 Katie Louise Raby1†
Katie Louise Raby1† Charalambos Michaeloudes2†
Charalambos Michaeloudes2† James Tonkin1,3†
James Tonkin1,3† Kian Fan Chung1,3
Kian Fan Chung1,3 Pankaj Kumar Bhavsar1,3*
Pankaj Kumar Bhavsar1,3*- 1National Heart and Lung Institute, Imperial College London, London, United Kingdom
- 2School of Medicine, European University Cyprus, Nicosia, Cyprus
- 3Department of Respiratory Medicine, Royal Brompton and Harefield Hospital, London, United Kingdom
The airway epithelium comprises of different cell types and acts as a physical barrier preventing pathogens, including inhaled particles and microbes, from entering the lungs. Goblet cells and submucosal glands produce mucus that traps pathogens, which are expelled from the respiratory tract by ciliated cells. Basal cells act as progenitor cells, differentiating into different epithelial cell types, to maintain homeostasis following injury. Adherens and tight junctions between cells maintain the epithelial barrier function and regulate the movement of molecules across it. In this review we discuss how abnormal epithelial structure and function, caused by chronic injury and abnormal repair, drives airway disease and specifically asthma and chronic obstructive pulmonary disease (COPD). In both diseases, inhaled allergens, pollutants and microbes disrupt junctional complexes and promote cell death, impairing the barrier function and leading to increased penetration of pathogens and a constant airway immune response. In asthma, the inflammatory response precipitates the epithelial injury and drives abnormal basal cell differentiation. This leads to reduced ciliated cells, goblet cell hyperplasia and increased epithelial mesenchymal transition, which contribute to impaired mucociliary clearance and airway remodelling. In COPD, chronic oxidative stress and inflammation trigger premature epithelial cell senescence, which contributes to loss of epithelial integrity and airway inflammation and remodelling. Increased numbers of basal cells showing deregulated differentiation, contributes to ciliary dysfunction and mucous hyperproduction in COPD airways. Defective antioxidant, antiviral and damage repair mechanisms, possibly due to genetic or epigenetic factors, may confer susceptibility to airway epithelial dysfunction in these diseases. The current evidence suggests that a constant cycle of injury and abnormal repair of the epithelium drives chronic airway inflammation and remodelling in asthma and COPD. Mechanistic understanding of injury susceptibility and damage response may lead to improved therapies for these diseases.
1 Introduction
The airway epithelium is a complex system of different cell types that works in conjunction with the immune system to form an intact barrier, which prevents foreign particles and pathogens from entering the submucosal compartment. The whole system works together as the first-line defence to protect and limit the damage caused by stress (1, 2). The mucociliary system traps and expels foreign particles from the respiratory tract. Furthermore, tight junctions and adherens junctions form cell to cell contacts, and regulate the flow of molecules across the basement membrane to the underlying sub-epithelium. However, defects in these intertwined processes and loss of barrier function can cause airway diseases, including chronic obstructive pulmonary disease (COPD) and asthma.
Chronic exposure to cigarette smoke, which is the main risk factor for COPD, as well as repeated infections, cause changes in the epithelium including loss of ciliated epithelial cells (3–6), increased goblet cell number (7), shortened cilia and reduced beat frequency (8, 9). Epithelial injury triggers a chronic immune response that drives alveolar destruction (emphysema), and airway remodelling that involves peribronchial fibrosis and possibly increased airway smooth muscle mass (1–5). Furthermore, the alterations in the cell type proportions, causes increased mucus production, a predominant feature of chronic bronchitis associated with COPD (10).
Severe asthma is a chronic inflammatory disease classified as heterogenous in relation to clinical phenotype, severity, and the response of the epithelium to injury. Epithelial injury in severe asthma, whether due to allergen, pollution, viral or bacterial insult, causes several changes in the response and integrity of the epithelium, leading to immune cell recruitment, inflammation, epithelial shedding (2, 11, 12). The asthmatic airway epithelial layer is disrupted as evidenced by the detachment of ciliated epithelial cells, and loss of cell-cell contacts that leads to reduced integrity and increased permeability (13–15). This allows for the release of pro-inflammatory cytokines, such as the alarmins IL-33 and IL-25 (16), and the Th2 cytokines IL-4 and IL-13 (14, 17). These mediators activate and recruit immune cells, which further damage the epithelial layer and contribute to patient exacerbations. Mediators produced by the injured epithelium and the local immune cells drive airway remodelling, which involves subepithelial fibrosis due to increased ECM deposition, and airway smooth muscle thickening caused by smooth muscle cell hyperplasia and/or hypertrophy (1).
In this review we will demonstrate how the structure and composition of the epithelium, and the basement membrane, and their interaction with the innate immune system facilitates homeostasis and protection of the airway. We will then discuss how structural and functional changes in the COPD and asthmatic epithelium further drive disease, exacerbations, and lung function decline (Table 1). Specifically, epithelial dysfunction in COPD is largely due to altered cell type composition and reduced mucociliary clearance in response to cigarette smoke exposure. On the other hand, in asthma, the reduced integrity and increased permeability is caused by loss of epithelial cell-cell contact and cell shedding due to chronic inflammation.
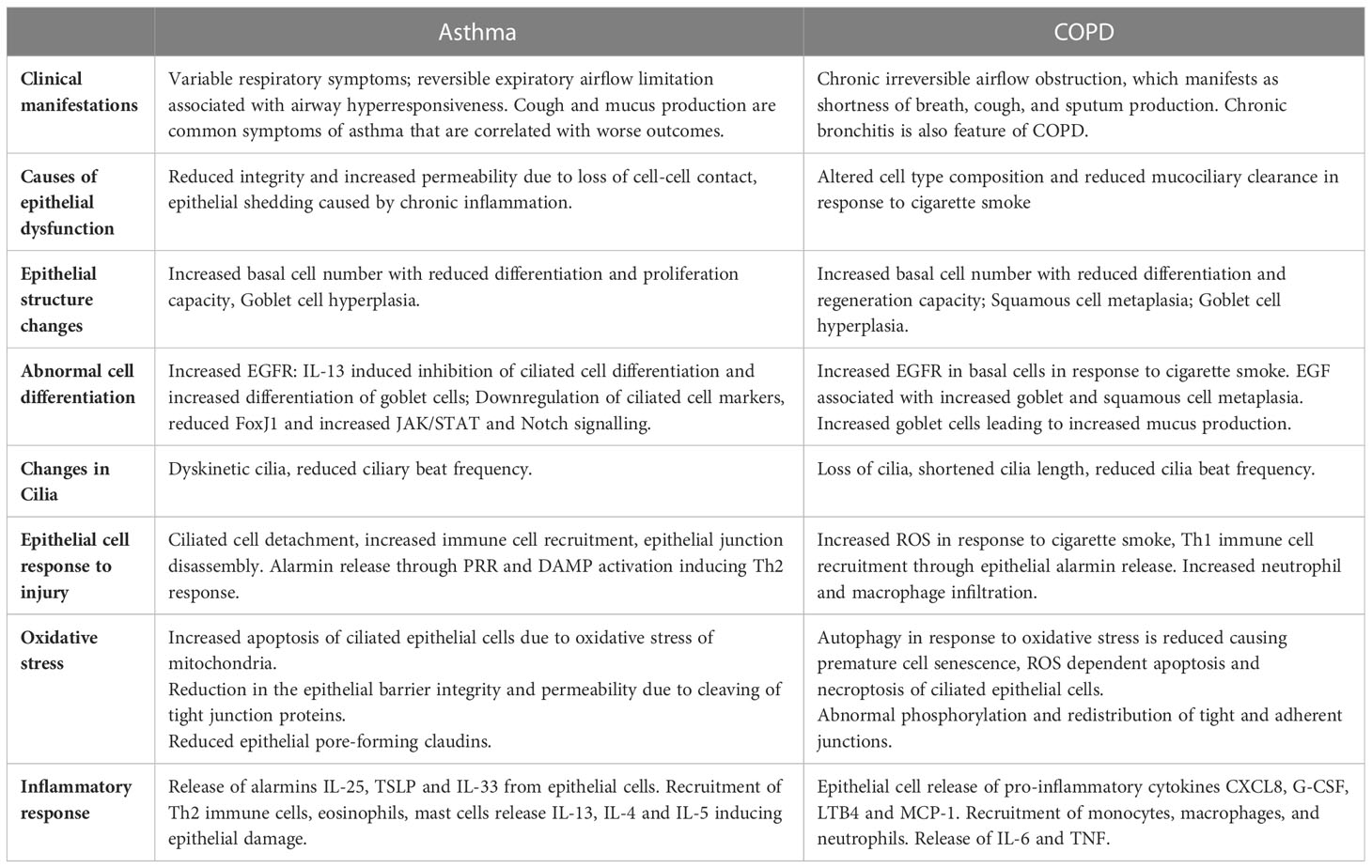
Table 1 Comparison of Asthma and COPD risk factors, pathogenesis and airway dysfunction manifestations.
2 Epithelial structure and cell types
The airway epithelium is a continuous sheet of epithelial cells, comprised of different cell types: predominantly ciliated, goblet, basal and club cells. These cells form a strong attachment to the basement membrane and their differing height of nuclei give it a characteristic pseudostratified appearance (14, 18, 19).
Basal cells are epithelial progenitor cells that are characterized by the expression of cytokeratins 5 and 13, and Tumour Protein 63 (TP63) that is necessary for their differentiation and formation of normal epithelium (17, 20). They are relatively undifferentiated and are anchored to the underlying basement membrane by hemidesmosomes. Basal cells have a capacity to migrate into areas of injury and differentiate into other cell types, which facilitate damage repair (17, 21).
Ciliated epithelial cells are the predominant cell type in the airway epithelium. They have characteristic projections on the apical surface (cilia) that provide forward and back-strokes in a coordinated manner to move mucus, and pathogens trapped within it, from the lower airways (22). The membrane of each cilium surrounds an axoneme which has a repeating ‘9 + 2’ structure: nine microtubule doublets which surround two central microtubules. These microtubules move relative to one another through force generated by dynein arms to move cilia in a coordinated manner (23). Ciliated epithelial cell differentiation occurs in the absence of Notch signalling. Without Notch suppression, Geminin coiled-coil containing nuclear protein (GMNC) induces Multiciliate Differentiation And DNA Synthesis Associated Cell Cycle Protein (MCIDAS), which then activates Forkhead box protein J1 (FOXJ1) (24). FOXJ1 is the primary transcriptional regulator of cilia formation, regulating the docking of basal bodies to the apical surface, the elongation of cilia and the generation of motile components (25).
The secretory cells in the epithelium are comprised of goblet cells and mucus-producing cells on the surface of submucosal glands. These cells produce mucus comprised of heavily glycosylated gel-forming mucins, which are highly water absorptive allowing them to trap and expel foreign pathogens. MUC5AC and MUC5B are the most expressed mucins, with MUC5AC predominantly expressed in goblet cells and MUC5B in submucosal glands (26, 27). Goblet cells facilitate constitutive mucus secretion while submucosal glands, controlled by autonomic innervation, can rapidly increase mucus production in response to stimulation. Notch signalling promotes goblet cell hyperplasia by supressing TP63 and GMNC and activating Sam-pointed domain Ets-like factor (SPDEF), a driver of goblet cell differentiation (24).
Club cells are found in the small airways and their gene expression is closely related to that of basal cells (28). They provide an immunomodulatory role, secreting club cell secretory protein (CCSP) which has antiprotease and anti-inflammatory activity (29, 30). In addition to their host defence and anti-inflammatory role, they have a progenitor role in the small airways with the capacity to differentiate into ciliated, secretory or alveolar cells (31, 32).
3 The basement membrane and cell-cell connections
The airway epithelium is a pseudo-stratified layer with clear apical and basolateral aspects that play a role in intercellular signalling (14, 15, 18). Epithelial cells sit on a basement membrane providing support and enabling communication with the underlying lamina propria. The basement membrane is an acellular structure formed of a basal lamina on top of the reticular basement membrane with proteins binding the two. The basal lamina is formed of collagen IV, laminin and proteoglycans synthesized by epithelial cells. The reticular basement membrane is formed of collagen I, III, IV and tenascin which are produced by fibroblasts in the underlying lamina propria. This functional and homeostatic complex is formed from tightly interwoven barrier and junctional complexes anchored to the basement membrane (1, 33). This binding and anchoring formed through integrin binding and hemidesmosomes stabilises the basal epithelial cell adhesion to the basement membrane (1, 33).
Adherens and tight junctions are complexes that regulate the airway epithelium’s permeability and maintain its barrier function against foreign antigens that are not cleared by the muco-ciliary system (Figure 1). The most apical of these complexes are the Tight Junctions (TJs), formed of multiple protein strands that maintain close opposition of adjacent cell membranes (1, 33). Tight junction proteins include the Zona occludin molecules (ZO-1, ZO-2, ZO-3), occludin, junctional adhesion molecules (JAMs) and claudins. These proteins maintain barrier function by binding to actin fibres and binding to each other in the cytoplasm (1, 33). Further functions of tight junctions include influencing cell morphology, cell proliferation and differentiation. The distribution and amount of specific pore-forming and barrier-forming claudins, play a role in determining the stability of the barrier (33).
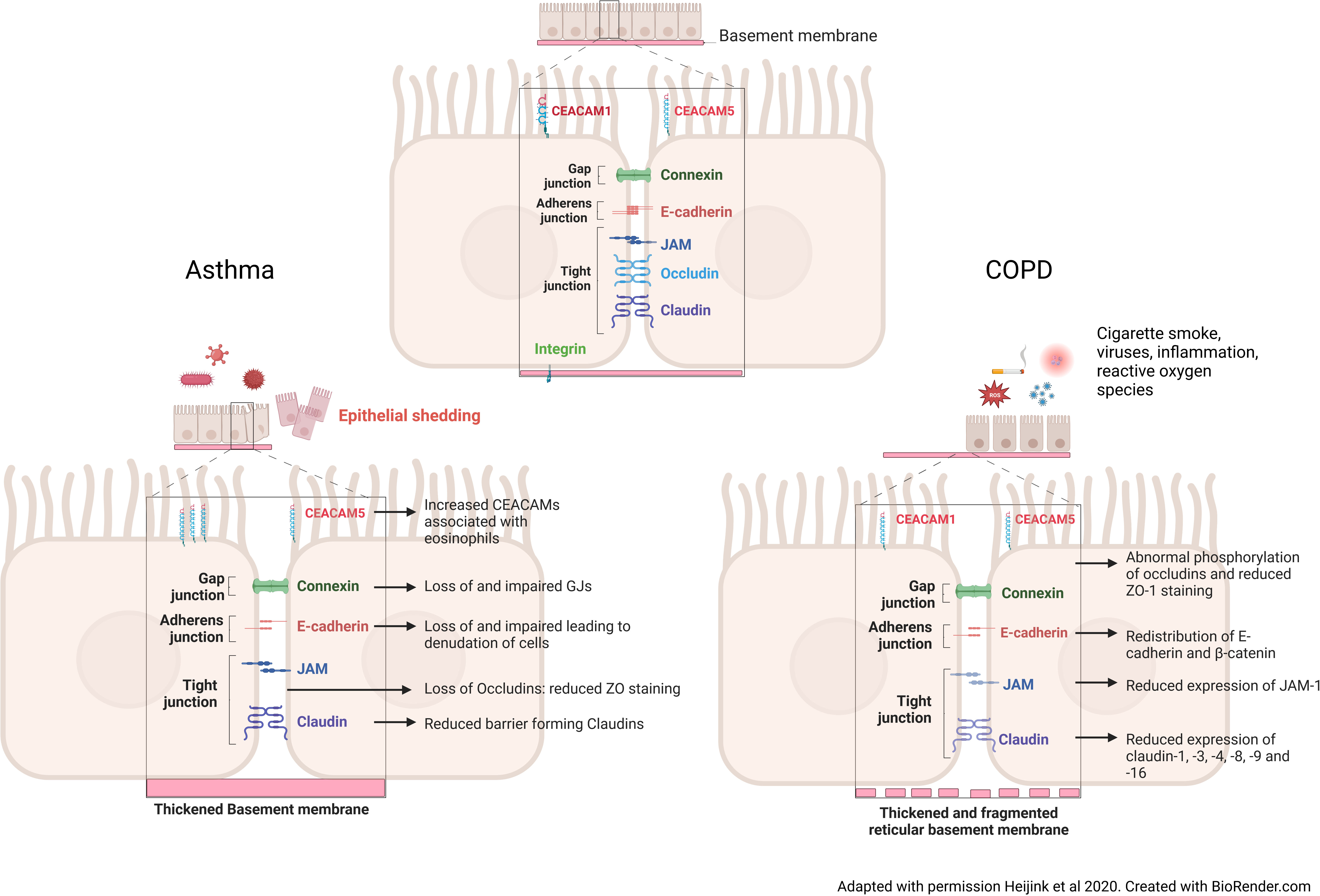
Figure 1 Changes to the intercellular junctions in asthma and COPD. In the asthmatic airway, after exposure to pathogens, inflammatory cytokines and the influx of inflammatory cells cause an increase in Carcinoembryonic Antigen-Related Cell Adhesion Molecule 5 (CEACAM5) expression on the apical of surface of the cells. These cytokines and inflammatory cells, also cause a loss of vital gap junction, adherens junction and of barrier forming tight junction proteins. A thickened basement membrane is a feature seen in the asthmatic airway, meaning that epithelial cell attachment to the membrane is not altered, but the cell-cell contact is reduced. Taken together this induces epithelial shedding and denudation, reduced barrier integrity and increased permeability in asthma. In the COPD airway, continued cigarette smoke exposure causes a redistribution of adherens junction proteins, namely E-cadherin and β-catenin, reduced tight junction protein expression through abnormal phosphorylation. Unlike the asthmatic airway, the basement membrane is fragmented, causing reduced binding of the cells to the basement membrane.
Adherens junctions form another junctional layer, which consists of the transmembrane protein E-cadherin bound to cadherin-catenin complexes in the cytoplasm of adjacent cells (15, 18, 34). They play an essential role in cell-cell adhesion, differentiation, proliferation and maturation of adjacent cells (33, 34). Desmosomes are junction complexes, found near adherens junctions, which interact with the cell’s intermediate filament cytoskeleton and maintain cell mechanical integrity and polarity (1, 15, 35). Finally, the gap junctions enable macromolecules and metabolite movement between cells and, through connexins, aid in the co-ordination of the epithelial ciliated cell beat frequency (18).
Despite forming a tight barrier, basement membrane pores are important for enabling cells to move between the epithelium and lamina propria and in reverse. In epithelial-mesenchymal transition (EMT), epithelial cells lose epithelial markers (cytokeratins and E-cadherin), migrate to the lamina propria and gain mesenchymal markers (vimentin, N-cadherin, S100A4, MMP-9 and α-smooth muscle actin). These cells synthesize extracellular matrix (ECM) to provide a framework for basal cells to infiltrate and differentiate to replace damaged epithelium.
4 Exposures to the epithelium, the acute and chronic inflammatory response
Apart from its important role as a barrier, the airway epithelium is also involved in the regulation of the innate immune response. Airway epithelial cells have pattern-recognition receptors that recognize inhaled irritants, such as allergens and pathogens, triggering the release of cytokines, chemokines and alarmins that regulate dendritic cell, T and B cell function and trigger an acute inflammatory response. In susceptible individuals, such as patients with asthma and COPD, prolonged activation of the epithelium by inhaled irritants leads to a chronic and exaggerated immune activation. This abnormal response leads to the production of high levels of cytokines, chemokines, growth factors, proteases and ROS, which exacerbate tissue injury and inflammation, and drive abnormal repair and airway remodelling (13, 16, 36, 37).
4.1 Epithelium-immune cell crosstalk
The interaction of immune and epithelial cells is necessary to maintain tissue homeostasis (1). The immune response is dependent upon the co-ordinated response and activation of pattern recognition receptors (PRRs) and damage-associated molecular patterns (DAMPs) that cause immune cell activation and recruitment (1, 2). In the healthy airway, there is an effective activation and resolution of the immune cell associated repair mechanisms that are induced by and in response to epithelial injury (1). The interaction of immune cells and the airway epithelium means there is a continuous influence of the epithelial cells on the immune cells and vice versa. As discussed previously, the airway epithelium is the first line defence against any foreign particles which are recognised by the PRRs and DAMPs. The main mechanism of defence activated by the PRRs, and DAMPs are epithelial alarmin release. Epithelial alarmins including IL-25, IL-33, TSLP and HMGB and chemokine release including CCL17, CCL-11 and CCL22, activates and recruits immune cells, both directly and indirectly (1–3).
5 Airway epithelial dysfunction in asthma
Asthma is a chronic inflammatory disease, characterized by a heightened, continued, and chronic immune response. Patients with asthma typically have a history of variable respiratory symptoms, with reversible expiratory airflow limitation that may be associated with airway hyperresponsiveness (38). In two thirds of cases, it is linked to a history of atopy but can also be associated with occupational and other exposures (39).
Cough and mucus production are common symptoms of asthma that are correlated with worse outcomes. Post-mortem studies have shown mucus plugging to be the primary cause of death in asthma (40). Mucus hypersecretion is associated with goblet cell hyperplasia and submucosal gland hypertrophy in the epithelium (41). Recent studies, using CT and MRI imaging, have shown an association between increased mucus and features of high Th2 inflammation, ventilation mismatch and lower FEV1 in patients with asthma (42–44). There has been a great expansion in the development of monoclonal antibody treatments for asthma that target mechanisms including the changes in the epithelium that contribute to mucus hypersecretion (45).
In asthma, the airway epithelium shows structural and functional abnormalities, which are possibly a result of chronic injury and abnormal repair. Epithelial injury, whether due to allergen, pollution, viral or bacterial insult, leads to several changes in the response and integrity of the epithelium, which is accompanied by immune cell recruitment, inflammation, and epithelial shedding and remodelling. In the normal healthy non-asthmatic airway this process resolves. However, the asthmatic airway epithelium shows exaggerated release of cytokines/chemokines, such as IL-6, CXCL8, and alarmins, including IL-25, TSLP, IL-33, in response to injury. These mediators activate and recruit immune cells such as eosinophils, Th2 cells, macrophages and neutrophils. Macrophages and neutrophils produce proteases and ROS, whilst eosinophils release cationic proteins, which further damage the epithelial layer, and contribute to symptom exacerbations (13, 15, 16, 37, 46, 47). Activated epithelial cells and innate immune cells also release pro-remodelling factors, such as epidermal growth factor receptor (EGFR) ligands, and transforming growth factor (TGF)-β, which trigger goblet cell hyperplasia, EMT, fibroblast activation, and airway smooth muscle cell proliferation and hypertrophy (1, 2).
5.1 Epithelial structural changes in asthma
Asthmatic airways show increased numbers of basal cells with an impaired proliferation and differentiation capacity, goblet cell hyperplasia and an imbalance in the production of mucins, with increased MUC5AC and reduced MUC5B (Figure 2) (15, 17, 18, 22, 48–50). These two mucins have different roles within the airways: MUC5B functions in normal mucus mucociliary clearance whereas MUC5AC exacerbates airway hyperresponsiveness and mucus plugging (51). Endobronchial biopsies from patients with asthma, imaged with scanning electron microscopy, also show reduced ciliation and increased areas of squamous epithelium (52). Functionally, ciliated cells sampled from patients with severe asthma show dyskinetic movement, slower ciliary beat frequency and microtubule defects (53). Club cell markers (CCSP) are also reduced in bronchial alveolar lavage fluid taken from patients with asthma (54). CCSP has also been shown to increase following allergen challenge in patients with allergic asthma, suggesting acute leakage due to club cell damage in the small airway epithelium (55).
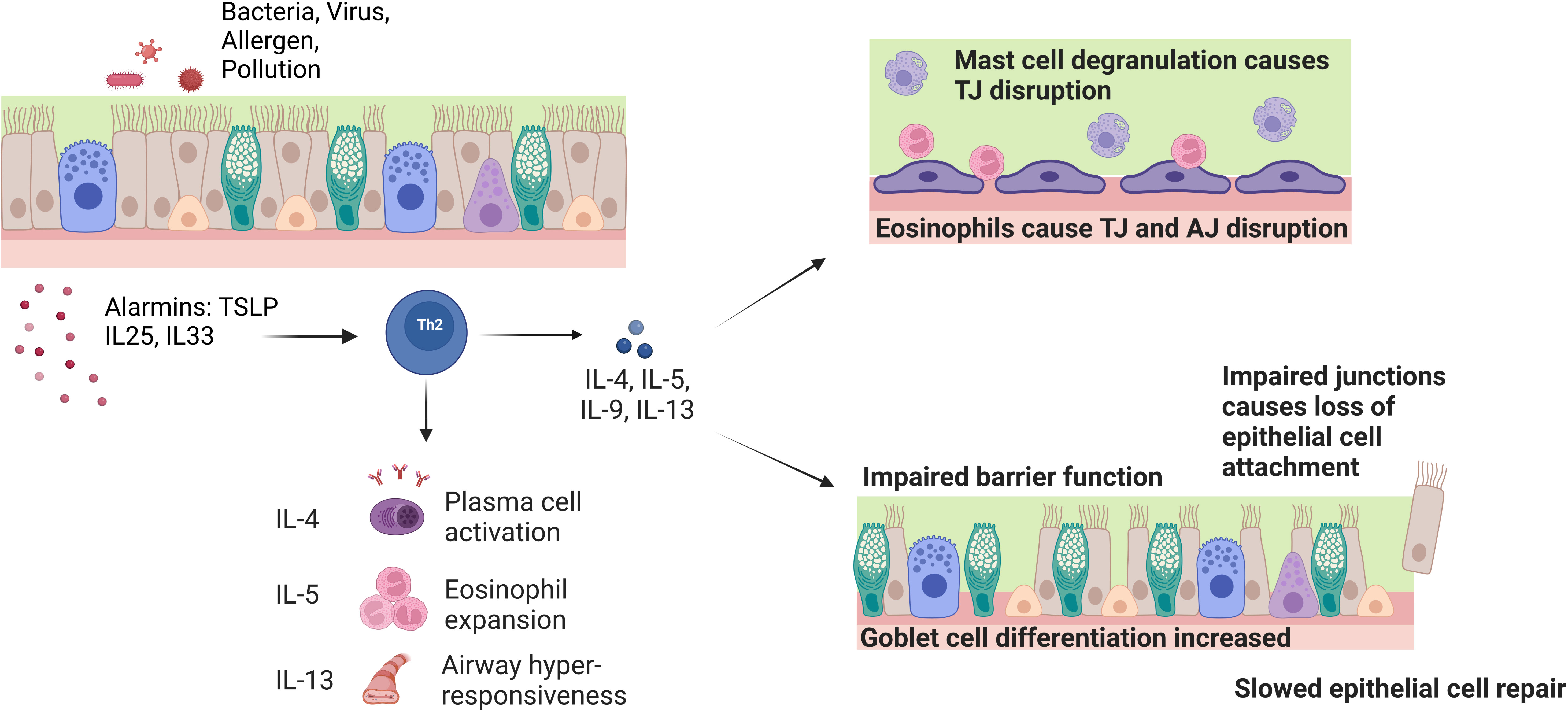
Figure 2 Effect of chronic inflammatory cells and cytokines on barrier function and integrity in asthma. Exposure of the airway to pathogens causes the release of epithelial cell alarmins such as TSLP, IL-25 and IL-33. The alarmins activate Th2 inflammatory cells that release the IL-4, IL-5 and IL-13 cytokines. These cytokines play a role in driving the symptoms seen in asthma such as plasma cell activation, eosinophil expansion and airway hyper-responsiveness. On a cellular level, the IL-5 induced eosinophil expansion interacts with the epithelial barrier and causes tight junction (TJ) disruption through reduction of barrier forming claudins and mast cell degranulation. IL-4 and IL-13 inhibit the expression of TJ and adherens junction (AJ) genes E-cadherin, occludin and ZO-1. These cytokines also induce goblet cell hyperplasia and defective basal cell repair causes impaired barrier function and loss of ciliated epithelial cells.
Abnormal cell differentiation may contribute to these abnormalities. EGFR, which is increased in the asthmatic epithelium, is a driver of abnormal cell differentiation and epithelial dysfunction (56–58). Furthermore, IL-13, elevated in allergic asthma, inhibits ciliated cell differentiation and causes reduced ciliary beat frequency and abnormal distribution of basal bodies, whilst enhancing goblet cell differentiation (17, 49). Specifically, IL-13 upregulates SPDEF and MUC5AC expression, whilst downregulating FOXJ1 and other cilia-related transcription factors via activation of JAK/STAT and Notch signalling and changes in histone methylation (59–61).
5.2 Epithelial integrity
In asthma, epithelial defects in the adherens, tight and gap junctions, as well as altered permeability and integrity, are present from the nose through to the lower airways (Figure 1). Junctional complex disruption is thought to occur through hyperphosphorylation and decreased expression of complex genes and proteins in asthmatic patients because of deeper penetration and increased immune response to particles (1, 17, 62). The disrupted and leaky barrier in asthma is well-documented and a positive feedback loop of IL-4 and IL-13 contributes to this phenotype seen in patients with asthma. Reduced expression of the barrier forming claudins -1, -3, -4 and -18 is associated with increased IL-13 production in asthma, from mild-moderate to severe asthma (14, 15, 33). Reduced ZO-1 and E-cadherin expression are evident in asthma where they cause denudation and detachment of ciliated epithelial cells (1, 14, 15, 62).
Ciliated epithelial cell detachment is paired with loss of cell-cell contacts and reduced integrity (12, 14, 15, 63, 64). This leads to pro-inflammatory cytokine release and recruitment of immune cells, which induce apoptosis and further damage the epithelial layer (12, 14, 15, 63, 64). Apoptosis and autophagy occur in the non-asthmatic airway epithelium and are crucial for airway epithelial homeostasis (21, 64). These mechanisms regulate the number of proliferating epithelial cells and remove damaged cells without the induction of a chronic inflammatory response (21, 64). However, in asthma there is increased apoptosis of ciliated epithelial cells leading to epithelial shedding and barrier dysfunction, which increases with disease severity.
Environmental pathogenic factors, including allergens, air pollutants and respiratory infections, and the associated immune response, cause airway epithelial injury (Figure 2). Allergen proteases, such as Der p1 in house dust mites, induce inflammation and disrupt the epithelial barrier either directly by cleaving tight junction proteins, or indirectly by activating protease activated receptor (PAR)-2 (14, 65, 66). PAR2-dependent signalling promotes epithelial junction disassembly through ZO-1 and occludin degradation, and EGFR-dependent redistribution of E-cadherin (67). Furthermore, ATP released from the injured epithelium may also promote loss of epithelial integrity through activation of purinergic receptors (68). EGFR activation by allergens also promotes EMT in airway epithelial cells (69). Allergens promote oxidative stress, which triggers epithelial cell death and inflammation through oxidative damage of DNA and mitochondria (70–74). Mitochondrial dysfunction has been shown to cause ROS-mediated disruption of barrier function in intestinal epithelium, whilst reduced E-cadherin expression and loss of barrier function are associated with attenuated mitochondrial biogenesis in airway epithelial cells (75, 76). Viral infections and inhaled pollutants can also cause epithelial injury by inducing cell death and disruption of tight junctions (77–80).
The asthmatic airway immune response also leads to loss of epithelial integrity. Mast cell degranulation, IL-5 and direct contact with eosinophils, leads to disruption of tight junctions and increased permeability (17, 37, 81). Furthermore, the T2 cytokines IL-4 and IL-13 inhibit the expression of the key tight and adherens junction genes ZO-1, E-cadherin and occludin, and drive goblet cell differentiation from basal cells (Figure 2) (13, 82).
Susceptibility to epithelial injury by environmental irritants, and abnormal repair mechanisms, are key factors in asthma pathogenesis. House dust mites, extracts from the allergenic fungus Alternaria Alternata and cigarette smoke were shown to induce greater barrier dysfunction in air-liquid-interface (ALI) cultures of asthmatic airway epithelial cells compared to those of healthy subjects (68, 83, 84). Inadequate antioxidant and antiviral mechanisms in asthmatic airways may also confer epithelial cells more susceptibility to oxidative and viral-induced damage (72, 85, 86). Epithelial cells from patients with asthma are also abnormally slow at repairing injury in in vitro models. This is possibly caused by asynchronous mitosis of epithelial cells and is associated with increased production of TGF-β which drives EMT and ECM production (87–89).
Taken together, the data suggests that there is a vicious cycle of epithelial cell injury and immune activation, which in conjunction with increased susceptibility and abnormal repair mechanisms promote an abnormal airway epithelium in asthma (13, 16, 37).
5.3 Epithelium-immune cell crosstalk in asthma
In the asthmatic airway, alarmin and chemokine release induced cytokine activation is a well-recognised disease driving mechanism. The release of alarmins primes the Th2 CD4 T-cell response through maturing dendritic cells (DCs) via CCL17 (42). DCs in asthma mount an uncontrolled Th2 response and acts to enhance ILC2, basophil and mast cell functionality (90). The epithelium response to injury enhances the Th2 release of pro-inflammatory cytokines such as IL-4 from mast cells and basophils increasing levels of IgE, IL-5 for the activation and maturation of eosinophils in the bone marrow and IL-13 that induces hyper-responsive smooth muscle contraction, goblet cell hyperplasia and the activation of macrophages (90).
The interaction between the epithelium and immune cells can influence many different parts of the airway through alarmin release; in the innate immune system, DCs cause increased activation of Th2 cells, basophils have increased histamine degranulation and ILC2 cells release increased pro-inflammatory cytokines (17). In the epithelial and endothelial cells, a feedback loop is induced, the epithelium becomes leakier due to cleaved junctions, smooth muscle cells become hyper-responsive and fibroblasts release increase CCL5, GM-CSF and CXCL8 (17).
6 Airway epithelial dysfunction in COPD
COPD is a respiratory disease characterised by chronic irreversible airflow obstruction, which manifests as shortness of breath, cough and sputum production. Breathlessness typically increases over time and is interspersed by periods of worsening respiratory symptoms known as exacerbations, which accelerate lung function decline.
Chronic bronchitis is a clinical phenotype of COPD which involves “chronic cough and sputum production for three months a year for two consecutive years” (91). Patients with chronic bronchitis have poorer quality of life, more frequent exacerbations, faster lung function decline and increased mortality (54, 91–93). COPD and chronic bronchitis are characterised by epithelial changes that contribute to chronic mucus secretion (Figure 3). A reduction in ciliated cells and an increase in secretory cells lead to more mucus, which is more difficult to clear and contributes to symptoms, repeated infections and faster lung function decline.
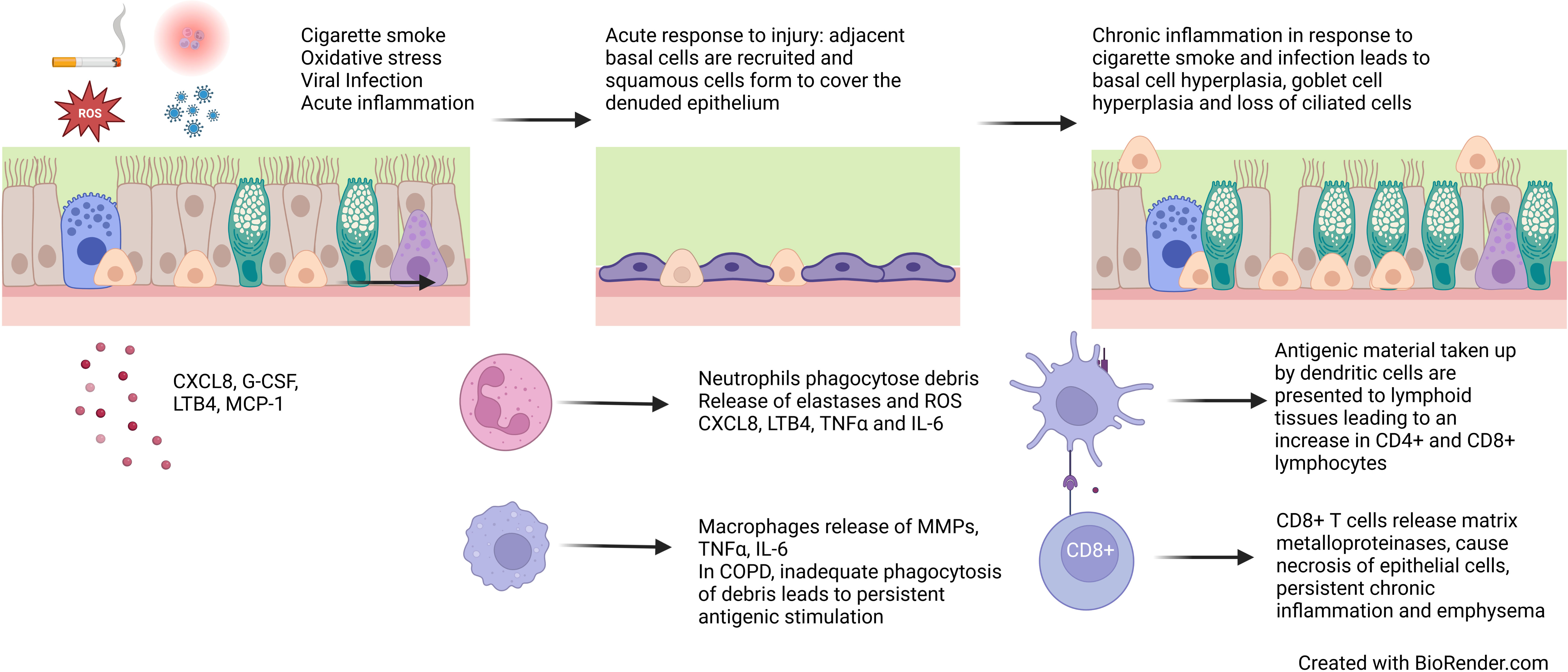
Figure 3 Epithelial changes in COPD. Cigarette smoke, reactive oxygen species, viral infection and acute inflammation leads to loss of ciliated epithelial cells and squamous metaplasia. Persistent exposures and chronic inflammation lead to basal cell hyperplasia, goblet cell hyperplasia and reduced ciliation.
6.1 Changes to the epithelial structure in COPD
There is evidence of increased numbers of basal cells in the airways of patients with COPD, which however show compromised capacity to regenerate and differentiate (94). EGFR is expressed in basal cells and can be activated by EGF produced by ciliated cells in response cigarette smoke and inflammatory mediators (5, 6). EGFR is increased in smokers and is associated with increased mucous and squamous metaplasia via Notch signalling (3).
Samples taken at bronchoscopy from people who smoke have shown a loss of cilia, shorter cilia and ultrastructural defects in the axoneme (8, 9, 90, 95–97). Gene expression of dynein arms (DNAH5, DNAH9 and DNAH11) that drive cilia beating, and intraflagellar transport proteins (IFT43, IFT 57, IFT144 and IFT172), which determine cilia length and development, have been shown to be reduced in smokers. Within COPD, similar features of loss of cilia, shorter cilia and slower cilia are seen when compared to healthy controls and healthy smokers (8, 98). Although inhaled therapies for COPD, β2-agonists and muscarinic antagonists, can increase ciliary movement these effects are blunted by high levels of inflammation (99, 100).
Goblet cells are increased in smokers and patients with COPD, which is thought to lead to an increase in baseline mucus production (7). Although submucosal gland density does not appear to be increased in COPD, gland inflammation is increased and may contribute to increased mucus release in response to infectious and non-infectious triggers (10). The primary mucins, MUC5AC and MUC5B are both increased in COPD (101–104). Furthermore, club cells are reduced in the small airway epithelium of patients with COPD (105). CCSP is reduced in smokers and patients with COPD and has been shown to correlate with FEV1 (106, 107).
6.2 Epithelial integrity and permeability
In COPD, there is a known and recognised defect in the epithelial barrier permeability and integrity in response to stressors (Figure 1). In epithelial cells from patients with COPD, reduced expression of occludins, claudins and JAM-1 is seen. Oxidative stress leads to abnormal phosphorylation of occludin and redistribution of occludins, ZO-1, E-cadherin and β-catenin throughout the junction (79). Increased permeability of the epithelium is seen in response to cigarette smoke and associated chronic inflammation, with a reduction in staining of ZO-1 (108, 109). Many viruses including rhinovirus also reduce ZO-1, and further lead to reduced epithelial resistance and increased permeability in ALI cultures (110), Epithelial cells exposed to TNFα and IFN-γ also show reduced ZO-1 and JAM staining and attenuated barrier function (111). This process of inflammation-induced loss of tight and adherens junctions predisposes the epithelium to further infection and exacerbates the persistent chronic inflammatory state seen in COPD.
The reticular basement membrane in COPD is thicker than in healthy controls but as not thick as seen in patients with asthma (112, 113). However, fragmented reticular basement membrane is seen in COPD and healthy smokers (114). In COPD, however, repeated trauma leads to persistent EMT activation through TGF-β activation and further basement membrane fragmentation (115, 116). EMT markers have been shown be increased in COPD and to correlate with the severity of airflow obstruction (117, 118).
6.3 Chronic inflammation in COPD
Chronic inflammation is a key factor in COPD pathogenesis resulting from repeated insults and oxidative stress, which cause cell death, senescence and abnormal cell differentiation and function. While there are many risk factors, cigarette smoke is the most common in COPD. Cigarette smoke contains reactive oxygen species (ROS) which can be directly cytotoxic to epithelial cells and trigger an acute inflammatory response, which is further exacerbated by bacterial and viral infections. Epithelial cells release pro-inflammatory cytokines CXCL8, G-CSF, LTB4 and MCP-1 resulting in the recruitment of monocytes, macrophages and neutrophils which in turn release IL-6 and TNFα (4). The inflammatory response and oxidative stress lead to necrosis and apoptosis of epithelial cells. In response to this trauma, cells adjacent to the area de-differentiate to cover the denuded area, forming a flattened area of squamous epithelium to protect against future damage (119). In health, if toxic exposure to the epithelium ceases, basal cells and CD45- stem cells differentiate for form a pseudostratified epithelium (120, 121). However, in COPD, persistent exposures and chronic inflammation prevent the formation of structurally and functionally normal epithelium (Figure 3).
In the COPD epithelium, the chronic inflammatory process occurs due to ongoing oxidative stress (usually from cigarette smoke), persistent activation of inflammatory cells through cell damage and impaired processes to fight infection. Toll-like receptors on epithelial cells are directly stimulated by cigarette smoke activating a cascade of inflammatory mediators, further tissue damage and release of antigenic material (122, 123). Antigens are also taken up by dendritic cells (which are increased in the COPD epithelium) and are presented to CD8+ T cells (124, 125). CD8+ T cells are increased in COPD and numbers inversely correlated with FEV1 (126). These cells are cytotoxic to epithelial cells and release proteolytic enzymes including matrix metalloproteases (MMPs) which can drive emphysema, a clinical feature of COPD. Phagocytes, in particular neutrophils and macrophages, are altered in COPD. Neutrophils and macrophages engulf microbes and apoptotic cells. Neutrophils and macrophages are increased in the epithelium and are associated with chronic bronchitis, more severe airflow obstruction and faster lung function decline (127–129). Despite being increased in number, they show impaired phagocytic ability which predispose to recurrent infection (130–132). Neutrophils and macrophages also release reactive oxygen species, pro-inflammatory cytokines, growth factors and MMPs, which propagate chronic inflammation, emphysema, and airway remodelling processes, such as EMT, fibroblast activation and airway smooth muscle proliferation (1) (Figure 3).
6.4 Epithelium-immune cell crosstalk in COPD
The continued interaction of the immune cells and epithelium (39, 133) in COPD is facilitated by the proximity of DCs to the airway epithelial cells. In COPD, DCs have been identified as upregulating the expression of chemokines CXCL10, CCL-20 and CCL-10 that cause the activation of T-cells, predominantly a Th1-pro inflammatory profile (39, 133). Epithelial alarmin release of IL-33 has also been identified as being upregulated in COPD; this has been shown to cause an increase in neutrophil and macrophage infiltration and activation. This drives a positive feedback loop on both epithelial cells and immune cell upregulation of IL-6 and CXCL8 release (39). The up-regulation of HGMB1 on epithelial cells in COPD stimulates increased IL-1b production and release from activated macrophages (39, 133).
6.5 The role of oxidative stress in epithelial dysfunction
ROS, from cigarette smoke or the inflammatory response, drive cell death, senescence, abnormal cell-cell connections and abnormal remodelling (134, 135). Epithelial cells have antioxidant mechanisms to mitigate the ill effects of ROS but these are impaired in COPD which renders these cells more susceptible to oxidative damage (72, 136–138). Epithelial cells in COPD, have a dysregulated redox state which promotes airway inflammation and remodelling by regulating the activity of ROS-dependent kinases, phosphatases, transcription factors and epigenetic effectors, and by drivingTGF- β expression and EGFR activation (139–141).
DNA damage occurs in response to oxidative stress and is associated with nucleotide base oxidation, and single and double-strand DNA breaks in COPD (142, 143). In addition to the increased insults, DNA repair processes are defective in patients with COPD (143, 144). These along with telomere damage, as a result of oxidative stress, lead to upregulation of the cell cycle inhibitor p16INK4 and mammalian target of rapamycin (mTOR)-mediated signalling to trigger premature senescence in COPD airway epithelial cells (145–149). Although acute senescence may play a role in damage repair, prolonged senescence may lead to impaired epithelial regeneration and damage repair by limiting the proliferative capacity of progenitor cells. Furthermore, although in cell cycle arrest, senescent cells remain metabolically active and secrete a multitude of proteases, cytokines, chemokines and growth factors, such as TGF-β (148). Therefore, accumulation of senescent cells may exacerbate lung inflammation and epithelial injury, and drive emphysema and fibrosis (150–155).
Mitochondria are major sites of oxidative damage in COPD epithelial cells. Cigarette smoke alters mitochondrial morphology and respiration in normal bronchial epithelial cells, whilst bronchial epithelial cells from COPD patients show abnormal mitochondrial morphology, possibly as a result of prolonged exposure to oxidative stress (156, 157). Mitochondrial dysfunction leads to increased production of ROS, which in turn cause further mitochondrial damage. This creates a vicious cycle of oxidative stress and mitochondrial dysfunction that drives pathology. Specifically, mitochondrial ROS promote apoptosis, senescence and inflammatory mediator release in airway epithelial cells, as well as impaired cellular function manifesting as reduced mucociliary clearance (158–161). Mitochondrial dysfunction also triggers ROS-dependent activation of necroptosis, a lytic form of cell death, that is accompanied with the release of immunogenic damage-associated molecular patterns (DAMPs). DAMPs, which include mitochondrial molecules such as ATP and cardiolipin, promote further inflammatory mediator release and result in increased MUC5AC expression in the airway epithelium (162–164).
Autophagy is a cellular process that is pivotal in the adaptation to oxidative stress. In this process, damaged molecules and organelles are engulfed into double-membrane bound vesicles called autophagosomes and fuse with lysosomes leading to their degradation. Reduced autophagic activity in COPD lungs, resulting from prolonged oxidative stress and ageing, may lead to insufficient removal of damaged cellular components and premature airway epithelial cell senescence (165–168). Specifically, impaired autophagic removal of mitochondria (mitophagy) has been reported in the airway epithelium of COPD patients, where it leads to accumulation of damaged mitochondria and epithelial cell senescence through ROS-mediated DNA damage (161, 169–171). However, other studies show excessive autophagic activity in COPD, leading to airway epithelial cell apoptosis, necroptosis and inflammatory mediator release (172–174). Cigarette smoke-induced autophagy has also been shown to cause degradation of ciliary proteins, and to promotes MUC5AC expression through activation of c-Jun N-terminal kinase (JNK) and activating protein (AP)-1 (175, 176). Therefore, dysregulated autophagy leads to the abnormal adaptation of airway epithelial cells to injury and contributes to airway remodelling.
6.6 Epigenetic and genetic determinants
Predisposing factors combined with exposures both in early life and adulthood likely contribute to epithelial pathogenesis seen in COPD. Gene polymorphisms, and epigenetic changes including DNA methylation, histone modifications and non-coding RNAs, may play a role in susceptibility and abnormal repair following epithelial injury in COPD (152, 177).
DNA methylation at gene promoters, a modification associated with reduced gene expression, is altered by cigarette smoking in airway epithelial cells (178–180). In ALI cultures derived from COPD bronchial epithelial cells, DNA hypomethylation at the SPDEF gene promoter leads to increased MUC5AC expression (178). In cigarette smoke extract-exposed COPD airway epithelial cells, gene promoter hypomethylation triggers increased expression of the aryl hydrocarbon receptor repressor (AHRR), an inhibitor of the cytoprotective transcription factor aryl hydrocarbon receptor (AHR). This leads to inhibition of AHR expression and increases susceptibility to CS-induced epithelial cell apoptosis and necroptosis (180). Conversely, reduced expression of the antioxidant transcription factor nuclear factor E2-related factor 2 (Nrf2) due to DNA hypermethylation in COPD, may increase the susceptibility of bronchial epithelial cells to oxidative stress-dependent cell death (181).
Acetylation of lysine residues on histones, which is catalysed by histone acetyltransferases and removed by histone deacetylases, is usually associated with open chromatin configuration and active gene transcription. Histone methylation on lysine and arginine residues, regulated by histone methyltransferases and demethylases, can activate or inhibit gene expression depending on the site and number of methyl groups added (182). An imbalance between histone acetylation and deacetylation, favouring acetylation, has also been observed in COPD lungs and has been associated with increased inflammatory gene expression (167, 179). Reduced expression of histone acetyltransferase binding to ORC1 (HBO1) in COPD leads to downregulation of anti-apoptotic protein Bcl-2, sensitising airway epithelial cells to oxidative stress-mediated apoptosis (183). Furthermore, reduced expression of the arginine methyltransferase coactivator-associated arginine methyltransferase 1 (CARM1) in COPD bronchial epithelial cells is associated with increased senescence, and impaired club cell regeneration and epithelial repair, possibly through altered histone methylation of cell cycle and differentiation genes (184, 185).
miRNAs are small non-coding RNAs that regulate gene expression through the induction of mRNA degradation and inhibition of translation. A number of miRNAs have been shown to be abnormally expressed in COPD airways where they are involved in airway epithelial inflammation, mucous hypersecretion, EMT and cellular senescence (152, 186–188).
Some of the gene polymorphisms identified by genome-wide association studies to be associated with COPD may increase susceptibility to epithelial injury. A gene variant of FAM13A, a protein expressed mainly in club cells and alveolar type 2 cells, is associated increased gene expression and reduced lung function in COPD. FAM13A is an inhibitor of Wnt signalling, a regulator of epithelial differentiation, therefore its upregulation may contribute to impaired club cell differentiation in COPD airways (189). Furthermore, FAM13A promotes fatty acid oxidation and mitochondrial respiration by driving the expression of carnitine palmitoyltransferase 1A (CPT1A), contributing to ROS-mediated bronchial epithelial apoptosis (190). However, FAM13A inhibits TGF-β-mediated EMT in airway epithelial cells, indicating it may also have protective effects against airway remodelling (191). Variants of the iron-regulatory protein 2 (IRP2) have also been associated with COPD susceptibility. IRP2 is found in airway epithelial cells, predominantly at the cilial surface. Increased IRP2 expression in COPD causes mitochondrial dysfunction due to iron overload, leading to increased airway epithelial cell death, inflammation and impaired mucociliary clearance (192, 193).
7 Discussion
The epithelium protects the airways against pathogens and inhaled irritants. It achieves this by acting as a physical barrier preventing them from entering into the submucosa, by trapping them in mucous and expelling them through ciliary clearance, and by orchestrating their clearance by the immune system. In asthma and COPD, inherent defects in protective and repair mechanisms in conjunction with repetitive injury, lead to significant structural and functional abnormalities in the epithelium. Specifically, mucous overproduction and ciliary dysfunction in COPD leads to defective clearance and increased entry of pathogens into the airway wall, triggering an immune response. In asthma, the disruption of tight junctions and adherens junctions between epithelial cells, epithelial shedding and deeper penetration of pathogens, again triggers a constant feedback of immune cell response. In both diseases, this leads to a continuous cycle of injury and abnormal repair that drives chronic airway inflammation and remodelling. Targeting the mechanisms of damage susceptibility and abnormal repair in the epithelium may lead to new and more effective therapies for asthma and COPD. Future work studying the effects of biologic therapies in asthma on barrier function needs to be explored. Studying how removing certain key cytokines such as IL-5, IL-4 and alarmins TSLP can alter the epithelial response to injury and how this could potentially change the epithelial-immune cell interaction. Studying key proteins and pathways that are altered in asthma and COPD epithelial barrier, such as Notch signalling, Wnt signalling, and gap junctions should be the future direction of studying epithelial barrier integrity alterations.
Author contributions
KR, CM, and JT wrote the manuscript. KR, CM, JT, KC, and PB conceived and finalised the manuscript.
Funding
Funding is WHRD_P18360.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Heijink IH, Kuchibhotla VNS, Roffel MP, Maes T, Knight DA, Sayers I, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. (2020) 75(8):1902–17. doi: 10.1111/all.14421
2. Carlier FM, de Fays C, Pilette C. Epithelial barrier dysfunction in chronic respiratory diseases. Front Physiol (2021) 12:691227. doi: 10.3389/fphys.2021.691227
3. Kurie JM, Shin HJ, Lee JS, Morice RC, Ro JY, Lippman SM, et al. Increased epidermal growth factor receptor expression in metaplastic bronchial epithelium. Clin Cancer Res (1996) 2(10):1787–93.
4. Shaykhiev R, Zuo WL, Chao I, Fukui T, Witover B, Brekman A, et al. EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype. Proc Natl Acad Sci (2013) 110(29):12102–7. doi: 10.1073/pnas.1303058110
5. Nadel JA, Burgel PR. The role of epidermal growth factor in mucus production. Curr Opin Pharmacol (2001) 1(3):254–8. doi: 10.1016/S1471-4892(01)00045-5
6. Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J (2004) 23(2):327–33. doi: 10.1183/09031936.03.00098803
7. Rogers DF. The airway goblet cell. Int J Biochem Cell Biol (2003) 35(1):1–6. doi: 10.1016/S1357-2725(02)00083-3
8. Leopold PL, O’Mahony MJ, Lian XJ, Tilley AE, Harvey BG, Crystal RG. Smoking is associated with shortened airway cilia. PLoS One (2009) 4(12):e8157. doi: 10.1371/journal.pone.0008157
9. Cipulo Ramos EM, De Toledo AC, Xavier RF, Fosco LC, Vieira RP, Ramos D, et al. Reversibility of impaired nasal mucociliary clearance in smokers following a smoking cessation programme. Respirology. (2011) 16(5):849–55. doi: 10.1111/j.1440-1843.2011.01985.x
10. Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell (2002) 13(9):3218–34. doi: 10.1091/mbc.e02-03-0134
11. Saetta M, Turato G, Facchini FM, Corbino L, Lucchini RE, Casoni G, et al. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med (1997) 156(5):1633–9. doi: 10.1164/ajrccm.156.5.9701081
12. Gon Y, Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int (2018) 67(1):12–7. doi: 10.1016/j.alit.2017.08.011
13. Oikonomou N, Schuijs MJ, Chatzigiagkos A, Androulidaki A, Aidinis V, Hammad H, et al. Airway epithelial cell necroptosis contributes to asthma exacerbation in a mouse model of house dust mite-induced allergic inflammation. Mucosal Immunol (2021) 14(5):1160–71. doi: 10.1038/s41385-021-00415-5
14. Georas SN, Rezaee F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol (2014) 134(3):509–20. doi: 10.1016/j.jaci.2014.05.049
15. Calvén J, Ax E, Rådinger M. The airway epithelium–a central player in asthma pathogenesis. Int J Mol Sci (2020) 21(23):8907. doi: 10.3390/ijms21238907
16. Noureddine N, Chalubinski M, Wawrzyniak P. The role of defective epithelial barriers in allergic lung disease and asthma development. J Asthma Allergy (2022) 15:487–504. doi: 10.2147/JAA.S324080
17. Whetstone CE, Ranjbar M, Omer H, Cusack RP, Gauvreau GM. The role of airway epithelial cell alarmins in asthma. Cells. (2022) 11(7):1105. doi: 10.3390/cells11071105
18. Hellings PW, Steelant B. Epithelial barriers in allergy and asthma. J Allergy Clin Immunol (2020) 145(6):1499–509. doi: 10.1016/j.jaci.2020.04.010
19. Erle DJ, Sheppard D. The cell biology of asthma. J Cell Biol (2014) 205(5):621–31. doi: 10.1083/jcb.201401050
20. Préfontaine D, Hamid Q. Airway epithelial cells in asthma. J Allergy Clin Immunol (2007) 120(6):1475–8. doi: 10.1016/j.jaci.2007.09.041
21. Warner SMB, Hackett TL, Shaheen F, Hallstrand TS, Kicic A, Stick SM, et al. Transcription factor p63 regulates key genes and wound repair in human airway epithelial basal cells. Am J Respir Cell Mol Biol (2013) 49(6):978–88. doi: 10.1165/rcmb.2012-0447OC
22. Dong X, Ding M, Zhang J, Ogülür I, Pat Y, Akdis M, et al. Involvement and therapeutic implications of airway epithelial barrier dysfunction in type 2 inflammation of asthma. Chin Med J (Engl) (2022) 135(05):519–31. doi: 10.1097/CM9.0000000000001983
23. Boateng E, Kovacevic D, Oldenburg V, Rådinger M, Krauss-Etschmann S. Role of airway epithelial cell miRNAs in asthma. Front Allergy (2022) 3:1–10. doi: 10.3389/falgy.2022.962693
24. Renthal R, Schneider BG, Miller MM, Ludueña RF. βIV is the major β-tubulin isotype in bovine cilia. Cell Motil. (1993) 25(1):19–29. doi: 10.1002/cm.970250104
25. Whitsett JA. Airway epithelial differentiation and mucociliary clearance. Ann Am Thorac Soc (2018) 15(Suppl 3):S143–8. doi: 10.1513/AnnalsATS.201802-128AW
26. Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harb Perspect Biol (2017) 9(4):a028241. doi: 10.1101/cshperspect.a028241
27. Hovenberg HW, Davies JR, Carlstedt I. Different mucins are produced by the surface epithelium and the submucosa in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem J (1996) 318(1):319–24. doi: 10.1042/bj3180319
28. Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers. (2013) 1(4):e24997. doi: 10.4161/tisb.24997
29. Zuo WL, Shenoy SA, Li S, O’Beirne SL, Strulovici-Barel Y, Leopold PL, et al. Ontogeny and biology of human small airway epithelial club cells. Am J Respir Crit Care Med (2018) 198(11):1375–88. doi: 10.1164/rccm.201710-2107OC
30. Barnes PJ. Club cells, their secretory protein, and COPD. CHEST. (2015) 147(6):1447–8. doi: 10.1378/chest.14-3171
31. Tokita E, Tanabe T, Asano K, Suzaki H, Rubin BK. Club cell 10-kDa protein attenuates airway mucus hypersecretion and inflammation. Eur Respir J (2014) 44(4):1002–10. doi: 10.1183/09031936.00080913
32. Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR. Clara Cell secretory protein–expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am J Respir Cell Mol Biol (2001) 24(6):671–81. doi: 10.1165/ajrcmb.24.6.4498
33. Dean CH, Snelgrove RJ. New rules for club development: new insights into human small airway epithelial club cell ontogeny and function. Am J Respir Crit Care Med (2018) 198(11):1355–6. doi: 10.1164/rccm.201805-0925ED
34. Nur Husna SM, Tan HTT, Md Shukri N, Mohd Ashari NS, Wong KK. Nasal epithelial barrier integrity and tight junctions disruption in allergic rhinitis: overview and pathogenic insights. Front Immunol (2021) 12:663626. doi: 10.3389/fimmu.2021.663626
35. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta BBA - Biomembr. (2008) 1778(3):660–9. doi: 10.1016/j.bbamem.2007.07.012
36. Nur Husna SM, Shukri NM, Ashari NSM, Wong KK. IL-4/IL-13 axis as therapeutic targets in allergic rhinitis and asthma. PeerJ. (2022) 10:e13444. doi: 10.7717/peerj.13444
37. Wang Y, Bai C, Li K, Adler KB, Wang X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir Med (2008) 102(7):949–55. doi: 10.1016/j.rmed.2008.01.017
38. Duan S, Han X, Jiao J, Wang M, Li Y, Wang Y, et al. Histone deacetylase activity is a novel target for epithelial barrier defects in patients with eosinophilic chronic rhinosinusitis with nasal polyps. Clin Exp Allergy (2023) 53(4). doi: 10.1111/cea.14258
39. Burgoyne RA, Fisher AJ, Borthwick LA. The role of epithelial damage in the pulmonary immune response. Cells. (2021) 10(10):2763. doi: 10.3390/cells10102763
40. Ruwanpura SM, McLeod L, Dousha LF, Seow HJ, West AC, West AJ, et al. Cross-talk between IL-6 trans-signaling and AIM2 inflammasome/IL-1β axes bridge innate immunity and epithelial apoptosis to promote emphysema. Proc Natl Acad Sci U S A. (2022) 119(36):e2201494119. doi: 10.1073/pnas.2201494119
41. Duchesne M, Okoye I, Lacy P. Epithelial cell alarmin cytokines: frontline mediators of the asthma inflammatory response. Front Immunol (2022) 13:975914. doi: 10.3389/fimmu.2022.975914
42. Chen YL, Chiang BL. Targeting TSLP with shRNA alleviates airway inflammation and decreases epithelial CCL17 in a murine model of asthma. Mol Ther Nucleic Acids (2016) 5(5):e316. doi: 10.1038/mtna.2016.29
43. Reddel HK, Bacharier LB, Bateman ED, Brightling CE, Brusselle GG, Buhl R, et al. Global initiative for asthma strategy 2021: executive summary and rationale for key changes. Am J Respir Crit Care Med (2022) 205(1):17–35. doi: 10.1164/rccm.202109-2205PP
44. Maskell N, Millar A, eds. Oxford Desk Reference: Respiratory Medicine. Oxford University Press (2009), 501. doi: 10.1093/med/9780199239122.001.0001
45. Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med (2009) 15(1):4–11. doi: 10.1097/MCP.0b013e32831da8d3
46. Rogers DF. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin Pharmacol (2004) 4(3):241–50. doi: 10.1016/j.coph.2004.01.011
47. Svenningsen S, Haider E, Boylan C, Mukherjee M, Eddy RL, Capaldi DPI, et al. CT and functional MRI to evaluate airway mucus in severe asthma. Chest. (2019) 155(6):1178–89. doi: 10.1016/j.chest.2019.02.403
48. Tang M, Elicker BM, Henry T, Gierada DS, Schiebler ML, Huang BK, et al. Mucus plugs persist in asthma, and changes in mucus plugs associate with changes in airflow over time. Am J Respir Crit Care Med (2022) 205(9):1036–45. doi: 10.1164/rccm.202110-2265OC
49. Chan R, Duraikannu C, Lipworth B. Clinical associations of mucus plugging in moderate to severe asthma. J Allergy Clin Immunol Pract (2023) 11(1):195–199.e2. doi: 10.1016/j.jaip.2022.09.008
50. Chung KF, Adcock IM. Precision medicine for the discovery of treatable mechanisms in severe asthma. Allergy. (2019) 74(9):1649–59. doi: 10.1111/all.13771
51. Andreev K, Graser A, Majer A, Mousset S, Finotto S. Therapeutical measures to control airway tolerance in asthma and lung cancer. Front Immunol (2012) 3:216. doi: 10.3389/fimmu.2012.00216
52. Kudo M, Ishigatsubo Y, Aoki I. Pathology of asthma. Front Microbiol (2013) 4:263. doi: 10.3389/fmicb.2013.00263
53. Ojiaku CA, Yoo EJ, Panettieri RA. Transforming growth factor β1 function in airway remodeling and hyperresponsiveness. Missing Link? Am J Respir Cell Mol Biol (2017) 56(4):432–42. doi: 10.1165/rcmb.2016-0307TR
54. Inoue H, Akimoto K, Homma T, Tanaka A, Sagara H. Airway epithelial dysfunction in asthma: relevant to epidermal growth factor receptors and airway epithelial cells. J Clin Med (2020) 9(11):3698. doi: 10.3390/jcm9113698
55. Fahy JV. Goblet cell and mucin gene abnormalities in asthma*. Chest. (2002) 122(6, Supplement):320S–6S. doi: 10.1378/chest.122.6_suppl.320S
56. Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, et al. IL-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest. (2001) 108(12):1817–24. doi: 10.1172/JCI200113557
57. Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG, et al. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol (2011) 45(5):1090–100. doi: 10.1165/rcmb.2011-0031OC
58. Lachowicz-Scroggins ME, Yuan S, Kerr SC, Dunican EM, Yu M, Carrington SD, et al. Abnormalities in MUC5AC and MUC5B protein in airway mucus in asthma. Am J Respir Crit Care Med (2016) 194(10):1296–9. doi: 10.1164/rccm.201603-0526LE
59. Lundgren R, Soderberg M, Horstedt P, Stenling R. Morphological studies of bronchial mucosal biopsies from asthmatics before and after ten years of treatment with inhaled steroids. Eur Respir J (1988) 1(10):883–9. doi: 10.1183/09031936.93.01100883
60. Thomas B, Rutman A, Hirst RA, Haldar P, Wardlaw AJ, Bankart J, et al. Ciliary dysfunction and ultrastructural abnormalities are features of severe asthma. J Allergy Clin Immunol (2010) 126(4):722–729.e2. doi: 10.1016/j.jaci.2010.05.046
61. Guerra S, Sherrill DL, Venker C, Ceccato CM, Halonen M, Martinez FD. Chronic bronchitis before age 50 years predicts incident airflow limitation and mortality risk. Thorax. (2009) 64(10):894–900. doi: 10.1136/thx.2008.110619
62. Stenberg H, Wadelius E, Moitra S, Åberg I, Ankerst J, Diamant Z, et al. Club cell protein (CC16) in plasma, bronchial brushes, BAL and urine following an inhaled allergen challenge in allergic asthmatics. Biomarkers. (2018) 23(1):51–60. doi: 10.1080/1354750X.2017.1375559
63. Polosa R, Puddicombe SM, Krishna MT, Tuck AB, Howarth PH, Holgate ST, et al. Expression of c-erbB receptors and ligands in the bronchial epithelium of asthmatic subjects. J Allergy Clin Immunol (2002) 109(1):75–81. doi: 10.1067/mai.2002.120274
64. Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax. (2005) 60(5):389–94. doi: 10.1136/thx.2004.030262
65. Gomperts BN, Kim LJ, Flaherty SA, Hackett BP. IL-13 regulates cilia loss and foxj1 expression in human airway epithelium. Am J Respir Cell Mol Biol (2007) 37(3):339–46. doi: 10.1165/rcmb.2006-0400OC
66. Yu L, Li N, Zhang J, Jiang Y. IL-13 regulates human nasal epithelial cell differentiation via H3K4me3 modification. J Inflammation Res (2017) 10, 10:181–8. doi: 10.2147/JIR.S149156
67. Gerovac BJ, Fregien NL. IL-13 inhibits multicilin expression and ciliogenesis via janus Kinase/Signal transducer and activator of transcription independently of notch cleavage. Am J Respir Cell Mol Biol (2016) 54(4):554–61. doi: 10.1165/rcmb.2015-0227OC
68. Ye C, Huang C, Zou M, Hu Y, Luo L, Wei Y, et al. The role of secreted Hsp90α in HDM-induced asthmatic airway epithelial barrier dysfunction. BMC Pulm Med (2019) 19(1):218. doi: 10.1186/s12890-019-0938-z
69. Jiang J, Liu JQ, Li J, Li M, Chen HB, Yan H, et al. Trek1 contributes to maintaining nasal epithelial barrier integrity. Sci Rep (2015) 5(1):9191. doi: 10.1038/srep09191
70. White SR. Apoptosis and the airway epithelium. J Allergy (2011) 2011:948406. doi: 10.1155/2011/948406
71. Jacquet A. The role of the house dust mite-induced innate immunity in development of allergic response. Int Arch Allergy Immunol (2011) 155(2):95–105. doi: 10.1159/000320375
72. Wan H, Winton HL, Soeller C, Gruenert DC, Thompson PJ, Cannell MB, et al. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen der p 1. Clin Exp Allergy (2000) 30(5):685–98. doi: 10.1046/j.1365-2222.2000.00820.x
73. Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J (2010) 36(5):1016–26. doi: 10.1183/09031936.00125809
74. Post S, Nawijn MC, Jonker MR, Kliphuis N, van den Berge M, van Oosterhout AJM, et al. House dust mite-induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy. (2013) 68(9):1117–25. doi: 10.1111/all.12202
75. Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite–promoted epithelial-to-Mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol (2010) 42(1):69–79. doi: 10.1165/rcmb.2008-0449OC
76. Mabalirajan U, Dinda AK, Kumar S, Roshan R, Gupta P, Sharma SK, et al. Mitochondrial structural changes and dysfunction are associated with experimental allergic Asthma1. J Immunol (2008) 181(5):3540–8. doi: 10.4049/jimmunol.181.5.3540
77. Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway Inflammation1. J Immunol (2009) 183(8):5379–87. doi: 10.4049/jimmunol.0900228
78. Michaeloudes C, Abubakar-Waziri H, Lakhdar R, Raby K, Dixey P, Adcock IM, et al. Molecular mechanisms of oxidative stress in asthma. Mol Aspects Med (2022) 85:101026. doi: 10.1016/j.mam.2021.101026
79. Chan TK, Tan WSD, Peh HY, Wong WSF. Aeroallergens induce reactive oxygen species production and DNA damage and dampen antioxidant responses in bronchial epithelial cells. J Immunol (2017) 199(1):39–47. doi: 10.4049/jimmunol.1600657
80. Lin CH, Hong YC, Kao SH. Aeroallergen der p 2 induces apoptosis of bronchial epithelial BEAS-2B cells via activation of both intrinsic and extrinsic pathway. Cell Biosci (2015) 5(1):71. doi: 10.1186/s13578-015-0063-5
81. Saito T, Ichikawa T, Numakura T, Yamada M, Koarai A, Fujino N, et al. PGC-1α regulates airway epithelial barrier dysfunction induced by house dust mite. Respir Res (2021) 22(1):63. doi: 10.1186/s12931-021-01663-6
82. Wang A, Keita ÅV, Phan V, McKay CM, Schoultz I, Lee J, et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am J Pathol (2014) 184(9):2516–27. doi: 10.1016/j.ajpath.2014.05.019
83. Gan H, Hao Q, Idell S, Tang H. Transcription factor Runx3 is induced by influenza a virus and double-strand RNA and mediates airway epithelial cell apoptosis. Sci Rep (2015) 5:17916. doi: 10.1038/srep17916
84. Linfield DT, Raduka A, Aghapour M, Rezaee F. Airway tight junctions as targets of viral infections. Tissue Barriers. (2021) 9(2):1883965. doi: 10.1080/21688370.2021.1883965
85. Petecchia L, Sabatini F, Varesio L, Camoirano A, Usai C, Pezzolo A, et al. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest. (2009) 135(6):1502–12. doi: 10.1378/chest.08-1780
86. Zhang Y, Zhang L, Chen W, Zhang Y, Wang X, Dong Y, et al. Shp2 regulates PM2.5-induced airway epithelial barrier dysfunction by modulating ERK1/2 signaling pathway. Toxicol Lett (2021) 350:62–70. doi: 10.1016/j.toxlet.2021.07.002
87. Furuta GT, Nieuwenhuis EES, Karhausen J, Gleich G, Blumberg RS, Lee JJ, et al. Eosinophils alter colonic epithelial barrier function: role for major basic protein. Am J Physiol-Gastrointest Liver Physiol (2005) 289(5):G890–7. doi: 10.1152/ajpgi.00015.2005
88. Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S, et al. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. (2013) 1(2):e24333. doi: 10.4161/tisb.24333
89. Leino MS, Loxham M, Blume C, Swindle EJ, Jayasekera NP, Dennison PW, et al. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PloS One (2013) 8(8):e71278. doi: 10.1371/journal.pone.0071278
90. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. (2015) 43(1):29–40. doi: 10.1016/j.immuni.2015.07.007
91. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol (2011) 128(3):549–556.e12. doi: 10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A6367
92. Wark PAB, Tooze M, Powell H, Parsons K. Viral and bacterial infection in acute asthma and chronic obstructive pulmonary disease increases the risk of readmission. Respirology. (2013) 18(6):996–1002. doi: 10.1111/resp.12099
93. Bucchieri F, Puddicombe SM, Lordan JL, Richter A, Buchanan D, Wilson SJ, et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol (2002) 27(2):179–85. doi: 10.1165/ajrcmb.27.2.4699
94. Stevens PT, Kicic A, Sutanto EN, Knight DA, Stick SM. Dysregulated repair in asthmatic paediatric airway epithelial cells: the role of plasminogen activator inhibitor-1. Clin Exp Allergy (2008) 38(12):1901–10. doi: 10.1111/j.1365-2222.2008.03093.x
95. Freishtat RJ, Watson AM, Benton AS, Iqbal SF, Pillai DK, Rose MC, et al. Asthmatic airway epithelium is intrinsically inflammatory and mitotically dyssynchronous. Am J Respir Cell Mol Biol (2011) 44(6):863–9. doi: 10.1165/rcmb.2010-0029OC
96. Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial–mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-β1. Am J Respir Crit Care Med (2009) 180(2):122–33. doi: 10.1164/rccm.200811-1730OC
97. Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2013) 187(3):228–37. doi: 10.1164/rccm.201210-1843CI
98. Kim V, Davey A, Comellas AP, Han MK, Washko G, Martinez CH, et al. Clinical and computed tomographic predictors of chronic bronchitis in COPD: a cross sectional analysis of the COPDGene study. Respir Res (2014) 15(1):52. doi: 10.1186/1465-9921-15-52
99. Burgel PR, Nesme-Meyer P, Chanez P, Caillaud D, Carré P, Perez T, et al. Cough and sputum production are associated with frequent exacerbations and hospitalizations in COPD subjects. Chest. (2009) 135(4):975–82. doi: 10.1378/chest.08-2062
100. Staudt MR, Buro-Auriemma LJ, Walters MS, Salit J, Vincent T, Shaykhiev R, et al. Airway basal Stem/Progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2014) 190(8):955–8. doi: 10.1164/rccm.201406-1167LE
101. Sisson JH, Papi A, Beckmann JD, Leise KL, Wisecarver J, Brodersen BW, et al. Smoke and viral infection cause cilia loss detectable by bronchoalveolar lavage cytology and dynein ELISA. Am J Respir Crit Care Med (1994) 149(1):205–13. doi: 10.1164/ajrccm.149.1.8111584
102. Auerbach O, Stout AP, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to sex, age, residence, smoking and pneumonia. N Engl J Med (1962) 267(3):111–9. doi: 10.1056/NEJM196207192670301
103. Lungarella G, Fonzi L, Ermini G. Abnormalities of bronchial cilia in patients with chronic bronchitis. Lung. (1983) 161(1):147–56. doi: 10.1007/BF02713856
104. Verra F, Escudier E, Lebargy F, Bernaudin JF, de Crémoux H, Bignon J. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am J Respir Crit Care Med (1995) 151(3_pt_1):630–4. doi: 10.1164/ajrccm/151.3_Pt_1.630
105. Stanley PJ, Wilson R, Greenstone MA, MacWilliam L, Cole PJ. Effect of cigarette smoking on nasal mucociliary clearance and ciliary beat frequency. Thorax. (1986) 41(7):519–23. doi: 10.1136/thx.41.7.519
106. Allen-Gipson DS, Romberger DJ, Forget MA, May KL, Sisson JH, Wyatt TA. IL-8 inhibits isoproterenol-stimulated ciliary beat frequency in bovine bronchial epithelial cells. J Aerosol Med (2004) 17(2):107–15. doi: 10.1089/0894268041457138
107. Evans CM, Koo JS. Airway mucus: the good, the bad, the sticky. Pharmacol Ther (2009) 121(3):332–48. doi: 10.1016/j.pharmthera.2008.11.001
108. Rusznak C, Mills PR, Devalia JL, Sapsford RJ, Davies RJ, Lozewicz S. Effect of cigarette smoke on the permeability and IL-1 β and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2000) 23(4):530–6. doi: 10.1165/ajrcmb.23.4.3959
109. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci (2017) 131(13):1541–58. doi: 10.1042/CS20160487
110. Hulbert WC, Walker DC, Jackson A, Hogg JC. Airway permeability to horseradish peroxidase in Guinea pigs: the repair phase after injury by cigarette smoke. Am Rev Respir Dis (1981) 123(3):320–6. doi: 10.1164/arrd.1981.123.3.320
111. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med (2008) 178(12):1271–81. doi: 10.1164/rccm.200801-136OC
112. Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC, et al. Epithelial mucin stores are increased in the Large airways of smokers with airflow obstruction. Chest. (2006) 130(4):1102–8. doi: 10.1378/chest.130.4.1102
113. Caramori G, Di Gregorio C, Carlstedt I, Casolari P, Guzzinati I, Adcock IM, et al. Mucin expression in peripheral airways of patients with chronic obstructive pulmonary disease. Histopathology. (2004) 45(5):477–84. doi: 10.1111/j.1365-2559.2004.01952.x
114. Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol (2008) 70:459–86. doi: 10.1146/annurev.physiol.70.113006.100702
115. Wickström C, Davies JR, Eriksen GV, Veerman ECI, Carlstedt I. MUC5B is a major gel-forming, oligomeric mucin from human salivary gland, respiratory tract and endocervix: identification of glycoforms and c-terminal cleavage. Biochem J (1998) 334(3):685–93. doi: 10.1042/bj3340685
116. Gamez AS, Gras D, Petit A, Knabe L, Molinari N, Vachier I, et al. Supplementing defect in club cell secretory protein attenuates airway inflammation in COPD. Chest. (2015) 147(6):1467–76. doi: 10.1378/chest.14-1174
117. Lumsden AB, McLean A, Lamb D. Goblet and Clara cells of human distal airways: evidence for smoking induced changes in their numbers. Thorax. (1984) 39(11):844–9. doi: 10.1136/thx.39.11.844
118. Park HY, Churg A, Wright JL, Li Y, Tam S, Man SFP, et al. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2013) 188(12):1413–9. doi: 10.1164/rccm.201305-0892OC
119. Herr C, Beisswenger C, Hess C, Kandler K, Suttorp N, Welte T, et al. Suppression of pulmonary innate host defence in smokers. Thorax. (2009) 64(2):144–9. doi: 10.1136/thx.2008.102681
120. Ibrahim HR, Aoki T, Pellegrini A. Strategies for new antimicrobial proteins and peptides: lysozyme and aprotinin as model molecules. Curr Pharm Des (2002) 8(9):671–93. doi: 10.2174/1381612023395349
121. Ellison RT, Giehl TJ. Killing of gram-negative bacteria by lactoferrin and lysozyme. J Clin Invest. (1991) 88(4):1080–91. doi: 10.1172/JCI115407
122. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol Mech Dis (2009) 4(1):435–59. doi: 10.1146/annurev.pathol.4.110807.092145
123. Baraldo S, Turato G, Badin C, Bazzan E, Beghé B, Zuin R, et al. Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax. (2004) 59(4):308–12. doi: 10.1136/thx.2003.012146
124. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med (2004) 350(26):2645–53. doi: 10.1056/NEJMoa032158
125. Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol (2007) 25:101–37. doi: 10.1146/annurev.immunol.25.022106.141647
126. Horiba K, Fukuda Y. Synchronous appearance of fibronectin, integrin α5β1, vinculin and actin in epithelial cells and fibroblasts during rat tracheal wound healing. Virchows Arch (1994) 425(4):425–34. doi: 10.1007/BF00189581
127. Rickard KA, Taylor J, Rennard SI, Spurzem JR. Migration of bovine bronchial epithelial cells to extracellular matrix components. Am J Respir Cell Mol Biol (1993) 8:63–3. doi: 10.1165/ajrcmb/8.1.63
128. Gochuico BR, Miranda KM, Hessel EM, De Bie JJ, Van Oosterhout AJM, Cruikshank WW, et al. Airway epithelial fas ligand expression: potential role in modulating bronchial inflammation. Am J Physiol-Lung Cell Mol Physiol (1998) 274(3):L444–9. doi: 10.1152/ajplung.1998.274.3.L444
129. Masubuchi T, Koyama S, Sato E, Takamizawa A, Kubo K, Sekiguchi M, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol (1998) 153(6):1903–12. doi: 10.1016/S0002-9440(10)65704-5
130. Dubar V, Gosset P, Aerts C, Voisin C, Wallaert B, Tonnel AB. In vitro acute effects of tobacco smoke on tumor necrosis factor a and interleukin-6 production by alveolar macrophages. Exp Lung Res (1993) 19(3):345–59. doi: 10.3109/01902149309064351
131. Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, et al. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2007) 175(10):998–1005. doi: 10.1164/rccm.200608-1113OC
132. Saetta M, Di Stefano A, Maestrelli P, Ferraresso A, Drigo R, Potena A, et al. Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitis. Am Rev Respir Dis (1993) 147(2):301–6. doi: 10.1164/ajrccm/147.2.301
133. Agrawal A, Agrawal S, Gupta S. Role of dendritic cells in inflammation and loss of tolerance in the elderly. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00896
134. van der Strate BWA, Postma DS, Brandsma CA, Melgert BN, Luinge MA, Geerlings M, et al. Cigarette smoke–induced emphysema. Am J Respir Crit Care Med (2006) 173(7):751–8. doi: 10.1164/rccm.200504-594OC
135. O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med (1997) 155(3):852–7. doi: 10.1164/ajrccm.155.3.9117016
136. Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghé B, et al. Airway inflammation in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2002) 166(1):105–10. doi: 10.1164/rccm.2111084
137. O’Donnell RA, Peebles C, Ward JA, Daraker A, Angco G, Broberg P, et al. Relationship between peripheral airway dysfunction, airway obstruction, and neutrophilic inflammation in COPD. Thorax. (2004) 59(10):837–42. doi: 10.1136/thx.2003.019349
138. Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, et al. CD8 + ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (1999) 160(2):711–7. doi: 10.1164/ajrccm.160.2.9812020
139. Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol (2003) 81(4):289–96. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x
140. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis (2018) 13:3341–8. doi: 10.2147/COPD.S176122
141. Ferrara F, D’Adda D, Falchi M, Dall’Asta L. The macrophagic activity of patients affected by pneumonia or chronic obstructive pulmonary disease. Int J Tissue React (1996) 18(4–6):109–14. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x
142. Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med (1996) 154(3):813–6. doi: 10.1164/ajrccm.154.3.8810624
143. Bartoli ML, Novelli F, Costa F, Malagrinò L, Melosini L, Bacci E, et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediators Inflamm (2011) 2011:e891752. doi: 10.1155/2011/891752
144. Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, Huyck HL, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol (2011) 187(2):987–98. doi: 10.4049/jimmunol.1001861
145. Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. (2005) 60(4):293–300. doi: 10.1136/thx.2004.027946
146. Mercado N, Thimmulappa R, Thomas CMR, Fenwick PS, Chana KK, Donnelly LE, et al. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun (2011) 406(2):292–8. doi: 10.1016/j.bbrc.2011.02.035
147. Gorowiec MR, Borthwick LA, Parker SM, Kirby JA, Saretzki GC, Fisher AJ. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic Biol Med (2012) 52(6):1024–32. doi: 10.1016/j.freeradbiomed.2011.12.020
148. Casalino-Matsuda SM, Monzón ME, Forteza RM. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am J Respir Cell Mol Biol (2006) 34(5):581–91. doi: 10.1165/rcmb.2005-0386OC
149. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J (2006) 28(1):219–42. doi: 10.1183/09031936.06.00053805
150. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight (2018) 3(3):e93203. doi: 10.1172/jci.insight.93203
151. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med (2019) 200(5):556–64. doi: 10.1164/rccm.201810-1975TR
152. Baker JR, Vuppusetty C, Colley T, Papaioannou AI, Fenwick P, Donnelly L, et al. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep (2016) 6(1):35871. doi: 10.1038/srep35871
153. Cottage CT, Peterson N, Kearley J, Berlin A, Xiong X, Huntley A, et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun Biol (2019) 2:307. doi: 10.1038/s42003-019-0532-1
154. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress–induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol (2015) 136(3):769–80. doi: 10.1016/j.jaci.2015.01.046
155. Haji G, Wiegman CH, Michaeloudes C, Patel MS, Curtis K, Bhavsar P, et al. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir Res (2020) 21(1):262. doi: 10.1186/s12931-020-01527-5
156. Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res (2013) 14(1):97. doi: 10.1186/1465-9921-14-97
157. Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am J Physiol-Lung Cell Mol Physiol (2013) 305(10):L737–46. doi: 10.1152/ajplung.00146.2013
158. Cui Y, Liang Y, Ip MSM, Mak JCW. Cigarette smoke induces apoptosis via 18 kDa translocator protein in human bronchial epithelial cells. Life Sci (2021) 265:118862. doi: 10.1016/j.lfs.2020.118862
159. Wang M, Zhang Y, Xu M, Zhang H, Chen Y, Chung KF, et al. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic Biol Med (2019) 134:229–38. doi: 10.1016/j.freeradbiomed.2019.01.0044
160. Bandela M, Suryadevara V, Fu P, Reddy SP, Bikkavilli K, Huang LS, et al. Role of lysocardiolipin acyltransferase in cigarette smoke-induced lung epithelial cell mitochondrial ROS, mitochondrial dynamics, and apoptosis. Cell Biochem Biophys (2022) 80(1):203–16. doi: 10.1007/s12013-021-01043-3
161. Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J (2015) 29(7):2912–29. doi: 10.1096/fj.14-268276
162. Pouwels SD, Hesse L, Faiz A, Lubbers J, Bodha PK, ten Hacken NHT, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol-Lung Cell Mol Physiol (2016) 311(5):L881–92. doi: 10.1152/ajplung.00135.2016
163. Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R, ten Hacken NHT, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol-Lung Cell Mol Physiol (2016) 310(4):L377–86. doi: 10.1152/ajplung.00174.2015
164. Kim K, Kim HJ, Binas B, Kang JH, Chung IY. Inflammatory mediators ATP and S100A12 activate the NLRP3 inflammasome to induce MUC5AC production in airway epithelial cells. Biochem Biophys Res Commun (2018) 503(2):657–64. doi: 10.1016/j.bbrc.2018.06.057
165. Tran I, Ji C, Ni I, Min T, Tang D, Vij N. Role of cigarette smoke–induced aggresome formation in chronic obstructive pulmonary disease–emphysema pathogenesis. Am J Respir Cell Mol Biol (2015) 53(2):159–73. doi: 10.1165/rcmb.2014-0107OC
166. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol-Cell Physiol (2018) 314(1):C73–87. doi: 10.1152/ajpcell.00110.2016
167. Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. OncoImmunology. (2012) 1(5):630–41. doi: 10.4161/onci.20297
168. Mercado N, Colley T, Baker JR, Vuppussetty C, Kono Y, Clarke C, et al. Bicaudal D1 impairs autophagosome maturation in chronic obstructive pulmonary disease. FASEB BioAdvances. (2019) 1(11):688–705. doi: 10.1096/fba.2018-00055
169. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. (2015) 11(3):547–59. doi: 10.1080/15548627.2015.1017190
170. Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. (2019) 15(3):510–26. doi: 10.1080/15548627.2018.1532259
171. Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PloS Biol (2007) 5(5):e110. doi: 10.1371/journal.pbio.0050110
172. Wang Y, Liu J, Zhou JS, Huang HQ, Li ZY, Xu XC, et al. MTOR suppresses cigarette smoke–induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J Immunol (2018) 200(8):2571–80. doi: 10.4049/jimmunol.1701681
173. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. (2014) 124(9):3987–4003. doi: 10.1172/JCI74985
174. Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PloS One (2008) 3(10):e3316. doi: 10.1371/journal.pone.0003316
175. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, et al. Histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. (2013) 123(12):5212–30. doi: 10.1172/JCI69636
176. Zhou JS, Zhao Y, Zhou HB, Wang Y, Wu YF, Li ZY, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol-Lung Cell Mol Physiol (2016) 310(11):L1042–52. doi: 10.1152/ajplung.00418.2015
177. Sundar IK, Rahman I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer. Am J Physiol-Lung Cell Mol Physiol (2016) 311(6):L1245–58. doi: 10.1152/ajplung.00253.2016
178. Song J, Heijink IH, Kistemaker LEM, Reinders-Luinge M, Kooistra W, Noordhoek JA, et al. Aberrant DNA methylation and expression of SPDEF and FOXA2 in airway epithelium of patients with COPD. Clin Epigenetics. (2017) 9(1):42. doi: 10.1186/s13148-017-0341-7
179. Buro-Auriemma LJ, Salit J, Hackett NR, Walters MS, Strulovici-Barel Y, Staudt MR, et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum Mol Genet (2013) 22(23):4726–38. doi: 10.1093/hmg/ddt326
180. Chen Q, Nwozor KO, van den Berge M, Slebos DJ, Faiz A, Jonker MR, et al. From differential DNA methylation in COPD to mitochondria: regulation of AHRR expression affects airway epithelial response to cigarette smoke. Cells. (2022) 11(21):3423. doi: 10.3390/cells11213423
181. Zhang Z, Fu C, Liu J, Sai X, Qin C, Di T, et al. Hypermethylation of the Nrf2 promoter induces ferroptosis by inhibiting the Nrf2-GPX4 axis in COPD. Int J Chron Obstruct Pulmon Dis (2021) 16:3347–62. doi: 10.2147/COPD.S340113
182. Sharma S, Yang IV, Schwartz DA. Epigenetic regulation of immune function in asthma. J Allergy Clin Immunol (2022) 150(2):259–65. doi: 10.1016/j.jaci.2022.06.002
183. Chen L, Luo L, Kang N, He X, Li T, Chen Y. The protective effect of HBO1 on cigarette smoke extract-induced apoptosis in airway epithelial cells. Int J Chron Obstruct Pulmon Dis (2020) 15:15–24. doi: 10.2147/COPD.S234634
184. Sarker RSJ, Conlon TM, Morrone C, Srivastava B, Konyalilar N, Verleden SE, et al. CARM1 regulates senescence during airway epithelial cell injury in COPD pathogenesis. Am J Physiol-Lung Cell Mol Physiol (2019) 317(5):L602–14. doi: 10.1152/ajplung.00441.2018
185. O’Brien KB, Alberich-Jordà M, Yadav N, Kocher O, DiRuscio A, Ebralidze A, et al. CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development. (2010) 137(13):2147–56. doi: 10.1242/dev.037150
186. Tasena H, Timens W, van den Berge M, van Broekhuizen J, Kennedy BK, Hylkema MN, et al. MicroRNAs associated with chronic mucus hypersecretion in COPD are involved in fibroblast–epithelium crosstalk. Cells. (2022) 11(3):526. doi: 10.3390/cells11030526
187. Di T, Yang Y, Fu C, Zhang Z, Qin C, Sai X, et al. Let-7 mediated airway remodelling in chronic obstructive pulmonary disease via the regulation of IL-6. Eur J Clin Invest. (2021) 51(4):e13425. doi: 10.1111/eci.13425
188. Pottelberge GRV, Mestdagh P, Bracke KR, Thas O, van DYMTA, GF J, et al. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2011) 183(7):898–906. doi: 10.1164/rccm.201002-0304OC
189. Lin X, Li Y, Gong L, Yun JH, Xu S, Tesfaigzi Y, et al. Tempo-spatial regulation of the wnt pathway by FAM13A modulates the stemness of alveolar epithelial progenitors. eBioMedicine. (2021) 69:103463. doi: 10.1016/j.ebiom.2021.103463
190. Jiang Z, Knudsen NH, Wang G, Qiu W, Naing ZZC, Bai Y, et al. Genetic control of fatty acid β-oxidation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2017) 56(6):738–48. doi: 10.1165/rcmb.2016-0282OC
191. Tam A, Leclair P, Li LV, Yang CX, Li X, Witzigmann D, et al. FAM13A as potential therapeutic target in modulating TGF-β-induced airway tissue remodeling in COPD. Am J Physiol-Lung Cell Mol Physiol (2021) 321(2):L377–91. doi: 10.1152/ajplung.00477.2020
192. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke–induced bronchitis and emphysema in mice. Nat Med (2016) 22(2):163–74. doi: 10.1038/nm.4021
Keywords: epithelium, injury, repair, asthma, COPD, barrier, cell junctions, permeability
Citation: Raby KL, Michaeloudes C, Tonkin J, Chung KF and Bhavsar PK (2023) Mechanisms of airway epithelial injury and abnormal repair in asthma and COPD. Front. Immunol. 14:1201658. doi: 10.3389/fimmu.2023.1201658
Received: 06 April 2023; Accepted: 19 June 2023;
Published: 13 July 2023.
Edited by:
Guirong Wang, Upstate Medical University, United StatesReviewed by:
Istvan Boldogh, University of Texas Medical Branch at Galveston, United StatesYahong Chen, Peking University Third Hospital, China
Copyright © 2023 Raby, Michaeloudes, Tonkin, Chung and Bhavsar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj Kumar Bhavsar, cC5iaGF2c2FyQGltcGVyaWFsLmFjLnVr
†These authors have contributed equally to this work