 Bimei Song
Bimei Song Yue Zeng
Yue Zeng Yaping Pan
Yaping Pan Jingbo Liu
Jingbo Liu- Department of Periodontics, School of Stomatology, China Medical University, Shenyang, China
Despite improvements in modern medical therapies, inflammatory diseases, such as atherosclerosis, diabetes, non-alcoholic fatty liver, chronic kidney diseases, and autoimmune diseases have high incidence rates, still threaten human health, and represent a huge financial burden. N6-methyladenosine (m6A) modification of RNA contributes to the pathogenesis of various diseases. As the most widely discussed m6A methyltransferase, the pathogenic role of METTL3 in inflammatory diseases has become a research hotspot, but there has been no comprehensive review of the topic. Here, we summarize the expression changes, modified target genes, and pathogenesis related to METTL3 in cardiovascular, metabolic, degenerative, immune, and infectious diseases, as well as tumors. In addition to epithelial cells, endothelial cells, and fibroblasts, METTL3 also regulates the function of inflammation-related immune cells, including macrophages, neutrophils, dendritic cells, Th17 cells, and NK cells. Regarding therapeutic applications, METTL3 serves as a target for the treatment of inflammatory diseases with natural plant drug components, such as emodin, cinnamaldehyde, total flavonoids of Abelmoschus manihot, and resveratrol. This review focuses on recent advances in the initiation, development, and therapeutic application of METTL3 in inflammatory diseases. Knowledge of the specific regulatory mechanisms involving METTL3 can help to deepen understanding of inflammatory diseases and lay the foundation for the development of precisely targeted drugs to address inflammatory processes.
1 Introduction
Inflammation is a protective homeostatic response, characterized by the activation of multiple immune and non-immune cells to eliminate harmful stimuli and promote tissue repair (1–3). At the cellular and molecular level, inflammation can be understood as a sophisticated regulatory network, composed of inducers, sensors, mediators, and effectors (2). Inducers can be divided into exogenous and endogenous inducers, where exogenous inducers include microbes and microbial products, allergens, foreign bodies, and toxic compounds (4), and endogenous inducers are derived from stressed or dead cells and damaged or dysfunctional tissues (2). Inducers can be detected by transmembrane or cytoplasmic sensors, such as Toll-like receptors (TLRs), C-type lectin receptors, retinoic acid-inducible gene (RIG)-I-like receptors, and NOD-like receptors (NLRs) (5), and trigger inflammatory mediators, including vasoactive amines, vasoactive peptides, lipid mediators, cytokines, chemokines, and proteolytic enzymes (2). These inflammatory mediators eventually affect the functional activities of effectors, including monocytes, macrophages, neutrophils, dendritic cells (DCs), lymphocytes, and mast cells, which are immune cells, as well as non-immune epithelial cells, fibroblasts, and endothelial cells (6).
Timely shutdown of inflammation is crucial for physiological homeostasis (6), and dysregulation of this process leads to chronic inflammation (3, 7). Although the regulatory mechanisms underlying chronic inflammation are not completely clear, it is established as associated with aging, chronic infection, unhealthy lifestyle, diet, and adverse environmental conditions (8). At the molecular level, chronic inflammation is usually triggered by endogenous danger signals (9) and causes tissue dysregulation, subsequently leading to various chronic inflammatory diseases, including cardiovascular diseases, type 2 diabetes, non-alcoholic fatty liver, rheumatoid arthritis, and neurodegenerative diseases, among others (3, 8, 10).
Recent studies indicate that RNA modification of N6-methyladenosine (m6A) is critical for inflammation (11); m6A describes methylation of internal adenosine residues in mRNA and is the most prevalent internal post-transcriptional modification of eukaryotic mRNA (12). m6A modification preferentially occurs at the consensus sequence, RRACH (R: purine, A: m6A site, H: non-guanine base), in mRNA (13, 14), and is preferentially located around stop codons, and enriched at 3’ untranslated regions and within long internal exons in human transcriptomes (15). The m6A methylation process is reversible and dynamically regulated by methyltransferases (writers), demethylases (erasers), and m6A-binding proteins (readers) (16). Methyltransferases (writers) add m6A to mRNA, and include METTL3 (17, 18), METTL14 (18), METTL16 (19), WTAP (20), KIAA1429 (21), RBM15 (22), and ZC3H13 (23). Further, m6A modification is removed by demethylases (erasers), including FTO (24) and ALKBH5 (25), while m6A-modified transcripts are recognized by m6A-binding proteins (readers), such as YTHDC1/2 (26, 27), YTHDF1/2/3 (28–30), HNRNPA2B1 (31), HNRNPC (32), and IGF2BP1/2/3 (33). Readers can bind to the m6A motif and affect mRNA metabolism, including mRNA splicing and export from the nucleus, stability, translation, and localization in the cytoplasm, thereby regulating gene expression at the post-transcriptional level (16).
METTL3 was the first m6A methyltransferase to be discovered. In 1997, Boker et al. isolated, purified, and sequenced METTL3 (also referred to as MT-A70) from HeLa cells, and confirmed that it is an important (N6-adenosine)-methyltransferase subunit (17). Together, METTL3, METTL14, and WTAP form the m6A methyltransferase complex (34), in which METTL3 is the core component that provides catalytic activity, while METTL14 functions as an RNA binding platform and WTAP is responsible for recruiting the complex to RNA targets (20, 35, 36). METTL3 functions in various pathophysiological processes, including the cell cycle, apoptosis, innate immunity, and inflammation, as well as cell proliferation, migration, invasion, and differentiation (37). As the m6A methylase most widely discussed in the context of inflammation and inflammatory diseases, the role of METTL3 in inflammatory diseases has not been systematically elucidated.
In this review, we describe the role of METTL3 in macrophages, neutrophils, DCs, T helper (Th)17 cells, and natural killer (NK) cells which play crucial pathological roles in chronic inflammation. We comprehensively summarize METTL3 expression changes and related pathogenesis in inflammatory diseases, including atherosclerosis, myocardial infarction (MI), non-alcoholic liver disease, hyperuricemia (HUA), diabetes nephropathy, osteoarthritis (OA), intervertebral disc degeneration (IDD), rheumatoid arthritis, psoriasis, inflammatory bowel disease (IBD), sepsis, pneumonia, and tumor. In conclusion, we focus on the therapeutic application of METTL3 in inflammatory diseases. Knowledge of the mechanisms by which METTL3 regulates inflammatory diseases will contribute to the development of improved targeted therapies and broaden clinical perspectives.
2 METTL3 regulates inflammation-related immune cell functions
2.1 METTL3 regulates macrophage function
Inflammation involves multiple immune cells, among which macrophages are key participants. Macrophages/monocytes are derived from progenitor cells in the bone marrow and enter the bloodstream (38). After being recruited, these cells can enter tissue through the vascular endothelium and become tissue-resident macrophages. Macrophages are involved in all aspects of the physiological and pathological processes of inflammation, including the perception of stimuli, phagocytosis of pathogens, removal of cell debris, presentation of antigens, secretion of cytokines, and promotion of tissue repair.
Macrophages are plastic and, when stimulated by the microenvironment, can polarize into different phenotypes, which are generally divided into classic activated or inflammatory (M1) and alternate activated or anti-inflammatory (M2) macrophages (39). M1 macrophages are activated by IFN-γ and TNF-α, or by lipopolysaccharide (LPS). They secret proinflammatory cytokines, such as TNF-α, IL-1α, IL1-β, IL-6, IL-12, IL-23, nitric oxide (NO), and cyclooxygenase-2 and produce reactive oxygen species (ROS). Functionally, M1 macrophages can remove pathogens and can also contribute to tissue damage (40). M2 macrophages are activated by IL-4, IL-13, and IL-10, and secrete IL-10 and TGF-β. M2 macrophages can clear apoptotic cells, promote angiogenesis and fibrosis, and coordinate tissue remodeling (41). Macrophages play a defensive role in acute inflammation but have strong pathological effects in chronic inflammation and inflammatory diseases (42, 43), and macrophage polarization is the pathological basis of many chronic diseases driven by inflammation (39, 40).
Recent studies have indicated that METTL3 can participate in regulating macrophage-mediated inflammation and M1 polarization (Figure 1). Li et al. found that m6A methylation and METTL3 levels were increased in macrophages stimulated by oxidized low-density lipoprotein (oxLDL) (44). Mechanistically, METTL3 promoted signal transducer and activator of transcription 1 (STAT1) expression by increasing the m6A level of the STAT1 transcript in macrophages, thereby elevating the levels of downstream inflammatory factors (44). In another study, Sun et al. found that, although oxLDL could mildly promote expression levels of METTL3 and METTL14, it decreased interaction between METTL3 and METTL14, leading to a reduction in overall m6A levels in macrophages (45). Further, Sun et al. found that Matrin-3 was reduced in macrophages stimulated by oxLDL, and involved in the formation of the METTL3-METTL14 complex, thus affecting its m6A modification of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) mRNA (45). METTL3 was proven to co-modify PGC-1 mRNA with YTHDF2 and inhibit PGC-1 expression, which reduced ATP production and increased ROS accumulation, eventually aggravating inflammatory responses (46). Guo found that circ_0029589 expression was down-regulated in peripheral blood mononuclear cell-derived macrophages from patients with acute coronary syndrome and that IRF-1 inhibited circ_0029589 through METTL3-mediated m6A modification, thereby facilitating macrophage pyroptosis and inflammation (47).
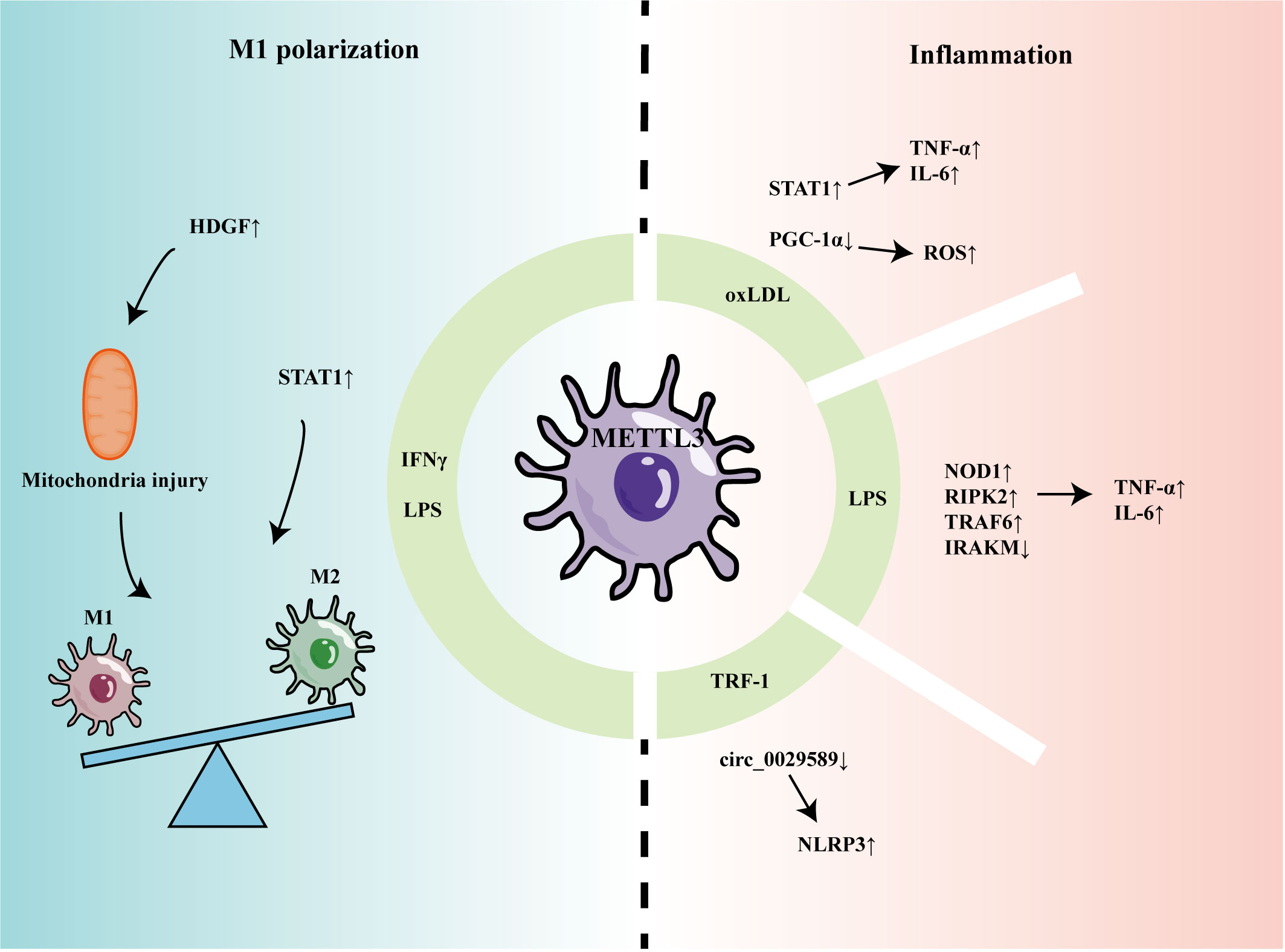
Figure 1 The role of METTL3 in regulating macrophage inflammation and polarization.
In another report of macrophage stimulation using LPS, levels of METTL3 and m6A modification were decreased (48). METTL3 knockdown promoted the stability of nucleotide-binding oligomerization domain containing 1 (NOD1) and receptor-interacting serine/threonine kinase 2 (RIPK2) mRNA, mediated by YTHDF2, thereby up-regulating levels of TNF-α, IL-6, and NO (48); however, Tong et al. found that METTL3-deficient macrophages produced less TNF-α and that METTL3 knockout mice had higher bacterial loads in the feces and cecum, indicating that METTL3 promotes the anti-infection activity of macrophages (49). Mechanistically, METTL3 deficiency reduced Irakm (a negative regulatory factor in the TLR4 signaling pathway) mRNA degradation and promoted its expression. Microglia are often referred to as tissue-resident macrophages of the central nervous system (50). When damage or infection is detected, microglia can transform into an activated phenotype with inflammatory functions, which is important in a variety of neuropathological conditions. METTL3 was increased in LPS-stimulated microglia and promoted TRAF6/NF-κB pathway expression by m6A modification of TRAF6, thereby inducing expression of inflammatory cytokines, including IL-1β, IL-6, TNF-α, and IL-18 (51).
METTL3 also promotes proinflammatory M1 polarization. METTL3 was increased in mouse bone marrow-derived M1 macrophages and methylated the mRNA encoding STAT1, a transcription factor that regulated M1 macrophage polarization, promoting its expression (52). Further, METTL3 promoted M1 polarization by regulating macrophage metabolism. METTL3 could upregulate hepatoma-derived growth factor (HDGF) expression in macrophages by enhancing its mRNA stability (53). Pro-inflammatory M1 macrophages mainly rely on the glycolysis pathway to generate energy (54), while HDGF promotes M1 macrophage polarization by inducing mitochondrial injury and thereby stimulating glycolysis (53).
2.2 METTL3 regulates neutrophil activation
Neutrophils are the first innate immune cells recruited to sites of infection. The anti-infection ability of neutrophils depends on migration, phagocytosis, degranulation, and formation of neutrophil extracellular traps (NETs). Neutrophils were considered to mainly be involved in acute inflammation, but it is now widely believed that neutrophils also have important roles in cancer, autoimmune diseases, and chronic inflammation (55). METTL3 levels were increased in bone marrow neutrophils from LPS-treated C57BL/6 mice and affected neutrophil migration, while METTL3 knockout reduced the release of both bone marrow and circulating neutrophils (56). Mechanistically, METTL3 mediated m6A modification of TLR4 mRNA, thus enhancing its expression and stimulating TLR4 pathway activation, leading to increased cytokine production and promoting CXCR2 expression (56).
2.3 METTL3 regulates DC activation
DCs are heterogeneous antigen-presenting cells that connect innate and adaptive immunity. DCs mediate immune defense and immune tolerance and disordered DC migration or activation can have pathological consequences associated with autoimmune and infectious diseases (57). METTL3 is involved in regulating DC activation. m6A modification and METTL3 levels were increased in mature bone marrow DCs from C57BL/6 mice (58), while METTL3 knockout could inhibit DC maturation and cytokine production (58, 59). METTL3 increased CD40 and CD80 mRNA m6A modification levels, thereby promoting CD40 and CD80 expression (58); these two membrane co-stimulatory molecules assist in antigen presentation and T-cell activation. In addition, METTL3 promoted Tirap expression by increasing its m6A modification level, which enhanced the activity of the TLR4/NF-κB signaling pathway and cytokine secretion (58).
2.4 METTL3 regulates Th17 cell function
Among Th cell subsets, Th17 cells are considered inflammatory, and their differentiation and pathogenic functions are related to various inflammatory and autoimmune diseases, such as psoriasis and rheumatoid arthritis (60). Particular cytokines and environmental stimuli induce Th17 cell differentiation and secretion of the pathogenic cytokine, IL-17 (61, 62). METTL3 was recently identified as involved in regulating the pathogenic function of Th17 cells. METTL3 and m6A modification were reduced in T cells from mice with experimental autoimmune uveitis (63), and METTL3 overexpression inhibited the differentiation of Th17 phenotype cells and reduced IL-17 secretion (63). Mechanistically, METTL3 increased ASH1L mRNA stability, which negatively regulates Th17 cell generation and function (63). In addition, elevated METTL3 and m6A modifications were found in bone samples from patients with periprosthetic joint infection (64); KEGG analysis demonstrated that differentially methylated genes are mainly enriched in Th17 cell differentiation and the IL-17 signaling pathway, indicating that Th17 cells and METTL3 may contribute to the pathogenesis of periprosthetic joint inflammation (64).
2.5 METTL3 regulates NK cell function
NK cells play an important role in virus and tumor immunity. NK cells can sense infected and tumor cells through natural cytotoxicity receptors, C-type lectin-like receptors, and co-activating receptors (65). Upon activation, NK cells release lytic granules and cytokines, and collaborate with other immune cells to kill target cells (66). In addition, NK cells can also activate death ligands, which induce target cell apoptosis (67). Recently, METTL3 has been reported to be associated with the function of infiltrating NK cells in tumors. Song et al. found that METTL3 was decreased in NK cells from patients with hepatocellular carcinoma (68). METTL3 knockdown inhibited NK cell infiltration and reactivity to IL-15, which is related to suppressed activation of AKT and MAPK pathways (68).
3 The role of METTL3 in inflammatory diseases
METTL3 plays important roles in both inflammation-related immune cells and other cell types, such as epithelial cells, endothelial cells, and fibroblasts. METTL3 expression levels can change under disease conditions, influencing expression of downstream target genes, and thereby participating in inflammatory disease pathogenesis. In the following sections, we elaborate on the mechanism of action of METTL3 and its effects on m6A reading proteins in cardiovascular, metabolic, degenerative, immune, infectious diseases, and tumors. The effects of METTL3 and the m6A reading proteins discussed in this review on RNA modification and metabolism are summarized in Figure 2. Expression changes and pathogenic mechanisms of METTL3 in various inflammatory diseases are shown in Tables 1, 2.
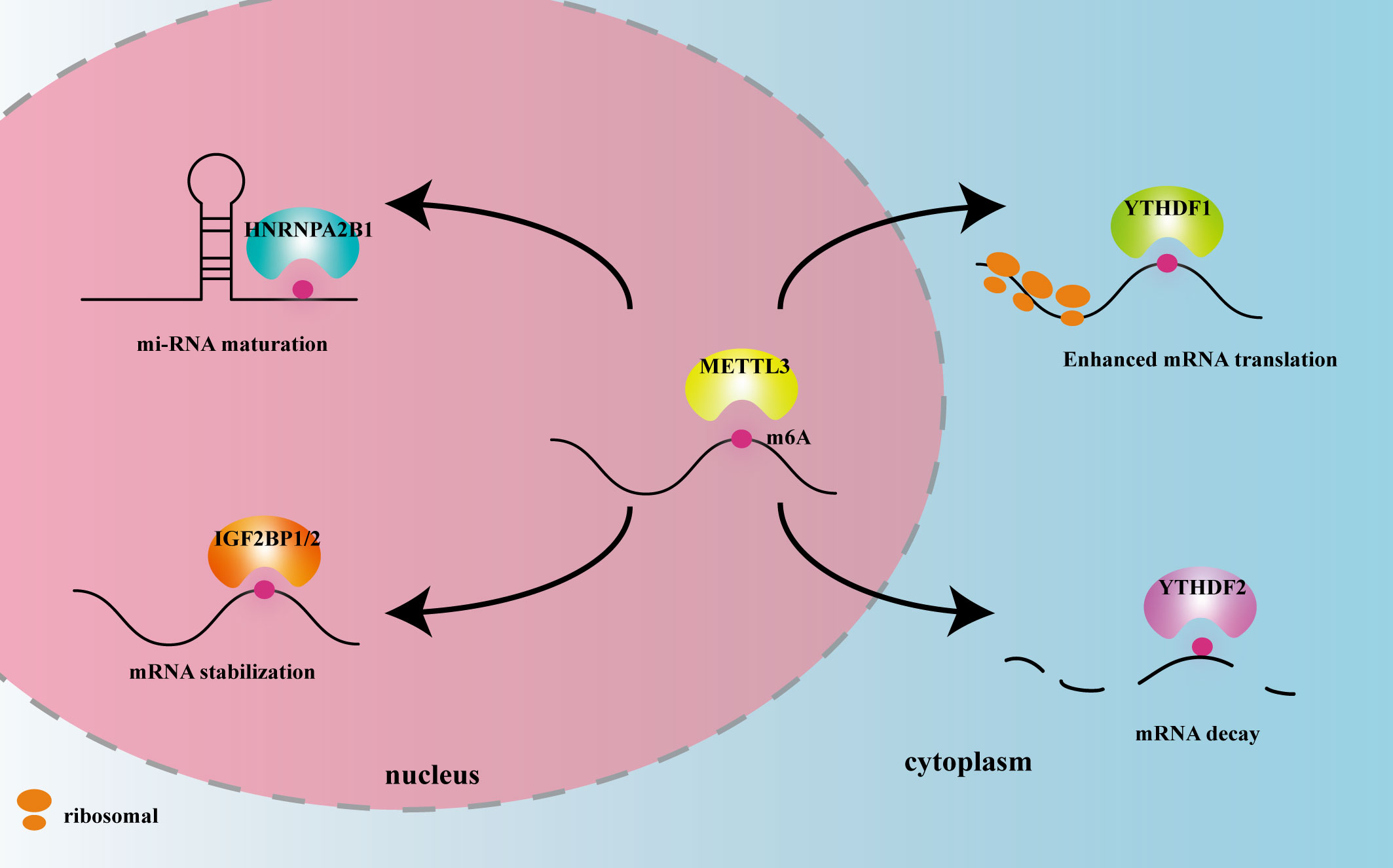
Figure 2 The effects of METTL3 and m6A reader proteins mentioned in this review on RNA modification and metabolism. Figure Legends: METTL3 adds the m6A motif to RNA. HNRNPA2B1 recognizes m6A in the nucleus and promotes miRNA maturation. IGF2BP1/2 recognizes m6A in the nucleus and promotes mRNA stability. YTHDF1 recognizes m6A in the cytoplasm and enhances mRNA translation. YTHDF2 recognizes m6A in the cytoplasm and promotes mRNA decay.
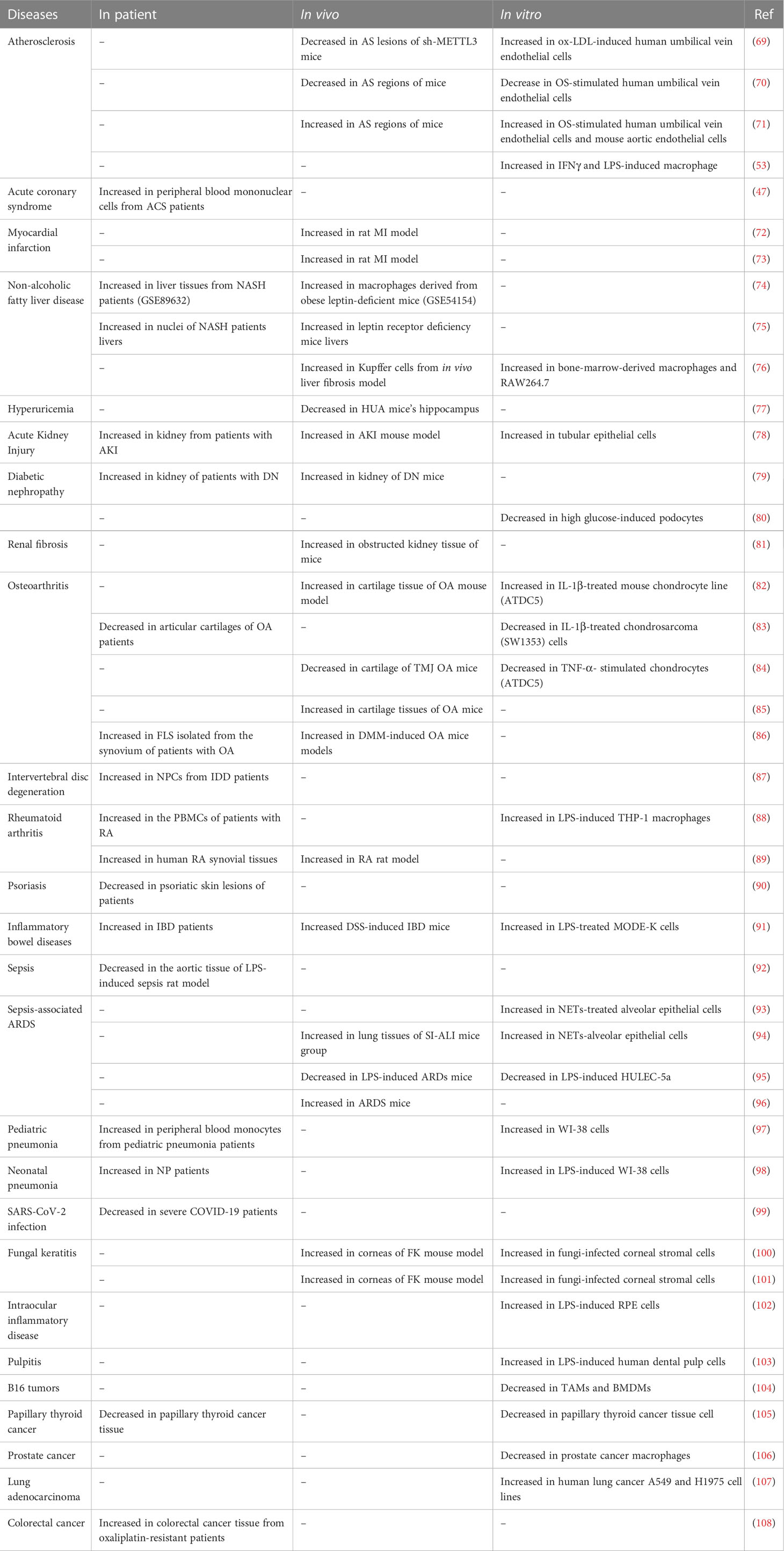
Table 1 Changes of METTL3 expression in inflammatory diseases.
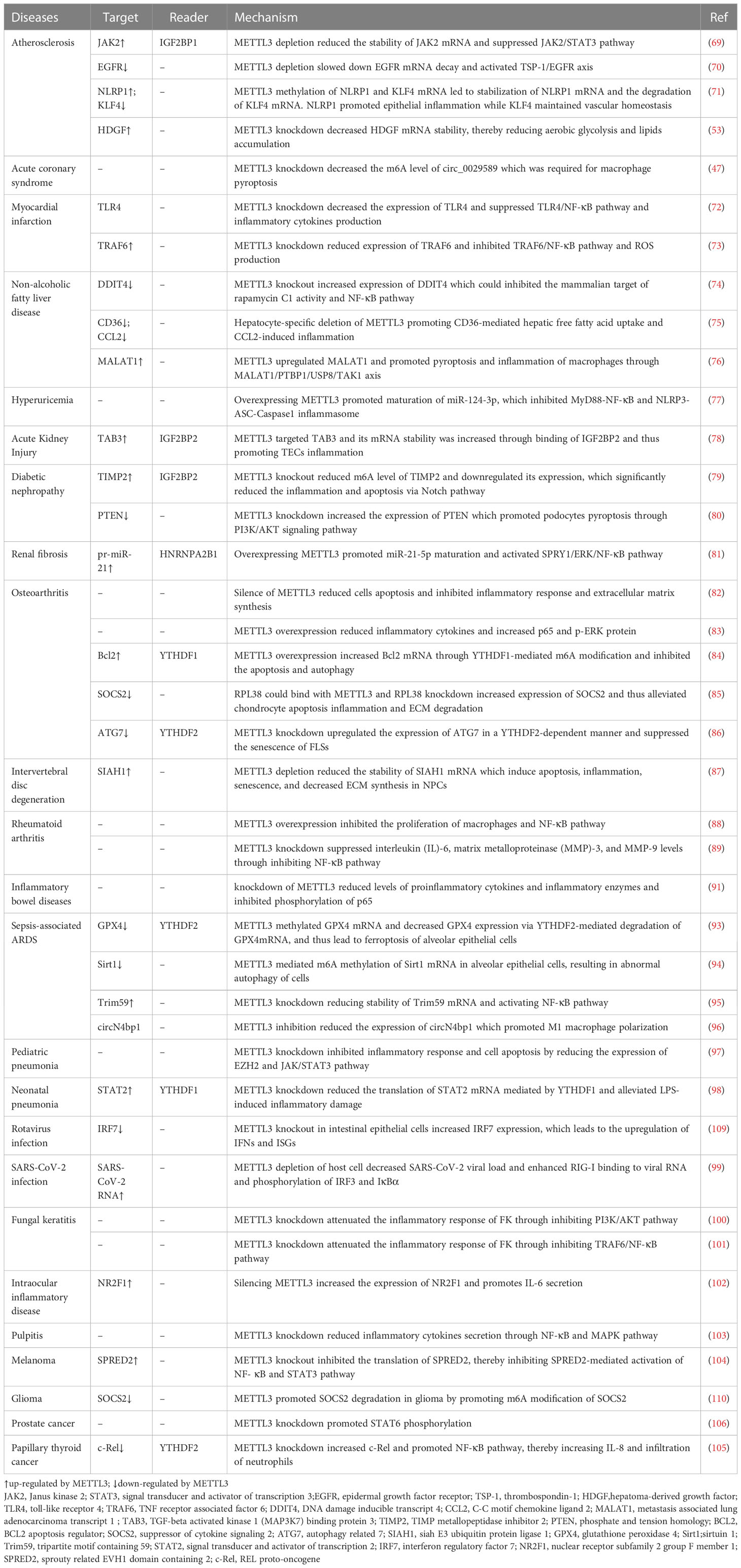
Table 2 METTL3-associated pathogenesis in inflammatory diseases.
3.1 Cardiovascular disease
3.1.1 Atherosclerosis
Atherosclerosis is an immunoinflammatory disease featuring the formation of lipid plaques in the intima of arteries, resulting in lumen stenosis (111). The process of atherosclerosis development includes the proliferation of smooth muscle cells and monocytes, increased synthesis and secretion of collagen fibers by smooth muscle cells, and lipid accumulation. Dong et al. found that METTL3 was up-regulated in ox-LDL-induced human umbilical vein endothelial cells and METTL3 knockdown prevented the development of atherosclerosis (69). Functionally, METTL3 m6A modifies JAK2 mRNA and IGF2BP1 enhances JAK2 mRNA stability, while METTL3 knockdown inhibits cell proliferation and angiogenesis by inhibiting the JAK2/STAT3 pathway (69). Stenosis caused by an atherosclerosis plaque will affect blood flow and generate oscillatory stress (OS) in the plaque area. OS causes epithelial activation and deterioration of atherosclerosis. Li et al. found that METTL3 expression was decreased in atherosclerotic regions with OS, leading to reduced m6A modification of epidermal growth factor receptor (EGFR), causing slower EGFR mRNA decay and increased EGFR expression (70). EGFR is associated with atherosclerosis progression (70); however, Chien et al. found that METTL3 was up-regulated under OS (71). METTL3 methylation stabilizes NLRP1 and leads to the degradation of KLF4 mRNA, where NLRP1 promotes epithelial inflammation and KLF4 maintains vascular homeostasis (71). Zheng et al. explored the pathogenesis of atherosclerosis from the perspective of macrophage polarization and energy metabolism (53) and found that HDGF was highly expressed in the aortas of patients with atherosclerosis and APOE knockout mice, as well as in M1 macrophages. HDGF knockdown alleviated inflammation, glycolysis, and lipid accumulation in M1 macrophages, while high METTL3 expression in macrophages increased HDGF levels through m6A modification, thereby aggravating atherosclerosis.
3.1.2 Myocardial infarction
Microglia inflammation in the paraventricular nucleus after MI results in excessive activation of the sympathetic nerve, leading to ventricular arrhythmia in patients with MI. Patients with MI have high mortality rates from sudden cardiac death caused by ventricular arrhythmia. Increased METTL3 elevates m6A modification of TLR4 and up-regulates its expression, which promotes sympathetic hyperactivity via the TLR4/NF-κB pathway and increases the incidence of ventricular arrhythmias post-MI (72). In another study, Qi et al. found that METTL3 expression increased in rats after MI, and METTL3 targeted TRAF6 and promoted inflammation after MI, through the TRAF6/NF-κB pathway (73).
3.2 Metabolic disease
3.2.1 Non-alcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD) describes conditions ranging from steatosis and non-alcoholic steatohepatitis (NASH), which is characterized by lobular inflammation and hepatocellular injury, through to cirrhosis and hepatocellular carcinoma, as the disease progresses (112). Liver-specific METTL3 knockout mice exhibit pathological features associated with NAFLD (113). Yet, in another study, myeloid lineage-restricted deletion of METTL3 protected against NAFLD development (74). The liver of myeloid-specific METTL3 knockout mice showed lower levels of lipid accumulation and monocyte infiltration, as well as low NAFLD activity inflammation scores (74). METTL3 appears to hinder NASH development. Li et al. showed that METTL3 overexpression alleviated NASH by suppressing the expression of CD36 and C-C motif chemokine ligand 2 (CCL2) (75); however, METTL3 regulated CD36 and CCL2 expression at the transcriptional level, rather than through m6A modification. METTL3 inhibited CD36 and CCL2 transcription by recruiting histone deacetylase 1/2 to induce deacetylation of H3K9 and H3K27 in the CD36 and CCL2 promoter regions. In contrast, in the liver fibrosis stage of NAFLD, METTL3 may become a promoter of disease. Shu et al. found that METTL3 was up-regulated in a liver fibrosis model and in macrophages, and aggravated liver fibrosis by stimulating macrophage pyroptosis and inflammation (76).
3.2.2 Hyperuricemia
HUA is a metabolic disease caused by raised serum uric acid (> 6.5–7 mg/dL) (114). Epidemiological evidence indicates that HUA is related to various cardiovascular diseases (115) and cognitive impairment (116). In an in vivo experiment, levels of inflammatory cytokines in the hippocampus of rats in the HUA group were significantly higher than those in the negative control group (117). Further, in a clinical study, serum proinflammatory cytokines and oxidative stress were increased in asymptomatic young patients with primary HUA, who were in a chronic inflammatory state, which may be the reason underlying systemic disease (118). The latest evidence suggests that METTL3 is a bridge linking HUA and neuroinflammation. Chen et al. found that METTL3 was down-regulated in HUA mice, while METTL3 knockdown aggravated mouse hippocampal neuronal apoptosis and microglia overactivation through the NF-κB and NLRP3 inflammasome pathway, thus aggravating neuroinflammation (77).
3.2.3 Diabetic complication
Type 2 diabetes mellitus results from the combination of two factors: defective insulin secretion by pancreatic β-cells and insulin resistance of peripheral target tissues (119). Diabetic kidney disease is the most common microvascular complication of diabetes. High glucose levels can elevate the expression of transforming growth factor β1, angiotensin II, cytokines, and advanced glycation end products, with adverse effects on a variety of renal cell types, including lipid oxidation, mitochondrial dysfunction, oxidation, endoplasmic reticulum stress, and fibrosis (120). Several epigenetic mechanisms, including DNA methylation, histone modification, and non-coding RNAs, have been implicated in the pathogenesis of diabetic kidney disease in recent years (120), and m6A modification is no exception. METTL3 is elevated in renal tubules from human biopsies and METTL3 silencing alleviated renal inflammation in tubular epithelial cells via the METTL3/TAB3 axis (78). METTL3 is also elevated in podocytes from renal biopsies from patients with diabetic nephropathy, while METTL3 knockout reduced the m6A levels of TIMP metallopeptidase inhibitor 2 (TIMP2) and downregulated its expression, which led to clear alleviation of inflammation and apoptosis via the Notch pathway in podocytes (79). Liu et al. found that m6A modification of phosphate and tension homology (PTEN) in podocytes was decreased under high glucose conditions (80). The underlying mechanism involved a high glucose-stimulated decrease in METTL3 expression in podocytes and decreased m6A modification of PTEN, which increased PTEN expression, and PTEN promoted podocyte pyroptosis through PI3K/Akt signaling. METTL3 contributes to the development of renal fibrosis by promoting inflammation. METTL3 facilitates miR-21-5p maturation, and miR-21-5p activates ERK/NF-κB signaling by suppressing sprouty RTK signaling antagonist 1 (SPRY1), which affects renal fibrosis progression (81).
3.3 Degenerative disease
3.3.1 Osteoarthritis
OA is a chronic joint disease characterized by progressive cartilage degeneration, subchondral bone sclerosis, and synovial inflammation (121). Sang et al. found that METTL3 was decreased in patients with OA (83); however, Liu et al. showed that METTL3 was increased in experimental OA (82). METTL3 silencing reduced chondrocyte apoptosis, decreased inflammatory cytokine secretion and promoted extracellular matrix (ECM) degradation (82). METTL3 overexpression increased BCL2 expression through YTHDF1-mediated m6A modification and inhibited chondrocyte apoptosis and autophagy (84). Ribosomal protein L38 (RPL38) can positively regulate METTL3, while RPL38 knockdown increased levels of suppressor of cytokine signaling 2 (SOCS2) through decreasing its m6A level, mediated by METTL3, thereby reducing inflammation and apoptosis of chondrocytes and inhibiting ECM degradation (85). Chen et al. investigated the role of fibroblast-like synoviocyte (FLS) senescence in the development of OA and found that METTL3 knockdown increased the expression of autophagy-related 7 (ATG7), which inhibited GATA binding protein 4 (GATA4) to ameliorate FLS senescence and OA progression (86).
3.3.2 Intervertebral disc degeneration
IDD is the main cause of back, neck, and radicular pain (122). The incidence rate of IDD rises with age, and its pathogenesis is related to senescence, apoptosis, inflammation levels, and ECM degradation of nucleus pulposus cells (NPCs) (123). Fang et al. found that METTL3 levels were increased in NPCs from patients with IDD (87), while METTL3 depletion resulted in the reduction of siah E3 ubiquitin protein ligase 1 (SIAH1) mRNA stability, which induced apoptosis, inflammation, and senescence, as well as decreasing ECM synthesis in NPCs (87).
3.4 Immune disease
3.4.1 Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease characterized by autoantibody production, synovial inflammation, cartilage damage, and bone erosion (124, 125). The etiology of rheumatoid arthritis is very complex. Current evidence shows that genotype, sex, environmental triggers, periodontal microbes, gastrointestinal microbes, and epigenetic modification are involved in the incidence and development of RA (125–127). Wang et al. found that METTL3 was significantly increased in peripheral blood mononuclear cells from patients with RA, and there were positive correlations between METTL3 levels and the markers of RA activity, C-reactive protein (CRP), and erythrocyte sedimentation rate (ESR) (88). Another in vivo experiment demonstrated that METTL3 was up-regulated in synovial tissue and FLSs from patients with RA and in an adjuvant-induced arthritis rat model (89). METTL3 silencing suppressed FLS proliferation, migration and invasion, and IL-6 expression, possibly by inhibiting the NF-κB pathway (89).
3.4.2 Psoriasis
Psoriasis is a chronic, immune-mediated disorder, which can be provoked by non-specific triggers, such as mild trauma, sunburn, stress, infection (particularly streptococcal), alcohol, or drugs (128). Briefly, triggers stimulate keratinocytes to produce antimicrobial peptides, which in turn activate Th cells to polarize and release IFN-γ, IL-17A/IL-17F, and IL-22; these cytokines result in excessive proliferation and abnormal differentiation of keratinocytes, which release pro-inflammatory cytokines, further aggravating psoriasis (129–131). Recent evidence suggests that m6A epigenetic modification also contributes to the pathogenesis of psoriasis. Lower levels of m6A methylation and METTL3 were detected in psoriatic skin lesions than in healthy human skin samples (90), and levels of m6A methylation and METTL3 were negatively correlated with psoriasis severity (90). Further, compared with mice heterozygous for METTL3 knockout, wild-type control mice showed ameliorated psoriasis-like clinical and pathological manifestations (90). Moreover, KEGG analysis suggests that METTL3 may play a role in the pathogenesis of psoriasis through the WNT pathway (90).
3.4.3 Inflammatory bowel disease
IBD describes chronic inflammatory disorders of the intestine (132). Maialen et al. used an online genome-wide association study and m6A transcription databases to screen five IBD-related single nucleotide polymorphisms in the UBE2L3, SLC22A4, TCF19, C6orf47, and SNAPC4 genes, suggesting that m6A methylation may contribute to IBD pathogenesis (133). Subsequently, METTL3 was found to be elevated in ulcerative colitis tissue from patients with IBD, a dextran sulfate sodium-induced IBD mouse model, and LPS-treated MODE-K cells, while in vivo and in vitro knockdown of METTL3 reduced levels of proinflammatory cytokines and inflammatory enzymes and inhibited p65 phosphorylation (91).
3.5 Infectious diseases
3.5.1 Sepsis
Sepsis is a life-threatening organ dysfunction resulting from dysregulated host responses to infection (134); it can be caused by almost any infectious organism, and the incidence rates of sepsis due to infection by gram-negative and gram-positive bacteria are almost equal (135). The most common infection sites leading to sepsis are the lung, abdomen, blood, kidney, and genitourinary tract (136). According to traditional understanding, sepsis is an overwhelming systemic inflammatory response to infection, accompanied by immunosuppression and secondary infection (137). Sepsis-induced organ dysfunction includes cardiomyopathy, encephalopathy, and cognitive impairment, as well as liver injury, acute kidney injury, and acute respiratory distress syndrome (ARDS). Although many studies have shown that sepsis can cause organ dysfunction by interfering with microcirculation, ischemia, hypoxia, mitophagy, and cell apoptosis, the exact mechanism involved is not fully understood (138).
Aortic injury during sepsis leads to insufficient organ perfusion, but the specific mechanism has not been fully elucidated. Shen et al. found that levels of m6A and METTL3 expression were significantly decreased in aortic tissue from an LPS-induced sepsis rat model (92). According to high-throughput sequencing, KEGG, and GO enrichment analyses, genes affected by m6A modification were related to cation channels, particularly TRP channels, indicating that METTL3 may aggravate vascular endothelial cell inflammation during sepsis by interfering with calcium ion transport (92).
NETs were increased in humans and mice with sepsis-associated ARDS and positively associated with disease severity level, indicating that NETs are harmful in sepsis-associated ARDS (93). Further, Zhang et al. found that NETs upregulated METTL3 expression, leading to methylation of glutathione peroxidase 4 (GPX4) mRNA and decreased GPX4 expression via accelerated degradation of GPX4 mRNA mediated by YTHDF2, with consequent ferroptosis of alveolar epithelial cells (93). Qu et al. showed that NETs could promote METTL3-mediated m6A methylation of sirtuin 1 (SIRT1) mRNA in alveolar epithelial cells, leading to abnormal cell autophagy and deterioration of ARDS (94). Chen et al. found that METTL3 expression was reduced in LPS-induced ARDS mice and LPS-stimulated human pulmonary vascular endothelial cells (95). METTL3 downregulation destroyed endothelial permeability and led to higher inflammatory cytokine expression by reducing the stability of Trim59 mRNA and activating the NF-κB pathway. CircN4bp1 up-regulated the expression of the enhancer of the zeste 2 polycomb repressive complex 2 subunit (EZH2) by combining with miR-138-5p, thus promoting M1 macrophage polarization and aggravating the degree of lung injury in ARDS mice (96). Further, Zhao et al. found that the m6A levels in circN4bp1 were increased in ARDS mice and that the inflammatory response could be blocked by inhibiting METTL3, suggesting that METTL3 might be involved in regulating circN4bp1 expression (96).
3.5.2 Pneumonia
Pneumonia is a common acute respiratory infection caused by many types of microorganisms, including bacteria, respiratory viruses, and fungi, and primarily occurs in children < 5 years old and elderly adults with underlying chronic conditions (139). Yang et al. found that METTL3 was increased in pediatric patients with pneumonia and WI-38 cells, while METTL3 knockdown inhibited the inflammatory response and apoptosis by reducing the expression of EZH2 and the JAK/STAT3 pathway (97). In another study, METTL3 knockdown reduced the translation of STAT2 mRNA mediated by YTHDF1 and alleviated LPS-induced inflammatory damage (98).
3.5.3 Other infectious diseases
When a virus invades, innate immune and epithelial cells can recognize viral structures, such as nucleic acids, peptides, and lipoproteins, leading to primary activation of interferon regulatory factor (IRF) 3/7 and the NF-κB signaling pathway, which induces the production of pro-inflammatory cytokines and interferon (140). Interferon induces interferon-stimulated gene (ISG) transcription by activating the JAK1/STAT1/STAT2 pathway (140). Some viruses can modify host cell genes through epigenetic strategies, to enhance replication and survival. Epstein–Barr virus infection can alter the m6A-modified epitranscriptome of host B cells and upregulate mRNA and protein levels of fas cell surface death receptor (FAS) but downregulate toll-like receptor 9 (TLR9), which are related to apoptosis and virus infection (141). Wang et al. found that METTL3-deficient mice had stronger resistance to rotavirus. METTL3 knockout in intestinal epithelial cells reduced m6A modification of IRF7 and increased its expression, leading to the upregulation of interferons and ISGs (109). Li et al. found that METTL3 depletion of host cells decreased m6A levels of SARS-CoV-2 and the host genome, subsequently increasing innate immune signaling and inflammatory gene expression (99).
Fungal keratitis (FK) is an aggressive infectious corneal disease caused by pathogenic fungi (142). METTL3 was increased in corneas of an FK mouse model and in fungi-infected corneal stromal cells, while METTL3 knockdown attenuated the FK inflammatory response by inhibiting the PI3K/AKT or TRAF6/NF-κB pathways (100, 101).
In addition, some studies of inflammation have been conducted using LPS-stimulated cells. METTL3 protein expression was elevated in LPS-induced retinal pigment epithelium cells, and silencing METTL3 increased the expression of nuclear receptor subfamily 2 group F member 1 (NR2F1) and promoted IL-6 secretion (102). Further, METTL3 was up-regulated in LPS-stimulated human dental pulp cells, and its knockdown reduced inflammatory cytokine secretion through the NF-κB and MAPK pathways (103).
3.6 Tumors
Chronic inflammation is a risk factor for tumor development, as it provides a microenvironment conducive to tumorigenesis, tumor development, and metastasis (143). A deep understanding of the regulatory mechanisms in the inflammatory tumor microenvironment will help to develop more effective immunotherapy. Most tumors recruit tumor-associated macrophages (TAMs) to create a microenvironment favorable for tumor growth. METTL3 has recently been shown to affect the recruitment and polarization of TAMs, with METTL3-deficient mice exhibiting increased infiltration of M1/M2-like TAMs and regulatory T cells into tumors compared with wild-type mice (104). METTL3 knockout promoted M1 and M2 polarization of macrophages by activating the NF-κB/STAT3 pathways (104). Co-culture of monocyte and glioma cells demonstrated that glioma cells overexpressing METTL3 inhibited SOCS2 expression and M1 polarization of monocytes (110), while SOCS2 overexpression could rescue this inhibition. Knockdown of METTL3 in TAMs infiltrating prostate cancer promotes M2 macrophage polarization by activating STAT6 (106). Further, M2-TAMs can promote lung adenocarcinoma immuno-resistance by enhancing METTL3-mediated m6A methylation (107), as well as inducing oxaliplatin resistance by increasing METTL3 in colorectal cancer cells (108).
In addition, METTL3 regulates the infiltration of tumor-associated neutrophils. Silencing METTL3 caused papillary thyroid cancer cells to secrete more pro-inflammatory factors, particularly IL-8, which can recruit infiltration of tumor-associated neutrophils. The NF-κB pathway inhibitor, BAY 11-7082, inhibits IL-8 production by tumor cells and neutrophil infiltration, as well as inhibiting tumor growth (105).
4 Therapeutic application of METTL3
The study of METTL3 expression changes and related pathogenic processes in inflammatory diseases provides clues for the development of effective drugs and informs novel diagnostic strategies (Table 3).
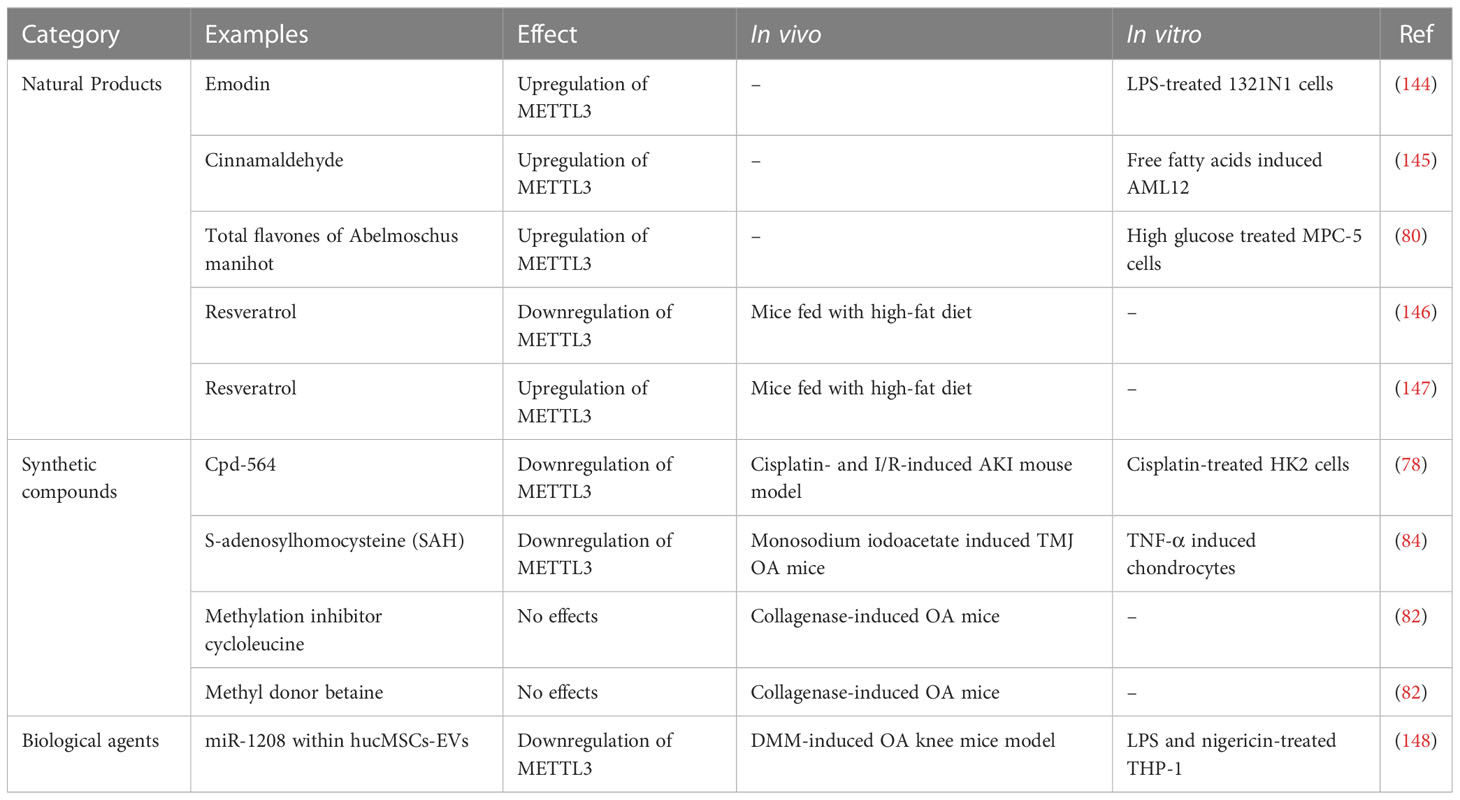
Table 3 Effects of natural products, synthetic compounds, and biological agents on METTL3.
The therapeutic potential of METTL3 is mainly as a pharmacological target for natural plant medicinal ingredients. Emodin, the main active component of rhubarb, exhibits significant anti-inflammatory activity (149) and can promote METTL3 expression, which downregulates NLRP3 by increasing its m6A levels in LPS-treated 1321N1 cells, thus suppressing pyroptosis and proinflammatory cytokine secretion (144). Cinnamaldehyde (CA) is a natural ingredient extracted from the herb, Ramulus cinnamomi, which has beneficial functions in lipid metabolism and inflammation of the liver (150). Xu et al. found that METTL3 was decreased in mouse hepatocytes stimulated by free fatty acids and aggravated cell steatosis by down-regulating CYP4F40, which could be compensated by CA (145), where CA can promote METTL3 expression and improve steatosis (145). Total flavones isolated from Abelmoschus manihot (TFA) have been used to treat kidney disease (151). Liu et al. found that TFA could upregulate METTL3 expression in podocytes stimulated by high glucose and that METTL3 overexpression promoted PTEN expression via m6A modification of PTEN, thereby inhibiting podocyte pyroptosis mediated by PI3K/Akt signaling (80). Resveratrol is a natural phytoalexin, present in grapes, peanuts, and their derivatives (152), which has a neuroprotective role (153). Izquierdo et al. found that adding resveratrol to the high-fat diet of mice could clearly up-regulate m6A levels in the hippocampus of their offspring and reduce expression of METTL3 and FTO, although the significance and impact of METTL3 reduction have not been studied (146). In another study, resveratrol intake in mice fed a high-fat diet could increase METTL3 in the liver tissue, reduce liver fat accumulation, and increase PPARα mRNA levels (147); however, the molecular mechanism underlying METTL3 activity in resveratrol-regulated liver lipid homeostasis remains to be determined.
The efficacy of METTL3 inhibitors has also been investigated. Cpd-564, a new METTL3 inhibitor identified by Wang et al., exerted reno-protective effects in cisplatin-treated HK2 cells and an acute kidney injury (AKI) mouse model (78). Cpd-564 inhibited TAB3 expression and inflammatory responses in HK2 cells tubule damage was alleviated in an AKI model after Cpd-564 administration; however, METTL3 inhibition may be harmful to temporomandibular joint (TMJ) OA. The METTL3 inhibitor, S-adenosylhomocysteine, promotes chondrocyte apoptosis and autophagy induced by TNF-α, and aggravated chondrocyte degeneration in TMJ OA mice (84). In addition, the methylation inhibitor, cycloleucine, can reduce IL-8 and IL-6 and promote collagen type II levels in OA cartilage tissue (82).
Moreover, METTL3 may be a target in OA treatment using extracellular vesicles derived from human umbilical cord mesenchymal stem cells (hucMSCs-EVs); miR-1208 in hucMSCs-EVs reduced m6A levels in NLRP3 mRNA by down-regulating METTL3 expression in macrophages, which prevented NLRP3 inflammasome activation and exerted anti-inflammatory effects (148).
In addition, METTL3 may serve as a novel biomarker for RA diagnosis. Wang et al. found that increased METTL3 in RA patients was positively correlated with CRP and ESR (88); however, its sensitivity and specificity require further verification.
5 Conclusion
The study of METTL3-mediated m6A modification brings new insights into the pathogenesis of inflammatory diseases. METTL3 is upregulated in most inflammatory diseases, indicating that it has pro-inflammatory effects; however, opposite expression trends in METTL3 expression have also been observed in the same diseases, including sepsis-related ARDS and OA, indicating that the effects of its expression may be related to cell type, modeling methods, disease status, or other potential regulatory factors. Moreover, METTL3 can regulate different target genes in the same disease, which challenges the accuracy and safety of drug development. In addition, METTL3 can regulate macrophage M1 polarization, implying that METTL3 is a potential therapeutic target in macrophage-related diseases. As METTL3 can regulate the infiltration and polarization of TAMs and tumor-associated neutrophils, it is meaningful to study the therapeutic application of METTL3 in anti-tumor immune responses. There have been promising studies of natural drugs targeting METTL3 in the treatment of inflammatory diseases, although their detailed pharmacological effects are not yet fully understood. Whether agents targeting METTL3 are natural plant-derived ingredients, chemically synthesized drugs, or biological agents, their therapeutic prospects warrant further research.
Author contributions
BS conceived and drafted the manuscript. BS and YZ designed and drew the figures. YC, JZ, and CX assisted in searching and sorting articles. YP and XZ reviewed and provided suggestions for the manuscript. JL reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Liaoning Province (2023-MS-165, 2022-BS-149).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140:771–6. doi: 10.1016/j.cell.2010.03.006
2. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi: 10.1038/nature07201
3. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell (2015) 160:816–27. doi: 10.1016/j.cell.2015.02.010
5. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
6. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol (2017) 18:826–31. doi: 10.1038/ni.3790
7. Headland SE, Norling LV. The resolution of inflammation: Principles and challenges. Semin Immunol (2015) 27:149–60. doi: 10.1016/j.smim.2015.03.014
8. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
9. Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol (2017) 17:208–14. doi: 10.1038/nri.2016.151
10. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
11. Luo J, Xu T, Sun K. N6-methyladenosine RNA modification in inflammation: roles, mechanisms, and applications. Front Cell Dev Biol (2021) 9:670711. doi: 10.3389/fcell.2021.670711
12. Tuck MT. The formation of internal 6-methyladenine residues in eucaryotic messenger RNA. Int J Biochem (1992) 24:379–86. doi: 10.1016/0020-711x(92)90028-y
13. Wei CM, Gershowitz A, Moss B. 5’-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry (1976) 15:397–401. doi: 10.1021/bi00647a024
14. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature (2012) 485:201–6. doi: 10.1038/nature11112
15. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
16. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol (2019) 20:608–24. doi: 10.1038/s41580-019-0168-5
17. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA (1997) 3:1233–47.
18. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mamMalian nuclear RNA N6-adenosine methylation. Nat Chem Biol (2014) 10:93–5. doi: 10.1038/nchembio.1432
19. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell (2017) 169:824–835.e14. doi: 10.1016/j.cell.2017.05.003
20. Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, et al. MamMalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res (2014) 24:177–89. doi: 10.1038/cr.2014.3
21. Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep (2014) 8:284–96. doi: 10.1016/j.celrep.2014.05.048
22. Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature (2016) 537:369–73. doi: 10.1038/nature19342
23. Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol Cell (2018) 69:1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015
24. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol (2011) 7:885–7. doi: 10.1038/nchembio.687
25. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, et al. ALKBH5 is a mamMalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
26. Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
27. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mamMalian spermatogenesis. Cell Res (2017) 27:1115–27. doi: 10.1038/cr.2017.99
28. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505:117–20. doi: 10.1038/nature12730
29. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
30. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA - PubMed. (Accessed February 10, 2023).
31. Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun (2018) 9:420. doi: 10.1038/s41467-017-02770-z
32. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature (2015) 518:560–4. doi: 10.1038/nature14234
33. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
34. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell (2017) 169:1187–200. doi: 10.1016/j.cell.2017.05.045
35. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature (2016) 534:575–8. doi: 10.1038/nature18298
36. Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, et al. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA N Y N (2018) 24:499–512. doi: 10.1261/rna.064063.117
37. Liu S, Zhuo L, Wang J, Zhang Q, Li Q, Li G, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res (2020) 10:1631–46.
38. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
39. Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. Sci World J (2011) 11:2391–402. doi: 10.1100/2011/213962
40. Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflammation Res (2016) 65:1–11. doi: 10.1007/s00011-015-0874-1
41. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MPJ, Donners MMPC. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
42. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
43. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili S-A, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
44. Li Z, Xu Q, Huangfu N, Chen X, Zhu J. Mettl3 promotes oxLDL-mediated inflammation through activating STAT1 signaling. J Clin Lab Anal (2022) 36:e24019. doi: 10.1002/jcla.24019
45. Sun Z, Chen W, Wang Z, Wang S, Zan J, Zheng L, et al. Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin Immunol Orlando Fla (2022) 245:109176. doi: 10.1016/j.clim.2022.109176
46. Zhang X, Li X, Jia H, An G, Ni J. The m(6)A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem (2021) 297:101058. doi: 10.1016/j.jbc.2021.101058
47. Guo M, Yan R, Ji Q, Yao H, Sun M, Duan L, et al. IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int Immunopharmacol (2020) 86:106800. doi: 10.1016/j.intimp.2020.106800
48. Cai Y, Yu R, Kong Y, Feng Z, Xu Q. METTL3 regulates LPS-induced inflammatory response via the NOD1 signaling pathway. Cell Signal (2022) 93:110283. doi: 10.1016/j.cellsig.2022.110283
49. Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m6A as a positive regulator of macrophage activation. Sci Adv (2021) 7:eabd4742. doi: 10.1126/sciadv.abd4742
50. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
51. Wen L, Sun W, Xia D, Wang Y, Li J, Yang S. The m6A methyltransferase METTL3 promotes LPS-induced microglia inflammation through TRAF6/NF-κB pathway. Neuroreport (2022) 33:243–51. doi: 10.1097/WNR.0000000000001550
52. Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am J Physiol Cell Physiol (2019) 317:C762–75. doi: 10.1152/ajpcell.00212.2019
53. Zheng L, Chen X, Yin Q, Gu J, Chen J, Chen M, et al. RNA-m6A modification of HDGF mediated by Mettl3 aggravates the progression of atherosclerosis by regulating macrophages polarization via energy metabolism reprogramming. Biochem Biophys Res Commun (2022) 635:120–7. doi: 10.1016/j.bbrc.2022.10.032
54. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
55. Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
56. Luo S, Liao C, Zhang L, Ling C, Zhang X, Xie P, et al. METTL3-mediated m6A mRNA methylation regulates neutrophil activation through targeting TLR4 signaling. Cell Rep (2023) 42:112259. doi: 10.1016/j.celrep.2023.112259
57. Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol (2021) 18:2461. doi: 10.1038/s41423-021-00726-4
58. Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat Commun (2019) 10:1898. doi: 10.1038/s41467-019-09903-6
59. Wu H, Xu Z, Wang Z, Ren Z, Li L, Ruan Y. Dendritic cells with METTL3 gene knockdown exhibit immature properties and prolong allograft survival. Genes Immun (2020) 21:193–202. doi: 10.1038/s41435-020-0099-3
60. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi: 10.1038/ni1254
61. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol (2019) 41:283–97. doi: 10.1007/s00281-019-00733-8
62. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
63. Zhao L, Liu Y, Ma B, Liu X, Wei R, Nian H. METTL3 inhibits autoreactive Th17 cell responses in experimental autoimmune uveitis via stabilizing ASH1L mRNA. FASEB J Off Publ Fed Am Soc Exp Biol (2023) 37:e22803. doi: 10.1096/fj.202201548R
64. Cai Y, Chen X, Huang C, Chen Y, Zhang C, Huang Z, et al. Alteration of m6A-tagged RNA profiles in bone originated from periprosthetic joint infection. J Clin Med (2023) 12:2863. doi: 10.3390/jcm12082863
65. Björkström NK, Strunz B, Ljunggren H-G. Natural killer cells in antiviral immunity. Nat Rev Immunol (2022) 22:112–23. doi: 10.1038/s41577-021-00558-3
66. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
67. Screpanti V, Wallin RPA, Grandien A, Ljunggren H-G. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol (2005) 42:495–9. doi: 10.1016/j.molimm.2004.07.033
68. Song H, Song J, Cheng M, Zheng M, Wang T, Tian S, et al. METTL3-mediated m(6)A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun (2021) 12:5522. doi: 10.1038/s41467-021-25803-0
69. Dong G, Yu J, Shan G, Su L, Yu N, Yang S. N6-Methyladenosine Methyltransferase METTL3 Promotes Angiogenesis and Atherosclerosis by Upregulating the JAK2/STAT3 Pathway via m6A Reader IGF2BP1. Front Cell Dev Biol (2021) 9:731810. doi: 10.3389/fcell.2021.731810
70. Li B, Zhang T, Liu M, Cui Z, Zhang Y, Liu M, et al. RNA N(6)-methyladenosine modulates endothelial atherogenic responses to disturbed flow in mice. eLife (2022) 11:e69906. doi: 10.7554/eLife.69906
71. Chien C-S, Li JY-S, Chien Y, Wang M-L, Yarmishyn AA, Tsai P-H, et al. METTL3-dependent N6-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci U.S.A. (2021) 118:e2025070118. doi: 10.1073/pnas.2025070118
72. Qi L, Hu H, Wang Y, Hu H, Wang K, Li P, et al. New insights into the central sympathetic hyperactivity post-myocardial infarction: Roles of METTL3-mediated m(6) A methylation. J Cell Mol Med (2022) 26:1264–80. doi: 10.1111/jcmm.17183
73. Qi L, Wang Y, Hu H, Li P, Hu H, Li Y, et al. m(6)A methyltransferase METTL3 participated in sympathetic neural remodeling post-MI via the TRAF6/NF-κB pathway and ROS production. J Mol Cell Cardiol (2022) 170:87–99. doi: 10.1016/j.yjmcc.2022.06.004
74. Qin Y, Li B, Arumugam S, Lu Q, Mankash SM, Li J, et al. m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep (2021) 37:109968. doi: 10.1016/j.celrep.2021.109968
75. Li X, Yuan B, Lu M, Wang Y, Ding N, Liu C, et al. The methyltransferase METTL3 negatively regulates nonalcoholic steatohepatitis (NASH) progression. Nat Commun (2021) 12:7213. doi: 10.1038/s41467-021-27539-3
76. Shu B, Zhou Y-X, Li H, Zhang R-Z, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discovery (2021) 7:368. doi: 10.1038/s41420-021-00756-x
77. Chen Y, Cao P, Xiao Z, Ruan Z. m(6)A methyltransferase METTL3 relieves cognitive impairment of hyPeruricemia mice via inactivating MyD88/NF-κB pathway mediated NLRP3-ASC-Caspase1 inflammasome. Int Immunopharmacol (2022) 113:109375. doi: 10.1016/j.intimp.2022.109375
78. Wang J-N, Wang F, Ke J, Li Z, Xu C-H, Yang Q, et al. Inhibition of METTL3 attenuates renal injury and inflammation by alleviating TAB3 m6A modifications via IGF2BP2-dependent mechanisms. Sci Transl Med (2022) 14:eabk2709. doi: 10.1126/scitranslmed.abk2709
79. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, et al. METTL3-mediated m(6)A modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol Ther J Am Soc Gene Ther (2022) 30:1721–40. doi: 10.1016/j.ymthe.2022.01.002
80. Liu B-H, Tu Y, Ni G-X, Yan J, Yue L, Li Z-L, et al. Total Flavones of Abelmoschus manihot Ameliorates Podocyte Pyroptosis and Injury in High Glucose Conditions by Targeting METTL3-Dependent m(6)A Modification-Mediated NLRP3-Inflammasome Activation and PTEN/PI3K/Akt Signaling. Front Pharmacol (2021) 12:667644. doi: 10.3389/fphar.2021.667644
81. Liu E, Lv L, Zhan Y, Ma Y, Feng J, He Y, et al. METTL3/N6-methyladenosine/ miR-21-5p promotes obstructive renal fibrosis by regulating inflammation through SPRY1/ERK/NF-κB pathway activation. J Cell Mol Med (2021) 25:7660–74. doi: 10.1111/jcmm.16603
82. Liu Q, Li M, Jiang L, Jiang R, Fu B. METTL3 promotes experimental osteoarthritis development by regulating inflammatory response and apoptosis in chondrocyte. Biochem Biophys Res Commun (2019) 516:22–7. doi: 10.1016/j.bbrc.2019.05.168
83. Sang W, Xue S, Jiang Y, Lu H, Zhu L, Wang C, et al. METTL3 involves the progression of osteoarthritis probably by affecting ECM degradation and regulating the inflammatory response. Life Sci (2021) 278:119528. doi: 10.1016/j.lfs.2021.119528
84. He Y, Wang W, Xu X, Yang B, Yu X, Wu Y, et al. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m(6)A modification. Bone (2022) 154:116182. doi: 10.1016/j.bone.2021.116182
85. Shi L, Hu H, Sun P, Li Z, Ji L, Liu S, et al. RPL38 knockdown inhibits the inflammation and apoptosis in chondrocytes through regulating METTL3-mediated SOCS2 m6A modification in osteoarthritis. Inflammation Res Off J Eur Histamine Res Soc Al (2022) 71:977–89. doi: 10.1007/s00011-022-01579-x
86. Chen X, Gong W, Shao X, Shi T, Zhang L, Dong J, et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis (2022) 81:87–99. doi: 10.1136/annrheumdis-2021-221091
87. Fang S, Zeng F, Chen R, Li M. SIAH1 promotes senescence and apoptosis of nucleus pulposus cells to exacerbate disc degeneration through ubiquitinating XIAP. Tissue Cell (2022) 76:101820. doi: 10.1016/j.tice.2022.101820
88. Wang J, Yan S, Lu H, Wang S, Xu D. METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-κB signaling pathway. Mediators Inflammation (2019) 2019:3120391. doi: 10.1155/2019/3120391
89. Shi W, Zheng Y, Luo S, Li X, Zhang Y, Meng X, et al. METTL3 promotes activation and inflammation of FLSs through the NF-κB signaling pathway in rheumatoid arthritis. Front Med (2021) 8:607585. doi: 10.3389/fmed.2021.607585
90. Wang Y, Huang J, Jin H. Reduction of methyltransferase-like 3-mediated RNA N6-methyladenosine exacerbates the development of psoriasis vulgaris in imiquimod-induced psoriasis-like mouse model. Int J Mol Sci (2022) 23:12672. doi: 10.3390/ijms232012672
91. Yang L, Wu G, Wu Q, Peng L, Yuan L. METTL3 overexpression aggravates LPS-induced cellular inflammation in mouse intestinal epithelial cells and DSS-induced IBD in mice. Cell Death Discovery (2022) 8:62. doi: 10.1038/s41420-022-00849-1
92. Shen Z-J, Han Y-C, Nie M-W, Wang Y-N, Xiang R-L, Xie H-Z. Genome-wide identification of altered RNA m(6)A profiles in vascular tissue of septic rats. Aging (2021) 13:21610–27. doi: 10.18632/aging.203506
93. Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci (2022) 18:3337–57. doi: 10.7150/ijbs.69141
94. Qu M, Chen Z, Qiu Z, Nan K, Wang Y, Shi Y, et al. Neutrophil extracellular traps-triggered impaired autophagic flux via METTL3 underlies sepsis-associated acute lung injury. Cell Death Discovery (2022) 8:375. doi: 10.1038/s41420-022-01166-3
95. Chen Y, Wu Y, Zhu L, Chen C, Xu S, Tang D, et al. METTL3-mediated N6-methyladenosine modification of trim59 mRNA protects against sepsis-induced acute respiratory distress syndrome. Front Immunol (2022) 13:897487. doi: 10.3389/fimmu.2022.897487
96. Zhao D, Wang C, Liu X, Liu N, Zhuang S, Zhang Q, et al. CircN4bp1 Facilitates Sepsis-Induced Acute Respiratory Distress Syndrome through Mediating Macrophage Polarization via the miR-138-5p/EZH2 Axis. Mediators Inflammation (2021) 2021:7858746. doi: 10.1155/2021/7858746
97. Yang Y, Yang X, Wu Y, Fu M. METTL3 promotes inflammation and cell apoptosis in a pediatric pneumonia model by regulating EZH2. Allergol Immunopathol (Madr) (2021) 49:49–56. doi: 10.15586/aei.v49i5.445
98. Li S-X, Yan W, Liu J-P, Zhao Y-J, Chen L. Long noncoding RNA SNHG4 remits lipopolysaccharide-engendered inflammatory lung damage by inhibiting METTL3 - Mediated m(6)A level of STAT2 mRNA. Mol Immunol (2021) 139:10–22. doi: 10.1016/j.molimm.2021.08.008
99. Li N, Hui H, Bray B, Gonzalez GM, Zeller M, Anderson KG, et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep (2021) 35:109091. doi: 10.1016/j.celrep.2021.109091
100. Huang L, Tang H, Hu J. METTL3 Attenuates Inflammation in Fusarium solani-Induced Keratitis via the PI3K/AKT Signaling Pathway. Invest Ophthalmol Vis Sci (2022) 63:20. doi: 10.1167/iovs.63.10.20
101. Tang H, Huang L, Hu J. Inhibition of the m6A Methyltransferase METTL3 Attenuates the Inflammatory Response in Fusarium solani-Induced Keratitis via the NF-κB Signaling Pathway. Invest Ophthalmol Vis Sci (2022) 63:2. doi: 10.1167/iovs.63.11.2
102. Meng J, Liu X, Tang S, Liu Y, Zhao C, Zhou Q, et al. METTL3 inhibits inflammation of retinal pigment epithelium cells by regulating NR2F1 in an m(6)A-dependent manner. Front Immunol (2022) 13:905211. doi: 10.3389/fimmu.2022.905211
103. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J Cell Mol Med (2018) 22:2558–68. doi: 10.1111/jcmm.13491
104. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun (2021) 12:1394. doi: 10.1038/s41467-021-21514-8
105. He J, Zhou M, Yin J, Wan J, Chu J, Jia J, et al. METTL3 restrains papillary thyroid cancer progression via m(6)A/c-Rel/IL-8-mediated neutrophil infiltration. Mol Ther J Am Soc Gene Ther (2021) 29:1821–37. doi: 10.1016/j.ymthe.2021.01.019
106. Jia G, Wang X, Wu W, Zhang Y, Chen S, Zhao J, et al. LXA4 enhances prostate cancer progression by facilitating M2 macrophage polarization via inhibition of METTL3. Int Immunopharmacol (2022) 107:108586. doi: 10.1016/j.intimp.2022.108586
107. Wu L, Cheng D, Yang X, Zhao W, Fang C, Chen R, et al. M2-TAMs promote immunoresistance in lung adenocarcinoma by enhancing METTL3-mediated m6A methylation. Ann Transl Med (2022) 10:1380. doi: 10.21037/atm-22-6104
108. Lan H, Liu Y, Liu J, Wang X, Guan Z, Du J, et al. Tumor-Associated Macrophages Promote Oxaliplatin Resistance via METTL3-Mediated m(6)A of TRAF5 and Necroptosis in Colorectal Cancer. Mol Pharm (2021) 18:1026–37. doi: 10.1021/acs.molpharmaceut.0c00961
109. Wang A, Tao W, Tong J, Gao J, Wang J, Hou G, et al. m6A modifications regulate intestinal immunity and rotavirus infection. eLife (2022) 11:e73628. doi: 10.7554/eLife.73628
110. Zhong C, Tao B, Yang F, Xia K, Yang X, Chen L, et al. Histone demethylase JMJD1C promotes the polarization of M1 macrophages to prevent glioma by upregulating miR-302a. Clin Transl Med (2021) 11:e424. doi: 10.1002/ctm2.424
111. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol (2006) 47:C7–12. doi: 10.1016/j.jacc.2005.09.068
112. Powell EE, Wong VW-S, Rinella M. Non-alcoholic fatty liver disease. Lancet Lond Engl (2021) 397:2212–24. doi: 10.1016/S0140-6736(20)32511-3
113. Barajas JM, Lin C-H, Sun H-L, Alencastro F, Zhu AC, Aljuhani M, et al. METTL3 regulates liver homeostasis, hepatocyte ploidy, and circadian rhythm-controlled gene expression in mice. Am J Pathol (2022) 192:56–71. doi: 10.1016/j.ajpath.2021.09.005
114. Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol (2016) 213:8–14. doi: 10.1016/j.ijcard.2015.08.109
115. Zhang S, Wang Y, Cheng J, Huangfu N, Zhao R, Xu Z, et al. HyPeruricemia and cardiovascular disease. Curr Pharm Des (2019) 25:700–9. doi: 10.2174/1381612825666190408122557
116. Afsar B, Elsurer R, Covic A, Johnson RJ, Kanbay M. Relationship between uric acid and subtle cognitive dysfunction in chronic kidney disease. Am J Nephrol (2011) 34:49–54. doi: 10.1159/000329097
117. Tian T, Liu X, Li T, Nie Z, Li S, Tang Y, et al. Detrimental effects of long-term elevated serum uric acid on cognitive function in rats. Sci Rep (2021) 11:6732. doi: 10.1038/s41598-021-86279-y
118. Zhou Y, Zhao M, Pu Z, Xu G, Li X. Relationship between oxidative stress and inflammation in hyPeruricemia: Analysis based on asymptomatic young patients with primary hyPeruricemia. Med (Baltimore) (2018) 97:e13108. doi: 10.1097/MD.0000000000013108
119. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci (2020) 21:6275. doi: 10.3390/ijms21176275
120. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol (2019) 15:327–45. doi: 10.1038/s41581-019-0135-6
121. Guilak F, Nims RJ, Dicks A, Wu C-L, Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol J Int Soc Matrix Biol (2018) 71–72:40–50. doi: 10.1016/j.matbio.2018.05.008
122. Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol (2014) 10:44–56. doi: 10.1038/nrrheum.2013.160
123. Francisco V, Pino J, González-Gay MÁ, Lago F, Karppinen J, Tervonen O, et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat Rev Rheumatol (2022) 18:47–60. doi: 10.1038/s41584-021-00713-z
124. Lieben L, Marshall L. Rheumatoid arthritis. Nat Rev Dis Primer (2018) 4:18002. doi: 10.1038/nrdp.2018.2
125. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
126. Baxter D, McInnes IB, Kurowska-Stolarska M. Novel regulatory mechanisms in inflammatory arthritis: a role for microRNA. Immunol Cell Biol (2012) 90:288–92. doi: 10.1038/icb.2011.114
127. Klein K, Gay S. Epigenetics in rheumatoid arthritis. Curr Opin Rheumatol (2015) 27:76–82. doi: 10.1097/BOR.0000000000000128
128. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker JNWN. Psoriasis. Lancet Lond Engl (2021) 397:1301–15. doi: 10.1016/S0140-6736(20)32549-6
129. Boehncke W-H, Schön MP. Psoriasis. Lancet Lond Engl (2015) 386:983–94. doi: 10.1016/S0140-6736(14)61909-7
130. Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating th17, th22, and th1 cells are increased in psoriasis. J Invest Dermatol (2010) 130:1373–83. doi: 10.1038/jid.2009.399
131. Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest (2007) 117:1119–27. doi: 10.1172/JCI31720
132. Chen C-J, Hu H, Liao W-T. Pathophysiology of inflammatory bowel diseases. N Engl J Med (2021) 384:1376–7. doi: 10.1056/NEJMc2101562
133. Sebastian-delaCruz M, Olazagoitia-Garmendia A, Gonzalez-Moro I, Santin I, Garcia-Etxebarria K, Castellanos-Rubio A. Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease. Epigenomes (2020) 4:16. doi: 10.3390/epigenomes4030016
134. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
135. Vincent J-L, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, et al. Sepsis in European intensive care units: Results of the SOAP study*. Crit Care Med (2006) 34:344–53. doi: 10.1097/01.ccm.0000194725.48928.3a
136. Karlsson S, Varpula M, Ruokonen E, Pettilä V, Parviainen I, Ala-Kokko TI, et al. Incidence, treatment, and outcome of severe sepsis in ICU-treated adults in Finland: the Finnsepsis study. Intensive Care Med (2007) 33:435–43. doi: 10.1007/s00134-006-0504-z
137. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med (2003) 348:138–50. doi: 10.1056/NEJMra021333
138. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (2018) 392:75–87. doi: 10.1016/S0140-6736(18)30696-2
139. Torres A, Cilloniz C, Niederman MS, Menéndez R, Chalmers JD, Wunderink RG, et al. Pneumonia. Nat Rev Dis Primer (2021) 7:25. doi: 10.1038/s41572-021-00259-0
140. Xu Q, Tang Y, Huang G. Innate immune responses in RNA viral infection. Front Med (2021) 15:333–46. doi: 10.1007/s11684-020-0776-7
141. Zheng X, Wang J, Zhang X, Fu Y, Peng Q, Lu J, et al. RNA m(6) A methylation regulates virus-host interaction and EBNA2 expression during Epstein-Barr virus infection. Immun Inflammation Dis (2021) 9:351–62. doi: 10.1002/iid3.396
142. Brown L, Leck AK, Gichangi M, Burton MJ, Denning DW. The global incidence and diagnosis of fungal keratitis. Lancet Infect Dis (2021) 21:e49–57. doi: 10.1016/S1473-3099(20)30448-5
143. Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncol Williston Park N (2002) 16:217–226, 229.
144. Wang B, Liu Y, Jiang R, Liu Z, Gao H, Chen F, et al. Emodin relieves the inflammation and pyroptosis of lipopolysaccharide-treated 1321N1 cells by regulating methyltransferase-like 3 -mediated NLR family pyrin domain containing 3 expression. Bioengineered (2022) 13:6740–9. doi: 10.1080/21655979.2022.2045836
145. Xu R, Xiao X, Zhang S, Pan J, Tang Y, Zhou W, et al. The methyltransferase METTL3-mediated fatty acid metabolism revealed the mechanism of cinnamaldehyde on alleviating steatosis. BioMed Pharmacother Biomed Pharmacother (2022) 153:113367. doi: 10.1016/j.biopha.2022.113367
146. Izquierdo V, Palomera-Ávalos V, Pallàs M, Griñán-Ferré C. Resveratrol Supplementation Attenuates Cognitive and Molecular Alterations under Maternal High-Fat Diet Intake: Epigenetic Inheritance over Generations. Int J Mol Sci (2021) 22:1453. doi: 10.3390/ijms22031453
147. Wu J, Li Y, Yu J, Gan Z, Wei W, Wang C, et al. Resveratrol attenuates high-fat diet induced hepatic lipid homeostasis disorder and decreases m6A RNA methylation. Front Pharmacol (2020) 11:568006. doi: 10.3389/fphar.2020.568006
148. Zhou H, Shen X, Yan C, Xiong W, Ma Z, Tan Z, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res Ther (2022) 13:322. doi: 10.1186/s13287-022-03005-9
149. Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: A review of its pharmacology, toxicity and pharmacokinetics. Phytother Res PTR (2016) 30:1207–18. doi: 10.1002/ptr.5631
150. Neto JGO, Boechat SK, Romão JS, Pazos-Moura CC, Oliveira KJ. Treatment with cinnamaldehyde reduces the visceral adiposity and regulates lipid metabolism, autophagy and endoplasmic reticulum stress in the liver of a rat model of early obesity. J Nutr Biochem (2020) 77:108321. doi: 10.1016/j.jnutbio.2019.108321
151. Li N, Tang H, Wu L, Ge H, Wang Y, Yu H, et al. Chemical constituents, clinical efficacy and molecular mechanisms of the ethanol extract of Abelmoschus manihot flowers in treatment of kidney diseases. Phytother Res (2021) 35:198–206. doi: 10.1002/ptr.6818
152. Li J, Zhang C-X, Liu Y-M, Chen K-L, Chen G. A comparative study of anti-aging properties and mechanism: resveratrol and caloric restriction. Oncotarget (2017) 8:65717–29. doi: 10.18632/oncotarget.20084
Keywords: m6A modification, METTL3, inflammation, inflammatory diseases, immune cell, therapy
Citation: Song B, Zeng Y, Cao Y, Zhang J, Xu C, Pan Y, Zhao X and Liu J (2023) Emerging role of METTL3 in inflammatory diseases: mechanisms and therapeutic applications. Front. Immunol. 14:1221609. doi: 10.3389/fimmu.2023.1221609
Received: 12 May 2023; Accepted: 27 July 2023;
Published: 21 August 2023.
Edited by:
Kishan Nyati, Osaka University, JapanCopyright © 2023 Song, Zeng, Cao, Zhang, Xu, Pan, Zhao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingbo Liu, amluZ2JvbGl1X2NtdUAxNjMuY29t