 Iwona T. Myszor
Iwona T. Myszor Gudmundur Hrafn Gudmundsson
Gudmundur Hrafn Gudmundsson- 1Faculty of Life and Environmental Sciences, Biomedical Center, University of Iceland, Reykjavik, Iceland
- 2Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
Innate immunity of the mucosal surfaces provides the first-line defense from invading pathogens and pollutants conferring protection from the external environment. Innate immune system of the airway epithelium consists of several components including the mucus layer, mucociliary clearance of beating cilia, production of host defense peptides, epithelial barrier integrity provided by tight and adherens junctions, pathogen recognition receptors, receptors for chemokines and cytokines, production of reactive oxygen species, and autophagy. Therefore, multiple components interplay with each other for efficient protection from pathogens that still can subvert host innate immune defenses. Hence, the modulation of innate immune responses with different inducers to boost host endogenous front-line defenses in the lung epithelium to fend off pathogens and to enhance epithelial innate immune responses in the immunocompromised individuals is of interest for host-directed therapy. Herein, we reviewed possibilities of modulation innate immune responses in the airway epithelium for host-directed therapy presenting an alternative approach to standard antibiotics.
Introduction
The airway epithelium of the respiratory tract is constantly exposed to particles and microbes inhaled with each breath that could possibly endanger the host. Hence, the highly specialized system of the host innate immune defenses is indispensable, as it keeps pathogens at bay and can limit the damaging effect of environmental pollutants. The airway epithelial cells together with stromal cells and tissue-residing immune cells shape immune responses in the local environment. Those immune responses protect the host from invading pathogens and maintain the local tissue homeostasis by producing signals for cell renewal and regeneration upon damage (1–4). In the first part of this review article, we provided an overview on innate immune functions of the airway epithelial cells covering recent developments like the identification of new cell types by single-cell transcriptomics. In the second part, we described pathogens strategies to subvert host front-line defenses followed by the third part, where we reviewed research on how pathogens subversion mechanisms can be circumvented through modulation of host epithelial innate immune responses by different inducers for host-directed therapy.
Innate immunity components of the human respiratory epithelium
The airway epithelial cells are central players in the communication between the host and the external environment and together with stromal and immune cells present in local tissues, shape immune responses during homeostasis and disease. This is possible because of a highly complex innate immune system consisting of several components that we review in this section. All those components work together to provide protection of the host mucosal surfaces from the external environment and tissue regeneration upon damage (1–4).
Human airway and alveolar epithelium
The airway epithelium lining the upper and lower respiratory tract is composed of different types of epithelial cells forming a single-cell layer of pseudostratified epithelium with tight physical links to the basal lamina and communicating with underlaying stromal cells, such as fibroblasts, airway smooth muscle cells, peri-endothelial (pericytes), and endothelial cells embedded in the connective tissue matrix (2, 4, 5). The four major types of cells in the airway epithelial layer are secretory club cells, goblet cells, ciliated cells, and basal cells (Figure 1) (3). Secretory club cells are columnar non-ciliated cells producing factors responsible for the neutralization of inhaled toxic substances and displaying immunomodulatory functions (6). In a mouse model, club cells were shown to act as self-renewing stem cells and as progenitors for ciliated cells that constitute the majority in the airway epithelium (7, 8). Club cells can also differentiate to mucus-producing goblet cells in response to allergens (9). Basal cells are small cuboidal cells that replenish different types of mature cells maintaining epithelial cell turnover during homeostasis and regenerating damaged epithelial barrier (10, 11). In respect to the important role in self-renewal and tissue regeneration, the basal cells seem to be protected from directed exposition to the lumen of the airways by other epithelial cells (Figure 1). Additional protection is provided by advanced innate host defense mechanisms. The proportion of each cell type occurring in human airways varies depending on the diameter of the airways, which results in diverse innate immune responses at different levels of the anatomical and histological organization of the respiratory system. For instance, mucus-secreting goblet cells occurring in submucosal glands of the human trachea and large bronchus are replaced by secretory club cells in terminal bronchioles (8, 12). The submucosal glands are composed of four major types of the airway epithelial cells and myoepithelial cells that together form a sophisticated machinery releasing fluids, mucus, and antimicrobial effectors into the luminal space of the airways (1).
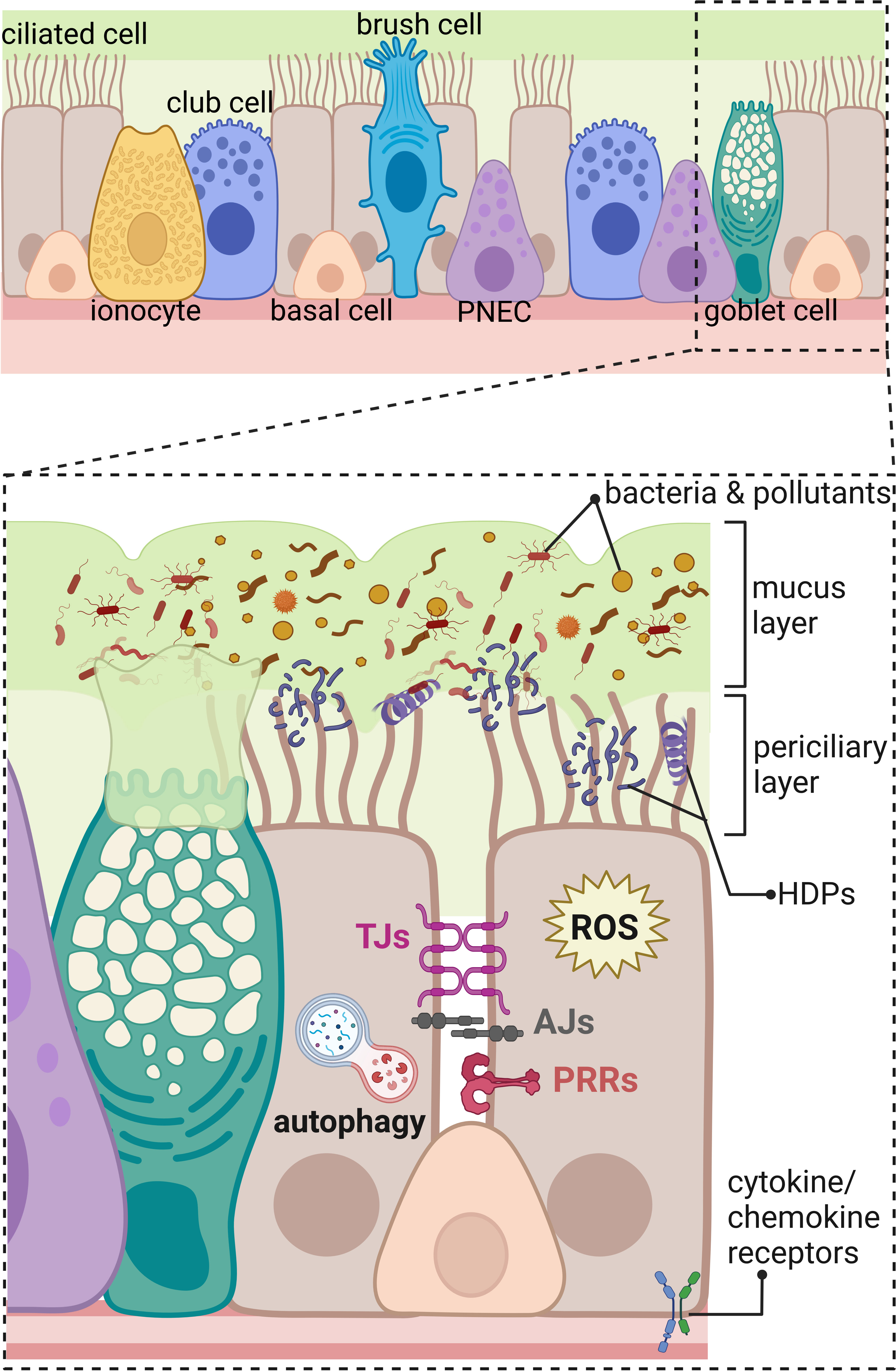
Figure 1 Innate immunity components of the human respiratory epithelium in the distal part of the respiratory tract. The upper panel shows an overview on the different types of cells composing respiratory epithelium of the distal part of the airways including ciliated cells, ionocytes, club cells, basal cells, brush (tuft) cells, pulmonary neuroendocrine cells (PNEC), and goblet cells. The lower panel represents magnified area showing a part of the respiratory epithelium with innate immunity components marked; beginning from the top: mucus layer with host defense peptides (HDPs) entrapping bacteria, and pollutants removed by beating cilia in the periciliary layer. The constant renewal of the mucus layer is provided by goblet cells. Transport of molecules through the paracellular space between neighboring cells is regulated by tight junctions (TJs) and adherens junctions (AJs). Danger signals are recognized by pathogen recognition receptors (PRRs) and cytokine/chemokine receptors. Intracellular components of the innate immunity in the respiratory epithelium include production of reactive oxygen species (ROS) and autophagy eliminating intracellular intruders and particles. Created with BioRender.com.
Additional types of the airway epithelial cells have been recently described (Figure 1) based on the increasing accessibility of the single-cell RNA sequencing, changing our view on the cellular landscape of the airway epithelium that is more heterogeneous than previously thought (3–5, 13). Rare types of airway epithelial cells were distinguished including brush cells (also called tuft cells) and solitary chemosensory cells identified in the human trachea and nasal cavity, respectively (14). The function of brush cells in humans remains largely uncharacterized and, based on the mouse studies, indicates regulation of type II immune responses, such as release of IL-25 in the nasal epithelium and protective reflexes, such as sneezing (14, 15). Interestingly, brush cells do not occur in the healthy human alveolar epithelium. Expansion of brush cells in the upper airways and their de novo formation in the lower airways were observed after viral infections with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and influenza A subtype H1N1, respectively (15). Chemosensory cells in the human nasal epithelium detect and mediate reflex reactions in response to irritants and pathogens to prevent their passage down to the lower airways (16). Another rare cell type in the human airway epithelium are pulmonary neuroendocrine cells (PNECs) clustered into neuroendocrine bodies (NEB) (2, 17). Apart from olfactory epithelium in the nasal cavity (18), PNECs are the only cells in the airways supplied with nerves; therefore, they participate in the neurotransmission, act as sensory cells for the oxygen level, and detect and respond to inhaled allergens (2, 17). Another recently identified rare cell types are the ionocytes, showing high expression level of cystic fibrosis transmembrane receptor (CFTR), regulating the ionic microenvironment of the mucus (19, 20). In addition, novel subtypes of progenitor cells were identified, such as precursors of ciliated cells—deuterosomal cells containing numerous centrioles and mucous-ciliated differentiation intermediate cells (3). Moreover, the population of basal cells is more heterogeneous than what was thought. The new intermediate cells were distinguished between basal and 1) club cells—suprabasal cells, 2) PNEC, ionocytes, and brush cells—parabasal cells, and 3) luminal secretory cells—Hillock cells (3, 5, 11).
The distal part of human airways ends with terminal bronchioles mainly composed of club and ciliated cells with very few rare types of cells and almost no basal cells present (8). One can argue that only few basal cells are present in the distal part of human airways because, in principle, the portion of air reaching alveoli should be warmed, moistened, and cleared from pollutants, as the vital function of the alveolar epithelium is the gas exchange provided by epithelial alveolar type 1 cells (also known as AT1, ATI, and type I pneumocytes) (2). Frequently, these defense mechanisms of the proximal part of the respiratory system are not sufficient. Therefore, alveolar innate immune defense mechanisms are activated to protect the host, such as surfactant proteins produced by alveolar type 2 cells (also known as AT2, ATII, and type II pneumocytes), that also function as progenitor cells for AT1 cells in the adult lung (1, 2, 21). Overall, the recent advances in transcriptomic and proteomic analyses opened a new avenue for further characterization of innate immune functions of new epithelial cells subtypes and the signaling pathways guarding their differentiation.
Host defense peptides
Numerous antimicrobial effectors are produced by the airway epithelial cells for effective host innate defenses. Among them are host defense peptides (HDPs) (Figure 1), previously commonly called antimicrobial peptides (AMPs), divided into two main families of cathelicidins and defensins (22–24). HDPs can be produced constitutively and induced upon stimuli. Their constitutive expression can be enhanced upon stress conditions, like infection or injury. Most of HDPs are stored in the cellular granules as a pro-form, which is further released and processed upon a danger signal (22, 25). Human cathelicidins have one dominant mature peptide LL-37 encoded by CAMP gene (cathelicidin antimicrobial peptide). Human cathelicidin is produced as a pro-form (hCAP18; pro-LL-37) containing N-terminal cathelin domain that is cleaved off by serine proteases, such as kallikreins in the skin and proteinase 3 from neutrophiles to release the mature LL-37 peptide (25–28). LL-37 has an α-helical structure and a cationic, amphipathic character and can be processed in the skin to shorter peptides (26, 29). Human cathelicidin is constitutively expressed at rather low protein levels in airway epithelial cells showing only the pro-form to be secreted when the cells are cultured in vitro. The expression of cathelicidin is enhanced upon stimuli, indicating a highly responsive inducible system for host defense (30–32). During inflammation, the migration of neutrophiles to the site of infection contributes to the processing of the pro-form hCAP18 to the active LL-37 peptide in the lung, as neutrophiles are loaded with proteinase 3 and other enzymes. However, whether the processing of constitutively expressed pro-form hCAP18 to the active LL-37 peptide takes place on the airway epithelial surfaces in physiological conditions during homeostasis remains to be further elucidated, as the in vitro studies with the airway epithelial cells do not closely recapitulate the in vivo environment. On the other hand, the analysis of the airway surface liquid (ASL) indicated the presence of mature LL-37 (33). However, one may argue about fact that the source of mature LL-37 peptide in ASL might come from immune cells present in lungs. Therefore, the dynamics of the cathelicidin processing to the mature LL-37 peptide in the homeostatic conditions of the lung epithelium remains to be further investigated, similar to the identification of potential lung tissue specific enzymes responsible for the cleavage and confirmation if LL-37 is processed to shorter peptides with characterization of their potential role in shaping local lung innate immune responses. In contrast to cathelicidins, human defensins have several members and are divided in α-, β-, and θ-defensins (34). Alpha- and beta-defensins are produced in the airway epithelium constitutively and inducible as inactive precursors that undergo further processing to the active form (21, 22, 35, 36). In human genome, there are genes encoding the third group of θ-defensins; however, proteins are not produced because of premature codon stop (37). The expression of antimicrobial effectors in each cell type present in the airway epithelium has been explored and is available in Human Protein Atlas (HPA). According to HPA, the main sources of cathelicidin in bronchus are basal respiratory cells (https://www.proteinatlas.org/ENSG00000164047-CAMP/single+cell+type/bronchus).
The primary function of the HDPs is their bactericidal effect at the range of micromolar concentrations on both Gram-positive and Gram-negative bacteria, but they are also effective against viruses and fungi (26, 35, 38). The common antimicrobial activity of HDPs is associated with their cationic and amphipathic properties. Positively charged HDPs can strongly interact through electrostatic and hydrophobic interactions with negatively charged phospholipids of the bacterial cell membrane and pathogen-associated molecular patterns (PAMPs), such as LPS of Gram-negative bacteria (39, 40). The interaction of HDPs with the bacterial membrane causes disruption, leakage of the intracellular components, and bacterial cell death (24). Another, less known antimicrobial mechanism of HDPs is the translocation of peptides through the membrane and binding to bacterial intracellular targets, for example, cardiolipin resulting in bacterial cell death (41). Furthermore, human β-defensin 3 has been shown to disrupt bacterial cell wall biosynthesis by binding to lipid II, which makes bacteria more vulnerable to damage (42). Hence, peptides have different activities against bacteria, but the membrane disruption seems of a major general importance. In case of the viral infections, HDPs bind and destabilize viral structures like viral envelope of influenza viruses, respiratory syncytial virus (RSV), Zika virus (38, 43, 44), and viral capsid of rhinoviruses (45). Similar mechanism takes place during fungal infections, like with Candida albicans, where HDPs permeabilize yeast cell membrane (46). Of note, at physiological concentrations, the HDPs do not damage human cell membranes due to its lack of negative charge in the outer leaflet of the membrane and presence of cholesterol (47). However, HDPs at higher concentrations might be able to damage host’s cells, for instance when granulocytes are recruited to the site of infection in the lung (48, 49). Interestingly, recently, it has been described that the fragments of LL-37 peptide (17–29 aa residues) can cluster together to form highly organized oligomers resembling fibril-like tubules of unknown function (50). Some pathogens, such as Staphylococcus aureus, produce similar tubules what is recently discussed as an example of the bacterial mimicry, perhaps to evade host antimicrobial responses potentially exerted by such HDPs fibril-like oligomers (51). However, the existence and physiological relevance of such LL-37 fibrils in the lung epithelial surfaces have not been described so far.
Apart from antimicrobial activity, HDPs display immunomodulatory functions (24). HDPs are chemoattractant for immune cells, e.g., human β-defensin 2 (hBD2) is a chemoattractant for mast cells (52) and LL-37 for neutrophils, monocytes, and T cells (53, 54). HDPs also indirectly recruits leukocytes to the local site of infection or injury by inducing release of chemokines and cytokines (24). For instance, defensins can induce expression of proinflammatory IL-8, a chemoattractant for neutrophils, in human A549 lung epithelial cell line and therefore display proinflammatory function in vitro (55). HDPs interfere with the cell signaling cascades while displaying at the same time a dual pro- and anti-inflammatory role, depending on the local environment and the phase of infection. During infection when nuclear factor kappa B (NF-κB) pathway is activated by bacterial LPS binding Toll-like receptor 4 (TLR4) receptor, LL-37 peptide can selectively inhibit production of proinflammatory tumor necrosis factor (TNF) and reactive oxygen species (ROS) while at the same time stimulate IL-8 production in epithelial cells to attract immune cells (56). HDPs have pro- and anti-inflammatory effects that seem to be exerted depending on the stage of infection. In the early stage of bacterial infection with Pseudomonas aeruginosa, LL-37 enhances proinflammatory response in airway epithelial cells (57). Cathelicidins are also able to prevent activation of TLR2 and TLR4 signaling in macrophages by non-viable bacteria and their products at later stage of infection, resulting in reduced production of proinflammatory response that might protect local tissue from the injury (58). Therefore, HDPs can play a dual role in shaping both pro- and anti-inflammatory response and maintaining tissue homeostasis. HDPs also display additional functions by enhancing phagocytosis, ROS production, and participation in neutrophil extracellular trap (NET) formation, contributing to enhancement of the bacterial clearance (24). HDPs also initiate T-cell response by promoting Th17 differentiation (59) and could, therefore, play a role in the intersection of innate and adaptive immunity. Furthermore, local environment also affects immunomodulatory function of HDPs by their post-translational modifications (PTMs). The citrullinated LL-37 peptide was detected in human bronchoalveolar lavage; however, citrullination affected the net charge of the peptide that lost the ability for bacterial killing (60). Another evidence is citrullination of LL-37 by peptidyl arginine deaminase, where the peptide loses its ability to enhance proinflammatory response in macrophages (61). These studies indicate that PTMs regulate the mode of action of HDPs for the maintenance of the local tissue homeostasis, and future exploration of how additional PTMs affect function of HDPs in lungs is of interest.
Apart from HDPs, there are also several antimicrobial proteins like lysozyme, degrading bacterial peptidoglycans, and bacteriostatic lipocalin 2 and lactotransferrin (21). The bacteriostatic effect of lipocalin 2 and lactoferrin are linked to the inhibition of iron uptake by bacteria from the local environment, as they bind bacterial iron-chelating molecules. Lipocalin 2 has been shown to be effective against Escherichia coli causing pneumonia (62), and mutant mice lacking lipocalin 2 were more susceptible to Klebsiella pneumoniae infections (63). Similarly, S100A7 (psoriasin) protein from S100 protein family containing several antimicrobial effectors (e.g., S100A8/9 protein known as calprotectin) has been shown to kill E. coli by Zn2+ chelation (64). Airway epithelial cells and alveolar macrophages constitutively express S100A7 that is enhanced upon S. aureus challenge (65). Interestingly, the mechanical strain generated during breathing enhances expression of S100A7 protein in alveolus-on-chip model through activation of mechanosensitive ion channel TRPV4 (transient receptor potential vanilloid-type 4). Moreover, when TRPV4 and the S100A7 target receptor—receptor for advanced glycation end products (RAGE)—were blocked by inhibitors, the viral load of H3N2 influenza was increased, demonstrating the importance of S100A7 for lung defenses (66). Furthermore, antiproteases like secretory leukocyte protease inhibitor (SLPI) and elafin exhibit antimicrobial properties against pathogens P. aeruginosa and S. aureus in lung epithelial cells (67–69) and have anti-inflammatory potential, e.g., by inhibition of NF-κB pathway through reducing degradation of IκBα in macrophages and endothelial cells (70). Ribonuclease 7 (RNase7) discovered in the skin where it displays antimicrobial effects against several pathogens has also been shown to be expressed in human respiratory tract (71). Interestingly, the primary source of RNase7 in human airways are basal cells that express RNase7 upon stimulation with inactivated H. influenzae and cigarette smoke, therefore indicated as a second line of front-line defenses in case of injury of mature differentiated epithelial cells (72). Similarly, RNase7 has been induced in airway epithelial cells upon infection with Mtb, where it marked intracellular bacteria to a limited extent. However, the direct effect of RNase7 on elimination of Mtb was not shown and requires further investigation (73). Additional components of the innate immunity in the lung epithelium are collectins, surfactant protein A and D (SP-A and SP-D), produced by alveolar type 2 cells (21). They tag bacteria for opsonization to increase phagocytosis by the alveolar macrophages (74). The immunomodulatory function of collectins can be exemplified by the inhibitory effect of the SP-A on the production of IL-8 by eosinophils present during allergic response (75). The antimicrobial and immunomodulatory functions are also displayed by plate-lung-nasal-clone (PLUNC) proteins shown to have antibacterial effect against Mycoplasma pneumoniae and reduce production of proinflammatory cytokine IL-8 (76). Of note, several chemokines like CXCL9 and CXCL11, present in the lung epithelium as a result of interferon gamma (IFNγ) stimulation, have antibacterial functions against E. coli and L. monocytogenes (77). Host defense effectors produced by club cells such as club cell protein 10 (CC-10) inhibit NF-κB signaling pathway and production of proinflammatory cytokines and chemokines in bronchial epithelial cells (78). Similar to CC-10, club cell secretory protein (CCSP) was shown to reduce inflammation and viral load during RSV infection (79). Next, the levels of secretoglobin A1A (SCGB1A1) also produced by club cells were reduced in the airways of patients with asthma in comparison to healthy individuals (80). Allergen-specific immunotherapy increased expression of SCGB1A1 considered as anti-inflammatory mediator in the lower airways (81). Secretoglobin A1A was also shown to affect alveolar macrophages by attenuation of the surge of inflammatory cytokines during activation of TLR receptors. Deficiency of secretoglobin A1A facilitated development of proinflammatory M1 phenotype of pulmonary macrophages, indicating the importance of physiological levels of SCGBA1A for innate immune defense and maintenance of local tissue homeostasis (82). Another important indirect link to HDP activity is the expression and function of CFTR and the non-gastric H+/K+ adenosine triphosphatase (ATP12A) in lung epithelial cells, including recently identified ionocytes. Both CFTR and ATP12A regulate pH of the airway surface liquid (ASL) by secretion of and H+, respectively, that is detrimental for antimicrobial activity of HDPs (83, 84).
A further important element of the innate immunity in the airway epithelial cells is the production of reactive oxygen species (ROS) (Figure 1). For many years, ROS were attributed to the lung tissue damage and tissue aging as a result of the oxidative stress (85, 86) but in fact ROS play an important function in the elimination of invading pathogens (21). Dual oxidases (DUOX) are key enzymes responsible for the generation of ROS in the lung epithelium, including superoxide and hydrogen peroxide, for effective pathogen elimination (87, 88). In addition, the protective role of NOX1 activity have been shown to limit inflammatory response and lung tissue damage exerted by influenza A at early stage of infection in mice (89) even though the NOX1 activity in different circumstances, such as hyperoxia, can cause tissue damage (85). The view on ROS generation and redox signaling in epithelial cells is developing to better understand their role in pathological situations and demonstrate that the physiological levels of ROS are important for cellular signaling during homeostasis (86, 90).
Mucus and mucociliary clearance
A physical component of the innate immune system is the airway surface liquid (ASL) composed of the mucus and periciliary layer on the airway epithelium (Figure 1) (91). The ASL provides a physical and chemical barrier for the invading pathogens and inhaled particles protecting the airway tissue from the damage (92). The chemical barrier of ASL is provided by the low salt content (93), pH maintained by and H+ (94, 95), and hydration maintained by Na+ and Cl− ion gradient (96). This environment facilitates the formation of MUC5B bundles (12) and keeps HDPs active for killing of entrapped pathogens in the mucus layer (83, 84) for their removal by mucociliary clearance of beating cilia (21, 93). The periciliary layer of ASL composed of transmembrane mucins and periciliary liquid reduces the friction of the constantly moving mucus that clears airways from bacteria and pollutants during homeostasis (12). The dynamic process of mucociliary clearance keeps the airways almost devoid of bacteria in healthy individuals (12, 93) causing the characterization of any existing lung microbiome very difficult and so far confirmed only by detection of bacterial nucleic acids (97, 98). In addition, the anaerobic bacterial fermentation products were detected in the lungs of patients with HIV (99, 100); however, those ground-breaking evidence shall be carefully interpreted and further confirmed by the characterization of additional microbiota metabolites and their original source of production. Impairment of the mucociliary clearance by primary ciliary dyskinesia (101) or defects in the function of ion channels in cystic fibrosis (CF) (92) cause clogging of the airways with mucus, creating a favorable environment for bacterial growth (12). Therefore, pre-existing bacteria during chronic respiratory diseases are sometimes referred in the literature as “lung microbiota” (102), although it should be emphasized that this terminology is used in the context of pathological conditions. In general, the studies on the mucus composition and physiology together with the interaction of innate immunity components with mucus are of interest and one of the future directions in the field investigating host defenses.
Pathogen recognition receptors
One of the initial functions of innate immunity in the airway epithelium is pathogen detection by pathogen recognition receptors (PRRs) (Figure 1), recognizing PAMPs. Upon stimulation, PRRs activate signaling cascade leading to the inflammation and clearance of the pathogens. Among different types of the PRRs, present in the airway epithelium are TLRs, NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin receptors (CLRs), and formyl-peptide receptors (FPRs) (21, 103–105).
In the airway epithelium, all types of the TLRs (TLR2/1, TLR2/6, TLR3, TLR4, TLR5, TLR7, TLR8, TLR9, and TLR11) are present, anchored in the cell membrane and endosomes (21). However, the mapping of TLRs with their exact spatial distribution within airway epithelial cell membranes at different regions of the respiratory tract, similar to the study done for the intestinal epithelium (106), is still missing. TLRs recognize bacterial lipopeptides, lipopolysaccharide (LPS), flagellin, DNA, and RNA by the leucine-rich repeat (LRR) motif that is linked through a single transmembrane domain to Toll/IL-1 receptor (TIR) intracellular motif. The activation of the receptor requires binding of the adaptor protein MyD88 or TIR-domain-containing adapter-inducing interferon-β (TRIF) to the TIR domain. MyD88 is an adaptor protein for all TLRs, except TLR3 and TRIF for TLR3 and TLR4. Other adaptor proteins like TIR domain containing adaptor protein (TIRAP), translocation associated membrane protein 1 (TRAM), sterile alpha and TIR motif containing (SARM) have been shown to be involved in the TLR signaling. The stimulation of TLRs initiates NF-κB and mitogen-activated protein kinase (MAPK) downstream signaling, leading to induction of proinflammatory cytokines and type I interferons (21, 107).
Unlike TLRs, apart from cell and endosomal membranes, NLRs occur as soluble receptors in the cytoplasm. In the airway epithelium, NLRs with caspase recruitment domain (CARD) are represented by NOD1 and NOD2 receptors, recognizing bacterial peptidoglycans and working in synergy with TLRs towards activation of NF-κB and MAPK pathways (108). The other types of NLRs, containing PYD (pyrin) and NLRB (baculoinhibitor of apoptosis protein) domains form inflammasome controlling cleavage of IL-1β and IL-18 pro-forms by caspase-1 (21, 103). For instance, in human lung epithelial cells NRLP3 receptor harboring pyrin domain forms NRLP3 inflammasome responsible for cleavage of IL-1β upon C. albicans or influenza A virus infection (109, 110).
Human airway epithelial cells have also cytoplasmic receptors for the detection of pathogen’s nucleic acids (21). Among them are RIG-I-like receptors (RLRs), recognizing viral ss/dsRNA of influenza and paramyxoviruses causing respiratory diseases, e.g., RSV (111, 112). The group of RLR receptors include retinoic acid inducible gene-I (RIG-I), melanoma differentiation-associated gene 5 (MDA5) and RIG-I-like receptor dsRNA helicase (LGP2) (103). With the involvement of the adaptor proteins, e.g., mitochondrial antiviral-signaling protein (MAVS), RIG-I and MDA5 receptors transduce signals activating NF-κB, interferon regulatory factor 3 and 7 (IRF3 and IRF7) pathways leading to the production of proinflammatory cytokines and interferons (108). Other cytoplasmic receptors recognize bacterial DNA and bacterial signaling molecules such as cyclic dinucleotides (CDNs) (21). A prominent member of this family is the stimulator of IFN genes (STING) located on the ER membrane. STING recognizes CDNs and is an adaptor protein for other cytoplasmic receptors like interferon gamma inducible protein 16 (IFI16) recognizing Streptococcus pneumoniae dsDNA (113). Moreover, STING can by activated by cyclic GMP-AMP (cGAMP) synthetized by cGAMP synthase (cGAS) in human airway epithelial cells upon detection of not only microbial/viral DNA in the cytoplasm but also by self-DNA coming from the nucleus or damaged mitochondria (114).
Additional PRRs are C-type lectin receptors (CLRs) on the airway epithelial cells recognizing carbohydrates present on the pathogens and activating proinflammatory response (21, 103). The CLRs occur as membrane-anchored receptors like Dectin-1 recognizing Haemophilus influenzae and Aspergillus fumigatus infections (115, 116). The second form of CLRs are soluble collectins containing C-type lectin domain like SP-A and SP-D exhibiting antimicrobial activity through opsonization of bacterial, viral, and fungal pathogens (21). Formyl-peptide receptors (FPRs) are also expressed on the epithelial cells of the respiratory tract, where they recognize not only bacterial formylated peptides but also host-derived stimulants, such as mitochondrial proteins from damaged cells that chemoattract leukocytes promoting inflammation. Host LL-37 has also been shown to activate FPR2 (aka FPRL1) and is considered as a proinflammatory stimulus in chronic obstructive pulmonary disease (COPD). Depending on the local tissue environment, activation of FPRs can have a dual pro- or anti-inflammatory role, and it has been associated with the tissue regeneration and wound healing (105, 117).
Interestingly, it has recently been shown that PTMs of PRRs can be considered as contributing to a fine-tuning mechanism limiting inflammation; for instance, palmitoylation of NLR family pyrin domain containing 3 (NLRP3) prevents activation of inflammasome (118). Studies on how different PTMs affect PRRs downstream signaling in response to pathogens and during inflammation is one of the future directions for research in the host–pathogen interactions field.
Cell junctional complexes
The structural elements of the airway epithelial barrier integrity are tight (TJs) and adherens junctions (AJs) that determine the polarization of the epithelial cells into the apical and basolateral site providing physical barrier regulating paracellular flux through epithelial layers (119). Hence, the epithelial barrier integrity provided by TJs and AJs can be considered as part of the epithelial defense system (Figure 1). TJs and AJs are composed of transmembrane proteins, such as occludin, tricellulin, claudins, and junctional adhesion molecules (JAMs) and many others present in TJs together with E-cadherin and nectins in AJs. Transmembrane proteins of TJs interact with the proteins of the intracellular junctional plaque containing multiple interaction domains, such as zonula occludens-1 (ZO-1)—a well-defined protein from the junctional plaque complex—and its deletion is lethal in the mouse embryos. The examples of AJs intracellular junctional plaque proteins are α- and β- and p120 catenins and afadin (AF6) displaying similar functions. The intracellular junctional plaque proteins are coupled to cytoskeleton proteins, for example to actin through C-terminal domain of ZO-1 (119). The importance of the functional epithelial barrier sealing the deeper tissues from the external environment for the maintenance of the local tissue homeostasis seems to be explained by the epithelial barrier hypothesis (120). It highlights that the leaky epithelial layer caused by disruption of TJs and AJs by detergents, pollutants, allergens, and pathogens (121–125) contributes to increased incidence of allergies, asthma, and autoimmune diseases (120). The exposure to such environmental insults creates a positive feedback loop of further epithelial layer destruction. This process includes a subsequent translocation of the microbiota and opportunistic pathogens through a disrupted epithelial layer to the lamina propria that activates macrophages and T cells, ultimately leading to inflammation (120). During inflammatory response, cytokines cause further disintegration of tight junctions in the airway epithelium (126, 127), exacerbating the inflammatory response that may become a chronic state. Therefore, the disruption of the net of TJs and AJs can be considered as one of the early onsets of the disease. Hence, the maintenance of the epithelial barrier integrity is crucial for balanced innate and adaptive immune responses in the airway epithelium.
Autophagy
Autophagy is a complex process involving interaction of several different proteins, leading to engulfment of the cytoplasmic cargo during autophagosome formation and its subsequent degradation upon fusion with lysosomes. Autophagy is one of the key cellular processes maintaining balance in cells exposed to a constantly changing environment. It plays a major “housekeeping” function on the cellular level where it is responsible for the degradation of damaged organelles and unfolded/misfolded proteins (128). An important function of the autophagy process in respect to innate immunity is the degradation of invading pathogens, when the initial innate immunity barrier—mucus, HDPs, and TJs—was not sufficient to halt the pathogen entering the cells (129). Pathogenic P. aeruginosa (130), conidia of Aspergillus fumigatus (131), and influenza A virus (132) can be effectively eliminated or restricted by autophagy in airway epithelial cells. Therefore, autophagy can be considered as part of cell autonomous innate immunity and an intracellular defense mechanism of the “last chance” for the prevention of pathogen dissemination and progression of infectious disease (133). The commonly used cell model to study the role of autophagy in innate immunity are phagocytic professional cells, such as macrophages. However, the importance of autophagy in epithelial cells should be highlighted due to the vital role of epithelial cells in the first line of the host defense and epithelial homeostasis (Figure 1). During homeostasis, autophagy can control ciliogenesis and ciliary function by regulating the length of motile cilia in airway cells (134). The biogenesis of primary cilia is regulated by autophagic degradation of centriole and centriolar satellite protein OFD1 (135) and the function of motile cilia by degradation of kinesin family member 19 (Kif19)—an essential protein controlling ciliary length that should be inhibited to maintain the correct length of cilia (136).
Moreover, the importance of autophagy in human lung is demonstrated in CF patients whose autophagy is impaired due to aggregating Beclin-1. In the cell and animal models of CF, this phenotype can be rescued upon Beclin-1 restoration, suggesting a key role of autophagy in the lung homeostasis (137). Autophagy was shown as a central contributor to IL-13-mediated mucus hypersecretion by airway epithelial cells in COPD and asthma (138). Moreover, autophagy regulates apical localization of DUOX1 in airway epithelial cells and ROS production in response to chronic IL-13 exposure (139). Furthermore, autophagy maintains the airway progenitor cells pool and regulates cell differentiation for epithelial regeneration (140). In addition, one of the clinical symptoms of the Hermansky–Pudlak syndrome type 1, a rare genetic disorder impairing vesicle trafficking, is lung fibrosis and impaired innate immune antimicrobial responses due to amplified mechanistic target of rapamycin kinase (mTOR) signaling resulting in reduced bacterial clearance, indicating autophagy as a key cellular process in physiological and pathological conditions (141, 142). Autophagy in the airway epithelium can be considered as a double-edge sword because in particulate-matter-induced airway inflammation, autophagy contributes to the epithelial injury (143). The basic research on the autophagy process and its regulation in different conditions remain future perspectives for the development of novel strategies for treating respiratory tract diseases.
Epigenetics and innate immunity
Innate immune responses in the airway epithelium are also regulated by epigenetic modifiers of DNA methylation and post-translational modifications of histones. The epigenetic regulation of gene expression by modulation leading to chromatin opening allows for a rapid response to environmental changes. These processes are tightly regulated by the equilibrium of epigenetic enzymes and their counterparts, for example by histone acetyltransferases (HATs)—histone deacetyltransferases (HDACs) and DNA methyltransferases (DMTs). The importance of the DNA methylation status seems to be highlighted by the studies showing that DNA methylation pattern is changed in respiratory tract diseases, such as increased DNA methylation in NLRP3 gene of COPD patients (144). On the other hand, decreased methylation in the promoter region of TLR2 contributed to enhanced inflammatory responses in the airway epithelium of CF patients in response to bacterial peptidoglycan (145). Different PTMs of histones can modulate the front-line innate immune defenses in response to pathogen invasion (Figure 2). For example, acetylation of histones allows for more permissive chromatin structure facilitating gene expression. Upon stimulation with LPS, the activation of MAPK pathway leads to phosphorylation of H3S10 and additional acetylation of H3S10K14 at the promoter of IL12, leading to chromatin opening for NF-κB and induction of IL-12 expression (146). Furthermore, an increased acetylation of H3K18 was observed in STAT6 locus in the airway epithelial cells of asthmatic patients (147). Although that study did not show increased STAT6 expression, it is known that STAT6 signaling is affected in asthma and, together with elevated levels of IL-4 and IL-13 driven by Th2 immune responses, leads to mucus hypersecretion in mice, which is a characteristic for asthma, COPD, and CF (148). Moreover, the treatment of asthmatic airway epithelial cells with IL-4 and IL-13 caused the impairment of the airway epithelial barrier integrity concomitant with enhanced expression of HDACs (1 and 9) and sirtuins (SIRT6 and 7). Interestingly, inhibition of HDACs in asthmatic airway epithelial cells by quisinostat (class I and II HDAC inhibitor) improved the epithelial barrier integrity by increasing the expression of TJ proteins (149). On the other hand, inhibition of HDACs for example, by deletion of Hdac1 in mice exposed to the allergen caused more stable Th2 immune responses resulting in mucus hypersecretion by goblet cells, demonstrating an important role of HDAC1 in allergic diseases (150). The enzyme HDAC2 can suppress NF-κB and AP-1 signaling leading to the inhibition of proinflammatory response caused by stimulation of TLR4. Interestingly, the downregulation of HDAC2 in lungs of COPD patients and rats exposed to cigarette smoke suggest that HDAC2 expression is modulated in response to environmental factors and protects from inflammation and mucus hypersecretion during homeostasis (148, 151, 152). Unlike HDAC2, exposition to cigarette smoke increased expression of HDAC6 enhancing autophagy. Rapid protein turnover due to increased autophagy led to shortening of the ciliary length that contributes to impaired mucociliary clearance observed in COPD. These effects were diminished in Hdac6−/Y mice exposed to cigarette smoke, suggesting the inhibition of HDAC6 as a potential therapeutic target for COPD treatment (153, 154). Apart from HDAC6, many epigenetic modifiers are suggested to regulate autophagy processes (155), such as EZH2 methyltransferase responsible for the trimethylation of H3K27 shown to inhibit autophagy (156). The importance of EZH2 was demonstrated to regulate differentiation of airway epithelial stem cells that is aberrant in Ezh2-deficient mice resembling an altered airway epithelial cell differentiation typical for COPD (157). Overall, the epigenetic regulation of innate immune responses is intensively investigated, and the studies showing epigenetic regulation of HDPs in the lung epithelium are ongoing (158).
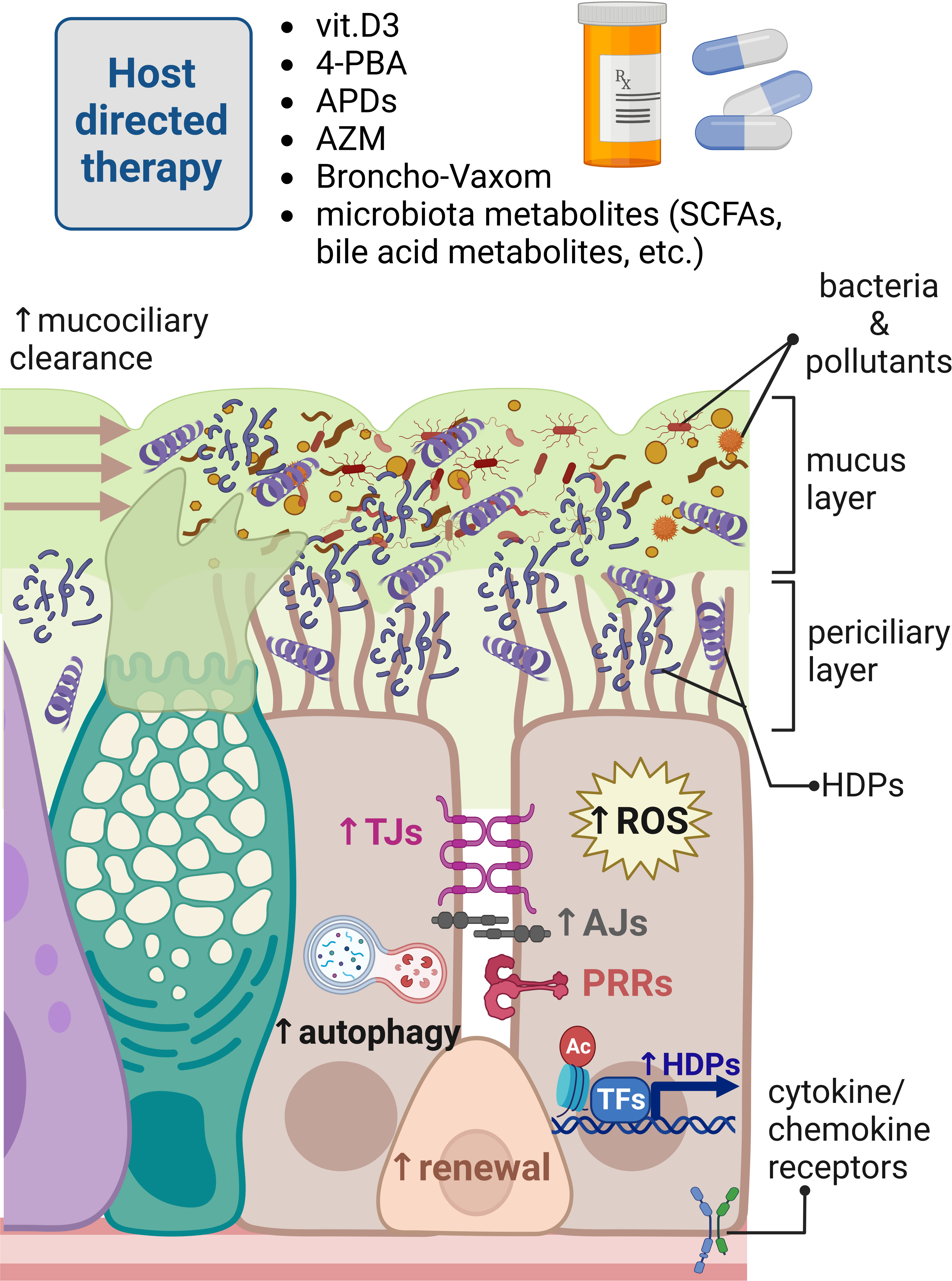
Figure 2 Modulation of innate immune responses in human respiratory epithelium for host-directed therapy. Different innate immunity inducers vitamin D3, 4-phenylbutyrate (4-PBA), aroylated phenylenediamines (APDs), Broncho-Vaxom (bacterial lysates), and microbiota metabolites can boost innate immune responses in the respiratory epithelium fending off pathogens invading mucosal surfaces of the respiratory tract. These responses are exerted by enhancement of the mucociliary clearance, removing dead bacteria and inhaled pollutants neutralized by increased expression of host defense peptides (HDPs) through the epigenetic modulation of histone epigenetic marks and transcriptional regulation. Innate immunity inducers and azithromycin (AZM) can also strengthen epithelial barrier integrity providing physical barrier for invading pathogens. Innate immunity modulators can increase autophagy and production of reactive oxygen species (ROS) for effective elimination of foreign molecules and microbes. Bacterial lysates, such as Broncho-Vaxom, can stimulate pathogen recognition receptors (PRRs), perhaps to train epithelial innate immunity through imprinting innate immune memory in renewing epithelial cells. Created with BioRender.com.
How pathogens subvert host innate immune defenses in lungs
Efficient innate immune responses in airway mucosa are essential for maintaining respiratory functions. The mucosal surface of the airways is also an initial site for pathogens interaction with the host. Many pathogens can subvert airway mucosal defense mechanisms and cause disease, especially in immunocompromised individuals. In this section, we will discuss those strategies based on the selected examples of respiratory pathogens that mainly concentrate on bacteria, as those were extensively studied throughout the years in comparison to viruses and fungi. Viral subversions mechanisms of host innate immune responses in lungs has recently gained more attention due to SARS-CoV-2 pandemic, while little is known about fungal strategies that seems to be relevant, especially in the development of severe fungal infections in immunocompromised individuals.
Bacteria
Pseudomonas aeruginosa (Pa) is an opportunistic pathogen usually harmless for healthy people; however, it can cause pneumonia in vulnerable immunocompromised individuals like patients with chronic respiratory diseases, such as CF, COPD, and hospitalized patients with supportive mechanical ventilation. Pa is known to use different strategies and virulence factors for the effective colonization of the host mucosal surfaces (159). The bacteria utilize flagellin for movement and pili for adhesion to the host cells. Once the host–pathogen adhesion is established, Pa injects bacterial toxins, such as ExoS to the host cells by type 3 secretion system (T3SS) causing acute infection (160). Furthermore, lung tissue damage in pneumonia is caused by disruption of the airway TJs and AJs integrity by Pa elastase that also degrades collectins (SP-A and SP-D), collagen, complement components, and LL-37 (159, 161). Pa rhamnolipids also disrupt TJs integrity in the airway epithelium facilitating Pa invasion (162). As an additional virulence factor, pyocyanin impairs ciliary function and inhibits host catalase, which contributes to the oxidative lung damage (163, 164). Furthermore, Pa competes for the iron source with host antimicrobial effectors, necessary for the activity of antimicrobial effector, lactoferrin, in the airway epithelium (165). Depending on the local environment of the host airway epithelium, especially during mucus clogging in CF and COPD, Pa can switch strategy of host colonization, from the invasive to a more adaptative one by formation of the biofilm. Quorum-sensing mechanisms allow for the communication of bacteria thriving in the biofilm and involve bacterial molecules, such as acyl homoserine lactones (AHL). At the same time, bacterial molecules involved in quorum sensing affect host cells; for example, AHL facilitates apoptosis of neutrophiles but not the host’s airway epithelial cells (166). In addition, 2-aminoacetophenone enhances the host’s HDAC1 activity suppressing proinflammatory response that facilitates bacterial survival (167). Moreover, Pa in biofilm produces alginate, a mucopolysaccharide used for bacterial encapsulation increasing bacterial fitness to persist on the mucosal surfaces of CF patients (168). These bacterial biofilm subversion and evasion mechanisms of the host’s innate immune defenses facilitate chronic Pa infection that can be very difficult to eliminate by antibiotics and contributes to development of antibiotic resistance. Many opportunistic pathogens have gained resistance to antibiotics routinely used for the treatment of infections. Therefore, Pa has been included in a group of ESKAPE pathogens (ESKAPE stands for the group of pathogens: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter spp.) that cause nosocomial infections (169).
Klebsiella pneumoniae (Kp) is another pathogen from the ESKAPE group causing pneumonia. Kp is well-known to contain several antibiotic resistance genes, including extended-spectrum beta-lactamase (ESBL), and carbapenemase encoding genes, which is referred to as a multidrug-resistant (MDR) Kp. Apart from gaining antibiotic resistance genes, Kp utilizes a variety of well-described strategies to evade host innate immune responses, especially affecting host innate immune cells (170), for instance, by interference with TLR signaling in macrophages exerted by targeting sterile alpha and TIR motif containing 1 (SARM1) protein to reduce inflammation and enhance IL-10 production (171). The interaction of Kp with the airway epithelial cells remains the first step of infection that is not well-characterized, and it is likely that Kp utilizes several strategies to compromise host epithelial front-line defenses. Kp secretes ytfL virulence factor that alters cytoskeleton organization in human and mouse airway epithelial cells by disassembling microtubules network (172). Kp translocation through airway epithelium mediated by induction of IFN-λ reduces the host airway epithelial barrier integrity that can facilitate bacterial invasion (173). Furthermore, the encapsulation of Kp with a polysaccharide coat has been considered as a determinant of bacterial virulence. Kp encapsulation limits bacterial binding by the complement component present on the airway mucosal surfaces and reduces bacterial clearance by the host epithelial cells (174). Recent studies showed that encapsulation helps Kp to overcome mouse innate immune defenses in the upper airways primed by microbiota through stimulation of IL-17A expression (175). Interestingly, a recent study indicates that asymptomatic colonization of the gut with MDR-Kp exacerbates pneumonia caused by Pa infection. Altered signaling on the gut–lung axis is caused by MDR-Kp-mediated dysbiosis resulting in reduced production of short-chain fatty acids (SCFAs) and reduced numbers of macrophages and DCs in the lung (176).
Streptococcus pneumoniae (Sp) is a respiratory pathogen frequently causing pneumonia (177). The virulence of Sp is associated with secretion of toxin, pneumolysin, exhibiting cytotoxic effect on the host cells accompanied with disintegration of TJs between host epithelial cells for Sp translocation (178). In addition, the cytotoxic effect of pneumolysin is caused by DNA damage associated with the cell cycle arrest (179). Another bacterial virulence factor is pyruvate oxidase SpxB responsible for H2O2 production and release of pneumolysin (180). Both virulence factors, pneumolysin with contribution of SpxB, were shown to reduce the host innate immune signaling through epigenetic mechanisms. Infection of mice and human airway epithelial cells with Sp led to the activation of the host phosphatase PP1 by its dephosphorylation, which further dephosphorylated histone 3 on serine 10 in the host cells promoting Sp intracellular expansion (181). Sp modifies a component of the cell wall, lipoteichoic acid, to increase its charge to resist host HDPs. Moreover, Sp serine protease PrtA is suggested to cleave HDPs (182, 183). The use of PrtA virulence factor as an antigen has been suggested to develop new vaccines in combination with other antigens to resolve problems of pneumococcal serotype specificity based on the fact that PrtA can also induce protective immunity in certain animal infection models (184). Another study by Biswas et al. showed that group A Streptococcus, mainly responsible for necrotizing fasciitis and rarely pneumonia, cleaves the host defense peptide LL-37 with ScpN protease into two shorter peptides that retained bactericidal properties. However, LL-37 cleavage products lost their immunomodulatory properties connected to the activation of P2X7 and EGFR signaling involved in neutrophil bacterial clearance and tissue regeneration, respectively (185, 186).
The respiratory pathogen Mycobacterium tuberculosis (Mtb) can bypass host immune defenses by hijacking host cell signaling in macrophages, neutrophiles, and DCs. This way, Mtb creates a favorable intracellular niche where bacteria can replicate and infect other cells or persist for decades in a latent state (187). In the initial step of bacterial colonization, Mtb infects respiratory epithelial cells and macrophages, while 7 days post-infection, the bacteria are present only in macrophages, indicating that bacteria do not replicate in epithelial cells (188). Interestingly, in the zebrafish model, a lipid component of Mycobacterium marinum outer membrane phthiocerol dimycocerosate (PDIM) spreads into the epithelial cell membrane and inhibits TLR/MyD88 signaling limiting recruitment of monocytes (189). In humans, Mtb selectively targets uptake by pulmonary macrophages that translocate to the lung interstitium and infect other types of cells. This process is mediated by Mtb virulence factor Esx-1 and the host IL-1β signaling (188). Despite activation of additional host innate immune components connected to the redox signaling, Mtb produces KatG and NuoG virulence proteins that neutralize and inhibit further production of ROS by macrophages and neutrophiles (190, 191). Moreover, CpsA allows Mtb to escape autophagic clearance in macrophages by blocking NADPH oxidase recruitment to the Mtb-containing phagosomes and activation of LC3-associated phagocytosis (192). Mtb creates its own intracellular niche for survival by altering host lipid metabolism, creating the formation of foamy macrophages with characteristic lipid droplets (193). Mechanisms of Mtb evasion and subversion of host immune responses in macrophages and other immune cells are thoroughly described in a recent review (187) that explains why only approximately three Mtb bacterial cells are sufficient for effective colonization of the host (194), indicating a highly advanced Mtb virulence system.
A better understanding of how bacteria breach the host innate defenses on the airway mucosal surfaces to further hijack the host cellular machinery for intracellular survival could result in the development of more efficient therapies. Studies on the molecular mechanism of bacterial virulence factors for host colonization are especially important in the context of immunocompromised individuals suffering from severe infections.
Viruses
Viruses use a variety of different strategies to compromise the host innate immune defenses and effectively replicate within the host cells or remain in the latent state. Due to the recent pandemic, the majority of research has concentrated on the strategies used by SARS-CoV-2 coronavirus to evade host innate immune responses (195). Yin et al. showed that the host IFN response mediated through intracellular PRRs—MDA5, LGP2, and NOD2—is delayed by several hours in comparison to the kinetics of the viral replication, indicating the viral-strategy-limiting effects of IFNs (196). Moreover, SARS-CoV-2 produces viral endonuclease EndoU that cleaves viral RNA and blocks phosphorylation of STAT1 and STAT2 and their nuclear translocation to inhibit host innate immune responses (197, 198). Subsequently, the virus produces ORF8 protein that disrupts PTMs of host histones promoting closed chromatin state and suppression of host innate anti-viral responses, allowing for viral replication (199). Another virus commonly causing respiratory tract infections, especially harmful for newborns and infants, is RSV, and there is no vaccine available (200). An effective RSV replication within host cells is achieved by initial induction of autophagy and then by blocking autophagosome–lysosome fusion in human airway epithelial cells. This strategy most likely allows to form a replication-favorable niche for the virus inside the autophagosome vesicle (201). The “mimicry” of host chemokines is used to promote infection by RSV, more precisely by the interaction of viral G protein binding receptor with CX3CL1 that facilitates RSV infection (202). The group of Rhinoviruses are the common group of pathogens attacking the host respiratory system causing cold (203). Human rhinoviruses C group (HRV-C) were shown to disrupt epithelial barrier integrity and to alter host metabolism towards glycolysis and fatty acid biosynthesis, facilitating viral replication (204). Rhinoviruses also target another RNA RLRs, such as STING trafficking into viral replication organelles through interaction with PI4P (205). Interestingly, recently, it has been shown that disruption of the circadian clock and expression of immune response genes encoding chemokine receptors (Ccr2, Ccr3, Ccr5, Ccr6, etc.) due to sleep deprivation make mice more susceptible to viral infections, highlighting the importance of environmental factors determining host immune responses against viral infections (206).
Fungi
Fungal pathogens have developed specific strategies to evade the host immune responses (207). Among them are C. albicans triggering oropharyngeal candidiasis, Aspergillus ssp. causing pulmonary aspergillosis, and Pneumocystis and Cryptococcus causing pneumonia (208). Candida albicans avoids host responses by neutralization of the host complement components by proteolytic cleavage with aspartyl proteases (209). Moreover, Aspergillus, Mucorales, and Candida spp. cause coronavirus disease (COVID)-associated fungal infections, and aspergillosis was the most prevalent in the group of patients treated with corticosteroids and tocilizumab (210). Cryptococcus neoformans evades host innate immune responses, for example, by encapsulation with polysaccharides glucuronoxylomannan (GXM) that inhibits NETs formation (211). Furthermore, C. neoformans also produces giant fungal cells, the so-called titan cells, to avoid phagocytosis and killing by ROS, hence considered as a fungal strategy used to establish pulmonary infections (212). Fungal infections are often a complication of the antibiotic treatment because of microbiota dysbiosis, where opportunistic fungal species, such as mentioned C. albicans, expand on the host mucosal surfaces forming a biofilm. Those biofilms composed of fungal and bacterial components are hard to eradicate, especially in CF and immunocompromised patients (24); therefore, development of new therapies against such biofilms is needed.
Modulation of lung innate immunity to overcome pathogen subversion mechanisms
Pathogens quickly adapt to the changing environment, and this includes the presence of antibiotics. The excessive use of antibiotics in the healthcare and animal husbandry has led to the selection for antimicrobial resistance (AMR), which resulted in a global problem that caused 4.95 million of AMR-associated deaths in 2019 (213). Antibiotic resistance occurs in most countries, and many opportunistic pathogens have gained resistance to antibiotics routinely used for the treatment of infections. It is estimated that 63.5% of all infections caused by multidrug-resistant strains were connected to the healthcare (214). Despite of the increasing social awareness on the proper antibiotic use, general reduction in prescribed antibiotics, and a better control of nosocomial infections, the spreading of AMR genes in some pathogens remained unchanged (215, 216). In addition, the development of new antibiotics is a long and costly process, taking several years until the new drug is introduced to the market resulting, in only few developed drugs in the last decades (217). Therefore, alternative strategies for the treatment of infectious diseases have gained more attention. By deciphering molecular pathways of host innate immunity and pathogen’s infection strategies, an opportunity of modulating these mechanisms might result in the development of novel alternative treatment approaches, limiting and/or reducing the use of antibiotics and thereby reducing the selection of AMR strains. Therefore, in our research, we postulate the concept of host-directed therapy (Figure 2) executed through the modulation of epithelial and macrophages innate immune responses by enhancing expression of HDPs, improvement of the epithelial barrier integrity, and stimulation of autophagy. Our concept of host-directed therapy can be extended to the restoration of mucociliary clearance and ion balance, training innate immune system via controlled TLR stimulation and epigenetic modulation of host innate immune responses. The approach of host-directed therapy does not anticipate replacement of the antibiotics—it is rather a supplementary treatment that can reduce time of antibiotic use in case of persistent infections requiring a long antibiotic treatment, such as tuberculosis and infections requiring prolonged treatment due to poor bioavailability of the drug in the tissue (218). In this section, we will discuss different angles of host-directed therapy for modulation of lung epithelial innate immune responses.
One of the directions in the field is the direct use of HDPs and their analogues, mainly for topical administration (219). Synthetic analogues of HDPs have also been tested in a mouse model of severe lung infection caused by highly virulent P. aeruginosa. Nasal instillation of IDR-1002 peptide followed by bacterial infection reduced bacterial count in bronchoalveolar fluid and inflammatory response, indicating therapeutic potential of preventive administration of HDP analogues (220). Although promising, this approach usually uses only one peptide monotherapy, which carries a risk of development bacterial resistance to HDPs, which has been claimed to be limited, however a possible scenario (221). Therefore, the use of HDPs mixture would be a better strategy than a single peptide monotherapy to avoid AMR development, which seems to be highlighted by the fact that in physiological conditions, pathogens usually encounter a mixture of different antimicrobial effectors present on mucosal surfaces. Another aspect is the cost of the production of synthetic peptides and their purification on a bigger scale. Therefore, we suggest an alternative approach by inducing expression of natural HDPs in the epithelial cells and tissue-residing immune cells, which has several advantages in comparison to using synthetic HDPs and their derivatives. First, several antimicrobial effectors are induced, at the same time limiting the risk for the selection of antibiotic resistance. Second, using different innate immune modulators allows for the precise regulation of the HDPs induction and its cessation if needed. Third, the use of different non-peptide inducers for host-directed therapy is expected to reduce the cost of production, making it more accessible for pharmacological use. Those innate immune inducers/modulators were explored by our research group and collaborators and initially included nutrients and bacterial metabolites (like vitamin D3 and butyrate) and further expanded to synthetic chemical compounds (such as aroylated phenylenediamines (APDs)) (Figure 2) (218). Among the nutrients, vitamin D3 was shown to induce cathelicidin expression via activating vitamin D receptor (VDR) from the group of nuclear receptors binding to the vitamin D responsive elements (VDRE) present in human cathelicidin gene promoter (222, 223). Lung epithelial cells were shown to express the enzyme CYP27B1 involved in conversion of a precursor 25-hydroxyvitamin D3 to an active 1α,25-dihydroxyvitamin D3 that induced cathelicidin expression, indicating the importance of the vitamin D3 levels for mucosal antimicrobial defenses (30). Interestingly, VDREs are absent in mouse CRAMP promoter, making the translation of research on vitamin D3 inducer to mouse models difficult. For that reason, the novel transgenic model of humanized mice was established recently and contains human VDRE in the promoter of mouse CRAMP gene, opening a new avenue for the studies on vitamin D3 (224). Another inducer of cathelicidin expression in lung epithelium and macrophages is butyrate, a short-chain fatty acid produced by microbiota commensal bacteria from Firmicutes phylum (225). Butyrate constitutes the primary energy source for colonocytes, exerting local immunomodulatory effects on the colonic epithelia, tissue residing immune cells, and gastrointestinal (GI)-distant organs because butyrate is distributed with blood similar to other microbiota metabolites (218, 226). Butyrate is known from its strong odor; therefore, 4-phenylbuturate (hereafter phenylbutyrate or 4-PBA) was used in the studies as an odorless butyrate derivative, which is an FDA-approved drug for treatment of the urea cycle disorders used as ammonia scavenger (227). Phenylbutyrate was shown to induce cathelicidin expression in the airway epithelium (Figure 2) and induced autophagy in Mtb-infected macrophages through induction of LL-37 acting via P2X7 receptor (228, 229). Phenylbutyrate also counteracted the downregulation of cathelicidin expression in lungs of Shigella-infected rabbits, mitigating pathogens strategy of effective host colonization (230, 231). Phenylbutyrate is an inhibitor of histone deacetylases (HDACs)—histone-modifying enzymes facilitating chromatin opening state by acetylation of histones. Although CAMP gene expression was induced by phenylbutyrate, the increased acetylation of histone 3 and histone 4 was not observed in the promoter of CAMP gene (228). Therefore, the detailed mechanism of CAMP induction by phenylbutyrate remains unknown, perhaps requiring analysis of more specific histone modifications in CAMP promoter. The combination of 1,25-dihydroxyvitamin D3 and 4-PBA is so far the most potent inducer of cathelicidin expression in the human model of the lung epithelium, most likely by combing transcriptional regulation of vitamin D3 and epigenetic modulation by 4-PBA. Importantly, the combination of vitamin D3 and 4-PBA has clinical translational potential and was shown to be effective in the clinical trial of the Mtb treatment combined with standard antibiotics. Patients with tuberculosis receiving phenylbutyrate and vitamin D3 showed faster clearance of Mtb in sputum samples than the group receiving placebo, indicating beneficial effects of host-directed therapy combined with antibiotics (232, 233).
Moreover, in respect to the lung epithelium, other inducers of HDPs were tested from a novel class of compounds called aroylated phenylenediamines (APDs) (Figure 2). Two synthetic APD-compounds, derivatives of Entinostat, which is another HDAC inhibitor known to induce CAMP gene expression, were shown to induce expression of antimicrobial effectors cathelicidin, calprotectin, lipocalin, and defensins. Of note, APDs were less toxic than Entinostat and did not induce proinflammatory responses (32, 234). Furthermore, innate immune inducers 4-PBA, vitamin D3, and APD compound were able to sensitize MDR K. pneumoniae to conventional antibiotics. The intracellular bacterial killing mechanism in infected macrophages was cathelicidin dependent for 4-PBA and vitamin D3 and ROS dependent for APD compound (235). This approach presents another angle on host-directed therapy, utilizing modulation of host innate immune responses and sensitization of multidrug-resistant pathogens to conventional antibiotics. Moreover, APD-compound induced autophagy in the differentiated lung epithelial cells through the epigenetic modulation of the H3K27 acetylation and AMP-activated protein kinase (AMPK) signaling (236). The preventive treatment of differentiated lung epithelial layers with APD compound enhanced the epithelial barrier integrity provided by tight junction’s proteins occludin and ZO-1 when differentiated monolayer cells were challenged with P. aeruginosa-conditioned medium (32). Interestingly, one of the most often prescribed antibiotics, azithromycin, was shown independently of its microbicidal properties to modulate host’s epithelial cell responses by strengthening epithelial integrity in the lung (Figure 2). This can be paradoxically considered as a positive side effect of azithromycin treatment that benefits cystic fibrosis patients by improving their condition (237). Azithromycin was shown to enhance epithelial barrier integrity by increasing trans-epithelial electrical resistance (TEER) and counteract the disruptive effect of P. aeruginosa-conditioned medium (238). Furthermore, azithromycin was shown to have a barrier protective effect and counteract proinflammatory response inflicted on the bronchial epithelia by cyclical mechanical stress during mechanical ventilation generated by a cyclical pressure air–liquid interface device (CPAD) (239). The supporting evidence suggest that the protective effect of azithromycin on lung epithelia is exerted by increased lamellar body formation and stimulation of epidermal differentiation (240). However, azithromycin is an antibiotic; therefore, prolonged use of azithromycin to strengthen the epithelial barrier is restricted because of the risk for antibiotic resistance development. Importantly, the mechanism behind strengthening of the lung epithelial barrier integrity by azithromycin and APDs remain unknown, and it is a subject of the ongoing research (32, 235, 236, 239, 240). On the contrary, vitamin D3 did not have this functional effect on the epithelial barrier strengthening while administered over the course of lung epithelial differentiation. Instead, it led to thickening of the lung epithelial layer in vitro displaying features of squamous metaplasia, indicating that this effect in the lung is tissue specific and is contrary to what has been observed in the gut (241). The group of Jun Sun showed that vitamin D3 regulates expression of a tight junction protein claudin-2, demonstrating the potential of VDR to regulate gut epithelial barrier integrity (242, 243). Different effects of innate immune inducers have been described depending on the tissue type, which may suggest possible epigenetic regulation of the epithelial barrier function. This concept seems to be additionally supported by the fact that many different respiratory tract diseases are associated with changes in the expression of histone-modifying enzymes, such as increased expression of histone deacetylases, HDACs (1 and 9) and sirtuins (SIRT6 and 7), in asthmatic bronchial epithelial cells. Inhibition of the HDACs with quisinostat (JNJ-26481585) in bronchial epithelial cells from asthmatic patients and allergic rhinitis improved epithelial barrier integrity by affecting expression and reorganization of TJs (149, 244). Recently, azithromycin was shown to attenuate wheezing in patients recovering from pulmonary inflammation. Azithromycin treatment helped to reduce time of wheezing for those patients, and these changes were associated with reduced expression of EZH2 (histone methyltransferase responsible for methylation of H3K27me3), reduced methylation of H3K27me3, and reduced expression of p65, suggesting that azithromycin exerts anti-inflammatory properties through epigenetic regulation (245). Overall, epigenetic modulation is part of the natural physiological regulation of innate immune responses in epithelial tissues and immune cells exerted by microbiota-produced metabolites of dietary products, highlighting the future directions for modulation of host innate immune responses through epigenetic therapies utilizing natural and synthetic epigenetic modulators. It is important to keep in mind that epigenetic therapies may exert off-target effects on other tissues, making the precise regulation difficult to control. However, transient epigenetic modulation over a short period of time with relatively rapidly degraded synthetic compounds and natural products, such as microbiota-produced butyrate, seems to be a reasonable approach.
In line with host-directed therapy is the concept of trained immunity responses for the treatment and prevention of respiratory tract infections. A type of immunotherapy with Broncho-Vaxom, a lyophilizate of the dead bacterial strains causing respiratory tract infections, such as K. pneumoniae, S. aureus, Streptococcus pyogenes, and Neisseria catarrhalis, is used to train immune responses in patients suffering from recurrent infections of upper and lower respiratory tract (Figure 2). Broncho-Vaxom showed efficiency in reducing recovery time and course of disease in pediatric patients with recurrent respiratory tract infections (246, 247). Beneficial effects of Broncho-Vaxom® (OM-85 BV) on human sinonasal epithelial cells were mediated through stimulation of the taste-receptor T2R signaling, leading to the production of nitric oxide (NO) responsible for direct bacterial killing and increased ciliary beating (248). Treatment of bronchial epithelial cells with bacterial lysates of Broncho-Vaxom protects epithelia from SARS-CoV-2 entry by reducing expression of host receptors used by the virus such as angiotensin-converting enzyme 2 (ACE2) (249, 250), suggesting that this form of host-directed therapy can be used as a preventive strategy to limit acute respiratory disease, for example in constantly exposed health workers (ClinicalTrials.gov Identifier: NCT04496245).
The concept of trained immunity refers to innate immunological memory previously attributed only to the adaptive immune responses. Trained immunity boosts secondary responses to infections or sterile inflammation after initial contact with the stimuli for the next faster and more efficient host responses. Although trained immunity responses are T- and B-cell independent, they complement the adaptive immune responses to maximize chances for the host survival. The innate immunological memory can be achieved by stimulation of innate immune cells, such as macrophages, NK cells, DCs, fibroblasts, and tissue-specific stem cells (251, 252). The primary example of trained immunity is Bacillus Calmette–Guérin (BCG) vaccination used routinely in vaccination against tuberculosis and shown to reprogram hematopoietic stem cells to differentiate towards monocytes/macrophages with enhanced capabilities of Mtb clearance. Unlike subcutaneous BCG vaccination, intravenous BCG administration enhanced myelopoiesis and rewired epigenetic program in bone-marrow-derived macrophages connected to the changes in the H3K27ac and H3K4me3 marks for more efficient Mtb clearance that was IFNγ dependent (253). Microbiota metabolites have also been identified as elicitors of trained immunity. One of them is butyrate that affects the trajectory of antimicrobial responses in macrophages by imprinting antimicrobial program in differentiating macrophages through HDAC3 inhibition, resulting in the induction of calprotectin and enhanced bacterial clearance through autophagy (254). Another microbiota metabolite, deoxycholic acid (DCA), a secondary bile acid detected in the bloodstream, is shown to enhance differentiation of granulocyte-monocyte precursors in the bone marrow through epigenetic alterations, providing innate protection against parasite E. histolytica. Interestingly, DCA in sera of children from Bangladesh who previously have documented history of amebiasis had lower levels of DCA than those who never suffered from infection (255). Trained immunity was also described in the lung in the context of the allergic inflammatory memory inflicted on basal lung stem cells by IL-4 and IL-13 exposure. The chronic exposure of respiratory epithelial progenitor cells to the inflammatory type 2 immune responses shifts their differentiation program causing epithelial barrier dysfunction observed in chronic allergic diseases, resulting in rhinosinusitis and taking more extreme form of nasal polyps (256). Overall, trained immunity can be achieved by exposition of tissue-specific stem cells and their progenitors to different stimuli imprinting innate immune responses through metabolic and epigenetic reprogramming (257).
Future perspectives
Innate immunity of the lung epithelial surfaces is a complex system working together with the adaptive immune responses for host defense and survival. Many aspects of the innate immune regulation and the link between innate and adaptive immune responses remain to be further elucidated. These include characterization of the role of new types of cells in the respiratory epithelium identified by single-cell RNA sequencing in shaping local innate immune responses and epigenetic modulation of such responses. An exciting avenue is the exploration of how different microbiota metabolites shape host immune responses in lung epithelial cells for better protection against pathogens with defining molecular mechanism that can be further extended to the development of the synthetic compounds for host-directed therapy. Defining pathogen’s effectors subverting host innate immune responses, especially in the context of compromised host innate immunity, include future steps of interest for the development of new treatment strategies. Furthermore, the concept of trained immunity mainly characterized in respect to immune cells remains to be further investigated in lung epithelial cells answering the questions on programming our epithelial cells, how innate immune memory confers to better host protection mechanisms, and whether there are any links to the development of chronic inflammatory diseases with defining environmental stimuli that shape such responses.
Author contributions
The conceptualization of the manuscript was based on ITM’s PhD dissertation thesis from year 2021. ITM wrote and edited the manuscript including figures created with BioRender.com under license agreement. GHG commented and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
GHG was supported by grants from Icelandic Research Fund (RANNÍS) and University of Iceland Research Fund.
Conflict of interest
GHG is a founder and stockholder in Akthelia Pharmaceuticals that holds a patent on APD compounds Patent No. US 9,957,226 B2.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol (2015) 16:27–35. doi: 10.1038/ni.3045
2. Zepp JA, Morrisey EE. Cellular crosstalk in the development and regeneration of the respiratory system. Nat Rev Mol Cell Biol (2019) 20:551–66. doi: 10.1038/s41580-019-0141-3
3. Hewitt RJ, Lloyd CM. Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol (2021) 21:347–62. doi: 10.1038/s41577-020-00477-9
4. Davis JD, Wypych TP. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol (2021) 14:978–90. doi: 10.1038/s41385-020-00370-7
5. Deprez M, Zaragosi LE, Truchi M, Becavin C, García SR, Arguel MJ, et al. A single-cell atlas of the human healthy airways. Am J Respir Crit Care Med (2020) 202:1636–45. doi: 10.1164/rccm.201911-2199OC
6. Reynolds SD, Malkinson AM. Clara Cell: progenitor for the bronchiolar epithelium. Int J Biochem Cell Biol (2010) 42:1–4. doi: 10.1016/j.biocel.2009.09.002
7. Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell (2009) 4:525–34. doi: 10.1016/j.stem.2009.04.002
8. Iwasaki A, Foxman EF, Molony RD. Early local immune defences in the respiratory tract. Nat Rev Immunol (2017) 17:7–20. doi: 10.1038/nri.2016.117
9. Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, et al. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest (2009) 119:2914–24. doi: 10.1172/JCI39731
10. Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U.S.A. (2009) 106:12771–5. doi: 10.1073/pnas.0906850106
11. Ruysseveldt E, Martens K, Steelant B. Airway basal cells, protectors of epithelial walls in health and respiratory diseases. Front Allergy (2021) 2:787128. doi: 10.3389/falgy.2021.787128
12. Hansson GC. Mucins and the microbiome. Annu Rev Biochem (2020) 89:769–93. doi: 10.1146/annurev-biochem-011520-105053
13. Garcıá SR, Deprez M, Lebrigand K, Cavard A, Paquet A, Arguel MJ, et al. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development (2019) 146:dev177428. doi: 10.1242/dev.177428
14. Ualiyeva S, Hallen N, Kanaoka Y, Ledderose C, Matsumoto I, Junger WG, et al. Airway brush cells generate cysteinyl leukotrienes through the ATP sensor P2Y2. Sci Immunol (2020) 43):eaax7224. doi: 10.1126/sciimmunol.aax7224
15. Strine MS, Wilen CB. Tuft cells are key mediators of interkingdom interactions at mucosal barrier surfaces. PloS Pathog (2022) 18:e1010318. doi: 10.1371/journal.ppat.1010318
16. Tizzano M, Gulbransen BD, Vandenbeuch A, Clapp TR, Herman JP, Sibhatu HM, et al. Nasal chemosensory cells use bitter taste signaling to detect irritants and bacterial signals. Proc Natl Acad Sci U.S.A. (2010) 107:3210–5. doi: 10.1073/pnas.0911934107
17. Branchfield K, Nantie L, Verheyden JM, Sui P, Wienhold MD, Sun X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science (2016) 351:707–10. doi: 10.1126/science.aad7969
18. Harkema JR, Carey SA, Wagner JG. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol (2006) 34:252–69. doi: 10.1080/01926230600713475
19. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature (2018) 560:319–24. doi: 10.1038/s41586-018-0393-7
20. Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature (2018) 560:377–81. doi: 10.1038/s41586-018-0394-6
21. Leiva-Juárez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol (2018) 11:21–34. doi: 10.1038/mi.2017.71
22. Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol (2009) 30:131–41. doi: 10.1016/J.IT.2008.12.003
23. Mansour SC, Pena OM, Hancock REW. Host defense peptides: front-line immunomodulators. Trends Immunol (2014) 35:443–50. doi: 10.1016/J.IT.2014.07.004
24. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discovery (2020) 19:311–32. doi: 10.1038/s41573-019-0058-8
25. Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem (1996) 238:325–32. doi: 10.1111/j.1432-1033.1996.0325z.x
26. Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U.S.A. (1995) 92:195–9. doi: 10.1073/PNAS.92.1.195
27. Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, et al. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J (2006) 20:2068–80. doi: 10.1096/fj.06-6075com
28. Sørensen OE, Follin P, Johnsen AH, Calafat J, Sandra Tjabringa G, Hiemstra PS, et al. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood (2001) 97:3951–9. doi: 10.1182/blood.V97.12.3951
29. Murakami M, Lopez-Garcia B, Braff M, Dorschner RA, Gallo RL. Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol (2004) 172:3070–7. doi: 10.4049/jimmunol.172.5.3070
30. Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, Hunninghake GW. Respiratory epithelial cells convert inactive vitamin d to its active form: potential effects on host defense. J Immunol (2008) 181:7090–9. doi: 10.4049/jimmunol.181.10.7090
31. Kulkarni NN, Yi Z, Huehnken C, Agerberth B, Gudmundsson GH. Phenylbutyrate induces cathelicidin expression via the vitamin d receptor: linkage to inflammatory and growth factor cytokines pathways. Mol Immunol (2015) 63:530–9. doi: 10.1016/J.MOLIMM.2014.10.007
32. Myszor IT, Parveen Z, Ottosson H, Bergman P, Agerberth B, Strömberg R, et al. Novel aroylated phenylenediamine compounds enhance antimicrobial defense and maintain airway epithelial barrier integrity. Sci Rep (2019) 9:7114. doi: 10.1038/s41598-019-43350-z
33. Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U.S.A. (1998) 95:9541. doi: 10.1073/PNAS.95.16.9541
35. Diamond G, Zasloff M, Eck H, Brasseur M, Lee Maloy W, Bevins CL. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: peptide isolation and cloning of a cDNA. Proc Natl Acad Sci U.S.A. (1991) 88:3952–6. doi: 10.1073/pnas.88.9.3952
36. Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, López-Boado YS, Stratman JL, et al. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science (1999) 286:113–7. doi: 10.1126/science.286.5437.113
37. Selsted M. θ-defensins: cyclic antimicrobial peptides produced by binary ligation of truncated α-defensins. Curr Protein Pept Sci (2005) 5:365–71. doi: 10.2174/1389203043379459
38. Currie SM, Findlay EG, McHugh BJ, Mackellar A, Man T, Macmillan D, et al. The human cathelicidin LL-37 has antiviral activity against respiratory syncytial virus. PloS One (2013) 8(8):e73659. doi: 10.1371/journal.pone.0073659
39. Sochacki KA, Barns KJ, Bucki R, Weisshaar JC. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc Natl Acad Sci U.S.A. (2011) 108:E77–81. doi: 10.1073/pnas.1101130108
40. Rosenfeld Y, Papo N, Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides: peptide properties and plausible modes of action. J Biol Chem (2006) 281:1636–43. doi: 10.1074/jbc.M504327200
41. Schneider VAF, Coorens M, Tjeerdsma-van Bokhoven JLM, Posthuma G, van Dijk A, Veldhuizen EJA, et al. Imaging the antistaphylococcal activity of CATH-2: mechanism of attack and regulation of inflammatory response. mSphere (2017) 2(6):e00370–17. doi: 10.1128/msphere.00370-17
42. Sass V, Schneider T, Wilmes M, Körner C, Tossi A, Novikova N, et al. Human β-defensin 3 inhibits cell wall biosynthesis in staphylococci. Infect Immun (2010) 78:2793–800. doi: 10.1128/IAI.00688-09
43. Tripathi S, Verma A, Kim E-J, White MR, Hartshorn KL. LL-37 modulates human neutrophil responses to influenza a virus. J Leukoc Biol (2014) 96:931–8. doi: 10.1189/jlb.4a1113-604rr
44. He M, Zhang H, Li Y, Wang G, Tang B, Zhao J, et al. Cathelicidin-derived antimicrobial peptides inhibit zika virus through direct inactivation and interferon pathway. Front Immunol (2018) 9:722. doi: 10.3389/fimmu.2018.00722
45. Sousa FH, Casanova V, Findlay F, Stevens C, Svoboda P, Pohl J, et al. Cathelicidins display conserved direct antiviral activity towards rhinovirus. Peptides (2017) 95:76–83. doi: 10.1016/j.peptides.2017.07.013
46. Ordonez SR, Amarullah IH, Wubbolts RW, Veldhuizen EJA, Haagsman HP. Fungicidal mechanisms of cathelicidins LL-37 and CATH-2 revealed by live-cell imaging. Antimicrob Agents Chemother (2014) 58:2240–8. doi: 10.1128/AAC.01670-13
47. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature (2002) 415:389–95. doi: 10.1038/415389a
48. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol (2003) 3:710–20. doi: 10.1038/nri1180
49. Zhang H, Porro G, Orzech N, Müllen B, Liu M, Slutsky AS. Neutrophil defensins mediate acute inflammatory response and lung dysfunction in dose-related fashion. Am J Physiol Lung Cell Mol Physiol (2001) 280(5):L947–54. doi: 10.1152/ajplung.2001.280.5.l947
50. Engelberg Y, Landau M. The human LL-37(17-29) antimicrobial peptide reveals a functional supramolecular structure. Nat Commun (2020) 11:1–10. doi: 10.1038/s41467-020-17736-x
51. Tayeb-Fligelman E, Tabachnikov O, Moshe A, Goldshmidt-Tran O, Sawaya MR, Coquelle N, et al. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science (2017) 355:831–3. doi: 10.1126/science.aaf4901
52. Niyonsaba F, Iwabuchi K, Matsuda H, Ogawa H, Nagaoka I. Epithelial cell-derived human b-defensin-2 acts as a chemotaxin for mast cells through a pertussis toxin-sensitive and phospholipase c-dependent pathway. Int Immunl (2002) 14:421–6. doi: 10.1093/intimm/14.4.421
53. De Yang B, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med (2000) 192:1069–74. doi: 10.1084/jem.192.7.1069
54. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, et al. The human antimicrobial and chemotactic peptides LL-37 and α-defensins are expressed by specific lymphocyte and monocyte populations. Blood (2000) 96:3086–93. doi: 10.1182/BLOOD.V96.9.3086
55. Van Wetering S, Mannesse-Lazeroms SPG, Van Sterkenburg MAJA, Daha MR, Dijkman JH, Hiemstra PS. Effect of defensins on interleukin-8 synthesis in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (1997) 272(5 Pt 1):L888–96. doi: 10.1152/ajplung.1997.272.5.l888
56. Mookherjee N, Brown KL, Bowdish DME, Doria S, Falsafi R, Hokamp K, et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J Immunol (2006) 176:2455–64. doi: 10.4049/jimmunol.176.4.2455
57. McHugh BJ, Wang R, Li H-N, Beaumont PE, Kells R, Stevens H, et al. Cathelicidin is a “fire alarm”, generating protective NLRP3-dependent airway epithelial cell inflammatory responses during infection with Pseudomonas aeruginosa. PloS Pathog (2019) 15:e1007694. doi: 10.1371/journal.ppat.1007694
58. Coorens M, Schneider VAF, de Groot AM, van Dijk A, Meijerink M, Wells JM, et al. Cathelicidins inhibit Escherichia coli –induced TLR2 and TLR4 activation in a viability-dependent manner. J Immunol (2017) 199:1418–28. doi: 10.4049/jimmunol.1602164
59. Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson-Jones L, et al. The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun (2021) 12:1–16. doi: 10.1038/s41467-021-21533-5
60. Al-Adwani S, Wallin C, Balhuizen MD, Veldhuizen EJA, Coorens M, Landreh M, et al. Studies on citrullinated LL-37: detection in human airways, antibacterial effects and biophysical properties. Sci Rep (2020) 10:1–14. doi: 10.1038/s41598-020-59071-7
61. Wong A, Bryzek D, Dobosz E, Scavenius C, Svoboda P, Rapala-Kozik M, et al. A novel biological role for peptidyl-arginine deiminases: citrullination of cathelicidin LL-37 controls the immunostimulatory potential of cell-free DNA. J Immunol (2018) 200:2327–40. doi: 10.4049/jimmunol.1701391
62. Wu H, Santoni-Rugiu E, Ralfkiaer E, Porse BT, Moser C, Høiby N, et al. Lipocalin 2 is protective against E. coli pneumonia. Respir Res (2010) 11:96. doi: 10.1186/1465-9921-11-96
63. Cramer EP, Dahl SL, Rozell B, Knudsen KJ, Thomsen K, Moser C, et al. Lipocalin-2 from both myeloid cells and the epithelium combats Klebsiella pneumoniae lung infection in mice. Blood (2017) 129:2813–7. doi: 10.1182/blood-2016-11-753434
64. Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder JM. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol (2004) 6:57–64. doi: 10.1038/ni1142
65. Andresen E, Lange C, Strodthoff D, Goldmann T, Fischer N, Sahly H, et al. S100A7/psoriasin expression in the human lung: unchanged in patients with COPD, but upregulated upon positive S. aureus detection. BMC Pulm Med (2011) 11:1–10. doi: 10.1186/1471-2466-11-10/FIGURES/7
66. Bai H, Si L, Jiang A, Belgur C, Zhai Y, Plebani R, et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Nat Commun (2022) 13:1–17. doi: 10.1038/s41467-022-29562-4
67. Williams SE, Brown TI, Roghanian A, Sallenave JM. SLPI and elafin: one glove, many fingers. Clin Sci (2006) 110:21–35. doi: 10.1042/CS20050115
68. Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, Dijkman JH. Antibacterial activity of antileukoprotease. Infect Immun (1996) 64:4520–4. doi: 10.1128/iai.64.11.4520-4524.1996
69. Simpson AJ, Maxwell AI, Govan JRW, Haslett C, Sallenave JM. Elafin (elastase-specific inhibitor) has anti-microbial activity against gram-positive and gram-negative respiratory pathogens. FEBS Lett (1999) 452:309–13. doi: 10.1016/S0014-5793(99)00670-5
70. Henriksen PA, Hitt M, Xing Z, Wang J, Haslett C, Riemersma RA, et al. Adenoviral gene delivery of elafin and secretory leukocyte protease inhibitor attenuates NF-κB-Dependent inflammatory responses of human endothelial cells and macrophages to atherogenic stimuli. J Immunol (2004) 172:4535–44. doi: 10.4049/jimmunol.172.7.4535
71. Harder J, Schröder JM. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J Biol Chem (2002) 277:46779–84. doi: 10.1074/JBC.M207587200
72. Amatngalim GD, van Wijck Y, de Mooij-Eijk Y, Verhoosel RM, Harder J, Lekkerkerker AN, et al. Basal cells contribute to innate immunity of the airway epithelium through production of the antimicrobial protein RNase 7. J Immunol (2015) 194:3340–50. doi: 10.4049/JIMMUNOL.1402169
73. Torres-Juarez F, Touqui L, Leon-Contreras J, Rivas-Santiago C, Enciso-Moreno JA, Hernández-Pando R, et al. RNase 7 but not psoriasin nor sPLA2-IIA associates with Mycobacterium tuberculosis during airway epithelial cell infection. Pathog Dis (2018) 76(2):1–8. doi: 10.1093/FEMSPD/FTY005
74. Han SH, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc (2015) 12:765–74. doi: 10.1513/AnnalsATS.201411-507FR
75. Cheng G, Ueda T, Nakajima H, Nakajima A, Kinjyo S, Motojima S, et al. Suppressive effects of SP-a on ionomycin-induced IL-8 production and release by eosinophils. Int Arch Allergy Immunol (1998) 117:59–62. doi: 10.1159/000053574
76. Chu HW, Thaikoottathil J, Rino JG, Zhang G, Wu Q, Moss T, et al. Function and regulation of SPLUNC1 protein in mycoplasma infection and allergic inflammation. J Immunol (2007) 179:3995–4002. doi: 10.4049/jimmunol.179.6.3995
77. Cole AM, Ganz T, Liese AM, Burdick MD, Liu L, Strieter RM. Cutting edge: IFN-inducible ELR – CXC chemokines display defensin-like antimicrobial activity. J Immunol (2001) 167:623–7. doi: 10.4049/jimmunol.167.2.623
78. Long X-B, Hu S, Wang N, Zhen H-T, Cui Y-H, Liu Z. Clara Cell 10-kDa protein gene transfection inhibits NF-κB activity in airway epithelial cells. PloS One (2012) 7:e35960. doi: 10.1371/journal.pone.0035960
79. Wang S-Z, Rosenberger CL, Bao Y-X, Stark JM, Harrod KS. Clara Cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J Immunol (2003) 171:1051–60. doi: 10.4049/jimmunol.171.2.1051
80. Zhu L, An L, Ran D, Lizarraga R, Bondy C, Zhou X, et al. The club cell marker SCGB1A1 downstream of FOXA2 is reduced in asthma. Am J Respir Cell Mol Biol (2019) 60(6):695–704. doi: 10.1165/rcmb.2018-0199OC
81. Zissler UM, Jakwerth CA, Guerth F, Lewitan L, Rothkirch S, Davidovic M, et al. Allergen-specific immunotherapy induces the suppressive secretoglobin 1A1 in cells of the lower airways. Allergy: Eur J Allergy Clin Immunol (2021) 76:2461–74. doi: 10.1111/all.14756
82. Xu M, Yang W, Wang X, Nayak DK. Lung secretoglobin Scgb1a1 influences alveolar macrophage-mediated inflammation and immunity. Front Immunol (2020) 11:584310. doi: 10.3389/fimmu.2020.584310
83. Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature (2012) 487:109–13. doi: 10.1038/nature11130
84. Shah VS, Meyerholz DK, Tang XX, Reznikov L, Alaiwa MA, Ernst SE, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science (2016) 351:503–7. doi: 10.1126/science.aad5589
85. Carnesecchi S, Deffert C, Pagano A, Garrido-Urbani S, Métrailler-Ruchonnet I, Schäppi M, et al. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med (2009) 180:972–81. doi: 10.1164/rccm.200902-0296OC
86. Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, Haigis MC. The aging lung: physiology, disease, and immunity. Cell (2021) 184:1990–2019. doi: 10.1016/j.cell.2021.03.005
87. Forteza R, Salathe M, Miot F, Forteza R, Conner GE. Regulated hydrogen peroxide production by duox in human airway epithelial cells. Am J Respir Cell Mol Biol (2005) 32:462–9. doi: 10.1165/rcmb.2004-0302OC
88. Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid Redox Signal (2009) 11:2453–65. doi: 10.1089/ars.2009.2558
89. Selemidis S, Seow HJ, Broughton BRS, Vinh A, Bozinovski S, Sobey CG, et al. Nox1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PloS One (2013) 8:e60792. doi: 10.1371/journal.pone.0060792
90. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol (2014) 15:411–21. doi: 10.1038/nrm3801
91. Haq IJ, Gray MA, Garnett JP, Ward C, Brodlie M. Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax (2016) 71:284–7. doi: 10.1136/thoraxjnl-2015-207588
92. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med (2010) 363:2233–47. doi: 10.1056/NEJMra0910061
93. Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest (2002) 109:571–7. doi: 10.1172/JCI15217
94. Chen EYT, Yang N, Quinton PM, Chin WC. A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol (2010) 299:L542. doi: 10.1152/ajplung.00180.2010
95. Zajac M, Dreano E, Edwards A, Planelles G, Sermet-gaudelus I. Airway surface liquid pH regulation in airway epithelium current understandings and gaps in knowledge. Int J Mol Sci (2021) 22(7):3384. doi: 10.3390/ijms22073384
96. Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol (2006) 68:543–61. doi: 10.1146/ANNUREV.PHYSIOL.68.072304.112754
97. Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med (2011) 184:957–63. doi: 10.1164/rccm.201104-0655OC
98. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, et al. Bacterial topography of the healthy human lower respiratory tract. mBio (2017) 8(1):e02287–16. doi: 10.1128/mBio.02287-16
99. Segal LN, Clemente JC, Li Y, Ruan C, Cao J, Danckers M, et al. Anaerobic bacterial fermentation products increase tuberculosis risk in antiretroviral-Drug-Treated HIV patients. Cell Host Microbe (2017) 21:530–537.e4. doi: 10.1016/j.chom.2017.03.003
100. Sulaiman I, Wu BG, Li Y, Tsay JC, Sauthoff M, Scott AS, et al. Functional lower airways genomic profiling of the microbiome to capture active microbial metabolism. Eur Respir J (2021) 58(1):2003434. doi: 10.1183/13993003.03434-2020
101. Kuek LE, Lee RJ. First contact: the role of respiratory cilia in host-pathogen interactions in the airways. Am J Physiol Lung Cell Mol Physiol (2020) 319:L603–19. doi: 10.1152/AJPLUNG.00283.2020
102. Chotirmall SH, Bogaert D, Chalmers JD, Cox MJ, Hansbro PM, Huang YJ, et al. Therapeutic targeting of the respiratory microbiome. Am J Respir Crit Care Med (2022) 206:535–44. doi: 10.1164/RCCM.202112-2704PP/SUPPL_FILE/DISCLOSURES.PDF
103. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
104. Hiemstra PS, McCray PB, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J (2015) 45:1150–62. doi: 10.1183/09031936.00141514
105. Jeong YS, Bae YS. Formyl peptide receptors in the mucosal immune system. Exp Mol Med (2020) 52:1694–704. doi: 10.1038/s12276-020-00518-2
106. Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL. Barton GM. a map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity (2018) 49:560–575.e6. doi: 10.1016/j.immuni.2018.07.016
107. De Nardo D. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine (2015) 74:181–9. doi: 10.1016/j.cyto.2015.02.025
108. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
109. Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza a virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi: 10.1016/j.immuni.2009.02.006
110. Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe (2009) 5:487–97. doi: 10.1016/j.chom.2009.05.002
111. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441:101–5. doi: 10.1038/nature04734
112. Bhoj VG, Sun Q, Bhoj EJ, Somers C, Chen X, Torres JP, et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc Natl Acad Sci U.S.A. (2008) 105:14046–51. doi: 10.1073/pnas.0804717105
113. Parker D, Prince A. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol (2011) 32:582–8. doi: 10.1016/j.it.2011.09.003
114. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17:1142–9. doi: 10.1038/ni.3558
115. Heyl KA, Klassert TE, Heinrich A, Müller MM, Klaile E, Dienemann H, et al. Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable Haemophilus influenzae. mBio (2014) 5:e01492–14. doi: 10.1128/mBio.01492-14
116. Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, et al. Requisite role for the dectin-1 β-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol (2009) 182:4938–46. doi: 10.4049/jimmunol.0804250
117. Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol (2002) 23:541–8. doi: 10.1016/S1471-4906(02)02316-5
118. Wang L, Cai J, Zhao X, Ma L, Zeng P, Zhou L, et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol Cell (2023) 83:281–297.e10. doi: 10.1016/J.MOLCEL.2022.12.002
119. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol (2016) 17:564–80. doi: 10.1038/nrm.2016.80
120. Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol (2021) 21:739–51. doi: 10.1038/s41577-021-00538-7
121. Wang M, Tan G, Eljaszewicz A, Meng Y, Wawrzyniak P, Acharya S, et al. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol (2019) 143:1892–903. doi: 10.1016/j.jaci.2018.11.016
122. Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci (2011) 68:877–92. doi: 10.1007/s00018-010-0500-x
123. Xian M, Ma S, Wang K, Lou H, Wang Y, Zhang L, et al. Akdis CA. particulate matter 2.5 causes deficiency in barrier integrity in human nasal epithelial cells. Allergy Asthma Immunol Res (2020) 12:56. doi: 10.4168/aair.2020.12.1.56
124. Heijink IH, Van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J (2010) 36:1016–26. doi: 10.1183/09031936.00125809
125. Sintobin I, Siroux V, Holtappels G, Pison C, Nadif R, Bousquet J, et al. Sensitisation to staphylococcal enterotoxins and asthma severity: a longitudinal study in the EGEA cohort. Eur Respir J (2019) 54(3):1900198. doi: 10.1183/13993003.00198-2019
126. Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Invest (2012) 92:1140–8. doi: 10.1038/labinvest.2012.67
127. Schmidt H, Braubach P, Schilpp C, Lochbaum R, Neuland K, Thompson K, et al. IL-13 impairs tight junctions in airway epithelia. Int J Mol Sci (2019) 20:3222. doi: 10.3390/ijms20133222
128. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
129. Kimmey JM, Stallings CL. Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol Med (2016) 22:1060–76. doi: 10.1016/j.molmed.2016.10.008
130. Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PloS One (2013) 8:e72263. doi: 10.1371/journal.pone.0072263
131. Croft CA, Culibrk L, Moore MM, Tebbutt SJ. Interactions of Aspergillus fumigatus conidia with airway epithelial cells: a critical review. Front Microbiol (2016) 7:472. doi: 10.3389/fmicb.2016.00472
132. Wang Y, Sharma P, Jefferson M, Zhang W, Bone B, Kipar A, et al. Non-canonical autophagy functions of ATG16L1 in epithelial cells limit lethal infection by influenza a virus. EMBO J (2021) 40:e105543. doi: 10.15252/embj.2020105543
133. Kinsella RL, Nehls EM, Stallings CL. Roles for autophagy proteins in immunity and host defense. Vet Pathol (2018) 55:366–73. doi: 10.1177/0300985818754967
134. Morleo M, Vieira HLA, Pennekamp P, Palma A, Bento-Lopes L, Omran H, et al. Crosstalk between cilia and autophagy: implication for human diseases. Autophagy (2022) 19(1):24–43. doi: 10.1080/15548627.2022.2067383
135. Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, et al. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature (2013) 502:254–7. doi: 10.1038/nature12606
136. Arora K, Lund JR, Naren NA, Zingarelli B, Naren AP. AC6 regulates the microtubule-depolymerizing kinesin KIF19A to control ciliary length in mammals. J Biol Chem (2020) 295:14250–9. doi: 10.1074/jbc.RA120.013703
137. Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol (2010) 12:863–75. doi: 10.1038/ncb2090
138. Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy (2016) 12:397–409. doi: 10.1080/15548627.2015.1056967
139. Dickinson JD, Sweeter JM, Warren KJ, Ahmad IM, De Deken X, Zimmerman MC, et al. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol (2018) 14:272–84. doi: 10.1016/j.redox.2017.09.013
140. Li K, Li M, Li W, Yu H, Sun X, Zhang Q, et al. Airway epithelial regeneration requires autophagy and glucose metabolism. Cell Death Dis (2019) 10:1–14. doi: 10.1038/s41419-019-2111-2
141. Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet (2008) 9:359–86. doi: 10.1146/annurev.genom.9.081307.164303
142. Cavounidis A, Pandey S, Capitani M, Friedrich M, Cross A, Gartner L, et al. Hermansky-pudlak syndrome type 1 causes impaired anti-microbial immunity and inflammation due to dysregulated immunometabolism. Mucosal Immunol (2022) 15:1431–46. doi: 10.1038/s41385-022-00572-1
143. Wu YF, Li ZY, Dong LL, Li WJ, Wu YP, Wang J, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy (2020) 16:435–50. doi: 10.1080/15548627.2019.1628536
144. Strom JE, Merid SK, Pourazar J, Blomberg A, Lindberg A, Ringh MV, et al. Chronic obstructive pulmonary disease is associated with epigenome-wide differential methylation in BAL lung cells. Am J Respir Cell Mol Biol (2022) 66:638–47. doi: 10.1165/rcmb.2021-0403OC
145. Shuto T, Furuta T, Oba M, Xu H, Li J, Cheung J, et al. Promoter hypomethylation of toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J (2006) 20:782–4. doi: 10.1096/fj.05-4934fje
146. Saccani S, Pantano S, Natoli G. p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat Immunol (2002) 3:69–75. doi: 10.1038/ni748
147. Stefanowicz D, Lee JY, Lee K, Shaheen F, Koo HK, Booth S, et al. Elevated H3K18 acetylation in airway epithelial cells of asthmatic subjects. Respir Res (2015) 16(1):95. doi: 10.1186/s12931-015-0254-y
148. Saco TV, Breitzig MT, Lockey RF, Kolliputi N. Epigenetics of mucus hypersecretion in chronic respiratory diseases. Am J Respir Cell Mol Biol (2018) 58:299–309. doi: 10.1165/rcmb.2017-0072TR
149. Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Rückert B, et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J Allergy Clin Immunol (2017) 139:93–103. doi: 10.1016/j.jaci.2016.03.050
150. Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol (2010) 185:3489–97. doi: 10.4049/jimmunol.0903610
151. Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol (2004) 31:633–42. doi: 10.1165/rcmb.2004-0006OC
152. Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. New Engl J Med (2005) 352:1967–76. doi: 10.1056/NEJMoa041892
153. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest (2013) 123:5212–30. doi: 10.1172/JCI69636
154. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Fah Sathirapongsasuti J, et al. Erratum: histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction (Journal of clinical investigation (2013) 123:12 (5212-5230) DOI: 10.1172/JCI69636). J Clin Invest (2020) 130:6189. doi: 10.1172/JCI143863
155. Baek SH, Kim K. Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell (2017) 65:781–5. doi: 10.1016/j.molcel.2016.12.027
156. Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy (2015) 11:2309–22. doi: 10.1080/15548627.2015.1117734
157. Byrd AL, Qu X, Lukyanchuk A, Liu J, Chen F, Naughton KJ, et al. Dysregulated polycomb repressive complex 2 contributes to chronic obstructive pulmonary disease by rewiring stem cell fate. Stem Cell Rep (2023) 18:289–304. doi: 10.1016/J.STEMCR.2022.11.009
158. Barrier ML, Myszor IT, Sahariah P, Sigurdsson S, Carmena-Bargueño M, Pérez-Sánchez H, et al. Aroylated phenylenediamine HO53 modulates innate immunity, histone acetylation and metabolism. Mol Immunol (2023) 155:153–64. doi: 10.1016/J.MOLIMM.2023.02.003
159. Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen–host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med (2005) 171:1209–23. doi: 10.1164/rccm.200408-1044SO
160. Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PloS Pathog (2012) 8:e1002616. doi: 10.1371/journal.ppat.1002616
161. Schmidtchen A, Frick I-M, Andersson E, Tapper H, Björck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol (2002) 46:157–68. doi: 10.1046/j.1365-2958.2002.03146.x
162. Zulianello L, Canard C, Köhler T, Caille D, Lacroix J-S, Meda P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect Immun (2006) 74:3134–47. doi: 10.1128/IAI.01772-05
163. Kanthakumar K, Taylor G T, Tsang KW, DR C, Rutman A, Smith S, et al. Mechanisms of action of Pseudomonas aeruginosa pyocyanin on human ciliary beat. In Vitro. (1993) 61(7):2848–53. doi: 10.1128/iai.61.7.2848-2853.1993
164. O’Malley YQ, Reszka KJ, Rasmussen GT, Abdalla MY, Denning GM, Britigan BE. The Pseudomonas secretory product pyocyanin inhibits catalase activity in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol (2003) 285(5):L1077–86. doi: 10.1152/ajplung.00198.2003
165. Cornelis P, Dingemans J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front Cell Infect Microbiol (2013) 3:75. doi: 10.3389/fcimb.2013.00075
166. Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, et al. The Pseudomonas aeruginosa autoinducer n-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun (2003) 71:5785–93. doi: 10.1128/IAI.71.10.5785-5793.2003
167. Bandyopadhaya A, Tsurumi A, Maura D, Jeffrey KL, Rahme LG. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat Microbiol (2016) 1:1–9. doi: 10.1038/nmicrobiol.2016.174
168. Rossi E, La Rosa R, Bartell JA, Marvig RL, Haagensen JAJ, Sommer LM, et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat Rev Microbiol (2021) 19:331–42. doi: 10.1038/s41579-020-00477-5
169. Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis (2008) 197:1079–81. doi: 10.1086/533452
170. Gonzalez-Ferrer S, Peñaloza HF, Budnick JA, Bain WG, Nordstrom HR, Lee JS, et al. Finding order in the chaos: outstanding questions in Klebsiella pneumoniae pathogenesis. Infect Immun (2021) 89(4):e00693–20. doi: 10.1128/IAI.00693-20
171. Feriotti C, Sá-Pessoa J, Calderón-González R, Gu L, Morris B, Sugisawa R, et al. Klebsiella pneumoniae hijacks the toll-IL-1R protein SARM1 in a type I IFN-dependent manner to antagonize host immunity. Cell Rep (2022) 40:111167. doi: 10.1016/j.celrep.2022.111167
172. Chua MD, Liou C-H, Bogdan AC, Law HT, Yeh K-M, Lin J-C, et al. Klebsiella pneumoniae disassembles host microtubules in lung epithelial cells. Cell Microbiol (2019) 21(3):e12977. doi: 10.1111/cmi.12977
173. Ahn D, Wickersham M, Riquelme S, Prince A. The effects of IFN-l on epithelial barrier function contribute to Klebsiella pneumoniae ST258 pneumonia. Am J Respir Cell Mol Biol (2019) 60:158–66. doi: 10.1165/rcmb.2018-0021OC
174. Cortés G, Álvarez D, Saus C, Albertí S. Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun (2002) 70:1075–80. doi: 10.1128/IAI.70.3.1075-1080.2002
175. Sequeira RP, McDonald JAK, Marchesi JR, Clarke TB. Commensal Bacteroidetes protect against Klebsiella pneumoniae colonization and transmission through IL-36 signalling. Nat Microbiol (2020) 5:304–13. doi: 10.1038/s41564-019-0640-1
176. Le Guern R, Grandjean T, Stabler S, Bauduin M, Gosset P, Kipnis É, et al. Gut colonisation with multidrug-resistant Klebsiella pneumoniae worsens Pseudomonas aeruginosa lung infection. Nat Commun (2023) 14(1):78. doi: 10.1038/s41467-022-35767-4
177. Lucas R, Czikora I, Sridhar S, Zemskov E, Gorshkov B, Siddaramappa U, et al. Mini-review: novel therapeutic strategies to blunt actions of pneumolysin in the lungs. Toxins (2013) 5:1244–60. doi: 10.3390/toxins5071244
178. Jacques LC, Panagiotou S, Baltazar M, Senghore M, Khandaker S, Xu R, et al. Increased pathogenicity of pneumococcal serotype 1 is driven by rapid autolysis and release of pneumolysin. Nat Commun (2020) 11:1–13. doi: 10.1038/s41467-020-15751-6
179. Rai P, He F, Kwang J, Engelward BP, Chow VTK. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci Rep (2016) 6:1–12. doi: 10.1038/srep22972
180. Bryant JC, Dabbs RC, Oswalt KL, Brown LR, Rosch JW, Seo KS, et al. Pyruvate oxidase of Streptococcus pneumoniae contributes to pneumolysin release. BMC Microbiol (2016) 16:1–12. doi: 10.1186/s12866-016-0881-6
181. Dong W, Rasid O, Chevalier C, Connor M, Eldridge MJG, Hamon MA. Streptococcus pneumoniae infection promotes histone H3 dephosphorylation by modulating host PP1 phosphatase. Cell Rep (2020) 30:4016–4026.e4. doi: 10.1016/J.CELREP.2020.02.116
182. Kovács M, Halfmann A, Fedtke I, Heintz M, Peschel A, Vollmer W, et al. A functional dlt operon, encoding proteins required for incorporation of d-alanine in teichoic acids in gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae. J Bacteriol (2006) 188:5797–805. doi: 10.1128/JB.00336-06
183. LaRock CN, Nizet V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim Biophys Acta Biomembr (2015) 1848:3047–54. doi: 10.1016/j.bbamem.2015.02.010
184. McKenna S, Huse KK, Giblin S, Pearson M, Shibar MSMAl, Sriskandan S, et al. The role of streptococcal cell-envelope proteases in bacterial evasion of the innate immune system. J Innate Immun (2022) 14:69–88. doi: 10.1159/000516956
185. Biswas D, Ambalavanan P, Ravins M, Anand A, Sharma A, Lim KXZ, et al. LL-37-mediated activation of host receptors is critical for defense against group a streptococcal infection. Cell Rep (2021) 34:108766. doi: 10.1016/J.CELREP.2021.108766
186. Crum NF, Russell KL, Kaplan EL, Wallace MR, Wu J, Ashtari P, et al. Pneumonia outbreak associated with group a Streptococcus species at a military training facility. Clin Infect Dis (2005) 40:511–8. doi: 10.1086/427502
187. Chandra P, Grigsby SJ, Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol (2022) 20(12):750–66. doi: 10.1038/s41579-022-00763-4
188. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
189. Cambier CJ, Banik SM, Buonomo JA, Bertozzi CR. Spreading of a mycobacterial cell surface lipid into host epithelial membranes promotes infectivity. Elife (2020) 9:1–68. doi: 10.7554/eLife.60648
190. Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol (2004) 52:1291–302. doi: 10.1111/j.1365-2958.2004.04078.x
191. Miller JL, Velmurugan K, Cowan MJ, Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-α-Mediated host cell apoptosis. PloS Pathog (2010) 6:e1000864. doi: 10.1371/journal.ppat.1000864
192. Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci U.S.A. (2017) 114:E8711–20. doi: 10.1073/pnas.1707792114
193. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for Mycobacterium tuberculosis persistence. PloS Pathog (2008) 4:e1000204. doi: 10.1371/journal.ppat.1000204
194. Donald PR, Diacon AH, Lange C, Demers AM, Von Groote-Biddlingmeier F, Nardell E. Droplets, dust and Guinea pigs: an historical review of tuberculosis transmission research, 1878-1940. Int J Tuberculosis Lung Dis (2018) 22:972–82. doi: 10.5588/ijtld.18.0173
195. Paludan SR, Mogensen TH. Innate immunological pathways in COVID-19 pathogenesis. Sci Immunol (2022) 7:5505. doi: 10.1126/sciimmunol.abm5505
196. Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep (2021) 34:108628. doi: 10.1016/j.celrep.2020.108628
197. Hackbart M, Deng X, Baker SC. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci U.S.A. (2020) 117:8094–103. doi: 10.1073/pnas.1921485117
198. Xia H, Cao Z, Xie X, Zhang X, Chen JYC, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep (2020) 33:108234. doi: 10.1016/j.celrep.2020.108234
199. Kee J, Thudium S, Renner DM, Glastad K, Palozola K, Zhang Z, et al. SARS-CoV-2 disrupts host epigenetic regulation via histone mimicry. Nature (2022) 610:381–8. doi: 10.1038/s41586-022-05282-z
200. Battles MB, McLellan JS. Respiratory syncytial virus entry and how to block it. Nat Rev Microbiol (2019) 17:233–45. doi: 10.1038/s41579-019-0149-x
201. Das S, St. Croix C, Good M, Chen J, Zhao J, Hu S, et al. Interleukin-22 inhibits respiratory syncytial virus production by blocking virus-mediated subversion of cellular autophagy. iScience (2020) 23:101256. doi: 10.1016/j.isci.2020.101256
202. Tripp RA, Jones LP, Haynes LM, Zheng HQ, Murphy PM, Anderson LJ. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol (2001) 2:732–8. doi: 10.1038/90675
203. Blacklow SC. Catching the common cold. Nat Struct Mol Biol (2004) 11:388–90. doi: 10.1038/nsmb0504-388
204. Michi AN, Yipp BG, Dufour A, Lopes F, Proud D. PGC-1α mediates a metabolic host defense response in human airway epithelium during rhinovirus infections. Nat Commun (2021) 12:1–19. doi: 10.1038/s41467-021-23925-z
205. Triantafilou M, Ramanjulu J, Booty LM, Jimenez-Duran G, Keles H, Saunders K, et al. Human rhinovirus promotes STING trafficking to replication organelles to promote viral replication. Nat Commun (2022) 13:1–16. doi: 10.1038/s41467-022-28745-3
206. Taylor L, Von Lendenfeld F, Ashton A, Sanghani H, Di Pretoro S, Usselmann L, et al. Sleep and circadian rhythm disruption alters the lung transcriptome to predispose to viral infection. iScience (2022) 26(2):105877. doi: 10.1016/j.isci.2022.105877
207. Hernández-Chávez MJ, Pérez-García LA, Niño-Vega GA, Mora-Montes HM. Fungal strategies to evade the host immune recognition. J Fungi (2017) 3(4):51. doi: 10.3390/jof3040051
208. Lionakis MS, Drummond RA, Hohl TM. Immune responses to human fungal pathogens and therapeutic prospects. Nat Rev Immunol (2023), 1–20. Online ahead of print. doi: 10.1038/s41577-022-00826-w
209. Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev (2003) 67:400–28. doi: 10.1128/mmbr.67.3.400-428.2003
210. Hoenigl M, Seidel D, Sprute R, Cunha C, Oliverio M, Goldman GH, et al. COVID-19-associated fungal infections. Nat Microbiol (2022) 7:1127–40. doi: 10.1038/s41564-022-01172-2
211. Rocha JDB, Nascimento MTC, Decote-Ricardo D, Côrte-Real S, Morrot A, Heise N, et al. Capsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci Rep (2015) 5:8008. doi: 10.1038/srep08008
212. Crabtree JN, Okagaki LH, Wiesner DL, Strain AK, Nielsen JN, Nielsen K. Titan cell production enhances the virulence of Cryptococcus neoformans. Infect Immun (2012) 80:3776–85. doi: 10.1128/IAI.00507-12
213. Murray CJ, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet (2022) 399:629–55. doi: 10.1016/S0140-6736(21)02724-0
214. Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European economic area in 2015: a population-level modelling analysis. Lancet Infect Dis (2019) 19:56–66. doi: 10.1016/S1473-3099(18)30605-4
215. Laxminarayan R, Van Boeckel T, Frost I, Kariuki S, Khan EA, Limmathurotsakul D, et al. The lancet infectious diseases commission on antimicrobial resistance: 6 years later. Lancet Infect Dis (2020) 20:e51–60. doi: 10.1016/s1473-3099(20)30003-7
216. Laxminarayan R, Van Boeckel T, Frost I, Kariuki S, Khan EA, Limmathurotsakul D, et al. Correction to lancet infect dis 2020. Lancet Infect Dis (2020) 20:e50. doi: 10.1016/S1473-3099(20)30146-8
217. Lewis K. The science of antibiotic discovery. Cell (2020) 181:29–45. doi: 10.1016/j.cell.2020.02.056
218. Bergman P, Raqib R, Rekha RS, Agerberth B, Gudmundsson GH. Host directed therapy against infection by boosting innate immunity. Front Immunol (2020) 11:1209. doi: 10.3389/fimmu.2020.01209
219. De Breij A, Riool M, Cordfunke RA, Malanovic N, De Boer L, Koning RI, et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci Transl Med (2018) 10(423):eaan4044. doi: 10.1126/scitranslmed.aan4044
220. Wuerth K, Lee AHY, Falsafi R, Gill EE, Hancock REW. Characterization of host responses during Pseudomonas aeruginosa acute infection in the lungs and blood and after treatment with the synthetic immunomodulatory peptide IDR-1002. Infect Immun (2019) 87(1):e00661-18. doi: 10.1128/IAI.00661-18/SUPPL_FILE/IAI.00661-18-S0001.PDF
221. Lazzaro BP, Zasloff M, Rolff J. Antimicrobial peptides: application informed by evolution. Science (2020) 368:eaau5480. doi: 10.1126/science.aau5480
222. Wang T-T, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol (2004) 173:2909–12. doi: 10.4049/JIMMUNOL.173.5.2909
223. Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin d receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J (2005) 19:1067–77. doi: 10.1096/fj.04-3284com
224. Lowry MB, Guo C, Zhang Y, Fantacone ML, Logan IE, Campbell Y, et al. A mouse model for vitamin d-induced human cathelicidin antimicrobial peptide gene expression. J Steroid Biochem Mol Biol (2020) 198:105552. doi: 10.1016/j.jsbmb.2019.105552
225. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol (2017) 19:29–41. doi: 10.1111/1462-2920.13589
226. Bilotta AJ, Cong Y. Gut microbiota metabolite regulation of host defenses at mucosal surfaces: implication in precision medicine. Precis Clin Med (2019) 2:110–9. doi: 10.1093/pcmedi/pbz008
227. Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D (2011) 11:227–49. doi: 10.2165/11591280-000000000-00000
228. Steinmann J, Halldórsson S, Agerberth B, Gudmundsson GH. Phenylbutyrate induces antimicrobial peptide expression. Antimicrob Agents Chemother (2009) 53:5127–33. doi: 10.1128/AAC.00818-09
229. Rekha RS, Rao Muvva SSVJ, Wan M, Raqib R, Bergman P, Brighenti S, et al. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy (2015) 11:1688–99. doi: 10.1080/15548627.2015.1075110
230. Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, et al. Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med (2001) 7:180–5. doi: 10.1038/84627
231. Sarker P, Ahmed S, Tiash S, Rekha RS, Stromberg R, Andersson J, et al. Phenylbutyrate counteracts Shigella mediated downregulation of cathelicidin in rabbit lung and intestinal epithelia: a potential therapeutic strategy. PloS One (2011) 6:e20637. doi: 10.1371/journal.pone.0020637
232. Mily A, Rekha RS, Kamal SMM, Arifuzzaman ASM, Rahim Z, Khan L, et al. Significant effects of oral phenylbutyrate and vitamin D3 adjunctive therapy in pulmonary tuberculosis: a randomized controlled trial. PloS One (2015) 10:e0138340. doi: 10.1371/journal.pone.0138340
233. Bekele A, Gebreselassie N, Ashenafi S, Kassa E, Aseffa G, Amogne W, et al. Daily adjunctive therapy with vitamin d 3 and phenylbutyrate supports clinical recovery from pulmonary tuberculosis: a randomized controlled trial in Ethiopia. J Intern Med (2018) 284:292–306. doi: 10.1111/joim.12767
234. Miraglia E, Nylén F, Johansson K, Arnér E, Cebula M, Farmand S, et al. Entinostat up-regulates the CAMP gene encoding LL-37 via activation of STAT3 and HIF-1α transcription factors. Sci Rep (2016) 6:33274. doi: 10.1038/srep33274
235. Sultana Rekha R, Karadottir H, Ahmed S, Gudmundsson GH, Agerberth B, Bergman P. Innate effector systems in primary human macrophages sensitize multidrug-resistant Klebsiella pneumoniae to antibiotics. Infect Immun (2020) 88(8):e00186–20. doi: 10.1128/IAI.00186-20
236. Myszor IT, Sigurdsson S, Viktorsdottir AR, Agerberth B, Eskelinen EL, Ogmundsdottir MH, et al. The novel inducer of innate immunity HO53 stimulates autophagy in human airway epithelial cells. J Innate Immun (2022) 14(5):477–92. doi: 10.1159/000521602
237. Principi N, Blasi F, Esposito S. Azithromycin use in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis (2015) 34:1071–9. doi: 10.1007/s10096-015-2347-4
238. Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol (2010) 42:62–8. doi: 10.1165/rcmb.2008-0357OC
239. Joelsson JP, Myszor IT, Sigurdsson S, Lehmann F, Page CP, Gudmundsson GH, et al. Azithromycin has lung barrier protective effects in a cell model mimicking ventilator-induced lung injury. ALTEX (2020) 37:545–60. doi: 10.14573/altex.2001271
240. Arason AJ, Joelsson JP, Valdimarsdottir B, Sigurdsson S, Gudjonsson A, Halldorsson S, et al. Azithromycin induces epidermal differentiation and multivesicular bodies in airway epithelia. Respir Res (2019) 20:129. doi: 10.1186/s12931-019-1101-3
241. Brockman-Schneider RA, Pickles RJ, Gern JE. Effects of vitamin d on airway epithelial cell morphology and rhinovirus replication. PloS One (2014) 9:e86755. doi: 10.1371/journal.pone.0086755
242. Zhang YG, Wu S, Lu R, Zhou D, Zhou J, Carmeliet G, et al. Tight junction CLDN2 gene is a direct target of the vitamin d receptor. Sci Rep (2015) 5:10642. doi: 10.1038/srep10642
243. Zhang Y, Garrett S, Carroll RE, Xia Y, Sun J. Vitamin d receptor upregulates tight junction protein claudin-5 against colitis-associated tumorigenesis. Mucosal Immunol (2022) 15(4):683–97. doi: 10.1038/s41385-022-00502-1
244. Steelant B, Wawrzyniak P, Martens K, Jonckheere AC, Pugin B, Schrijvers R, et al. Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. J Allergy Clin Immunol (2019) 144:1242–1253.e7. doi: 10.1016/j.jaci.2019.04.027
245. Wu S, Tian X, Mao Q, Peng C. Azithromycin attenuates wheezing after pulmonary inflammation through inhibiting histone H3K27me3 hypermethylation mediated by EZH2. Clin Epigenet (2023) 15:12. doi: 10.1186/s13148-023-01430-y
246. Berber AC, Del-Rio-Navarro BE. Use of broncho-vaxom® in private practice: phase IV trial in 587 children. Clin Ther (1996) 18:1068–79. doi: 10.1016/S0149-2918(96)80062-2
247. Yin J, Xu B, Zeng X, Shen K. Broncho-vaxom in pediatric recurrent respiratory tract infections: a systematic review and meta-analysis. Int Immunopharmacol (2018) 54:198–209. doi: 10.1016/j.intimp.2017.10.032
248. Triantafillou V, Workman AD, Patel NN, Maina IW, Tong CCL, Kuan EC, et al. Broncho-vaxom ®R (OM-85 BV) soluble components stimulate sinonasal innate immunity. Int Forum Allergy Rhinol (2019) 9:370–7. doi: 10.1002/alr.22276
249. Fang L, Zhou L, Tamm M, Roth M. OM-85 broncho-vaxom®, a bacterial lysate, reduces SARS-CoV-2 binding proteins on human bronchial epithelial cells. Biomedicines (2021) 9:1544. doi: 10.3390/biomedicines9111544
250. Pivniouk V, Pivniouk O, DeVries A, Uhrlaub JL, Michael A, Pivniouk D, et al. The OM-85 bacterial lysate inhibits SARS-CoV-2 infection of epithelial cells by downregulating SARS-CoV-2 receptor expression. J Allergy Clin Immunol (2022) 149:923–933.e6. doi: 10.1016/j.jaci.2021.11.019
251. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
252. Hamada A, Torre C, Drancourt M, Ghigo E. Trained immunity carried by non-immune cells. Front Microbiol (2019) 9:3225. doi: 10.3389/fmicb.2018.03225
253. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell (2018) 172:176–190.e19. doi: 10.1016/j.cell.2017.12.031
254. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity (2019) 50:432–445.e7. doi: 10.1016/j.immuni.2018.12.018
255. Burgess SL, Leslie JL, Uddin J, Oakland DN, Gilchrist C, Moreau GB, et al. Gut microbiome communication with bone marrow regulates susceptibility to amebiasis. J Clin Invest (2020) 140:4019–24. doi: 10.1172/JCI133605
256. Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature (2018) 560:649–54. doi: 10.1038/s41586-018-0449-8
Keywords: innate immunity, airway epithelium, microbiota metabolites, epigenetics, innate immune memory
Citation: Myszor IT and Gudmundsson GH (2023) Modulation of innate immunity in airway epithelium for host-directed therapy. Front. Immunol. 14:1197908. doi: 10.3389/fimmu.2023.1197908
Received: 31 March 2023; Accepted: 24 April 2023;
Published: 12 May 2023.
Edited by:
Bruno Rivas-Santiago, Unidad de Investigación Biomédica de Zacatecas (IMSS), MexicoReviewed by:
Gill Diamond, University of Louisville, United StatesCesar Rivas-Santiago, National Council of Science and Technology (CONACYT), Mexico
Copyright © 2023 Myszor and Gudmundsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gudmundur Hrafn Gudmundsson, Z2hyYWZuQGhpLmlz; Iwona T. Myszor, aXRtQGhpLmlz