with sepsis (288). Another potential therapeutic target was shown to be SQSTM1, a stress response protein that was released from monocytes and macrophages upon LPS stimulation (290). Interestingly, an inhibitory antibody against SQSTM1 attenuated DIC in bacteria-induced sepsis (290).
Promising results have also been demonstrated in preclinical studies examining the effect of NET inhibition on sepsis-induced thrombosis (108, 138, 296). DNase is an enzyme capable of digesting DNA. Two types of DNases have been found: DNase 1, which degrades free DNA, and DNase 1-like 3 (DNase1L3), which degrades DNA bound to proteins, such as histones (140). Patients with sepsis were shown to have decreased levels of DNase 1 compared to healthy controls (302). Moreover, mice with a combined deficiency of DNase1 and DNase1L3 demonstrated increased thrombosis in pulmonary vessels compared to mice capable of producing at least one of these two enzymes when both were infected either with LPS or heat-killed E. coli (140). Exogenous DNase administration promoted NET degradation and resulted in reduced intravascular thrombin activity as well as decreased microthrombus formation in liver sinusoids (108). The timing of the DNase infusion seems to affect its beneficial effect, with delayed infusion resulting in a significant decrease in TAT complexes compared to the insignificant effect observed with early infusion (296). Importantly, DNase administration, aiming to decrease LPS-induced NETosis, prior to S. aureus infection, did not negatively affect pathogen capture and clearance (138). However, the impact of DNase infusion after bacterial infection was not examined in that study. In contrast, mice treated with DNase at seven consecutive time points, beginning 1 h after cecal ligation and puncture (CLP), showed increased bacterial dissemination and higher serum IL-6 levels 6 h after CLP and higher mortality at 24 h (303). However, it should be noted that pathogens’ spread and levels of systemic inflammation 24 h after CLP, as well as overall survival, did not increase in DNase-treated mice compared to controls (303). Besides the evaluation of DNase treatment in preclinical sepsis models, a clinical trial (NCT05453695) is going to assess intravenous DNase safety and tolerability as well as its effect on organ dysfunction, duration of ICU hospitalization, mortality, and blood coagulation in patients with sepsis (304). Regarding inhibition of NET formation, reparixin, an inhibitor of interleukin-8 CXC chemokine receptor type 1/2 (CXCR1/2), resulted in decreased NET formation and subsequently reduced fibrin deposition in the lungs of septic mice (284). Similarly, infusion of platelets pretreated with stachydrine, an herbal product, attenuated NET formation, platelet–neutrophil interaction, and subsequent thrombosis (136).
Complement activation, despite being an essential component of the innate immune response, is associated with increased mortality in sepsis (305, 306), while it also contributes to immunothrombosis (307, 308). Blockade of C3a or C5a led to decreased DIC in E. coli or (CLP)-induced sepsis animal models (91, 93, 94). Noteworthy, combined inhibition of C5 activation and CD14 attenuated more robustly hypercoagulability in sepsis (295, 297).
Conclusion
Immunothrombosis is a key pathophysiological mechanism of sepsis, arising from the complex interplay between innate immune and endothelial cells on one side and platelets and the coagulation cascade on the other. Although initially beneficial for the host, uncontrolled and systemic activation of this process in sepsis can lead to thrombotic and bleeding complications, ranging from subclinical abnormalities in coagulation tests to severe clinical manifestations, such as DIC. Endothelial dysfunction, characterized by glycocalyx degradation, increased vascular permeability, as well as proinflammatory and procoagulant properties of endothelial cells, further facilitates immunothrombosis progression. Despite the high burden of coagulation disorders that have a negative impact on sepsis patients’ prognosis, there is no effective treatment aiming to target the immunothrombotic process. Although a variety of anticoagulants and antiplatelet drugs have been evaluated in clinical trials, their beneficial effects are inconsistent, and they are also characterized by a high rate of bleeding complications. However, there is still ongoing research assessing the effect of new molecules on thrombosis in sepsis, with agents targeting intracellular inflammatory pathways, NET formation, and complement components demonstrating promising results. Future studies are called to unravel the pathophysiologic mechanisms involved in immunothrombosis in sepsis, to identify potential prognostic biomarkers, to develop risk scores to predict the outcomes of sepsis, and to test novel therapeutics against immunothrombosis in sepsis.
Author contributions
EM and EA researched data for the article, contributed to the interpretation of the published studies, and drafted all display items. KSte developed the concept, discussed the content, jointly conceived the display items of the article, and edited the article. ST-C, BEV, AG, and KSta provided conceptual advice, participated in the interpretation of the data, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors’ work is supported by the Biotechnology and Biological Sciences Research Council (BBSRC) of the UK Research and Innovation (UKRI) to KSte and AG, by a Wellcome Trust Institutional Strategic Support Fund to ST-C, by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (MODVASC, grant agreement No. 759248), and by the German Research Foundation DFG (SFB1366, project number 394046768) to KSte. This work is supported by the Health + Life Science Alliance Heidelberg Mannheim and received state funds approved by the State Parliament of Baden-Württemberg.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
2. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (2018) 392(10141):75–87. doi: 10.1016/s0140-6736(18)30696-2
3. Patel P, Michael JV, Naik UP, McKenzie SE. Platelet fcγriia in immunity and thrombosis: Adaptive immunothrombosis. J Thromb Haemost (2021) 19(5):1149–60. doi: 10.1111/jth.15265
4. Kanneganti TD. Intracellular innate immune receptors: Life inside the cell. Immunol Rev (2020) 297(1):5–12. doi: 10.1111/imr.12912
5. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
6. Levi M, van der Poll T. Coagulation and sepsis. Thromb Res (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
7. Zeerleder S, Schroeder V, Lämmle B, Wuillemin WA, Hack CE, Kohler HP. Factor xiii in severe sepsis and septic shock. Thromb Res (2007) 119(3):311–8. doi: 10.1016/j.thromres.2006.02.003
8. Hoppensteadt D, Tsuruta K, Cunanan J, Hirman J, Kaul I, Osawa Y, et al. Thrombin generation mediators and markers in sepsis-associated coagulopathy and their modulation by recombinant thrombomodulin. Clin Appl Thromb Hemost (2014) 20(2):129–35. doi: 10.1177/1076029613492875
9. LaRosa SP, Opal SM, Utterback B, Yan SC, Helterbrand J, Simpson AJ, et al. And antithrombin levels are predictive of poor outcome in gram-negative sepsis caused by burkholderia pseudomallei. Int J Infect Dis (2006) 10(1):25–31. doi: 10.1016/j.ijid.2005.06.001
10. Panigada M, Zacchetti L, L'Acqua C, Cressoni M, Anzoletti MB, Bader R, et al. Assessment of fibrinolysis in sepsis patients with urokinase modified thromboelastography. PloS One (2015) 10(8):e0136463. doi: 10.1371/journal.pone.0136463
11. Shaw AD, Vail GM, Haney DJ, Xie J, Williams MD. Severe protein c deficiency is associated with organ dysfunction in patients with severe sepsis. J Crit Care (2011) 26(6):539–45. doi: 10.1016/j.jcrc.2011.05.006
12. Costello RA, Nehring SM. Disseminated intravascular coagulation. In: Statpearls. Treasure Island (FL: StatPearls Publishing LLC (2022).
13. Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med (2021) 47(11):1181–247. doi: 10.1007/s00134-021-06506-y
14. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med (2008) 359(9):938–49. doi: 10.1056/NEJMra0801082
15. Grover SP, Mackman N. Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol (2019) 39(3):331–8. doi: 10.1161/atvbaha.118.312130
16. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol (2007) 27(8):1687–93. doi: 10.1161/atvbaha.107.141911
17. Broze GJ Jr. Tissue factor pathway inhibitor and the revised theory of coagulation. Annu Rev Med (1995) 46:103–12. doi: 10.1146/annurev.med.46.1.103
18. Mammen EF. Antithrombin: Its physiological importance and role in dic. Semin Thromb Hemost (1998) 24(1):19–25. doi: 10.1055/s-2007-995819
19. Van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein c-epcr system: Integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol (2004) 24(8):1374–83. doi: 10.1161/01.Atv.0000134298.25489.92
20. Walker FJ. Regulation of activated protein c by protein s. The role of phospholipid in factor va inactivation. J Biol Chem (1981) 256(21):11128–31.
21. Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role Fibrin J Biol Chem (1982) 257(6):2912–9. doi: 10.1016/S0021-9258(19)81051-7
22. Sprengers ED, Kluft C. Plasminogen activator inhibitors. Blood (1987) 69(2):381–7. doi: 10.1182/blood.V69.2.381.381
23. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med (2020) 202(3):361–70. doi: 10.1164/rccm.201910-1911TR
24. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood (2019) 133(9):906–18. doi: 10.1182/blood-2018-11-882993
25. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. Il-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U.S.A. (2020) 117(36):22351–6. doi: 10.1073/pnas.2010229117
26. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res (2005) 96(12):1233–9. doi: 10.1161/01.RES.0000171805.24799.fa
27. Vlachogiannis NI, Sachse M, Georgiopoulos G, Zormpas E, Bampatsias D, Delialis D, et al. Adenosine-to-Inosine alu rna editing controls the stability of the pro-inflammatory long noncoding rna Neat1 in atherosclerotic cardiovascular disease. J Mol Cell Cardiol (2021) 160:111–20. doi: 10.1016/j.yjmcc.2021.07.005
28. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
29. Yuki K, Koutsogiannaki S. Pattern recognition receptors as therapeutic targets for bacterial, viral and fungal sepsis. Int Immunopharmacol (2021) 98:107909. doi: 10.1016/j.intimp.2021.107909
30. Amalakuhan B, Habib SA, Mangat M, Reyes LF, Rodriguez AH, Hinojosa CA, et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine (2016) 88:267–73. doi: 10.1016/j.cyto.2016.08.028
31. Kayal S, Jaïs JP, Aguini N, Chaudière J, Labrousse J. Elevated circulating e-selectin, intercellular adhesion molecule 1, and Von willebrand factor in patients with severe infection. Am J Respir Crit Care Med (1998) 157(3 Pt 1):776–84. doi: 10.1164/ajrccm.157.3.9705034
32. van Poelgeest EP, Dillingh MR, de Kam M, Malone KE, Kemper M, Stroes ESG, et al. Characterization of immune cell, endothelial, and renal responses upon experimental human endotoxemia. J Pharmacol Toxicol Methods (2018) 89:39–46. doi: 10.1016/j.vascn.2017.10.004
33. Zullo JA, Fan J, Azar TT, Yen W, Zeng M, Chen J, et al. Exocytosis of endothelial lysosome-related organelles hair-triggers a patchy loss of glycocalyx at the onset of sepsis. Am J Pathol (2016) 186(2):248–58. doi: 10.1016/j.ajpath.2015.10.001
34. Lukasz A, Hillgruber C, Oberleithner H, Kusche-Vihrog K, Pavenstädt H, Rovas A, et al. Endothelial glycocalyx breakdown is mediated by angiopoietin-2. Cardiovasc Res (2017) 113(6):671–80. doi: 10.1093/cvr/cvx023
35. Yang CH, Guan XD, Chen J, Ouyang B, Chen MY, Li LF, et al. [the study of the mechanism of the effect of heparin on tissue perfusion of sepsis patients]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue (2008) 20(9):550–2.
36. Iba T, Levy JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost (2019) 17(2):283–94. doi: 10.1111/jth.14371
37. Yang X, Meegan JE, Jannaway M, Coleman DC, Yuan SY. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc Res (2018) 114(13):1752–63. doi: 10.1093/cvr/cvy167
38. Dolmatova EV, Wang K, Mandavilli R, Griendling KK. The effects of sepsis on endothelium and clinical implications. Cardiovasc Res (2021) 117(1):60–73. doi: 10.1093/cvr/cvaa070
39. Fernández-Sarmiento J, Molina CF, Salazar-Pelaez LM, Flórez S, Alarcón-Forero LC, Sarta M, et al. Biomarkers of glycocalyx injury and endothelial activation are associated with clinical outcomes in patients with sepsis: A systematic review and meta-analysis. J Intensive Care Med (2023) 38(1):95–105. doi: 10.1177/08850666221109186
40. Piotti A, Novelli D, Meessen J, Ferlicca D, Coppolecchia S, Marino A, et al. Endothelial damage in septic shock patients as evidenced by circulating syndecan-1, sphingosine-1-Phosphate and soluble ve-cadherin: A substudy of albios. Crit Care (2021) 25(1):113. doi: 10.1186/s13054-021-03545-1
41. Saoraya J, Wongsamita L, Srisawat N, Musikatavorn K. Plasma syndecan-1 is associated with fluid requirements and clinical outcomes in emergency department patients with sepsis. Am J Emerg Med (2021) 42:83–9. doi: 10.1016/j.ajem.2021.01.019
42. Mangat M, Amalakuhan B, Habib S, Reyes LF, Hinojosa CA, Rodriguez AH, et al. High endocan levels are associated with the need for mechanical ventilation among patients with severe sepsis. Eur Respir J (2017) 50(1). doi: 10.1183/13993003.00013-2017
43. Scherpereel A, Depontieu F, Grigoriu B, Cavestri B, Tsicopoulos A, Gentina T, et al. Endocan, a new endothelial marker in human sepsis. Crit Care Med (2006) 34(2):532–7. doi: 10.1097/01.ccm.0000198525.82124.74
44. Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, et al. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest (2018) 128(4):1471–84. doi: 10.1172/jci97488
45. Fisher J, Douglas JJ, Linder A, Boyd JH, Walley KR, Russell JA. Elevated plasma angiopoietin-2 levels are associated with fluid overload, organ dysfunction, and mortality in human septic shock. Crit Care Med (2016) 44(11):2018–27. doi: 10.1097/ccm.0000000000001853
46. Statz S, Sabal G, Walborn A, Williams M, Hoppensteadt D, Mosier M, et al. Angiopoietin 2 levels in the risk stratification and mortality outcome prediction of sepsis-associated coagulopathy. Clin Appl Thromb Hemost (2018) 24(8):1223–33. doi: 10.1177/1076029618786029
47. Yu WK, McNeil JB, Wickersham NE, Shaver CM, Bastarache JA, Ware LB. Angiopoietin-2 outperforms other endothelial biomarkers associated with severe acute kidney injury in patients with severe sepsis and respiratory failure. Crit Care (2021) 25(1):48. doi: 10.1186/s13054-021-03474-z
48. Davis JS, Yeo TW, Piera KA, Woodberry T, Celermajer DS, Stephens DP, et al. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Crit Care (2010) 14(3):R89. doi: 10.1186/cc9020
49. Engelberger RP, Pittet YK, Henry H, Delodder F, Hayoz D, Chioléro RL, et al. Acute endotoxemia inhibits microvascular nitric oxide-dependent vasodilation in humans. Shock (2011) 35(1):28–34. doi: 10.1097/SHK.0b013e3181ec71ab
50. Jonkam CC, Bansal K, Traber DL, Hamahata A, Maybauer MO, Maybauer DM, et al. Pulmonary vascular permeability changes in an ovine model of methicillin-resistant staphylococcus aureus sepsis. Crit Care (2009) 13(1):R19. doi: 10.1186/cc7720
51. Soriano AO, Jy W, Chirinos JA, Valdivia MA, Velasquez HS, Jimenez JJ, et al. Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit Care Med (2005) 33(11):2540–6. doi: 10.1097/01.ccm.0000186414.86162.03
52. Singh J, Lee Y, Kellum JA. A new perspective on no pathway in sepsis and adma lowering as a potential therapeutic approach. Crit Care (2022) 26(1):246. doi: 10.1186/s13054-022-04075-0
53. Bampatsias D, Mavroeidis I, Tual-Chalot S, Vlachogiannis NI, Bonini F, Sachse M, et al. Beta-Secretase-1 antisense rna is associated with vascular ageing and atherosclerotic cardiovascular disease. Thromb Haemost (2022) 122(11):1932–42. doi: 10.1055/a-1914-2094
54. Lambrinoudaki I, Delialis D, Georgiopoulos G, Tual-Chalot S, Vlachogiannis NI, Patras R, et al. Circulating amyloid beta 1-40 is associated with increased rate of progression of atherosclerosis in menopause: A prospective cohort study. Thromb Haemost (2021) 121(5):650–8. doi: 10.1055/s-0040-1721144
55. Tektonidou MG, Kravvariti E, Vlachogiannis NI, Georgiopoulos G, Mantzou A, Sfikakis PP, et al. Clinical value of amyloid-Beta1-40 as a marker of thrombo-inflammation in antiphospholipid syndrome. Rheumatol (Oxford) (2021) 60(4):1669–75. doi: 10.1093/rheumatology/keaa548
56. Stakos DA, Stamatelopoulos K, Bampatsias D, Sachse M, Zormpas E, Vlachogiannis NI, et al. The alzheimer's disease amyloid-beta hypothesis in cardiovascular aging and disease: Jacc focus seminar. J Am Coll Cardiol (2020) 75(8):952–67. doi: 10.1016/j.jacc.2019.12.033
57. Stamatelopoulos K, Pol CJ, Ayers C, Georgiopoulos G, Gatsiou A, Brilakis ES, et al. Amyloid-beta (1-40) peptide and subclinical cardiovascular disease. J Am Coll Cardiol (2018) 72(9):1060–1. doi: 10.1016/j.jacc.2018.06.027
58. Iba T, Levi M, Levy JH. Intracellular communication and immunothrombosis in sepsis. J Thromb Haemost (2022) 20(11):2475–84. doi: 10.1111/jth.15852
59. Wang SC, Wang XY, Liu CT, Chou RH, Chen ZB, Huang PH, et al. The dipeptidyl peptidase-4 inhibitor linagliptin ameliorates endothelial inflammation and microvascular thrombosis in a sepsis mouse model. Int J Mol Sci (2022) 23(6). doi: 10.3390/ijms23063065
60. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in covid-19 immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/jci141374
61. Liberale L, Akhmedov A, Vlachogiannis NI, Bonetti NR, Nageswaran V, Miranda MX, et al. Sirtuin 5 promotes arterial thrombosis by blunting the fibrinolytic system. Cardiovasc Res (2021) 117(10):2275–88. doi: 10.1093/cvr/cvaa268
62. Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, et al. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler Thromb Vasc Biol (2018) 38(8):1901–12. doi: 10.1161/atvbaha.118.311150
63. Stojkovic S, Kaun C, Basilio J, Rauscher S, Hell L, Krychtiuk KA, et al. Tissue factor is induced by interleukin-33 in human endothelial cells: A new link between coagulation and inflammation. Sci Rep (2016) 6:25171. doi: 10.1038/srep25171
64. Kushak RI, Nestoridi E, Lambert J, Selig MK, Ingelfinger JR, Grabowski EF. Detached endothelial cells and microparticles as sources of tissue factor activity. Thromb Res (2005) 116(5):409–19. doi: 10.1016/j.thromres.2005.01.013
65. Nestoridi E, Tsukurov O, Kushak RI, Ingelfinger JR, Grabowski EF. Shiga toxin enhances functional tissue factor on human glomerular endothelial cells: Implications for the pathophysiology of hemolytic uremic syndrome. J Thromb Haemost (2005) 3(4):752–62. doi: 10.1111/j.1538-7836.2005.01205.x
66. Muth H, Maus U, Wygrecka M, Lohmeyer J, Grimminger F, Seeger W, et al. Pro- and antifibrinolytic properties of human pulmonary microvascular versus artery endothelial cells: Impact of endotoxin and tumor necrosis factor-alpha. Crit Care Med (2004) 32(1):217–26. doi: 10.1097/01.Ccm.0000104941.89570.5f
67. Parry GC, Mackman N. Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler Thromb Vasc Biol (1995) 15(5):612–21. doi: 10.1161/01.atv.15.5.612
68. Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, et al. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci U.S.A. (1990) 87(18):7095–9. doi: 10.1073/pnas.87.18.7095
69. van Hinsbergh VW, Kooistra T, van den Berg EA, Princen HM, Fiers W, Emeis JJ. Tumor necrosis factor increases the production of plasminogen activator inhibitor in human endothelial cells in vitro and in rats in vivo. Blood (1988) 72(5):1467–73. doi: 10.1182/blood.V72.5.1467.1467
70. Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA Jr. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: Characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci U.S.A. (1986) 83(12):4533–7. doi: 10.1073/pnas.83.12.4533
71. Lupu C, Westmuckett AD, Peer G, Ivanciu L, Zhu H, Taylor FB Jr., et al. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of escherichia coli sepsis. Am J Pathol (2005) 167(4):1161–72. doi: 10.1016/s0002-9440(10)61204-7
72. Song D, Ye X, Xu H, Liu SF. Activation of endothelial intrinsic nf-{Kappa}B pathway impairs protein c anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood (2009) 114(12):2521–9. doi: 10.1182/blood-2009-02-205914
73. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial nf-kappab in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med (2008) 205(6):1303–15. doi: 10.1084/jem.20071393
74. Cone JB, Ferrer TJ, Wallace BH, Wang J, Hauer-Jensen M. Alterations in endothelial thrombomodulin expression in zymosan-induced lung injury. J Trauma (2003) 54(4):731–6. doi: 10.1097/01.Ta.0000054652.38788.5a
75. Drake TA, Cheng J, Chang A, Taylor FB Jr. Expression of tissue factor, thrombomodulin, and e-selectin in baboons with lethal escherichia coli sepsis. Am J Pathol (1993) 142(5):1458–70.
76. Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, et al. Dysfunction of endothelial protein c activation in severe meningococcal sepsis. N Engl J Med (2001) 345(6):408–16. doi: 10.1056/nejm200108093450603
77. Ikeda M, Matsumoto H, Ogura H, Hirose T, Shimizu K, Yamamoto K, et al. Circulating syndecan-1 predicts the development of disseminated intravascular coagulation in patients with sepsis. J Crit Care (2018) 43:48–53. doi: 10.1016/j.jcrc.2017.07.049
78. Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med (1997) 185(9):1619–27. doi: 10.1084/jem.185.9.1619
79. Chen Y, Li X, Lin X, Liang H, Liu X, Zhang X, et al. Complement C5a induces the generation of neutrophil extracellular traps by inhibiting mitochondrial Stat3 to promote the development of arterial thrombosis. Thromb J (2022) 20(1):24. doi: 10.1186/s12959-022-00384-0
80. Fattahi F, Grailer JJ, Jajou L, Zetoune FS, Andjelkovic AV, Ward PA. Organ distribution of histones after intravenous infusion of fitc histones or after sepsis. Immunol Res (2015) 61(3):177–86. doi: 10.1007/s12026-015-8628-2
81. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol (2015) 67(10):2780–90. doi: 10.1002/art.39239
82. Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, et al. Alternative pathway activation of complement by shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol (2011) 187(1):172–80. doi: 10.4049/jimmunol.1100491
83. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol (2006) 177(7):4794–802. doi: 10.4049/jimmunol.177.7.4794
84. Wojta J, Kaun C, Zorn G, Ghannadan M, Hauswirth AW, Sperr WR, et al. C5a stimulates production of plasminogen activator inhibitor-1 in human mast cells and basophils. Blood (2002) 100(2):517–23. doi: 10.1182/blood.v100.2.517
85. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost (1997) 77(2):394–8. doi: 10.1055/s-0038-1655974
86. Peerschke EI, Reid KB, Ghebrehiwet B. Platelet activation by C1q results in the induction of alpha Iib/Beta 3 integrins (Gpiib-iiia) and the expression of p-selectin and procoagulant activity. J Exp Med (1993) 178(2):579–87. doi: 10.1084/jem.178.2.579
87. Ando B, Wiedmer T, Hamilton KK, Sims PJ. Complement proteins C5b-9 initiate secretion of platelet storage granules without increased binding of fibrinogen or Von willebrand factor to newly expressed cell surface gpiib-iiia. J Biol Chem (1988) 263(24):11907–14. doi: 10.1016/S0021-9258(18)37872-4
88. Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood (1986) 68(4):875–80. doi: 10.1182/blood.V68.4.875.875
89. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med (1983) 158(2):603–15. doi: 10.1084/jem.158.2.603
90. Muhlfelder TW, Niemetz J, Kreutzer D, Beebe D, Ward PA, Rosenfeld SI. C5 chemotactic fragment induces leukocyte production of tissue factor activity: A link between complement and coagulation. J Clin Invest (1979) 63(1):147–50. doi: 10.1172/jci109269
91. Keshari RS, Silasi R, Popescu NI, Patel MM, Chaaban H, Lupu C, et al. Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of escherichia coli sepsis. Proc Natl Acad Sci U.S.A. (2017) 114(31):E6390–e9. doi: 10.1073/pnas.1706818114
92. Mizuno T, Yoshioka K, Mizuno M, Shimizu M, Nagano F, Okuda T, et al. Complement component 5 promotes lethal thrombosis. Sci Rep (2017) 7:42714. doi: 10.1038/srep42714
93. Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of escherichia coli sepsis. Blood (2010) 116(6):1002–10. doi: 10.1182/blood-2010-02-269746
94. Laudes IJ, Chu JC, Sikranth S, Huber-Lang M, Guo RF, Riedemann N, et al. Anti-C5a ameliorates Coagulation/Fibrinolytic protein changes in a rat model of sepsis. Am J Pathol (2002) 160(5):1867–75. doi: 10.1016/s0002-9440(10)61133-9
95. Liebman HA, Feinstein DI. Thrombosis in patients with paroxysmal noctural hemoglobinuria is associated with markedly elevated plasma levels of leukocyte-derived tissue factor. Thromb Res (2003) 111(4-5):235–8. doi: 10.1016/j.thromres.2003.09.018
96. Hou PC, Filbin MR, Wang H, Ngo L, Huang DT, Aird WC, et al. Endothelial permeability and hemostasis in septic shock: Results from the process trial. Chest (2017) 152(1):22–31. doi: 10.1016/j.chest.2017.01.010
97. Johansen ME, Johansson PI, Ostrowski SR, Bestle MH, Hein L, Jensen AL, et al. Profound endothelial damage predicts impending organ failure and death in sepsis. Semin Thromb Hemost (2015) 41(1):16–25. doi: 10.1055/s-0034-1398377
98. Katayama S, Nunomiya S, Koyama K, Wada M, Koinuma T, Goto Y, et al. Markers of acute kidney injury in patients with sepsis: The role of soluble thrombomodulin. Crit Care (2017) 21(1):229. doi: 10.1186/s13054-017-1815-x
99. Zhang J, Xue M, Chen Y, Liu C, Kuang Z, Mu S, et al. Identification of soluble thrombomodulin and tissue plasminogen activator-inhibitor complex as biomarkers for prognosis and early evaluation of septic shock and sepsis-induced disseminated intravascular coagulation. Ann Palliat Med (2021) 10(10):10170–84. doi: 10.21037/apm-21-2222
100. Mosad E, Elsayh KI, Eltayeb AA. Tissue factor pathway inhibitor and p-selectin as markers of sepsis-induced non-overt disseminated intravascular coagulopathy. Clin Appl Thromb Hemost (2011) 17(1):80–7. doi: 10.1177/1076029609344981
101. Hoshino K, Kitamura T, Nakamura Y, Irie Y, Matsumoto N, Kawano Y, et al. Usefulness of plasminogen activator inhibitor-1 as a predictive marker of mortality in sepsis. J Intensive Care (2017) 5:42. doi: 10.1186/s40560-017-0238-8
102. Kinasewitz GT, Yan SB, Basson B, Comp P, Russell JA, Cariou A, et al. Universal changes in biomarkers of coagulation and inflammation occur in patients with severe sepsis, regardless of causative micro-organism [Isrctn74215569]. Crit Care (2004) 8(2):R82–90. doi: 10.1186/cc2459
103. Ostrowski SR, Haase N, Müller RB, Møller MH, Pott FC, Perner A, et al. Association between biomarkers of endothelial injury and hypocoagulability in patients with severe sepsis: A prospective study. Crit Care (2015) 19(1):191. doi: 10.1186/s13054-015-0918-5
104. Zegeye MM, Lindkvist M, Fälker K, Kumawat AK, Paramel G, Grenegård M, et al. Activation of the Jak/Stat3 and Pi3k/Akt pathways are crucial for il-6 trans-Signaling-Mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun Signal (2018) 16(1):55. doi: 10.1186/s12964-018-0268-4
105. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, et al. Hemolytic uremic syndrome-associated shiga toxins promote endothelial-cell secretion and impair Adamts13 cleavage of unusually Large Von willebrand factor multimers. Blood (2005) 106(13):4199–209. doi: 10.1182/blood-2005-05-2111
106. Abrams ST, Morton B, Alhamdi Y, Alsabani M, Lane S, Welters ID, et al. A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. Am J Respir Crit Care Med (2019) 200(7):869–80. doi: 10.1164/rccm.201811-2111OC
107. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet Tlr4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
108. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129(10):1357–67. doi: 10.1182/blood-2016-09-741298
109. Zhang H, Zhou Y, Qu M, Yu Y, Chen Z, Zhu S, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol (2021) 11:677902. doi: 10.3389/fcimb.2021.677902
110. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
111. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol (2016) 99(1):153–62. doi: 10.1189/jlb.3A0415-161R
112. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12(12):2074–88. doi: 10.1111/jth.12710
113. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet Hmgb1 through release of neutrophil-extracellular traps and DNA. Sci Rep (2018) 8(1):2068. doi: 10.1038/s41598-018-20479-x
114. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood (2014) 123(16):2573–84. doi: 10.1182/blood-2013-07-516484
115. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced netosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
116. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3ar-dependent netosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun (2016) 7:11037. doi: 10.1038/ncomms11037
117. Chen Z, Zhang H, Qu M, Nan K, Cao H, Cata JP, et al. Review: The emerging role of neutrophil extracellular traps in sepsis and sepsis-associated thrombosis. Front Cell Infect Microbiol (2021) 11:653228. doi: 10.3389/fcimb.2021.653228
118. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188(7):3522–31. doi: 10.4049/jimmunol.1102404
119. Yu S, Liu J, Yan N. Endothelial dysfunction induced by extracellular neutrophil traps plays important role in the occurrence and treatment of extracellular neutrophil traps-related disease. Int J Mol Sci (2022) 23(10). doi: 10.3390/ijms23105626
120. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, et al. Neutrophil extracellular traps promote angiogenesis: Evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol (2016) 36(10):2078–87. doi: 10.1161/atvbaha.116.307634
121. Boufenzer A, Carrasco K, Jolly L, Brustolin B, Di-Pillo E, Derive M, et al. Potentiation of nets release is novel characteristic of trem-1 activation and the pharmacological inhibition of trem-1 could prevent from the deleterious consequences of nets release in sepsis. Cell Mol Immunol (2021) 18(2):452–60. doi: 10.1038/s41423-020-00591-7
122. Jin J, Qiao S, Liu J, Li W, Wang F, Gao X, et al. Neutrophil extracellular traps promote thrombogenicity in cerebral venous sinus thrombosis. Cell Biosci (2022) 12(1):114. doi: 10.1186/s13578-022-00845-z
123. Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells Via tlr and activation of nf-κb and ap-1. Thromb Res (2016) 137:211–8. doi: 10.1016/j.thromres.2015.10.012
124. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PloS One (2012) 7(2):e32366. doi: 10.1371/journal.pone.0032366
125. Liang Y, Pan B, Alam HB, Deng Q, Wang Y, Chen E, et al. Inhibition of peptidylarginine deiminase alleviates lps-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur J Pharmacol (2018) 833:432–40. doi: 10.1016/j.ejphar.2018.07.005
126. Meegan JE, Yang X, Beard RS Jr., Jannaway M, Chatterjee V, Taylor-Clark TE, et al. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem Biophys Res Commun (2018) 503(3):1498–502. doi: 10.1016/j.bbrc.2018.07.069
127. Bianchini EP, Razanakolona M, Helms J, Zouiti F, Couteau-Chardon A, Marin-Esteban V, et al. The proteolytic inactivation of protein z-dependent protease inhibitor by neutrophil elastase might promote the procoagulant activity of neutrophil extracellular traps in sepsis. Thromb Haemost (2022) 122(4):506–16. doi: 10.1055/a-1530-3980
128. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol (2014) 34(9):1977–84. doi: 10.1161/atvbaha.114.304114
129. Cruz DBD, Helms J, Aquino LR, Stiel L, Cougourdan L, Broussard C, et al. DNA-Bound elastase of neutrophil extracellular traps degrades plasminogen, reduces plasmin formation, and decreases fibrinolysis: Proof of concept in septic shock plasma. FASEB J (2019) 33(12):14270–80. doi: 10.1096/fj.201901363RRR
130. Yang S, Qi H, Kan K, Chen J, Xie H, Guo X, et al. Neutrophil extracellular traps promote hypercoagulability in patients with sepsis. Shock (2017) 47(2):132–9. doi: 10.1097/shk.0000000000000741
131. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet Tlr2 and Tlr4. Blood (2011) 118(7):1952–61. doi: 10.1182/blood-2011-03-343061
132. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein c activation. J Thromb Haemost (2011) 9(9):1795–803. doi: 10.1111/j.1538-7836.2011.04422.x
133. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol (2015) 35(12):2544–53. doi: 10.1161/atvbaha.115.306035
134. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U.S.A. (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
135. Kambas K, Markiewski MM, Pneumatikos IA, Rafail SS, Theodorou V, Konstantonis D, et al. C5a and tnf-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol (2008) 180(11):7368–75. doi: 10.4049/jimmunol.180.11.7368
136. Sun X, Zhou M, Pu J, Wang T. Stachydrine exhibits a novel antiplatelet property and ameliorates platelet-mediated thrombo-inflammation. BioMed Pharmacother (2022) 152:113184. doi: 10.1016/j.biopha.2022.113184
137. Arroyo AB, Fernández-Pérez MP, Del Monte A, Águila S, Méndez R, Hernández-Antolín R, et al. Mir-146a is a pivotal regulator of neutrophil extracellular trap formation promoting thrombosis. Haematologica (2021) 106(6):1636–46. doi: 10.3324/haematol.2019.240226
138. Carestia A, Davis RP, Davis L, Jenne CN. Inhibition of immunothrombosis does not affect pathogen capture and does not promote bacterial dissemination in a mouse model of sepsis. Platelets (2020) 31(7):925–31. doi: 10.1080/09537104.2019.1704711
139. Wang Y, Luo L, Braun O, Westman J, Madhi R, Herwald H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation Via the intrinsic pathway of coagulation in mice. Sci Rep (2018) 8(1):4020. doi: 10.1038/s41598-018-22156-5
140. Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host dnases prevent vascular occlusion by neutrophil extracellular traps. Science (2017) 358(6367):1202–6. doi: 10.1126/science.aam8897
141. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity Via neutrophil serine proteases. Nat Med (2010) 16(8):887–96. doi: 10.1038/nm.2184
142. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15(11):1318–21. doi: 10.1038/nm.2053
143. Abdeen S, Bdeir K, Abu-Fanne R, Maraga E, Higazi M, Khurram N, et al. Alpha-defensins: Risk factor for thrombosis in covid-19 infection. Br J Haematol (2021) 194(1):44–52. doi: 10.1111/bjh.17503
144. Delabranche X, Stiel L, Severac F, Galoisy AC, Mauvieux L, Zobairi F, et al. Evidence of netosis in septic shock-induced disseminated intravascular coagulation. Shock (2017) 47(3):313–7. doi: 10.1097/shk.0000000000000719
145. Schoergenhofer C, Schwameis M, Hobl EL, Ay C, Key NS, Derhaschnig U, et al. Potent irreversible P2y12 inhibition does not reduce lps-induced coagulation activation in a randomized, double-blind, placebo-controlled trial. Clin Sci (Lond) (2016) 130(6):433–40. doi: 10.1042/cs20150591
146. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
147. Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood (2017) 129(8):1021–9. doi: 10.1182/blood-2016-06-722298
148. Maruchi Y, Tsuda M, Mori H, Takenaka N, Gocho T, Huq MA, et al. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit Care (2018) 22(1):176. doi: 10.1186/s13054-018-2109-7
149. Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood (2012) 120(10):2133–43. doi: 10.1182/blood-2012-06-437772
150. Maugeri N, Brambilla M, Camera M, Carbone A, Tremoli E, Donati MB, et al. Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J Thromb Haemost (2006) 4(6):1323–30. doi: 10.1111/j.1538-7836.2006.01968.x
151. Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis-the journey of tf through nets. Front Immunol (2012) 3:385. doi: 10.3389/fimmu.2012.00385
152. Osterud B, Rao LV, Olsen JO. Induction of tissue factor expression in whole blood: Lack of evidence for the presence of tissue factor expression in granulocytes. Thromb Haemost (2000) 83(6):861–7. doi: 10.1055/s-0037-1613934
153. Ho J, Zhang L, Liu X, Wong SH, Wang MHT, Lau BWM, et al. Pathological role and diagnostic value of endogenous host defense peptides in adult and neonatal sepsis: A systematic review. Shock (2017) 47(6):673–9. doi: 10.1097/shk.0000000000000815
154. Abu-Fanne R, Stepanova V, Litvinov RI, Abdeen S, Bdeir K, Higazi M, et al. Neutrophil α-defensins promote thrombosis in vivo by altering fibrin formation, structure, and stability. Blood (2019) 133(5):481–93. doi: 10.1182/blood-2018-07-861237
155. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PloS One (2012) 7(9):e45427. doi: 10.1371/journal.pone.0045427
156. Bajaj MS, Ghosh M, Bajaj SP. Fibronectin-adherent monocytes express tissue factor and tissue factor pathway inhibitor whereas endotoxin-stimulated monocytes primarily express tissue factor: Physiologic and pathologic implications. J Thromb Haemost (2007) 5(7):1493–9. doi: 10.1111/j.1538-7836.2007.02604.x
157. Chen F, Zhao Z, Zhou J, Lu Y, Essex DW, Wu Y. Protein disulfide isomerase enhances tissue factor-dependent thrombin generation. Biochem Biophys Res Commun (2018) 501(1):172–7. doi: 10.1016/j.bbrc.2018.04.207
158. Del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue-Factor-Bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood (2005) 106(5):1604–11. doi: 10.1182/blood-2004-03-1095
159. Rauch U, Bonderman D, Bohrmann B, Badimon JJ, Himber J, Riederer MA, et al. Transfer of tissue factor from leukocytes to platelets is mediated by Cd15 and tissue factor. Blood (2000) 96(1):170–5. doi: 10.1182/blood.V96.1.170
160. Zhou H, Wolberg AS, Roubey RA. Characterization of monocyte tissue factor activity induced by igg antiphospholipid antibodies and inhibition by dilazep. Blood (2004) 104(8):2353–8. doi: 10.1182/blood-2004-01-0145
161. Pawlinski R, Wang JG, Owens AP 3rd, Williams J, Antoniak S, Tencati M, et al. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood (2010) 116(5):806–14. doi: 10.1182/blood-2009-12-259267
162. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin d-dependent phosphatidylserine exposure. Immunity (2019) 51(6):983–96.e6. doi: 10.1016/j.immuni.2019.11.005
163. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity (2019) 50(6):1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
164. Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B, Mann KG. Tissue factor activity in whole blood. Blood (2005) 105(7):2764–70. doi: 10.1182/blood-2004-09-3567
165. Kälsch T, Elmas E, Nguyen XD, Suvajac N, Klüter H, Borggrefe M, et al. Endotoxin-induced effects on platelets and monocytes in an in vivo model of inflammation. Basic Res Cardiol (2007) 102(5):460–6. doi: 10.1007/s00395-007-0667-y
166. Ryan TAJ, Preston RJS, O'Neill LAJ. Immunothrombosis and the molecular control of tissue factor by pyroptosis: Prospects for new anticoagulants. Biochem J (2022) 479(6):731–50. doi: 10.1042/bcj20210522
167. Wang J, Pendurthi UR, Rao LVM. Acid sphingomyelinase plays a critical role in lps- and cytokine-induced tissue factor procoagulant activity. Blood (2019) 134(7):645–55. doi: 10.1182/blood.2019001400
168. Ryan TAJ, O'Neill LAJ. Innate immune signaling and immunothrombosis: New insights and therapeutic opportunities. Eur J Immunol (2022) 52(7):1024–34. doi: 10.1002/eji.202149410
169. Schoergenhofer C, Schwameis M, Gelbenegger G, Buchtele N, Thaler B, Mussbacher M, et al. Inhibition of protease-activated receptor (Par1) reduces activation of the endothelium, coagulation, fibrinolysis and inflammation during human endotoxemia. Thromb Haemost (2018) 118(7):1176–84. doi: 10.1055/s-0038-1655767
170. Htun P, Fateh-Moghadam S, Tomandl B, Handschu R, Klinger K, Stellos K, et al. Course of platelet activation and platelet-leukocyte interaction in cerebrovascular ischemia. Stroke (2006) 37(9):2283–7. doi: 10.1161/01.Str.0000236638.75591.61
171. Langer HF, Daub K, Braun G, Schönberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells Via mac-1/Jam-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol (2007) 27(6):1463–70. doi: 10.1161/atvbaha.107.141515
172. Bakogiannis C, Sachse M, Stamatelopoulos K, Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine (2019) 122:154157. doi: 10.1016/j.cyto.2017.09.013
173. Vardon Bounes F, Mémier V, Marcaud M, Jacquemin A, Hamzeh-Cognasse H, Garcia C, et al. Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci Rep (2018) 8(1):13536. doi: 10.1038/s41598-018-31910-8
174. Tatsidou PT, Chantzichristos VG, Tsoumani ME, Sidiropoulou S, Ntalas IV, Goudevenos JA, et al. Circulating progenitor cells and their interaction with platelets in patients with an acute coronary syndrome. Platelets (2019) 30(3):314–21. doi: 10.1080/09537104.2018.1430355
175. Rath D, Chatterjee M, Borst O, Müller K, Stellos K, Mack AF, et al. Expression of stromal cell-derived factor-1 receptors Cxcr4 and Cxcr7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. Eur Heart J (2014) 35(6):386–94. doi: 10.1093/eurheartj/eht448
176. Stellos K, Bigalke B, Borst O, Pfaff F, Elskamp A, Sachsenmaier S, et al. Circulating platelet-progenitor cell coaggregate formation is increased in patients with acute coronary syndromes and augments recruitment of Cd34+ cells in the ischaemic microcirculation. Eur Heart J (2013) 34(32):2548–56. doi: 10.1093/eurheartj/eht131
177. Geisler T, Fekecs L, Wurster T, Chiribiri A, Schuster A, Nagel E, et al. Association of platelet-Sdf-1 with hemodynamic function and infarct size using cardiac Mr in patients with Ami. Eur J Radiol (2012) 81(4):e486–90. doi: 10.1016/j.ejrad.2011.06.019
178. Stellos K, Bigalke B, Langer H, Geisler T, Schad A, Kögel A, et al. Expression of stromal-Cell-Derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of Cd34+ progenitor cells. Eur Heart J (2009) 30(5):584–93. doi: 10.1093/eurheartj/ehn566
179. Stellos K, Bigalke B, Stakos D, Henkelmann N, Gawaz M. Platelet-bound p-selectin expression in patients with coronary artery disease: Impact on clinical presentation and myocardial necrosis, and effect of diabetes mellitus and anti-platelet medication. J Thromb Haemost (2010) 8(1):205–7. doi: 10.1111/j.1538-7836.2009.03659.x
180. Stellos K, Panagiota V, Kögel A, Leyhe T, Gawaz M, Laske C. Predictive value of platelet activation for the rate of cognitive decline in alzheimer's disease patients. J Cereb Blood Flow Metab (2010) 30(11):1817–20. doi: 10.1038/jcbfm.2010.140
181. Geisler T, Mueller K, Aichele S, Bigalke B, Stellos K, Htun P, et al. Impact of inflammatory state and metabolic control on responsiveness to dual antiplatelet therapy in type 2 diabetics after pci: Prognostic relevance of residual platelet aggregability in diabetics undergoing coronary interventions. Clin Res Cardiol (2010) 99(11):743–52. doi: 10.1007/s00392-010-0179-x
182. Geisler T, Anders N, Paterok M, Langer H, Stellos K, Lindemann S, et al. Platelet response to clopidogrel is attenuated in diabetic patients undergoing coronary stent implantation. Diabetes Care (2007) 30(2):372–4. doi: 10.2337/dc06-1625
183. Wegrzyn G, Walborn A, Rondina M, Fareed J, Hoppensteadt D. Biomarkers of platelet activation and their prognostic value in patients with sepsis-associated disseminated intravascular coagulopathy. Clin Appl Thromb Hemost (2021) 27. doi: 10.1177/1076029620943300
184. McDonald B, Dunbar M. Platelets and intravascular immunity: Guardians of the vascular space during bloodstream infections and sepsis. Front Immunol (2019) 10:2400. doi: 10.3389/fimmu.2019.02400
185. Garcia C, Compagnon B, Poëtte M, Gratacap MP, Lapébie FX, Voisin S, et al. Platelet versus megakaryocyte: Who is the real bandleader of thromboinflammation in sepsis? Cells (2022) 11(9). doi: 10.3390/cells11091507
186. Ghuman H, Shepherd-Roberts A, Watson S, Zuidscherwoude M, Watson SP, Voelz K. Mucor circinelloides induces platelet aggregation through integrin αiibβ3 and fcγriia. Platelets (2019) 30(2):256–63. doi: 10.1080/09537104.2017.1420152
187. Matus V, Valenzuela JG, Hidalgo P, Pozo LM, Panes O, Wozniak A, et al. Human platelet interaction with e. coli O111 promotes tissue-Factor-Dependent procoagulant activity, involving toll like receptor 4. PloS One (2017) 12(9):e0185431. doi: 10.1371/journal.pone.0185431
188. Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, et al. Amplification of bacteria-induced platelet activation is triggered by fcγriia, integrin αiibβ3, and platelet factor 4. Blood (2014) 123(20):3166–74. doi: 10.1182/blood-2013-11-540526
189. Tilley DO, Arman M, Smolenski A, Cox D, O'Donnell JS, Douglas CW, et al. Glycoprotein ibα and fcγriia play key roles in platelet activation by the colonizing bacterium, streptococcus oralis. J Thromb Haemost (2013) 11(5):941–50. doi: 10.1111/jth.12175
190. Vanassche T, Kauskot A, Verhaegen J, Peetermans WE, van Ryn J, Schneewind O, et al. Fibrin formation by staphylothrombin facilitates staphylococcus aureus-induced platelet aggregation. Thromb Haemost (2012) 107(6):1107–21. doi: 10.1160/th11-12-0891
191. Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mrna in human platelets. J Thromb Haemost (2011) 9(4):748–58. doi: 10.1111/j.1538-7836.2011.04208.x
192. Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, et al. Invasive streptococcus pneumoniae trigger platelet activation Via toll-like receptor 2. J Thromb Haemost (2010) 8(12):2757–65. doi: 10.1111/j.1538-7836.2010.04093.x
193. Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, Foster TJ. Direct interaction of iron-regulated surface determinant isdb of staphylococcus aureus with the Gpiib/Iiia receptor on platelets. Microbiol (Reading) (2010) 156(Pt 3). doi: 10.1099/mic.0.036673-0
194. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation Via Tlr4/Myd88 and the cgmp-dependent protein kinase pathway. J Immunol (2009) 182(12):7997–8004. doi: 10.4049/jimmunol.0802884
195. Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, et al. Role of streptococcus gordonii surface proteins Sspa/Sspb and hsa in platelet function. Infect Immun (2007) 75(12):5740–7. doi: 10.1128/iai.00909-07
196. O'Brien L, Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, et al. Multiple mechanisms for the activation of human platelet aggregation by staphylococcus aureus: Roles for the clumping factors clfa and clfb, the serine-aspartate repeat protein sdre and protein a. Mol Microbiol (2002) 44(4):1033–44. doi: 10.1046/j.1365-2958.2002.02935.x
197. Kerrigan SW, Douglas I, Wray A, Heath J, Byrne MF, Fitzgerald D, et al. A role for glycoprotein ib in streptococcus sanguis-induced platelet aggregation. Blood (2002) 100(2):509–16. doi: 10.1182/blood.v100.2.509
198. Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, et al. Activation of protease-activated receptors by gingipains from porphyromonas gingivalis leads to platelet aggregation: A new trait in microbial pathogenicity. Blood (2001) 97(12):3790–7. doi: 10.1182/blood.v97.12.3790
199. Landoni VI, Pittaluga JR, Carestia A, Castillo LA, Nebel MC, Martire-Greco D, et al. Neutrophil extracellular traps induced by shiga toxin and lipopolysaccharide-treated platelets exacerbate endothelial cell damage. Front Cell Infect Microbiol (2022) 12:897019. doi: 10.3389/fcimb.2022.897019
200. Surewaard BGJ, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, et al. α-toxin induces platelet aggregation and liver injury during staphylococcus aureus sepsis. Cell Host Microbe (2018) 24(2):271–84.e3. doi: 10.1016/j.chom.2018.06.017
201. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with covid-19. Blood (2020) 136(11):1317–29. doi: 10.1182/blood.2020007214
202. Stellos K, Sauter R, Fahrleitner M, Grimm J, Stakos D, Emschermann F, et al. Binding of oxidized low-density lipoprotein on circulating platelets is increased in patients with acute coronary syndromes and induces platelet adhesion to vascular wall in vivo–brief report. Arterioscler Thromb Vasc Biol (2012) 32(8):2017–20. doi: 10.1161/atvbaha.111.244707
203. Galgano L, Guidetti GF, Torti M, Canobbio I. The controversial role of lps in platelet activation in vitro. Int J Mol Sci (2022) 23(18). doi: 10.3390/ijms231810900
204. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of nf-κb linking inflammation and thrombosis. Front Immunol (2019) 10:85. doi: 10.3389/fimmu.2019.00085
205. van Geffen JP, Swieringa F, Heemskerk JW. Platelets and coagulation in thrombus formation: Aberrations in the Scott syndrome. Thromb Res (2016) 141(Suppl 2):S12–6. doi: 10.1016/s0049-3848(16)30355-3
206. Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell (2009) 139(6):1143–56. doi: 10.1016/j.cell.2009.11.001
207. Li J, Kim K, Jeong SY, Chiu J, Xiong B, Petukhov PA, et al. Platelet protein disulfide isomerase promotes glycoprotein ibα-mediated platelet-neutrophil interactions under thromboinflammatory conditions. Circulation (2019) 139(10):1300–19. doi: 10.1161/circulationaha.118.036323
208. Liu XL, Wang XZ, Liu XX, Hao D, Jaladat Y, Lu F, et al. Low-dose heparin as treatment for early disseminated intravascular coagulation during sepsis: A prospective clinical study. Exp Ther Med (2014) 7(3):604–8. doi: 10.3892/etm.2013.1466
209. Jaimes F, de la Rosa G, Morales C, Fortich F, Arango C, Aguirre D, et al. Unfractioned heparin for treatment of sepsis: A randomized clinical trial (the hetrase study). Crit Care Med (2009) 37(4):1185–96. doi: 10.1097/CCM.0b013e31819c06bc
210. Shorr AF, Williams MD. Venous thromboembolism in critically ill patients. observations from a randomized trial in sepsis. Thromb Haemost (2009) 101(1):139–44.
211. Levi M, Levy M, Williams MD, Douglas I, Artigas A, Antonelli M, et al. Prophylactic heparin in patients with severe sepsis treated with drotrecogin Alfa (Activated). Am J Respir Crit Care Med (2007) 176(5):483–90. doi: 10.1164/rccm.200612-1803OC
212. Pernerstorfer T, Hollenstein U, Hansen J, Knechtelsdorfer M, Stohlawetz P, Graninger W, et al. Heparin blunts endotoxin-induced coagulation activation. Circulation (1999) 100(25):2485–90. doi: 10.1161/01.cir.100.25.2485
213. Hofmann M, Rest A, Hafner G, Tanner B, Brockerhoff P, Weilemann LS. D-dimer, thrombin-antithrombin iii-complex (Tat) and prothrombin fragment 1+2 (Ptf). parameters for monitoring therapy with low molecular-weight heparin in coagulation disorders. Anaesthesist (1997) 46(8):689–96. doi: 10.1007/s001010050455
214. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. heparin therapy and survival rate. Scand J Infect Dis (1971) 3(2):127–33. doi: 10.3109/inf.1971.3.issue-2.06
215. Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE, et al. Platelet P2y12 inhibitors reduce systemic inflammation and its prothrombotic effects in an experimental human model. Arterioscler Thromb Vasc Biol (2015) 35(12):2562–70. doi: 10.1161/atvbaha.115.306528
216. Berthelsen RE, Ostrowski SR, Bestle MH, Johansson PI. Co-Administration of iloprost and eptifibatide in septic shock (Co-ilepss)-a randomised, controlled, double-blind investigator-initiated trial investigating safety and efficacy. Crit Care (2019) 23(1):301. doi: 10.1186/s13054-019-2573-8
217. Derhaschnig U, Pachinger C, Schweeger-Exeli I, Marsik C, Jilma B. Blockade of Gpiib/Iiia by eptifibatide and tirofiban does not alter tissue factor induced thrombin generation in human endotoxemia. Thromb Haemost (2003) 90(6):1054–60. doi: 10.1160/th03-04-0233
218. Galstian GM, Krechetova AV, Troitskaia VV, Makarova PM, Shutova NA, Gemdzhian ÉG, et al. Therapy with high-doses of antithrombin iii in patients with septic shock and agranulocytosis. Anesteziol Reanimatol (2014) 59(4):39–45.
219. Gando S, Saitoh D, Ishikura H, Ueyama M, Otomo Y, Oda S, et al. A randomized, controlled, multicenter trial of the effects of antithrombin on disseminated intravascular coagulation in patients with sepsis. Crit Care (2013) 17(6):R297. doi: 10.1186/cc13163
220. Hoffmann JN, Wiedermann CJ, Juers M, Ostermann H, Kienast J, Briegel J, et al. Benefit/Risk profile of high-dose antithrombin in patients with severe sepsis treated with and without concomitant heparin. Thromb Haemost (2006) 95(5):850–6. doi: 10.1160/TH05-07-0530
221. Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, et al. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost (2006) 4(1):90–7. doi: 10.1111/j.1538-7836.2005.01697.x
222. Leitner JM, Firbas C, Mayr FB, Reiter RA, Steinlechner B, Jilma B. Recombinant human antithrombin inhibits thrombin formation and interleukin 6 release in human endotoxemia. Clin Pharmacol Ther (2006) 79(1):23–34. doi: 10.1016/j.clpt.2005.10.003
223. Gonano C, Sitzwohl C, Meitner E, Weinstabl C, Kettner SC. Four-day antithrombin therapy does not seem to attenuate hypercoagulability in patients suffering from sepsis. Crit Care (2006) 10(6):R160. doi: 10.1186/cc5098
224. Hoffmann JN, Mühlbayer D, Jochum M, Inthorn D. Effect of long-term and high-dose antithrombin supplementation on coagulation and fibrinolysis in patients with severe sepsis. Crit Care Med (2004) 32(9):1851–9. doi: 10.1097/01.ccm.0000139691.54108.1f
225. Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. high-dose antithrombin iii in severe sepsis: A randomized controlled trial. Jama (2001) 286(15):1869–78. doi: 10.1001/jama.286.15.1869
226. Eisele B, Lamy M, Thijs LG, Keinecke HO, Schuster HP, Matthias FR, et al. Antithrombin iii in patients with severe sepsis. a randomized, placebo-controlled, double-blind multicenter trial plus a meta-analysis on all randomized, placebo-controlled, double-blind trials with antithrombin iii in severe sepsis. Intensive Care Med (1998) 24(7):663–72. doi: 10.1007/s001340050642
227. Inthorn D, Hoffmann JN, Hartl WH, Mühlbayer D, Jochum M. Effect of antithrombin iii supplementation on inflammatory response in patients with severe sepsis. Shock (1998) 10(2):90–6. doi: 10.1097/00024382-199808000-00002
228. Inthorn D, Hoffmann JN, Hartl WH, Mühlbayer D, Jochum M. Antithrombin iii supplementation in severe sepsis: Beneficial effects on organ dysfunction. Shock (1997) 8(5):328–34. doi: 10.1097/00024382-199711000-00003
229. Fourrier F, Chopin C, Huart JJ, Runge I, Caron C, Goudemand J. Double-blind, placebo-controlled trial of antithrombin iii concentrates in septic shock with disseminated intravascular coagulation. Chest (1993) 104(3):882–8. doi: 10.1378/chest.104.3.882
230. Schoergenhofer C, Buchtele N, Gelbenegger G, Derhaschnig U, Firbas C, Kovacevic KD, et al. Defibrotide enhances fibrinolysis in human endotoxemia - a randomized, double blind, crossover trial in healthy volunteers. Sci Rep (2019) 9(1):11136. doi: 10.1038/s41598-019-47630-6
231. Murao A, Kato T, Yamane T, Honda G, Eguchi Y. Benefit profile of thrombomodulin Alfa combined with antithrombin concentrate in patients with sepsis-induced disseminated intravascular coagulation. Clin Appl Thromb Hemost (2022) 28:10760296221077096. doi: 10.1177/10760296221077096
232. Kuroda H, Masuda Y. Comparison of protective effects of recombinant antithrombin gamma and plasma-derived antithrombin on sepsis-induced disseminated intravascular coagulation and multiple organ failure. Clin Appl Thromb Hemost (2020) 26. doi: 10.1177/1076029620981630
233. Kim YJ, Ko BS, Park SY, Oh DK, Hong SB, Jang S, et al. Effect of high-dose antithrombin supplementation in patients with septic shock and disseminated intravascular coagulation. Sci Rep (2019) 9(1):16626. doi: 10.1038/s41598-019-52968-y
234. Hayakawa M, Yamakawa K, Kudo D, Ono K. Optimal antithrombin activity threshold for initiating antithrombin supplementation in patients with sepsis-induced disseminated intravascular coagulation: A multicenter retrospective observational study. Clin Appl Thromb Hemost (2018) 24(6):874–83. doi: 10.1177/1076029618757346
235. Tagami T, Matsui H, Fushimi K, Yasunaga H. Supplemental dose of antithrombin use in disseminated intravascular coagulation patients after abdominal sepsis. Thromb Haemost (2015) 114(3):537–45. doi: 10.1160/th15-01-0053
236. Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: An observational nationwide study. J Thromb Haemost (2014) 12(9):1470–9. doi: 10.1111/jth.12643
237. Eid A, Wiedermann CJ, Kinasewitz GT. Early administration of high-dose antithrombin in severe sepsis: Single center results from the kybersept-trial. Anesth Analg (2008) 107(5):1633–8. doi: 10.1213/ane.0b013e318184621d
238. Kreuz WD, Schneider W, Nowak-Göttl U. Treatment of consumption coagulopathy with antithrombin concentrate in children with acquired antithrombin deficiency–a feasibility pilot study. Eur J Pediatr (1999) 158(Suppl 3):S187–91. doi: 10.1007/pl00014353
239. Gtc Biotherapeutics, Inc. Antithrombin (Recombinant). Food and Drug Administration Website (2009). Available at: https://www.fda.gov/media/75529/download.
240. Vincent JL, Francois B, Zabolotskikh I, Daga MK, Lascarrou JB, Kirov MY, et al. Effect of a recombinant human soluble thrombomodulin on mortality in patients with sepsis-associated coagulopathy: The scarlet randomized clinical trial. Jama (2019) 321(20):1993–2002. doi: 10.1001/jama.2019.5358
241. Hagiwara A, Tanaka N, Uemura T, Matsuda W, Kimura A. Can recombinant human thrombomodulin increase survival among patients with severe septic-induced disseminated intravascular coagulation: A single-centre, open-label, randomised controlled trial. BMJ Open (2016) 6(12):e012850. doi: 10.1136/bmjopen-2016-012850
242. Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, et al. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, art-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med (2013) 41(9):2069–79. doi: 10.1097/CCM.0b013e31828e9b03
243. Pappalardo F, Crivellari M, Di Prima AL, Agracheva N, Celinska-Spodar M, Lembo R, et al. Protein c zymogen in severe sepsis: A double-blinded, placebo-controlled, randomized study. Intensive Care Med (2016) 42(11):1706–14. doi: 10.1007/s00134-016-4405-5
244. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin Alfa (Activated) in adults with septic shock. N Engl J Med (2012) 366(22):2055–64. doi: 10.1056/NEJMoa1202290
245. Dhainaut JF, Antonelli M, Wright P, Desachy A, Reignier J, Lavoue S, et al. Extended drotrecogin Alfa (Activated) treatment in patients with prolonged septic shock. Intensive Care Med (2009) 35(7):1187–95. doi: 10.1007/s00134-009-1436-1
246. Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, et al. Drotrecogin Alfa (Activated) in children with severe sepsis: A multicentre phase iii randomised controlled trial. Lancet (2007) 369(9564):836–43. doi: 10.1016/s0140-6736(07)60411-5
247. Abraham E, Laterre PF, Garg R, Levy H, Talwar D, Trzaskoma BL, et al. Drotrecogin Alfa (Activated) for adults with severe sepsis and a low risk of death. N Engl J Med (2005) 353(13):1332–41. doi: 10.1056/NEJMoa050935
248. van der Poll T, Levi M, Nick JA, Abraham E. Activated protein c inhibits local coagulation after intrapulmonary delivery of endotoxin in humans. Am J Respir Crit Care Med (2005) 171(10):1125–8. doi: 10.1164/rccm.200411-1483OC
249. Kalil AC, Coyle SM, Um JY, LaRosa SP, Turlo MA, Calvano SE, et al. Effects of drotrecogin Alfa (Activated) in human endotoxemia. Shock (2004) 21(3):222–9. doi: 10.1097/01.shk.0000116778.27924.79
250. Ely EW, Angus DC, Williams MD, Bates B, Qualy R, Bernard GR. Drotrecogin Alfa (Activated) treatment of older patients with severe sepsis. Clin Infect Dis (2003) 37(2):187–95. doi: 10.1086/375775
251. Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein c for severe sepsis. N Engl J Med (2001) 344(10):699–709. doi: 10.1056/nejm200103083441001
252. Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, et al. Efficacy and safety of tifacogin (Recombinant tissue factor pathway inhibitor) in severe sepsis: A randomized controlled trial. Jama (2003) 290(2):238–47. doi: 10.1001/jama.290.2.238
253. Abraham E, Reinhart K, Svoboda P, Seibert A, Olthoff D, Dal Nogare A, et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: A multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med (2001) 29(11):2081–9. doi: 10.1097/00003246-200111000-00007
254. de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, et al. Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood (2000) 95(4):1124–9. doi: 10.1182/blood.V95.4.1124.002k20_1124_1129
255. Mayr FB, Firbas C, Leitner JM, Spiel AO, Reiter RA, Beyer D, et al. Effects of the pan-selectin antagonist bimosiamose (Tbc1269) in experimental human endotoxemia. Shock (2008) 29(4):475–82. doi: 10.1097/shk.0b013e318142c4e8
256. Derhaschnig U, Bergmair D, Marsik C, Schlifke I, Wijdenes J, Jilma B. Effect of interleukin-6 blockade on tissue factor-induced coagulation in human endotoxemia. Crit Care Med (2004) 32(5):1136–40. doi: 10.1097/01.ccm.0000126265.08175.be
257. Verbon A, Meijers JC, Spek CA, Hack CE, Pribble JP, Turner T, et al. Effects of Ic14, an anti-Cd14 antibody, on coagulation and fibrinolysis during low-grade endotoxemia in humans. J Infect Dis (2003) 187(1):55–61. doi: 10.1086/346043
258. Jilma B, Marsik C, Mayr F, Graninger MT, Taylor FB Jr., Ribel MC, et al. Pharmacodynamics of active site-inhibited factor viia in endotoxin-induced coagulation in humans. Clin Pharmacol Ther (2002) 72(4):403–10. doi: 10.1067/mcp.2002.127740
259. Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A, et al. Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood (1997) 89(8):2701–5. doi: 10.1182/blood.V89.8.2701
260. Mitaka C, Kawagoe I, Satoh D, Hayashida M. Effect of recombinant human soluble thrombomodulin on coagulation-related variables in patients with sepsis-induced disseminated intravascular coagulation: A retrospective observational study. Clin Appl Thromb Hemost (2021) 27. doi: 10.1177/10760296211050356
261. Levi M, Vincent JL, Tanaka K, Radford AH, Kayanoki T, Fineberg DA, et al. Effect of a recombinant human soluble thrombomodulin on baseline coagulation biomarker levels and mortality outcome in patients with sepsis-associated coagulopathy. Crit Care Med (2020) 48(8):1140–7. doi: 10.1097/ccm.0000000000004426
262. Ogura T, Eguchi T, Amano M, Sano T, Nishioka N, Miyano A, et al. Multicenter clinical experience with recombinant soluble thrombomodulin for disseminated intravascular coagulation associated with severe acute cholecystitis. Thromb Res (2019) 176:74–8. doi: 10.1016/j.thromres.2018.12.025
263. Yoshihiro S, Sakuraya M, Hayakawa M, Ono K, Hirata A, Takaba A, et al. Recombinant human-soluble thrombomodulin contributes to reduced mortality in sepsis patients with severe respiratory failure: A retrospective observational study using a multicenter dataset. Shock (2019) 51(2):174–9. doi: 10.1097/shk.0000000000001148
264. Hayakawa M, Yamakawa K, Saito S, Uchino S, Kudo D, Iizuka Y, et al. Recombinant human soluble thrombomodulin and mortality in sepsis-induced disseminated intravascular coagulation. A multicentre retrospective study. Thromb Haemost (2016) 115(6):1157–66. doi: 10.1160/th15-12-0987
265. Yasuda N, Goto K, Ohchi Y, Abe T, Koga H, Kitano T. The efficacy and safety of antithrombin and recombinant human thrombomodulin combination therapy in patients with severe sepsis and disseminated intravascular coagulation. J Crit Care (2016) 36:29–34. doi: 10.1016/j.jcrc.2016.06.008
266. Yamakawa K, Ogura H, Fujimi S, Morikawa M, Ogawa Y, Mohri T, et al. Recombinant human soluble thrombomodulin in sepsis-induced disseminated intravascular coagulation: A multicenter propensity score analysis. Intensive Care Med (2013) 39(4):644–52. doi: 10.1007/s00134-013-2822-2
267. Yamakawa K, Fujimi S, Mohri T, Matsuda H, Nakamori Y, Hirose T, et al. Treatment effects of recombinant human soluble thrombomodulin in patients with severe sepsis: A historical control study. Crit Care (2011) 15(3):R123. doi: 10.1186/cc10228
268. Rimmer E, Kumar A, Doucette S, Marshall J, Dial S, Gurka D, et al. Activated protein c and septic shock: A propensity-matched cohort study*. Crit Care Med (2012) 40(11):2974–81. doi: 10.1097/CCM.0b013e31825fd6d9
269. Dalton HJ, Carcillo JA, Woodward DB, Short MA, Williams MD. Biomarker response to drotrecogin Alfa (Activated) in children with severe sepsis: Results from the resolve clinical trial*. Pediatr Crit Care Med (2012) 13(6):639–45. doi: 10.1097/PCC.0b013e318250ad48
270. Decembrino L, D'Angelo A, Manzato F, Solinas A, Tumminelli F, De Silvestri A, et al. Protein c concentrate as adjuvant treatment in neonates with sepsis-induced coagulopathy: A pilot study. Shock (2010) 34(4):341–5. doi: 10.1097/SHK.0b013e3181e7623e
271. Crivellari M, Della Valle P, Landoni G, Pappalardo F, Gerli C, Bignami E, et al. Human protein c zymogen concentrate in patients with severe sepsis and multiple organ failure after adult cardiac surgery. Intensive Care Med (2009) 35(11):1959–63. doi: 10.1007/s00134-009-1584-3
272. Vincent JL, Angus DC, Artigas A, Kalil A, Basson BR, Jamal HH, et al. Effects of drotrecogin Alfa (Activated) on organ dysfunction in the prowess trial. Crit Care Med (2003) 31(3):834–40. doi: 10.1097/01.Ccm.0000051515.56179.E1
273. Yamakawa K, Umemura Y, Hayakawa M, Kudo D, Sanui M, Takahashi H, et al. Benefit profile of anticoagulant therapy in sepsis: A nationwide multicentre registry in Japan. Crit Care (2016) 20(1):229. doi: 10.1186/s13054-016-1415-1
274. Egi M, Ogura H, Yatabe T, Atagi K, Inoue S, Iba T, et al. The Japanese clinical practice guidelines for management of sepsis and septic shock 2020 (J-sscg 2020). J Intensive Care (2021) 9(1):53. doi: 10.1186/s40560-021-00555-7
275. Wiedermann CJ. Antithrombin concentrate use in disseminated intravascular coagulation of sepsis: Meta-analyses revisited. J Thromb Haemost (2018) 16(3):455–7. doi: 10.1111/jth.13950
276. Valeriani E, Squizzato A, Gallo A, Porreca E, Vincent JL, Iba T, et al. Efficacy and safety of recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy: A systematic review and meta-analysis. J Thromb Haemost (2020) 18(7):1618–25. doi: 10.1111/jth.14812
277. Yamakawa K, Murao S, Aihara M. Recombinant human soluble thrombomodulin in sepsis-induced coagulopathy: An updated systematic review and meta-analysis. Thromb Haemost (2019) 119(1):56–65. doi: 10.1055/s-0038-1676345
278. Martí-Carvajal AJ, Solà I, Lathyris D, Cardona AF. Human recombinant activated protein c for severe sepsis. Cochrane Database Syst Rev (2012) 3:Cd004388. doi: 10.1002/14651858.CD004388.pub5
279. Fan Y, Jiang M, Gong D, Zou C. Efficacy and safety of low-Molecular-Weight heparin in patients with sepsis: A meta-analysis of randomized controlled trials. Sci Rep (2016) 6:25984. doi: 10.1038/srep25984
280. Fu S, Yu S, Wang L, Ma X, Li X. Unfractionated heparin improves the clinical efficacy in adult sepsis patients: A systematic review and meta-analysis. BMC Anesthesiol (2022) 22(1):28. doi: 10.1186/s12871-021-01545-w
281. Li X, Liu Z, Luo M, Xi Y, Li C, Wang S, et al. Therapeutic effect of low-Molecular-Weight heparin on adult sepsis: A meta-analysis. Ann Palliat Med (2021) 10(3):3115–27. doi: 10.21037/apm-21-169
282. Eisen DP, Leder K, Woods RL, Lockery JE, McGuinness SL, Wolfe R, et al. Effect of aspirin on deaths associated with sepsis in healthy older people (Antisepsis): A randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir Med (2021) 9(2):186–95. doi: 10.1016/s2213-2600(20)30411-2
283. He W, Xi Q, Cui H, Zhang P, Huang R, Wang T, et al. Forsythiaside b ameliorates coagulopathies in a rat model of sepsis through inhibition of the formation of Pad4-dependent neutrophil extracellular traps. Front Pharmacol (2022) 13:1022985. doi: 10.3389/fphar.2022.1022985
284. Alsabani M, Abrams ST, Cheng Z, Morton B, Lane S, Alosaimi S, et al. Reduction of netosis by targeting Cxcr1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br J Anaesth (2022) 128(2):283–93. doi: 10.1016/j.bja.2021.10.039
285. Keshari RS, Silasi R, Popescu NI, Regmi G, Chaaban H, Lambris JD, et al. Cd14 inhibition improves survival and attenuates thrombo-inflammation and cardiopulmonary dysfunction in a baboon model of escherichia coli sepsis. J Thromb Haemost (2021) 19(2):429–43. doi: 10.1111/jth.15162
286. Zheng B, Yang H, Zhang J, Wang X, Sun H, Hu F, et al. Lidocaine alleviates sepsis-induced acute lung injury in mice by suppressing tissue factor and matrix metalloproteinase-2/9. Oxid Med Cell Longev (2021) 2021:3827501. doi: 10.1155/2021/3827501
287. Vital SA, Senchenkova EY, Ansari J, Gavins FNE. Targeting Anxa1/Formyl peptide receptor 2 pathway affords protection against pathological thrombo-inflammation. Cells (2020) 9(11). doi: 10.3390/cells9112473
288. Cornelius DC, Travis OK, Tramel RW, Borges-Rodriguez M, Baik CH, Greer M, et al. Nlrp3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PloS One (2020) 15(6):e0234039. doi: 10.1371/journal.pone.0234039
289. Yang X, Cheng X, Tang Y, Qiu X, Wang Z, Fu G, et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood (2020) 135(14):1087–100. doi: 10.1182/blood.2019002282
290. Zhou B, Liu J, Zeng L, Zhu S, Wang H, Billiar TR, et al. Extracellular Sqstm1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol (2020) 5(12):1576–87. doi: 10.1038/s41564-020-00795-7
291. Wang D, Yang Y, Wang Y, Proulle V, Andreasen PA, Hong W, et al. Embelin ameliorated sepsis-induced disseminated intravascular coagulation intensities by simultaneously suppressing inflammation and thrombosis. BioMed Pharmacother (2020) 130:110528. doi: 10.1016/j.biopha.2020.110528
292. Xia BT, Beckmann N, Winer LK, Kim Y, Goetzman HS, Veile RE, et al. Amitriptyline treatment mitigates sepsis-induced tumor necrosis factor expression and coagulopathy. Shock (2019) 51(3):356–63. doi: 10.1097/shk.0000000000001146
293. Steven S, Jurk K, Kopp M, Kröller-Schön S, Mikhed Y, Schwierczek K, et al. Glucagon-like peptide-1 receptor signalling reduces microvascular thrombosis, nitro-oxidative stress and platelet activation in endotoxaemic mice. Br J Pharmacol (2017) 174(12):1620–32. doi: 10.1111/bph.13549
294. Wake H, Mori S, Liu K, Morioka Y, Teshigawara K, Sakaguchi M, et al. Histidine-rich glycoprotein prevents septic lethality through regulation of immunothrombosis and inflammation. EBioMedicine (2016) 9:180–94. doi: 10.1016/j.ebiom.2016.06.003
295. Egge KH, Thorgersen EB, Pischke SE, Lindstad JK, Pharo A, Bongoni AK, et al. Organ inflammation in porcine escherichia coli sepsis is markedly attenuated by combined inhibition of C5 and Cd14. Immunobiology (2015) 220(8):999–1005. doi: 10.1016/j.imbio.2015.04.002
296. Mai SH, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with dnase reduces organ damage and improves outcome in a murine model of sepsis. Shock (2015) 44(2):166–72. doi: 10.1097/shk.0000000000000396
297. Barratt-Due A, Thorgersen EB, Egge K, Pischke S, Sokolov A, Hellerud BC, et al. Combined inhibition of complement C5 and Cd14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. J Immunol (2013) 191(2):819–27. doi: 10.4049/jimmunol.1201909
298. Thorgersen EB, Hellerud BC, Nielsen EW, Barratt-Due A, Fure H, Lindstad JK, et al. Cd14 inhibition efficiently attenuates early inflammatory and hemostatic responses in escherichia coli sepsis in pigs. FASEB J (2010) 24(3):712–22. doi: 10.1096/fj.09-140798
299. Tsai WH, Shih CH, Yu YB, Hsu HC. Plasma levels in sepsis patients of annexin A1, lipoxin A4, macrophage inflammatory protein-3a, and neutrophil gelatinase-associated lipocalin. J Chin Med Assoc (2013) 76(9):486–90. doi: 10.1016/j.jcma.2013.05.004
300. Cornelius DC, Baik CH, Travis OK, White DL, Young CM, Austin Pierce W, et al. Nlrp3 inflammasome activation in platelets in response to sepsis. Physiol Rep (2019) 7(9):e14073. doi: 10.14814/phy2.14073
301. Swanson KV, Deng M, Ting JP. The Nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
302. Sohrabipour S, Muniz VS, Sharma N, Dwivedi DJ, Liaw PC. Mechanistic studies of dnase I activity: Impact of heparin variants and Pad4. Shock (2021) 56(6):975–87. doi: 10.1097/shk.0000000000001804
303. Meng W, Paunel-Görgülü A, Flohé S, Hoffmann A, Witte I, MacKenzie C, et al. Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care (2012) 16(4):R137. doi: 10.1186/cc11442
304. National Library of Medicine (U.S.). Intravenous dnase I for the treatment of sepsis (Idealsepsisi) (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT?term=NCT&draw=2&rank=1.
305. Hack CE, Nuijens JH, Felt-Bersma RJ, Schreuder WO, Eerenberg-Belmer AJ, Paardekooper J, et al. Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am J Med (1989) 86(1):20–6. doi: 10.1016/0002-9343(89)90224-6
306. Robin M, Intrator L, Rapin M. Letter: Complement activation in septic shock. N Engl J Med (1975) 293(24):1261–2. doi: 10.1056/nejm197512112932416
307. Keragala CB, Draxler DF, McQuilten ZK, Medcalf RL. Haemostasis and innate immunity - a complementary relationship: A review of the intricate relationship between coagulation and complement pathways. Br J Haematol (2018) 180(6):782–98. doi: 10.1111/bjh.15062
Keywords: immunothrombosis, endothelial dysfunction, neutrophil extracellular traps, tissue factor, sepsis
Citation: Maneta E, Aivalioti E, Tual-Chalot S, Emini Veseli B, Gatsiou A, Stamatelopoulos K and Stellos K (2023) Endothelial dysfunction and immunothrombosis in sepsis. Front. Immunol. 14:1144229. doi: 10.3389/fimmu.2023.1144229
Received: 14 January 2023; Accepted: 13 March 2023;
Published: 04 April 2023.
Edited by:
Madhumita Chatterjee, Department of Pharmacology, Experimental Therapy and Toxicology, GermanyReviewed by:
Patrick Münzer, University of Tübingen, GermanyBirte Weber, University Hospital Frankfurt, Germany
Copyright © 2023 Maneta, Aivalioti, Tual-Chalot, Emini Veseli, Gatsiou, Stamatelopoulos and Stellos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleni Maneta, ZWxlbjM1MkBnbWFpbC5jb20=; Konstantinos Stellos, a29uc3RhbnRpbm9zLnN0ZWxsb3NAbmV3Y2FzdGxlLmFjLnVr; S29uc3RhbnRpbm9zLlN0ZWxsb3NAbWVkbWEudW5pLWhlaWRlbGJlcmcuZGU=
†These authors have contributed equally to this work