 Meiying Wang1,2,3
Meiying Wang1,2,3 Snehin Rajkumar3
Snehin Rajkumar3 Yupeng Lai1Xingjiao Liu1
Yupeng Lai1Xingjiao Liu1 Jing He1,4Tatsuya Ishikawa3
Jing He1,4Tatsuya Ishikawa3 Dhiraj Nallapothula3
Dhiraj Nallapothula3 Ram Raj Singh3,5,6,7*
Ram Raj Singh3,5,6,7*- 1Department of Rheumatology and Immunology, Shenzhen Second People’s Hospital, The First Affiliated Hospital of Shenzhen University, Shenzhen, China
- 2Peking University Shenzhen Hosiptal, Shenzhen, China
- 3Autoimmunity and Tolerance Laboratory, Division of Rheumatology, Department of Medicine, David Geffen School of Medicine at University of California Los Angeles (UCLA), Los Angeles, CA, United States
- 4Department of Nephrology, Peking University Shenzhen Hospital, Shenzhen, China
- 5Department of Pathology and Laboratory Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
- 6Molecular Toxicology Interdepartmental Program, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
- 7Jonsson Comprehensive Cancer Center, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
In response to inflammatory stimuli in conditions such as autoimmune disorders, infections and cancers, immune cells organize in nonlymphoid tissues, which resemble secondary lymphoid organs. Such immune cell clusters are called tertiary lymphoid structures (TLS). Here, we describe the potential role of TLS in the pathogenesis of autoimmune disease, focusing on lupus nephritis, a condition that incurs major morbidity and mortality. In the kidneys of patients and animals with lupus nephritis, the presence of immune cell aggregates with similar cell composition, structure, and gene signature as lymph nodes and of lymphoid tissue-inducer and -organizer cells, along with evidence of communication between stromal and immune cells are indicative of the formation of TLS. TLS formation in kidneys affected by lupus may be instigated by local increases in lymphorganogenic chemokines such as CXCL13, and in molecules associated with leukocyte migration and vascularization. Importantly, the presence of TLS in kidneys is associated with severe tubulointerstitial inflammation, higher disease activity and chronicity indices, and poor response to treatment in patients with lupus nephritis. TLS may contribute to the pathogenesis of lupus nephritis by increasing local IFN-I production, facilitating the recruitment and supporting survival of autoreactive B cells, maintaining local production of systemic autoantibodies such as anti-dsDNA and anti-Sm/RNP autoantibodies, and initiating epitope spreading to local autoantigens. Resolution of TLS, along with improvement in lupus, by treating animals with soluble BAFF receptor, docosahexaenoic acid, complement inhibitor C4BP(β-), S1P1 receptor modulator Cenerimod, dexamethasone, and anti-CXCL13 further emphasizes a role of TLS in the pathogenesis of lupus. However, the mechanisms underlying TLS formation and their roles in the pathogenesis of lupus nephritis are not fully comprehended. Furthermore, the lack of non-invasive methods to visualize/quantify TLS in kidneys is also a major hurdle; however, recent success in visualizing TLS in lupus-prone mice by photon emission computed tomography provides hope for early detection and manipulation of TLS.
1 Introduction
Tertiary lymphoid structures (TLS), also known as tertiary lymphoid organs, ectopic lymphoid neogenesis, or ectopic lymphoid tissues, are organized clusters of immune cells that form in non-lymphoid tissues after birth. TLS are neither stable structures nor present in embryonic life, but instead can be induced by chronic inflammatory stimuli and tissue injury (1, 2) in autoimmune diseases such as Sjogren syndrome, rheumatoid arthritis, systemic lupus erythematosus (SLE), and myositis (3–5), allograft rejection (2, 6), chronic infections (5), and cancers (7–9).
Impaired tolerance to self-antigens via various mechanisms marks the early step in the pathogenesis of autoimmune diseases (10, 11), resulting in the initial activation of autoreactive cells. These activated autoimmune cells may then infiltrate the target organs, where the persistent exposure to antigens in chronically inflamed environments keeps them activated. Such chronically activated immune cells can serve as a substitute for lymphoid tissue inducer (LTi) cells and initiate the formation of TLS in tissues. TLS may execute tissue-specific immune responses, thereby providing communications between immune cells and local resident cells (12). This process typically aids in the clearance or neutralization of pathogens through the local induction of plasma cells that generate specific antibodies (13). TLS often resolve after successful antigen clearance or upon resolution of inflammation. However, TLS resolution might not happen in the context of persistent antigen presentation in autoimmune-prone backgrounds, thereby enabling the local induction and expansion of autoreactive T and B cells, which may cause increased autoantibody production and contribute to local pathology as in SLE.
SLE is a highly heterogeneous disease, with different patients exhibiting different manifestations. Kidneys may be involved in up to 50% of adults and up to 80% of children with SLE (14, 15). Kidney disease in SLE, called lupus nephritis (LN), continues to be a major contributor of morbidity and mortality in SLE (16, 17). Current treatments for LN may cause systemic immune suppression with many adverse effects. Hence, understanding local mechanisms of disease in kidneys may open new avenues to develop organ-targeted treatments for LN, thereby avoiding systemic toxicity. In this article, we will review the current understanding of TLS as it pertains to LN.
2 Secondary lymphoid organs
TLS share similarities with secondary lymphoid organs (SLO) with regard to their structure and function (18–21). Understanding the development of SLO may provide clues to mechanisms underlying TLS formation. SLO consist of lymph nodes, spleen, tonsils, Peyer’s patches, and mucosa-associated lymphoid tissues distributed throughout the body. These structures play a role in initiating and organizing adaptive immune responses by facilitating the interaction between antigens and immune cells. The development and formation of SLO, particularly lymph nodes and Peyer’s patches, involve CD34+ hematopoietic stem cells and TNF-related activation-induced cytokine (TRANCE+) stromal cells, as well as cytokines, chemokines, adhesion molecules, and specialized vasculature (22). SLO formation, which occurs during embryogenesis or in the first few weeks after birth, involves hematopoietic lymphoid tissue inducer (LTi) cells, a subtype of innate lymphoid cells (CD45+CD4+CD3−) that express RORγt and Id2. The process also involves the interactions of LTi cells with TRANCE and CXC-chemokine ligand 13 (CXCL13) through their receptors, TRANCE-R and CXCR5, respectively (23). Once LTi cells aggregate, their function is sustained by IL-7. This induces the expression of lymphotoxin α1β2 which binds to the lymphotoxin β receptor expressed on mesenchymal lymphoid tissue organizer (LTo) cells. This binding promotes LTo cells to express adhesion molecules, including vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), mucosal addressin cell-adhesion molecule 1 (MAdCAM1), and peripheral node addressin (PNAd), and to generate lymphoid chemokines, such as CC-chemokine ligand 19 (CCL19), CCL21, and CXCL13. Then, the arrangement of lymphocytes expressing CCR7 and CXCR5 receptors, which contributes to the formation of T cell and B cell zones within SLO, is facilitated by follicular reticular cells that express CCL19 and/or CCL21 and follicular dendritic cells (FDCs) that express CXCL13 (24).
Germinal center (GC) formation in SLO is regulated by antigen-driven interactions between B cells and follicular T helper (Tfh) or follicular T regulatory cells (Tfr). Tfh cells support the GC reaction through their expression of CD40L that engages CD40 on GC B cells, activates the NF-κB pathway, and triggers B cell proliferation. Tfh cells can also upregulate the Tfr–associated transcription factor Foxp3, which dampens Tfh cells’ expression of CD40L and IL-21 and contributes to GC extinction (25). The transcription factor BCL6 is essential to the differentiation of Tfh cells and the formation of GCs. In the absence of T cell–expressed BCL6, B cell responses to protein antigens do not occur and GCs fail to form (26). GCs are crucial for generating high-affinity antibodies against pathogens. GC B cells are positively selected for clonal expansion based on affinity of their cell surface receptors for an antigen – high affinity for an antigen favors differentiation of GC B cells into proliferative plasmablast cells. Some of these plasmablasts subsequently enter the quiescent plasma cells compartment, ultimately resulting in the generation of high-affinity, long-lived plasma cells and memory B cells within the GC (27). Somatic hypermutation has the potential to generate self-reactive immunoglobulins and humoral autoimmunity, whereas negative selection in GC proper entails elimination by apoptosis of GC B cells that acquire self-reactivity through somatic hypermutation (25).
The distinct microenvironmental niches within SLO that harbor different sets of innate and adaptive immune cell populations are formed by the structural scaffold of follicular reticular cells, which also generate conduit networks for cell communication (28). In addition, specialized blood vessels known as high endothelial venules (HEVs) play a role in guiding lymphocytes to SLO. They facilitate the extravasation of lymphocytes from the bloodstream, a necessary step for the initiation of adaptive immune responses in lymphoid tissues (29–31). HEVs are critical components of the immune system that facilitate lymphocyte recirculation and immune surveillance, playing a role in recognizing and responding to pathogens as well as in chronic inflammatory diseases (32).
Patients with SLE exhibit many abnormalities of SLO, including follicular hyperplasia, coagulative necrosis, diverse histological lesions, and increased 18-F-fluoro-2-deoxy-D-glucose (18F-FDG) uptake (33, 34). A higher 18F-FDG accumulation was also found in organs with higher immune cell compositions such as the lymph nodes, spleen, and thymus as compared to organs like the brain, liver, and lungs in NZB/NZW F1 mouse model of lupus (35). These observations suggest that immune cells in SLO in humans and animals with lupus are in an active state. Although the exact pathogenic contributions of these abnormalities are not fully understood, SLO abnormalities may contribute to autoimmune pathogenesis in multiple ways including initial breakdown of tolerance and activation of autoreactive T and B cells (10, 11, 36). For example, tissue-resident dendritic cells that carry local antigens to tissue-draining lymph nodes to induce tolerance do not migrate well to lymph nodes in lupus-prone mice, thus contributing to loss of tolerance and inflammation (37, 38).
3 Tertiary lymphoid structures: initiation and formation
After the initial activation in SLO, activated immune cells may migrate to the target organs where the persistent exposure to antigens in chronically inflamed environments may keep immune cells activated. Such activated cells in tissues may serve as primary regulators of local TLS formation, analogous to the role of classical LTi cells in SLO. Activated immune cells produce CXCL13 and IL-7, and recruit LTi cells to the site of inflammation (39) in a positive feedback loop. T helper 17 (Th17) cells (40), B cells (41), and M1-polarized macrophages (42) have been shown to possess the ability to replace LTi cells and initiate TLS formation in various pathological conditions (Figure 1).
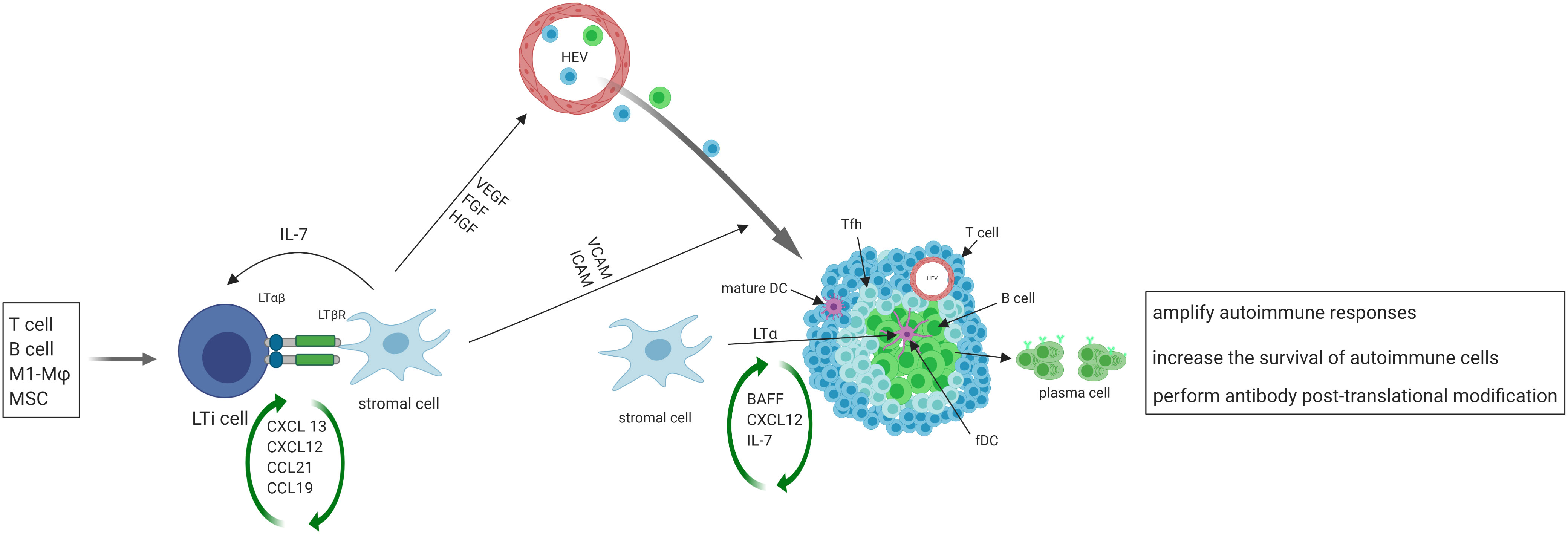
Figure 1 The initiation, formation and function of TLS. Activated immune cells produce CXCL13 and IL-7, recruit LTi cells to the site of inflammation. LTi cells secrete lymphotoxin (LT) αβ, which binds to lymphotoxin β receptor (LTβR) expressed on local stromal cells, inducing the production of chemokines such as CXCL13, CXCL12, CCL21, and CCL19. These chemokines then trigger the recruitment of lymphocytes, which initiate the development of TLS. The interaction of LTβ and LTβR also leads to the secretion of VEGF, FGF and hepatocyte growth factor (HGF) by stromal cells, as well as the secretion of vascular cell adhesion molecule 1 (VCAM1) and ICAM1. VEGF, FGF, and HGF play a crucial role in promoting the development of high endothelial venules (HEVs), while ICAM and VCAM expressed on local stromal cells facilitate the recruitment of T and B cells from nearby HEVs and promote their eventual organization into TLS. Some local stromal cells differentiate into FDCs upon stimulation with LTα, and FDCs aid in the development and sustenance of TLS by producing chemokines and providing a cellular network that facilitates B cell migration. A subset of CD4+ T cells polarizes into T follicular helper (Tfh, CXCR5hiPD-1hiICOShi) cells that promote B cell activation and antibody production. TLS facilitate the process of affinity maturation and differentiation of naïve B cells into germinal center B cells and plasma cells that produce antibodies.
On the other hand, stromal cells play the same role in TLS formation as classical LTo in SLO. At sites of inflammation, LTi cells aggregate and release lymphotoxin αβ, which binds to the lymphotoxin β receptor expressed on stromal cells residing in the area (43). The resident stromal cells express the classical LTo cell phenotype and respond to the accumulation of LTi cells by inducing the production of homeostatic chemokines such as CXCL13, CXCL12, CCL21, and CCL19. These chemokines recruit lymphocytes to the site of inflammation and initiate TLS formation (18–20, 44). Upon activation of the lymphotoxin β–lymphotoxin β receptor signaling pathway in stromal cells and surrogate LTi cells, the secretion of several growth factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and hepatocyte growth factor are induced (45). Additionally, adhesion molecules such as VCAM1 and intercellular adhesion molecule 1 (ICAM1) are also secreted (46). VEGF, FGF, and hepatocyte growth factor are critical for promoting the development of HEVs. Additionally, the presence of ICAM and VCAM on stromal cells in the vicinity aids in the recruitment of T and B cells from nearby HEVs and promotes the organization of these cells into TLS (47).
Following the organization of TLS by cytokines/chemokines, some local stromal cells differentiate into FDCs upon stimulation with lymphotoxin α. FDCs play a role in supporting the development and maintenance of TLS by producing chemokines and providing a cellular network for B cell migration (13). A subset of CD4+ T cells polarizes into T follicular helper (Tfh, CXCR5hiPD-1hiICOShi) cells that promote B cell activation and antibody production (48). In addition to CD4+ T follicular helper (Tfh) cells, T cell compartments also contain CD8+ cytotoxic T cells, CD4+ T helper 1 (Th1) cells, and regulatory T cells (Tregs) (49). TLS display similar organization to SLO, where B cells aggregate around FDCs expressing the CD21 antigen, and T cells areas contain dendritic cells expressing the marker MIDC-8 (50). Subsequently, TLS facilitate the process of affinity maturation and differentiation of naïve B cells into GC B cells and plasma cells that produce antibodies (51). As in SLO, follicular reticular cells form a dense network to anchor TLS at the site of chronic inflammation (1). At the same time, vasculature comprised of HEVs and lymphatic vessels forms the surrounding compartments to facilitate ingress and egress of immune cells.
Taken together, activated immune cells can function as primary regulators of TLS formation, substituting for LTi cells. Resident stromal cells function as LTo cells to stimulate the production of cytokines and chemokines, creating a positive feedback loop that induces the formation of GCs by lymphoid cells. The development of TLS can be divided into three sequential stages. The first stage, known as the ‘immature’ or ‘early’ TLS, is composed of dense lymphocytic aggregates with mostly T cells and fibroblasts, but no FDCs and without segregated T and B cell zones. The second stage, referred to as the ‘premature’ or ‘primary follicle-like’ TLS, contains FDCs and small B cell follicle but lacks GC reaction. The third stage, considered as the ‘mature’ or ‘secondary follicle-like’ TLS, is characterized by a GC that appears as well-organized aggregates of T and B cells with segregated T and B cell zones, presence of FDC networks, development of HEVs, and specialized lymphoid fibroblasts (49, 52).
TLS formation relies on inducible inflammatory triggers, such as CXCL13 and CCL19, as well as lymphorganogenic cytokines like lymphotoxin αβ. Transcription factor BCL-6, which is essential for Tfh differentiation (52), was overexpressed in TLS in skin lesions (53). Importantly, reducing B cell activating factor (BAFF) decreased TLS formation in an inducible model of LN (54), suggesting a role of BAFF in TLS formation. The processes of B cell selection, activation, affinity maturation, isotype switching, and plasma cell formation can occur in GCs of TLS in a manner similar to that in GCs of SLO (55). Studies in patients and animal models of autoimmune diseases, such as autoimmune diabetes, myasthenia gravis and Wegener’s granulomatosis, have demonstrated the process of selection and affinity maturation of B cells in TLS. For example, granulomas of upper respiratory tract in patients with Wegener’s granulomatosis contain clusters of B lymphocytes, dendritic cells and plasma cells with evidence of initial selection and affinity maturation of B cells, which is potentially driven by self-antigen proteinase-3 (56). B cell selection, activation, affinity maturation, isotypic switching and plasma cell formation have also been demonstrated in TLS associated with tumors, resulting in an “in situ” anti-tumor antibody production (57). Thus, mature TLS with GCs are active sites of generation of autoantibodies and antibodies against tumor antigens. Additionally, molecular and cellular interaction in the TLS promotes the local inflammatory response in the organs targeted by autoimmune diseases, as described below for kidneys in LN.
4 TLS in LN
TLS are found in autoimmune diseases’ target tissues such as kidneys (lupus), salivary glands (Sjögren’s syndrome), and synovial joints (rheumatoid arthritis) (58). TLS have been reported in aged human kidneys as well as in kidneys from patients and animals with various kidney diseases including IgA nephropathy, renal allograft rejection, and pyelonephritis (59, 60). In many conditions, the extent and stage of TLS appear to correlate with disease severity (59, 60). These observations suggest that TLS formation may be a common pathological process, which may progress with disease such as LN.
4.1 Evidence for the formation of TLS in LN
The first demonstration of TLS in lupus likely came from an induced model, where lipogranulomas picked from the peritoneal wall of BALB/cJ mice injected with hydrocarbon oil TMPD exhibited organized structures with HEV and discrete areas of B and CD4+ T cells, and activated DCs, and lymphoid chemokines (61). The TLS in these lipogranulomas recapitulated many functional characteristics of SLO (62). Subsequent studies in spontaneous and induced models, including NZB/NZW F1, MRL-lpr, BXSB, anti-nucleosome IgG passive transfer, silica instillation and TLR8-knockout mice, demonstrated the formation of TLS (Table 1). TLS in lupus mouse kidneys had similar cell composition, structure, and gene signature as lymph nodes (50), and therefore may function as a kidney-specific type of lymph node. A comparison with human LN gene expression revealed similar up-regulated genes as observed during the development of murine LN and TLS (50). In animal models, TLS formation was associated with the development of systemic autoimmune disease (65) and with local autoantibody production (66).
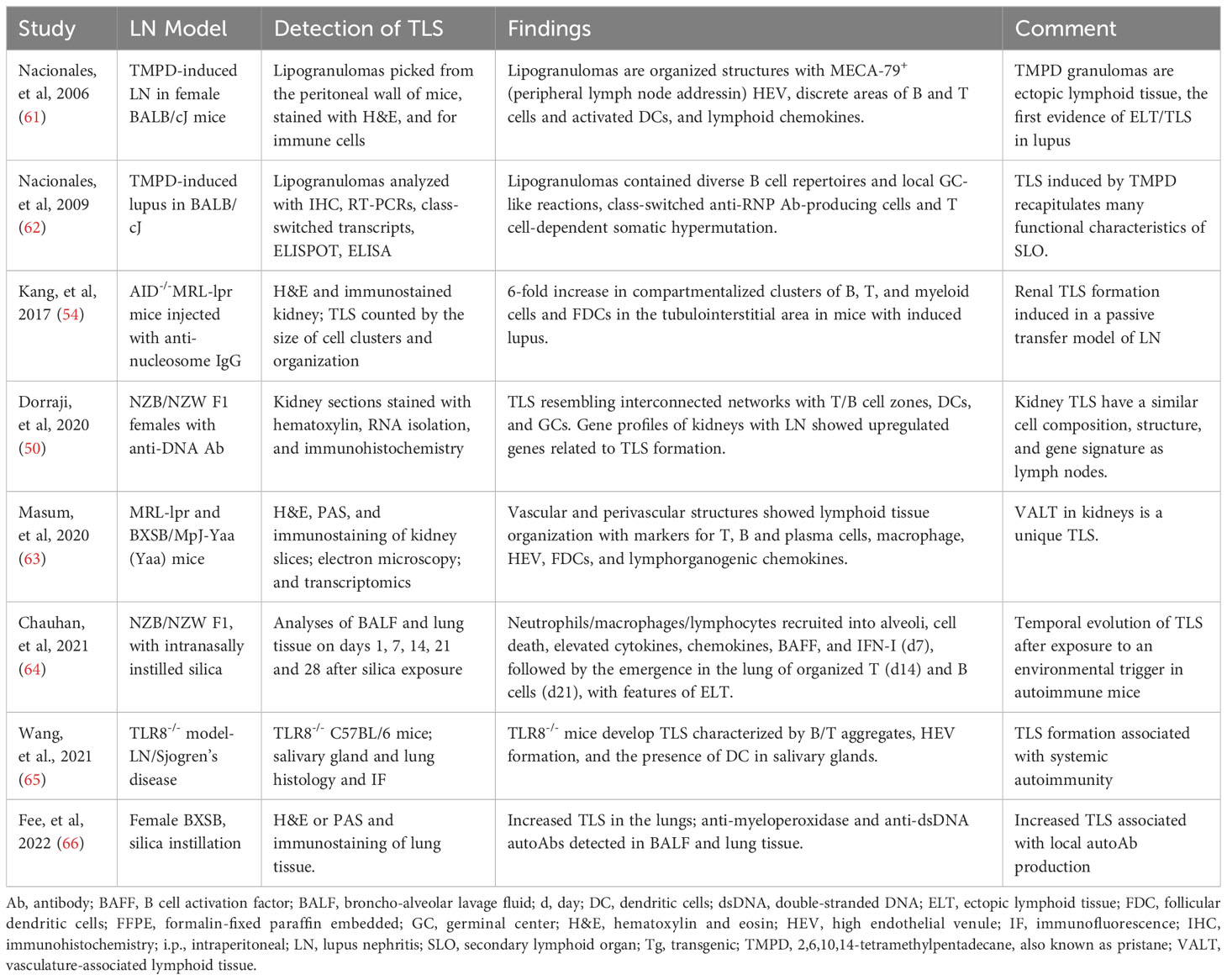
Table 1 Evidence supporting the formation of TLS in LN: Animal model studies.
A timed exposure to an environmental trigger allowed the investigators to assess a temporal evolution of TLS in lupus. Silica instillation in NZB/NZW F1 mice induced the recruitment of neutrophils, macrophages and lymphocytes into alveoli, with local cell death, elevated cytokines, chemokines, BAFF, and IFN-I by day 7, followed by the emergence in the lung of organized T (day 14) and B cells (day 21), with features of TLS (64).
Soon after the description of TLS in an animal model of lupus (61), Steinmetz and colleagues identified four organizational levels of intrarenal aggregates in patients with LN, ranging from scattered B cells to highly structured clusters with central FDCs (67) (Table 2). Majority of the organized tubulointerstitial infiltrates in LN kidneys were well-circumscribed T:B cell aggregates, also considered as class II ectopic lymphoid tissue, which were identified in 46% to 57% of LN biopsies (68–70). Most of the remaining organized infiltrates were described as class I ectopic lymphoid tissue and less than 10% had class III that were GCs containing FDCs with B cell clonal expansion and somatic hypermutation. GCs contained CD138–CD20+ centroblasts, whereas T:B aggregates had CD138+CD20low/2 plasmablasts (68). Most patients with class IV LN had TLS in kidney biopsies, up to 94% of cases in one study. Thus, animal model and human studies have demonstrated the formation of TLS in LN.
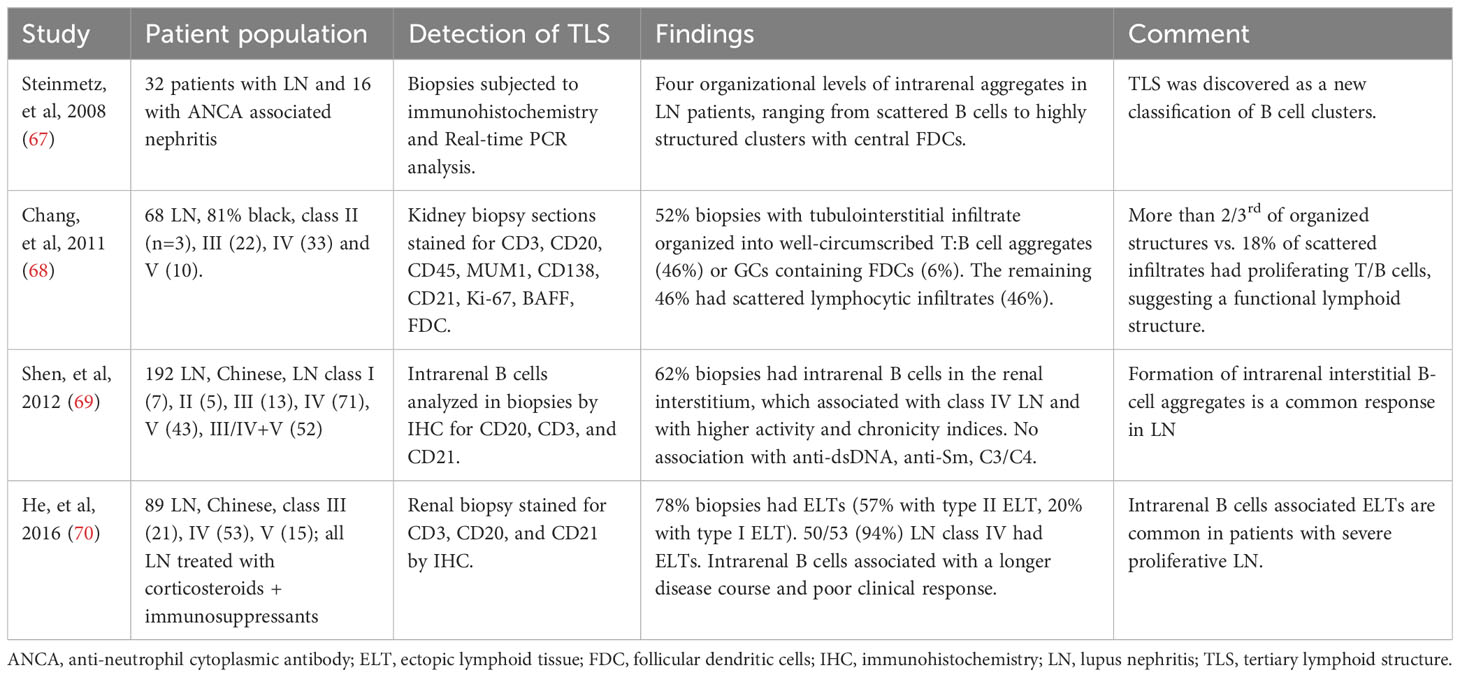
Table 2 Evidence supporting the formation of TLS in LN: Human studies.
4.2 Potential mechanisms underlying TLS formation in LN
In kidney biopsies from patients with LN, CXCL13 expressed in cells with dendritic morphology in areas of infiltration with B cells that expressed the cognate receptor CXCR5 (67). Furthermore, the presence of TLS in human kidney biopsies correlated with increased B cell hyperactivity and serum levels of CXCL13 (70). Subsequently, transcriptomic studies of LN kidneys in MRL/lpr and BXSB models showed increased cxcl13/cxcr5 along with upregulated expression signature associated with TLS formation (63), suggesting a role of CXCL13/CXCR5 in TLS formation in LN. In animal studies, BAFF, TLR7 signaling, and Fli-1 have also been suggested to play a role in TLS formation (54, 65, 71). These and other molecular and cellular mechanisms associated with TLS formation in lupus are described in Table 3. In kidney injury models, genetic deletion or antibody neutralization suggested a role of IL-17A in TLS formation in kidneys (74). Among cellular mechanisms, mesenchymal stem cells have been suggested to act as LTi cells in kidneys through interaction with T cells, thus inducing TLS formation in LN (72). Other studies suggest a possible role of innate lymphoid cells type 3 in perivascular tissues and impaired tissue-resident dendritic cells in TLS formation (37, 73). Identification of molecules associated with TLS formation (Table 4) and their possible roles in TLS formation and LN pathogenesis would be critical for therapeutic targeting of TLS.
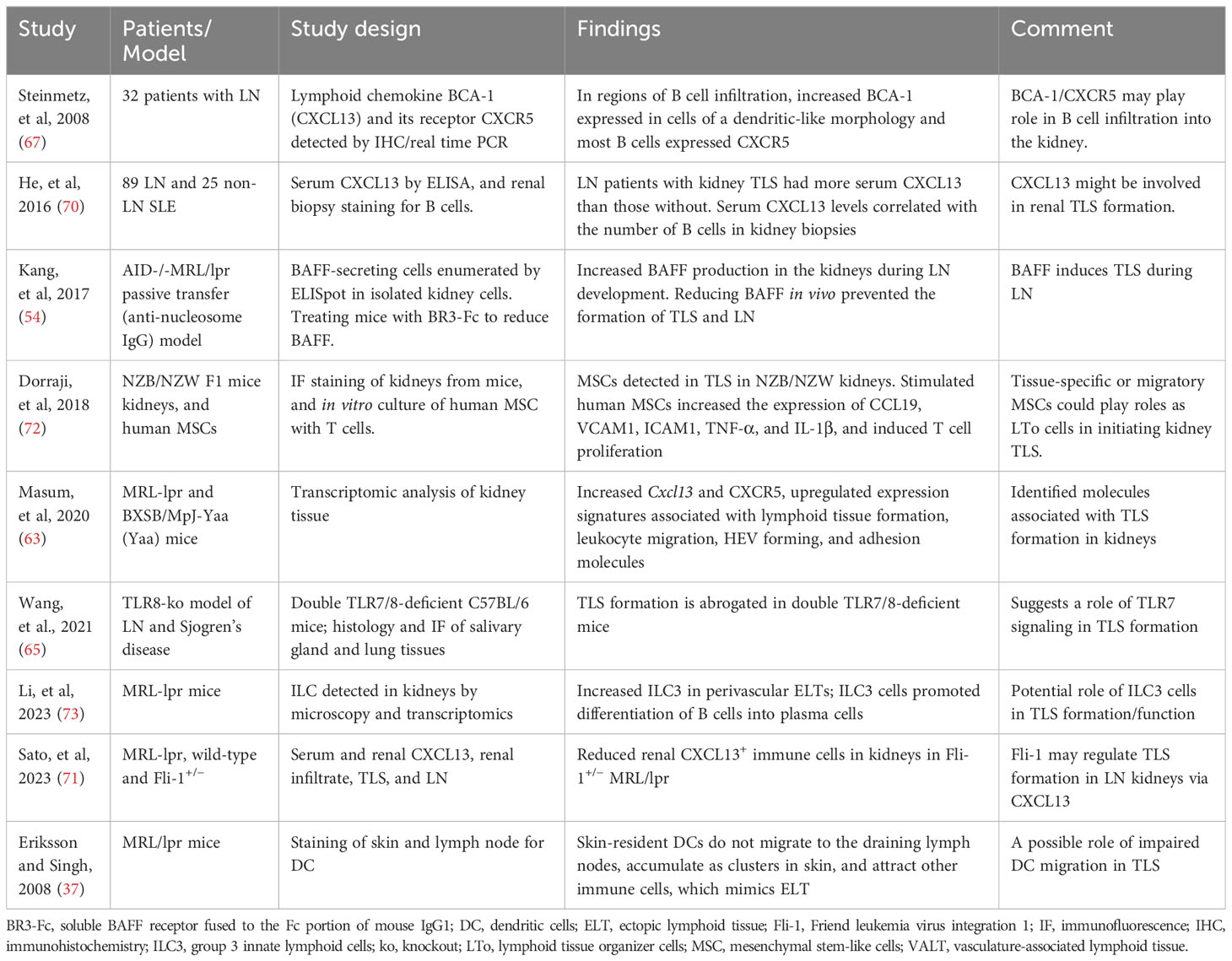
Table 3 Potential mechanisms that may promote TLS formation in lupus.
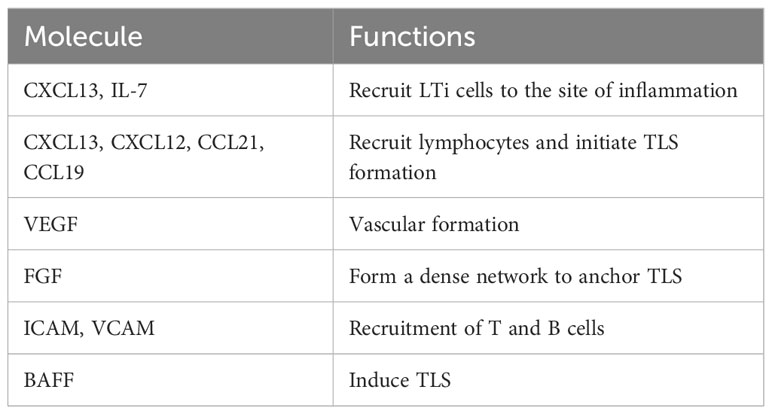
Table 4 Molecules associated with TLS formation.
4.3 Possible contributions of TLS to LN
Although the principal lesion within the kidney in LN is glomerulonephritis, tubulointerstitial inflammation is common. Severity of tubulointerstitial disease on renal biopsy, rather than severity of glomerular disease, predicts progression to renal failure (75, 76). Tubulointerstitial lesions commonly have TLS containing T:B cell aggregates, plasmablast foci, and GCs (68). These TLS are sites of in situ antigen-driven selection of B cells. Thus, human LN appears to arise from both systemic and in situ autoimmune responses, with the latter more closely associated with a poor prognosis (6, 7, 12). Consistently, TLS was associated with higher disease activity and chronicity indices and poor treatment response in LN (68–70). TLS was also associated with the progression of kidney damage in other models of kidney injury (74).
In lupus-prone mice, anti-dsDNA antibody positive animals developed TLS in their kidneys, and when they became proteinuric, a large and organized TLS with GC-like structures formed within the kidney (50, 54, 72). These animals had a higher ex vivo FDG accumulation in the kidney of anti-dsDNA antibody positive group compared to antibody-negative mice, which is consistent with the presence of large population of metabolic active or activated immune cells (35). Thus, TLS may contribute to LN in multiple ways (Table 5), such as by being a local source of activated immune cells (35), local IFN-I production (61), maintaining systemic autoantibody production (66, 78), site for intermolecular epitope spreading (80, 81) to local autoantigens such as to locally overexpressed vimentin (79), and facilitating tubulointerstitial inflammation (63, 68).
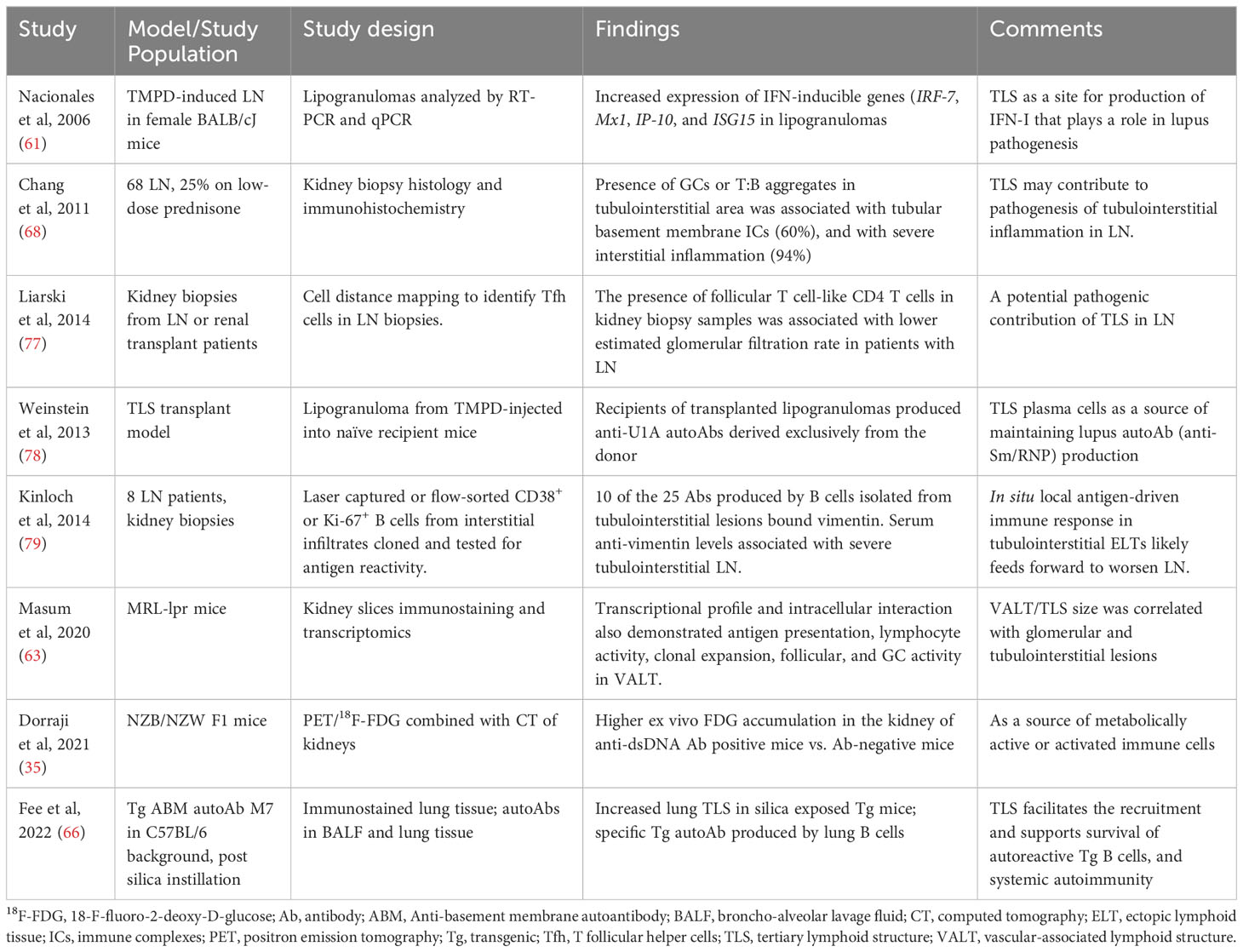
Table 5 Evidence supporting potential contributions of TLS to LN.
4.4 Potential mechanism whereby TLS might promote LN development.
Animal and human studies suggest several mechanisms by which TLS might contribute to lupus disease (Table 6). First, TLS may promote high-affinity autoantibody production. In lupus mice, B cell proliferation, somatic hypermutation, and class switch recombination are found within TLS (62). In human studies, most kidney biopsies from LN patients had well-defined T:B cell aggregates or GCs containing FDCs within tubulointerstitial infiltrates, which correlated with intrarenal B cell clonal expansion and ongoing somatic hypermutation (68). Dendritic cells in lupus mice infiltrate the kidney, present antigens, and locally amplify inflammation signals (84), thus promoting TLS. FDCs express Fcγ receptors and complement receptors, allowing for the binding of antigen-antibody complexes and presentation of antigens to B cells. This leads to the selection of B cells that produce high-affinity antibodies (85). Furthermore, TLS GCs harbor proliferating B lymphocytes expressing activation-induced deaminase, a key enzyme in somatic hypermutation and class switch recombination, resulting in the generation of high-affinity, class-switched antibodies (86).

Table 6 Summary of possible mechanisms by which TLS might contribute to LN.
Second, TLS may contribute to LN development by promoting the longevity of autoreactive lymphocytes. In the TMPD-SLE model, autoreactive memory B cells and anti-Sm/RNP autoantibody-producing plasma cells home to TLS (78), which may allow them to escape from normal immune censoring mechanisms in this location. TLS can provide survival signals for incoming lymphocytes and differentiated long-lived plasma cells through the secretion of IL-7, CXCL12, and BAFF (87). In support of the latter, transgenic overexpression in mice of BAFF, a critical B cell survival factor, induces autoimmune nephritis (88). An inflammatory environment could also promote mesenchymal stem cells’ production of lymphocyte survival cytokines, IL-7 and CXCL12, which can increase T cell recruitment and CD4+ T cell proliferation and differentiation in kidney-TLS in lupus mice (72).
Third, the site of antibody synthesis and the local microenvironment can influence the post-translational modifications of immunoglobulins (87). Thus, TLS’ effect on post-translational modifications, such as glycosylation and sialylation, may influence the function of antibody, and their pathogenic potential in the context of autoimmune disease. For example, altered glycosylation of IgG in the kidneys of MRL-lpr mice has been associated with LN (89), whereas the transfer of sialylated autoantigen-reactive IgG antibody into lupus-prone FcγRIIB-deficient mice attenuated LN (90). Although, the data directly demonstrating the regulation of post-translational modification of autoantibody by TLS are lacking in LN patients, there have been several reports on the association between altered glycosylation and inflammation in patients with SLE (91).
Fourth, a human study analyzed the antigen specificity of antibodies produced by B cells isolated by laser-capture or flow sorting from tubulointerstitial lesions in kidney biopsies from LN patients (79). Ten of the 25 antibodies thus identified bound vimentin which was overexpressed in these kidneys. Thus, in situ local antigen-driven immune response in tubulointerstitial TLS may lead to autoimmune spreading, which might lead to worsening of LN.
5 GC vs. extrafollicular responses in the context of TLS in LN
The kidney infiltrates in patients with LN may range from scattered B and T cells to the fully organized TLS with GCs containing FDCs (67, 68, 77) (Table 2). Among LN kidney biopsies with organized infiltrates, about 10% exhibited TLS with typical features of GCs containing FDCs with B cell clonal expansion and somatic hypermutation (68), about 50% of patients had well-circumscribed T:B cell aggregates, and the remaining patients had dense lymphocytic aggregates without segregated T and B cell zone and without FDCs (68–70). The latter have been suggested to be ‘immature’ or ‘early’ TLS or class I ectopic lymphoid tissue, while others have argued that these organized kidney infiltrates are distinct from TLS (92). Chronic autoimmune responses in these organized lympho-myeloid infiltrates have been suggested to be extrafollicular (92, 93). Animal studies have also suggested that spontaneous or induced responses in TLS can, but do not have to, include GCs (62). In fact, extensive intraclonal somatic V region diversification was observed in a persistent non-canonical extrafollicular response in lupus-prone mice (94). The same autoreactive B cell clonotype proliferating and mutating extrafollicularly and in GCs was found in lupus-prone NZM.2410 mice (95). Furthermore, the mutation rate in autoreactive extrafollicular responses in MRL/lpr mice was similar to that seen in peak GCs (96). Somatic hypermutation and isotype switching were also observed in synovial lymphoid infiltrates without a GC-like structure of rheumatoid arthritis patients (97). In these and other studies in autoimmune diseases’ target tissues, at least some of the microdissected B cell clusters that yielded mutated V region sequences did not resemble GC structures and were most likely an extrafollicular-like response (92).
There is some evidence to suggest that certain infections and immune inflammatory states favor the GC or the extrafollicular response (92). It has been suggested that the extrafollicular response may occur in the context of severe infections with high pathogen burden (92). Corollary to this, the abundant antigenic target of the immune response, such as DNA or DNA-containing immune complexes as in SLE, may trigger an extrafollicular response, whereas the localized antigen, such as insulin as in type I diabetes, may trigger a GC response. The degree of inflammation and cytokine response may also dictate the GC vs. extrafollicular response. It is also likely that autoimmune responses exist along a spectrum from GC- to extrafollicular-dominant, and may even evolve over time with the severity of disease in an individual.
In SLE, B cells in the GC are influenced by the expression of Absent in Melanoma 2 (AIM2). Reduced AIM2 expression can decrease B cells in the GC and alleviate lupus symptoms (98), highlighting the role of GC in lupus disease. However, blocking the GC pathway by knocking out the transcription factor Bcl6 in GC B cells did not protect animals from anti-dsDNA antibody production, plasma cell output, and immune complex deposition in glomeruli in the R848 model (99). Understanding these different autoimmune scenarios might help to stratify patients within a given autoimmune disease (100), which might help guide the selection of targeted therapies.
6 Clinical implications and therapeutic targeting of TLS
Persistence of TLS in autoimmune conditions is associated with a poor prognosis and with a failure to respond to B cell-depleting therapies (3, 101). Therefore, identifying mechanisms underlying the formation and persistence of TLS could present new therapeutic options for autoimmunity. For example, treatment of mice with BR3-Fc (soluble BAFF receptor fused to the Fc portion of mouse IgG1) reduced BAFF levels and prevented the formation of TLS in kidneys and development of LN in an inducible model of SLE (54). In recent clinical trials, adding belimumab that blocks BAFF to standard therapy in people with active LN improved renal function, decreased flare rate and allowed glucocorticoid withdrawal (102, 103). It remains to be determined whether belimumab reduced TLS in the kidneys of patients with LN.
In animal studies, several treatments have been shown to reduce TLS and autoimmune disease (Table 7). For example, treating MRL-lpr mice with a blocking antibody against CXCL13 that plays a role in TLS formation improves neuropsychiatric lupus (108). Treatment with a classical pathway complement inhibitor C4BP(β-) or with the selective S1P1 receptor modulator, Cenerimod, also reduced TLS and autoimmune disease in lupus-prone mice (105, 106). Additionally, dexamethasone was highly effective in preventing TLS formation in MR/lpr mice (63). These observations further emphasize the role of TLS in the development and progression of lupus and open avenues for modulating TLS as a treatment option.
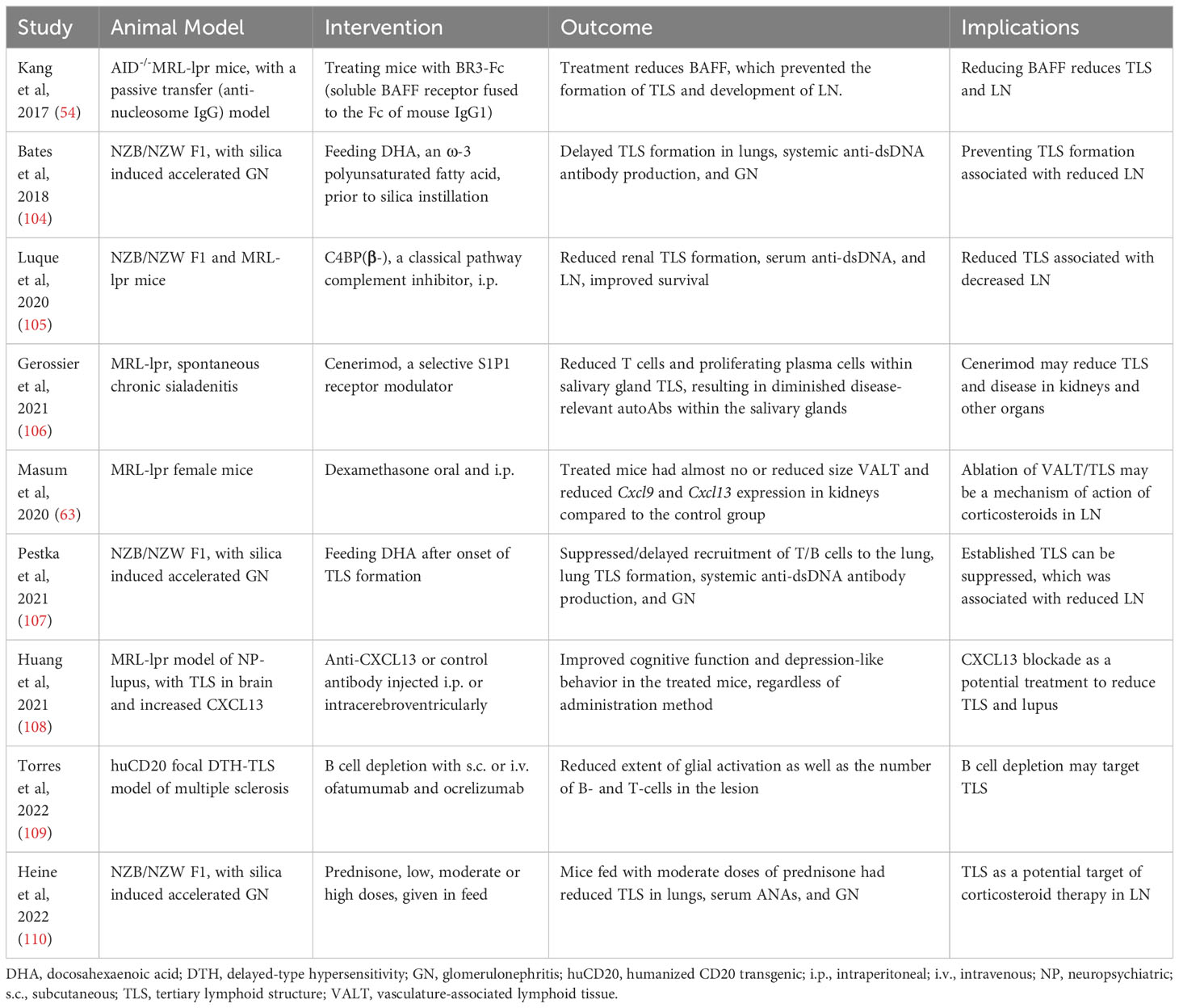
Table 7 Potential therapeutic targeting of TLS.
TLS can also serve as a prognostic marker, as the presence of TLS in the kidneys of lupus-prone mice and patients with LN (68, 111) is often associated with poor prognosis and increased autoantibody production (3). Furthermore, analysis of TLS densities in LN biopsies, their cell composition and cytokine/chemokine secretions, and the antibody produced by plasma cells educated in TLS might help in predicting therapeutic responses to immune modulating drugs. Further work is needed to determine if the persistence of TLS correlates with a poor response to treatment in LN.
7 Challenges and future directions for TLS in LN
Many questions remain regarding TLS as well as relative contributions of TLS among the cascade of pathogenic events that lead to LN. TLS studies thus far have used variable evaluation strategies. A comprehensive analysis of TLS phenotypes, density and spatial distribution using a standardized, reproducible, quantitative approach is needed. For example, a study in human melanoma used 7-color multiplex immunostaining of whole tissue sections from 103 human melanoma samples to characterize TLS phenotypes along the expression of established TLS-defining molecular and cellular components (112). Such integrated qualitative, quantitative, and spatial analysis of TLS at different stages of LN using a large human sample size can inform the relative contributions of TLS in LN.
The lack of non-invasive methods to visualize/quantify TLS in kidneys is a major hurdle in using TLS in the clinical assessment. However, Dorraji and colleagues were able to visualize TLS in the pancreas of lupus-prone, anti-dsDNA antibody positive, NZB/NZW F1 mice by single photon emission computed tomography (SPECT) using 99mTC labeled Albumin Nanocoll (35). Further advancement in SPECT and in vivo imaging technologies may make it possible to detect and longitudinally monitor TLS in LN.
Further research is needed to understand the composition and anatomy of TLS and roles of different cell types in TLS function. TLS develop in near proximity to large veins and arteries and contain lymph vessels and micro capillaries (50, 72). The larger renal drainage lymphatics in mice can be found in the same hilar area (50, 113). However, it is unknown if the blood flow in TLS is connected to the larger vessels. Little is known about the lymphatic flow within TLS (114). There is also limited research on the role of certain immune cell types, such as Treg cells, within TLS.
The presence of TLS may also pose challenges to treatment in LN. For example, local B cells in TLS may not be easily inhibited by systemic therapy, such as rituximab. In a study of chronic renal allograft rejection, systemic rituximab failed to cause TLS regression in kidney despite the depletion of peripheral B cells (115). This finding suggests that the local TLS may facilitate B cell survival and allow the evasion of systemic rituximab-mediated depletion. A case series reported three cases with recurrent and refractory oral pemphigus vulgaris. Two of the three had negative circulating desmoglein antibodies, whereas intralesional rituximab was effective in the treatment of the two (116). Intralesional rituximab likely killed the lesional B cells in the TLS and enhanced the lesion healing. The presence of TLS may also explain the relapse and treatment resistance of local lesions in the absence of circulating autoantibodies.
8 Conclusions
TLS recapitulate the functions of SLO at local chronic inflammatory sites. After the initial loss of tolerance, the activated autoimmune cells may infiltrate the target organs, where they may induce the formation of TLS that, in turn, may maintain the autoimmune response by facilitating interactions among different immune cell types and between various macromolecules and immune cells. TLS may facilitate a local activation of T and B cells in tissues resulting in a rapid response to local antigens, which may resemble the systemic adaptive immune response that occurs in SLO. Additionally, TLS may provide a platform for encounters between local autoantigens, antigen-presenting cells and lymphocytes, thus promoting an efficient antigen processing and presentation leading to the spreading of autoimmune response to additional autoantigens. In fact, animal studies have demonstrated the induction of high affinity antibodies in TLS. Furthermore, locally activated immune cells as well as tissue-resident cells may become a source of inflammatory mediators, thus exacerbating kidney injury. Both animal model and human studies support the role of TLS in the kidney to amplify autoimmune responses as well as cause the progression of renal damage in LN. These observations provide a strong rationale for developing new therapies that target events that initiate TLS formation, molecules that promote the maturation and organization of TLS, and by depleting the locally activated immune cells in the TLS of kidneys in LN. However, there are still many hurdles to resolve before realizing the potential of anti-TLS therapies. For example, the lack of non-invasive methods to visualize/quantify TLS in kidneys is a major hurdle in using TLS for clinical assessment, but recent success in visualizing TLS in the pancreas of lupus-prone mice by SPECT provides hope for early detection of TLS in LN using in vivo imaging.
Author contributions
MW and RS developed the outline and objective of the article, MW and YL wrote the draft of the article, TI and DN assisted with literature review, preparation of figures, and writing, and MW and RS revised it critically for important intellectual content. XL, SR, and JH contributed to the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the Science and Technology Program for Basic Research in Shenzhen (No. JCYJ20190809095811254, No. JCYJ20200109140412476), the Clinical Research Project in Shenzhen (20213357002 and 20213357028), NIH R01 AR050797, R01 AI080778, and R01 AR056465 grants.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol (2016) 7:477. doi: 10.3389/fimmu.2016.00477
2. Dieudé M, Turgeon J, Rimbaud AK, Beillevaire D, Qi S, Patey N, et al. Extracellular vesicles derived from injured vascular tissue promote the formation of tertiary lymphoid structures in vascular allografts. Am J Transplant (2020) 20(3):726–38. doi: 10.1111/ajt.15707
3. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol (2017) 13(3):141–54. doi: 10.1038/nrrheum.2016.217
4. Manzo A, Bombardieri M, Humby F, Pitzalis C. Secondary and ectopic lymphoid tissue responses in rheumatoid arthritis: from inflammation to autoimmunity and tissue damage/remodeling. Immunol Rev (2010) 233(1):267–85. doi: 10.1111/j.0105-2896.2009.00861.x
5. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol (2012) 33(6):297–305. doi: 10.1016/j.it.2012.04.006
6. Thaunat O, Patey N, Caligiuri G, Gautreau C, Mamani-Matsuda M, Mekki Y, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J Immunol (2010) 185(1):717–28. doi: 10.4049/jimmunol.0903589
7. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature (2020) 577(7791):561–5. doi: 10.1038/s41586-019-1914-8
8. Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol (2016) 7:407. doi: 10.3389/fimmu.2016.00407
9. Colbeck EJ, Ager A, Gallimore A, Jones GW. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or bystander sentinels in disease? Front Immunol (2017) 8:1830. doi: 10.3389/fimmu.2017.01830
10. Singh RR, Dubey S, Pinkhasov J. Immune tolerance defects in lupus. In: Wallace DJ, Hahn BH, editors. Dubois' Lupus Erythematosus and Related Syndromes, 8th ed. Philadelphia: Elsevier Saunders (2013). p. 256–72.
11. Wang M, Ishikawa T, Lai Y, Nallapothula D, Singh RR. Diverse roles of NETosis in the pathogenesis of lupus. Front Immunol (2022) 13:895216. doi: 10.3389/fimmu.2022.895216
12. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol (2012) 189(7):3767–76. doi: 10.4049/jimmunol.1201216
13. Jing F, Choi EY. Potential of cells and cytokines/chemokines to regulate tertiary lymphoid structures in human diseases. Immune Netw (2016) 16(5):271–80. doi: 10.4110/in.2016.16.5.271
14. Hanly JG, O'Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, et al. The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatol (Oxford) (2016) 55(2):252–62. doi: 10.1093/rheumatology/kev311
15. Maria NI, Davidson A. Protecting the kidney in systemic lupus erythematosus: from diagnosis to therapy. Nat Rev Rheumatol (2020) 16(5):255–67. doi: 10.1038/s41584-020-0401-9
16. Yen EY, Rajkumar S, Sharma R, Singh RR. Lupus nephritis mortality in the United States, 1999-2019: profound disparities by race/ethnicity and place of residence and a recent worsening trend. Arthritis Rheumatol (2021) 73(suppl 9):938-9.
17. Saran R, Robinson B, Abbott KC, Agodoa LYC, Bhave N, Bragg-Gresham J, et al. US renal data system 2017 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis (2018) 71(3 Suppl 1):A7. doi: 10.1053/j.ajkd.2018.03.001
18. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol (2008) 26:627–50. doi: 10.1146/annurev.immunol.26.021607.090257
19. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol (2003) 3(4):292–303. doi: 10.1038/nri1054
20. Cupedo T, Mebius RE. Role of chemokines in the development of secondary and tertiary lymphoid tissues. Semin Immunol (2003) 15(5):243–8. doi: 10.1016/j.smim.2003.08.002
21. Blum KS, Pabst R. Keystones in lymph node development. J Anat (2006) 209(5):585–95. doi: 10.1111/j.1469-7580.2006.00650.x
22. Bar-Ephraïm YE, Mebius RE. Innate lymphoid cells in secondary lymphoid organs. Immunol Rev (2016) 271(1):185–99. doi: 10.1111/imr.12407
23. Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, et al. Tyrosine kinase receptor RET is a key regulator of Peyer's patch organogenesis. Nature (2007) 446(7135):547–51. doi: 10.1038/nature05597
24. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol (Baltimore Md 1950) (2002) 169(1):424–33. doi: 10.4049/jimmunol.169.1.424
25. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408
26. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
27. Pahwa S. Searching for the origin of the enigmatic circulating T follicular helper cells. J Clin Invest (2019) 129(8):3048–51. doi: 10.1172/JCI130311
28. Acton SE, Onder L, Novkovic M, Martinez VG, Ludewig B. Communication, construction, and fluid control: lymphoid organ fibroblastic reticular cell and conduit networks. Trends Immunol (2021) 42(9):782–94. doi: 10.1016/j.it.2021.07.003
29. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol (2006) 7(4):344–53. doi: 10.1038/ni1330
30. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. (2014) 157(4):832–44. doi: 10.1016/j.cell.2014.04.016
31. Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor α, and CCR7 ligands in lymph node development. J Exp Med (2003) 197(9):1191–8. doi: 10.1084/jem.20021294
32. Blanchard L, Girard JP. High endothelial venules (HEVs) in immunity, inflammation and cancer. Angiogenesis (2021) 24(4):719–53. doi: 10.1007/s10456-021-09792-8
33. Kojima M, Motoori T, Asano S, Nakamura S. Histological diversity of reactive and atypical proliferative lymph node lesions in systemic lupus erythematosus patients. Pathol Res Pract (2007) 203(6):423–31. doi: 10.1016/j.prp.2007.03.002
34. Makis W, Ciarallo A, Gonzalez-Verdecia M, Probst S. Systemic lupus erythematosus associated pitfalls on (18)F-FDG PET/CT: reactive follicular hyperplasia, kikuchi-fujimoto disease, inflammation and lymphoid hyperplasia of the spleen mimicking lymphoma. Nucl Med Mol Imaging (2018) 52(1):74–9. doi: 10.1007/s13139-017-0471-z
35. Dorraji ES, Oteiza A, Kuttner S, Martin-Armas M, Kanapathippillai P, Garbarino S, et al. Positron emission tomography and single photon emission computed tomography imaging of tertiary lymphoid structures during the development of lupus nephritis. Int J Immunopathol Pharmacol (2021) 35:20587384211033683. doi: 10.1177/20587384211033683
36. Pellefigues C, Dema B, Lamri Y, Saidoune F, Chavarot N, Lohéac C, et al. Prostaglandin D2 amplifies lupus disease through basophil accumulation in lymphoid organs. Nat Commun (2018) 9(1):725. doi: 10.1038/s41467-018-03129-8
37. Eriksson AU, Singh RR. Cutting edge: migration of langerhans dendritic cells is impaired in autoimmune dermatitis. J Immunol (2008) 181(11):7468–72. doi: 10.4049/jimmunol.181.11.7468
38. King JK, Philips RL, Eriksson AU, Kim PJ, Halder RC, Lee DJ, et al. Langerhans cells maintain local tissue tolerance in a model of systemic autoimmune disease. J Immunol (2015) 195(2):464–76. doi: 10.4049/jimmunol.1402735
39. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26(5):643–54. doi: 10.1016/j.immuni.2007.04.009
40. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35(6):986–96. doi: 10.1016/j.immuni.2011.10.015
41. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med (2011) 208(1):125–34. doi: 10.1084/jem.20100052
42. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, et al. M1 macrophages act as LTbetaR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res (2014) 101(3):434–43. doi: 10.1093/cvr/cvt263
43. Upadhyay V, Fu YX. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat Rev Immunol (2013) 13(4):270–9. doi: 10.1038/nri3406
44. Vondenhoff MF, Greuter M, Goverse G, Elewaut D, Dewint P, Ware CF, et al. LTbetaR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J Immunol (2009) 182(9):5439–45. doi: 10.4049/jimmunol.0801165
45. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci USA (2007) 104(12):5026–31. doi: 10.1073/pnas.0606697104
46. Peduto L, Dulauroy S, Lochner M, Spath GF, Morales MA, Cumano A, et al. Inflammation recapitulates the ontogeny of lymphoid stromal cells. J Immunol (2009) 182(9):5789–99. doi: 10.4049/jimmunol.0803974
47. Fleige H, Ravens S, Moschovakis GL, Bolter J, Willenzon S, Sutter G, et al. IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med (2014) 211(4):643–51. doi: 10.1084/jem.20131737
48. Yan L, de Leur K, Hendriks RW, van der Laan LJW, Shi Y, Wang L, et al. T follicular helper cells as a new target for immunosuppressive therapies. Front Immunol (2017) 8:1510. doi: 10.3389/fimmu.2017.01510
49. Jamaly S, Rakaee M, Abdi R, Tsokos GC, Fenton KA. Interplay of immune and kidney resident cells in the formation of tertiary lymphoid structures in lupus nephritis. Autoimmun Rev (2021) 20(12):102980. doi: 10.1016/j.autrev.2021.102980
50. Dorraji SE, Kanapathippillai P, Hovd AK, Stenersrod MR, Horvei KD, Ursvik A, et al. Kidney tertiary lymphoid structures in lupus nephritis develop into large interconnected networks and resemble lymph nodes in gene signature. Am J Pathol (2020) 190(11):2203–25. doi: 10.1016/j.ajpath.2020.07.015
51. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol (2016) 7:401. doi: 10.3389/fimmu.2016.00401
52. Zhao H, Wang H, Zhou Q, Ren X. Insights into tertiary lymphoid structures in the solid tumor microenvironment: anti-tumor mechanism, functional regulation, and immunotherapeutic strategies. Cancer Biol Med (2021) 18(4):981–91. doi: 10.20892/j.issn.2095-3941.2021.0029
53. Zhou S, Liu Z, Yuan H, Zhao X, Zou Y, Zheng J, et al. Autoreactive B cell differentiation in diffuse ectopic lymphoid-like structures of inflamed pemphigus lesions. J Invest Dermatol (2020) 140(2):309–18 e8. doi: 10.1016/j.jid.2019.07.717
54. Kang S, Fedoriw Y, Brenneman EK, Truong YK, Kikly K, Vilen BJ. BAFF Induces Tertiary Lymphoid Structures and Positions T Cells within the Glomeruli during Lupus Nephritis. J Immunol (2017) 198(7):2602–11. doi: 10.4049/jimmunol.1600281
55. Fridman WH, Siberil S, Pupier G, Soussan S, Sautes-Fridman C. Activation of B cells in Tertiary Lymphoid Structures in cancer: Anti-tumor or anti-self? Semin Immunol (2023) 65:101703. doi: 10.1016/j.smim.2022.101703
56. Voswinkel J, Assmann G, Held G, Pitann S, Gross WL, Holl-Ulrich K, et al. Single cell analysis of B lymphocytes from Wegener's granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol (2008) 154(3):339–45. doi: 10.1111/j.1365-2249.2008.03775.x
57. Garaud S, Buisseret L, Solinas C, Gu-Trantien C, de Wind A, Van den Eynden G, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight (2019) 5(18):e129641. doi: 10.1172/jci.insight.129641
58. Sato Y, Silina K, van den Broek M, Hirahara K, Yanagita M. The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol (2023) 19(8):525–37. doi: 10.1038/s41581-023-00706-z
59. Sato Y, Boor P, Fukuma S, Klinkhammer BM, Haga H, Ogawa O, et al. Developmental stages of tertiary lymphoid tissue reflect local injury and inflammation in mouse and human kidneys. Kidney Int (2020) 98(2):448–63. doi: 10.1016/j.kint.2020.02.023
60. Robson KJ, Kitching AR. Tertiary lymphoid tissue in kidneys: understanding local immunity and inflammation. Kidney Int (2020) 98(2):280–3. doi: 10.1016/j.kint.2020.04.026
61. Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (Pristane). Am J Pathol (2006) 168(4):1227–40. doi: 10.2353/ajpath.2006.050125
62. Nacionales DC, Weinstein JS, Yan XJ, Albesiano E, Lee PY, Kelly-Scumpia KM, et al. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J Immunol (2009) 182(7):4226–36. doi: 10.4049/jimmunol.0800771
63. Otani Y, Ichii O, Masum MA, Kimura J, Nakamura T, Elewa YHA, et al. BXSB/MpJ-Yaa mouse model of systemic autoimmune disease shows increased apoptotic germ cells in stage XII of the seminiferous epithelial cycle. Cell Tissue Res (2020) 381(1):203–16. doi: 10.1007/s00441-020-03190-0
64. Chauhan PS, Wagner JG, Benninghoff AD, Lewandowski RP, Favor OK, Wierenga KA, et al. Rapid induction of pulmonary inflammation, autoimmune gene expression, and ectopic lymphoid neogenesis following acute silica exposure in lupus-prone mice. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.635138
65. Wang Y, Roussel-Queval A, Chasson L, Hanna Kazazian N, Marcadet L, Nezos A, et al. TLR7 signaling drives the development of sjögren’s syndrome. Front Immunol (2021) 12:676010. doi: 10.3389/fimmu.2021.676010
66. Fee L, Kumar A, Tighe RM, Foster MH. Autoreactive B cells recruited to lungs by silica exposure contribute to local autoantibody production in autoimmune-prone BXSB and B cell receptor transgenic mice. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.933360
67. Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int (2008) 74(4):448–57. doi: 10.1038/ki.2008.191
68. Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol (2011) 186(3):1849–60. doi: 10.4049/jimmunol.1001983
69. Shen Y, Sun C-Y, Wu F-X, Chen Y, Dai M, Yan Y-C, et al. Association of intrarenal B-cell infiltrates with clinical outcome in lupus nephritis: A study of 192 cases. Clin Dev Immunol (2012) 2012:1–7. doi: 10.1155/2012/967584
70. He N, Chen WL, Long KX, Zhang X, Dong GF. Association of serum CXCL13 with intrarenal ectopic lymphoid tissue formation in lupus nephritis. J Immunol Res (2016) 2016:4832543. doi: 10.1155/2016/4832543
71. Sato S, Zhang XK, Matsuoka N, Sumichika Y, Saito K, Yoshida S, et al. Transcription factor Fli-1 impacts the expression of CXCL13 and regulates immune cell infiltration into the kidney in MRL/lpr mouse. Lupus Sci Med (2023) 10(1). doi: 10.1136/lupus-2022-000870
72. Dorraji SE, Hovd AK, Kanapathippillai P, Bakland G, Eilertsen GO, Figenschau SL, et al. Mesenchymal stem cells and T cells in the formation of Tertiary Lymphoid Structures in Lupus Nephritis. Sci Rep (2018) 8(1):7861. doi: 10.1038/s41598-018-26265-z
73. Li F, Liang Z, Zhong H, Hu X, Tang Z, Zhu C, et al. Group 3 innate lymphoid cells exacerbate lupus nephritis by promoting B cell activation in kidney ectopic lymphoid structures. Kidney Int Rep (2023) 8:S207–S8. doi: 10.1016/j.ekir.2023.02.466
74. Luo R, Cheng Y, Chang D, Liu T, Liu L, Pei G, et al. Tertiary lymphoid organs are associated with the progression of kidney damage and regulated by interleukin-17A. Theranostics (2021) 11(1):117–31. doi: 10.7150/thno.48624
75. Esdaile JM, Levinton C, Federgreen W, Hayslett JP, Kashgarian M. The clinical and renal biopsy predictors of long-term outcome in lupus nephritis: a study of 87 patients and review of the literature. Q J Med (1989) 72(269):779–833.
76. Hsieh C, Chang A, Brandt D, Guttikonda R, Utset TO, Clark MR. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res (Hoboken) (2011) 63(6):865–74. doi: 10.1002/acr.20441
77. Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med (2014) 6(230):230ra46. doi: 10.1126/scitranslmed.3008146
78. Weinstein JS, Delano MJ, Xu Y, Kelly-Scumpia KM, Nacionales DC, Li Y, et al. Maintenance of anti-Sm/RNP autoantibody production by plasma cells residing in ectopic lymphoid tissue and bone marrow memory B cells. J Immunol (2013) 190(8):3916–27. doi: 10.4049/jimmunol.1201880
79. Kinloch AJ, Chang A, Ko K, Henry Dunand CJ, Henderson S, Maienschein-Cline M, et al. Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol (2014) 66(12):3359–70. doi: 10.1002/art.38888
80. Singh RR, Hahn BH, Tsao BP, Ebling FM. Evidence for multiple mechanisms of polyclonal T cell activation in murine lupus. J Clin Invest (1998) 102(10):1841–9. doi: 10.1172/JCI3872
81. Singh RR, Hahn BH. Reciprocal T-B determinant spreading develops spontaneously in murine lupus: implications for pathogenesis. Immunol Rev (1998) 164:201–8. doi: 10.1111/j.1600-065X.1998.tb01221.x
82. Tang H, Zhu M, Qiao J, Fu Y-X. Lymphotoxin signalling in tertiary lymphoid structures and immunotherapy. Cell Mol Immunol (2017) 14(10):809–18. doi: 10.1038/cmi.2017.13
83. Cui W, Yoneda R, Ueda N, Kurokawa R. Arginine methylation of translocated in liposarcoma (TLS) inhibits its binding to long noncoding RNA, abrogating TLS-mediated repression of CBP/p300 activity. J Biol Chem (2018) 293(28):10937–48. doi: 10.1074/jbc.RA117.000598
84. Sahu R, Bethunaickan R, Singh S, Davidson A. Structure and function of renal macrophages and dendritic cells from lupus-prone mice. Arthritis Rheumatol (2014) 66(6):1596–607. doi: 10.1002/art.38410
85. El Shikh ME, El Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol (2010) 31(6):205–11. doi: 10.1016/j.it.2010.03.002
86. Teillaud JL, Dieu-Nosjean MC. Tertiary lymphoid structures: an anti-tumor school for adaptive immune cells and an antibody factory to fight cancer? Front Immunol (2017) 8:830. doi: 10.3389/fimmu.2017.00830
87. Pipi E, Nayar S, Gardner DH, ColaFrancesco S, Smith C, Barone F. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol (2018) 9:1952. doi: 10.3389/fimmu.2018.01952
88. Fletcher CA, Groom JR, Woehl B, Leung H, Mackay C, Mackay F. Development of autoimmune nephritis in genetically asplenic and splenectomized BAFF transgenic mice. J Autoimmun (2011) 36(2):125–34. doi: 10.1016/j.jaut.2010.12.002
89. Hashii N, Kawasaki N, Itoh S, Nakajima Y, Kawanishi T, Yamaguchi T. Alteration of N-glycosylation in the kidney in a mouse model of systemic lupus erythematosus: relative quantification of N-glycans using an isotope-tagging method. Immunology (2009) 126(3):336–45. doi: 10.1111/j.1365-2567.2008.02898.x
90. Bartsch YC, Rahmoller J, Mertes MMM, Eiglmeier S, Lorenz FKM, Stoehr AD, et al. Sialylated autoantigen-reactive igG antibodies attenuate disease development in autoimmune mouse models of lupus nephritis and rheumatoid arthritis. Front Immunol (2018) 9:1183. doi: 10.3389/fimmu.2018.01183
91. Tomana M, Schrohenloher RE, Reveille JD, Arnett FC, Koopman WJ. Abnormal galactosylation of serum IgG in patients with systemic lupus erythematosus and members of families with high frequency of autoimmune diseases. Rheumatol Int (1992) 12(5):191–4. doi: 10.1007/BF00302151
92. Elsner RA, Shlomchik MJ. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity (2020) 53(6):1136–50. doi: 10.1016/j.immuni.2020.11.006
93. Clark MR, Trotter K, Chang A. The pathogenesis and therapeutic implications of tubulointerstitial inflammation in human lupus nephritis. Semin Nephrol (2015) 35(5):455–64. doi: 10.1016/j.semnephrol.2015.08.007
94. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science (2002) 297(5589):2066–70. doi: 10.1126/science.1073924
95. Sang A, Niu H, Cullen J, Choi SC, Zheng YY, Wang H, et al. Activation of rheumatoid factor-specific B cells is antigen dependent and occurs preferentially outside of germinal centers in the lupus-prone NZM2410 mouse model. J Immunol (2014) 193(4):1609–21. doi: 10.4049/jimmunol.1303000
96. Kleinstein SH, Louzoun Y, Shlomchik MJ. Estimating hypermutation rates from clonal tree data. J Immunol (2003) 171(9):4639–49. doi: 10.4049/jimmunol.171.9.4639
97. Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA (1996) 93(1):221–5. doi: 10.1073/pnas.93.1.221
98. Yang M, Long D, Hu L, Zhao Z, Li Q, Guo Y, et al. AIM2 deficiency in B cells ameliorates systemic lupus erythematosus by regulating Blimp-1–Bcl-6 axis-mediated B-cell differentiation. Signal Transduct Target Ther (2021) 6(1):341. doi: 10.1038/s41392-021-00725-x
99. Voss LF, Howarth AJ, Wittenborn TR, Hummelgaard S, Juul-Madsen K, Kastberg KS, et al. The extrafollicular response is sufficient to drive initiation of autoimmunity and early disease hallmarks of lupus. Front Immunol (2022) 13:1021370. doi: 10.3389/fimmu.2022.1021370
100. Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I. Extrafollicular responses in humans and SLE. Immunol Rev (2019) 288(1):136–48. doi: 10.1111/imr.12741
101. Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis (2007) 66(5):700–3. doi: 10.1136/ard.2006.060772
102. Furie R, Rovin BH, Houssiau F, Amoura Z, Santiago M, Contreras G, et al. BLISS-LN: A randomized, double-blind, placebo-controlled phase 3 trial of intravenous belimumab in patients with active lupus nephritis. Ann Rheum Dis (2020) 79(Supplement 1):103. doi: 10.1136/annrheumdis-2020-eular.3881
103. Binda V, Trezzi B, Del Papa N, Beretta L, Frontini G, Porata G, et al. Belimumab may decrease flare rate and allow glucocorticoid withdrawal in lupus nephritis (including dialysis and transplanted patient). J Nephrol (2020) 33(5):1019–25. doi: 10.1007/s40620-020-00706-3
104. Bates MA, Akbari P, Gilley KN, Wagner JG, Li N, Kopec AK, et al. Dietary docosahexaenoic acid prevents silica-induced development of pulmonary ectopic germinal centers and glomerulonephritis in the lupus-prone NZBWF1 mouse. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02002
105. Luque A, Serrano I, Ripoll E, Malta C, Gomà M, Blom AM, et al. Noncanonical immunomodulatory activity of complement regulator C4BP(β-) limits the development of lupus nephritis. Kidney Int (2020) 97(3):551–66. doi: 10.1016/j.kint.2019.10.016
106. Gerossier E, Nayar S, Froidevaux S, Smith CG, Runser C, Iannizzotto V, et al. Cenerimod, a selective S1P1 receptor modulator, improves organ-specific disease outcomes in animal models of Sjögren’s syndrome. Arthritis Res Ther (2021) 23(1):289. doi: 10.1186/s13075-021-02673-x
107. Pestka JJ, Akbari P, Wierenga KA, Bates MA, Gilley KN, Wagner JG, et al. Omega-3 polyunsaturated fatty acid intervention against established autoimmunity in a murine model of toxicant-triggered lupus. Front Immunol (2021) 12:653464. doi: 10.3389/fimmu.2021.653464
108. Huang MW, Stock AD, Putterman C. CXCL13 neutralization attenuates neuropsychiatric manifestations in lupus-prone mice. Front Immunol (2021) 12:763065. doi: 10.3389/fimmu.2021.763065
109. Torres JB, Roodselaar J, Sealey M, Ziehn M, Bigaud M, Kneuer R, et al. Distribution and efficacy of ofatumumab and ocrelizumab in humanized CD20 mice following subcutaneous or intravenous administration. Front Immunol (2022) 13:814064. doi: 10.3389/fimmu.2022.814064
110. Heine LK, Benninghoff AD, Ross EA, Rajasinghe LD, Wagner JG, Lewandowski RP, et al. Comparative effects of human-equivalent low, moderate, and high dose oral prednisone intake on autoimmunity and glucocorticoid-related toxicity in a murine model of environmental-triggered lupus. Front Immunol (2022) 13:972108. doi: 10.3389/fimmu.2022.972108
111. Heller F, Lindenmeyer MT, Cohen CD, Brandt U, Draganovici D, Fischereder M, et al. The contribution of B cells to renal interstitial inflammation. Am J Pathol (2007) 170(2):457–68. doi: 10.2353/ajpath.2007.060554
112. Werner F, Wagner C, Simon M, Glatz K, Mertz KD, Laubli H, et al. A standardized analysis of tertiary lymphoid structures in human melanoma: disease progression- and tumor site-associated changes with germinal center alteration. Front Immunol (2021) 12:675146. doi: 10.3389/fimmu.2021.675146
113. Russell PS, Hong J, Windsor JA, Itkin M, Phillips ARJ. Renal lymphatics: anatomy, physiology, and clinical implications. Front Physiol (2019) 10:251. doi: 10.3389/fphys.2019.00251
114. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest (2014) 124(3):953–9. doi: 10.1172/JCI71611
115. Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux-Bacchi V, Dieu-Nosjean MC, et al. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation (2008) 85(11):1648–53. doi: 10.1097/TP.0b013e3181735723
Keywords: autoimmune disease, ectopic lymphoid tissue, lupus nephritis, systemic lupus erythematosus, tertiary lymphoid structure
Citation: Wang M, Rajkumar S, Lai Y, Liu X, He J, Ishikawa T, Nallapothula D and Singh RR (2023) Tertiary lymphoid structures as local perpetuators of organ-specific immune injury: implication for lupus nephritis. Front. Immunol. 14:1204777. doi: 10.3389/fimmu.2023.1204777
Received: 12 April 2023; Accepted: 17 October 2023;
Published: 31 October 2023.
Edited by:
Steven O’Reilly, STipe Therapeutics, DenmarkReviewed by:
Ola Grimsholm, Medical University of Vienna, AustriaJames J. Pestka, Michigan State University, United States
Copyright © 2023 Wang, Rajkumar, Lai, Liu, He, Ishikawa, Nallapothula and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ram Raj Singh, UlJTaW5naEBtZWRuZXQudWNsYS5lZHU=