 Sven Vanselow
Sven Vanselow Volker Wahn2
Volker Wahn2 Catharina Schuetz
Catharina Schuetz- 1Infill Healthcare Communication, Königswinter, Germany
- 2Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine at Charité University Hospital Berlin, Berlin, Germany
- 3Medical Faculty of The Technical University (TU) Dresden, Department of Pediatrics, University Hospital Carl Gustav Carus, Dresden, Germany
- 4University Center for Rare Diseases, University Hospital Carl Gustav Carus, Dresden, Germany
Activated PI3Kδ syndrome (APDS) is a rare inborn error of immunity (IEI) characterized primarily by frequent infections, lymphoproliferation and autoimmunity. Since its initial description in 2013, APDS has become part of the growing group of nearly 500 IEIs affecting various components of the immune system. The two subtypes of APDS - APDS1 and APDS2 - are caused by variants in the PIK3CD and PIK3R1 genes, respectively. Due to the rarity of the disease and the heterogeneous clinical picture, many patients are not diagnosed until years after symptom onset. Another challenge is the large number of PIK3CD and PIK3R1 variants whose functional significance for developing APDS is inconclusive. Treatment of APDS has so far been mostly symptom-oriented with immunoglobulin replacement therapy, immunosuppressive therapies and antibiotic or antiviral prophylaxes. Additionally, allogeneic stem cell transplantation as well as new targeted therapies are options targeting the root cause that may improve patients’ quality of life and life expectancy. However, the clinical course of the disease is difficult to predict which complicates the choice of appropriate therapies. This review article discusses diagnostic procedures and current and future treatment options, and highlights the difficulties that physicians, patients and their caretakers face in managing this complex disease. This article is based on cohort studies, the German and US guidelines on the management of primary immunodeficiencies as well as on published experience with diagnosis and compiled treatment experience for APDS.
Introduction
The group of inborn errors of immunity (IEI) currently comprises nearly 500 heritable disorders of immune function and/or regulation (1). While individual IEI disorders are rare, IEIs as a group account for a significant disease and financial burden for healthcare systems (2–4). Current estimates are that around 1 in 2000 people are affected by IEI, with more individuals diagnosed due to increasing awareness amongst healthcare providers (5, 6). The International Union of Immunological Societies (IUIS) classifies IEIs into 10 groups according to the part of the immune system predominantly affected or associated features, e.g. into combined immunodeficiencies (affecting T- and B cell function), predominantly antibody deficiencies (affecting primarily B cell function), immune dysregulatory disorders or others (1).
An important milestone was the introduction of newborn screening for severe T-cell lymphopenia/severe combined immunodeficiencies (SCID). SCID is invariably lethal if left untreated, but can be cured with early hematopoietic stem cell transplantation (7). Some IEIs are included in umbrella terms such as common variable immunodeficiency (CVID), while others have been characterized as distinct entities including X-linked agammaglobulinemia (XLA) (8), chronic granulomatous disease (CGD) (9) or activated PI3Kδ syndrome (APDS) (10, 11). These diseases are not being routinely tested for in newborns, although a few countries include testing for XLA. For many patients, there is a significant delay between symptom onset and diagnosis, oftentimes leading to irreversible organ damage (12).
APDS is a very rare disease. It may be caused by point mutations or deletions in one of the two genes encoding the two phosphoinositide 3-kinase δ (PI3Kδ) subunits. Both gain-of-function (GOF) in PIK3CD and loss-of-function (LOF) in PIK3R1 contribute to the clinical phenotype (13), i.e. the two mechanisms are represented in the pathogenesis of APDS. The disease pattern of APDS has been largely deciphered and may serve as an example to explain how GOF and LOF mutations in a signaling pathway cause disease. The spectrum of symptoms and their severity greatly vary among APDS patients. APDS often manifests early in life with a combination of recurrent infections, lymphoproliferation, and autoimmune disease, and - later in life - with deteriorating lung function or lymphoma. However, many patients may be oligosymptomatic. Indeed, even asymptomatic individuals have been described in literature (14). Consequently, the first hurdle in patient care is diagnosis of affected individuals, which requires attentive general practitioners and efficient diagnostic algorithms. The heterogeneity of the APDS patient collective also poses a challenge for choosing the right therapeutic approach. While symptom-oriented therapy may be appropriate for some patients, others develop life-threatening infections or lymphoma. Accordingly, the consideration of whether to proceed with potentially curative hematopoietic stem cell transplantation (HSCT), associated with potential risks, is not straightforward. APDS patients thus have an unmet need for therapeutic options which address immunodeficiency and lymphoproliferation alike with limited side effects.
Whole exome sequencing (WES) and whole genome sequencing (WGS) for genetic testing will soon be within reach of (almost) every patient. This has helped identify distinct gene variants underlying APDS and many other IEIs. Genetic diagnosis may be of considerable importance for a patient’s therapeutic management: opening the possibility for targeted therapies, e.g. pharmacological inhibition of an overactivated enzyme such as PI3Kδ (15, 16).
The aim of this review is to describe the molecular mechanism underlying APDS and update the reader on the current state of diagnosis and treatment.
Demographics
APDS is a very rare disease with an estimated prevalence of 1-2 affected people per million. The APDS registry of the European Society for Immunodeficiencies (ESID) has registered around 200 patients from Europe and the Middle East (17, 18). The exact number of APDS patients is unknown, as many patients are not registered or not diagnosed as having an IEI. APDS can be subdivided into APDS1 and APDS2, two very similar conditions which are caused by different point mutations or deletions in either of the two genes encoding PI3Kδ subunits: PIK3CD and PIK3R1 respectively (19).
Symptoms and long-term consequences
APDS1 and APDS2 are characterized by similar symptoms, although some differences are observed, e.g. for susceptibility to certain infections or the age at onset of lymphoma (19–22). Frequent infections and autoimmunity limit patients’ quality of life while severe infections and lymphoma lower the average life expectancy of APDS patients (21, 23–25). An overview of major signs and symptoms is provided in Figure 1.
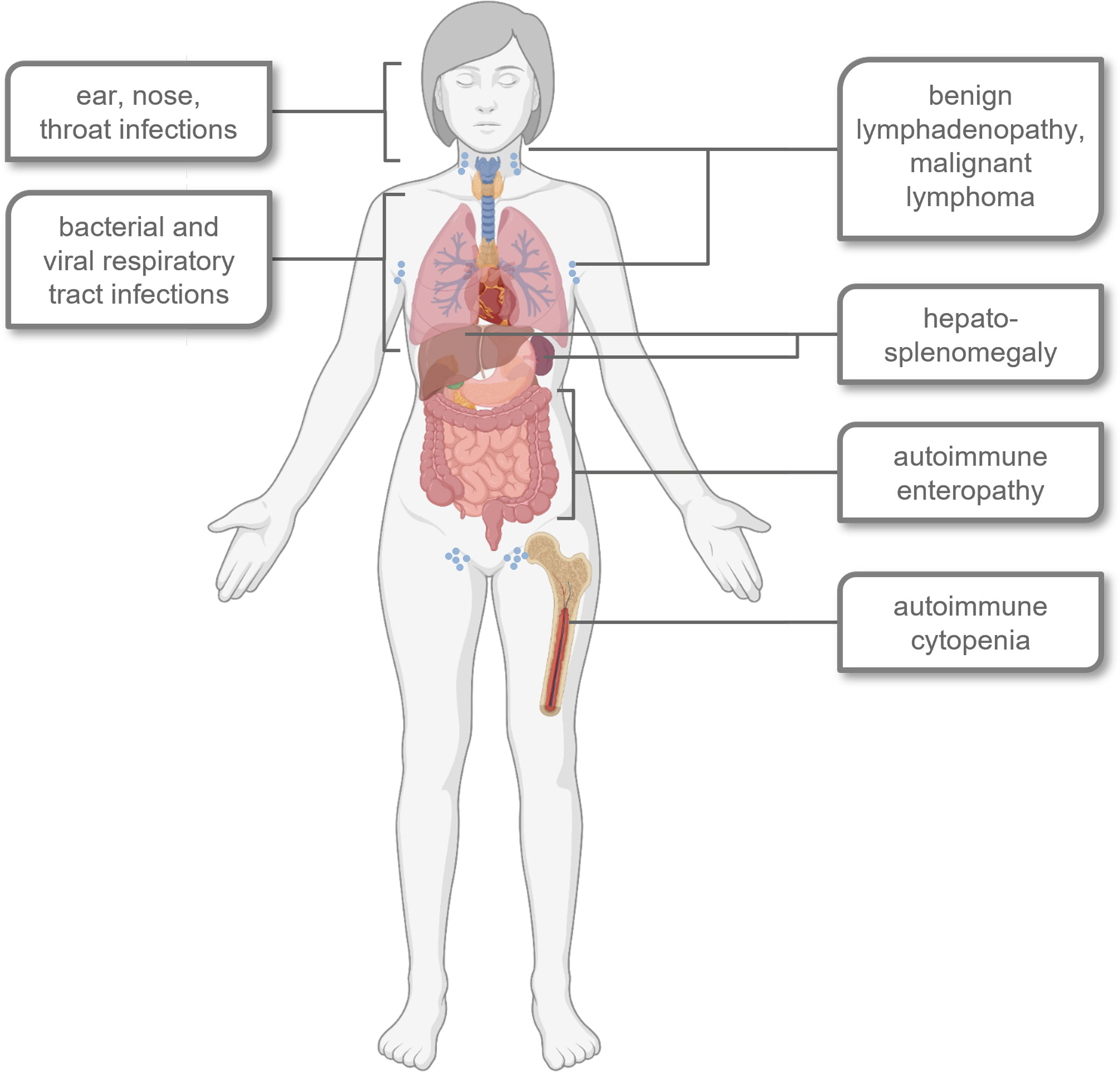
Figure 1 Schematic overview of typical signs, symptoms and affected organ systems in most APDS patients. Created with BioRender.com©.
Immunodeficiency
Alterations in the PI3K/Akt signaling pathway affect the function of T- and B lymphocytes. Several studies have shown a lack of naïve B cells - and a shift towards transitional B cells and plasmablasts - which can be attributed to increased rates of apoptosis as well as impaired class switch to IgG antibodies (26). In most patients, the increased susceptibility to infections manifests before the age of five years (14, 21). Since the production of plasma cells and memory B cells is also affected, patients often show hypogammaglobulinemia, a Hyper-IgM phenotype, and a selective lack of response to T cell independent vaccine stimuli (10, 20, 23, 27). Due to their inability to produce sufficient levels of neutralizing IgG and IgA antibodies, patients suffer from recurrent bacterial infections - in particular with Streptococcus pneumoniae (10). This often results in bronchiectases, chronic productive cough and deteriorating lung function (28).
Hyperactivation of PI3Kδ leads to a decrease in CD4+ T cells, while CD8+ T-cell levels are usually normal or increased (23), leading to an inverse CD4+/CD8+ ratio. CD8+ T cells often show signs of senescence (CD57+ expression and short telomers) and exhaustion (increased PD-1, CD160 and CD244 expression). At the same time, reservoirs of naïve CD4+ and CD8+ T cells are depleted (29, 30). Consequently, patients are often unable to eliminate viral infections, resulting in chronic infections with Herpes viruses such as Epstein-Barr-Virus and Cytomegalovirus (EBV, CMV) (29, 31). APDS is classified as a “predominantly antibody deficiency” by the IUIS (1), but since both humoral and cell-mediated immune defenses are affected, APDS could also be classified as a combined immunodeficiency.
Although APDS1 and APDS2 share several features like impaired B-cell class-switch and T follicular helper cell function, there are also differences observed in the degree of defective B cell expansion and affinity maturation, as well as intracellular signaling (32).
Bronchiectases and disease burden
Bronchiectases primarily result from recurrent lung infections, but might also be driven by an interplay between infection and TNF-α induced p110δ expression in lung epithelium during inflammation (23). Since p110δ is constitutively active, this would also explain why APDS1 patients are more prone to developing bronchiectases compared to APDS2 patients, who carry variants in p85α. Irreversible lung damage such as bronchiectases further aggravate the problem of recurrent and chronic inflammatory lung disease. Bronchiectases significantly contribute to poor quality of life.
As APDS patients require permanent treatment and medical attention, this also creates a significant burden on the healthcare system (33). Early diagnosis and treatment may slow the development of bronchiectases and thus preserve both lung function and quality of life (25). Therefore, APDS and other IEI are an important differential diagnosis when bronchiectases occur early in life (23).
Lymphoproliferation
Lymphoproliferation in APDS may manifest as lymphadenopathy, splenomegaly and hepatomegaly, increasing the risk for lymphomagenesis. Benign lymphoproliferation is usually the second most frequent clinical sign of APDS following repeated infections (14). Lymphadenopathy and splenomegaly in APDS patients can be massive and mimic malignant lymphoma. Besides peripheral lymph nodes mucosa-associated lymphoid tissues may be affected. Histologically, affected lymph nodes can display high rates of B-cell and T-cell proliferation (34, 35). Hemophagocytic lymphohistiocytosis (HLH)-like clinical pictures have been described, in one case followed by Hodgkin’s lymphoma. One case study reported an association of APDS2 with Kikuchi-Fujimoto syndrome (36), another presented with gut-associated T-cell lymphoproliferative disease (37, 38). Thus, careful histological examination of lymph nodes by an experienced pathologist is required to avoid unnecessary administration of cytostatic drugs (34, 35).
Recurrent adenoid hypertrophy often requires ENT surgery as patients suffer from obstructive sleep apnea. The risk for lymphoma is particularly high in APDS2 patients, with the cumulative risk estimated at 78% at the age of 40 years (21). PI3Kδ is predominantly - but not exclusively - expressed in leukocytes, which may explain why until now no predisposition for solid tumors had been observed in APDS patients (23). Long-term data in larger patient cohorts, however, seems to indicate a slightly increased risk for solid tumors as well (Maccari et al., 2023, accepted). The risk for APDS patients to develop lymphoma is aggravated by their inability to control infections with oncogenic viruses, e.g. EBV (20, 21, 30).
Autoimmunity/autoinflammation
Overlap between immunodeficiency and autoimmunity is frequently observed in patients with various IEIs (39). Mechanisms preventing adequate defense against pathogens also affect self-tolerance and immune regulation (40, 41). Chronic activation of PI3Kδ enhances B-cell survival and increases the number of plasma cells producing autoreactive antibodies (42, 43). Studies in a PI3KδE1020K mouse model indicate that polyclonal and self-reactive B cells expand at the expense of antigen-specific clones (26). Chronic infection with EBV promoting polyclonal B-cell proliferation may be an additional risk factor for autoimmunity (44). The predisposition for autoimmunity or autoinflammation can manifest as autoimmune cytopenias (45), chronic organ inflammation, colitis, arthritis (20, 21, 23) and granulomatous skin lesions (22, 39). Although common, frequencies of autoimmune phenomena in APDS patients vary, and may be absent (14).
Growth retardation, syndromic features and developmental delay
Short stature and a general failure to thrive are not uncommon in APDS patients. Short stature may be a secondary effect of frequent infections and gastrointestinal inflammation. It is seen more frequently in APDS2 patients, indicating tissue-specific involvement of p110δ and p85α in somatic development and immune regulation. Indeed, other PIK3R1 variants are associated with SHORT syndrome (short stature, hyperextensibility of joints, and/or inguinal hernia, ocular depression, Rieger anomaly, and teething delay), a condition affecting - among other things - stature and metabolism (46). In addition, neurodevelopmental delay and syndromic features are observed specifically in APDS2 patients (21, 23).
Genetics and molecular mechanism of APDS
APDS1 is caused by heterozygous GOF variants in the PIK3CD gene (p110δ protein) (10, 30, 47), and APDS2 by heterozygous LOF variants in the PIK3R1 gene (p85α protein) (11, 20). These variants likely occur sporadically but may be passed on in families for several generations or else occur de novo (19). A founder effect is unlikely since APDS1-affected families do not share long-range haplotypes or other markers indicating such an effect (10). APDS inheritance follows an autosomal dominant pattern (20, 30), and is characterized by high penetrance (23). Furthermore, LOF mutations of the phosphatase and tensin homolog gene (PTEN) cause a syndrome with similar characteristics as APDS (APDS-like) (48).
PI3Ks are a family of lipid kinases with important roles in all eukaryotic cells. They synthesize phosphatidylinositol 3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). PIP3 in turn activates downstream proteins like Akt, PDK1 and mTOR. While PI3Kα and -β are active in all cells of the body, PI3Kγ and -δ are mostly - though not exclusively - active in leukocytes. PI3Kδ and its two subunits are highly relevant for the function of the adaptive immune system (49–52). p110δ is catalytically active, while p85α serves as a regulatory subunit (53). Together, these core components of the PI3K/Akt signaling pathway regulate the growth and differentiation of lymphocytes in concert with other molecules (Figure 2). In B cells, p110δ can be activated via the B-cell receptor or via cytokine signals. In T cells, the main activator for p110δ is T-cell receptor signaling. p85α interacts with p110δ in its resting state, inhibiting catalytic activity. Most PI3Kδ-activating variants in PIK3CD affect the C-terminal part of its gene product, primarily amino acid E1021 (Figures 3A, B) (10, 52). These variants increase baseline activity by either impairing inhibitory interactions with p85α, or increasing the affinity of p110δ for the plasma membrane, or both (55). Interaction between the nSH2 and iSH2 (SH2 = Src-homology 2) domains of p85α and between the core domain, helical domain and catalytic domain of p110δ is necessary for the inhibition of p110δ in its resting state (Figure 3C). Moreover, the p85α iSH2 domain is needed for establishing a solid interaction with the N-terminal p110δ adaptor binding domain (ABD) (49, 55, 56). Virtually all APDS2 patients carry a variant in the splice donor site for intron 10 of PIK3R1, leading to deletion of exon 10 (aa434–475) (11, 52) and to LOF in p85α (Figures 3A, C). As a dedicated regulator of p110δ, such LOF effectively results in GOF of the PI3Kδ complex which is the unifying characteristic of all APDS-associated gene variants. Thus, APDS elegantly illustrates how GOF in effector enzymes and LOF in their regulators have similar effects. Accordingly, LOF in regulators with primary inhibitory effects are considered GOF mutations by some authors (13).
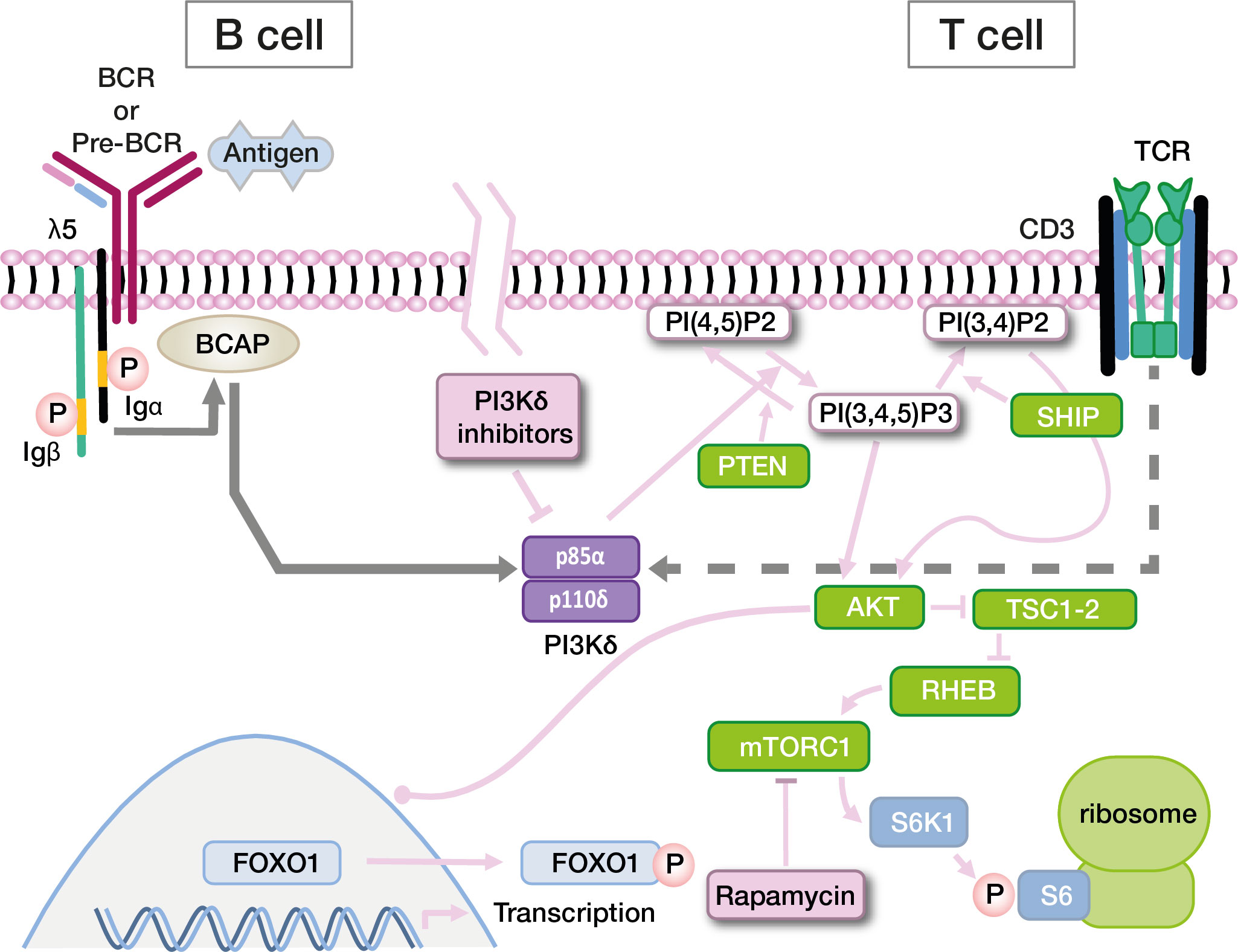
Figure 2 Simplified diagram of the PI3K signaling pathway in B cells and T cells, and targets for directed drug therapy. Other pathways also involving Pi3K (via IL-4R or CXCR5) are not depicted. Signaling events are extensively discussed in the text. Abbreviations: BCR, B cell receptor; TCR, T-cell receptor; Igα, CD79a; Igβ, CD79b; BCAP PI(*)P2, Phosphatidylinositol-*-bisphosphate; PI(*)P3, Phosphatidylinositol-*-trisphosphate; SH2, Src homology 2 domain; SHIP, SH2 domain-containing inositol-5-phosphatase; PTEN, phosphatase and tensin homologue; AKT, a SH2 containing serine/threonine kinase; TSC1-2 RHEB, RAS homologue enriched in brain; mTORC1, mTOR complex 1; S6K1, Ribosomal protein S6 kinase beta-1; FOXO1, Forkhead Box O1.
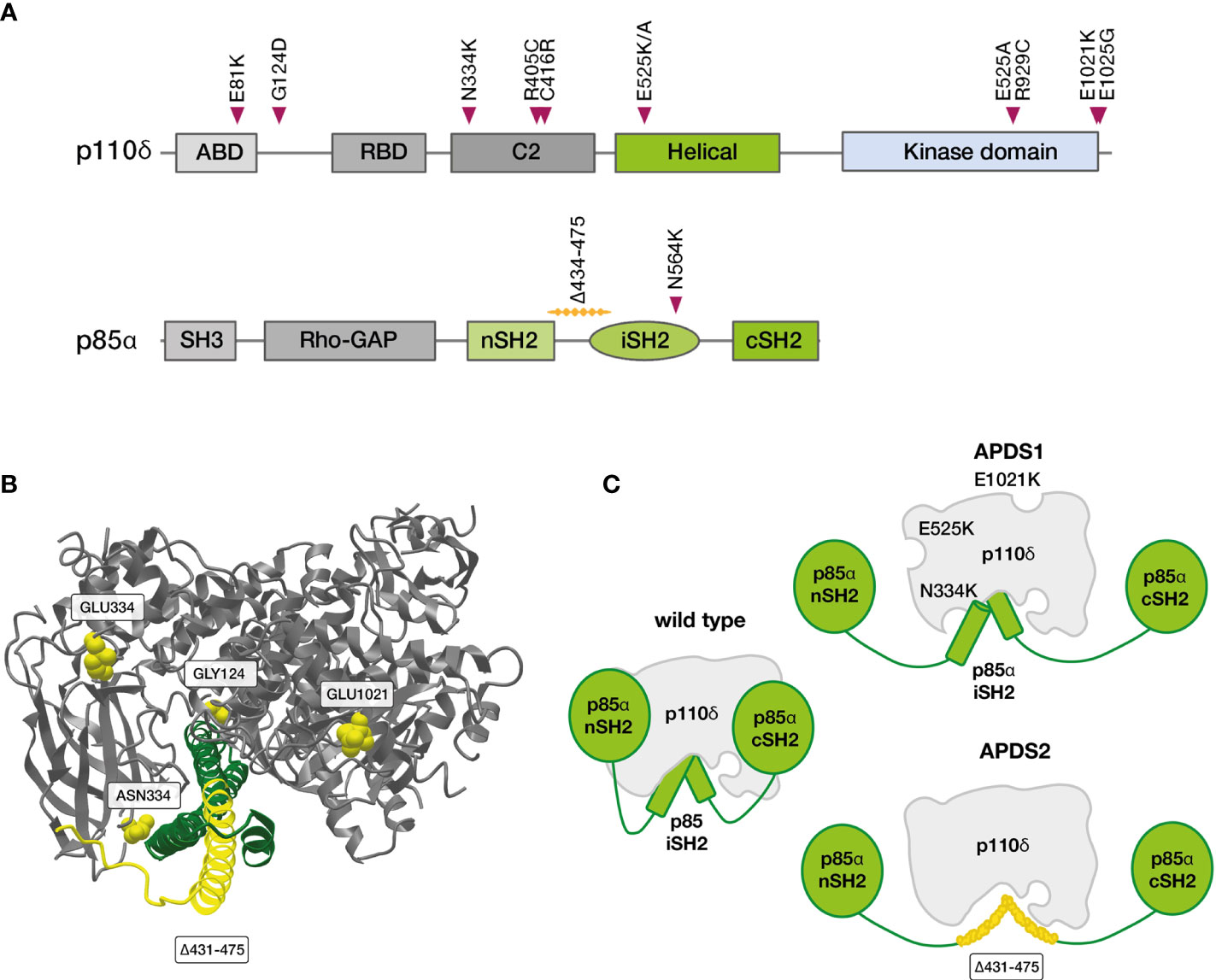
Figure 3 Variants in PIK3CD and PIK3R1 linked to APDS (52). (A) Linear depiction of p110δ and p85α proteins with confirmed, APDS-causing variants (B) Partial crystal structure of Homo sapiens p110δ (grey, positions commonly affected in APDS1 in yellow) in complex with part of the SH2 domains of Bos taurus p85α (green, positions commonly deleted in APDS2 in yellow), frequent APDS variants are indicated in yellow (MMDB-ID: 5DXU; image created with iCn3D webtool) (54). (C) Consequences of gene variants in p110δ (APDS1) and p85α (APDS2) on the PI3Kδ complex. iSH2, inter SH2 domain; cSH2, C-terminal SH2; nSH2, N-terminal SH2 domain; SH2, Src homology 2 domain.
Diagnostic algorithm in APDS
Early diagnosis of IEI, including APDS, is essential to define suitable treatments that may delay or prevent irreversible organ damage. Unfortunately, diagnosis in IEI patients is often delayed. For example, analyses of the ESID and United Kingdom Primary Immunodeficiency (UKPID) registries suggested minimum prevalences of 8.8/100000 for the Island of Ireland in 2022, and 4.4/100000 in France in 2017 (57, 58), which are well below the estimated ratio of 1/2000 in the general population. A total of 3100 IEI patients were recorded in the German immunodeficiency register PID-NET in 2019, and 4700 in 2021 (59, 60). In Germany, APDS and other IEIs are most likely underdiagnosed, however, the exact prevalence is unknown. Additionally, some patients may have been diagnosed but have not been reported to the registry (61). As a first step in IEI diagnosis, the treating pediatrician or general practitioner must appraise an unusual pattern of symptoms in their patient (Figure 4). Although so-called “warning signs” of IEI (62) may help identify immunologic disorders, awareness of IEI among doctors may be a limiting factor (63, 64). Such typical warning signs were proposed by the North American Jeffrey Modell Foundation (https://info4pi.org), some of which have been integrated into diagnostic guidelines. Unfortunately, these do not include signs like autoimmunity or lymphoproliferation. Expertise in diagnosing IEIs is mostly confined to physicians specialized in managing primary immunodeficiencies, and diagnosing immune-related disorders may take up to ten years, as is the case with APDS (12, 14, 65, 66), the discovery of the causative gene variants underlying APDS has helped shorten this delay. Given the sparsity and geographical distribution of clinical immunologists, there is a high probability of patients not being directly referred to a specialist. In Germany, only 220 clinical immunologists practice compared to 16.100 pediatricians, 1900 hemato-oncologists, and 900 rheumatologists (67). The General Medical Council of the United Kingdom currently (early 2023) lists 182 immunologists, as opposed to 1954 hemato-oncologists and 1426 rheumatologists (68). Patients with signs of lymphoproliferation may see an oncologist, a gastroenterologist when either enteropathy or failure to thrive are the leading symptoms, or else a rheumatologist when autoimmunity prevails. Oftentimes, patients see multiple subspecialists, and the underlying primary immunodeficiency remains unrecognized for long periods of time.
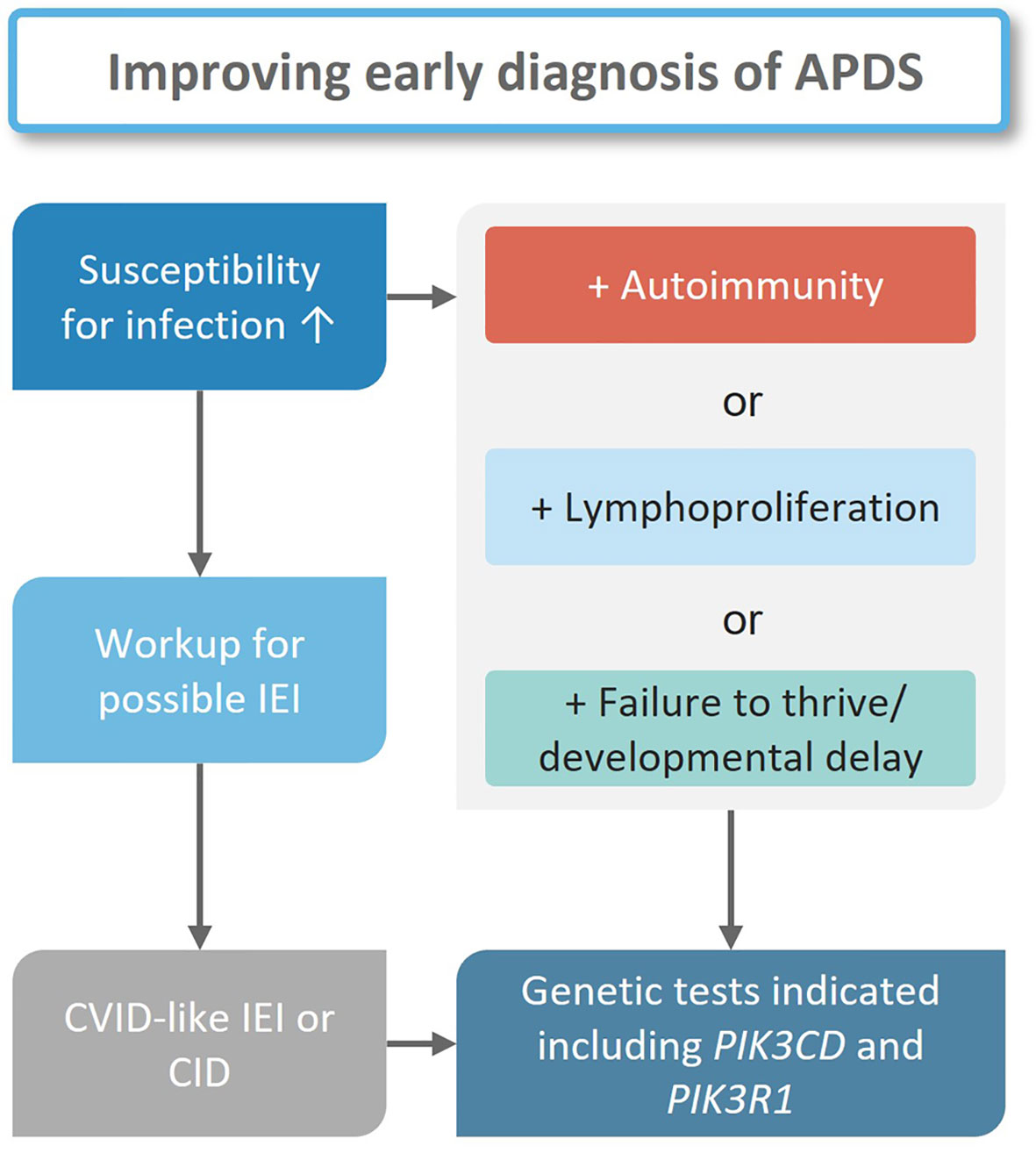
Figure 4 Diagnostic workflow for suspected APDS patients. In all cases with increased susceptibility to infections an appropriate workup for a possible IEI is indicated. If a diagnosis is established genetic tests should follow. If one or more other clinical findings are present in addition to infections, genetic tests should follow independent from an IEI diagnosis.
Centers specialized in IEI will ask individuals with suspected IEI for a detailed history including a family tree. Patients will then undergo a comprehensive physical examination before laboratory assays are initiated. The results of this basic assessment may help decide how likely an IEI is, and whether in-depth analyses are necessary to determine the exact nature of a suspected IEI (Figure 4) (64, 69–71). Phenotypical overlaps are common in IEI as is the case for APDS, which may fit under the umbrella entity of common variable immunodeficiency (CVID), although APDS is probably better classified as a combined immunodeficiency (CID). Only in exceptional cases would the clinical presentation (symptoms and lymphocyte phenotyping) suggest APDS to the experienced immunologist prior to genetic testing.
Current guidelines recommend performing advanced immunological diagnostics of suspected IEI patients prior to molecular testing (69, 70). Genetic sequencing is the accepted confirmatory step for most suspected IEIs. The treating physician may choose between gene panels, where predefined sets of genes are sequenced, or whole exome sequencing (WES). Additionally, whole genome sequencing (WGS) will complement - or even supersede - both panel and exome approaches. Given the heterogeneous clinical presentation of APDS patients, there is a potential risk of selecting the “wrong” gene panel: at worst missing the mutated gene. WES and WGS have become more affordable and are now being covered by some health insurances in developed countries, meaning many specialists may directly choose WES - or, a few, WGS - in search of a monogenic cause of the suspected IEI. It should be noted however, that only a limited number of genes from WES/WGS may be analyzed by geneticists (referred to as panel-in-exome or virtual panel). It is therefore of great value that the treating physician and the geneticist directly communicate before the genetic analysis is finalized to allow for re-evaluation of the sequencing data, and targeted filtering according to the human phenotype ontology terms (e.g. when no pathogenic variant has been identified in a given panel to explain the phenotype) (72). If a specific gene variant is identified, targeted Sanger sequencing may help segregate the variant within the family, possibly identifying other carriers or mildly affected family members.
Identification of gene variants and variant databases
Thanks to the availability and affordability of gene and genome sequencing, an increasing number of IEIs are being mapped to the underlying genetic causes. The first variant known to cause APDS1, the E1021K variant in p110δ, was first published in 2006 (73), but APDS1 was not described as a distinct disease entity until 2013 (10). Soon thereafter, the aa434–475 deletion in p85α was recognized to cause a similar condition (11). Extensive databases on disease-causing variants, including those associated with IEI are maintained on platforms such as OMIM (www.omim.org), OrphaNET (www.orpha.net), ClinVar (www.ncbi.nlm.nih.gov/clinvar), and DisGeNET (disgenet.org) (74). However, identifying a disease-causing gene variant in the depths of the human genome remains challenging for many patients. For the identification of disease-causing variants, experts cross-check variant databases for neutral variants and use pathogenic variant prediction algorithms (75). Besides the few variants in the PIK3CD and PIK3R1 genes functionally proven to be pathogenic (52), there are hundreds of variants of unknown significance (VUS), whose causality for an immunodeficiency has not yet been determined. Patients with an APDS-like presentation and abnormal PI3K/Akt signalling but without a pathogenic variant in the two APDS genes (e.g. PTEN deficiency) require further investigations. Databases like Franklin (https://franklin.genoox.com) and varsome (https://varsome.com) collect information on the pathogenicity of variants. The online tool LitVar (www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/LitVar) offers assistance with literature research for previously reported variants. However, geneticists should not solely rely on published information as the field is constantly evolving and new information is being added to the various databases. In APDS - as is the case generally for rare hereditary diseases - the exchange between experts and the use of common platforms will greatly facilitate linking gene variants to patient phenotypes, and furthermore enrich the currently known spectrum of IEI (76). Genetic tests have the potential of making diagnostics for monogenetic diseases accessible to patients who do not yet benefit fully from modern medicine, for example due to the economic situation in their home country.
Functional testing for PI3K activity
A number of variants have been functionally ascertained to underlie APDS, based on the correlation between genetics and symptomatology of patients (10, 11). However, to confirm a direct link between genotype and clinical phenotype, a functional proof of pathogenicity in the affected molecular pathomechanism is crucial. The primary characteristic of APDS-causing variants lies in increased activity of PI3Kδ and downstream proteins, including Akt and mTOR. Confirming a suspected diagnosis of APDS requires matching of the clinical phenotype and a (likely) pathogenic variant. This would leave patients who do not have one of the confirmed APDS variants without a diagnosis, regardless of their phenotype. Therefore in some academic settings, the overactivation of the PI3K/Akt pathway may be investigated in order to characterize the biology of APDS patient cells. This includes analysis of phosphorylation levels of downstream proteins like Akt, FOXO1, and of the ribosomal protein S6 (11, 20, 30). However, these functional tests have not found their way into clinical practice yet, which would require fresh patient samples to be shipped to a designated diagnostic center (30). The phospho-S6 ribosomal protein, however, has been proposed as a predictive marker in sarcoma (77), so it is conceivable that a phospho-S6 assay could be standardized as one possible confirmatory test for APDS. Developing functional testing for VUS in APDS could serve as a proof-of principle for IEIs characterized by GOF or LOF variants in other genes. In the future, functional testing could help assess the efficacy of PI3Kδ inhibitors in APDS and other diseases with abnormal PI3K/Akt signaling, e.g. PTEN deficiency, which could help provide existing therapy options for more patients and to monitor therapy.
Treatment options and unmet need in APDS
APDS in the context of other IEI
Many monogenic immunodeficiencies such as APDS are rare diseases. The disease burden of APDS patients varies greatly: from family members incidentally diagnosed with relatively mild symptoms, to patients with life-threatening or chronic airway infections, severe colitis or early-onset lymphoma (14, 24). Although it affects humoral and cellular immunity, APDS may be misclassified as CVID. Like in CVID, APDS patients suffer from frequent upper and lower airway disease, autoimmunity, lymphoproliferation and a B-cell deficiency (78). The increased risks of sepsis and lymphoma result in an inferior life expectancy (21, 23). Unlike in CVID, disease symptoms usually manifest early in childhood. Early symptom-onset is a common feature of many other monoallelic combined immunodeficiencies with similarities to APDS like CTLA-4 deficiency, NFKB1 deficiency, autoimmune lymphoproliferative syndrome variants (ALPS) or STAT3 gain-of-function (27, 28, 79–81). In terms of phenotypic severity, APDS presents as a spectrum somewhere between CVID and CID, with additional immune deregulatory features.
Symptomatic treatment
APDS is an IEI of moderate-to-high severity with great heterogeneity in the patient population, which is reflected in the various strategies employed in managing the disease. PI3Kδ hyperactivity has multiple effects on the immune functions which makes it difficult to treat patients in a symptom-oriented fashion. Recurrent infections can be managed with immunoglobulin replacement therapy (IRT) and antibiotic prophylaxis. Indeed, most patients (70-80%, see Table 1) receive both IRT and antibiotics regularly or against acute infection, with some benefit observed in decreasing the frequency of respiratory tract infections (RTI) (21, 23, 24, 52). However, these options are inadequate in addressing virus reactivation or chronic viral infections, as well as in preventing lung damage. Two APDS1 and APDS2 patient cohorts documented by Coulter et al. and Elkaim et al. are reported with chronic viral infection and with progressive bronchiectases despite IRT (21, 23).
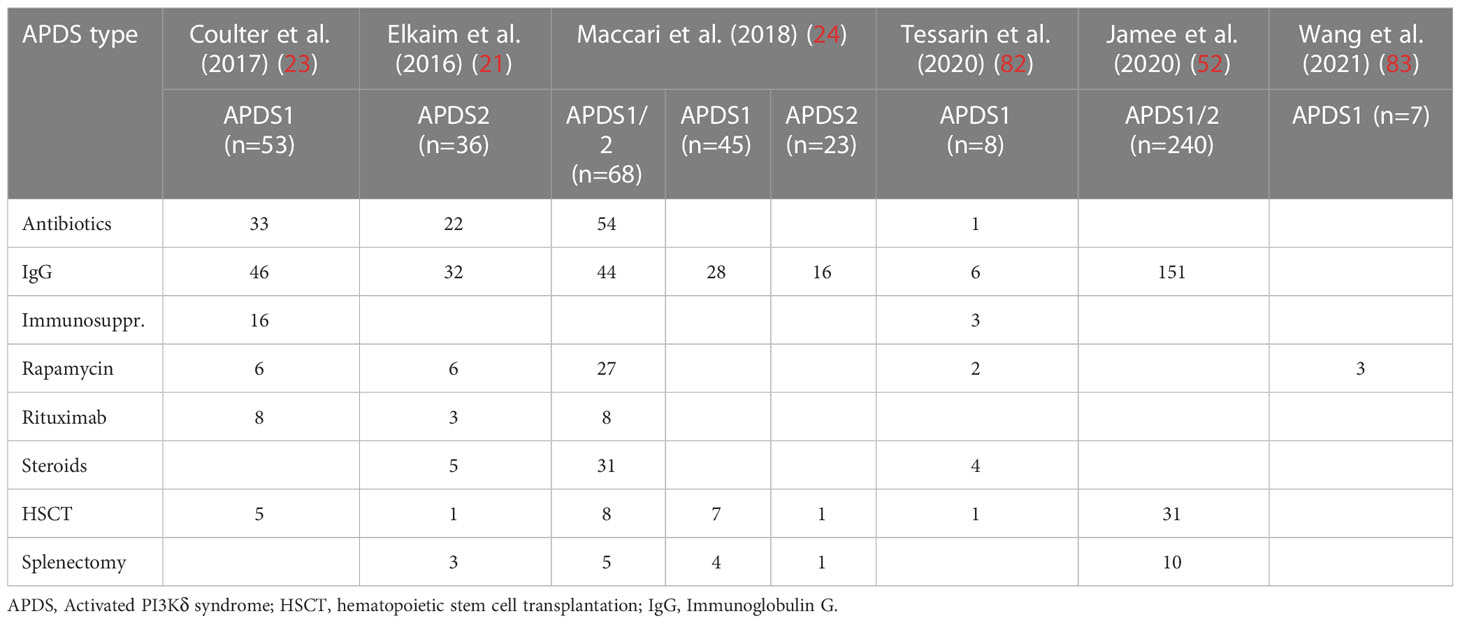
Table 1 Reported treatment choices for APDS patients. It should be noted that some individuals are included in more than one publication.
Immunosuppression
To address autoimmunity and lymphoproliferation, immunosuppressants are frequently used in APDS patients, including corticosteroids and the mTOR inhibitor rapamycin (see Table 1). The targeted inhibitory effect of rapamycin on mTOR, which is located downstream of PI3Kδ in the PI3K/Akt pathway, provides a rationale for its use in APDS. While rapamycin appears to be particularly effective in the treatment of lymphoproliferation, its efficacy with respect to inflammation and autoimmune hemolysis is limited. It has been reported that natural antibody production may improve under rapamycin (24, 84). The drug is tolerated by most patients, but treatment interruptions or permanent discontinuation due to side effects have been documented (23, 24). The B-cell-depleting antibody rituximab has been used in APDS patients with good efficacy against hemolysis and lymphoproliferation, but invariably entails profound and long-lasting hypogammaglobulinemia. Some APDS patients (<10%) were splenectomized for autoimmune cytopenia or for diagnostic purposes (21, 23, 52).
Stem cell transplantation
As treatment with approved drugs is often insufficient to control the debilitating and life-threatening symptoms in APDS patients, HSCT is considered a viable option with curative potential. According to the literature across all cohorts analyzed, between 10-20% of APDS patients have been treated with HSCT (see Table 1). HSCT is mostly offered to APDS1 patients (up to 20%) compared to APDS2 patients (<5%) (21, 23, 24, 52, 82). Life-threatening lymphoproliferation and recurrent infections despite high-dose IRT were most frequently cited as indications for HSCT (85). Conforming to data on patients with other genetically defined CID, mortality after HSCT – usually from infection – was 10-20% (85, 86). Frequent complications were graft-versus-host-disease (GvHD) and low or declining donor-type chimerism. A recent study reported a rate of 14% of patients who required a second transplantation, with one patient requiring a third (87). While HSCT has the potential to lower the frequency of infections or severity of lymphoproliferation (85, 86), manifestations in non-hematopoietic organs are likely to persist. To date, the outcome of HSCT is superior in younger patients as reported in a number of other IEIs (88). However, since the course of disease is unpredictable in young children there is a potential risk of subjecting patients with a possibly mild course of disease to potential risks of HSCT. In the literature, HSCT-related events represent the second most common cause of death in APDS patients (25).
Targeted therapies
As discussed above, immunomodulation with the mTOR-targeting molecule rapamycin is more effective and has fewer side effects compared to the long-term use of broad-acting immunosuppressants such as corticosteroids or azathioprine. Nonetheless, rapamycin is associated with a number of side effects including diabetes-like symptoms and lung toxicity. This is because its target - mTOR - is not only involved in BCR and TCR signaling via PI3Kδ, but also the insulin and growth hormone signaling pathways (89). In contrast, specific inhibitors of PI3Kδ directly target the protein whose hyperactivity is at the root of APDS. Idelalisib was the first PI3Kδ inhibitor allowing for PI3Kδ activity to be regulated to normal levels in in vitro in samples from leukemia and lymphoma patients, but had considerable side effects in vivo (31, 55). Three other PI3Kδ inhibitors, nemiralisib, seletalisib and leniolisib are being tested in APDS patients. Nemiralisib showed favourable results in an animal model, decreasing mortality in S. pneumoniae infected mice with p110δE1020K (90). However, a study on 5 human patients treated with inhaled nemiralisib over 3 months showed good tolerance, but no measurable anti-inflammatory effects (NCT02593539) (91). Seletalisib was tested in an open label phase Ib study and demonstrated encouraging results modulating disease activity, but serious adverse events occurred in three of seven patients. The safety profile was still considered acceptable for this group of patients (92). Leniolisib, an oral inhibitor of PI3Kδ, showed promising results in an open-label trial with six APDS1 patients (15), later confirmed in a randomized, placebo-controlled phase III trial (NCT02435173). The drug was well tolerated by all patients and improved symptoms of lymphadenopathy, immune cell derangement and cytopenias. Moreover, it achieved reduction in lymph node and spleen volumes, normalized mean IgM levels, increased the number of naïve B cells and decreased the rate of transitional B cells (93). The observational period was too short for an assessment of infection load, lung function or risk of lymphoma. Currently, an extension study for leniolisib is underway (NCT02859727), in which its long-term safety and effect on patient health and quality of life are being assessed. Preliminary results confirm the favorable safety profile and long-term reconstitution of immune function, including a significant reduction of infections, shown by discontinuation of IRT in a number of probands (94). Thus, leniolisib has the potential to become a standard treatment for APDS patients from 12 years and older. Targeted therapies have the potential to normalize PI3K pathway activity in all somatic cells. In order to have a positive influence on the physical development of APDS patients, immune-modulating therapies must be initiated as early as possible. A study with leniolisib in patients aged 4-11 is recruiting patients (NCT05438407), and another with patients aged 1-6 (NCT05693129) is in preparation. Further evaluation of the long-term efficacy and safety of leniolisib is needed to assess its capability to modify future management of APDS, such as potential use as a bridging therapy prior to HSCT. Clear prerequisites for the indication of leniolisib, e.g. the genotype-phenotype correlation allowing prognosis of the expected course, remain to be determined.
Summary and outlook
The present review has addressed the clinical, immunological, and molecular characteristics of APDS, and discussed the current challenges in the diagnosis and treatment of this complex disease. Based on our review of literature, we have identified the following key challenges in the diagnosis and treatment of APDS:
1. Underdiagnosis of APDS as a result of the heterogeneous clinical picture and lack of awareness for IEI warning signs among health care providers.
2. Limited availability and lack of standardized functional testing for APDS to identify patients where genetics remain inconclusive, e.g. variants of unknown significance
3. Limitation of conventional drug therapies to control APDS symptoms
4. Lack of consensus for predicting a severe disease course to guide decisions in personalized management including HSCT
5. Need for therapeutic options that address immunological and neurological symptoms without significant side effects
There is a need for continuing education across specialties caring for IEI patients. Multidisciplinary cooperation is essential in order to a) overcome current underdiagnosis of IEI and b) spread awareness that early and precise diagnosis usually has a direct impact on treatment decisions, including targeted therapies. In light of an increasing number of molecules for targeted treatment of specific IEI, defining the genotype of IEI should be part of diagnostic routine. Still, patients may present with typical signs of APDS but have inconclusive genetics. To characterize so-called VUS, and to identify more variants that result in PI3K pathway hyperactivity, functional testing procedures within reach of specialty clinics need to be validated. Traditional management of APDS is based on antibiotic or antiviral treatment and prophylaxis for recurrent infections, IRT as well as immunosuppressive agents to control symptoms of immune dysregulation (21, 23, 52). Since the current therapeutic options are sometimes insufficient to adequately control symptoms, HSCT as a potentially curative treatment could be an alternative for selected patients. Especially in younger patients at risk for severe course of disease, HSCT can improve immune function, and reduce frequency of infections and severity of lymphoproliferation (85, 86). However, markers predicting severity of disease and individual risk to develop lymphoma are currently missing, so risk vs. benefit assessment remains a true challenge. Large observational studies such as the ESID APDS registry are best suited to identify such markers. The promising results for targeted treatment with leniolisib suggest that targeted therapy could become a viable option in APDS and other monogenetic IEI. Leniolisib is a convincing example of a targeted disease-modifying drug developed for the treatment of a distinct IEI. The drug was approved by the FDA (95) and is awaiting approval by the EMA.
Since both HSCT and targeted therapies have advantages and shortcomings, both approaches need to be discussed depending on a patient’s condition and disease severity. Better understanding of the pathomechanisms underlying other IEIs holds the prospect of making novel or repurposed targeted therapeutics available for patients, either newly diagnosed or with progressive disease. The relative simplicity of their application could also make them a reasonable option for patients living in countries where facilities for stem cell transplantation are not yet available. Targeted treatments may prevent or delay disease progression and contribute to a better quality of life in individuals with IEI.
Author contributions
SV is the first author and was the main person responsible for the literature search and writing of the manuscript text. Furthermore, he was responsible for drafting the outline. VW supported the first author in the conceptualization of the article, contributed content ideas, checked the content for scientific correctness, and assisted in the structuring of the article. CS contributed her expert clinical and scientific knowledge to the content of the article, provided additional literature sources, and helped harmonize the structure of the content. Furthermore, she provided critical feedback on the messaging of the article regarding methods of diagnosis and treatment options for APDS.
Funding
Funding for the writing of this article and the submission processing fee comes from Pharming Group N.V. Catharina Schütz’ research is in part funded by the Rosemarie-Germscheid Stiftung (funding goes to Klinisches Studienzentrum Pädiatrie – Universitätsklinikum Carl-Gustav-Carus).
Acknowledgments
We thank Dr. rer. nat. Anna Stittrich from the BIH Center for Regenerative Therapies (BCRT) for her help with the section on genetic testing.
Conflict of interest
Author SV was employed by Infill Healthcare Communication. Author VW received a honorarium from Pharming Group N.V. for the supervision of the writing of this article. His homepage www.immundefekt.de is supported by CSL Behring. Author CS has declined a honorarium for her contribution to this manuscript. She was PI for Phase 3 Study CCDZ173X2201 Novartis and PI for the open extension study CCDZ173X2201E1 Novartis/Pharming.
The authors declare that this study received funding from Pharming Group N.V.. The funder had the following involvement in the study: writing of the article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. Springer US (2022) 42(7):1473–507. doi: 10.1007/s10875-022-01289-3
2. Quinn J, Modell V, Orange JS, Modell F. Growth in diagnosis and treatment of primary immunodeficiency within the global Jeffrey modell centers network. Allergy Asthma Clin Immunol (2022) 18:1–19. doi: 10.1186/s13223-022-00662-6
3. Modell V, Gee B, Lewis DB, Orange JS, Roifman CM, Routes JM, et al. Global study of primary immunodeficiency diseases (PI)-diagnosis, treatment, and economic impact: an updated report from the Jeffrey modell foundation. Immunol Res (2011) 51:61–70. doi: 10.1007/s12026-011-8241-y
4. Anderson JT, Cowan J, Condino-Neto A, Levy D, Prusty S. Health-related quality of life in primary immunodeficiencies: impact of delayed diagnosis and treatment burden. Clin Immunol (2022) 236:108931. doi: 10.1016/j.clim.2022.108931
5. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol (2007) 27:497–502. doi: 10.1007/s10875-007-9103-1
6. Rubin Z, Pappalardo A, Schwartz A, Antoon JW. Prevalence and outcomes of primary immunodeficiency in hospitalized children in the United States. J Allergy Clin Immunol Pract (2018) 6:1705–1710.e1. doi: 10.1016/j.jaip.2017.12.002
7. Quinn J, Orange JS, Modell V, Modell F. The case for severe combined immunodeficiency (SCID) and T cell lymphopenia newborn screening: saving lives…one at a time. Immunol Res (2020) 68:48–53. doi: 10.1007/s12026-020-09117-9
8. Smith CIE, Berglöf A. X-Linked agammaglobulinemia. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® (2020). Seattle (WA): University of Washington, Seattle; 1993–2023.
9. Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am (2013) 27:89–99. doi: 10.1016/j.hoc.2012.11.002
10. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Sci (80- ) (2013) 342:866–71. doi: 10.1126/science.1243292
11. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124:3923–8. doi: 10.1172/JCI75746
12. Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European society for immunodeficiency (ESID) registry data 11 medical and health sciences 1117 public health and health services. Orphanet J Rare Dis (2018) 13:1–17. doi: 10.1186/s13023-018-0941-0
13. Boisson B, Quartier P, Casanova JL. Immunological loss-of-function due to genetic gain-of-function in humans: autosomal dominance of the third kind. Curr Opin Immunol (2015) 32:90–105. doi: 10.1016/j.coi.2015.01.005
14. Bloomfield M, Klocperk A, Zachova R, Milota T, Kanderova V, Sediva A. Natural course of activated phosphoinositide 3-kinase delta syndrome in childhood and adolescence. Front Pediatr (2021) 9:697706. doi: 10.3389/fped.2021.697706
15. Rao VK, Webster S, Dalm VASH, Šedivá A, Van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome”–targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130:2307–16. doi: 10.1182/blood-2017-08-801191
16. Burke JE, Triscott J, Emerling BM, Hammond GRV. Beyond PI3Ks: targeting phosphoinositide kinases in disease. Nat Rev Drug Discovery (2022) 14:1–30. doi: 10.1038/s41573-022-00582-5
17. ESID APDS registry Freiburg. (2022). Available at: https://www.uniklinik-freiburg.de/cci/studien/apds-registry.html (Accessed March 15, 2023).
18. Condliffe AM, Ehl S, Kracker S, Maccari ME. APDS registry an ESID registry level 3 project. (2021). pp. 1–2. Available at: https://www.esid.org/content/download/18305/491868/file/APDS_NEWSLETTER3_01-2021.pdf (accessed March 15, 2023)
19. Michalovich D, Nejentsev S. Activated PI3 kinase delta syndrome: from genetics to therapy. Front Immunol (2018) 9:369. doi: 10.3389/fimmu.2018.00369
20. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211:2537–47. doi: 10.1084/jem.20141759
21. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138:210–218.e9. doi: 10.1016/j.jaci.2016.03.022
22. Ewertowska M, Grześk E, Urbańczyk A, Dąbrowska A, Bąbol-Pokora K, Łęcka M, et al. Activated phosphoinositide 3-kinase delta syndrome 1 and 2 (APDS 1 and APDS 2): similarities and differences based on clinical presentation in two boys. Allergy Asthma Clin Immunol (2020) 16:22. doi: 10.1186/s13223-020-00420-6
23. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
24. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: the European society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol (2018) 9:543. doi: 10.3389/fimmu.2018.00543
25. Hanson J, Bonnen PE. Real-world evidence of mortality and survival rates in 256 individuals with APDS (2022). Available at: https://www.medrxiv.org/content/10.1101/2022.12.05.22283110v1 (Accessed March 15, 2023).
26. Preite S, Cannons JL, Radtke AJ, Vujkovic-Cvijin I, Gomez-Rodriguez J, Volpi S, et al. Hyperactivated PI3Kδ promotes self and commensal reactivity at the expense of optimal humoral immunity. Nat Immunol (2018) 19:986–1000. doi: 10.1038/s41590-018-0182-3
27. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Schölvinck L, Kalina T, et al. Genetic defects in PI3Kδ affect b-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77–86. doi: 10.1016/j.clim.2017.01.004
28. Condliffe AM, Chandra A. Respiratory manifestations of the activated phosphoinositide 3-kinase delta syndrome. Front Immunol (2018) 5:338(9). doi: 10.3389/fimmu.2018.00338
29. Wentink MWJ, Mueller YM, Dalm VASH, Driessen GJ, van Hagen PM, van Montfrans JM, et al. Exhaustion of the CD8+ T cell compartment in patients with mutations in phosphoinositide 3-kinase delta. Front Immunol (2018) 9:446. doi: 10.3389/fimmu.2018.00446
30. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi: 10.1038/ni.2771
31. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3K δ regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U.S.A. (2017) 114:1982–7. doi: 10.1073/pnas.1617244114
32. Nguyen T, Lau A, Bier J, Cooke KC, Lenthall H, Ruiz-Diaz S, et al. Human PIK3R1 mutations disrupt lymphocyte differentiation to cause activated PI3Kδ syndrome 2. J Exp Med (2023) 220:e20221020. doi: 10.1084/jem.20221020
33. Diel R, Chalmers JD, Rabe KF, Nienhaus A, Loddenkemper R, Ringshausen FC. Economic burden of bronchiectasis in Germany. Eur Respir J (2019) 53:1802033. doi: 10.1183/13993003.02033-2018
34. Baleydier F, Ranza E, Schäppi M, Rougemont AL, Merlini L, Ansari M, et al. Activated phosphoinositide 3 kinase delta syndrome (APDS): a primary immunodeficiency mimicking lymphoma. J Pediatr Hematol Oncol (2019) 41:E521–4. doi: 10.1097/MPH.0000000000001328
35. Pham MN, Cunningham-Rundles C. Evaluation of lymphoproliferative disease and increased risk of lymphoma in activated phosphoinositide 3 kinase delta syndrome: a case report with discussion. Front Pediatr (2018) 8:402(6). doi: 10.3389/fped.2018.00402
36. Abulaila KJ, Sabha MA, Misk MR, Alhalabeye AA, Jobran AWM, Abunejma FM, et al. Activated phosphoinositide 3-kinase delta syndrome 2 associated with kikuchi – fujimoto disease: a rare Palestinian case report. Ann Med Surg (Lond) (2023) 85(5):2064–7. doi: 10.1097/MS9.0000000000000476
37. Teranishi H, Ishimura M, Koga Y, Eguchi K, Sonoda M, Kobayashi T, et al. Activated phosphoinositide 3-kinase δ syndrome presenting with gut-associated T-cell lymphoproliferative disease. Rinsho Ketsueki (2017) 58(1):20–5. doi: 10.11406/rinketsu.58.20
38. Luo Y, Xia Y, Wang W, Li Z, Jin Y, Gong Y, et al. Identification of a novel de novo gain-of-function mutation of PIK3CD in a patient with activated phosphoinositide 3-kinase δ syndrome. Clin Immunol (2018) 197:60–7. doi: 10.1016/j.clim.2018.08.007
39. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol (2018) 14:7–18. doi: 10.1038/nrrheum.2017.198
40. Azizi G, Yazdani R, Rae W, Abolhassani H, Rojas M, Aghamohammadi A, et al. Monogenic polyautoimmunity in primary immunodeficiency diseases. Autoimmun Rev (2018) 17:1028–39. doi: 10.1016/j.autrev.2018.05.001
41. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary immunodeficiency and autoimmunity: a comprehensive review. J Autoimmun (2019) 99:52–72. doi: 10.1016/j.jaut.2019.01.011
42. Lau A, Avery DT, Jackson K, Lenthall H, Volpi S, Brigden H, et al. Activated PI3Kd breaches multiple B-cell tolerance checkpoints and causes autoantibody production. J Exp Med (2020) 217:e20191336. doi: 10.1084/jem_20191336
43. Preite S, Gomez-Rodriguez J, Cannons JL, Schwartzberg PL. T And b-cell signaling in activated PI3K delta syndrome: from immunodeficiency to autoimmunity. Immunol Rev (2019) 291:154–73. doi: 10.1111/imr.12790
44. Houen G, Trier NH. Epstein-Barr Virus and systemic autoimmune diseases. Front Immunol (2021) 11:587380. doi: 10.3389/fimmu.2020.587380
45. Schworer SA, Francis OL, Johnson SM, Smith BD, Gold SH, Smitherman AB, et al. Autoimmune cytopenia as an early and initial presenting manifestation in activated PI3 kinase delta syndrome (APDS): case report and review. J Pediatr Hematol Oncol (2021) 43:281–7. doi: 10.1097/MPH.0000000000002214
46. Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet (2013) 93:150–7. doi: 10.1016/j.ajhg.2013.05.023
47. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34:272–6. doi: 10.1007/s10875-014-0012-9
48. Tsujita Y, Mitsui-sekinaka K, Imai K, Yeh T. Phosphatase and tensin homolog ( PTEN ) mutation can cause activated phosphatidylinositol 3-kinase d syndrome – like immunodeficiency + immunode fi ciency. J Allergy Clin Immunol (2016) 138:1672–1680.e10. doi: 10.1016/j.jaci.2016.03.055
49. Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. p110δ, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U.S.A. (1997) 94:4330–5. doi: 10.1073/pnas.94.9.4330
50. Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, et al. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity (2006) 25:545–57. doi: 10.1016/j.immuni.2006.08.015
52. Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, Jadidi-Niaragh F, et al. Clinical, immunological, and genetic features in patients with activated PI3Kδ syndrome (APDS): a systematic review. Clin Rev Allergy Immunol (2020) 59:323–33. doi: 10.1007/s12016-019-08738-9
53. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol (2012) 13:195–203. doi: 10.1038/nrm3290
54. Heffron TP, Heald RA, Ndubaku C, Wei B, Augistin M, Do S, et al. The rational design of selective benzoxazepin inhibitors of the α-isoform of phosphoinositide 3-kinase culminating in the identification of ( s )-2-((2-(1-Isopropyl-1 h -1,2,4-triazol-5-yl)-5,6-dihydrobenzo[ f ]imidazo[1,2- d ][1,4]oxazepin-9-yl)oxy)propa. J Med Chem (2016) 59(3):985–1002. doi: 10.1021/acs.jmedchem.5b01483
55. Lucas CL, Chandra A, Nejentsev S, Condliffe AM. PI3K δ and primary immunodeficiencies. (2017) Nat Rev Immunol 16:702–14. doi: 10.1038/nri.2016.93.PI3K
56. Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci (2015) 40:88–100. doi: 10.1016/j.tibs.2014.12.003
57. Ryan P, Redenbaugh V, McGucken J, Kindle G, Devlin LA, Coulter T, et al. Inborn errors of immunity on the island of Ireland — a cross-jurisdictional UKPID/ESID registry report. J Clin Immunol (2022) 42:1293–9. doi: 10.1007/s10875-022-01274-w
58. Mahlaoui N, Jais JP, Brosselin P, Mignot C, Beaurain B, Brito C, et al. Prevalence of primary immunodeficiencies in France is underestimated. J Allergy Clin Immunol (2017) 140:1731–3. doi: 10.1016/j.jaci.2017.06.020
59. Ehl S, Farmand S. Primäre Immundefekte: Wenn ein Infekt auf den anderen folgt. Aerzteblatt (2019) 116:27–8.
60. Proimmun e.V. Unterstützung des PID-NET registers 2021/2022 (2021). Available at: https://www.proimmun.uni-freiburg.de/unterstuetzung-des-pid-net-register-2021/ (Accessed March 15, 2023).
61. El-Helou SM, Biegner AK, Bode S, Ehl SR, Heeg M, Maccari ME, et al. The German national registry of primary immunodeficiencies (2012–2017). Front Immunol (2019) 10:1272. doi: 10.3389/fimmu.2019.01272
62. Lehman H, Hernandez-Trujillo V, Ballow M. Diagnosing primary immunodeficiency: a practical approach for the non-immunologist. Curr Med Res Opin (2015) 31:697–706. doi: 10.1185/03007995.2014.1001063
63. Modell V. The impact of physician education and public awareness on early diagnosis of primary immunodeficiencies: Robert a. Good Immunol Symposium. Immunol Res (2007) 38:43–7. doi: 10.1007/s12026-007-0048-5
64. Tavakol M, Jamee M, Azizi G, Sadri H, Bagheri Y, Zaki-Dizaji M, et al. Diagnostic approach to the patients with suspected primary immunodeficiency. Endocr Metab Immune Disord Drug Targets (2020) 20:157–71. doi: 10.2174/1871530319666190828125316
65. Slade CA, Bosco JJ, Giang TB, Kruse E, Stirling RG, Cameron PU, et al. Delayed diagnosis and complications of predominantly antibody deficiencies in a cohort of Australian adults. Front Immunol (2018) 9:694. doi: 10.3389/fimmu.2018.00694
66. Cabañero-Navalon MD, Garcia-Bustos V, Nuñez-Beltran M, Císcar Fernández P, Mateu L, Solanich X, et al. Current clinical spectrum of common variable immunodeficiency in Spain: the multicentric nationwide GTEM-SEMI-CVID registry. Front Immunol (2022) 13:1033666. doi: 10.3389/fimmu.2022.1033666
67. Ärztekammer D. Ärztestatistik zum 31. December 2021. (2021). Available at: https://www.bundesaerztekammer.de/baek/ueber-uns/aerztestatistik/2021 (accessed March 2023)
68. UK GMC. GNC UK report on medical specialists (2023). Available at: https://data.gmc-uk.org/gmcdata/home/#/reports/TheRegister/Stats/report (Accessed March 15, 2023).
69. Farmand S, Baumann U, von Bernuth H, Borte M, Borte S, Boztug K, et al. Leitlinie “Diagnostik auf vorliegen eines primären immundefekts.” (2017). AWMF-Reg-Nr 112/001 Klasse S2k. Available at: https://www.awmf.org/leitlinien/detail/ll/112-001.html (Accessed March 15, 2023).
70. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol (2014) 136:1186–1205.e78. doi: 10.1016/j.jaci.2015.04.049
71. Cordero E, Goycochea-Valdivia W, Mendez-Echevarria A, Allende LM, Alsina L, Bravo García-Morato M, et al. Executive summary of the consensus document on the diagnosis and management of patients with primary immunodeficiencies. J Allergy Clin Immunol Pract (2020) 8:3342–7. doi: 10.1016/j.jaip.2020.05.008
72. Taylor A, Alloub Z, Tayoun AA. A simple practical guide to genomic diagnostics in a pediatric setting. Genes (Basel) (2021) 12:818. doi: 10.3390/genes12060818
73. Jou ST, Chien YH, Yang YH, Wang TC, Shyur SD, Chou CC, et al. Identification of variations in the human phosphoinositide 3-kinase p110δ gene in children with primary b-cell immunodeficiency of unknown aetiology. Int J Immunogenet (2006) 33:361–9. doi: 10.1111/j.1744-313X.2006.00627.x
74. Piñero J, Saüch J, Sanz F, Furlong LI. The DisGeNET cytoscape app: exploring and visualizing disease genomics data. Comput Struct Biotechnol J (2021) 19:2960–7. doi: 10.1016/j.csbj.2021.05.015
75. Ehrhart F, Willighagen EL, Kutmon M, Van Hoften M, Curfs LMG, Evelo CT. A resource to explore the discovery of rare diseases and their causative genes. Sci Data (2021) 8:124. doi: 10.1038/s41597-021-00905-y
76. Bienstock RJ. Data sharing advances rare and neglected disease clinical research and treatments. ACS Pharmacol Transl Sci (2019) 2:491–6. doi: 10.1021/acsptsci.9b00034
77. Iwenofu OH, Lackman RD, Staddon AP, Goodwin DG, Haupt HM, Brooks JSJ. Phospho-S6 ribosomal protein: a potential new predictive sarcoma marker for targeted mTOR therapy. Mod Pathol (2008) 21:231–7. doi: 10.1038/modpathol.3800995
78. Filion CA, Taylor-Black S, Maglione PJ, Radigan L, Cunningham-Rundles C. Differentiation of common variable immunodeficiency from IgG deficiency. J Allergy Clin Immunol Pract (2019) 7:1277–84. doi: 10.1016/j.jaip.2018.12.004
79. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190:1–7. doi: 10.1111/cei.12997
80. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
81. Vogel TP, Leiding JW, Cooper MA, Forbes Satter LR. STAT3 gain-of-function syndrome. Front Pediatr (2023) 9:770077(10). doi: 10.3389/fped.2022.770077
82. Tessarin G, Rossi S, Baronio M, Gazzurelli L, Colpani M, Benvenuto A, et al. Activated phosphoinositide 3-kinase delta syndrome 1: clinical and immunological data from an italian cohort of patients. J Clin Med (2020) 9:1–16. doi: 10.3390/jcm9103335
83. Wang Y, Yang Q, Chen X, Tang W, Zhou L, Chen Z, et al. Phenotypic characterization of patients with activated PI3K d syndrome 1 presenting with features of systemic lupus erythematosus. Genes Dis (2021) 8(6):907–17. doi: 10.1016/j.gendis.2020.04.012
84. Kang J, Kim SK, Kim D, Choi SR, Lim YJ, Kim SK, et al. Successful sirolimus treatment for Korean patients with activated phosphoinositide 3-kinase δ syndrome 1: the first case series in Korea. Yonsei Med J (2020) 61:542–6. doi: 10.3349/ymj.2020.61.6.542
85. Okano T, Imai K, Tsujita Y, Mitsuiki N, Yoshida K, Kamae C, et al. Hematopoietic stem cell transplantation for progressive combined immunodeficiency and lymphoproliferation in patients with activated phosphatidylinositol-3-OH kinase δ syndrome type 1. J Allergy Clin Immunol (2019) 143:266–75. doi: 10.1016/j.jaci.2018.04.032
86. Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol (2017) 139:1046–9. doi: 10.1016/j.jaci.2016.09.040
87. Dimitrova D, Nademi Z, Maccari ME, Ehl S, Uzel G, Tomoda T, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol (2022) 149:410–421.e7. doi: 10.1016/j.jaci.2021.04.036
88. Bakunina K, Markovitch B, Albert MH, Slatter MA, Gennery AR, Tayfun G, et al. Hematopoietic stem cell transplantation for wiskott-Aldrich syndrome: an EBMT inborn errors working party analysis. Blood (2022) 139(13):2066–79. doi: 10.1182/blood.2021014687
89. Dominguez-Villar M. PI3K and AKT isoforms in immunity. Curr Topics Microbiol Immunol (2022) 436:158. doi: 10.1007/978-3-031-06566-8
90. Stark AK, Chandra A, Chakraborty K, Alam R, Carbonaro V, Clark J, et al. PI3Kδ hyper-activation promotes development of b cells that exacerbate streptococcus pneumoniae infection in an antibody-independent manner. Nat Commun (2018) 9(1):3174. doi: 10.1038/s41467-018-05674-8
91. Begg M, Amour A, Jarvis E, Tang T, Franco SS, Want A, et al. An open label trial of nemiralisib, an inhaled PI3 kinase delta inhibitor for the treatment of activated PI3 kinase delta syndrome. Pulm Pharmacol Ther (2023) 79:102201. doi: 10.1016/j.pupt.2023.102201
92. Diaz N, Juarez M, Cancrini C, Heeg M, Soler-Palacín P, Payne A, et al. Seletalisib for activated PI3Kδ syndromes: open-label phase 1b and extension studies. J Immunol (2020) 205:2979–87. doi: 10.4049/jimmunol.2000326
93. Rao VK, Webster S, Šedivá A, Plebani A, Schuetz C, Shcherbina A, et al. Randomized, placebo-controlled, phase 3 trial of PI3Kδ inhibitor leniolisib for activated PI3Kδ syndrome. Blood (2022) 141:971–83. doi: 10.1182/blood.2022018546
94. Rao VK, Webster S, Šedivá A, Plebani A, Schuetz C, Shcherbina A, et al. Interim analysis of safety and hematological parameters of an ongoing long-term open-label extension study of investigational PI3Kδ inhibitor leniolisib for patients with activated PI3K delta syndrome (APDS) through December 2021, in: 64th American Society of Hematology Annual Meeting and Exposition. (2022) Dec 10-13. pp. 1643–5, Blood 140. doi: 10.1182/blood-2022-160334
95. Food and Drug Administration. Approval of leniolisib in the USA (2023). Available at: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-activated-phosphoinositide-3-kinase-delta-syndrome (Accessed March 15, 2023).
Keywords: APDS, IEI, inborn errors of immunity, targeted therapy, stem cell transplantation, rare disease, PID, primary immunodeficiency
Citation: Vanselow S, Wahn V and Schuetz C (2023) Activated PI3Kδ syndrome – reviewing challenges in diagnosis and treatment. Front. Immunol. 14:1208567. doi: 10.3389/fimmu.2023.1208567
Received: 19 April 2023; Accepted: 04 July 2023;
Published: 20 July 2023.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Eleonora Gambineri, University of Florence, ItalyAnita Chandra, University of Cambridge, United Kingdom
Copyright © 2023 Vanselow, Wahn and Schuetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Vanselow, dmFuc2Vsb3dAaW5maWxsLmNvbQ==