 Mingyu Xie1†
Mingyu Xie1† Jingjing Wan1†Xin Zheng2Xian Zou1Wanting Chen1Kanglin Zhang1Huiting Yuan1
Jingjing Wan1†Xin Zheng2Xian Zou1Wanting Chen1Kanglin Zhang1Huiting Yuan1 Zhenhong Zhang1Haisheng Zeng1*
Zhenhong Zhang1Haisheng Zeng1*- 1Department of Pediatric Rheumatology and Immunology, Dongguan Children’s Hospital, Dongguan, Guangdong, China
- 2Department of Pediatric Rheumatology and Immunology, Huizhou Central People’s Hospital, Huizhou, Guangdong, China
Background: Cryopyrin-associated periodic syndromes (CAPS) have been considered autoinflammatory diseases resulting from NLRP3 gene mutations. In recent years, these conditions have been redefined as NLRP3-associated autoinflammatory diseases (NLRP3-AID). Our previous study highlighted a case of a Chinese individual carrying the de novo NLRP3 mutation.
Results: A female child carrying a de novo variant (c.1718T>G, p. L573W) in the NLRP3 gene was presented in this work. The patient manifested various symptoms, including recurrent fever, a rash resembling urticaria, arthritis, physical growth retardation, a notable prominence of the forehead, and a flat nose bridge. Additionally, inflammatory markers, like WBC count, PLT count, CRP, ESR, and IL-6 showed elevated levels. Additionally, we observed interstitial pulmonary disease in the patient, which is not frequently mentioned in previous studies. Notably, the proband did not present with any ocular, auditory, or neurological symptoms. After 12 weeks of subcutaneous canakinumab injection, there was a clear improvement in the patient’s clinical manifestations and inflammatory markers.
Conclusion: Our study contributes to broadening the clinical spectrum of established pathogenic variants of NLRP3 gene, which are related to NLRP3-AID.
Background
Autoinflammatory diseases (AIDs) encompass various diseases resulting from innate immune system dysregulation, causing systemic effects (1). AIDs was first described by McDermott et al. in 1999 (2). As per the 2022 Classification of Monogenic Immune Disorders issued by the International Union of Immunological Societies (IUIS), there exists a total of 59 autoinflammatory disorder types resulting from mutations of 56 different genes, and this number is continuously expanding (3). Notably, among these conditions, NLRP3-AID was initially recognized in 2001 as the underlying factor leading to familial cold autoinflammatory syndrome (FCAS) (4). NLRP3 gene is responsible for encoding cryopyrin protein, and its mutations commonly lead to three distinct periodic fever syndromes with varying severity: FCAS (OMIM 120100), Muckle-Wells syndrome (MWS) (OMIM 191900), as well as chronic infantile neurologic cutaneous articular syndrome (CINCA) (OMIM 607115) (5). To align with the 2018 Consensus Proposal for Taxonomy and Definition of Autoinflammatory Diseases, the term NLRP3-AID was recommended to encompass the spectrum of CAPS (6).
NLRP3 variants can activate caspase-1, causing IL-1β (a pro-inflammatory cytokine) overexpression. Consequently, patients exhibit nonspecific clinical signs like urticaria-like rash, periodic fever, sensorineural deafness, arthritis, uveitis, and aseptic meningitis (5). Currently, over 170 NLRP3 variants are identified, most of them are located within exon 3 that is responsible for encoding an oligomerization domain in cryopyrin protein (7). This research article presents a case study of an infant with NLRP3-AID who received treatment at our medical center. The clinical manifestations, medical progression, and subsequent monitoring of the affected individual are detailed herein.
Case presentation
The proband, a second-born female child of Han Chinese parents, was delivered via cesarean section at 37 weeks gestation due to cloudy amniotic fluid. The birth weight and length were 2.2 kg and 48 cm separately.
Shortly after birth, the proband experienced fever, accompanied by a red maculopapular rash that would fade upon pressure (Figure 1A). Neonatal pediatricians suspected a possible sepsis infection. The laboratory analyses revealed increased levels of acute-phase reactants during febrile episodes, with C-reactive protein (CRP) at 45.2 mg/L, procalcitonin (PCT) at 1.64 ng/mL, and interleukin-6 (IL-6) at 9.41 pg/mL. However, cultures of blood, urine, and stool yielded no growth. The proband’s body temperature would spontaneously return to normal, leading to a resolution of the rash. Without any obvious cause for the rash and fever, the parents opted to discharge the proband from the hospital. The proband continued to experience occasional rashes on a daily basis, with her weight and height measuring under the average for her age. When she was 1 years old, she also developed additional arthritis in her knees.
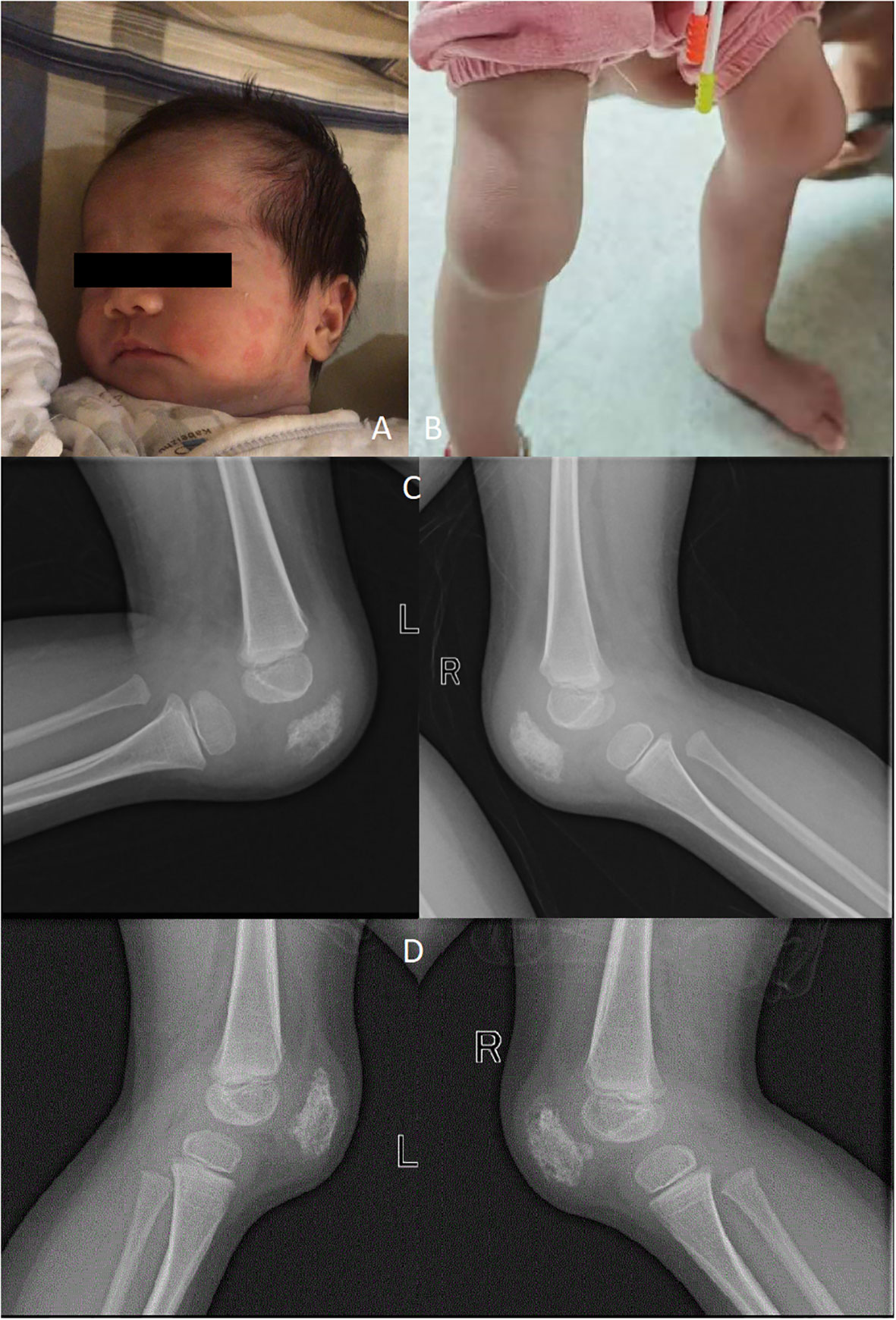
Figure 1 Rash (A); Deformities of the knee (B); X-ray showing destruction of patella (C); X-ray of the knee were taken half a year after Canakinumab treatment (D).
When she was 2 years and 9 months old, she was admitted into Dongguan Children’s Hospital due to bilateral knee joint swelling and pain. Her height and weight were 82 cm and 8.7 kg (under the 3rd percentile separately). In addition, the patient exhibited features such as a prominent forehead, flat nose bridge, rough skin, an unclosed anterior fontanelle, and enlarged knee joints (Figure 1B). The proband demonstrated normal intellectual and speech development. Laboratory tests revealed neutrophilic leukocytosis, thrombocytosis, increased acute phase reactants, and moderate microcytic hypochromic anemia. The white blood cell count (WBC) was 22.55 × 109/L, with a neutrophil granulocyte count of 12.17 × 109/L, hemoglobin (HGB) level of 98 g/L, and blood platelet (PLT) count of 466 × 109/L. CRP levels were 66 mg/L, whereas the erythrocyte sedimentation rate (ESR) was 99 mm/h. Examination of peripheral blood cytokines demonstrated heightened levels of IL-6 at 32.02 pg/mL (range, 0–5.3 pg/mL), with IL-1β at 0.13 pg/mL (range, 0–3.4 pg/mL), IL-10 at 0.83 pg/mL (range, 0–4.91 pg/mL), and IL-12 at 1.62 pg/mL (range, 0–3.2 pg/mL). Rheumatoid factors (RF), antinuclear antibody profile (ANAs), human leukocyte antigen B27 (HLA-B27), as well as cyclic citrullinated peptide (CCP) were negative. Immunoglobulin levels showed IgA at 2.41 g/L, IgM at 17.7 g/L, and IgG at 2.33 g/L. Lymphocyte subset analysis indicated B and T cell activation. Ferritin levels were 43.34 ng/mL. The DR of the knees revealed patella bone destruction (Figure 1C). A chest CT scan showed interstitial lung disease in the left lung (Figure 2A), but she had no respiratory symptoms. Echocardiographic findings were normal. Nervous system MRI results were normal. The proband’s parents and older sister did not present similar clinical manifestations. The affected individual was given the diagnosis of juvenile idiopathic arthritis and subsequently received a treatment regimen of prednisone and methotrexate, as prescribed by a rheumatologist. However, her clinical symptoms exhibited only marginal improvement.
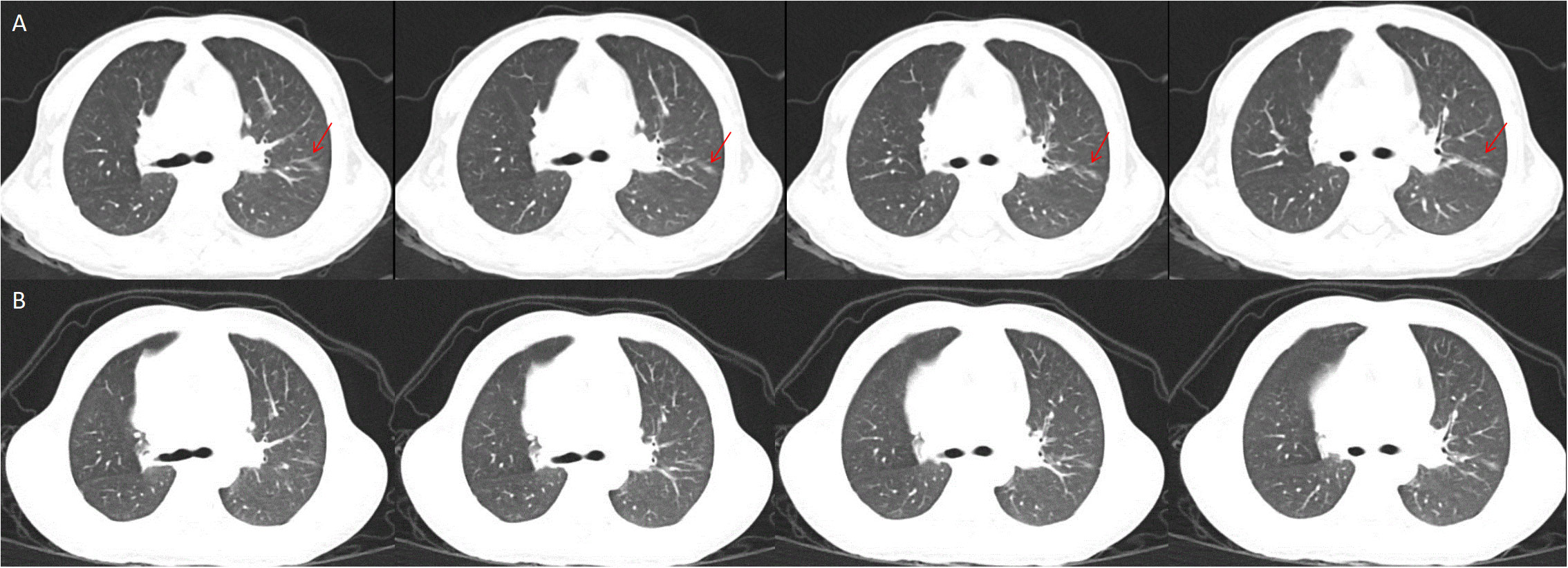
Figure 2 Interstitial lung disease in the left lung, the red snip point (A), Pulmonary CT after eight months of Canakinumab treatment (B).
Whole exome sequencing (WES) analysis on this patient detected the de novo missense mutation (c.1718T>G, p. L573W) in exon 4 of NLPR3 gene (NM_004895). Neither of the proband’s parents carried NLRP3 gene mutations. Based on these findings, clinicians suggested that the proband was affected by an autoinflammatory disease caused by the NLRP3 genetic defect. Canakinumab (a monoclonal antibody against IL-1β), obtained from Hong Kong, was administered every 4 weeks to treat the autoinflammatory disease, guided by genetic testing. After one month of Canakinumab treatment, the fever, rash, and arthritis significantly improved. Inflammatory markers, including CRP, ESR, and IL-6, returned to normal levels after 12 weeks (Table 1). However, patellar bone destruction did not improve after half a year of Canakinumab treatment, and it merely showed a reduction in the soft tissue swelling around the knee joint (Figure 1D). Pulmonary CT showed that the interstitial changes of the left lung were slightly reduced after eight months of Canakinumab treatment (Figure 2B). The patient did not require the use of steroids or antirheumatic drugs at present. Her weight and height showed improvement, and she did not experience any adverse effects during the observation period.

Table 1 Changes in inflammatory markers before and after treatment with Canakinumab.
Materials and methods
Study patient
This work was conducted at Dongguan Children’s Hospital, where demographic data, genetic results, and clinical presentations were documented using a Standardized Case Report Form.
DNA extraction and WES
Peripheral blood was sampled in the affected individual together with her parents to extract genomic DNA with a Magen SolPure Blood DNA kit, in line with specific guidelines. Next-generation sequencing was employed to sequence the proband’s DNA, while first-generation sequencing was used to confirm the parents’ DNA based on the proband’s genetic findings.
Bioinformatic analysis of the variants
Sequence variant annotation was carried out with various literature and population databases, which included 1000 Genomes (http://www.1000genomes.org/), gnomAD (http://gnomad.broadinstitute.org/), dbSNP (http://www.ncbi.nlm.nih.gov/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), OMIM (https://www.omim.org/) and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php). Protein structure analysis, prediction of functional and conserved domains, along with multiple sequence alignment were completed using online software (https://www.uniprot.org/). Variant interpretation followed the guidelines provided by the American College of Medical Genetics (ACMG) (8). Additionally, web tools such as PolyPhen-2, MutationTaster, MutationAssessor, and SIFT, were utilized to predict potential pathogenicity of the variants.
Results
Genetic analysis
The WES results demonstrated the missense mutation, c.1718T>G (p.L573W), within exon 4 in NLPR3 gene (NM_004895) of this patient. The particular mutation, c.1718T>G (p.L573W), was absent within gnomAD, ExAC, as well as 1000 Genomes Project databases. Furthermore, NLRP3 gene mutation was not detected in her parents (Figure 3). The missense mutation, c.1718T>G (p.L573W), was located adjacent to NACHT-NTPase domain in NLRP3 protein (Figure 4). Furthermore, our analysis revealed that the amino acid residue affected by the mutation, L573, was highly conserved across different species. Prediction tools indicated that the variant NLRP3 was classified as “disease causing” by MutationTaster, “affecting protein function” by SIFT, and “probably damaging”, and the PolyPhen-2 score was 0.999 (sensitivity and specificity of 0.00 and 1.00 separately). Additionally, MutationAssessor categorized the mutation as having a “high” impact on protein function.
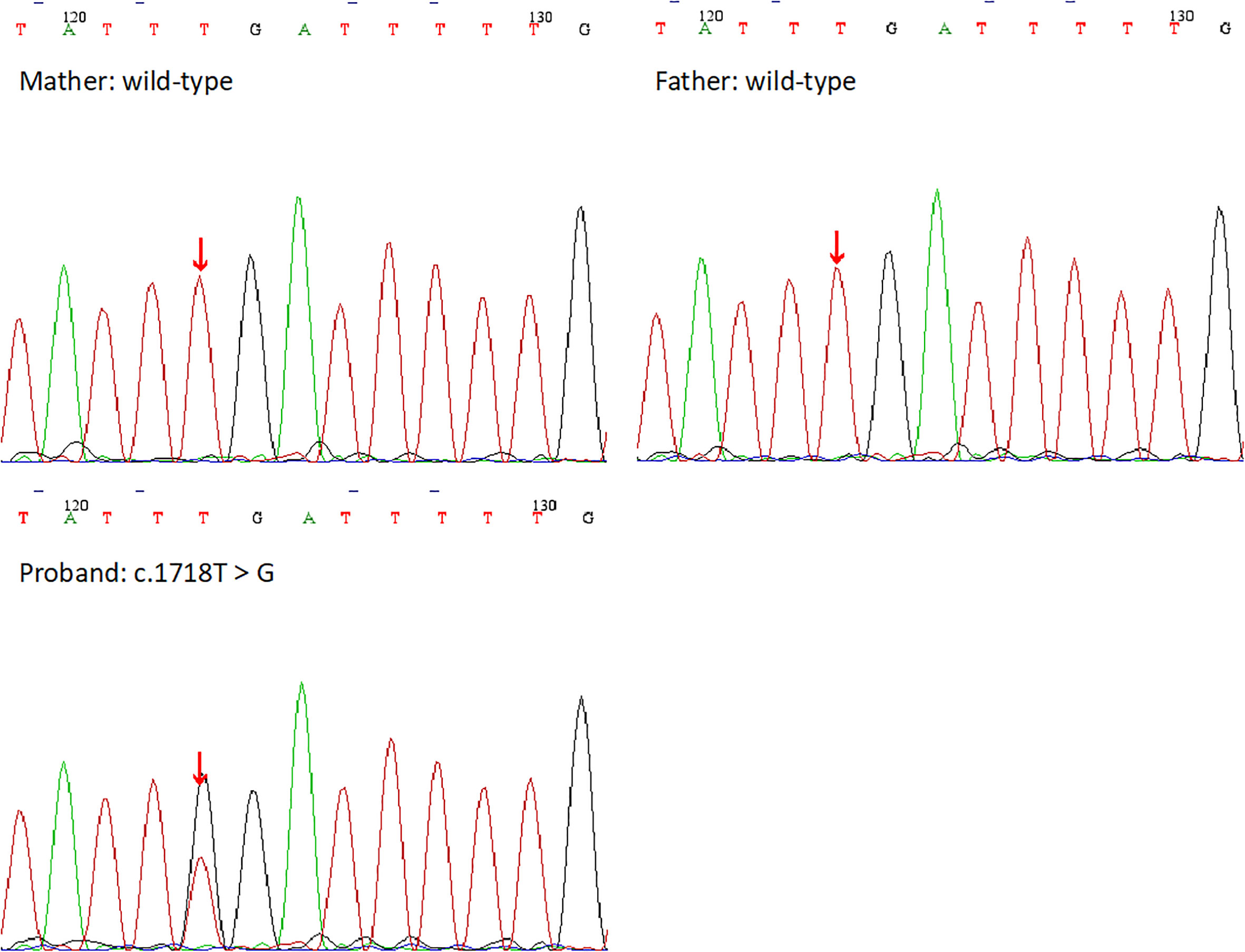
Figure 3 Schematic representation of the NLRP3 protein. Most previously reported mutations are located in NACHT domain, and the novel mutation is in a non-functional domain.
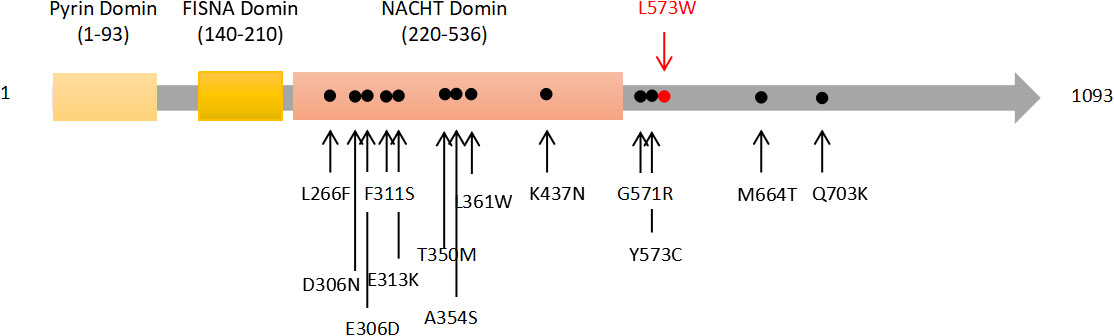
Figure 4 The DNA sequencing results of the proband and her family. Her parents showed the wild-type at the base of c.1718T, but she showed a heterozygous mutation c.1718T>G.
Discussion and conclusions
As of now, around 124 pathogenic or possibly pathogenic NLRP3 gene variants have been recognized (www.ncbi.nlm.nih.gov/clinvar/). Most of these variants are situated within exon 3, responsible for encoding “NLR-binding” domain in cryopyrin protein. These variants have been linked to three primary clinical syndromes: FCAS, MWS, and CINCA. FCAS shows the features of rash and fever without eye or hearing involvement. In contrast, MWS demonstrates the features of an urticaria-like rash unrelated to cold exposure, prominent skeletal manifestations, and severe hearing loss. CINCA is diagnosed when a newborn presents with rash, chronic meningitis, arthropathy, fever, and inflammation (9). The most common clinical signs observed in patients with NLRP3-associated autoinflammatory diseases are fever, rash, and elevated levels of ESR, CRP, and IL-6 (Table 2). Previous study indicated that half of children’s patients with CAPS exhibited neurologic manifestations with varying degrees of severity, including headache, papilledema, sensorineural deafness, epilepsy, hypophrenia, hydrocephalus or aseptic meningitis (14). Headache was the most common neurological symptom (15). However, in our study, we identified a de novo NLRP3 variant, c.1718T>G (p.L573W), in a patient who did not exhibit any neurological symptoms other than physical growth retardation. Of course, we couldn’t not exclude the possibility that the proband was unable to accurately describe her symptoms because of her younger age. The characteristic of lower frequency neurological symptom in younger patients has also been reflected in previous reports (15). We speculated that neurological symptoms might be associated with the time of diagnosis and clinical intervention. Patients with higher frequency of neurological symptoms were older age at diagnosis (9, 15).
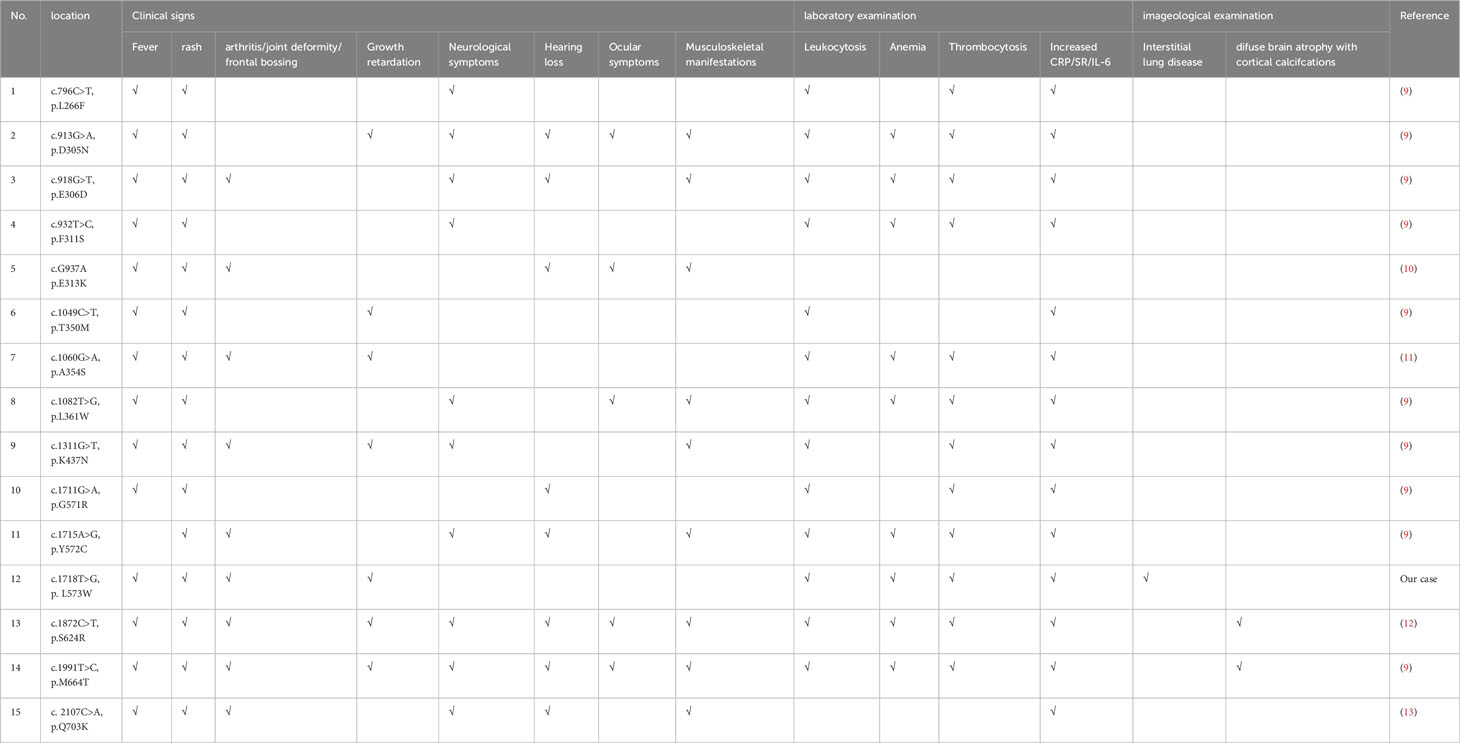
Table 2 Clinical manifestations of previously reported patients with various NLRP3 mutations and the novel NLRP3-AID.
Although an accurate genotype-phenotype correlation has not yet been established, de novo mutations can be often related to serous CINCA phenotype (16). In our study, the proband who did not exhibit the typical CINCA phenotype, such as hearing loss, optic neuritis or severe central nervous system manifestations. Previous reports have described patients with de novo NLRP3 variants, such as c.932T>C (p.F311S), c.796C>T (p.L266F), c.1311G>T (p.K437N), c.1711G>A (p.G571R), and c.1049C>T (p.T350M), who showed milder phenotypes resembling FCAS and MWS (9). This difference in phenotype may be attributed to earlier diagnosis or intervention with appropriate treatment. Our findings indicate that the clinical phenotype of NLRP3-associated autoinflammatory diseases is not solely determined by the specific domain or location of the mutated amino acid in NLRP3. Although most documented NLRP3 mutations are situated in the NACHT domain, they can give rise to various clinical phenotypes, encompassing FCAS, MWS, or CINCA. Additionally, it is worth noting that the same variant can lead to different clinical phenotypes (9). This highlights the importance of a comprehensive evaluation by clinicians when assessing patients with NLRP3-AID.
NLRP3-AID is often misdiagnosed as juvenile idiopathic arthritis due to shared symptoms such as fever, rash, and joint inflammation. However, most patients with NLRP3-AID do not exhibit increased levels of ferritin which can help clinicians differentiate between the two conditions (9). NLRP3-AID may sometimes be misdiagnosed as other febrile cycle syndromes, such as Familial Mediterranean fever (FMF) and periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome. FMF represents the monogenic autoinflammatory disorder resulting from MEFV variants. Initially considered an autosomal recessive disorder, recent studies have indicated about 25% dominant inheritance (17). FMF shares a similar pathogenesis with NLRP3-AID, as both conditions involve pyrin deficiency causing caspase-1 activation together with proinflammatory factor generation like IL-1β (18). However, FMF shows the typical clinical features of fever, serositis, erysipelas-like erythema and arthritis (19). For our case, DNA sequencing results ruled out the presence of any MEFV mutation in the proband. On the other hand, PFAPA is a polygenic disease strongly associated with genetic variants close to IL12A, IL10, STAT4, as well as CCR1–3 genes (20). The clinical features of PFAPA generally include periodic high fever (ranging from 39°C to 40°C), pharyngitis, cervical adenitis, aphthous ulcers, and mild abdominal/muscle pain (21). Clearly, these clinical manifestations do not match the symptoms observed in our case. Furthermore, we observed a rare presentation in our case study. A chest CT scan revealed mild interstitial lung disease, which has not been previously reported in similar case studies.
IL-1 Receptor antagonists, such as canakinumab, anakinra, and rilonacept, are safe and effective for long-term treatment (5). In this instance, our case received canakinumab therapy at 2 mg/kg every 28 days. Canakinumab accounts for the monoclonal antibody targeting IL-1β by specifically binding to soluble IL-1β (5). Notably, four weeks following the administration of canakinumab injections, the patient exhibited substantial improvement in their clinical symptoms. By week 12, the inflammatory markers had returned to near-normal levels. The patient’s height and weight also improved with proper control of these inflammatory markers. Nonetheless, an extended observation period should be necessary for assessing the long-time effect and potential adverse effects of the treatment.
The NLRP3 variant c.1718T>G (p.L573W) identified in our study may contribute to the development of NLRP3-AID. This novel variant adds to the existing spectrum of known NLRP3 mutations in human populations. However, further mechanistic studies are crucial for developing novel treatments that target NLRP3-AID.
Data availability statement
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Ethics Committee of Dongguan Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from another research group. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
MX and JW were in charge of data collection and manuscript drafting. XZ, XAZ, WC, KZ, ZZ, and HZ were responsible for study conception, design, manuscript writing, checking and revision. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Krainer J, Siebenhandl S, Weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun (2020) 109:102421. doi: 10.1016/j.jaut.2020.102421
2. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell (1999) 97(1):133–44. doi: 10.1016/s0092-8674(00)80721-7
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2022) 42(7):1473–507. doi: 10.1007/s10875-022-01289-3
4. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
5. Welzel T, Kuemmerle-Deschner JB. Diagnosis and management of the cryopyrin-associated periodic syndromes (CAPS): what do we know today? J Clin Med (2021) 10(1):128. doi: 10.3390/jcm10010128
6. Ben-Chetrit E, Gattorno M, Gul A, Kastner DL, Lachmann HJ, Touitou I, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis (2018) 7(11):1558–65. doi: 10.1136/annrheumdis-2017-212515
7. Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat (2008) 29(6):803–8. doi: 10.1002/humu.20720
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster F, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Zhou Y, Wang W, Zhong L, Wang L, Ma M, Tang X, et al. Clinical and genetic spectrum of 14 cases of NLRP3-associated autoinflammatory disease (NLRP3-AID) in China and a review of the literature. Orphanet J Rare Dis (2022) 7(1):214. doi: 10.1186/s13023-022-02364-z
10. Chen P, He L, Pang X, Wang X, Yang T, Wu H. NLRP3 is expressed in the spiral ganglion neurons and associated with both syndromic and nonsyndromic sensorineural deafness. Neural Plast (2016) 2016:3018132. doi: 10.1155/2016/3018132
11. Vahedi M, Parvaneh N, Vahedi S, Shahrooei M, Ziaee V. Identification of a new variant in NLRP3 gene by whole exome sequencing in a patient with cryopyrin-associated periodic syndrome. Case Rep Immunol (2021) 2021:2023119. doi: 10.1155/2021/2023119
12. Ducharme-Bénard S, Roberge G, Chapdelaine H. A unique presentation of NLRP3-associated autoinflammatory disease: case report. BMC Rheumatol (2022) 6(1):91. doi: 10.1186/s41927-022-00321-8
13. Naselli A, Penco F, Cantarini L, Insalaco A, Alessio M, Tommasini A, et al. Clinical characteristics of patients carrying the Q703K variant of the NLRP3 gene: A 10-year multicentric national study. J Rheumatol (2016) 43(6):1093–100. doi: 10.3899/jrheum.150962
14. Kilic H, Sahin S, Duman C, Adrovic A, Barut K, Turanli ET, et al. Spectrum of the neurologic manifestations in childhood-onset cryopyrin-associated periodic syndrome. Eur J Paediatr Neurol (2019) 23(3):466–72. doi: 10.1016/j.ejpn.2019.03.006
15. Parker T, Keddie S, Kidd D, Lane T, Maviki M, Hawkins PN, et al. Neurology of the cryopyrin-associated periodic fever syndrome. Eur J Neurol (2016) 23(7):1145–51. doi: 10.1111/ene.12965
16. Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis (2015) 74(11):2043–9. doi: 10.1136/annrheumdis-2013-204991
17. Ozdogan H, Ugurlu S. Familial mediterranean fever. Presse Med (2019) 48(1 Pt 2):e61–76. doi: 10.1016/j.lpm.2018.08.014
18. Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The pyrinInflammasome in health and disease. Front Immunol (2019) 7:1745(10). doi: 10.3389/fimmu.2019.01745
19. Rigante D, Lopalco G, Tarantino G, Compagnone A, Fastiggi M, Cantarini L. Non-canonical manifestations of familial Mediterranean fever: a changing paradigm. Clin Rheumatol (2015) 34(9):1503–11. doi: 10.1007/s10067-015-2916-z
20. Manthiram K, Preite S, Dedeoglu F, Demir S, Ozen S, Edwards KM, et al. Common genetic susceptibility loci link PFAPA syndrome, Behçet’s disease, and recurrent aphthous stomatitis. Proc Natl Acad Sci U S A (2020) 117(25):14405–11. doi: 10.1073/pnas.2002051117
Keywords: autoinflammatory disease, NLRP3-AID, NLRP3, mutation, newborn
Citation: Xie M, Wan J, Zheng X, Zou X, Chen W, Zhang K, Yuan H, Zhang Z and Zeng H (2023) Case Report: A de novo NLRP3 variant resulting in autoinflammatory disease in a Chinese newborn. Front. Immunol. 14:1238551. doi: 10.3389/fimmu.2023.1238551
Received: 29 June 2023; Accepted: 31 August 2023;
Published: 03 October 2023.
Edited by:
Ozgur Kasapcopur, Istanbul University-Cerrahpasa, TurkeyReviewed by:
Sezgin Sahin, Istanbul University-Cerrahpasa, TurkeyMikhail Kostik, Saint Petersburg State Pediatric Medical University, Russia
Copyright © 2023 Xie, Wan, Zheng, Zou, Chen, Zhang, Yuan, Zhang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haisheng Zeng, aGFpc2hlbmcyMDA2QDEyNi5jb20=
†These authors have contributed equally to this work