 Yaxin Li
Yaxin Li Haiyan Wang
Haiyan Wang Zhen Zhao
Zhen Zhao- Department of Blood Transfusion, The Affiliated Hospital of Qingdao University, Qingdao, China
It has been confirmed that platelets play a key role in tumorigenesis. Tumor-activated platelets can recruit blood cells and immune cells to migrate, establish an inflammatory tumor microenvironment at the sites of primary and metastatic tumors. On the other hand, they can also promote the differentiation of mesenchymal cells, which can accelerate the proliferation, genesis and migration of blood vessels. The role of platelets in tumors has been well studied. However, a growing number of studies suggest that interactions between platelets and immune cells (e.g., dendritic cells, natural killer cells, monocytes, and red blood cells) also play an important role in tumorigenesis and tumor development. In this review, we summarize the major cells that are closely associated with platelets and discuss the essential role of the interaction between platelets with these cells in tumorigenesis and tumor development.
1 Introduction
Tumorigenesis and tumor development are not only manifested in the malignant proliferation of tumor cells, but also include their immune escape and metastasis. The tumor microenvironment has an extremely important influence on it. The mechanism of platelet action in the tumor microenvironment has been well explored, but the exact underlying mechanisms and implications of platelet interactions with other cells in tumor development and progression remain unknown. We endeavored to summarize the current literature in order to dissect the specific role of platelets in tumors.
Platelets are the main players in primary and secondary hemostasis. It has been discovered that platelets also play an crucial role in tumorigenesis and tumor development. On the one hand, tumor cells induce platelet aggregation, which promotes the release of angiogenic factors and therefore further facilitating cancer progression (1). In contrast, platelets that have been “educated” by cancer cells can promote cancer development by interacting with other cells. Specifically, the platelets that activated by tumor cells can recruit immune cells and blood cells to migrate to the tumor site, modulate the inflammatory environment at the primary and metastatic tumor sites, and promote proliferation and metastasis of tumor cells (2). Platelets also regulate the effector functions of immune cells, promote antigen presentation in dendritic cells, recruit and differentiate monocytes, promote the formation of neutrophil extracellular traps, activates immune responses, and reduce the barrier effect of natural killer (NK) cells (3–6). Moreover, tumor-activated platelets stimulate the division of vascular endothelial cells; enhance the permeability of vascular endothelial cells and promote angiogenesis and migration of tumor cells (7). Finally, platelets can also promote thrombosis by interacting with red blood cells and facilitating the initiation of the clotting cascade, thereby increasing the risk of death in cancer patients (8).
2 Platelet-tumor cell interactions in tumor development
Platelets are small fragments of anucleated cells which are released from megakaryocytes (MKs). Beyond their traditional roles in thrombosis and hemostasis, platelets are also identified as key mediators of malignancy. Cancer patients are usually accompanied by excessive thrombocytosis and platelet activation (9–11).Tumor cells activate platelets through different stimuli, while platelets promote tumor cell proliferation, intracellular signaling and EMT through various mediators. Platelet-tumor cell interactions play an important role in tumor growth, thrombosis, angiogenesis, dissemination, metastasis and immune escape (12, 13). Platelets have been shown to exert their impact in multiple ways, and tumors, in turn, can also influence the platelet number, behavior, and even phenotype by a process that is called platelet “education” (14). Platelets have the following main roles in tumor microenvironment:(1) Contributes to the growth of tumors. Platelets have a large number of growth factors in their alpha granules (15–18). They can be present in the tumor microenvironment and contact with tumor cells directly (19, 20). When activated, platelets secrete vascular endothelial growth factor (VEGF) (21, 22), transforming growth factor β (TGF-β)and platelet-derived growth factor (PDGF) (23) promoting induction of tumor growth, angiogenesis and tumor neovascularization (24–26). (2) facilitation of the tumor thrombosis. Armand Trousseau first described in 1865 that tumor cells could induce thrombosis formation (named Trousseau’s syndrome) (27). Tumor cells can promote platelet activation by stimulating platelet aggregation, leads to a phenomenon that is known as tumor cell-induced platelet aggregation (TCIPA) (28). TCIPA is not only associated with a very high risk of thrombosis, but is also thought to promote angiogenesis and metastasis and is thus considered to be inversely associated with prognosis and survival (29, 30). During TCIPA, platelets express many selectins and integrins that attach to Circulating tumor cells (CTCs) to protect tumor cells to achieve immune escape (31–33). (3)Induction of tumor angiogenesis. Platelets have the ability to induce tumor angiogenesis. Platelets can deliver a variety of pro-angiogenic factors to tumors as well as stimulate tumor cells to express pro-angiogenic factors, including PDGF, VEGF, MMP and FGF (34). Platelets are actively recruited by cancer cells via P-selectin (35). When platelets arrive at the tumor microenvironment they will adhere to the cancer cell membrane and stimulate VEGF secretion, promote cancer cell proliferation and angiogenesis (36). Not only that, animal models confirm that tumor-educated platelets stimulate angiogenesis more efficient than normal platelets, by delivering pro-angiogenic factors more efficiently (37). (4)Promotes tumor cell invasion and metastasis and escape immune surveillance. An important strategy for cancer cell survival in the circulation is interaction with platelets, and almost all cancer metastatic processes seem to be mediated through tumor cell-platelet interactions (38). In tumor microenvironment, expression of PD-1-PD-L1 ligand by tumor cells and other stromal cells inhibit T cells and helps tumor cells evade immune surveillance. Tumor cells with high PD-L1 expression are able to fight cancer immunosurveillance (39, 40). PD-L1 expression in tumors is associated with poorer prognosis in various cancers (41). It has been suggested that PD-L1 is reported to be highly expressed on platelets from cancer patients and is lacking on normal platelets (42–44), which is thought to be the result of PD-L1 transfered from cancer cells to platelets (39). However, more studies have demonstrated that normal platelets also express PD-L1. Zaslavsky et al. demonstrated the presence of PD-L1 in platelets from both healthy individuals and patients with advanced cancer using Western blot and confirmed the contribution of PD-L1-expressing platelets to overall PD-L1 expression in tumors (45, 46). They demonstrated that platelet attach to PD-L1-negative tumor cells resulted in PD-L1 protein expression and platelets can modulate tumor cells to evade immune surveillance by expressing PD-L1 (47). They also observed that in vitro, anti-platelet drugs such as aspirin can neutralize the inhibitory effect of platelets on T cell-mediated cytotoxicity (45). Rolfes et al. also detected PD-L1 on platelets isolated from patients with head and neck squamous cell carcinoma (HNSCC) and confirmed that platelets from healthy donors also express small amount PD-L1. Not only that, they observed significantly higher PD-L1 expression in platelets of cancer-free individuals who smoked regularly (42) and suggested that PD-L1 may predict early lung cancer (48). These studies have confirmed that platelets express PD-L1 and can promote PD-L1 expression in tumors. Platelets increase the expression of PD-L1 in cancer cells mainly through a number of signaling pathways: Platelets and platelet secretion products can activate NF-κB, TGF-β/Smad and JAK/STAT pathways in tumor cells (49), which also increase the expression of PD-L1 in cancer cells exposured to platelets directly through Smad2/3 and NF-κB signaling pathways, and increases PD-L1 mostly through the TGFβR1/Smad signaling pathway exposured to platelets indirectly (45). Platelets also stimulate the release of metalloproteinases from tumor cells, and in turn contributes to tumor cell invasion by accelerating extracellular matrix degradation (50, 51). In addition, activated platelets secrete lysophosphatidic acid, which upregulates matrix metalloproteinase 2 (MMP2), MMP7, and MMP9 activities in cancer cells, then promote cancer progression by facilitating the detachment of cancer cells from their primary site and into the circulation (52). When tumor cells leave tumor microenvironment and reach the circulation, they are subject to high shear stress of the immune system (53). At this phase, tumor cells express megakaryocyte genes and platelet surface markers to mimic platelets (54, 55), and platelets can transfer major histocompatibility complex (MHC) class I to the tumor cells, giving tumor cells a false “pseudo-normal” appearance and thus evading natural killer cell (56). The capacity of platelets to prevent circulating tumor cells from attacking by the immune system may contribute significantly to the metastatic process (57, 58).
3 Platelet- neutrophil interactions in tumors
Tumor development is a complicated process that depends on several pivotal hallmarks, such as the induction of angiogenesis, promotionof inflammation, evasion of immune destruction, and activation of invasion and metastasis. Herein, platelet-leukocyte interactions play a crucial role (59).Platelets play a critical role in promoting thrombosis and inflammation progression, linking hemostasis and immune responses (60, 61). They can also be viewed as an extension of the cellular immune system (3, 62–65)as they play a crucial role in modulating leukocyte function and the release of inflammatory signals.
3.1 Platelet- neutrophil interactions promote inflammation in the tumor microenvironment
Tumors are not simply clusters of malignant cells, but complex structures composed of malignant,non-transformed cells, and the tumor microenvironment (66–71). It was found that the TEM contains a large number of different types of cells, including fibroblasts, vascular endothelial cells, adipocytes, stromal cells and immune cells such as B lymphocytes, T lymphocytes, tumor-associated macrophages and NK cells. Most cells in the TEM can produce cytokines and play an important role in promoting or suppressing tumors (72). The tumor microenvironment contains a large number of inflammatory cells and pro-inflammatory mediators (such as cytokines, chemokines and prostaglandins) that are capable of participating in a variety of inflammatory responses and acting on malignant cells in an autocrine and/or paracrine manner (73). Platelet- neutrophil interactions act as pro-inflammatory agents in the tumor microenvironment through various mechanisms (Figure 1). First, platelet- neutrophil interactions are mediated by P-selectin (or CD62P)-P-selectin glycoprotein ligand 1 (PSGL-1) and further stabilized by platelet glycoprotein (GP) Ib, glycoprotein (GP)IIb/IIIa, intercellular adhesion molecule-2 (ICAM-2), and the CD40 ligand (CD40L). Platelets also interact with leukocytes to form platelet- neutrophil aggregates that circulate in the blood, promoting leukocyte recruitment, activation, extravasation, phenotypic switching, and other functional changes (2, 3). Furthermore, platelets are activated upon injury and directly contact pathogens via their Toll-like receptors (TLRs) (2). TLR binding triggers the expression of several adapter proteins and downstream kinases induces the expression of essential pro-inflammatory mediators, activates the innate immune response, and promotes the maturation of adaptive immune components (74). Moreover, activated platelets release granule contents that regulate the tumor microenvironment, such as pro-inflammatory cytokines (30). These platelet-released pro-inflammatory cytokines are powerful activators and recruiters of leukocytes which in turn contribute to the immunomodulatory effects of platelets (75, 76). Examples of these chemokines include chemokine ligand 1 (CXCL1),CXCL4,CXCL5,CXCL7, CXCL12,interleukin-8 (IL-8),and transforming growth factorβ (TGF-β) (30, 77).
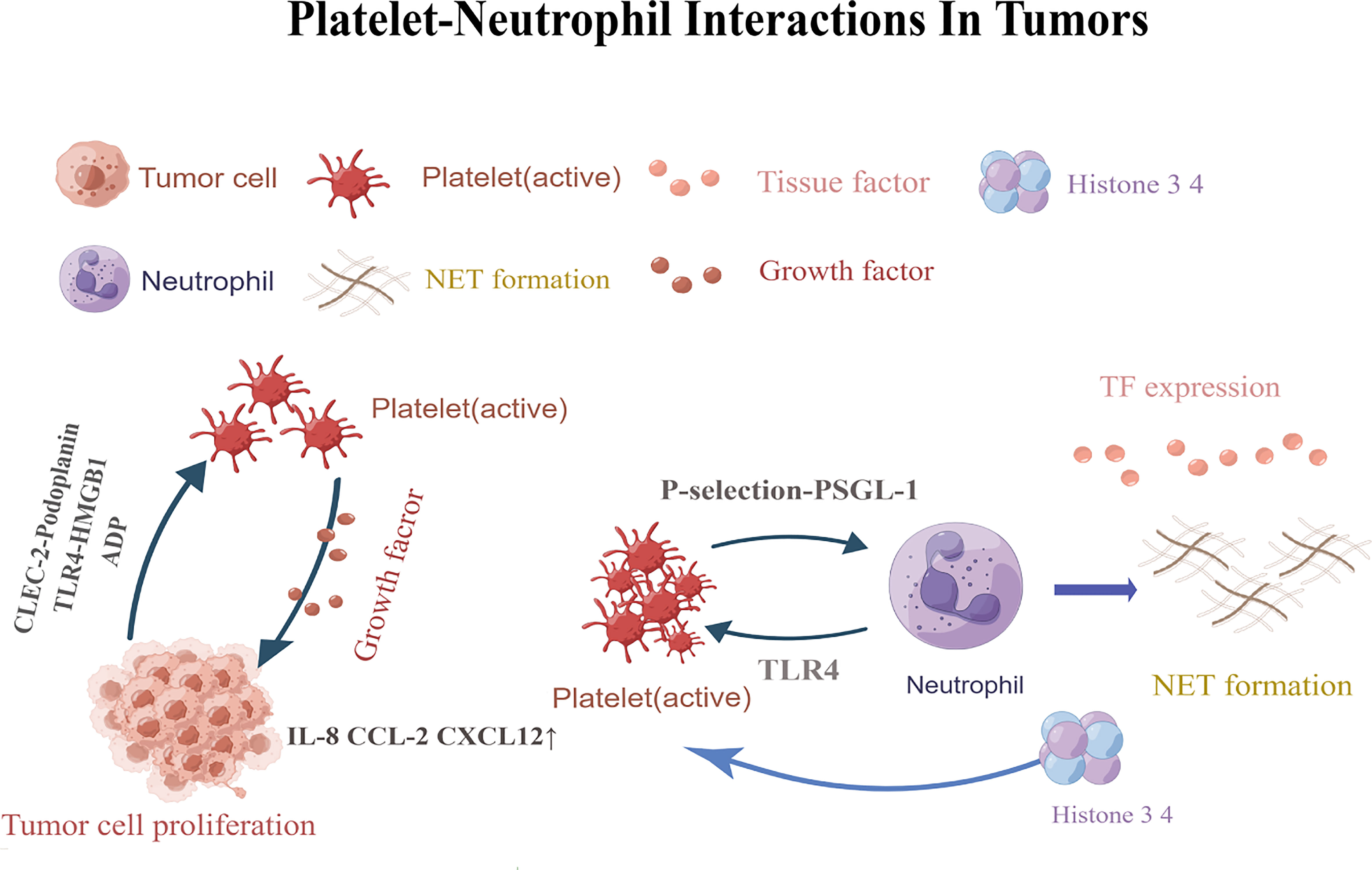
Figure 1 Platelets are activated by CLEC-2-podoplanin and TLR4-HMGB1.Activated platelets release factors that promote adhesion and angiogenesis, and increaseIL-8, CXCL12,and CCL2 levels. Eventually leading to tumor cell angiogenesis and tumor proliferation. P-selectin-PSGL-1 promotes the formation of neutrophil traps. TLR4-induced platelet activation can induce platelets to bind to neutrophils and form robust NETs, releasing histones 3 and 4, which further activate circulating platelets and promote the formation of more NETs.CLEC-2,C-type lectin-like immune receptor 2; TLR4,Toll-like receptor; HMGB1,high-mobility group box 1; PSGL-1,P-selectin glycoprotein ligand 1; NETs, Neutrophil extracellular traps. This figure is drawn by Figdraw (https://figdraw.com/). The "↑" arrows refer to the increased expression of IL-8, CXCL12, and CCL2.
3.2 Platelet- neutrophil cell interactions promote tumor thrombosis and angiogenesis
The strong association between thrombosis and cancer has been known since 1865 when Armand Trousseau first confirmed that localized cancers could lead to venous thrombosis at distant sites (27, 78). The risk of venous thromboembolism in patients with cancer is four- to seven-fold more than in healthy individuals. Cancer-related thrombosis is a cause of high cancer morbidity and mortality (79). The main reason of thrombosis in cancer patients is t platelets activated by cancer cells (78, 80). The interaction between platelets and leukocytes, such as the activation of tissue factor (TF) and G proteins and the promotion of neutrophil trap formation and platelet activation, plays a crucial role in promoting tumor thrombosis. The hypercoagulable state is caused by multiple factors, including changes in the expression of circulating pro-coagulation proteins and systemic inflammation, including TF, fibrinogen, factor V (FV), FVI, FVIII, and FX (81, 82). First, TF is normally expressed in an inactive form on the cell membrane of tumor cells, and platelets play a key role in activating TF. Direct interaction with TF-expressing leukocytes and platelets can strengthen TF activity and promote coagulation (83). Additionally, tumor cells show increased vascular permeability and upregulated TF expression (84). Once it comes in contact with activator factor VIIa in the bloodstream, TF is arguably the primary activator of the coagulation cascade (15, 85, 86). It is commonly expressed in cancer cells and cancer-derived particles (15, 16, 85–87). The origin and significance of blood-derived tissue factor (TF) is widely debated. Monocytes have long been considered as the main source of blood-derived TF (88–91). Some studies have identified TF proteins in platelets by flow cytometry, ELISA, Western blotting, immunocytochemistry and immunoelectron microscopy (92–94). TF in platelets is present in α-granules and also in the open tubule system (95, 96) and is hopefully output to the plasma membrane upon activation (95). Reports strongly suggest that the initiation of platelet PCA is dependent on platelet activation-induced ab initio synthesis of TF. TF expressed by platelets is probably through three proven mechanisms, namely: (1) FusionTF-containing microparticles (MPs) with activated platelet membranes; (2) stored in platelet α-granules and extravasates to the cell surface after platelet activation;(3) Activated platelets synthesize surface TF from de novo after splicing the pre-TF mRNA (97). Human platelets, a new type of TF-synthesizing cell, will act as “TF-bearing cells”, ensuring that TF is localized at the right time and place, thus promoting coagulation with greater rational precision (98).
In addition to the TF, neutrophil extracellular traps (NETs) have also been shown to have a strong relationship with thrombosis. The association between thrombosis and neutrophil extracellular traps(NETs) was first demonstrated several years ago by Fuchs et al (82). Platelets play a huge role in the formation of NETs. Firstly, the pro-thrombotic effect of NETs can be explained by their high histone and nucleic acid content, rendering them highly pro-coagulant with the capacity to activate and aggregate platelets (82). Interaction between platelet P-selectin and neutrophil P-selectin glycoprotein ligand 1 (PSGL-1) promotes neutrophil activation and migration, leading to IL-8 release and the formation of neutrophil extracellular traps (NETs) (5, 99). NETs, in turn, recruit more platelets by presenting histones at platelet TLR4,leading to its activation, andre-initiation of a pro-coagulant environment that promotes thrombosis (81, 100). Besides, one key damage-associated molecular pattern (DAMP) expressed and released by platelets is High Mobility Group Box 1 (HMGB1). HMGB1 is a highly conserved, non-histone chromosomal protein that has pro-inflammatory and pro-coagulant functions and mediates NET formation in venous thrombosis (101–103) and tumor (104). HMGB1 induces NETosis through C-X-C motif chemokine receptor 4 (CXCR4) and Toll-like receptor 4 (TLR4),and can be inhibited by neutralizing HMGB1 antibodies (105, 106). HMGB1 induces NETosis and activates platelets; it is also released from activated platelets and extruded from NETosed neutrophils. Extruded HMGB1 in turn, induces platelet activation, thrombosis and further aggravates NETosis (107). Therefore, HMGB1 acts as the center of the crosstalk between thrombosis and NETosis and mediates the exacerbation cascade caused by this crosstalk. Moreover,HMGB1 signals is thought to exert tumor-promoting effects in malignant tumors via the tumor HMGB1/RAGE axis through agonist receptors, such as receptors for advanced glycosylation end products (RAGE), and other pattern recognition receptors, like TLR2, TLR4, and TLR9 (108, 109). It has been demonstrated that tumor growth is accompanied by elevated HMGB which promotes cancer cell survival and protects cells from the cytotoxic effects of adriamycin in homozygous mice (110). Finally, neutrophil- derived extracellular DNA provides scaffolding for platelet activation (82) and is indispensable for the spread of deep-vein thrombosis due to its ability to bind and activate FXII (111). In addition, tumor cells and its derived micro-particles are able to trigger NET formation directly or indirectly by activating platelets, which leads to a further activation of platelet and release of TF (112). Recent clinical evidence suggests a definite link between NET formation and thrombosis in in tumor patients. These data indicate that NETs promote the development of venous thromboembolism (VTE) in these patients with tumor (113).
Platelet-neutrophil interactions promote not only tumor thrombosis, but also tumor angiogenesis. Platelets are locally activated through the interaction of podoplanin with C-type lectin-like immune receptor 2 (CLEC-2),high-mobility group box 1 (HMGB1) with TLR4 (13, 114). Activated platelets release factors that promote cell adhesion and angiogenesis, leading to tumor proliferation. They also release growth factors and upregulate cytokine release from leukocytes, endothelial cells, and tumor cells, which increases the local tissue IL-8,CXCL12andCCL2levels, promoting cell proliferation and tumor angiogenesis (86).
3.3 Platelet- neutrophil interactions promote tumor metastasis
In addition to their role in thrombosis, NETs are also involved in tumor metastasis. Research has shown that the deposition of neutrophil traps in vivo increased the adhesion of tumor cells to the liver and lung microvasculature, leading to tumor cell migration and invasion (87). In contrast, it was found that NETs formed in the absence of systemic inflammation increased tumor adhesion in vivo (82, 100). Finally, it has also been documented that NETs are responsible for awakening dormant cancers, promoting a pro-cancer microenvironment that leads to tumor metastasis (115). Not only that, the interaction between platelets and neutrophils enhances their cytotoxicity by increasing reactive oxygen species production (116). At later stages of tumor development, neutrophils promote tumor cell metastasis by inhibiting NK cells (78) and cytotoxic T-cells (83).
4 Platelet-erythrocyte interactions in tumors
Erythrocytes, or red blood cells (RBCs), have a major influence on blood coagulation, hemostasis, and thrombosis and have been a major focus of hematology, hemostasis, and thrombosis research (117). The role of erythrocyte-platelet interactions in tumors has also received increasing attention. Anemia is an independent poor prognostic factor for patients with various malignancies, adversely affecting their survival and quality of life (118). Studies have shown that tumor embolization, vascular endothelial damage, and the activation of coagulation factors and platelets cause fibrin deposition in the microvasculature. These ultimately lead to the fragmentation of RBCs and the development of tumor-associated microvascular pathologic hemolytic anemia. The lack of responsiveness of bone marrow erythrocytes to erythropoietin (EPO) in the presence of cytokines, such as tissue necrosis factor α (TNF-α) and interferon γ (IFN-γ),also leads to the development of tumor-associated anemia (119). Anemia reduces the number of RBCs in the central column of the blood vessel, resulting in fewer platelets coming into contact with the endothelial surface surrounding the flowing blood column. As a result, impaired primary hemostasis and bleeding could occur (119). Furthermore, tumor-associated anemia can reduce the oxygen-carrying capacity of the blood, contributing to tumor hypoxia (120). Persistent tumor hypoxia can enhance malignant progression and increase tumor cell aggressiveness through clonal selection and genomic changes (121, 122).
As previously mentioned, cancer patients are more likely to have thromboembolism than healthy people. Thromboembolism is reported to be the leading cause of death in cancer patients who are receiving chemotherapy (123). In addition, the activation and hypercoagulability of platelets induced by cancer cells positively affect the development and spread of tumors. Patients with abnormally high hematocrit levels are at an increased risk for thrombotic disorders (124). There are several mechanisms involving erythrocyte-related platelet activation. Firstly, RBCs enhance platelet activation by releasing adenosine diphosphate (ADP). RBCs can directly modulate platelet reactivity through chemical signaling (125). RBCs can release ADP into the bloodstream under low pH, low oxygen pressure, hemolysis, RBC deformation, permanent RBC damage, shear stress, and as a response to mechanical deformation, causing platelet activation. Furthermore, activated platelets release granules containing ADP, which further promotes platelet activation (126–129). Ex vivo studies involving blood transfusion have also demonstrated that the release of ADP from RBCs promotes platelet activation and aggregation (130). Secondly, RBCs enhances platelet activation by reducing the bioavailability of Nitric Oxide (NO).Hemoglobin released from damaged RBCs enhances platelet activation and accelerates thrombus formation by decreasing NO bioavailability, preventing the suppressive effect of NO on platelet activation (131, 132). Damaged RBCs can also release arginase, which cleaves l-arginine and reduces NO release (131). Although the hemoglobin released during erythrocyte injury has been recognized as a transporter and a scavenger of NO, it has recently been shown that hemoglobin can also preserve the function of NO through the formation of S-nitrosothiols (133). S-nitrosothiols have anti-platelet activity similar to NO, so it can be concluded that injured erythrocytes may enhance platelet aggregation by scavenging NO and inhibit platelet activation by releasing functional NO equivalents (134). In addition to elucidating the role of hemoglobin(Hb) reactions with NO in platelet activation, Hb has also been shown to directly activate platelets (135), demonstrating that its platelet aggregation-promoting effect is due to the activation of platelets produced by ROSfrom the iron in Hb (136). And thirdly, RBCs also promote the marginalization of platelets. Under normal conditions, RBCs preferentially move down the center of blood vessels, promoting platelet margination, increasing platelet-endothelium interactions, and enhancing platelet adhesion and activation (137).In the disease state, abnormal erythrocytes and erythrocyte-derived microvesicles may also adhere to the extracellular matrix or endothelium, activate platelets and other cells, and enhance local thrombin production during thrombosis (117, 138).Furthermore, they can also interact with platelets, leading to the formation of erythrocyte-platelet aggregates (117, 139). RBCs also bind to platelets through ICAM-4 and fibrinogen to form erythrocyte-platelet aggregates that promote tumor thrombosis and stabilization (8, 140, 141). On the one hand, this interaction can further activate platelets and promote thrombosis. On the other hand, it can lead to the release of biochemical messengers and signal transduction via erythrocyte deformation, further activating platelets to promote their high expression of P-selectin,and enhancing platelet adhesion to tumor cells (142). In conclusion, platelet function can be amplified by the interaction of activated platelets with intacted red blood cells (142).
5 Platelet-monocyte interactions in tumors
Monocytes, the innate immune cells of the mononuclear phagocyte system, have emerged as important regulators of tumor development and progressions (143–145). When inflammation or tumors occur in vivo, monocytes and neutrophils are first attracted from the peripheral blood stream to the site of injury (146). Monocytes can then be recruited to pre-metastatic sites, interact with tumor cells, inhibit their adhesion, and promote natural killer (NK) cells to eliminate tumor cells by secreting chemokines including CCL3, CCL4, and CCL5 (147, 148).
5.1 Platelet-monocyte interactions and inflammation
Platelet-monocyte interactions have a tremendous impact on the inflammatory responses because such interactions are not only pro-inflammatory but also anti-inflammatory. In humans, three distinct monocyte populations were first classified through their morphology and differential expression of cluster of differentiation (CD) 14 and 16on their membrane surface (149). Classical monocytes express high levels of CD14 and no CD16 (CD14++CD16−), intermediate monocytes express high levels of both CD14 and CD16(CD14++CD16++),while non-classical monocytes express low levels of CD14 but high levels of CD16+ (CD14+CD16++) (149, 150). CD16+ monocytes are traditionally referred to as “pro-inflammatory” monocytes (151–153). Platelets are considered to be an immune regulator (3, 154) that can regulate immune system function by interacting with immune cells (2, 3, 60, 155, 156) or by releasing soluble mediators IL-1β, (TGF-β, PF4, MIP1α or RANTES). Normally, PLT preferentially binds monocytes (157–160), mainly by P-selectin (CD62P)-PSGL-1 (161). Platelets preferentially bind to intermediate and non-classical monocyte subtypes that express CD16. Furthermore, activated platelets are able to release large amounts of TGF-β, leading to upregulation of CD16 on monocytes and consequently inducing a transition from classical monocytes to intermediate and/or non-classical monocytes (162, 163).
Several factors, such as infection or injury, can contribute to the activation of both monocytes and platelets. Once activated, monocytes and platelets can interact directly through receptors expressed on their surfaces, then the soluble factors released from platelets can further modulate monocyte activity. This interaction can promote the recruitment and adhesion of monocytes or lead to platelet-monocyte complex formation (164). It has been proven that thrombin- activated platelets bind to circulating monocytes, thereby inducing the production of pro-inflammatory cytokines such as IL-8, TNFα, and IL-6,further promoting a pro-inflammatory phenotype (2, 165). In vitro, platelet release of TGF-β activates the p38 MAPK pathway, which induces de novo synthesis of cyclooxygenase-2 to promote inflammatory monocyte responses (166). Additionally, platelet-derivedHMGB1promotes the migration of monocytes to inflammatory tissues (101, 167). In addition, activated platelets release ADP from granules that bind to ADP receptors(or P2Y12receptors), prompting the rapid transfer of P-selectin to the platelet membrane and increasing the recruitment of neutrophils, monocytes, and lymphocytes to the tumor site (168). Gil-Bernabé found that platelet-tumor cell conjugates were also able to recruit monocytes to early metastatic niches by chemokines produced from platelets (e.g. RANTES) (169, 170). After this, recruited monocytes produce VEGF to improve the extravasation of tumor cells, thereby increasing vascular permeability (171) and promoting the formation of metastases (170, 172). Moreover,platelet factor 4 (PF4) drives monocyte migration by binding to the CCR1 receptor (173). Platelet-monocyte interactions also play a vital role in locally restricting inflammation. PF4 not only acts as an inflammatory mediator but also downregulates the chemotactic receptors CCR1, CCR5, and CCR2 on isolated human monocytes to interfere with monocyte migration (174). Contrary to their pro-inflammatory effect, platelets have also been shown to dampen pro-inflammatory responses by triggering IL-10 expression and downregulating TNF-α and IL-6 release by monocytes (2)and macrophages (175). These opposing effects of platelets on innate immune cells may be determined via the activation status of platelets (176, 177) or by the blood flow microenvironment (178). The underlying mechanisms for this need to be explored further in future studies.
5.2 Platelet-monocyte aggregation in tumors
Monocyte-platelet aggregates (MPA), are hallmarks of platelet and monocyte activation and play a potential important role in promoting inflammatory responses and thrombosis. It serves as a bridge between inflammation and thrombosis (151, 179, 180). PSGL-1-P-selectin interactions appear to be the initiating signal in monocyte-platelet aggregation and are further consolidated by enhancing its interactions with other receptorsand other downstream signals (2, 116, 181, 182). MPA recruits monocytes to tumor sites, regulates the inflammatory environment at primary and metastatic tumor sites, and influences tumorigenesis and cancer progression (4, 183, 184). In addition, MPA formation upregulates CD16 expression on monocytes, resulting in an enhanced pro-inflammatory effect (165). In contrast, additional data demonstrate that MPA enhances the expression of the anti-inflammatory cytokine IL-1,inhibiting the progression of inflammation (147, 178). However, future studies need to explore further the exact mechanisms underlying this opposing effect.Monocytes are the primary source of tissue factors in the blood and are a key element of the extrinsic coagulation cascade reaction. Monocytes actively bind to platelets, forming MPAs that are highly susceptible to thrombosis (185). Activated platelets rapidly trigger the surface exposure of TF in monocytes through the interaction of P-selectin with monocyte PSGL1,rapidly contributing to intravascular coagulation and thrombosis, and promoting tumor development and metastasis (186). Monocytes account for 16% of platelet thrombus-bound leukocytes, almost four times the proportion of monocytes in the circulating blood; this demonstrates the importance of MPAs in thrombosis (185). Platelets also interact directly with monocytes, facilitating increased expression of PSGL-1, CD40,CD11b, and CCR2 on the surface of monocytes (4, 163), which in turn enhances the formation of MPAs and promotes the recruitment of more monocytes to the endothelium (4, 181, 182, 187). In summary, MPA is involved in the inflammatory response, maintains blood in a hypercoagulable state, and play an vital role in tumorigenesis, cancer progression, and metastasis. Platelets and their interactions with monocytes also affect various stages of tumor development. However, the exact mechanisms and effects of MPA on tumorigenesis and cancer progression remain unclear and need to be explored further.
6 Platelet-macrophage interactions in tumors
Macrophages are immune cells that play a key role in innate immunity and are also involved in tissue repair and remodeling (188). Depending on the prevalent stimuli in inflammatory microenvironments, human macrophages can be differentiated from circulating monocytes in vitro and polarized towards a pro- or anti-inflammatory phenotype (typically recognized as classically activated type 1 [M1] and alternatively activated type 2 [M2] macrophages, respectively).
6.1 Types of macrophages and their role in tumors
M1 macrophages are induced by IFN-γ, bacteria-derived lipopolysaccharides (LPS),and TNF-αand are known to release cytokines that can promote inflammation and tissue damage (189–191). M1 macrophages also produce high levels of IL-12, IL-1, IL-23, TNF-α, CXCL10, and nitric oxide synthase (iNOS), which play important roles in the inhibition and killing of tumor cells. M1 macrophages also present tumor-specific antigens, indirectly inhibiting tumor growth (192).
In contrast, M2 macrophages are induced by IL-13 and IL-4and are well-known to release anti-inflammatory cytokines that promote tissue remodeling and inhibit inflammation (193, 194). M2 macrophages release epidermal growth factor, chemokines (such as CCL24and CCL22), and cytokines (such as TNF-α and IL-6) that directly promote tumor growth and metastasis and promote tumor progression. Meanwhile, several potent immunosuppressive factors produced by M2 macrophages (such as IL-10 and prostaglandin E2 [PGE2]) suppress anti-tumor immune responses and promote angiogenesis and stromal remodeling in the tumor microenvironment, ultimately promoting tumor development and metastasis (195). Tumor-associated macrophages mostly exhibit the M2 phenotype, and their significant contribution to tumor progression and metastasis has been noted (196). Platelets also play a key role in promoting macrophage polarization and consequently play a significant role in tumor progression.
6.2 Activated platelets induce monocyte differentiation into anti-inflammatory M2 macrophages
It has been found that monocyte incubation with activated platelets results in a biased differentiation into M2-type macrophages (197). Activated platelets produce TGF-β, which can convert M1 macrophages into M2 macrophages. Activated M2 macrophages produce small amounts of IL-12 and large amounts of IL-10, which suppress the inflammatory state of the body, attenuates the antineoplastic immune response, promote tumor angiogenesis, and accelerate tumorigenesis, development, and metastasis (198–205). ActivatedM2 macrophages also affect the T-cell inactivation, which severely reduces the body’s ability to resist cancer development and progression (205). In addition, platelet activation synthesizes lipid mediators such as thromboxane A2 (TXA2) (65, 206, 207) or PGE (208–210). PGE2directly induces IL-10 release from macrophages, decreases IL-6 and TNF-α release, controls tumor-associated inflammation, and decreases the body’s anti-tumor immune response (147, 176). In addition, activated platelets release PF4, which increases the levels of macrophage nuclear factor-kappa B (NF-κB), promotes MMP-9 gene transcription, and facilitates tumor vascular remodeling (210, 211). The release of vascular endothelial growth factor (VEGF) during PLT activation can also promote the recruitment of circulating macrophages to tumor sites (212–214).
6.3 Activated platelets also induce monocyte differentiation into pro-inflammatory M1 macrophages
Controversially, some studies have illustrated that platelets are able to modulate not only the pro-inflammatory but also the anti-inflammatory response of macrophages. Angiogenic factors and chemokines (e.g., CXCL4) may induce or modulate the pro-inflammatory response of macrophages during platelet activation (215). Increased levels of the pro-inflammatory cytokines IL-6,TNF-α, and IL-23, were observed after co-incubation of LPS- activated human monocyte-derived macrophages with activated autologous platelets, suggesting that the presence of activated platelets at the sites of inflammation may intensify pro-inflammatory macrophage activation (164). Other studies have shown that the presence of platelets skewed monocytes towards the M1phenotype in a cell-contact-dependent manner via the GPIb-CD11b axis in mice (196). However, given the differences between mouse and human macrophages, the biological relevance of platelet-macrophage interactions in human innate immunity remains uncertain.
7 Platelet- dendritic cell interactions in tumors
Dendritic cells (DCs), also known as “professional” antigen-presenting cells (APCs), can process and cross-present antigens to induce effective antigen-specific T-cell responses and are principal modulators of adaptive immunity (216). The role of dendritic cells in tumors has been well-investigated, and the effects of the interactions between platelets and dendritic cells on tumors have attracted significant attention.
7.1 Platelets promote DC maturation and activation
Platelets not only enhance the activity of macrophages and neutrophils but also affect the maturation and activation of DCs (217). Activated platelet-derived soluble CD40L (sCD40L or CD154) stimulates DC maturation and enhances antigen presentation capability. Platelets contribute to adaptive immunity by interacting with and activating DCs via the CD40-CD40L axis. The platelet-mediated activation of DCs results in antigen presentation to T-cells (3, 218, 219). Platelet-DC interactions also promote the over expression of co-stimulatory molecules on DCs and increase the production of cytokines such as TNF-α, IL-12, and IL-6.These cytokines promote the maturation of DCs to target T lymphocyte antigens that help activate type I helper T-cells (TH1 cells) and CD8+ T-cells to kill tumor cells (220). In addition, platelet-DC interactions may be regulated through the P-selectin/PSGL1 axis, and then platelets firmly adhere to CD11b/CD18 (MAC-1) on DCs via their junctional adhesion molecule C (JAM-C), resulting in DC activation and platelet phagocytosis (220).
7.2 Platelets inhibit DC maturation and activation
In contrast, some platelet-derived factors have the opposite effect, such as VEGF, serotonin/5-HT, PF4, TGF-β, and PGE2. Platelet-derived VEGF can inhibit antigen presentation in mature dendritic cells, thereby suppressing their immune surveillance function (221). In addition, 5-HT decreases the expression of DC co-stimulatory molecules and increases IL-10 levels, thereby reducing the ability of DCs to stimulate T-cells, suppress cellular immunity, and promote tumor cell evasion of immune cell killing (222). PF4 increases the responsiveness of DCs to TLR ligands (223) and reduced ability of dendritic cells to present antigens (224). TGF-β1is also a factor secreted by platelets that inhibits the activation and maturation of DCs in vitro. TGF-β1 inhibits the upregulation of key T-cell co-stimulatory molecules on the DC surface and reduces their antigen-presenting capacity (220). Moreover, platelets and platelet-derived soluble mediators inhibit the pro-inflammatory properties of DCs and even induce an anti-inflammatory phenotype, decreasing the T-cell priming capacity of DCs. This allows tumor cells to evade the killing effect of T-cells and promote tumor progression (225, 226).
Platelets exhibit complex interactions with DCs. These complex and sometimes contradictory interactions appear to directly affect tumor development. However, while platelets have been well-investigated in the context of thrombosis, hemostasis, and vascular inflammation, the potential mechanism and impactions of the interaction between platelets and dendritic cells in tumor development and progression are still unknown. Therefore, further studies are necessary to clarify the exact mechanism underlying platelet-DC interactions and their impact on tumor development.
8 Platelet-T-cell and platelet-B-cell interactions in tumors
T-cells mainly include CD8+ and CD4+cells. CD8+T-cells are also recognized to be cytotoxic. They recognize antigens presented in major histocompatibility complex class I molecules and cause tumor cell death through cytokine secretion. CD4+ cells are mainly responsible for regulating the immune response (60). They are further divided into various Th types, with Th1, Th2,and Th17 cells acting as immune effectors and T regulatory cells as immune suppressors.
8.1 Platelet-CD4+T-cell interactions in tumors
The communication between platelets and CD4+T-cells is similar to that between pre- and post-synaptic neurons. Platelets store large amounts of mediators in their vesicles that can be released rapidly, similar to the release of neurotransmitters from pre-synaptic neurons (3, 227, 228). Moreover, similar to post-synaptic neurons, CD4+T-cells also have multiple neurotransmitter receptors (e.g., dopamine and serotonin receptors) that provide a direct pathway through which platelets can immediately communicate (229–232). In addition to platelet-derived neurotransmitters, platelets release multiple soluble factors, integrins, and adhesion molecules during activation, affectingCD4+T-cell function.
Studies have shown that serotonin(5HT), CCL5,and platelet-activating factor(PAF) play important roles in activatingCD4+T-cells. First,CXCL4 found in platelets recruits and activates T-cells, and T cells can also activate platelets through CD40L interaction with the CD40 on platelets, leading to the release of plateletsCCL5 and further recruitment of T cells to prevent tumor progression (233, 234). Platelet-derived factors such asCCL5 and CXCL4 can also stimulate the proliferation and differentiation of enhance Th1 and Th17cells (235). In addition, platelet-derived 5HTcan stimulate CD4+T-cell proliferation and produce IFN-γ (232), promoting the activation and proliferation of naïve T-cells, enhancing cellular immunity, and increasing tumor cell cytotoxicity (236, 237). Furthermore, epinephrine has been shown to stimulate both Th17 and Th2 cells and produce CXCL1, which has been demonstrated to stimulate Th17 cells and may substitute or enhance the effect of other platelet-derived factors (238).
Research has confirmed that platelet-derived TGF-β inhibits the differentiation of immature T-cells to Th1 cells and promotes their differentiation into regulatory T lymphocyte (Treg) subpopulations (239). Tregs express glycoprotein A repetitions predominant (GARP), which binds and activates latent TGF-β (240–243) involved in enhancing the suppressive ability of Tregs and maintaining Treg-mediated peripheral tolerance (241, 244, 245). Studies have shown that the GARP-TGF-β complex, together with platelet-secreted lactate, inhibits T-cell responses against melanoma and colorectal cancer, thus exerting suppressive effects on anti-tumor immunity (239, 246). Platelet-derived microvesicles have been shown to inhibit IL-17 production by Treg cells through P-selectin. Moreover, IL-17 is considered to promote neovascularization, tumor cell proliferation, and immunosuppression (247). Therefore, platelets modulate the immunosurveillance processes during cancer development.
Besides stimulating CD4+T-cells via soluble factors, platelets also directly contact T-cells, thereby regulating the T-cell immune response. Studies have shown that when platelets have a reduced ability to produce pro-inflammatory factors and stimulate CD4+T-cells, their ability to form aggregates with CD4+T-cells is significantly increased. Platelets bind to CD4+T-cells through multiple receptors, including P-selectin (CD62P) and integrins (αIIb and βIII), forming platelet-CD4+T-cell aggregates that interfere with the interactions between CD4+T-cells and APCs (232, 248).
8.2 Platelet-CD8+T-cell interactions in tumor
The influence of interactions between platelets and CD8T cells on tumors is twofold. On the one hand, some studies have observed that metastatic tumors have lower numbers of CD8 T cells and higher frequencies of platelets than non-metastatic tumors, indicating that the interplay between platelets and CD8 T cells influences metastasis, and also confirmed the important role of platelet-MDSC-CD8 T-cell axis in it. Platelets secrete CXCL4 to induce Myeloid Derived Suppressor Cells(MDSC) production, and MDSCs negatively inhibits the function of CD8 T cells in return (249).. This facilitates the escape of tumor cells from the primary tumor site and into circulation resulting the establishment of metastasis. On the other hand, RANTES upregulates the cytotoxic function of CD8 + T-helper cells as well as cytokine production (250). Besides APCs attributed to their receptors and high expression of MHC-I on their surface (251), platelets are able to present antigens to naïve T-cells, effectively stimulate CD8+T-cells and promote them to exert cellular immunity and kill tumor cells (232, 252). Additionally, it has been demonstrated that in patients with lung cancer, platelets can attach to T-cells (more platelets attach to CD8+ cells than to CD4+ cells, and these platelets express P-selectin) and form platelet-T-cell aggregates to promote thrombosis, increasing the risk of death in these patients (253).
8.3 Platelet-B-cellinteractions in tumors
Platelets activate B-cells by releasing soluble CD40 and CD40L;increasing the production of IgG1, IgG2, and IgG3; inducing B-cell proliferation, differentiation, and allotype switching; memory B-cell production, and enhancing humoral immunity (254). In addition, platelets contain kappa-light chain enhancers of members of the activated B-cell NF-κB family of proteins (255) that influence inflammation and thrombosis (256). Moreover, platelet-lymphocyte interactions can promote lymphocyte recruitment to sites of inflammation infection or tumors and further mediate lymphocyte killing of tumors (257).
9 Platelet-natural killer cell interactions in tumors
NK cells play a central role in tumor immunosurveillance (258). NK cells are activated through the following mechanisms. First, the activation of NK cells is triggered by the recognition of the”missing self” of abnormal cells, where the “self” mainly refers to MHC class I molecules. “Missing self” means that NK cells preferentially destroy target cells with low or no expression of MHC class I molecules (259). Another mechanism is mediated by the “induced self”recognition of abnormal cells. “Induced self” refers to the expression of ligands used to activate NK receptors, especially NK group 2D (NKG2D), which enhances the cytotoxicity of NK cells (260). Platelets can inhibit NK cell recognition of abnormal cells by highly expressing MHC class I molecules and down regulating the expression and release of NKG2DL(NKG2D ligand) (6) (Figure 2).
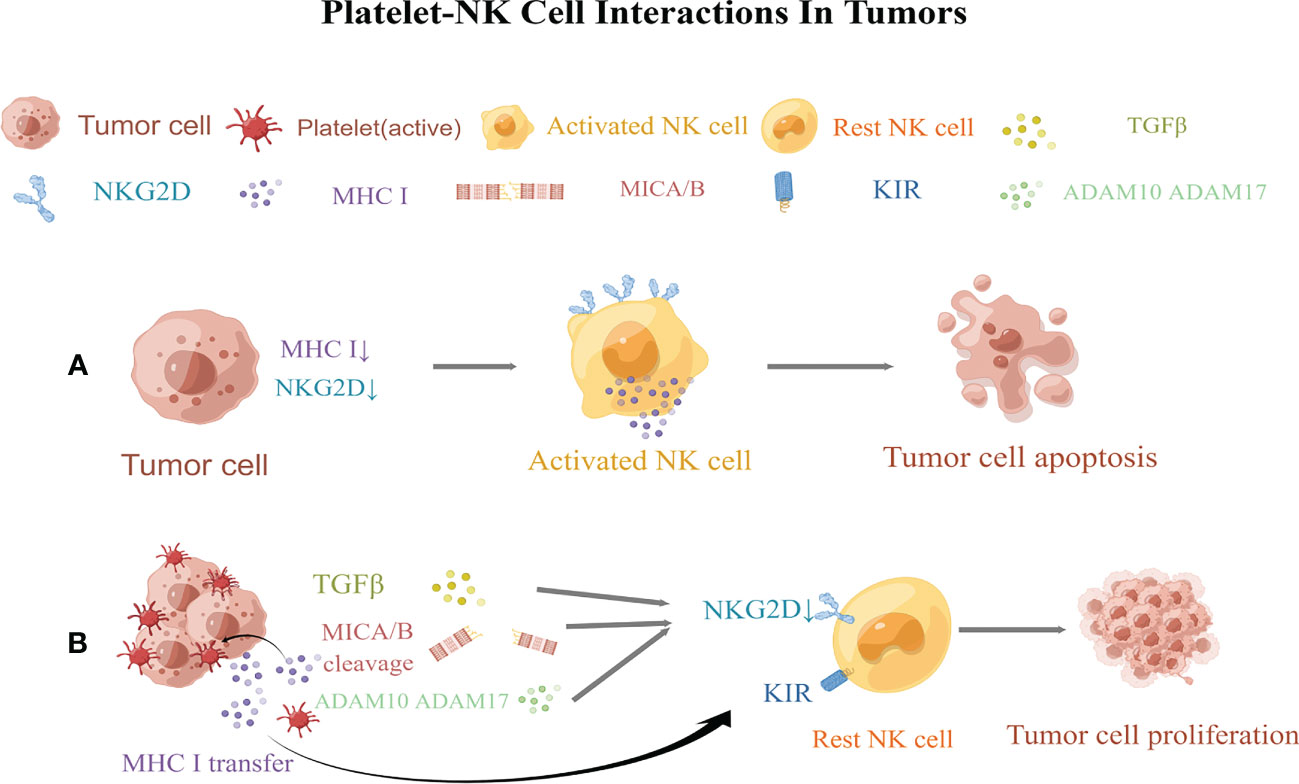
Figure 2 Regulation of natural killer (NK) cellsl responsiveness by platelets. (A) Normal NK cells kill tumor cells via the “missing self” and “induced self.” (B) Platelets obstruct NK cells, allowing tumor cells to proliferate by evading immune surveillance. Activated platelets then wrap around tumor cells, promote the transfer of MHC class I molecules to the surface of cancer cells, and decreases the “missing self” recognition of abnormal cells by NK cells. Platelets then reduce the expression of MICA and MICB on tumor cells and impair the “induced self” recognition of NK cells. Moreover, platelets also express the enzymes ADAM10 and ADAM17, which shed NKG2D on tumor cells. In turn, platelets release TGF-β to downregulate NKG2D and induce the expression of killer cell immunoglobulin-like receptors (KIRs) on NK cells. MICA and MICB, MHC class I chain-related protein (A,B) ADAM10andADAM17, a disintegrin and metalloproteinase domain-containing protein10 and 17; NKG2D,NK group 2D; KIRs, killer cell immunoglobulin-like receptors. This figure is drawn by Figdraw (https://figdraw.com/). The "↓" arrows refer to the decreased expression of MHCI and NKG2D.
9.1 Platelets weaken the “missing self” of NK cells
In the later stages of tumor development, malignant cells often metastasize via the bloodstream (261). However, owing to the abundance of NK cells in the blood (262), it is a hostile environment for tumor cell growth and metastasis. To resist this harsh environment, tumor cells release large amounts of cytotoxic factors to inhibit the killing effect of NK cells. More importantly, when tumor cells enter the circulation, platelets may provide an alternative mechanism to evade NK cell surveillance and increase the metastatic potential of tumor cells (57). The quantity and quality of platelets are directly proportional to the number of tumor cells that metastasize (263, 264). The depletion of platelets after intravenous inoculation of fibrosarcoma, lymphoma, and melanoma cell lines has been shown to directly inhibit metastasis in the lungs of mice with normal immune function, and this effect was reversed after the depletion of NK cells. This further indicates that platelets help circulating tumor cells evade immune surveillance by NK cells (58).Palumbo and his colleagues showed that tumor cells escape NK cell-mediated surveillance through fibrin deposition, thereby strengthening platelet aggregation on the surface of tumor cells (57). The ability of tumor cells to trigger platelet aggregation is related to the enhanced metastatic potential (58). Tumor cells can bind to platelet P-selectin via tumor cell-induced platelet aggregation, forming aggregates to protect themselves from circulation and to “hide” in the presence of NK cells (31). As reported previously, tumor cells were rapidly encapsulated in the presence of platelets in vitro, and the formation of a “platelet envelope” leads to the transfer of MHC class I to the surface of tumor cells, resulting in high levels of platelet-derived normal class I MHC expression on tumor cells. This “pseudoexpression” weakens the “missing self” recognition of abnormal cells by NK cells and reduces NK cell cytotoxicity (6, 56).
9.2 Platelets weaken the “induced self” of NK cells
In addition, the platelet coating on tumor cells reduces the surface expression of NKG2D ligands, including MHC class I chain–related proteins A and B(MICA and MICB) on tumor cells, impairing the “induced self” recognition of NK cells (265). It is widely known that tumor cells divide their own NKG2DL by expressing “a disintegrin and metalloproteinase domain-containing protein10 and 17” (ADAM10 and ADAM17) (266–268). Platelets can express both ADAM10 and ADAM17that can induce the shedding of NKG2D from tumor cells, thereby suppressing NK cell anti-tumor immune function (269–275). Additional studies have shown that the platelet-mediated release of TGF-β mediates NKG2DL downregulation and presents MHC class I molecules to NK cells via killer cell immunoglobulin-like receptor (KIR),which may result in the loss of cytotoxicity and IFN-γ-producing capacity of human NK cells (276). Platelets also express NKG2DL, specifically UL2-binding protein (ULBP2), which may be released in a soluble form. The bioactivity of soluble ULBP2 is unclear, however, its shedding might inhibit the NKG2D-inducedrecognition of platelet-cancer aggregates (277).
10 Platelet-endothelial cellinteractions in tumors
Endothelial cells (ECs) are situated on the inner surface of blood vessels, therefore in immediate contact with blood components and cells. ECs are also important for controlling blood fluidity; leukocyte activation, adhesion and migration; and platelet adhesion and aggregation (278).
10.1 Platelet-endothelial cell interactions promote thrombosis
Healthy ECs release prostacyclin (PGI2) and NO that counteract platelet activation, prevent clotting and thrombosis, and maintain unobstructed blood flow and tissue perfusion (279, 280). After endothelial injury, platelets were gathered in the damaged area of endothelium, quickly adhered and activated, resulting in the release of α-granules and the activation of integrin αIIbβ3 (281, 282). The von Willebrand factor (VWF) is essential for platelet adhesion. It is a multimeric glycoprotein synthesized by ECs and stored in Weibel-Palade bodies. VWF is located in α-granules of platelets and is also synthesized by megakaryocytes (283). Injured ECs release VWF, which acts as a bridge between platelets and subendothelial collagen via glycoprotein (GP)-Ibαon the platelet surface; it is also involved in platelet aggregation and thrombosis (283–285). Another function of the VWF is to act as a carrier and stabilizer of plasma FVII in circulation and to protect it from degradation by activated protein C by forming the VWF-FVII complex (286–288). VWF also interacts with platelet GPIbα, enhancing integrin β2-mediated interactions between platelets and neutrophils (278, 289) and coordinating the activation of neutrophils, platelets, and coagulation cascades that promote tumorigenesis and progression. In addition, collagen interacts with platelets via integrin α2β1 and GPVI to induce intracellular signaling and enhance platelet adhesion. The platelet fibrinogen receptor integrin αIIbβ3 strengthens platelet adhesion and promotes thrombosis (290). Moreover, platelets and ECs synthesize PAF, which promotes platelet adhesion to ECs as well as their activation and aggregation (291).
10.2 Platelet-endothelial cell interactions promote tumor angiogenesis
Growing tumors require large amounts of nutrients and oxygen, but a lack of contact with their internal vascular network often leads to hypoxia. Thus, angiogenesis is a crucial stage of cancer progression. The α-particles of platelets are rich in vasoactive substances and chemokines, such as serotonin,TXA2, PAF, platelet-derived growth factor-BB (PDGF-BB) and VEGF (292, 293). When platelets are activated, it can not only release PDGF-BB to promotes the upregulation of VEGFgeneexpression in vascular ECs, but also directly releases VEGF promoting the proliferation, differentiation, and migration of vascular ECs, enhances the permeability of vascular ECs, promotes neovascularization, provides oxygen and nutrients to ischemic and hypoxic tissues, and causes the rapid growth of tumor cells (7, 294, 295). The role of VEGF in neoplastic diseases also leads to enhanced TF expression and increased VWF release in ECs, thereby promoting thrombus formation and development at tumor sites (296). Platelets also indirectly promote leukocyte migration by activating ECs and inducing the secretion of Weibel-Palade vesicles from EC, leading to CD62P expression and IL-8 release. In turn, the release of these molecules leads to neutrophil rolling, adhesion, and extravasation and promotes the tumor inflammatory microenvironment and tumor development (289).
11 Summary
Platelets play an important role in hemostasis and thrombosis, and they also play a key role in tumorigenesis and tumor development by interacting with all cells in the tumor microenvironment (Figure 3). It affects various biological processes in the disease state. Studies have shown that platelets are involved in the immune response, regulate the tumor microenvironment, and are closely associated with the pathophysiology of thrombosis, inflammation, and tumor progression. The mechanisms of platelets interacting with cells in the tumor microenvironment are complex and appear to have a dual behavior, pro- cancer and anti-cancer. Various platelet-cell interactions, in turn, lead to continuous platelet activation, further promote tumor development.
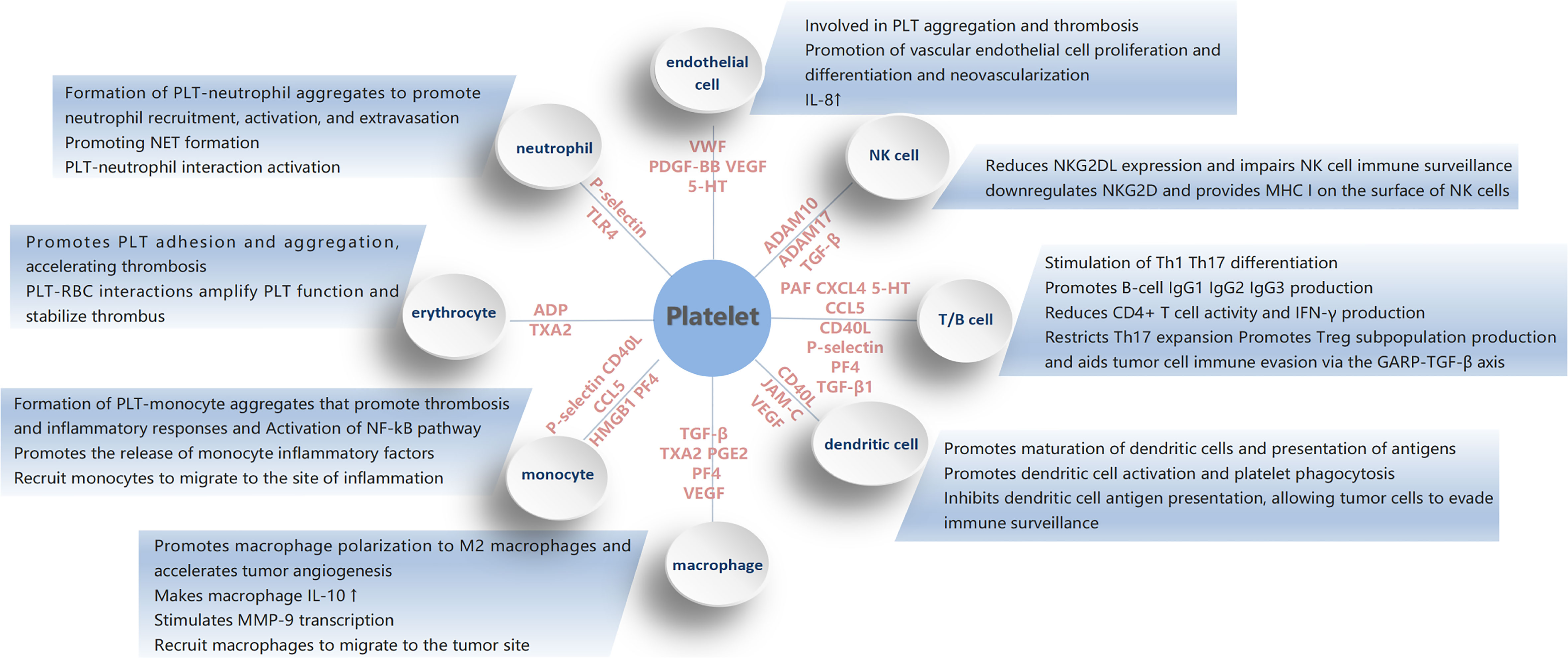
Figure 3 The figure shows the mechanism of platelet action with various immune cells.
In conclusion, better understanding of the interaction between platelets and cells in the tumor microenvironment is helpful to understanding the mechanisms of the dual role of platelets in promoting and suppressing cancer, and future studies concerning these questions will provide a new direction for cancer prevention and therapy.
Author contributions
ZZ and YY performed the scientific literature search, collected and analyzed data, designed the review structure and wrote the manuscript. ZM and LQ prepared the tables and figures. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the Joint Fund for Innovation and Development of Shandong Natural Science Foundation (Grant No.ZR2022LSW024), the Clinical Medicine +X Project of Qingdao University (Grant No.2020018).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discovery (2007) 6(4):273–86. doi: 10.1038/nrd2115
2. Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother (2016) 43(2):78–88. doi: 10.1159/000444807
3. Semple JW, Italiano JE Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11(4):264–74. doi: 10.1038/nri2956
4. Singh MV, Davidson DC, Jackson JW, Singh VB, Silva J, Ramirez SH, et al. Characterization of platelet–monocyte complexes in HIV–1–Infected individuals: possible role in HIV–associated neuroinflammation. J Immunol (2014) 192(10):4674–84. doi: 10.4049/jimmunol.1302318
5. Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of p–selectin glycoprotein ligand–1 enhances tyrosine phosphorylation and activates mitogen–activated protein kinases in human neutrophils. J Biol Chem (1997) 272(45):28750–6. doi: 10.1074/jbc.272.45.28750
6. Maurer S, Ferrari de Andrade L. NK cell interaction with platelets and myeloid cells in the tumor milieu. Front Immunol (2020) 11:608849. doi: 10.3389/fimmu.2020.608849
7. Jang HY, Myoung SM, Choe JM, Kim T, Cheon YP, Kim YM, et al. Effects of autologous platelet–rich plasma on regeneration of damaged endometrium in female rats. Yonsei Med J (2017) 58(6):1195–203. doi: 10.3349/ymj.2017.58.6.1195
8. Sirachainan N. Thalassemia and the hypercoagulable state. Thromb Res (2013) 132(6):637–41. doi: 10.1016/j.thromres.2013.09.029
9. Bambace NM, Holmes CE. The platelet contribution to cancer progression. J Thromb Haemost (2011) 9(2):237–49. doi: 10.1111/j.1538-7836.2010.04131.x
10. Josa V, Krzystanek M, Eklund AC, Salamon F, Zarand A, Szallasi Z, et al. Relationship of postoperative thrombocytosis and survival of patients with colorectal cancer. Int J Surg (2015) 18:1–6. doi: 10.1016/j.ijsu.2015.03.005
11. Zhao JM, Wang YH, Yao N, Wei KK, Jiang L, Hanif S, et al. Poor prognosis significance of pretreatment thrombocytosis in patients with colorectal cancer: a meta–analysis. Asian Pac J Cancer Prev (2016) 17(9):4295–300.
12. Haemmerle M, Stone RL, Menter DG, Afshar-Kharghan V, Sood AK. The platelet lifeline to cancer: Challenges and opportunities. Cancer Cell (2018) 33(6):965–83. doi: 10.1016/j.ccell.2018.03.002
13. Yu LX, Yan L, Yang W, Wu FQ, Ling Y, Chen SZ, et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell–released high–mobility group box1 protein. Nat Commun (2014) 5:5256. doi: 10.1038/ncomms6256
14. López JA. Introduction to a review series on platelets and cancer. Blood (2021) 137(23):3151–2. doi: 10.1182/blood.2020010237
15. Holinstat M. Normal platelet function. Cancer Metastasis Rev (2017) 36(2):195–8. doi: 10.1007/s10555-017-9677-x
16. Machlus KR, Italiano JE Jr. The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol (2013) 201(6):785–96. doi: 10.1083/jcb.201304054
17. Linden MD. Platelet physiology. Methods Mol Biol (2013) 992:13–30. doi: 10.1007/978-1-62703-339-8_2
18. Pagel O, Walter E, Jurk K, Zahedi RP. Taking the stock of granule cargo: Platelet releasate proteomics. Platelets (2017) 28(2):119–28. doi: 10.1080/09537104.2016.1254762
19. Li N. Platelets in cancer metastasis: To help the "villain" to do evil. Int J Cancer (2016) 138(9):2078–87. doi: 10.1002/ijc.29847
20. Karpatkin S, Ambrogio C, Pearlstein E. The role of tumor–induced platelet aggregation, platelet adhesion and adhesive proteins in tumor metastasis. Prog Clin Biol Res (1988) 283:585–606.
21. Verheul HM, Hoekman K, Luykx-de Bakker S, Eekman CA, Folman CC, Broxterman HJ, et al. Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res (1997) 3(12 Pt 1):2187–90.
22. Pinedo HM, Verheul HM, D'Amato RJ, Folkman J. Involvement of platelets in tumour angiogenesis? Lancet (1998) 352(9142):1775–7. doi: 10.1016/S0140-6736(98)05095-8
23. Wang S, Li Z, Xu R. Human cancer and platelet interaction, a potential therapeutic target. Int J Mol Sci (2018) 19(4):1246. doi: 10.3390/ijms19041246
24. Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev (2017) 36(2):249–62. doi: 10.1007/s10555-017-9673-1
25. Cross MJ, Claesson–Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci (2001) 22(4):201–7. doi: 10.1016/S0165-6147(00)01676-X
26. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med (2003) 9(6):669–76. doi: 10.1038/nm0603-669
27. Varki A. Trousseau's syndrome: multiple definitions and multiple mechanisms. Blood (2007) 110(6):1723–9. doi: 10.1182/blood-2006-10-053736
28. Jurasz P, Alonso–Escolano D, Radomski MW. Platelet–cancer interactions: mechanisms and pharmacology of tumour cell–induced platelet aggregation. Br J Pharmacol (2004) 143(7):819–26. doi: 10.1038/sj.bjp.0706013
29. Ferroni P, Martini F, Portarena I, Grenga I, Riondino S, La Farina F, et al. An activated protein c–dependent thrombin generation assay predicts chemotherapy–associated venous thromboembolism in cancer patients. Thromb Haemost (2011) 105(5):931–2. doi: 10.1160/TH10-11-0757
30. Olsson AK, Cedervall J. The pro–inflammatory role of platelets in cancer. Platelets (2018) 29(6):569–73. doi: 10.1080/09537104.2018.1453059
31. Egan K, Cooke N, Kenny D. Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis (2014) 31(6):697–704. doi: 10.1007/s10585-014-9660-7
32. Jiang X, Wong KHK, Khankhel AH, Zeinali M, Reategui E, Phillips MJ, et al. Microfluidic isolation of platelet–covered circulating tumor cells. Lab Chip (2017) 17(20):3498–503. doi: 10.1039/C7LC00654C
33. Palacios-Acedo AL, Mège D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L. Platelets, thrombo–inflammation, and cancer: Collaborating with the enemy. Front Immunol (2019) 10:1805. doi: 10.3389/fimmu.2019.01805
34. Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer (2005) 113(5):752–60. doi: 10.1002/ijc.20657
35. Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell–derived microparticles bearing p–selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med (2009) 206(9):1913–27. doi: 10.1084/jem.20082297
36. Qi C, Li B, Guo S, Wei B, Shao C, Li J, et al. P–Selectin–Mediated adhesion between platelets and tumor cells promotes intestinal tumorigenesis in Apc(Min/+) mice. Int J Biol Sci (2015) 11(6):679–87. doi: 10.7150/ijbs.11589
37. Scherer .Pietramaggiori G, Cervi SS, Klement D, Orgill G, D P. Tumors stimulate platelet delivery of angiogenic factors in vivo: an unexpected benefit. Am J Pathol (2008) 173(6):1609–16. doi: 10.2353/ajpath.2008.080474
38. Faria AVS, Andrade SS, Peppelenbosch MP, Ferreira-Halder CV, Fuhler GM. Platelets in aging and cancer–"double–edged sword". Cancer Metastasis Rev (2020) 39(4):1205–21. doi: 10.1007/s10555-020-09926-2
39. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discovery (2018) 8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367
40. Zou W, Wolchok JD, Chen L. PD–L1 (B7–H1) and PD–1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8(328):328rv4. doi: 10.1126/scitranslmed.aad7118
41. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor–infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U.S.A. (2007) 104(9):3360–5. doi: 10.1073/pnas.0611533104
42. Rolfes V, Idel C, Pries R, Plötze-Martin K, Habermann J, Gemoll T, et al. PD–L1 is expressed on human platelets and is affected by immune checkpoint therapy. Oncotarget (2018) 9(44):27460–70. doi: 10.18632/oncotarget.25446
43. Darga EP, Dolce EM, Fang F, Kidwell KM, Gersch CL, Kregel S, et al. PD–L1 expression on circulating tumor cells and platelets in patients with metastatic breast cancer. PloS One (2021) 16(11):e0260124. doi: 10.1371/journal.pone.0260124
44. Hinterleitner C, Strähle J, Malenke E, Hinterleitner M, Henning M, Seehawer M, et al. Platelet PD–L1 reflects collective intratumoral PD–L1 expression and predicts immunotherapy response in non–small cell lung cancer. Nat Commun (2021) 12(1):7005. doi: 10.1038/s41467-021-27303-7
45. Cho MS, Lee H, Gonzalez-Delgado R, Li D, Sasano T, Carlos-Alcalde W, et al. Platelets increase the expression of PD–L1 in ovarian cancer. Cancers (Basel) (2022) 14(10):2498. doi: 10.3390/cancers14102498
46. Asgari A, Lesyk G, Poitras E, Govindasamy N, Terry K, To R, et al. Platelets stimulate programmed death–ligand 1 expression by cancer cells: Inhibition by anti–platelet drugs. J Thromb Haemost (2021) 19(11):2862–72. doi: 10.1111/jth.15478
47. Zaslavsky AB, Adams MP, Cao X, Maj T, Choi JE, Stangl-Kremser J, et al. Platelet PD–L1 suppresses anti–cancer immune cell activity in PD–L1 negative tumors. Sci Rep (2020) 10(1):19296. doi: 10.1038/s41598-020-76351-4
48. Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA–Seq of tumor–educated platelets enables blood–based pan–cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell (2015) 28(5):666–76. doi: 10.1016/j.ccell.2015.09.018
49. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial–mesenchymal–like transition and promotes metastasis. Cancer Cell (2011) 20(5):576–90. doi: 10.1016/j.ccr.2011.09.009
50. Medina C, Jurasz P, Santos-Martinez MJ, Jeong SS, Mitsky T, Chen R, et al. Platelet aggregation–induced by caco–2 cells: regulation by matrix metalloproteinase–2 and adenosine diphosphate. J Pharmacol Exp Ther (2006) 317(2):739–45. doi: 10.1124/jpet.105.098384
51. Radziwon-Balicka A, Santos-Martinez MJ, Corbalan JJ, O'Sullivan S, Treumann A, Gilmer JF, et al. Mechanisms of platelet–stimulated colon cancer invasion: role of clusterin and thrombospondin 1 in regulation of the P38MAPK–MMP–9 pathway. Carcinogenesis (2014) 35(2):324–32. doi: 10.1093/carcin/bgt332
52. Lou XL, Sun J, Gong SQ, Yu XF, Gong R, Deng H. Interaction between circulating cancer cells and platelets: clinical implication. Chin J Cancer Res (2015) 27(5):450–60. doi: 10.3978/j.issn.1000-9604.2015.04.10
53. Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I–5–Iodo–2'–deoxyuridine. J Natl Cancer Inst (1970) 45(4):773–82.
54. Lin RJ, Afshar–Kharghan V, Schafer AI. Paraneoplastic thrombocytosis: the secrets of tumor self–promotion. Blood (2014) 124(2):184–7. doi: 10.1182/blood-2014-03-562538
55. Tímár J, Tóvári J, Rásó E, Mészáros L, Bereczky B, Lapis K. Platelet–mimicry of cancer cells: epiphenomenon with clinical significance. Oncology (2005) 69(3):185–201. doi: 10.1159/000088069
56. Placke T, Örgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, et al. Platelet–derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res (2012) 72(2):440–8. doi: 10.1158/0008-5472.CAN-11-1872
57. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell–mediated elimination of tumor cells. Blood (2005) 105(1):178–85. doi: 10.1182/blood-2004-06-2272
58. Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res (1999) 59(6):1295–300.
59. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
60. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood (2014) 123(18):2759–67. doi: 10.1182/blood-2013-11-462432
61. Dib PRB, Quirino-Teixeira AC, Merij LB, Pinheiro MBM, Rozini SV, Andrade FB, et al. Innate immune receptors in platelets and platelet–leukocyte interactions. J Leukoc Biol (2020) 108(4):1157–82. doi: 10.1002/JLB.4MR0620-701R
62. Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol (2004) 25(9):489–95. doi: 10.1016/j.it.2004.07.003
63. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res (2007) 100(1):27–40. doi: 10.1161/01.RES.0000252802.25497.b7
64. Huang HS, Chang HH. Platelets in inflammation and immune modulations: functions beyond hemostasis. Arch Immunol Ther Exp (Warsz) (2012) 60(6):443–51. doi: 10.1007/s00005-012-0193-y
65. Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol (2013) 35(3):254–61. doi: 10.1111/ijlh.12084
66. Yan Q, Zeng Z, Gong Z, Zhang W, Li X, He B, et al. EBV–miR–BART10–3p facilitates epithelial–mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget (2015) 6(39):41766–82. doi: 10.18632/oncotarget.6155
67. Xiao K, Yu Z, Li X, Li X, Tang K, Tu C, et al. Genome–wide analysis of Epstein–Barr virus (EBV) integration and strain in C666–1 and raji cells. J Cancer (2016) 7(2):214–24. doi: 10.7150/jca.13150
68. Song Y, Li X, Zeng Z, Li Q, Gong Z, Liao Q, et al. Epstein–Barr Virus encoded miR–BART11 promotes inflammation–induced carcinogenesis by targeting FOXP1. Oncotarget (2016) 7(24):36783–99. doi: 10.18632/oncotarget.9170
69. He B, Li W, Wu Y, Wei F, Gong Z, Bo H, et al. Epstein–Barr Virus–encoded miR–BART6–3p inhibits cancer cell metastasis and invasion by targeting long non–coding RNA LOC553103. Cell Death Dis (2016) 7(9):e2353. doi: 10.1038/cddis.2016.253
70. Tu C, Zeng Z, Qi P, Li X, Yu Z, Guo C, et al. Genome–wide analysis of 18 Epstein–Barr viruses isolated from primary nasopharyngeal carcinoma biopsy specimens. J Virol (2017) 91(17):e00301–17. doi: 10.1128/JVI.00301-17
71. Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, et al. The emerging role of Epstein–Barr virus encoded microRNAs in nasopharyngeal carcinoma. J Cancer (2018) 9(16):2852–64. doi: 10.7150/jca.25460
72. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
73. Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer–related inflammation. Cytokine (2008) 43(3):374–9. doi: 10.1016/j.cyto.2008.07.014
74. Kerr BA, Miocinovic R, Smith AK, Klein EA, Byzova TV. Comparison of tumor and microenvironment secretomes in plasma and in platelets during prostate cancer growth in a xenograft model. Neoplasia (2010) 12(5):388–96. doi: 10.1593/neo.10166
75. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet–mediated modulation of adaptive immunity. a communication link between innate and adaptive immune compartments. Immunity (2003) 19(1):9–19. doi: 10.1016/S1074-7613(03)00177-8
76. Nishat S, Wuescher LM, Worth RG. Platelets enhance dendritic cell responses against staphylococcus aureus through CD40–CD40L. Infect Immun (2018) 86(9):e00186–18. doi: 10.1128/IAI.00186-18
77. Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol (2008) 28(11):1920–7. doi: 10.1161/ATVBAHA.108.169417
78. Schlesinger M. Role of platelets and platelet receptors in cancer metastasis. J Hematol Oncol (2018) 11(1):125. doi: 10.1186/s13045-018-0669-2
79. Ünlü B, Versteeg HH. Cancer–associated thrombosis: The search for the holy grail continues. Res Pract Thromb Haemost (2018) 2(4):622–9. doi: 10.1002/rth2.12143
80. Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci (2016) 53(6):409–30. doi: 10.1080/10408363.2016.1200008
81. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P–selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
82. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U.S.A. (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
83. Stoiber D, Assinger A. Platelet–leukocyte interplay in cancer development and progression. Cells (2020) 9(4):855. doi: 10.3390/cells9040855
84. Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, et al. Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol (1998) 152(2):399–411.
85. Lindemann S, Krämer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost (2007) 5 Suppl 1:203–11. doi: 10.1111/j.1538-7836.2007.02517.x
86. Menter DG, Tucker SC, Kopetz S, Sood AK, Crissman JD, Honn KV. Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev (2014) 33(1):231–69. doi: 10.1007/s10555-014-9498-0
87. Mezouar S, Frère C, Darbousset R, Mege D, Crescence L, Dignat-George F, et al. Role of platelets in cancer and cancer–associated thrombosis: Experimental and clinical evidences. Thromb Res (2016) 139:65–76. doi: 10.1016/j.thromres.2016.01.006
88. Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, et al. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood (1998) 91(11):4216–23. doi: 10.1182/blood.V91.11.4216
89. Del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue–factor–bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood (2005) 106(5):1604–11. doi: 10.1182/blood-2004-03-1095
90. Brand K, Fowler BJ, Edgington TS, Mackman N. Tissue factor mRNA in THP–1 monocytic cells is regulated at both transcriptional and posttranscriptional levels in response to lipopolysaccharide. Mol Cell Biol (1991) 11(9):4732–8. doi: 10.1128/mcb.11.9.4732-4738.1991
91. Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle p–selectin glycoprotein ligand 1 and platelet p–selectin. J Exp Med (2003) 197(11):1585–98. doi: 10.1084/jem.20021868
92. Camera M, Frigerio M, Toschi V, Brambilla M, Rossi F, Cottell DC, et al. Platelet activation induces cell–surface immunoreactive tissue factor expression, which is modulated differently by antiplatelet drugs. Arterioscler Thromb Vasc Biol (2003) 23(9):1690–6. doi: 10.1161/01.ATV.0000085629.23209.AA
93. Engelmann B. Initiation of coagulation by tissue factor carriers in blood. Blood Cells Mol Dis (2006) 36(2):188–90. doi: 10.1016/j.bcmd.2005.12.020
94. Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, et al. Signal–dependent splicing of tissue factor pre–mRNA modulates the thrombogenicity of human platelets. J Exp Med (2006) 203(11):2433–40. doi: 10.1084/jem.20061302
95. Müller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, et al. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J (2003) 17(3):476–8. doi: 10.1096/fj.02-0574fje
96. Lopez-Vilchez I, Escolar G, Diaz-Ricart M, Fuste B, Galan AM, White JG. Tissue factor–enriched vesicles are taken up by platelets and induce platelet aggregation in the presence of factor VIIa. Thromb Haemost (2007) 97(2):202–11. doi: 10.1160/TH06-04-0216
97. Key NS. Platelet tissue factor: how did it get there and is it important? Semin Hematol (2008) 45(2 Suppl 1):S16–20. doi: 10.1053/j.seminhematol.2008.03.012
98. Panes O, Matus V, Sáez CG, Quiroga T, Pereira J, Mezzano D. Human platelets synthesize and express functional tissue factor. Blood (2007) 109(12):5242–50. doi: 10.1182/blood-2006-06-030619
99. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med (2017) 23(3):279–87. doi: 10.1038/nm.4294
100. Cedervall J, Hamidi A, Olsson AK. Platelets, NETs and cancer. Thromb Res (2018) 164 Suppl 1:S148–s152. doi: 10.1016/j.thromres.2018.01.049
101. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet–derived HMGB1 is a critical mediator of thrombosis. J Clin Invest (2015) 125(12):4638–54. doi: 10.1172/JCI81660
102. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil–extracellular traps and DNA. Sci Rep (2018) 8(1):2068. doi: 10.1038/s41598-018-20479-x
103. Wu H, Li R, Pei LG, Wei ZH, Kang LN, Wang L, et al. Emerging role of high mobility group box–1 in thrombosis–related diseases. Cell Physiol Biochem (2018) 47(4):1319–37. doi: 10.1159/000490818
104. Wang Z, Yang C, Li L, Jin X, Zhang Z, Zheng H, et al. Tumor–derived HMGB1 induces CD62L(dim) neutrophil polarization and promotes lung metastasis in triple–negative breast cancer. Oncogenesis (2020) 9(9):82. doi: 10.1038/s41389-020-00267-x
105. Otxoa-de-Amezaga A, Gallizioli M, Pedragosa J, Justicia C, Miró-Mur F, Salas-Perdomo A, et al. Location of neutrophils in different compartments of the damaged mouse brain after severe Ischemia/Reperfusion. Stroke (2019) 50(6):1548–57. doi: 10.1161/STROKEAHA.118.023837
106. Tadie JM, Bae HB, Deshane JS, Bell CP, Lazarowski ER, Chaplin DD, et al. Toll–like receptor 4 engagement inhibits adenosine 5'–monophosphate–activated protein kinase activation through a high mobility group box 1 protein–dependent mechanism. Mol Med (2012) 18(1):659–68. doi: 10.2119/molmed.2011.00401
107. Kim SW, Lee JK. Role of HMGB1 in the interplay between NETosis and thrombosis in ischemic stroke: A review. Cells (2020) 9(8):1794. doi: 10.3390/cells9081794
108. Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple toll–like receptors. Am J Physiol Cell Physiol (2006) 290(3):C917–24. doi: 10.1152/ajpcell.00401.2005
109. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll–like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem (2004) 279(9):7370–7. doi: 10.1074/jbc.M306793200
110. Wang JD, Wang YY, Lin SY, Chang CY, Li JR, Huang SW, et al. Exosomal HMGB1 promoted cancer malignancy. Cancers (Basel) (2021) 13(4):877. doi: 10.3390/cancers13040877
111. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
112. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type–specific roles of NF–κB linking inflammation and thrombosis. Front Immunol (2019) 10:85. doi: 10.3389/fimmu.2019.00085
113. Mauracher LM, Posch F, Martinod K, Grilz E, Däullary T, Hell L, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost (2018) 16(3):508–18. doi: 10.1111/jth.13951
114. Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC–2. J Clin Invest (2019) 129(1):12–23. doi: 10.1172/JCI122955
115. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
116. Schrottmaier WC, Kral JB, Badrnya S, Assinger A. Aspirin and P2Y12 inhibitors in platelet–mediated activation of neutrophils and monocytes. Thromb Haemost (2015) 114(3):478–89. doi: 10.1160/TH14-11-0943
117. Weisel JW, Litvinov RI. Red blood cells: the forgotten player in hemostasis and thrombosis. J Thromb Haemost (2019) 17(2):271–82. doi: 10.1111/jth.14360
118. Shrivastava S, Mahantshetty U, Engineer R, Chopra S, Hawaldar R, Hande V, et al. Cisplatin chemoradiotherapy vs radiotherapy in FIGO stage IIIB squamous cell carcinoma of the uterine cervix: A randomized clinical trial. JAMA Oncol (2018) 4(4):506–13. doi: 10.1001/jamaoncol.2017.5179
119. Thakar S, Gabarin N, Gupta A, Radford M, Warkentin TE, Arnold DM. Anemia–induced bleeding in patients with platelet disorders. Transfus Med Rev (2021) 35(3):22–8. doi: 10.1016/j.tmrv.2021.06.001
120. Vaupel P, Mayer A, Höckel M. Impact of hemoglobin levels on tumor oxygenation: the higher, the better? Strahlenther Onkol (2006) 182(2):63–71. doi: 10.1007/s00066-006-1543-7
121. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26(2):225–39. doi: 10.1007/s10555-007-9055-1
122. Vaupel P, Kelleher DK, Höckel M. Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol (2001) 28(2 Suppl 8):29–35. doi: 10.1016/S0093-7754(01)90210-6
123. Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost (2007) 5(3):632–4. doi: 10.1111/j.1538-7836.2007.02374.x
124. Kroll MH, Michaelis LC, Verstovsek S. Mechanisms of thrombogenesis in polycythemia vera. Blood Rev (2015) 29(4):215–21. doi: 10.1016/j.blre.2014.12.002
125. Klatt C, Krüger I, Zey S, Krott KJ, Spelleken M, Gowert NS, et al. Platelet–RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J Clin Invest (2018) 128(9):3906–25. doi: 10.1172/JCI92077
126. Reimers RC, Sutera SP, Joist JH. Potentiation by red blood cells of shear–induced platelet aggregation: relative importance of chemical and physical mechanisms. Blood (1984) 64(6):1200–6. doi: 10.1182/blood.V64.6.1200.1200
127. Born GV, Bergquist D, Arfors KE. Evidence for inhibition of platelet activation in blood by a drug effect on erythrocytes. Nature (1976) 259(5540):233–5. doi: 10.1038/259233a0
128. Bergqvist D, Arfors KE. Haemostatic platelet plug formation in the isolated rabbit mesenteric preparation–an analysis of red blood cell participation. Thromb Haemost (1980) 44(1):6–8. doi: 10.1055/s-0038-1650069
129. Alkhamis TM, Beissinger RL, Chediak JR. Red blood cell effect on platelet adhesion and aggregation in low–stress shear flow. myth or fact? ASAIO Trans (1988) 34(3):868–73.
130. Silvain J, Pena A, Cayla G, Brieger D, Bellemain-Appaix A, Chastre T, et al. Impact of red blood cell transfusion on platelet activation and aggregation in healthy volunteers: results of the TRANSFUSION study. Eur Heart J (2010) 31(22):2816–21. doi: 10.1093/eurheartj/ehq209
131. Helms CC, Marvel M, Zhao W, Stahle M, Vest R, Kato GJ, et al. Mechanisms of hemolysis–associated platelet activation. J Thromb Haemost (2013) 11(12):2148–54. doi: 10.1111/jth.12422
132. Tran PL, Pietropaolo MG, Valerio L, Brengle W, Wong RK, Kazui T, et al. Hemolysate–mediated platelet aggregation: an additional risk mechanism contributing to thrombosis of continuous flow ventricular assist devices. Perfusion (2016) 31(5):401–8. doi: 10.1177/0267659115615206
133. Gladwin MT, Lancaster JR Jr, Freeman BA, Schechter AN. Nitric oxide's reactions with hemoglobin: a view through the SNO–storm. Nat Med (2003) 9(5):496–500. doi: 10.1038/nm0503-496
134. Dubovoy T, Engoren M. Thrombotic risks in red blood cell transfusions. Semin Thromb Hemost (2016) 42(2):102–11. doi: 10.1055/s-0035-1569069
135. Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis–associated pulmonary hypertension, and nitric oxide scavenging by cell–free hemoglobin. Blood (2007) 110(6):2166–72. doi: 10.1182/blood-2006-12-061697
136. Iuliano L, Violi F, Pedersen JZ, Praticò D, Rotilio G, Balsano F. Free radical–mediated platelet activation by hemoglobin released from red blood cells. Arch Biochem Biophys (1992) 299(2):220–4. doi: 10.1016/0003-9861(92)90267-Z
137. Flamm MH, Diamond SL. Multiscale systems biology and physics of thrombosis under flow. Ann BioMed Eng (2012) 40(11):2355–64. doi: 10.1007/s10439-012-0557-9
138. Gil–Bernabé AM, Lucotti S, Muschel RJ. Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol (2013) 162(4):433–41. doi: 10.1111/bjh.12381
139. Walton BL, Lehmann M, Skorczewski T, Holle LA, Beckman JD, Cribb JA, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood (2017) 129(18):2537–46. doi: 10.1182/blood-2016-10-746479
140. Goel MS, Diamond SL. Adhesion of normal erythrocytes at depressed venous shear rates to activated neutrophils, activated platelets, and fibrin polymerized from plasma. Blood (2002) 100(10):3797–803. doi: 10.1182/blood-2002-03-0712
141. White J, Lancelot M, Sarnaik S, Hines P. Increased erythrocyte adhesion to VCAM–1 during pulsatile flow: Application of a microfluidic flow adhesion bioassay. Clin Hemorheol Microcirc (2015) 60(2):201–13. doi: 10.3233/CH-141847
142. Vallés J, Santos MT, Aznar J, Martínez M, Moscardó A, Piñón M, et al. Platelet–erythrocyte interactions enhance alpha(IIb)beta(3) integrin receptor activation and p–selectin expression during platelet recruitment: down–regulation by aspirin ex vivo. Blood (2002) 99(11):3978–84. doi: 10.1182/blood.V99.11.3978
143. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen–presenting functions. Nat Rev Immunol (2017) 17(6):349–62. doi: 10.1038/nri.2017.28
144. Ugel S, Canè S, De Sanctis F, Bronte V. Monocytes in the tumor microenvironment. Annu Rev Pathol (2021) 16:93–122. doi: 10.1146/annurev-pathmechdis-012418-013058
145. Orozco SL, Canny SP, Hamerman JA. Signals governing monocyte differentiation during inflammation. Curr Opin Immunol (2021) 73:16–24. doi: 10.1016/j.coi.2021.07.007
146. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol (2016) 37(1):41–52. doi: 10.1016/j.it.2015.11.008
147. Gudbrandsdottir S, Hasselbalch HC, Nielsen CH. Activated platelets enhance IL–10 secretion and reduce TNF–α secretion by monocytes. J Immunol (2013) 191(8):4059–67. doi: 10.4049/jimmunol.1201103
148. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science (2015) 350(6263):985–90. doi: 10.1126/science.aac9407
149. Passlick B, Flieger D, Ziegler–Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood (1989) 74(7):2527–34. doi: 10.1182/blood.V74.7.2527.2527
150. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood (2011) 118(5):e16–31. doi: 10.1182/blood-2010-12-326355
151. Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, et al. Circulating monocyte–platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis (2019) 282:11–8. doi: 10.1016/j.atherosclerosis.2018.12.029
152. Merah-Mourah F, Cohen SO, Charron D, Mooney N, Haziot A. Identification of novel human monocyte subsets and evidence for phenotypic groups defined by interindividual variations of expression of adhesion molecules. Sci Rep (2020) 10(1):4397. doi: 10.1038/s41598-020-61022-1
153. Gjelstrup MC, Stilund M, Petersen T, Møller HJ, Petersen EL, Christensen T. Subsets of activated monocytes and markers of inflammation in incipient and progressed multiple sclerosis. Immunol Cell Biol (2018) 96(2):160–74. doi: 10.1111/imcb.1025
154. Tunjungputri RN, van der Ven AJ, Riksen N, Rongen G, Tacke S, van den Berg TN, et al. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2– and TLR4–mediated inflammatory responses. Thromb Haemost (2015) 113(5):1035–45. doi: 10.1160/TH14-07-0579
155. Gros A, Ollivier V, Ho–Tin–Noé B. Platelets in inflammation: regulation of leukocyte activities and vascular repair. Front Immunol (2014) 5:678. doi: 10.3389/fimmu.2014.00678
156. Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost (2014) 12(11):1764–75. doi: 10.1111/jth.12730
157. Ahn KC, Jun AJ, Pawar P, Jadhav S, Napier S, McCarty OJ, et al. Preferential binding of platelets to monocytes over neutrophils under flow. Biochem Biophys Res Commun (2005) 329(1):345–55. doi: 10.1016/j.bbrc.2005.01.146
158. Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet–leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol (2001) 115(2):451–9. doi: 10.1046/j.1365-2141.2001.03101.x
159. Li N, Hu H, Lindqvist M, Wikström-Jonsson E, Goodall AH, Hjemdahl P. Platelet–leukocyte cross talk in whole blood. Arterioscler Thromb Vasc Biol (2000) 20(12):2702–8. doi: 10.1161/01.ATV.20.12.2702
160. Zamora C, Riudavets M, Anguera G, Alserawan L, Sullivan I, Barba A, et al. Circulating leukocyte–platelet complexes as a predictive biomarker for the development of immune–related adverse events in advanced non–small cell lung cancer patients receiving anti–PD–(L)1 blocking agents. Cancer Immunol Immunother (2021) 70(6):1691–704. doi: 10.1007/s00262-020-02793-4
161. Zarbock A, Müller H, Kuwano Y, Ley K. PSGL–1–Dependent myeloid leukocyte activation. J Leukoc Biol (2009) 86(5):1119–24. doi: 10.1189/jlb.0209117
162. Phillips JH, Chang CW, Lanier LL. Platelet–induced expression of fc gamma RIII (CD16) on human monocytes. Eur J Immunol (1991) 21(4):895–9. doi: 10.1002/eji.1830210406
163. Badrnya S, Schrottmaier WC, Kral JB, Yaiw KC, Volf I, Schabbauer G, et al. Platelets mediate oxidized low–density lipoprotein–induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol (2014) 34(3):571–80. doi: 10.1161/ATVBAHA.113.302919
164. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: Platelet innate immune interactions and thromboinflammation. Int J Mol Sci (2022) 23(7):3868. doi: 10.3390/ijms23073868
165. Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte–platelet interaction induces a pro–inflammatory phenotype in circulating monocytes. PloS One (2011) 6(10):e25595. doi: 10.1371/journal.pone.0025595
166. Eligini S, Barbieri SS, Arenaz I, Tremoli E, Colli S. Paracrine up–regulation of monocyte cyclooxygenase–2 by platelets: role of transforming growth factor–beta1. Cardiovasc Res (2007) 74(2):270–8. doi: 10.1016/j.cardiores.2006.12.013
167. Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, et al. Platelet HMGB1 is required for efficient bacterial clearance in intra–abdominal bacterial sepsis in mice. Blood Adv (2018) 2(6):638–48. doi: 10.1182/bloodadvances.2017011817
168. Liverani E, Rico MC, Yaratha L, Tsygankov AY, Kilpatrick LE, Kunapuli SP. LPS–induced systemic inflammation is more severe in P2Y12 null mice. J Leukoc Biol (2014) 95(2):313–23. doi: 10.1189/jlb.1012518
169. Gil-Bernabé AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of monocytes/macrophages by tissue factor–mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood (2012) 119(13):3164–75. doi: 10.1182/blood-2011-08-376426
170. Läubli H, Spanaus KS, Borsig L. Selectin–mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood (2009) 114(20):4583–91. doi: 10.1182/blood-2008-10-186585
171. Noy R, Pollard JW. Tumor–associated macrophages: from mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
172. Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, et al. Blockade of a chemokine, CCL2, reduces chronic colitis–associated carcinogenesis in mice. Cancer Res (2009) 69(19):7884–92. doi: 10.1158/0008-5472.CAN-09-1451
173. Fox JM, Kausar F, Day A, Osborne M, Hussain K, Mueller A, et al. CXCL4/Platelet factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci Rep (2018) 8(1):9466. doi: 10.1038/s41598-018-27710-9
174. Schwartzkopff F, Petersen F, Grimm TA, Brandt E. CXC chemokine ligand 4 (CXCL4) down–regulates CC chemokine receptor expression on human monocytes. Innate Immun (2012) 18(1):124–39. doi: 10.1177/1753425910388833
175. Ando Y, Oku T, Tsuji T. Platelets attenuate production of cytokines and nitric oxide by macrophages in response to bacterial endotoxin. Platelets (2016) 27(4):344–50. doi: 10.3109/09537104.2015.1103369
176. Linke B, Schreiber Y, Picard-Willems B, Slattery P, Nüsing RM, Harder S, et al. Activated platelets induce an anti–inflammatory response of Monocytes/Macrophages through cross–regulation of PGE(2) and cytokines. Mediators Inflamm 2017 (2017) p:1463216. doi: 10.1155/2017/1463216
177. Bournazos S, Rennie J, Hart SP, Dransfield I. Choice of anticoagulant critically affects measurement of circulating platelet–leukocyte complexes. Arterioscler Thromb Vasc Biol (2008) 28(1):e2–3. doi: 10.1161/ATVBAHA.107.153387
178. Takeda Y, Marumo M, Nara H, Feng ZG, Asao H, Wakabayashi I. Selective induction of anti–inflammatory monocyte–platelet aggregates in a model of pulsatile blood flow at low shear rates. Platelets (2016) 27(6):583–92. doi: 10.3109/09537104.2016.1153616
179. Zaldivia MT, Rivera J, Hering D, Marusic P, Sata Y, Lim B, et al. Renal denervation reduces monocyte activation and monocyte–platelet aggregate formation: An anti–inflammatory effect relevant for cardiovascular risk. Hypertension (2017) 69(2):323–31. doi: 10.1161/HYPERTENSIONAHA.116.08373
180. Liang H, Duan Z, Li D, Li D, Wang Z, Ren L, et al. Higher levels of circulating monocyte–platelet aggregates are correlated with viremia and increased sCD163 levels in HIV–1 infection. Cell Mol Immunol (2015) 12(4):435–43. doi: 10.1038/cmi.2014.66
181. Armstrong PC, Kirkby NS, Chan MV, Finsterbusch M, Hogg N, Nourshargh S, et al. Novel whole blood assay for phenotyping platelet reactivity in mice identifies ICAM–1 as a mediator of platelet–monocyte interaction. Blood (2015) 126(10):e11–8. doi: 10.1182/blood-2015-01-621656
182. Schulz C, von Brühl ML, Barocke V, Cullen P, Mayer K, Okrojek R, et al. EMMPRIN (CD147/basigin) mediates platelet–monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost (2011) 9(5):1007–19. doi: 10.1111/j.1538-7836.2011.04235.x
183. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PloS Biol (2016) 14(5):e1002459. doi: 10.1371/journal.pbio.1002459
184. Gawaz M, Neumann FJ, Dickfeld T, Koch W, Laugwitz KL, Adelsberger H, et al. Activated platelets induce monocyte chemotactic protein–1 secretion and surface expression of intercellular adhesion molecule–1 on endothelial cells. Circulation (1998) 98(12):1164–71. doi: 10.1161/01.CIR.98.12.1164
185. Shantsila E, Lip GY. The role of monocytes in thrombotic disorders. insights from tissue factor, monocyte–platelet aggregates and novel mechanisms. Thromb Haemost (2009) 102(5):916–24. doi: 10.1160/TH09-01-0023
186. Ivanov II, Apta BHR, Bonna AM, Harper MT. Platelet p–selectin triggers rapid surface exposure of tissue factor in monocytes. Sci Rep (2019) 9(1):13397. doi: 10.1038/s41598-019-49635-7
187. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with mac–1. Circulation (2007) 115(12):1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201
188. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
189. Arnold CE, Gordon P, Barker RN, Wilson HM. The activation status of human macrophages presenting antigen determines the efficiency of Th17 responses. Immunobiology (2015) 220(1):10–9. doi: 10.1016/j.imbio.2014.09.022
190. Utomo L, Bastiaansen-Jenniskens YM, Verhaar JA, van Osch GJ. Cartilage inflammation and degeneration is enhanced by pro–inflammatory (M1) macrophages in vitro, but not inhibited directly by anti–inflammatory (M2) macrophages. Osteoarthritis Cartilage (2016) 24(12):2162–70. doi: 10.1016/j.joca.2016.07.018
191. Lee HW, Choi HJ, Ha SJ, Lee KT, Kwon YG. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim Biophys Acta (2013) 1835(2):170–9. doi: 10.1016/j.bbcan.2012.12.007
192. Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti–tumor macrophages in tumor microenvironment. Cell Immunol (2017) 316:1–10. doi: 10.1016/j.cellimm.2017.04.005
193. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
194. Zhang S, Chuah SJ, Lai RC, Hui JHP, Lim SK, Toh WS. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials (2018) 156:16–27. doi: 10.1016/j.biomaterials.2017.11.028
195. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer (2017) 117(11):1583–91. doi: 10.1038/bjc.2017.356
196. Carestia A, Mena HA, Olexen CM, Ortiz Wilczyñski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro–inflammatory phenotype and increase survival of septic mice. Cell Rep (2019) 28(4):896–908.e5. doi: 10.1016/j.celrep.2019.06.062
197. Lee SJ, Yoon BR, Kim HY, Yoo SJ, Kang SW, Lee WW, et al. Activated platelets convert CD14(+)CD16(–) into CD14(+)CD16(+) monocytes with enhanced FcγR–mediated phagocytosis and skewed M2 polarization. Front Immunol (2020) 11:611133. doi: 10.3389/fimmu.2020.611133
198. Li H, Hicks JJ, Wang L, Oyster N, Philippon MJ, Hurwitz S, et al. Customized platelet–rich plasma with transforming growth factor β1 neutralization antibody to reduce fibrosis in skeletal muscle. Biomaterials (2016) 87:147–56. doi: 10.1016/j.biomaterials.2016.02.017
199. Okizaki S, Ito Y, Hosono K, Oba K, Ohkubo H, Amano H, et al. Suppressed recruitment of alternatively activated macrophages reduces TGF–β1 and impairs wound healing in streptozotocin–induced diabetic mice. BioMed Pharmacother (2015) 70:317–25. doi: 10.1016/j.biopha.2014.10.020
200. Kim R, Song BW, Kim M, Kim WJ, Lee HW, Lee MY, et al. Regulation of alternative macrophage activation by MSCs derived hypoxic conditioned medium, via the TGF–β1/Smad3 pathway. BMB Rep (2020) 53(11):600–4. doi: 10.5483/BMBRep.2020.53.11.177
201. Saha B, Kodys K, Szabo G. Hepatitis c virus–induced monocyte differentiation into polarized M2 macrophages promotes stellate cell activation via TGF–β. Cell Mol Gastroenterol Hepatol (2016) 2(3):302–316.e8. doi: 10.1016/j.jcmgh.2015.12.005
202. Li Z, Xiao J, Xu X, Li W, Zhong R, Qi L, et al. M–CSF, IL–6 and TGF–β promote generation of a new subset of tissue repair macrophage for traumatic brain injury recovery. Sci Adv (2021) 7(11):eabb6260. doi: 10.1126/sciadv.abb6260
203. Qin S, Zheng JH, Xia ZH, Qian J, Deng CL, Yang SL. CTHRC1 promotes wound repair by increasing M2 macrophages via regulating the TGF–β and notch pathways. BioMed Pharmacother (2019) 113:108594. doi: 10.1016/j.biopha.2019.01.055
204. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett (2008) 267(2):204–15. doi: 10.1016/j.canlet.2008.03.028
205. Dwyer AR, Greenland EL, Pixley FJ. Promotion of tumor invasion by tumor–associated macrophages: The role of CSF–1–Activated phosphatidylinositol 3 kinase and src family kinase motility signaling. Cancers (Basel) (2017) 9(6):68. doi: 10.3390/cancers9060068
206. Pierre S, Linke B, Suo J, Tarighi N, Del Turco D, Thomas D, et al. GPVI and thromboxane receptor on platelets promote proinflammatory macrophage phenotypes during cutaneous inflammation. J Invest Dermatol (2017) 137(3):686–95. doi: 10.1016/j.jid.2016.09.036
207. Dütting S, Bender M, Nieswandt B. Platelet GPVI: a target for antithrombotic therapy? Trends Pharmacol Sci (2012) 33(11):583–90. doi: 10.1016/j.tips.2012.07.004
208. Hubertus K, Mischnik M, Timmer J, Herterich S, Mark R, Moulard M, et al. Reciprocal regulation of human platelet function by endogenous prostanoids and through multiple prostanoid receptors. Eur J Pharmacol (2014) 740:15–27. doi: 10.1016/j.ejphar.2014.06.030
209. Waehre T, Damås JK, Yndestad A, Taskén K, Pedersen TM, Smith C, et al. Effect of activated platelets on expression of cytokines in peripheral blood mononuclear cells – potential role of prostaglandin E2. Thromb Haemost (2004) 92(6):1358–67. doi: 10.1160/TH04-03-0146
210. Siasos G, Tousoulis D, Kioufis S, Oikonomou E, Siasou Z, Limperi M, et al. Inflammatory mechanisms in atherosclerosis: the impact of matrix metalloproteinases. Curr Top Med Chem (2012) 12(10):1132–48. doi: 10.2174/1568026611208011132
211. Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of e–selectin is induced by the platelet–specific chemokine platelet factor 4 through LRP in an NF–kappaB–dependent manner. Blood (2005) 105(9):3545–51. doi: 10.1182/blood-2004-07-2617
212. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor–associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology (2013) 2(12):e26968. doi: 10.4161/onci.26968
213. Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor–induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol (2012) 227(1):17–28. doi: 10.1002/path.3989
214. Johnson KE, Wilgus TA. Multiple roles for VEGF in non–melanoma skin cancer: angiogenesis and beyond. J Skin Cancer 2012 (2012) p:483439. doi: 10.1155/2012/483439
215. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte–derived macrophages. J Immunol (2010) 184(9):4810–8. doi: 10.4049/jimmunol.0901368
216. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol (2012) 30:1–22. doi: 10.1146/annurev-immunol-100311-102839
217. Czapiga M, Kirk AD, Lekstrom–Himes J. Platelets deliver costimulatory signals to antigen–presenting cells: a potential bridge between injury and immune activation. Exp Hematol (2004) 32(2):135–9. doi: 10.1016/j.exphem.2003.11.004
218. Langer HF, Daub K, Braun G, Schönberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells via mac–1/JAM–C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol (2007) 27(6):1463–70. doi: 10.1161/ATVBAHA.107.141515
219. Nording H, Sauter M, Lin C, Steubing R, Geisler S, Sun Y, et al. Activated platelets upregulate β(2) integrin mac–1 (CD11b/CD18) on dendritic cells, which mediates heterotypic cell–cell interaction. J Immunol (2022) 208(7):1729–41. doi: 10.4049/jimmunol.2100557
220. Strobl H, Knapp W. TGF–beta1 regulation of dendritic cells. Microbes Infect (1999) 1(15):1283–90. doi: 10.1016/S1286-4579(99)00256-7
221. Huang Q, Hu X, He W, Zhao Y, Hao S, Wu Q, et al. Fluid shear stress and tumor metastasis. Am J Cancer Res (2018) 8(5):763–77.
222. Katoh N, Soga F, Nara T, Tamagawa-Mineoka R, Nin M, Kotani H, et al. Effect of serotonin on the differentiation of human monocytes into dendritic cells. Clin Exp Immunol (2006) 146(2):354–61. doi: 10.1111/j.1365-2249.2006.03197.x
223. Xia CQ, Kao KJ. Effect of CXC chemokine platelet factor 4 on differentiation and function of monocyte–derived dendritic cells. Int Immunol (2003) 15(8):1007–15. doi: 10.1093/intimm/dxg100
224. Silva-Cardoso SC, Affandi AJ, Spel L, Cossu M, van Roon JAG, Boes M, et al. CXCL4 exposure potentiates TLR–driven polarization of human monocyte–derived dendritic cells and increases stimulation of T cells. J Immunol (2017) 199(1):253–62. doi: 10.4049/jimmunol.1602020
225. Saris A, Steuten J, Schrijver DP, van Schijndel G, Zwaginga JJ, van Ham SM, et al. Inhibition of dendritic cell activation and modulation of T cell polarization by the platelet secretome. Front Immunol (2021) 12:631285. doi: 10.3389/fimmu.2021.631285
226. Tešić N, Pekle Simonič I, Roškar K, Rožman P, Švajger U. Dendritic cells generated in the presence of platelet lysate have a reduced type 1 polarization capacity. Immunol Invest (2020) 49(3):215–31. doi: 10.1080/08820139.2019.1624768
227. Blair P, Flaumenhaft R. Platelet alpha–granules: basic biology and clinical correlates. Blood Rev (2009) 23(4):177–89. doi: 10.1016/j.blre.2009.04.001
228. Yun SH, Sim EH, Goh RY, Park JI, Han JY. Platelet activation: The mechanisms and potential biomarkers. BioMed Res Int 2016 (2016) p:9060143. doi: 10.1155/2016/9060143
229. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med (2017) 4:48. doi: 10.3389/fcvm.2017.00048
230. Levite M. Dopamine and T cells: dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases. Acta Physiol (Oxf) (2016) 216(1):42–89. doi: 10.1111/apha.12476
231. Xu XR, Carrim N, Neves MA, McKeown T, Stratton TW, Coelho RM, et al. Platelets and platelet adhesion molecules: novel mechanisms of thrombosis and anti–thrombotic therapies. Thromb J (2016) 14(Suppl 1):29. doi: 10.1186/s12959-016-0100-6
232. Starossom SC, Veremeyko T, Yung AW, Dukhinova M, Au C, Lau AY, et al. Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ Res (2015) 117(9):779–92. doi: 10.1161/CIRCRESAHA.115.306847
233. Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe (2008) 4(2):179–87. doi: 10.1016/j.chom.2008.07.003
234. Danese S, de la Motte C, Reyes BM, Sans M, Levine AD, Fiocchi C. Cutting edge: T cells trigger CD40–dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J Immunol (2004) 172(4):2011–5. doi: 10.4049/jimmunol.172.4.2011
235. Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, et al. Platelets regulate CD4+ T–cell differentiation via multiple chemokines in humans. Thromb Haemost (2011) 106(2):353–62. doi: 10.1160/TH11-01-0020
236. León–Ponte M, Ahern GP, O'Connell PJ. Serotonin provides an accessory signal to enhance T–cell activation by signaling through the 5–HT7 receptor. Blood (2007) 109(8):3139–46. doi: 10.1182/blood-2006-10-052787
237. O'Connell PJ, Wang X, Leon-Ponte M, Griffiths C, Pingle SC, Ahern GP. A novel form of immune signaling revealed by transmission of the inflammatory mediator serotonin between dendritic cells and T cells. Blood (2006) 107(3):1010–7. doi: 10.1182/blood-2005-07-2903
238. Ponomarev ED. Fresh evidence for platelets as neuronal and innate immune cells: Their role in the activation, differentiation, and deactivation of Th1, Th17, and tregs during tissue inflammation. Front Immunol (2018) 9:406. doi: 10.3389/fimmu.2018.00406
239. Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP–TGFβ axis. Sci Immunol (2017) 2(11):eaai7911. doi: 10.1126/sciimmunol.aai7911
240. Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFβ. Mol Biol Cell (2012) 23(6):1129–39. doi: 10.1091/mbc.e11-12-1018
241. Cuende J, Liénart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, et al. Monoclonal antibodies against GARP/TGF–β1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med (2015) 7(284):284ra56. doi: 10.1126/scitranslmed.aaa1983
242. Stockis J, Dedobbeleer O, Lucas S. Role of GARP in the activation of latent TGF–β1. Mol Biosyst (2017) 13(10):1925–35. doi: 10.1039/C7MB00251C
243. Metelli A, Wu BX, Fugle CW, Rachidi S, Sun S, Zhang Y, et al. Surface expression of TGFβ docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res (2016) 76(24):7106–17. doi: 10.1158/0008-5472.CAN-16-1456
244. Meyer-Martin H, Hahn SA, Beckert H, Belz C, Heinz A, Jonuleit H, et al. GARP inhibits allergic airway inflammation in a humanized mouse model. Allergy (2016) 71(9):1274–83. doi: 10.1111/all.12883
245. Hahn SA, Stahl HF, Becker C, Correll A, Schneider FJ, Tuettenberg A, et al. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4+ T cells. Blood (2013) 122(7):1182–91. doi: 10.1182/blood-2012-12-474478
246. Metelli A, Salem M, Wallace CH, Wu BX, Li A, Li X, et al. Immunoregulatory functions and the therapeutic implications of GARP–TGF–β in inflammation and cancer. J Hematol Oncol (2018) 11(1):24. doi: 10.1186/s13045-018-0570-z
247. Dinkla S, van Cranenbroek B, van der Heijden WA, He X, Wallbrecher R, Dumitriu IE, et al. Platelet microparticles inhibit IL–17 production by regulatory T cells through p–selectin. Blood (2016) 127(16):1976–86. doi: 10.1182/blood-2015-04-640300
248. Sotnikov I, Veremeyko T, Starossom SC, Barteneva N, Weiner HL, Ponomarev ED. Platelets recognize brain–specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PloS One (2013) 8(3):e58979. doi: 10.1371/journal.pone.0058979
249. Joseph R, Soundararajan R, Vasaikar S, Yang F, Allton KL, Tian L, et al. CD8(+) T cells inhibit metastasis and CXCL4 regulates its function. Br J Cancer (2021) 125(2):176–89. doi: 10.1038/s41416-021-01338-5
250. Rosenberg SA. Decade in review–cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol (2014) 11(11):630–2. doi: 10.1038/nrclinonc.2014.174
251. Razavi AS, Mohtashami M, Razi S, Rezaei N. TGF–β signaling and the interaction between platelets and T–cells in tumor microenvironment: Foes or friends? Cytokine (2022) 150:155772. doi: 10.1016/j.cyto.2021.155772
252. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, et al. Platelets present antigen in the context of MHC class I. J Immunol (2012) 189(2):916–23. doi: 10.4049/jimmunol.1200580
253. Meikle CK, Meisler AJ, Bird CM, Jeffries JA, Azeem N, Garg P, et al. Platelet–T cell aggregates in lung cancer patients: Implications for thrombosis. PloS One (2020) 15(8):e0236966. doi: 10.1371/journal.pone.0236966
254. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogné M, Richard Y, et al. Human platelets can activate peripheral blood b cells and increase production of immunoglobulins. Exp Hematol (2007) 35(9):1376–87. doi: 10.1016/j.exphem.2007.05.021
255. Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase a from an NFkappaB–IkappaB complex. J Biol Chem (2010) 285(24):18352–63. doi: 10.1074/jbc.M109.077602
256. Fuentes E, Rojas A, Palomo I. NF–κB signaling pathway as target for antiplatelet activity. Blood Rev (2016) 30(4):309–15. doi: 10.1016/j.blre.2016.03.002
257. Hu H, Zhu L, Huang Z, Ji Q, Chatterjee M, Zhang W, et al. Platelets enhance lymphocyte adhesion and infiltration into arterial thrombus. Thromb Haemost (2010) 104(6):1184–92. doi: 10.1160/TH10-05-0308
258. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell (2017) 32(2):135–54. doi: 10.1016/j.ccell.2017.06.009
259. Gasser S, Raulet DH. Activation and self–tolerance of natural killer cells. Immunol Rev (2006) 214:130–42. doi: 10.1111/j.1600-065X.2006.00460.x
260. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science (1999) 285(5428):730–2. doi: 10.1126/science.285.5428.730
261. Paoletti C, Hayes DF. Circulating tumor cells. Adv Exp Med Biol (2016) 882:235–58. doi: 10.1007/978-3-319-22909-6_10
262. Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK cell development, function, and residence. Cell (2020) 180(4):749–763.e13. doi: 10.1016/j.cell.2020.01.022
263. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease–activated receptors, and fibrinogen in hematogenous metastasis. Blood (2004) 104(2):397–401. doi: 10.1182/blood-2004-02-0434
264. Bakewell SJ, Nestor P, Prasad S, Tomasson MH, Dowland N, Mehrotra M, et al. Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proc Natl Acad Sci U.S.A. (2003) 100(24):14205–10. doi: 10.1073/pnas.2234372100
265. Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol (2013) 78(2):120–9. doi: 10.1111/sji.12072
266. Salih HR, Rammensee HG, Steinle A. Cutting edge: down–regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169(8):4098–102. doi: 10.4049/jimmunol.169.8.4098
267. Groh V, Wu J, Yee C, Spies T. Tumour–derived soluble MIC ligands impair expression of NKG2D and T–cell activation. Nature (2002) 419(6908):734–8. doi: 10.1038/nature01112
268. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
269. Colciaghi F, Marcello E, Borroni B, Zimmermann M, Caltagirone C, Cattabeni F, et al. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology (2004) 62(3):498–501. doi: 10.1212/01.WNL.0000106953.49802.9C
270. Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, et al. Tumor necrosis factor–alpha–converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res (2004) 95(7):677–83. doi: 10.1161/01.RES.0000143899.73453.11
271. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor–associated MICA is shed by ADAM proteases. Cancer Res (2008) 68(15):6368–76. doi: 10.1158/0008-5472.CAN-07-6768
272. Boutet P, Agüera-González S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, et al. Cutting edge: the metalloproteinase ADAM17/TNF–alpha–converting enzyme regulates proteolytic shedding of the MHC class I–related chain b protein. J Immunol (2009) 182(1):49–53. doi: 10.4049/jimmunol.182.1.49
273. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Fürst D, et al. Shedding of endogenous MHC class I–related chain molecules a and b from different human tumor entities: heterogeneous involvement of the "a disintegrin and metalloproteases" 10 and 17. Int J Cancer (2013) 133(7):1557–66. doi: 10.1002/ijc.28174
274. Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma–initiating cells. Neuro Oncol (2014) 16(3):382–91. doi: 10.1093/neuonc/not232
275. Maurer S, Kropp KN, Klein G, Steinle A, Haen SP, Walz JS, et al. Platelet–mediated shedding of NKG2D ligands impairs NK cell immune–surveillance of tumor cells. Oncoimmunology (2018) 7(2):e1364827. doi: 10.1080/2162402X.2017.1364827
276. Kopp HG, Placke T, Salih HR. Platelet–derived transforming growth factor–beta down–regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res (2009) 69(19):7775–83. doi: 10.1158/0008-5472.CAN-09-2123
277. Nowbakht P, Ionescu MC, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for natural killer cell–activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood (2005) 105(9):3615–22. doi: 10.1182/blood-2004-07-2585
278. Krüger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: An update. Int J Mol Sci (2019) 20(18):4411. doi: 10.3390/ijms20184411
279. Wolinsky H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ Res (1980) 47(3):301–11. doi: 10.1161/01.RES.47.3.301
280. Mackman N. Triggers, targets and treatments for thrombosis. Nature (2008) 451(7181):914–8. doi: 10.1038/nature06797
281. Vermeersch E, Denorme F, Maes W, De Meyer SF, Vanhoorelbeke K, Edwards J, et al. The role of platelet and endothelial GARP in thrombosis and hemostasis. PloS One (2017) 12(3):e0173329. doi: 10.1371/journal.pone.0173329
282. Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci (2004) 117(Pt 16):3415–25. doi: 10.1242/jcs.01325
283. Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost (2003) 1(7):1335–42. doi: 10.1046/j.1538-7836.2003.00260.x
284. Kalagara T, Moutsis T, Yang Y, Pappelbaum KI, Farken A, Cladder-Micus L, et al. The endothelial glycocalyx anchors von willebrand factor fibers to the vascular endothelium. Blood Adv (2018) 2(18):2347–57. doi: 10.1182/bloodadvances.2017013995
285. Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiol (Bethesda) (2017) 32(2):162–77. doi: 10.1152/physiol.00020.2016
286. Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von willebrand factor strings under fluid shear stress. Blood (2009) 113(7):1589–97. doi: 10.1182/blood-2008-05-158584
287. Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin–initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest (2007) 117(4):953–60. doi: 10.1172/JCI30537
288. Peyvandi F, Garagiola I, Baronciani L. Role of von willebrand factor in the haemostasis. Blood Transfus (2011) 9 Suppl 2(Suppl 2):s3–8. doi: 10.2450/2011.002S
289. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood (2013) 121(6):1008–15. doi: 10.1182/blood-2012-06-437392
290. Goto S, Tamura N, Handa S, Arai M, Kodama K, Takayama H. Involvement of glycoprotein VI in platelet thrombus formation on both collagen and von willebrand factor surfaces under flow conditions. Circulation (2002) 106(2):266–72. doi: 10.1161/01.CIR.0000021427.87256.7E
291. Agarwal KC, Clarke E, Rounds S, Parks RE Jr, Huzoor A. Platelet–activating factor (PAF)–induced platelet aggregation. modulation by plasma adenosine and methylxanthines. Biochem Pharmacol (1994) 48(10):1909–16. doi: 10.1016/0006-2952(94)90589-4
292. Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, et al. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res (2002) 91(1):25–31. doi: 10.1161/01.RES.0000026420.22406.79
293. Italiano JE Jr., Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro– and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood (2008) 111(3):1227–33. doi: 10.1182/blood-2007-09-113837
294. Gao Y, Childers EP, Becker ML. L–Leucine–Based poly(ester urea)s for vascular tissue engineering. ACS Biomater Sci Eng (2015) 1(9):795–804. doi: 10.1021/acsbiomaterials.5b00168
295. Kieb M, Sander F, Prinz C, Adam S, Mau-Möller A, Bader R, et al. Platelet–rich plasma powder: A new preparation method for the standardization of growth factor concentrations. Am J Sports Med (2017) 45(4):954–60. doi: 10.1177/0363546516674475
Keywords: platelets, immune cells, interaction, tumorigenesis, tumor development
Citation: Li Y, Wang H, Zhao Z, Yang Y, Meng Z and Qin L (2023) Effects of the interactions between platelets with other cells in tumor growth and progression. Front. Immunol. 14:1165989. doi: 10.3389/fimmu.2023.1165989
Received: 14 February 2023; Accepted: 31 March 2023;
Published: 17 April 2023.
Edited by:
Mariana Aris, FUCA, ArgentinaReviewed by:
Gloria Riitano, Department of Experimental Medicine, Sapienza University of Rome, ItalyAlexander Zaslavsky, Department of Urology, University of Michigan, United States
Copyright © 2023 Li, Wang, Zhao, Yang, Meng and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyan Wang, d2h5X3BoZEAxNjMuY29t