 Jeffrey A. Bluestone
Jeffrey A. Bluestone Brent S. McKenzie
Brent S. McKenzie- Sonoma Biotherapeutics, South San Francisco, CA, United States
Regulatory T (Treg) cells are essential for maintaining peripheral tolerance, preventing autoimmunity, and limiting chronic inflammatory diseases. This small CD4+ T cell population can develop in the thymus and in the peripheral tissues of the immune system through the expression of an epigenetically stabilized transcription factor, FOXP3. Treg cells mediate their tolerogenic effects using multiple modes of action, including the production of inhibitory cytokines, cytokine starvation of T effector (e.g., IL-2), Teff suppression by metabolic disruption, and modulation of antigen-presenting cell maturation or function. These activities together result in the broad control of various immune cell subsets, leading to the suppression of cell activation/expansion and effector functions. Moreover, these cells can facilitate tissue repair to complement their suppressive effects. In recent years, there has been an effort to harness Treg cells as a new therapeutic approach to treat autoimmune and other immunological diseases and, importantly, to re-establish tolerance. Recent synthetic biological advances have enabled the cells to be genetically engineered to achieve tolerance and antigen-specific immune suppression by increasing their specific activity, stability, and efficacy. These cells are now being tested in clinical trials. In this review, we highlight both the advances and the challenges in this arena, focusing on the efforts to develop this new pillar of medicine to treat and cure a variety of diseases.
1 History and biology of T regulatory cells
The demonstration of immune tolerance dates to Owens, Burnet, Medawar, and others in the 1950s, including the awarding of a Nobel Prize in 1960 (reviewed in (1). Importantly, over the next two decades, tolerance was shown to be transferable with leukocytes from tolerant animals, suggesting that a specialized cell population was responsible. Although the concept of suppressive T cells was around for many decades starting with the work of Kondo and Gershon (2), the field was littered with phenomenology, lack of reliable molecular markers of the suppressor cell population, and conflicting data; yet, studies by Nishizuka and colleagues showed that mice thymectomized at 3 days after birth achieved systemic organ-specific autoimmunity (3). Critically, these diseases could be prevented or even reversed with the adoptive transfer of CD4+CD25+ T cells (4). This was reminiscent of the Medawar work demonstrating the importance of tolerance induction early in life and implicated a small subset of T cells in the biology.
In the late 1990s, studies of a mouse strain with multi-system autoimmunity (scurfy mice) led to the identification of the transcription factor Foxp3 (5). The scurfy mouse, which was originally developed at Oak Ridge National Labs during the Manhattan (atomic bomb) project, displayed many of the same attributes of the disease seen in neonatally thymectomized mice (6). Furthermore, a disease in male children—immune-mediated, polyendocrinopathy, X-linked—resembled the pathology found in scurfy mice, and it was shown that these patients also possessed mutations in FOXP3 (7). Further studies by multiple groups demonstrated that FOXP3 is expressed primarily in the CD4+CD25+ subset of T cells that were shown to be capable of controlling autoimmunity in mice (8–10). Thus, by the mid-2000s, T regulatory (Treg) cells could be defined at the molecular level in both mice and humans, and their critical role in maintaining immunological tolerance was unequivocally established.
Over the last 20 years, immunological tolerance has been linked to multiple mechanisms (1, 11). However, thymically derived Treg cells represent the dominant mechanism for maintaining normal immune homeostasis and preventing fatal multi-organ autoimmunity. These cells possess a broad repertoire of self-reactivity and are found in every tissue of the body. Preclinical studies have demonstrated that Treg cells from the periphery can traffic to many tissues and can treat many different models of autoimmune disease, and their persistence within the tissue depends on the presence of self-antigens and commensal bacteria (12–14). Importantly, there is strong evidence that the local tissue and the infiltrating Tregs have a selective activity for proteins present in the specific inflamed tissue and, in some instances, the local microbiota (15–17). While Treg cells from the blood are primarily naïve-like cells, tissue-resident Tregs are “tuned” to the specific environment within a given tissue. They can display different chemokine receptors, transcription factors, and metabolic properties.
In addition to the dominant population of thymically derived FOXP3+ Treg cells, in the periphery, factors such as TGFβ can convert conventional T cells to FOXP3+ Tregs, especially in the gut (18). Such peripherally “induced” treg cells are not as clearly defined at the molecular level as the thymically derived FOXP3+ Treg cells, and in pre-clinical models, they do not appear to be as stable (19, 20). In this regard, Dijke et al. performed an examination using discarded human thymuses as a source of therapeutic Tregs. They concluded that thymic Tregs are superior to Tregs derived from peripheral or cord blood (21).
There are multiple mechanisms by which Treg cells have been shown to inhibit immune function (22, 23). Tregs directly suppress antigen presentation directly through the engagement of checkpoint receptors, CTLA-4, and stripping major histocompatibility complex proteins of the cell surface of antigen-presenting cells (24). In addition, Tregs function indirectly and mediate bystander suppression through the production of suppressive cytokines (including IL-10, IL-35, and TGFβ), consumption of T cell growth factors (including IL-2), and altering the local metabolic environment to limit Teff cell activity (11). It is important to note that Treg activation is dependent on activation through the T cell receptor and CD28 engagement as a co-stimulatory signal. In fact, the ability to respond to IL-2 is also TCR/CD28 dependent, supporting a critical interplay of these signaling pathways in determining the ability of these cells to mediate bystander suppression (25–27). Finally, Tregs have also been shown to mediate what is called infectious tolerance (28–31). Gershon and Kondo were the first to coin the phrase “infectious tolerance” in the 1970s (2, 28). The term was based on the observation, by this group and others (29–31), that long-term tolerance to allogeneic skin grafts and other immune settings was induced by adoptively transferred Tregs but, once established, remained durable by the recruitment and development of other immunosuppressive cell populations. This ability of Tregs to promote other regulatory cells is likely to provide more robust and durable tolerance. In addition to inhibition of inflammation, Treg cells also promote the repair of damage by releasing tissue and stem cell factors (32, 33). The multifaceted activity of Treg cells provides a means to reduce inflammation with one cell type but via many different mechanisms, and thus Treg and Treg-friendly immune therapies have therapeutic potential in a variety of autoimmune, inflammatory, and transplant-related diseases.
One major concern with any potential tolerogenic therapy is the durability of the treatment. For most therapies, patients must take their medications on a daily, weekly, or monthly basis. In many instances, the inflammatory mechanisms can become resistant to the pathway modulators, leading to a continued reduction in original efficacy. Living drugs, such as Treg cell therapies, hold the prospect of a one-and-done therapy due to the potential of the drug to be long-lived—for instance, in the case of CAR-T cell therapies in cancer, June and colleagues have shown that the cells can survive for more than 10 years (34). However, it is yet to be shown that Tregs have the same longevity, that is, where infectious tolerance may be a distinct advantage. Several pre-clinical studies have shown that one consequence of the multifunctional activity of Tregs is their ability to recruit and influence other immune cells (such as myeloid and naive T cells) to differentiate into regulatory cell populations (MDSCs, Tr1 cells, and additional Tregs), leading to an amplification of the immunoregulation such that the long-term effects of Treg therapy can be durable. In the end, the depth and the breadth of the therapeutic effect of Tregs will be crucial as new modalities of treatments for people with chronic and debilitating diseases. These therapies must, by necessity, lead ultimately to more than a modest step-wise improvement to the standard of care. In addition, a combination of precision medicine using biomarkers to identify specific patient segments that are most likely to respond to the therapy, combined with an ability to take advantage of the multiple immune regulatory activities or even manipulate the cells to maximize the therapeutic impact, will be required to fulfil the promise of these new therapies.
2 The tolerogenic potential of Treg therapies
Treg cells provide unique opportunities for meaningful therapeutic differentiation that can be envisioned to tackle the complex pathobiologies that underlie most autoimmune and inflammatory diseases. In contrast to other approaches, Treg therapies have been shown to induce immune tolerance, which means the ability to treat for a short period of time resulting in long-term disease-free existence. Moreover, Tregs can exert their activities in both lymphoid and non-lymphoid tissues due to the multiple mechanisms that the cells use to control the immunological responses (35). Indeed, in a variety of preclinical studies, Treg adoptive immunotherapy has been shown to effectively ameliorate systemic inflammation and specific organ injury. These model systems include a wide variety of autoimmune models, including inflammatory bowel diseases (36), type 1 diabetes (T1D) (37), experimental autoimmune encephalomyelitis (EAE) (38), collagen-induced arthritis (39), as well as organ transplant rejection (40) and non-immune diseases, such as stroke (41) and amyotrophic lateral sclerosis (ALS) (42), where inflammation plays a role in disease pathogenesis. Thus, Treg therapies have the potential to significantly improve a variety of autoimmune and inflammatory disease activities given the broad applicability of the therapeutic approach.
However, given the bystander properties of Tregs and the recognition that organ-specific Tregs play a critical role in controlling autoimmunity in the local tissue environment, it was hypothesized that Tregs with single-antigen specificities might have an increased specific activity and a greater safety profile. In fact, data in multiple autoimmune (43) and transplant settings (44) showed that Treg cells expressing a single T cell receptor (TCR) suppressed autoreactive, multi-specific Teff cells and led to long-term tolerance. In fact, preliminary data in humans suggest that alloantigen-specific Treg cells can be effective in the transplantation setting (45). More recent studies have utilized new synthetic biology approaches to provide singular antigen specificity to Tregs, including the introduction of specific TCR (46) and chimeric antigen receptors (CAR) (47) to target specific inflamed tissues. These studies have shown not only efficacy in multiple pre-clinical models but also 10–25-fold increased activity when compared with polyclonal Treg populations. Thus, given the polypharmacy nature of Tregs, their activity in mechanistically different diseases, and their ability to be engineered, Treg therapeutics represent a new approach to creating deep and durable treatments.
3 Challenges and key features to be addressed in developing Treg therapeutics
3.1 Will Treg therapy promote broad immunosuppression?
The advantage of CAR-engineered Treg therapy is that it is directed to a single antigen that is not dependent on HLA presentation and thus can be focused on antigens expressed in inflamed tissues. In contrast, TCR-engineered Treg cells engage HLA peptide-expressing cells which can be expressed in both the draining lymph node and the inflammatory sites. Although there are concerns that broad immunosuppression might occur in the presence of an acute infection or even in the context of cancer, healthy individuals routinely combat pathogens and cancer in the presence of antigen-specific Treg activity. There are examples where the deletion of Tregs in pre-clinical models can improve effector immunity, but, in general, Tregs are part of the homeostatic balance of a healthy immune response (48, 49). Data in the NOD mouse model of T1D, in which Treg therapy prevents the autoimmune destruction of pancreatic beta cells, demonstrate that viral immunity is preserved. In adoptive Treg therapy for clinical bone marrow transplant, immunity toward the cancer is likewise maintained. In fact, studies have shown that Treg activity at sites of pathogenic infection can limit the overall tissue damage, and uncontrolled chronic inflammation can be a driver for cancer in several settings (50). Finally, in recent years, low-dose IL-2 and IL-2 muteins have been tested as therapeutics in a variety of clinical settings given their ability to expand Tregs in vivo (51). In many of these studies, there has been two- to fivefold increases in Treg numbers in the circulation, even over a long-term treatment regimen, yet there have been no reported increases in pathogen infections or cancer, suggesting that Tregs are not overly immunosuppressive in the context of infections. Finally, as discussed above, Tregs need to constantly engage antigens to survive—that is, without ongoing TCR and CD28 engagement, Tregs fail to persist. Thus, the current preclinical and human data suggest that Treg therapy does not inhibit the natural immunity to pathogens or cancer.
However, despite the early results suggesting that Tregs will be a safe therapy, the potential liability of a broad anti-inflammatory activity is also being addressed in a variety of ways. First, several of the companies are incorporating tags and kill switches in therapeutic Treg products that might allow the patient to be treated with clinically approved monoclonal antibodies and small molecules to eliminate the adoptively transferred Treg. Additionally, efforts are underway to create regulated CARs and TCRs that allow the specificity to be modulated in case of adverse effects of the treatment. In addition, as in the CAR-T space in cancer, the use of mRNA or short-lived DNA to deliver the specificity will lead to a transient expression of the CAR or TCR. This might be especially useful in conditions in which a severe pathology is driven in a large part by inflammation and, once resolved, does not require ongoing Treg persistence. Examples include acute respiratory distress syndrome (ARDS) or stroke, among others. Such settings would benefit from or perhaps require the development of an off-the-shelf Treg cell product that might be short-lived.
3.2 What will be the potential adverse events following Treg therapies?
Assuming that the immunosuppressive activity of Tregs can be harnessed effectively to inhibit a disease-specific inflammation, several other concerns must be addressed in considering a robust cell therapy. Most importantly, the Tregs must maintain their phenotype and function stably over time, even in the harshest inflammatory settings. There are some studies of mouse Tregs showing that, under certain inflammatory conditions, a subset of Treg cells can lose Foxp3 expression and produce proinflammatory cytokines (52, 53). However, other studies suggest that Tregs are quite stable (54). Similar conflicting results have been seen in patients with autoimmune diseases, usually at the site of inflammation. This has raised concerns related to cytokine release syndrome (CRS) as seen in some cancer cell therapy settings using CAR-T cells. However, in these settings, CRS primarily occurs in the context of hematological malignancies that carry large antigen loads within the blood. Moreover, in many settings (such as transplantation and autoimmunity), the patients are on other immunosuppressive drugs known to moderate the effector cytokine production. In fact, the human clinical data suggests that Treg therapy does not promote CRS in a fashion like that of CAR Teff cells. The stability of Tregs is maintained, and Treg function has been shown to reduce inflammation and have less infection events compared with immunosuppression. That said, as highlighted below, there are multiple academic and company efforts to ensure the stability of these cells (Table 1).
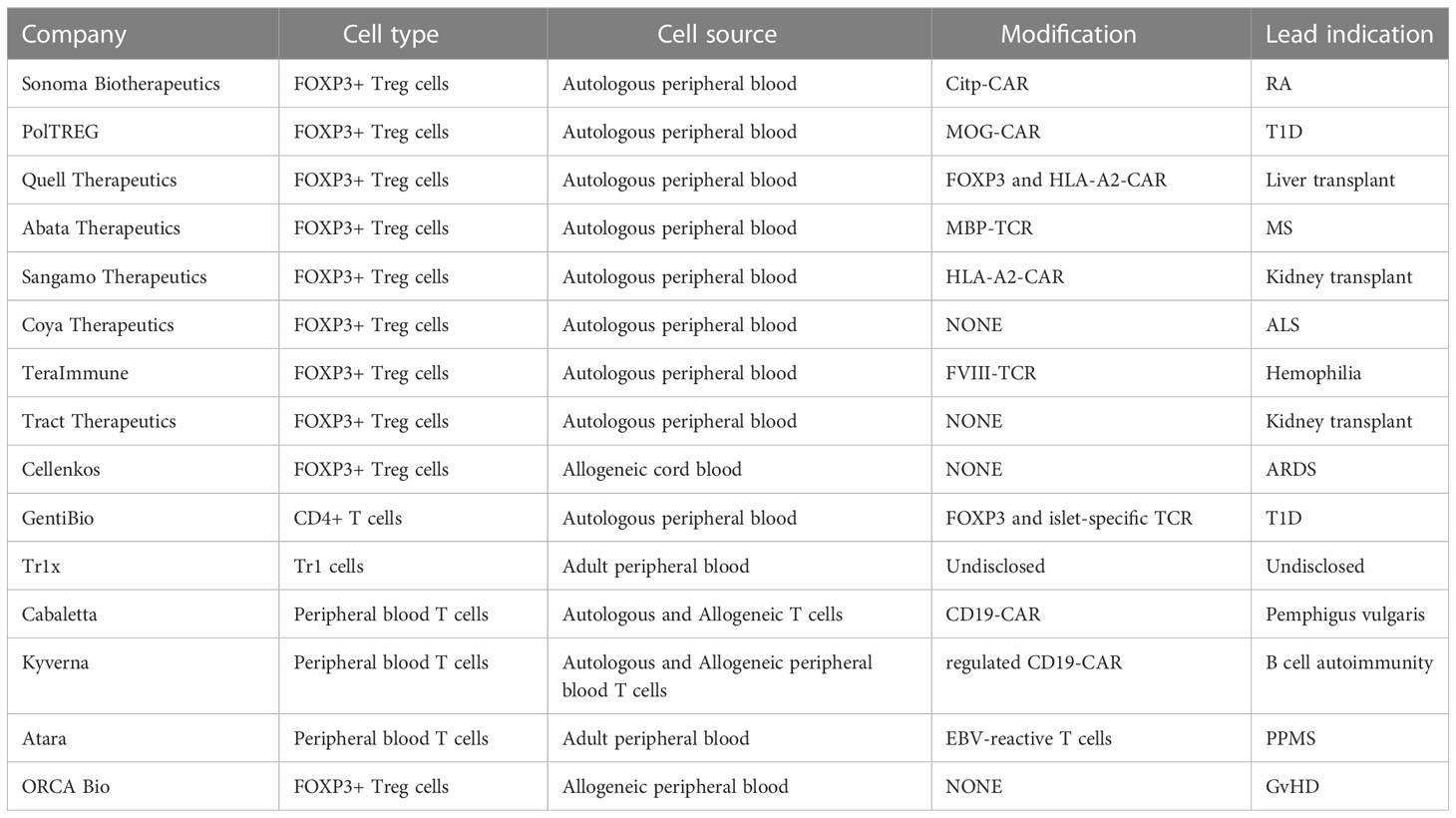
Table 1 Companies involved in regulatory cell adoptive immunotherapy.
4 Lessons learned from clinical trials
Over the last 15 years, there have been more than 25 early-stage clinical trials conducted, mostly in an academic setting, to investigate the safety and biological activity of Tregs in diseases ranging from autoimmunity, organ transplantation, graft versus host, and other inflammatory diseases. An overall summary of a small “subset” of clinical trials is found in Table 2. These trials have led to clear evidence that the therapy is feasible, can be administered safely, and is tolerated for a long term. To date, there have been no life-threatening adverse events, including no observed increase in infections or evidence of cancer development (either the administered cells or in the patients). In fact, as confidence in the therapy has grown, more than 10 companies have initiated Treg programs using both conventional and engineered Treg cells. The first industry-sponsored trials are underway, with efficacy studies on the horizon, to assess the therapeutic potential of these novel cell therapies. That said, there are key lessons that have been learned from past studies that inform future efforts and have created potential opportunities to improve the therapies.
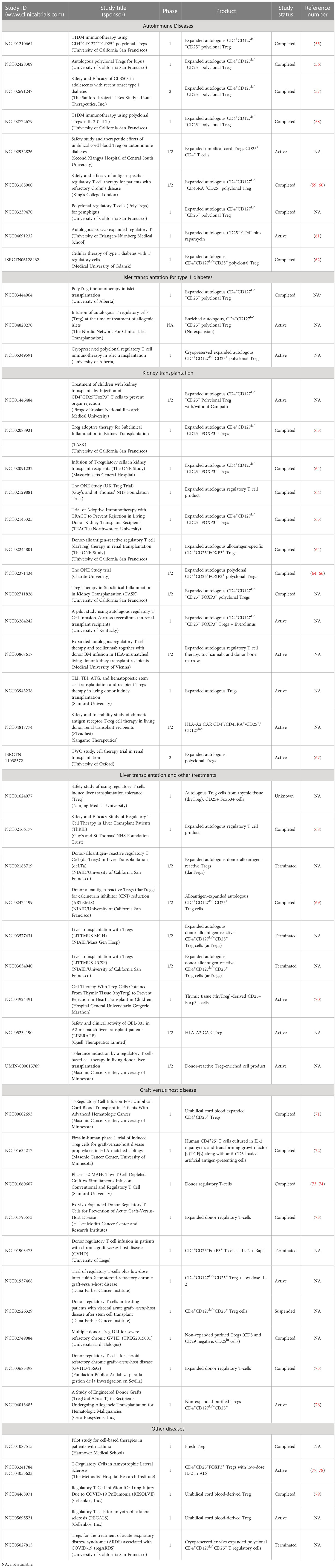
Table 2 Summary of a subset of clinical trials with Tregs in multiple diseases.
4.1 Experiences in Treg expansion
Fundamental research on Tregs demonstrated that the cells signal in a similar fashion as conventional T cells, requiring both a TCR and CD28 co-stimulatory signals for maximal expansion. In addition, Tregs have been shown to be exquisitely dependent on the IL-2 growth factor for both expansion and survival. Moreover, Tregs coming from individuals with autoimmune or other inflammatory diseases can be dysfunctional, leading to differences in expansion capabilities and potential phenotype changes, such as increased interferon production, when compared with Treg derived from healthy individuals. Thus, investigators employed similar tools as those used for T conventional cell expansion, namely, the use of anti-CD3/anti-CD28-coated beads in combination with IL-2 to expand Tregs. In previously published studies, the cells are routinely expanded 300- to 500-fold in 14 days. The purity of the expanded Treg populations is usually over 90% FOXP3+, and high levels of multiple phenotypic and functional markers are expressed, including CTLA-4, GITR, LAP, and CD25 (55, 80).
Most importantly, the expanded Tregs have equal or better phenotypic and functional activity when compared with freshly isolated Tregs (55). This is especially evident in Tregs isolated from patients with autoimmune diseases, where genetic and/or environmental influences often result in Tregs that are less effective in multiple suppressive activities and, in many cases, less stable as read out by reduced FOXP3, CD25, and CTLA-4 expression as well as less ability to respond to growth factors such as IL-2. However, the process of expansion generally leads to a population expressing higher levels of key functional markers and “repair” of functional defects. In fact, in early studies of IL-2 therapies, the repair of endogenous Treg’s inability to respond effectively to IL-2 was not only reversed but found to be stable even a year after the discontinuation of cytokine therapy (81). Most importantly, the expansion of the Tregs derived from peripheral blood as well as cord blood was highly stable when examined over time after Treg transfer into patients—for instance, an analysis of circulating adoptively transferred Treg cells demonstrated the continued expression of key functional and phenotypic markers for at least a year post-transfer (55). The results of an extensive bulk and single-cell T cell receptor analysis suggest that the expanded population of Tregs remains broad after expansion. Thus, it is not surprising that the current efforts to develop Treg populations for therapy have begun to employ novel genetic approaches to homogenize and maximize Treg phenotype and functional activity. This includes the use of antigen-specific receptors, including CARs or TCRs, to focus the tissue specificity of the Treg product.
4.2 Experiences in Treg stability in vitro and in vivo
Despite the previously described limited animal studies, to date, there is no evidence of Treg instability in any human clinical studies. This result may be due to multiple reasons. First, the expansion process is much more robust in human versus mouse, and the likely preferential expansion of thymus-derived CD45RA naïve Tregs using current approaches may selectively result in the expansion of the most stable Treg populations. In fact, the literature suggests that the majority of “unstable” Tregs are likely derived from induced peripheral Tregs, generated in the periphery in vivo upon encounter with TGFβ and other Treg-promoting cytokines (19, 22). Moreover, in vitro studies suggest that human Tregs isolated and expanded under the conditions noted above are “resistant” to certain inflammatory cytokines suspected of promoting instability in rodent Tregs—for instance, culturing human Tregs with IL-6 or TNFα (both present in highly inflamed tissues) routinely results in increased expansion and function, which are likely due to the upregulation of the TNFR2 receptor on the expanded Tregs (52, 82, 83). Finally, it cannot be ruled out that unstable Tregs are only present in tissues subject to extreme inflammatory milieu; however, as will be noted below, in several clinical settings, biopsy analysis post-transfer showed that Tregs in target tissues expressed higher levels of FOXP3 and CTLA-4 at 3 months post-transfer of Treg. Thus, it is likely that current expansion technologies result in the production of highly purified Treg populations that are stable in disease settings (including autoimmune lesions and inflamed transplanted organs).
At present, the majority of Treg products generated for clinical application are autologous. For autoimmune diseases, this means that the expansion protocol needs to consider the possibility of contaminated activated Teff cells, which express high levels of CD25 like Tregs, as well as genetically and environmentally induced Treg dysfunction. One approach to avoid these potential issues has been to use allogeneic Tregs derived from healthy individuals or umbilical cord blood (UCB) (72, 79). UCB Tregs are overwhelmingly naïve and have a very broad repertoire (84). In some cases, such as in the suppression of graft versus host disease, this has the added advantage of 5%–15% of the Tregs having alloreactivity and thus recognize the class II MHC of the host. However, there are multiple risks associated with this approach. First, should the Treg cells become unstable in patients, there is an expanded repertoire that can target the allogeneic tissue, be it host or transplanted tissues. Secondly, the host immune system can recognize and destroy the adoptively transferred Treg product. This was seemingly apparent in the first cord blood-derived Treg study in GvHD, where the cell product had a shorter duration in vivo when compared with autologous cells in other settings (71, 72). However, there are circumstances where an off-the-shelf allogeneic product may be required, such as in the treatment of acute ARDS, such as COVID, where the cells need to be administered immediately (79). As will be highlighted elsewhere, new approaches to both genetically engineered Tregs as well as potential stem cell-derived Treg products may enable the disruption of core allogeneic proteins, such as MHC antigens, and avoid rejection. Interestingly, the inherent alloreactivity of Tregs has been exploited in the organ and islet transplant setting, where investigators have either relied on the resident alloreactive Tregs in the polyclonal Treg population or developed approaches to selectively expand the alloreactive cells prior to transfer. This approach has great potential but may be superseded by the genetic engineering approaches described below.
It should be noted that, in some settings, the challenge of high purity and stability has required additional precautions in the development of Treg products—for instance, Teff cell contamination has been an issue due to inadequate separation technology and/or patient cell surface phenotype variation. As noted previously, preclinical studies suggest that peripherally derived Tregs, especially Helios- Tregs, are less stable than their thymus-derived counterparts. Thus, depending on the source of Tregs cells, the potential for Teff or Teff-like activity is possible. To avoid some of the challenges to isolating and expanding highly stable and pure Tregs, investigators have taken advantage of the selective ability of rapamycin to suppress effector T cell growth while preserving Treg expansion. The differential mechanistic role of mTORC1 and mTORC2 in Treg versus Teff cells results in the rapamycin-induced increased stability of Tregs, selectively inhibiting Teff cell expansion (85, 86). In the future, new genetic engineering approaches that are now being developed will both increase the ability to isolate highly pure populations of Tregs as well as provide safety switches and a more stable Treg population in settings where instability remains a risk.
4.3 Experiences in early phase 1/2 trials of Tregs
As noted in Table 2, there have been over 25 phase 1/2 clinical trials to test the efficacy of autologous and allogeneic polyclonal adult and UCB-derived Tregs in a variety of diseases. However, there have been only a limited number of presentations and no published phase 2 studies using polyclonal or antigen-specific engineered Tregs. In the case of the T1D study, NCT02691247, the company, Caladrius, released a press release saying that the trial did not meet its primary endpoint. In the case of the ALS study, recruitment was impacted by COVID-19, and no meaningful evaluation could be made. The early Treg studies were performed in patients experiencing acute or chronic GvHD, using either cord blood allogeneic or adult-derived autologous Tregs, based on supportive pre-clinical mouse data. In the first study reported out of Poland, ex vivo-expanded CD4+CD127lo/-CD25+ Treg cells were infused into patients with active GvHD (87). A similar study was performed with cord blood-derived Tregs by Wagner and colleagues (71, 72). The studies showed that pure Tregs could be isolated, expanded, and transferred into patients with the disease. It is also worth noting that one of the impetuses for using cord blood-derived Tregs was the greater ease in purifying the population due to the distinctly high levels of CD25 on these cells. However, although the typical expansion time—14 days—of adult-derived autologous Tregs is sufficient to generate target doses, in the case of cord blood, the small number of Treg at the start of culture required more extensive expansion, often three to four cycles (71, 72). This can lead to increased instability of the product and, in some cases, diversion to a Th2 (IL-4)-producing Treg functional state. In the cord blood-derived allogeneic Treg study, there was an early suggestion of efficacy. This approach in GvHD is now being examined, with or without low-dose IL-2, in a formal phase II study to determine the efficacy and biomarker activity.
4.4 Autologous Tregs have been tested in a variety of clinical settings
The first clinical studies of autologous Tregs in autoimmunity were conducted in patients with T1D. These studies included children and adults recently diagnosed with the disease and were based on extensive preclinical data that suggested that Tregs were able to reverse diabetes in a spontaneous animal model of T1D, the NOD mouse, if given soon after the diagnosis. The protocol for Treg therapy in T1D patients required the transfer of ex vivo-expanded autologous polyclonal CD4+CD127lo/-CD25+ Treg cells within 6 weeks of diagnosis (55, 58, 62). In these studies, the c-peptide levels, as a readout of insulin production, were monitored for up to 2 years.
While there was a suggestion of efficacy, the studies were small and uncontrolled. In the study conducted in Poland, the treated children had higher c-peptide levels and lower insulin requirements than the historical data in untreated children (62). In the first phase I clinical trial conducted in the United States, ex vivo-expanded polyclonal Treg cells were infused into 14 patients with new-onset T1D in doses ranging from 5 × 106 to 2.6 × 109 cells. The infusions were well tolerated, and the c-peptide levels in most patients remained stable for 1 year, although efficacy could not be conclusively shown (55). Notably, the cell pharmacokinetic analysis of Treg cells labeled with deuterium during ex vivo expansion showed that a subset of the infused Treg cells persisted in peripheral blood for at least 1 year and that the Treg cells remained phenotypically stable after the infusion. A phase II clinical trial performed by Caladrius Biosciences, in which 113 newly diagnosed (less than 100 days post-T1D diagnosis) adolescents with T1D were either untreated or infused with autologous ex vivo polyclonally expanded Treg cells (56). The treated group failed to show a clinical effect of the therapy, i.e., preservation of c-peptide production 1 year after the infusion. While the highly enrolled study was found to be safe, with no major serious adverse events reported, the study highlighted the need to consider a more targeted antigen-specific Treg population to gain true efficacy as seen in the pre-clinical studies.
Recent years have seen an increased number of polyclonal Tregs trials in multiple different clinical settings. There have been multiple studies of autologous Treg therapy in patients receiving kidney, liver, and, most recently, islet transplantation (Table 2). Once again, the therapies have been deemed safe, and in several instances, there has been biomarker evidence (such as reduced effector cytokine production in the urine of patients treated at the time of protocol biopsy proven inflammation) suggesting biologic activity. One biomarker study, conducted in the liver transplant setting, showed that the therapy was safe, and there was an altered effector cytokine profile in the blood. Another example of a small safety trial was a study conducted by Appel and colleagues looking at the effect of Treg therapy in patients with ALS. The pre-clinical results had suggested that, even in this non-immune-mediated disease, the control of the associated inflammation could impact disease progression. In the study of Appel et al., three patients with highly advanced ALS were treated with multiple courses of autologous Tregs along with low-dose IL-2 and monitored for disease progression (77, 78). There were some suggestions that the disease progression slowed during immunotherapy; however, it was not clear what role the IL-2 was playing in the therapeutic effects.
One of the challenges in the early Treg interventional studies in autoimmunity has been the small number of patients enrolled in any given trial and the limited biomarker analyses. Although biopsies were possible in the transplant studies, any effects were difficult to interpret given the concomitant treatment of patients with multiple immune-suppressive drugs. Furthermore, in many settings, biopsies were not obtained due to logistical and safety reasons. However, several phase 1 studies in autoimmune settings have been performed where serial biopsies could be performed. In one published study, a patient with cutaneous lupus erythematosus was treated with 1 × 108 ex vivo-expanded CD4+CD127lo/-CD25+ Treg cells. The affected skin was biopsied both on day 0 (prior to Treg injection) and at 12 weeks. The biopsy was assessed for changes in Tregs and Teff cells. As noted above, there was a significant increase in the number of Tregs when the 12-week biopsy was compared with that of day 0. More importantly, the level of FOXP3 was significantly increased in the cells isolated from the biopsy at week 12. In contrast, at the same time point, there was an approximately 75% reduction in interferon-producing CD4+ Teff cells when compared with the day 0 biopsy. Interestingly, in some patients, IL-17-producing T cells were preserved. Thus, a shift from IFNγ to IL-17 may lead to reduced inflammation and increased tissue repair. Similar results were observed in a phase 1 trial in patients with pemphigus (unpublished results/manuscript in preparation). In one patient with paired samples, the percentage of Tregs cells increased in the skin biopsy at week 12, similar to that observed in the lupus patient (56). There was a significant decrease in both the number of Teff cells and their ability to produce interferon at 12 weeks. These results together suggest that the Treg therapy had an impact on the microenvironment of the pathogenic lesion, resulting in diminished inflammatory processes.
There have been several studies looking at alloantigen-reactive Tregs based on preclinical efficacy studies. Overall, Treg cell infusions have been safe and well tolerated. A challenge in these studies, however, has been in manufacturing the cells, potentially because these patients are chronically immunosuppressed, which alters the Tregs both quantitatively and qualitatively. In addition, it appears that the alloreactive Tregs can be sequestered in the target tissue—in the cited instance, the liver—resulting in a reduced precursor frequency in the blood (69).
5 Opportunities to enhance Treg cell therapy
5.1 Enhanced antigen-specific activation
Controlling the local activation and proliferation of Tregs is key to maximize their therapeutic potential. Because Treg cells require engagement through the TCR/CD28 for Treg cell survival and regulatory function, harnessing these physiological signals is an attractive option for controlling Treg activation. TCR or CAR engineering has the potential to direct Treg activation in an antigen- or tissue-restricted way, thus leading to the control of Treg activation, and only in the tissues of particular interest. For TCR-Treg development, the identification of tissue-specific antigen/peptide targets is critical. With CD4+ restriction, class II peptides are the most obvious choice, but the tools to identify and characterize class II-restricted peptides fall far behind those of class I. For class I-restricted TCRs, it is possible to isolate receptors that are independent of CD8 co-receptor activity or the Tregs can be genetically modified to express CD8 which would allow MHC class I-restricted antigens to become viable targets for TCR-engineered Tregs. From an engineering perspective, endogenous TCRs can be left intact if engineered TCRs are still able to preferentially pair and out compete endogenous receptor complexes for TCR signaling components needed to transduce the signals required. Forcing a very high level of expression of the engineered TCRs on the cell surface, utilizing known mutations to induce preferential chain pairing, capturing, or tethering them to the cell surface, and enhancing recycling are all viable methods being explored in the field.
While using TCRs for Treg recognition is appropriate in many settings—particularly those with a strong HLA association, another approach is the use of chimeric antigen receptors (CARs) which can avoid HLA restriction completely. One advantage is that the CAR affinities can be finely tuned to change both the level of antigen-specific Treg activation as well as the amount of off-target “tonic” signaling. Moreover, the CARs can be engineered to alter the site and timing of activation. Whereas TCR-restricted specificities would need cell–cell contact, CAR activation can also be driven by soluble factors, tissue matrix, or exogenous ligands. Advances in high throughput binder generation with exquisite specificity and selectivity have enabled the generation of novel CARs with confirmational epitopes, overcoming some of the challenges to the identification of tissues and disease-specific reactivities. CARs can be re-engineered to have TCR-like properties and even recognize specific peptide/MHC complexes, bypassing the need for TCR engineering while capturing the natural site of Treg activation at the cell surface of antigen-presenting cells (88).
5.2 Enhancing efficacy
Most new anti-inflammatory drugs fail because of two main reasons: lack of clinical safety and lack of clinical efficacy. If the early clinical work with polyclonal Tregs continues to be a guide, Treg therapies are likely to be safe. As such, the lack of clinical efficacy remains the biggest unknown in the field today. Harnessing the natural homing and tissue-specific regulatory mechanisms of Tregs as well as engineering new mechanisms of immune regulation may be the key to achieving a meaningful and durable clinical effect. Here are some key areas of focus for the field.
5.2.1 Cytokines
As potent immune regulators, cytokines are particularly important controllers of Treg-mediated effects and provide many opportunities for engineered products. Enhancing or stabilizing the secretion of anti-inflammatory cytokines (such as IL-10 and TGFβ) may, in certain indications, enhance the anti-inflammatory actions and better control the magnitude of tissue damage. Tregs could also be engineered to express dominant negative or decoy receptors (cell surface or secreted) negating the effects of local and systemic pro-inflammatory cytokines. The remarkable ability of Tregs to home to sites of inflammation could also be harnessed to deliver locally acting anti-inflammatory molecules to the right cell at the right time. This may give new life to dozens of therapies whose off-tissue pharmacology precluded further clinical development. Tregs could also be engineered to respond to certain cytokine stimuli in new ways—for instance, there may be an opportunity to control Treg activation, persistence, and responsiveness to local inflammatory signals. Logic gated systems have gained popularity in the CAR-T effector space in the past few years by harnessing tissue-specific signals (including cytokines) to turn on (or shut down) proliferation or effector functions (89, 90).
5.2.2 Tissue-protective and repair factors
In addition to the well-documented anti-inflammatory effects of Tregs, it is becoming increasingly clear just how important these cells can be in maintaining tissue homeostasis (32, 33)—for example, amphiregulin (shown to be secreted by Tregs) can halt tissue damage and encourage tissue repair and the reestablishment of normal tissue function. Creating Tregs capable of producing or enhancing the natural tissue-protective and repair factors within a specific tissue will increase the potential that Treg therapies will lead to transformative, durable treatments—for example, logic gated systems could be used to “switch” Treg cells from an anti-inflammatory function to a repair function once inflammation has subsided.
5.2.2.1 Homing
As mentioned above, Treg cells have the capacity to enter most tissues of the body, but they most efficiently enter sites of inflammation. In some instances, this may lead to unwanted off-disease effects and/or reduce the number of transferred cells homing to important sites of action. To induce tissue homing bias, it is possible to select for unique tissue homing subsets or to engineer homing capabilities to bulk populations of Tregs to send them to specific locations—for example, in treating inflammatory bowel diseases, it may be advantageous to select for α4β7-expressing Tregs capable of homing to MAdCAM-1-expressing endothelium in sites of tissue inflammation and in Peyer’s patches and mesenteric lymph nodes (91). This may enhance the total tissue load of Treg and enhance both the anti-inflammatory and the pro-repair mechanisms. Alternatively, one could select against or modify homing receptors that aid the transit of Tregs to other tissues and sites of cell clearance—for example, in the liver (a major site of T cell clearance). Homing receptors are interesting targets for logic gated systems as described above. A homing receptor-mediated logic gate could be used to selectively kill cells homing to unwanted sites of action (for example, the CNS) and avoid any consequences of tissue-specific immunosuppression in sites of ongoing infection or increased tumor risk. In this regard, Hoeppli et al. demonstrated that the migration capacity of human Tregs can be tailored by adding cytokines (IL-12 and IFNγ) and or/metabolites (retinoic acid) to culture conditions during in vitro expansion (92).
5.3 Durability and stability
5.3.1 Enhanced growth factor independence
As noted above, Treg cells are highly dependent on growth factors for survival, activation, and proliferation. Although many cytokines and growth factors can influence Tregs, IL-2 is a critical target for advanced genetic engineering. As discussed above, in proinflammatory conditions, particularly those mediated by T cells, IL-2 is in abundance, and Treg are readily able to harness IL-2 through their high-affinity IL-2 receptor complexes. This has a twofold effect: it stimulates Treg expansion (and function) while capturing IL-2 and thus prevent Teff cells in the environment from receiving a needed growth factor, leading to activation-induced cell death. In those indications where there is insufficient IL-2—for example, in non-T cell driven inflammation—it may be necessary to provide IL-2 signals directly. As noted previously, in the ALS clinical trials, exogenous IL-2 was provided as support for expanded Treg cells following infusion. Tregs may be engineered to provide a cell-intrinsic IL-2 signal in several ways, including IL-2 secretion, IL-2 tethering, and modulation of intracellular signaling pathways. Given the central importance of IL-2 signaling in Tregs, coupled with the inconsistent presence of IL-2 in inflammatory disease of interest, it is likely that IL-2 will remain a key focus for Treg growth and persistence.
5.3.2 Enhancing stability
Although data surrounding the plasticity of adoptively transferred human Tregs is lacking, in certain disease settings, Tregs can become destabilized (as shown by changes in FOXP3 expression and genome methylation status). As Treg therapies utilize TCRs and CARs to confer disease and/or tissue specificity, ensuring the stability of any Treg product becomes paramount. As noted previously, existing clinical trials, even those using alloantigen-specific expanded Treg cells, have proven safe without evidence of a destabilized Treg product. Nonetheless, due to the lack of relevant long-term model systems for using human Treg cells, there is considerable interest in maximizing Treg product stability. There are several approaches to further enhance Treg stability, ranging from overt over-expression of FOXP3 to modification of endogenous genomic loci. One such approach is epigenetic modification of the FOXP3 locus to stabilize Treg phenotype and functional activity (93). By targeting the CAR or TCR specificity to the FOXP3 locus, the stability of the Tregs would be linked to the expression of an introduced TCR or CAR. Other pathways (including IL-2 modulation) may also be effective at promoting FOXP3 expression and lineage stability. In all cases, stability will need to be assessed in the context of a pro-inflammatory environment.
5.3.3 Allogenic Treg products
There is an increasing interest in the use of allogenic cell products, including gene-modified mature Tregs and iPSC-derived off-the-shelf cell platforms. Beyond the advantages from a cost-of-goods and deliverability perspective, a key advantage is the ability to ensure product consistency and limit patient-to-patient variability. Additionally, the ability to use healthy donor cells would eliminate any patient segment considerations on Treg quality or quantity. Finally, as noted earlier, an off-the-shelf product would be more amenable to rapid dosing for acute indications such as ARDS or stroke in which it would be impossible to make autologous Tregs quickly enough to impact pathology.
Another significant advantage to an allogeneic product, particularly based on iPSC technology, would be the ability to make several (maybe even dozens) of genetic modifications in a serial manner. It is conceivable to generate “panels” of engineered cells with, for example, differing specificities and then match a particular antigen-specific Treg with a patient segment. Each modification can be thoroughly characterized in isolation or in conjunction with other modifications before clinical studies. Allogenic products may be more amenable to repeat dosing as well as exploring combination therapies. Despite the promise of allogenic products, CAR-T effector clinical development is just beginning. Both engineering and validation have proven more difficult than expected.
6 Concluding remarks
Autoimmune disorders arise from defects in immune tolerance and affect more than 50 million people in the United States and more than 4% of the world’s population. Autoimmune disorders have a high impact on an individual’s morbidity and mortality as well as their quality of life, given that their chronicity, various organ manifestations, and association with co-morbidities are common and devastating for many sufferers. Despite several transformative medicines that have improved many autoimmune diseases, most patients do not adequately respond to existing therapies. Long-term drug therapy is required to maintain long-term efficacy; thus, there remains a major unmet medical need. Importantly, the goal of achieving true immune tolerance by re-establishing immune homeostasis remains unrealized.
As summarized in this review, Tregs, which constitute only a small percentage of circulating T cells, play a pivotal role in establishing and maintaining peripheral tolerance, preventing autoimmune diseases, and limiting chronic inflammatory diseases (94–97). In fact, Treg cells are now widely regarded as the primary cells involved in the persistence of peripheral tolerance. Their essential role is based on the cells’ multiple functionalities ranging from the production of key immunosuppressive cytokines and metabolites to the cell surface expression of key checkpoint molecules to control antigen presentation and metabolism. Importantly, the cells exhibit broad bystander suppression and can mediate infectious tolerance amplifying the impact of the cells, thus resulting in robust and durable efficacy.
As cell therapies have flourished in the cancer space as a new pillar of medicine, it is not surprising that Treg therapies, which have the capacity to control inflammation and autoimmunity, have become the latest approach to treat these devastating diseases. A wealth of pre-clinical data has shown that adoptive Treg cell therapy can be effective in the treatment of diseases ranging from autoimmunity and prevention of organ transplant rejection to cancer-related GvHD and neurological diseases such as ALS and stroke. Efforts are underway to exploit these cells as immunotherapies to treat and potentially prevent these diseases (97). Early clinical studies suggest that Treg adoptive immunotherapy is safe and can lead to biological and molecular changes that alter disease biology. In the oncology setting, however, it is likely that modifications to Treg cells may be required to fully achieve durable and long-lasting effects. Making use of synthetic biology, efforts are underway to genetically manipulate these cells to incorporate novel antigen specificities, altered cell characteristics including the reliance of certain growth factors and a carrier of payloads that can modify local inflamed tissues.
The future for exploiting Treg therapies will depend on solving key questions regarding process development, the assessment of Treg stability and durability, and the development of novel techniques to build new and enhanced activities. Although the field of Treg cell therapies is only at the “end of the beginning”, many have suggested that this living drug may finally enable achieving immune tolerance—the holy grail of immunotherapy.
Author contributions
JAB, BM, JB and FR contributed to the writing, revision, reading, and approval of the submitted manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors wish to acknowledge the contributions of many colleagues who have pioneered the work summarized in this review.
Conflict of interest
All the authors are employed by Sonoma Biotherapeutics.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schwartz RH. Historical overview of immunological tolerance. Cold Spring Harb Perspect Biol (2012) 4:a006908. doi: 10.1101/cshperspect.a006908
2. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunol (1970) 18:723–37.
3. Kojima A, Tanaka-Kojima Y, Sakakura T, Nishizuka Y. Spontaneous development of autoimmune thyroiditis in neonatally thymectomized mice. Lab Invest (1976) 34:550–57.
4. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64. doi: 10.4049/jimmunol.155.3.1151
5. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi: 10.1038/83784
6. Appleby MW, Ramsdell F. Scurfy, the Foxp3 locus, and the molecular basis of peripheral tolerance. Curr Top Microbiol Immunol (2008) 321:151–68. doi: 10.1007/978-3-540-75203-5_7
7. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
8. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi: 10.1038/ni909
9. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–36. doi: 10.1038/ni904
10. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
11. Waldmann H. Immunological tolerance. Elsevier Reference Module Biomed Sci (2014) 1–7. doi: 10.1016/B978-0-12-801238-3.00116-1
12. Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol (2015) 195:2507–13. doi: 10.4049/jimmunol.1500801
13. Pearce NW, Spinelli A, Gurley KE, Hall BM. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. v. dependence of CD4+ suppressor cells on the presence of alloantigen and cytokines, including interleukin 2. Transplantation (1993) 55:374–80. doi: 10.1097/00007890-199302000-00027
14. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
15. Samy ET, Wheeler KM, Roper RJ, Teuscher C, Tung KS. Cutting edge: Autoimmune disease in day 3 thymectomized mice is actively controlled by endogenous disease- specific regulatory T cells. J Immunol (2008) 180:4366–70. doi: 10.4049/jimmunol.180.7.4366
16. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
17. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature (2011) 478:250–54. doi: 10.1038/nature10434
18. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol (2013) 4:152. doi: 10.3389/fimmu.2013.00152
19. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209:1713–22. doi: 10.1084/jem.20120822
20. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus- derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ treg cells. J Exp Med (2012) 209:1723–42. doi: 10.1084/jem.20120914
21. Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant (2016) 16:58–71. doi: 10.1111/ajt.13456
22. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259:88–102. doi: 10.1111/imr.12160
23. Shevach EM. Foxp3+ T regulatory cells: Still many unanswered questions - a perspective after 20 years of study. Front Immunol (2018) 9:1048. doi: 10.3389/fimmu.2018.01048
24. Nakayama M, Hori A, Toyoura S, Yamaguchi SI. Shaping of T cell functions by trogocytosis. Cells (2021) 10:1155. doi: 10.3390/cells10051155
25. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in treg cell function. Nat Immunol (2016) 17:1322–33. doi: 10.1038/ni.3540
26. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15:1070–78. doi: 10.1038/ni.3004
27. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40. doi: 10.1016/S1074-7613(00)80195-8
28. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature (1953) 172(4379):603–6. doi: 10.1038/172603a0
29. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. "Infectious" transplantation tolerance. Science (1993) 259:974–77. doi: 10.1126/science.8094901
30. Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev (2006) 212:301–13. doi: 10.1111/j.0105-2896.2006.00406.x
31. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med (2002) 196:255–60. doi: 10.1084/jem.20020394
32. Zhang C, Li L, Feng K, Fan D, Xue W, Lu J. 'Repair' treg cells in tissue injury. Cell Physiol Biochem (2017) 43:2155–69. doi: 10.1159/000484295
33. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162:1078– 89. doi: 10.1016/j.cell.2015.08.021
34. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature (2022) 602:503–09. doi: 10.1038/s41586-021-04390-6
35. Akkaya B, Shevach EM. Regulatory T cells: Master thieves of the immune system. Cell Immunol (2020) 355:104160. doi: 10.1016/j.cellimm.2020.104160
36. Uhlig HH, Coombes J, Mottet C, Izcue A, Thompson C, Fanger A, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol (2006) 177:5852–60. doi: 10.4049/jimmunol.177.9.5852
37. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199:1455–65. doi: 10.1084/jem.20040139
38. Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin- reactive Foxp3+ treg. Eur J Immunol (2009) 39:1108–17. doi: 10.1002/eji.200839073
39. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum (2005) 52:2212–21. doi: 10.1002/art.21195
40. Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med (2008) 14:88–92. doi: 10.1038/nm1688
41. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med (2009) 15:192–99. doi: 10.1038/nm.1927
42. Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PloS One (2008) 3:e2740. doi: 10.1371/journal.pone.0002740
43. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
44. Pilat N, Steiner R, Sprent J. Treg therapy for the induction of immune tolerance in transplantation–not lost in translation? Intl J Mol Sci (2023) 24:1752. doi: 10.3390/ijms24021752
45. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell- based cell therapy in living donor liver transplantation. Hepatology (2016) 64:632–43. doi: 10.1002/hep.28459
46. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology (2008) 134:2014–24. doi: 10.1053/j.gastro.2008.02.060
47. Pierini A, Iliopoulou BP, Peiris H, Pérez-Cruz M, Baker J, Hsu K, et al. T Cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight (2017) 2:e92865. doi: 10.1172/jci.insight.92865
48. Veiga-Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev (2013) 255:182–96. doi: 10.1111/imr.12085
49. Belkaid Y. Paradoxical roles of Foxp3+ T cells during infection: from regulators to regulators. Cell Host Microbe (2008) 3:341–43. doi: 10.1016/j.chom.2008.05.011
50. Multhoff G, Molls M, Radons J. Chronic inflammation in cancer development. Front Immunol (2012) 2:98. doi: 10.3389/fimmu.2011.00098
51. Graßhoff H, Comdühr S, Monne LR, Müller A, Lamprecht P, Riemekasten G, et al. Low-dose IL-2 therapy in autoimmune and rheumatic diseases. Front Immunol (2021) 12:648408. doi: 10.3389/fimmu.2021.648408
52. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory t. cell phenotype in the inflammatory environment. J Immunol (2013) 190:1076–84. doi: 10.4049/jimmunol.1202659
53. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
54. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329:1667–71. doi: 10.1126/science.1191996
55. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
56. Dall'Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol (2019) 71:431–40. doi: 10.1002/art.40737
57. Caladrius Biosciences. Caladrius biosciences reports top-line data for the phase 2a Sanford project: T-Rex trial of CLBS03 for recent onset type 1 diabetes (2019). Available at: https://www.globenewswire.com/en/news-release/2019/02/13/1725052/18623/en/Caladrius-Biosciences-Reports-Top-Line-Data-for-the-Phase-2a-Sanford-Project-T-Rex-Trial-of-CLBS03-for-Recent-Onset-Type-1-Diabetes.html.
58. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low- dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6:e147474. doi: 10.1172/jci.insight.147474
59. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory crohn's disease. Gastroenterology (2012) 143:1207–17. doi: 10.1053/j.gastro.2012.07.116
60. Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for crohn's disease. Gut (2016) 65:584–94. doi: 10.1136/gutjnl-2014-306919
61. Voskens C, Stoica D, Rosenberg M, Vitali F, Zundler S, Ganslmayer M, et al. Autologous regulatory T-cell transfer in refractory ulcerative colitis with concomitant primary sclerosing cholangitis. Gut (2023) 72:49–53. doi: 10.1136/gutjnl-2022-327075
62. Marek-Trzonkowska N, Myśliwec M, Siebert J, Trzonkowski P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes (2013) 14:322–32. doi: 10.1111/pedi.12029
63. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant (2017) 17:2945–54. doi: 10.1111/ajt.14415
64. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (2020) 395:1627–39. doi: 10.1016/S0140-6736(20)30167-7
65. Mathew JM, H-Voss J, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep (2018) 8:7428. doi: 10.1038/s41598-018-25574-7
66. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. BMJ (2020) 371:m3734. doi: 10.1136/bmj.m3734
67. Brook MO, Hester J, Petchey W, Rombach I, Dutton S, Bottomley MJ, et al. Transplantation without overimmunosuppression (TWO) study protocol: A phase 2b randomised controlled single-centre trial of regulatory T cell therapy to facilitate immunosuppression reduction in living donor kidney transplant recipients. BMJ Open (2022) 12:e061864. doi: 10.1136/bmjopen-2022-061864
68. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7:7563–77. doi: 10.18632/oncotarget.6927
69. Tang Q, Leung J, Peng Y, Sanchez-Fueyo A, Lozano JJ, Lam A, et al. Selective decrease of donor-reactive tregs after liver transplantation limits treg therapy for promoting allograft tolerance in humans. Sci Transl Med (2022) 14:eabo2628. doi: 10.1126/scitranslmed.abo2628
70. Bernaldo-de-Quirós E, Cózar B, López-Esteban R, Clemente M, Gil-Jaurena JM, Pardo C, et al. A novel GMP protocol to produce high-quality treg cells from the pediatric thymic tissue to be employed as cellular therapy. Front Immunol (2022) 13:893576. doi: 10.3389/fimmu.2022.893576
71. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
72. MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, et al. First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA- matched siblings. Blood Adv (2021) 5:1425–36. doi: 10.1182/bloodadvances.2020003219
73. Hamilton BK. Current approaches to prevent and treat GVHD after allogeneic stem cell transplantation. Hematol Am Soc Hematol Educ Program (2018) 2018:228–35. doi: 10.1182/asheducation-2018.1.228
74. Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, Sahaf B, et al. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight (2019) 4:e127244. doi: 10.1172/jci.insight.127244
75. Andújar-Sánchez F, Lopes-Ramos T, Bejarano-García JA, García-Guerrero E, Calderón-Cabrera C, Caballero-Velázquez T, et al. Combined treatment of graft versus host disease using donor regulatory T cells and ruxolitinib. Sci Rep (2022) 12:8348. doi: 10.1038/s41598-022-12407-x
76. Meyer E, ORCA team. (2021). Available at: https://www.cancernetwork.com/view/orca-t-improves-outcomes-over-standard-of-care-for-serious-hematologic-malignancies.
77. Beers DR, Thonhoff JR, Faridar A, Thome AD, Zhao W, Wen S, et al. Tregs attenuate peripheral oxidative stress and acute phase proteins in ALS. Ann Neurol (2022) 92:195–200. doi: 10.1002/ana.26375
78. Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in- human study. Neurol Neuroimmunol Neuroinflamm (2018) 5:e465. doi: 10.1212/NXI.0000000000000465
79. Lyu MA, Huang M, Zeng K, Li L, Khoury JD, Nishimoto M, et al. Allogeneic cord blood regulatory T cells can resolve lung inflammation. Cytotherapy (2023) 25:245–53. doi: 10.1016/j.jcyt.2022.10.009
80. Balcerek J, Shy BR, Putnam AL, Masiello LM, Lares A, Dekovic F, et al. Polyclonal regulatory T cell manufacturing under cGMP: A decade of experience. Front Immunol (2021) 12:744763. doi: 10.3389/fimmu.2021.744763
81. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments tregs yet transiently impairs β-cell function. Diabetes (2012) 61:2340–48. doi: 10.2337/db12-0049
82. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood (2016) 128:1651–59. doi: 10.1182/blood-2016-02-700849
83. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. IL- 6 and TNFα drive extensive proliferation of human tregs without compromising their lineage stability or function. Front Immunol (2021) 12:783282. doi: 10.3389/fimmu.2021.783282
84. Motwani K, Peters LD, Vliegen WH, El-Sayed AG, Seay HR, Lopez MC, et al. Human regulatory T cells from umbilical cord blood display increased repertoire diversity and lineage stability relative to adult peripheral blood. Front Immunol (2020) 11:611. doi: 10.3389/fimmu.2020.00611
85. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
86. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A rapamycin- based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
87. Trzonkowski P, Dukat-Mazurek A, Bieniaszewska M, Marek-Trzonkowska N, Dobyszuk A, Juścińska J, et al. Treatment of graft-versus-host disease with naturally occurring T regulatory cells. BioDrugs (2013) 27:605–14. doi: 10.1007/s40259-013-0050-5
88. Poorebrahim M, Mohammadkhani N, Mahmoudi R, Gholizadeh M, Fakhr E, Cid-Arregui A. TCR-like CARs and TCR-CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther (2021) 28:581–9. doi: 10.1038/s41417-021-00307-7
89. Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science (2022) 378:eaba1624. doi: 10.1126/science.aba1624
90. Nirschl CJ, Brodkin HR, Hicklin DJ, Ismail N, Morris K, Seidel-Dugan C, et al. Discovery of a conditionally activated IL-2 that promotes antitumor immunity and induces tumor regression. Cancer Immunol Res (2022) 10:581–96. doi: 10.1158/2326-6066.CIR-21-0831
91. Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol (2014) 7:6–19. doi: 10.1038/mi.2013.73
92. Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplant (2019) 19:62–76. doi: 10.1111/ajt.14936
93. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular mechanisms controlling Foxp3 expression in health and autoimmunity: From epigenetic to post-translational regulation. Front Immunol (2020) 10:3136. doi: 10.3389/fimmu.2019.03136
94. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science (2021) 373:510–16. doi: 10.1126/science.abh1654
95. Terry LV, Oo YH. The next frontier of regulatory T cells: Promising immunotherapy for autoimmune diseases and organ transplantations. Front Immunol (2020) 11:565518. doi: 10.3389/fimmu.2020.565518
96. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
Keywords: Treg, immune tolerance, Foxp3, autoimmunity, immune regulation
Citation: Bluestone JA, McKenzie BS, Beilke J and Ramsdell F (2023) Opportunities for Treg cell therapy for the treatment of human disease. Front. Immunol. 14:1166135. doi: 10.3389/fimmu.2023.1166135
Received: 14 February 2023; Accepted: 22 March 2023;
Published: 19 April 2023.
Edited by:
Lesley Ann Smyth, University of East London, United KingdomReviewed by:
Carolina Isabel Rojas, University of Chile, ChileOksana Kehoe, Keele University, United Kingdom
Copyright © 2023 Bluestone, McKenzie, Beilke and Ramsdell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey A. Bluestone, amJsdWVzdG9uZUBzb25vbWFiaW8uY29t