 Daniel H. Zimmerman1*
Daniel H. Zimmerman1* Zoltan Szekanecz2
Zoltan Szekanecz2 Adrienn Markovics3
Adrienn Markovics3 Kenneth S. Rosenthal4
Kenneth S. Rosenthal4 Roy E. Carambula1
Roy E. Carambula1 Katalin Mikecz5
Katalin Mikecz5- 1CEL-SCI Corporation, Vienna, VA, United States
- 2Department of Rheumatology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary
- 3Department of Orthopedic Surgery and Department of Internal Medicine, Division of Rheumatology, Rush University Medical Center, Chicago, IL, United States
- 4Department of Basic Sciences, Augusta University/University of Georgia Medical Partnership, Athens, GA, United States
- 5Department of Orthopedic Surgery, Rush University Medical Center, Chicago, IL, United States
Rheumatoid arthritis (RA) is recognized as an autoimmune joint disease driven by T cell responses to self (or modified self or microbial mimic) antigens that trigger and aggravate the inflammatory condition. Newer treatments of RA employ monoclonal antibodies or recombinant receptors against cytokines or immune cell receptors as well as small-molecule Janus kinase (JAK) inhibitors to systemically ablate the cytokine or cellular responses that fuel inflammation. Unlike these treatments, a therapeutic vaccine, such as CEL-4000, helps balance adaptive immune homeostasis by promoting antigen-specific regulatory rather than inflammatory responses, and hence modulates the immunopathological course of RA. In this review, we discuss the current and proposed therapeutic products for RA, with an emphasis on antigen-specific therapeutic vaccine approaches to the treatment of the disease. As an example, we describe published results of the beneficial effects of CEL-4000 vaccine on animal models of RA. We also make a recommendation for the design of appropriate clinical studies for these newest therapeutic approaches, using the CEL-4000 vaccine as an example. Unlike vaccines that create or boost a new immune response, the clinical success of an immunomodulatory therapeutic vaccine for RA lies in its ability to redirect autoreactive pro-inflammatory memory T cells towards rebalancing the “runaway” immune/inflammatory responses that characterize the disease. Human trials of such a therapy will require alternative approaches in clinical trial design and implementation for determining safety, toxicity, and efficacy. These approaches include adaptive design (such as the Bayesian optimal design (BOIN), currently employed in oncological clinical studies), and the use of disease-related biomarkers as indicators of treatment success.
1 Introduction
Currently used targeted or biological therapies in rheumatoid arthritis (RA) are effective and safe in most patients but increase the risk to certain infections. There is an unmet need for novel antigen-specific therapeutic approaches. In an earlier article, we looked at therapeutic vaccines for RA (1), and later we compared advanced targeted approaches (2) that are currently being used or proposed for the treatment of RA, including our Ligand Epitope Antigen Presentation System (LEAPS) technology (Figure 1) (6, 7).
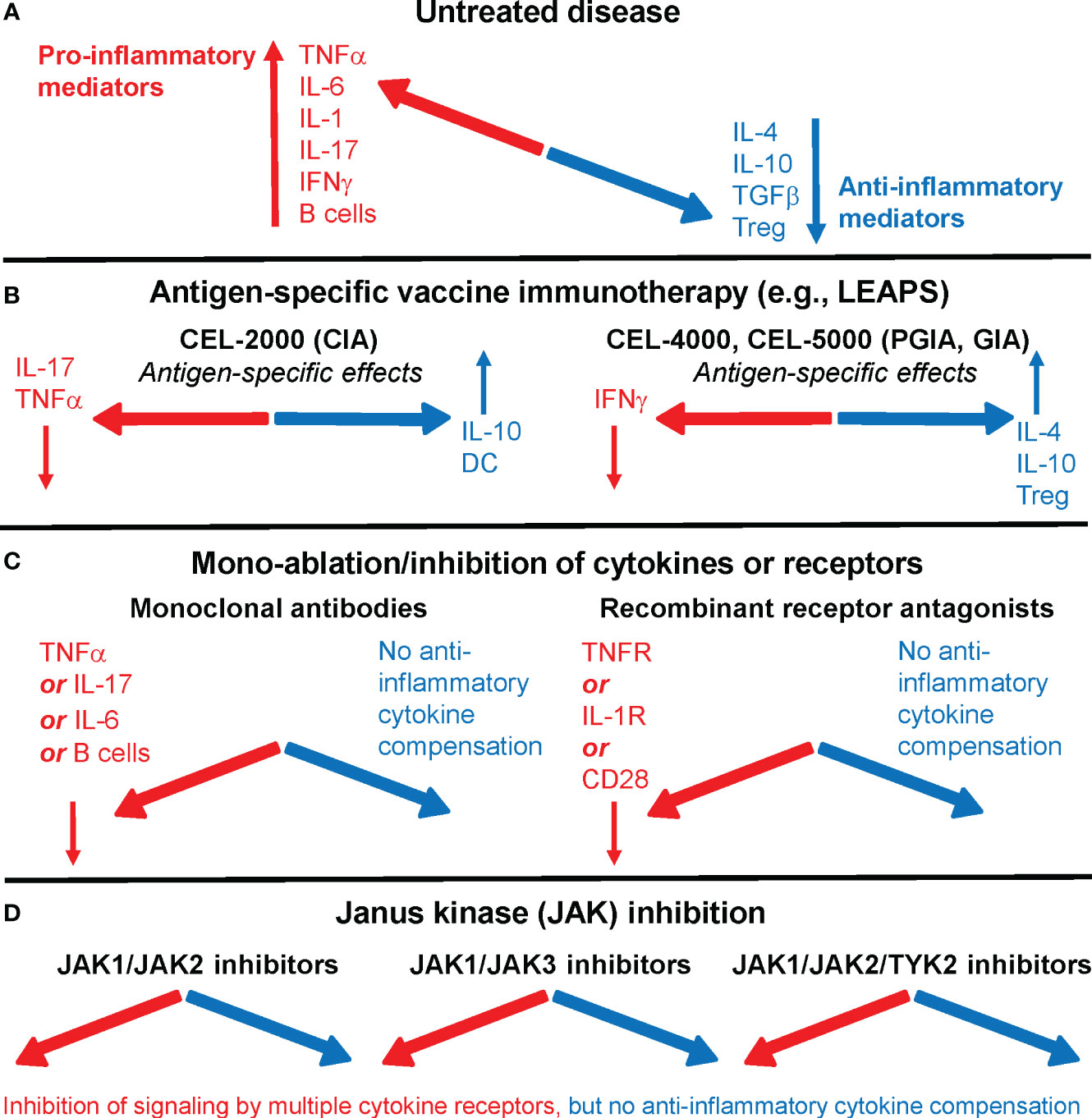
Figure 1 Immune system imbalance in rheumatoid arthritis (RA) or animal models of the disease and the effects of therapies on pro-inflammatory or anti-inflammatory mediators. (A) In untreated RA, pro-inflammatory mediators (red) overwhelm anti-inflammatory/regulatory mediators (blue). (B) Antigen-specific vaccine immunotherapy, as represented by Ligand Epitope Antigen Presentation System (LEAPS) immunotherapy in animal models of RA, restores immune balance. CEL-2000 LEAPS vaccine treatment of the collagen-induced arthritis (CIA) model of RA (left-side panel); CEL-4000 and CEL-5000 LEAPS vaccine treatments of the cartilage proteoglycan-induced and proteoglycan G1 domain-induced arthritis (PGIA and GIA) models (right-side panel) act in an antigen-specific fashion in these animal models of RA. (C) Current RA therapeutics using mono-ablation or inhibition of pro-inflammatory mediators, while reducing disease severity in most patients, introduce a different kind of immune imbalance. Monoclonal antibody treatment (left-side panel) directed against TNFα (e.g., infliximab), IL-6 receptor (tocilizumab), or B cells (rituximab), and recombinant receptor antagonists (right-side panel) induce systemic immune suppression without increasing the output of anti-inflammatory mediators. (D) Oral treatments with Janus kinase (JAK) inhibitors prevent intracellular pro-inflammatory cytokine signaling by using different classes of JAK inhibitors (e.g., tofacitinib, filgotinib, or baricitinib), but do not correct the deficiency of anti-inflammatory mediator production in RA. For more details, see references (2–5).
Many of the current approaches use monoclonal antibodies (mAbs) or soluble cytokine receptor antagonists to ablate a single cytokine, such as tumor necrosis factor (TNF)α, interleukin (IL)-1, or IL-6, or interfere with a specific T helper cell response (Th1 or Th17) by targeting the cytokines that initiate or are being produced during autoimmune/inflammatory responses in RA. Another ablative approach targets a cell surface marker (CD20) of B cells, to reduce pathological antibody production and stop the cycle of B cell-driven antigen-specific T cell activation, via elimination of B cells by an anti-CD20 mAb. These treatments are referred to as biological Disease Modifying Anti-Rheumatic Drugs (DMARDs).
More recent therapies target intracellular second messenger pathways that are activated by inflammatory cytokines by inhibiting one or more Janus kinase (JAK) family protein tyrosine kinases. The JAKs phosphorylate signal transducer and activator of transcription (STAT) proteins, which then dimerize, translocate to the nucleus, and activate genes coding for cytokines and other inflammatory mediators. The JAK inhibitors are orally administered small molecules (3).
To gain deeper insight into the rationale of current therapeutic approaches, the reader is directed to recent reviews on RA in general (8), the role of biomarkers in the disease (9), and preventative strategies in at-risk individuals for RA (10), RA vaccines (11), and RA animal models (12). For currently approved RA therapeutics and recommendations for their clinical use, see published guidelines by the European Alliance of Associations for Rheumatology (EULAR, formerly the European League Against Rheumatism) and the American College of Rheumatology (ACR) for the management of RA using synthetic and biological DMARDs (13, 14). We also direct the reader to publicly available online sources but urge caution as some sites may include “off-label” use of drugs intended for RA patients.
Immunologically, RA is driven by antigens, multiple cytokines (such as TNFα, IL-6, IL-17, IL-23, IFNγ, IL-1), and a variety of cells including T cells, B cells, macrophages, neutrophils, osteoclasts, and synovial fibroblasts. Current United States Food and Drug Administration (FDA)-approved treatments mainly focus on one component of this multicomponent disease. All of these existing therapeutic approaches are systemic and as such, they compromise immune responses that are important for preventing certain new infections or the recurrence of chronic infections, especially tuberculosis, herpes viruses, and hepatitis B and C (15). In addition, treatment with certain JAK inhibitors might be associated with an increased risk of malignancy compared to anti-TNFα therapy (16), and can also increase the risk of venous thromboembolic disease (17).
Numerous autoantigens have been implicated as potential targets for antigen-specific therapeutic interventions in RA. As the immune-mediated damage of joint cartilage is a universal feature of RA, initial studies have identified articular cartilage molecules such as type II collagen (CII), the large aggregating proteoglycan (PG, aggrecan), and chondrocyte glycoprotein 39 as autoantigens [reviewed in (18, 19)]. The autoantigen repertoire was extended to proteins present not only in the joints, but also at extraarticular locations, including autologous serum IgG-antigen immune complexes, heat shock proteins, glucose-6-phosphate isomerase (GPI) and some others (18, 19). It is important to note that arthritis can be induced in experimental animals by immunization with cartilage CII or PG [reviewed in (12)] but not by immunization with other putative RA autoantigens. However, passive transfer of GPI-reactive serum from arthritic KBxN mice to non-diseased naïve animals can induce arthritis in the recipients (12). Nonetheless, the relationship between adaptive immune reactivity to these autoantigens and joint pathology in RA (or animal models) has not been completely understood (18). A paradigm shift occurred in the field of autoimmunity associated with RA with the discovery of anti-citrullinated protein antibodies (ACPAs) in the serum of a large proportion of RA patients (20). While serum ACPA helped develop an important diagnostic tool for RA, current immunological views (21) suggest that the majority of citrullinated proteins represent neoantigens (instead of autoantigens) that have undergone extensive post-translational modification (citrullination) and then triggered the production of widely cross-reactive ACPAs, and likely T cell responses, in RA patients. Thus far, attempts to identify Th cells that promote the production of singular antigen-specific ACPAs by B cells of RA patients have been met with limited success (21).
2 Antigen-specific and immunomodulatory therapies in RA and animal models
As an alternative to ablative therapies, immunomodulatory therapy represents an approach to rebalancing the immune system. Modulation of the functions of the adaptive immune system as a treatment of RA has been attempted using cell-based therapies or autoantigen-loaded liposomes (Table 1).
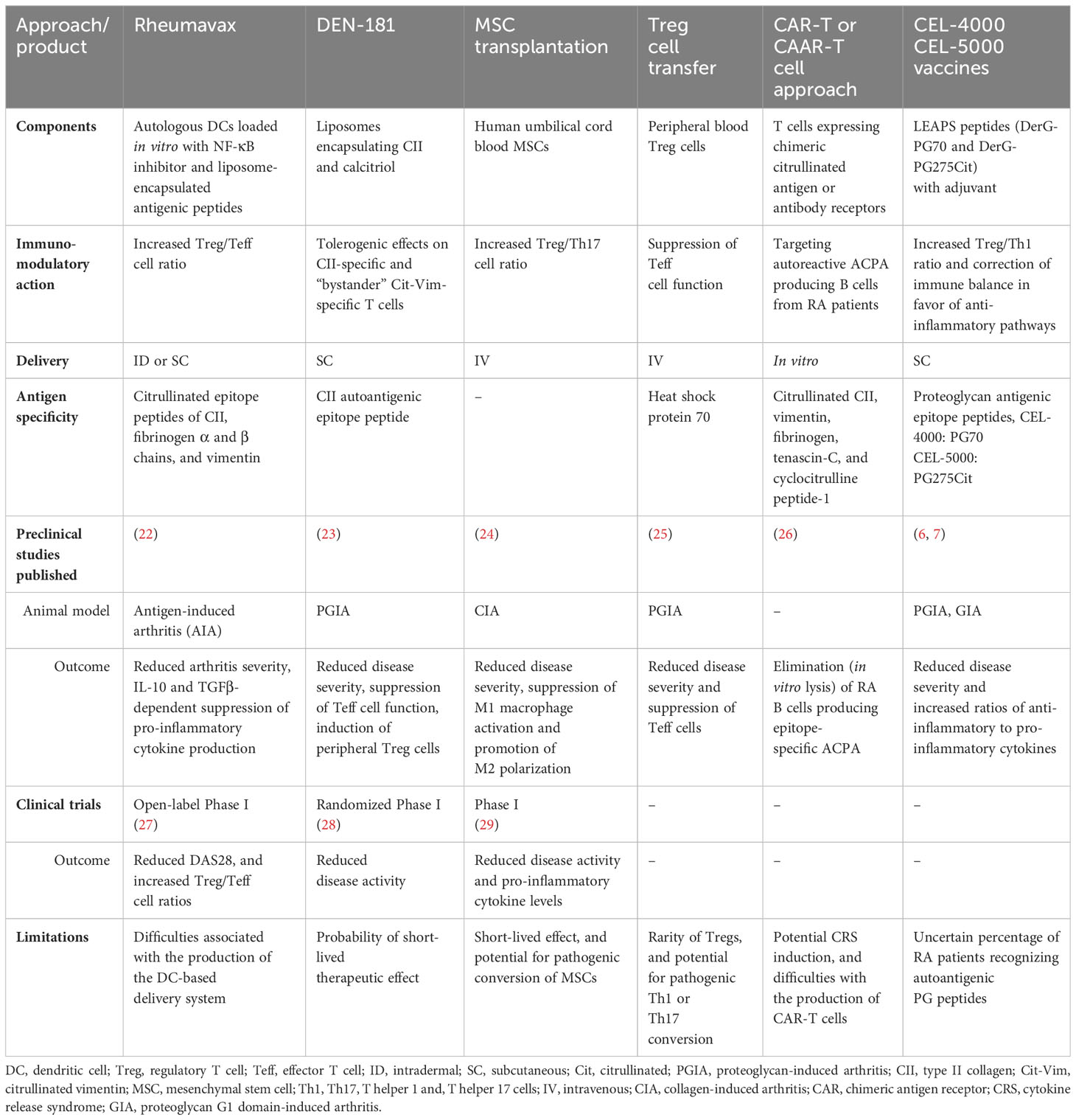
Table 1 Antigen-specific and immunomodulatory therapeutic approaches in RA.
Several preclinical and clinical trials have investigated the potential benefit of mesenchymal stem cell (MSC) transplantation in RA. For example, human MSCs have been reported to significantly attenuate collagen antibody-induced arthritis (CAIA) in mice (30). In clinical studies with RA patients, MSC therapy resulted in an increased T regulatory (Treg)/Th17 cell ratio (31, 32) and improved immune balance as indicated by decreased IL-1β, IL-6, IL-8 and TNFα, as well as increased production of IL-10 and transforming growth factor (TGF)β (29, 33). However, the potential of MSCs to differentiate into pathogenic cells in vivo is a serious concern in these studies (Table 1).
Based on the immunomodulatory effects of Treg cells, adoptive Treg cell transfer has gained attention in recent years. This approach requires the isolation, activation, and expansion of autologous or allogeneic Tregs prior to infusion back into the patient (34). In the collagen-induced arthritis (CIA) mouse model, adoptively transferred Tregs decreased disease severity and progression (35). Despite promising results, the clinical application of adoptive Treg transfer is hindered by the low occurrence of Tregs in peripheral blood, the probability of conversion into pathogenic T cells, and difficulties in processing the cells for therapy (36, 37) (Table 1). As an alternative, the safety and efficacy of a mAb that selectively activates Tregs has been investigated in a phase IIb, randomized, placebo-controlled clinical trial in patients with active RA, but did not demonstrate efficacy (38).
The clinical success of chimeric antigen receptor (CAR)-T cells in haemato-oncology boosted interest towards their application in autoimmune diseases. Recently, patients with refractory systemic lupus erythematosus (SLE) were treated successfully with CD19-directed CAR-T cell therapy to ablate B cells (39). In this promising study, CD19-targeting CAR-T cell treatment led to drug-free remission in SLE patients even after B cell recovery occurred post-treatment. One specific CAR-T cell approach proposed for RA is represented by the chimeric autoantibody receptor-T (CAAR-T) cells. These cells express an extracellular autoantigen recognized by the B cell receptor. The T cell-expressing autoantigen promotes binding to autoreactive B cells, subsequent CAAR-T cell activation and then lysis of the pathogenic B cells (40). It has been hypothesized that development of CAAR-T cells, expressing citrullinated antigens, would allow selective deletion of B cells producing anti-citrullinated protein Abs (ACPA, a hallmark of RA), while saving other B cells (41). In an RA study, four citrullinated peptide epitopes were selected as ligands for targeting autoreactive ACPA-producing B cells. Engineered T cells expressing a fixed anti-fluorescein isothiocyanate (FITC) CAR were constructed and tested for their ability to eliminate ACPA-specific autoreactive B cells. The anti-FITC CAR-T cells demonstrated the potential to recognize the corresponding FITC-labeled citrullinated peptide epitope and lyse autoreactive B cell subsets from RA patients in vitro (26). Nevertheless, the potential application of CAR- or CAAR-T cells in autoimmune diseases is limited by the various types of autoreactive responses driving RA, the different immune cells involved in RA, and serious side effects such as Cytokine Release Syndrome (CRS, also known as “cytokine storm”) as well as by the difficulty in preparing CAR- or CAAR-T cells (42) (Table 1).
Dendritic cell (DC)-based immunomodulatory approaches showed initial success in both animal models (22) and clinical trials in ACPA positive RA patients genetically predisposed to disease by possessing human leukocyte antigen-shared epitope (HLA-SE) risk alleles (27). Rheumavax, an intradermally administered vaccine was created from autologous DCs that were loaded with an irreversible NF-κB inhibitor (BAY11-7082) and exposed to liposomes containing four citrullinated peptide antigens (collagen type II1237-1249-Cit1240, fibrinogen α chain717-725-Cit720, fibrinogen β chain433-441-Cit436, and vimentin447-455-Cit450), respectively. Rheumavax (27) was administered once intradermally at two dose levels to HLA-SE and ACPA positive RA patients. This open-label phase I trial demonstrated a good safety and biological activity profile for Rheumavax as indicated by an increased ratio of Treg to T effector (Teff) cells and decreases in disease activity score in 28 joints (DAS28) within 1 month in Rheumavax-treated patients (Table 1). Again, although autologous cell therapies have been commercially available for almost a decade (43), to date, they have not been proven to be economically successful due to the extremely high cost of generating these patient-specific cells.
Another DC-targeting approach was tested in a double-blind, placebo-controlled exploratory phase I trial (28). DEN-181 vaccine, comprised of liposomes encapsulating a self-peptide collagen II259-273 (an autoantigen) and a NF-κB inhibitor 1,25-dihydroxycholecalciferol (calcitriol), was tested in RA patients in this trial. A single ascending dose of DEN-181 was administered subcutaneously to ACPA positive, HLA-SE positive RA patients treated with methotrexate only. DEN-181 was well tolerated and showed dose-associated immunomodulatory effects (28). In a pre-clinical animal study, the same group used a cartilage proteoglycan (PG, aggrecan)-derived peptide aggrecan89-103 as an autoantigenic epitope encapsulated into liposomes with or without calcitriol, and the liposomes were tested in the PG-induced arthritis (PGIA) model of RA. The authors reported that disease severity significantly decreased in an antigen-specific manner in mice receiving aggrecan89-103/calcitriol-containing liposomes every four days after disease onset (23) (Table 1).
As an easier-to-deliver mechanism for treating RA, we reported promising results with a peptide conjugate-based, antigen-specific immunomodulatory approach utilizing the LEAPS technology-based CEL-4000 vaccine (see Figure 1; Table 1). This approach promotes the rebalancing of pathogenic immune responses by inducing immunomodulation, not just by inducing tolerance or immune suppression, (44–47). CEL-4000 modulates the production of pro-inflammatory and anti-inflammatory/regulatory cytokines in a downward and an upward direction, respectively, and most importantly, in an antigen-specific manner. CEL-4000 incorporates the immunogenic PG70 epitope (from cartilage PG aggrecan) into a conjugate with the DerG LEAPS immune cell binding ligand (ICBL). The DerG ICBL peptide within the conjugate binds to, and acts on, CD4+ T cells. As illustrated in Figure 2, the disease (RA)-related epitope peptide moiety (PG70) of the LEAPS conjugate is presented to cognate CD4+ T cells through the engagement of MHC II on the surface of antigen-presenting cells (APC) and the TCR of the T cell while the DerG ICBL modulates the T cell activity through CD4 (48). Using fluorescence-labeled tetramers of the DerG-PG70 (CEL-4000) peptide for flow cytometry, our earlier study (7) demonstrated preferential binding of this conjugate to CD4+ T cells isolated from a PG-induced murine arthritis model of RA.
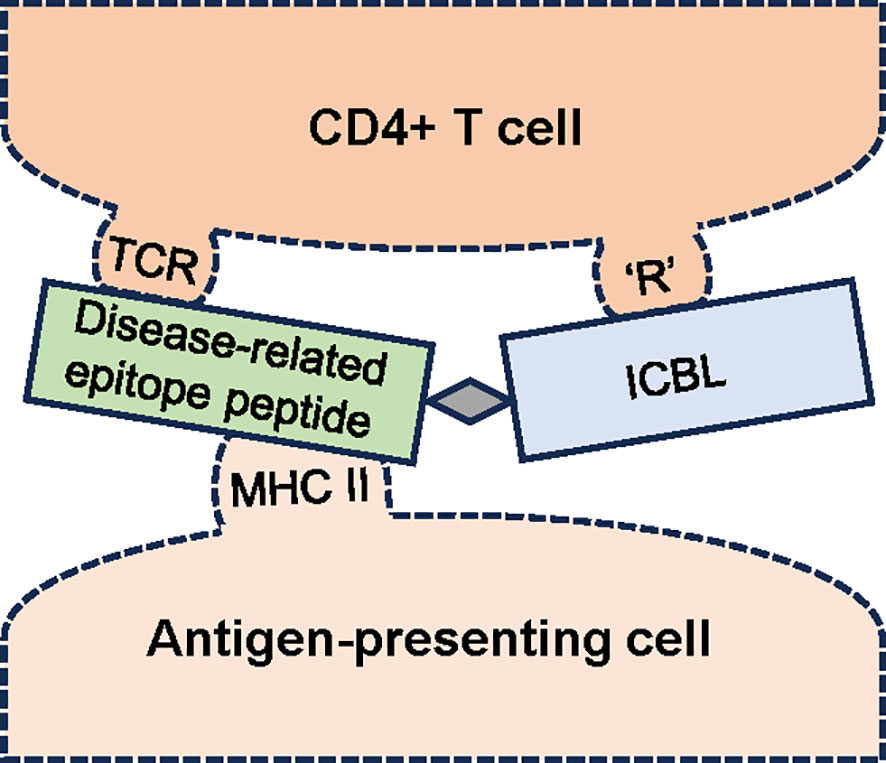
Figure 2 Schematic diagram of the interaction of an antigen-specific ligand epitope antigen presentation system (LEAPS) peptide conjugate with cognate CD4+ T cells. The LEAPS peptide is composed of an immune cell binding ligand (ICBL, blue rectangle), in this case, a DerG peptide derived from the β chain of human major histocompatibility complex (MHC) II. The ICBL moiety can bind to CD4 or another receptor (‘R’) on the T cell surface. DerG binding to CD4 modulates cellular activity. The disease-related epitope peptide (green rectangle), in this case, PG70 derived from human cartilage proteoglycan, is connected to the ICBL via a 3 amino acid spacer (grey diamond). The PG70 peptide is presented in the context of MHC II on the surface of the antigen-presenting cell (APC) to the T cell receptor (TCR). The binding of DerG-PG70 LEAPS peptide conjugate to cognate CD4+ T cells and APCs has been demonstrated in our earlier study (7).
Thus, by engaging CD4+ T cells, CEL-4000 induces reduction or conversion of Th1 and Th17 cytokine production, resulting in a dominance of Th2 and Treg responses (6, 7). CEL-4000 was tested in the PGIA mouse model of RA (developed by Tibor T Glant and his colleagues) (49, 50). The PGIA and recombinant proteoglycan G1 domain-induced arthritis (GIA) models were used for the LEAPS studies for several reasons: (i) The diseased mice best resemble the pathology of RA; (ii) Like seropositive RA patients, the mice with PGIA or GIA produce ACPA and rheumatoid factor (RF); (iii) Arthritis development is more frequent in females than males; and (iv) Disease incidence increases with age. These features are not readily observed in other RA models, such as CIA or adjuvant/pristane-induced arthritis (AIA/PIA). Mice in our preclinical studies received three PG or recombinant human PG G1 domain (rhG1) antigen injections to initiate an early RA-like disease (PGIA or GIA) before treatment with the CEL-4000 vaccine. The first vaccine was administered subcutaneously right after the initial development of arthritis symptoms was noted in the fore- and hindlimbs, and a second dose was delivered in a similar way two weeks later. Improvement in disease severity, as assessed by visual arthritis scores (VAS), was noted at three weeks after the initiation of vaccine treatment, and the mice receiving CEL-4000 ended up with lower VAS as well as milder histological signs of joint inflammation than the controls receiving adjuvant only (7). CEL-4000 also modulated the animals’ immune responses by upregulating the production of anti-inflammatory/regulatory cytokines and downregulating the secretion of pro-inflammatory cytokines (Table 1). This was evident by detecting increases in IL-4, IL-10, and TGFβ levels and Foxp3+ Treg cells as well as decreased production of TNFα, IL-17 and IFNγ (6, 7). Presumably, such a rebalancing of the immune response cannot be observed in non-diseased (non-immunized, naïve) animals because we hypothesize that the CEL-4000 vaccine acts only on established, disease-driving and antigen-specific T cells (6, 7), which are not present or not active in naïve non-diseased animals or their healthy human counterparts. The antigen-specificity and immunomodulatory mechanism of the LEAPS vaccine has an advantage over the systemic ablative or inhibitory effects of DMARDs (mAbs or receptor antagonists, or the JAK inhibitors), as these currently used approaches compromise overall immunity and none of these therapies amend the dysregulation of the immune system that is observed in RA.
Additional developments have occurred for other forms of vaccines, including DNA vaccines, exosomes, Poly-lactic-co-glycolic acid (PLGP) particles, and epitope-modified (i.e., altered peptide ligand, APL) peptides, which have been examined in animal models of RA or other autoimmune diseases. A mRNA vaccine was tested in the autoimmune mouse model of multiple sclerosis (51) but similar tests have not yet been reported in animal models of RA. This is likely to change, based on the successes of mRNA vaccines against COVID-19. However, a mRNA vaccine requires a target antigen(s), but only a limited number of new RA-specific antigenic epitopes have been investigated, and the protective cytokines and immune responses in disease models of human RA (23, 27, 28, 44–47, 52–60) have not been recently revisited.
3 Preclinical studies of the CEL-4000 vaccine in mouse models of RA
For a vaccine therapy for RA to be effective, it must be able to act on the established pro-inflammatory and disease-driving immune responses, primarily on T cell-mediated immunity. As mentioned earlier, CEL-4000 vaccination successfully curtailed disease progression in mice already showing arthritis symptoms and immune reactions to PG (PGIA model) or to the recombinant G1 domain of PG (GIA model) (6, 7). The imbalance of immune responses in the diseased mice was demonstrated by increased (>1) ratios of pro-inflammatory cytokines such as IFNγ, IL-17, and TNFα, to anti-inflammatory/regulatory cytokines such as IL-4, IL-10, and TGFβ. This cytokine imbalance in the PGIA and GIA models of RA favored the pro-inflammatory Th1 and Th17 T cell responses over the Th2 and Treg regulatory responses. Disease progression was curtailed and the ratios were flipped in favor of the Th2 and Treg cytokines in mice treated with CEL-4000 (DerG-PG70, a conjugate of the DerG ICBL and the PG70 epitope peptides) but disease continued to progress in those treated with a LEAPS vaccine composed of a J ICBL and PG70 (J-PG70) (6, 7). Our recent studies showed that CEL-4000 (DerG-PG70), directed to the appropriate immune cells by the DerG ICBL, modulated the ongoing inflammatory immune response in an antigen (PG70)-specific manner, thus correcting the dysregulated immunological state involved in the disease. Since CEL-4000 is an antigen-specific vaccine, other immune responses should remain intact due to the absence of systemic immune suppression.
In a different RA model (CIA), driven by a different antigen, and a Th17 CD4 T cell response, a J-CII conjugate vaccine (CEL-2000) also reduced disease severity and serum levels of IL-17 in mice (61). This highlights the importance of determining which Th pro-inflammatory cytokines (Th17 in CIA) are driving the disease in order to utilize the appropriate LEAPS therapy to rebalance the immune system and obtain a favorable response to LEAPS therapy.
Studies were also performed on a second DerG-LEAPS vaccine that incorporated a different PG peptide representing a citrullinated PG epitope (DerG-PG275Cit; CEL-5000) (Table 2). Citrullinated proteins are considered to be neoantigens that, through elicitation of autoAbs and Th cells, may trigger the onset of disease in RA patients (62). The PG275Cit alternative peptide was also investigated to provide an additional epitope for individuals with a different T cell receptor (TCR) specificity. Although initial human studies are likely to be performed on CEL-4000 only, having two therapeutic LEAPS vaccines offers an opportunity to switch therapies for an individual or to combine the therapies to have broader antigenic coverage in a future immunomodulatory treatment of RA. CEL-5000 was tested alone and in combination with CEL-4000 in mice with GIA. The progression of arthritis was curtailed in mice treated with either CEL-5000 alone or in combination with CEL-4000. Interestingly, the humoral immune response to CEL-5000 alone was different from that to CEL-4000 (6).
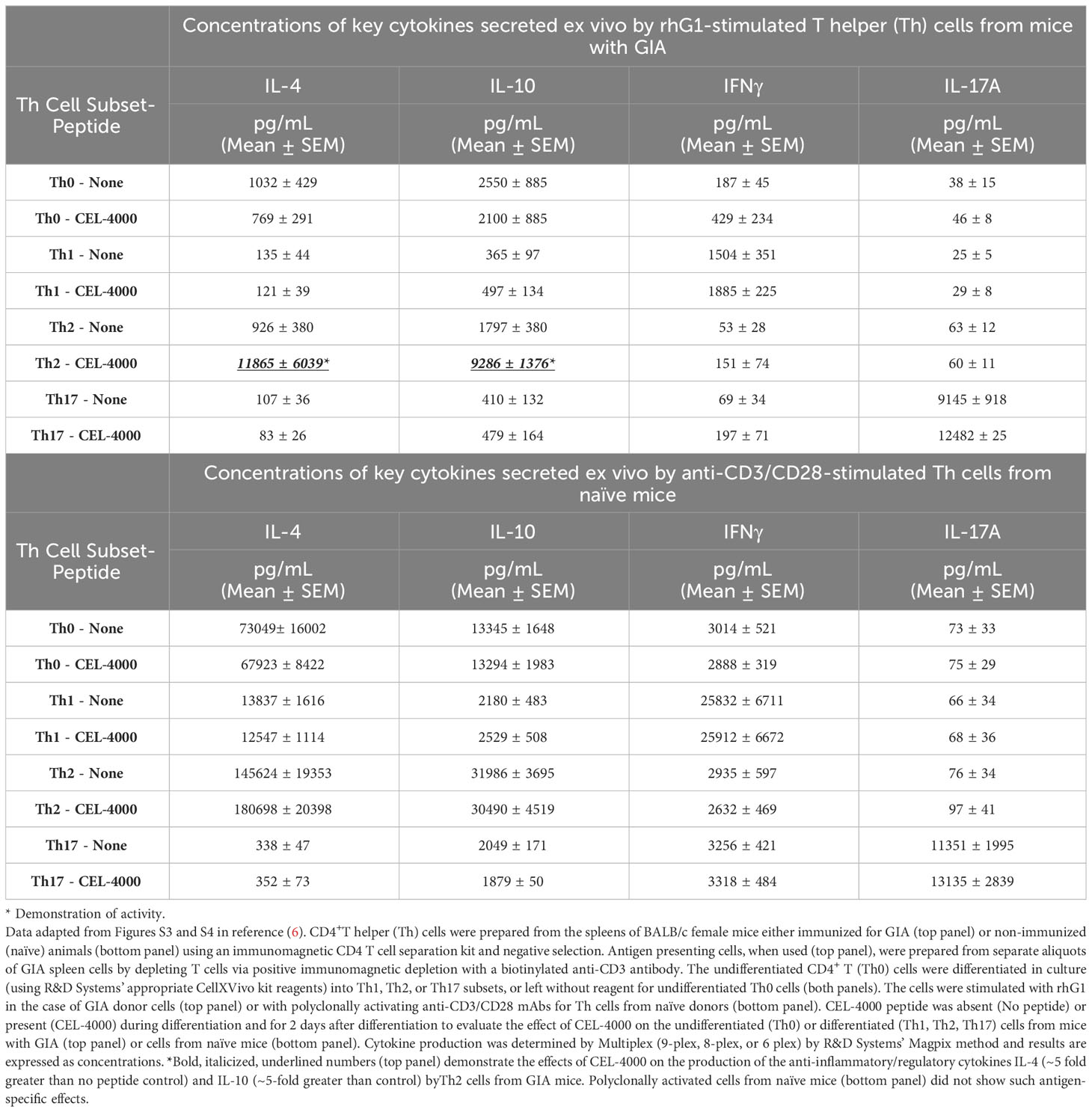
Table 2 Effects of CEL-4000 peptide on ex vivo differentiated and stimulated CD4+ T cells from mice with GIA or from naive mice.
As we stated in a previous PG epitope-related study (63), the PG70 epitope sequence differs, but the PG275 epitope sequence is identical, between humans and mice. Mice with either PGIA or GIA recognize PG70 as a “foreign” epitope, but poorly recognize the PG275 “self” epitope. This was also reflected in the serum Ab titers, as the GIA mice did not produce much antibody against either the native or the citrullinated form of the PG275 epitope but made Abs to the PG70 epitope (6) (Figure 3). Even though mice with GIA produced Abs only in response to vaccination with CEL-4000 (or with the combination of CEL-4000 and CEL-5000), both therapies were effective because the immune modulation was through T cells and the anti-CEL-4000 Abs (shown in Figure 3) probably did not neutralize the CEL-4000 vaccine. In a previous PG epitope-related in vitro study on RA patient cell and serum samples, most of the patient samples showed reactivity (T cell and Ab responses) to both the PG70 and PG275 epitopes, especially to the citrullinated version of PG275 (63), suggesting that treatment with CEL-4000 and CEL-5000 vaccines (alone or in combination) would be effective in a certain proportion of patients with RA.
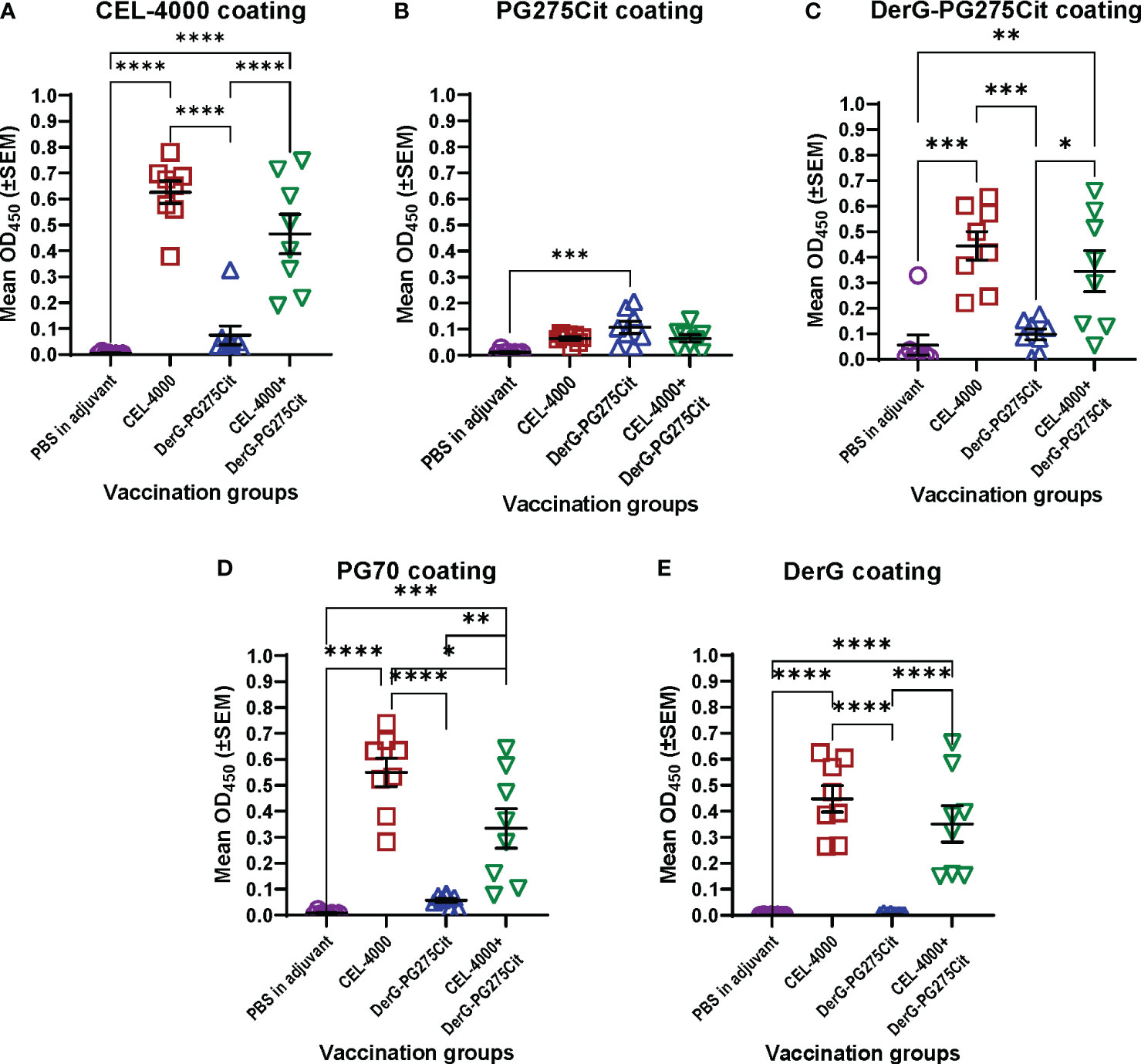
Figure 3 Serum antibody responses of mice with GIA after vaccination with adjuvant (control) or CEL-4000, DerG-PG275Cit (CEL-5000) or the combination of CEL-4000 and CEL-5000. Mouse serum samples (at 1:1000 dilution) were reacted with plate-immobilized CEL-4000 (DerG-PG70) (A), PG275Cit (B), DerG-PG275Cit (C), PG70 (D) or DerG (E) peptides. IgG antibodies bound to the peptides were detected with anti-mouse IgG antibody. The optical density (OD) was determined at 450 nm using an ELISA reader. Data are expressed as the mean ± SEM (n = 8 mice/group; data analyzed using one-way ANOVA followed by Tukey`s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). Data adapted from Figure 6 in reference (6).
The classical approach to demonstrating an immune response to most vaccines [see the guidance document including the references by US FDA (64)] is to examine anti-vaccine Ab titers. CEL4000 was tested for safety and anti-vaccine responses in healthy non-human primates (NHP, cynomolgus monkeys) by Altasciences, Co, Inc, (Laval, Quebec, Canada, and Everette, WA, USA), an independent research agency. Altasciences is a Good Laboratory Practice (GLP) practicing Contract Research Organization (CRO). They reported detection of anti-CEL-4000 Abs, but no T cell immune responses to vaccination (data available from Altasciences and on file), as would be expected for a nondiseased animal. This demonstrates that, for the LEAPS vaccines, anti-peptide Abs may actually be inappropriate and unrevealing as indicators of therapeutic efficacy. Better surrogate markers for both the CEL-4000 or a combined CEL-4000/CEL-5000 vaccine treatment efficacy would be changes in cytokine profiles indicative of restored immune regulation as well as reduction of disease symptoms.
As an example of how surrogate markers can be used, the immunomodulatory effects of CEL-4000 or the combination of CEL-4000 and CEL-5000 vaccines were demonstrated on established effector/memory T (Teff/Tm) cell responses. T cells of mice with GIA were treated in vitro with the immunizing antigen (rhG1) to expand antigen-specific Teff and Tm cell populations in culture, then treated to promote their differentiation into Th1, Th2 or Th17 cells or left untreated (for Th0 undifferentiated cells) prior to in vitro treatment with the CEL-4000 peptide (Table 2). As expected from earlier studies on the DerG (or G) ICBL (65–68), CEL-4000 had an effect on Th2 and Treg cells in vivo (6, 7), which was demonstratable herein in vitro on antigen-experienced differentiated Th2 cells by the increased production of IL-10 and IL-4 (a regulatory and an anti-inflammatory cytokine, respectively), but no similar effect was noted for the Th0, Th1, or Th17 cells obtained from mice with GIA (Table 2, top panel). In addition, as shown in Table 2 (bottom panel), CEL-4000 treatment did not affect similarly differentiated T cells from naïve mice (of the same sex and strain) when these cells were treated with the polyclonal activator anti-CD3/anti-CD28 mAbs in vitro (6). Antigen specificity of the PG70 peptide and the importance of the DerG ICBL in CEL-4000 was demonstrated by the lack of response to a PG70 conjugate incorporating a different ICBL (J- PG70) and to a DerG peptide attached to an irrelevant influenza virus epitope, respectively (6). By acting on Th2 (and possibly Treg) cells in these in vitro experiments, CEL-4000 appeared to provide an enhanced and antigen-specific regulatory mechanism by modulating the profile and function of Tm/Treg cells obtained from mice with GIA.
4 Recommendation for adaptive design clinical studies of CEL-4000 vaccine
Designing or planning, and possibly conducting, phase I studies for vaccine-based immunomodulation (i.e., immunotherapy) for ongoing RA requires a different approach than a preventive vaccine. For a preventive vaccine administered to an “immunonaive” individual for protection against infection, once safety has been determined, efficacy can be indicated by the presence and titers of neutralizing anti-microbial Abs, which can be readily assayed. T cell responses and associated cytokines are secondary findings even if they are important for protection. For a therapeutic vaccine administered to a patient with RA, the relevant findings, in addition to lessening of the disease symptoms, are the changes in T cell responses and/or specific cytokine production, the values of which will be presumably different in each patient.
Evaluation of the efficacy of therapies for RA (as for cancer, for example) is only relevant in disease-bearing animals or individuals. In all cases, efficacy of the treatment is indicated by curtailment of the progression or by accelerated resolution of the disease. Therefore, relevant human testing of the CEL-4000 vaccine must be performed on individuals with RA. Ideally, testing would be performed on patients with early-stage RA by demonstrating reduced disease progression as well as changes in immunological parameters as surrogate indicators of a successful provision of therapy. For such a vaccine, adaptive design studies, commonly used for testing cancer therapies on diseased patients (even for phase I First in Human/First in Man (FIH/FIM) studies), would be most appropriate, as they are considered more ethical and efficient than standard phase I trials with healthy volunteers.
In designing Phase 1 studies for CEL-4000, we reviewed several studies and websites for guidance, especially those that were carried out for RA, and those employing adaptive design efficacy-based studies (69–76). As shown in Figure 4 (as modified from Pallman et al. (69)), an adaptive design study proceeds with concurrent analysis of safety and efficacy based on agreed-upon rules to allow progression of the study (determined prior to trial initiation) and implemented by an independent drug safety monitoring board. For example, with a low number of subjects treated with a single dose of vaccine, specific steps (Design, Conduct, Analyze, Review and Adapt) are established, and data is analyzed and used to determine the optimal treatment regime for the next round or cycle. Adaptive design studies allow a change in dose during the course of a trial, based on safety or efficacy parameters. If a dose is judged to be unsafe, based on a > 2 serious adverse event (SAE) score, then it is futile to continue, and a futility designation is made to discontinue treatments at the given or higher doses. However, if a dose is judged as safe i.e., having a ≤ 2 SAE score, then the study can proceed to examine the parameters of efficacy or surrogates of efficacy in the same subjects. Notably, futility consideration can also occur with acceptable safety profiles if immune response or efficacy criteria are not met, but this scenario would allow an increase in dosing or frequency of administration within safety limits, instead of study termination.
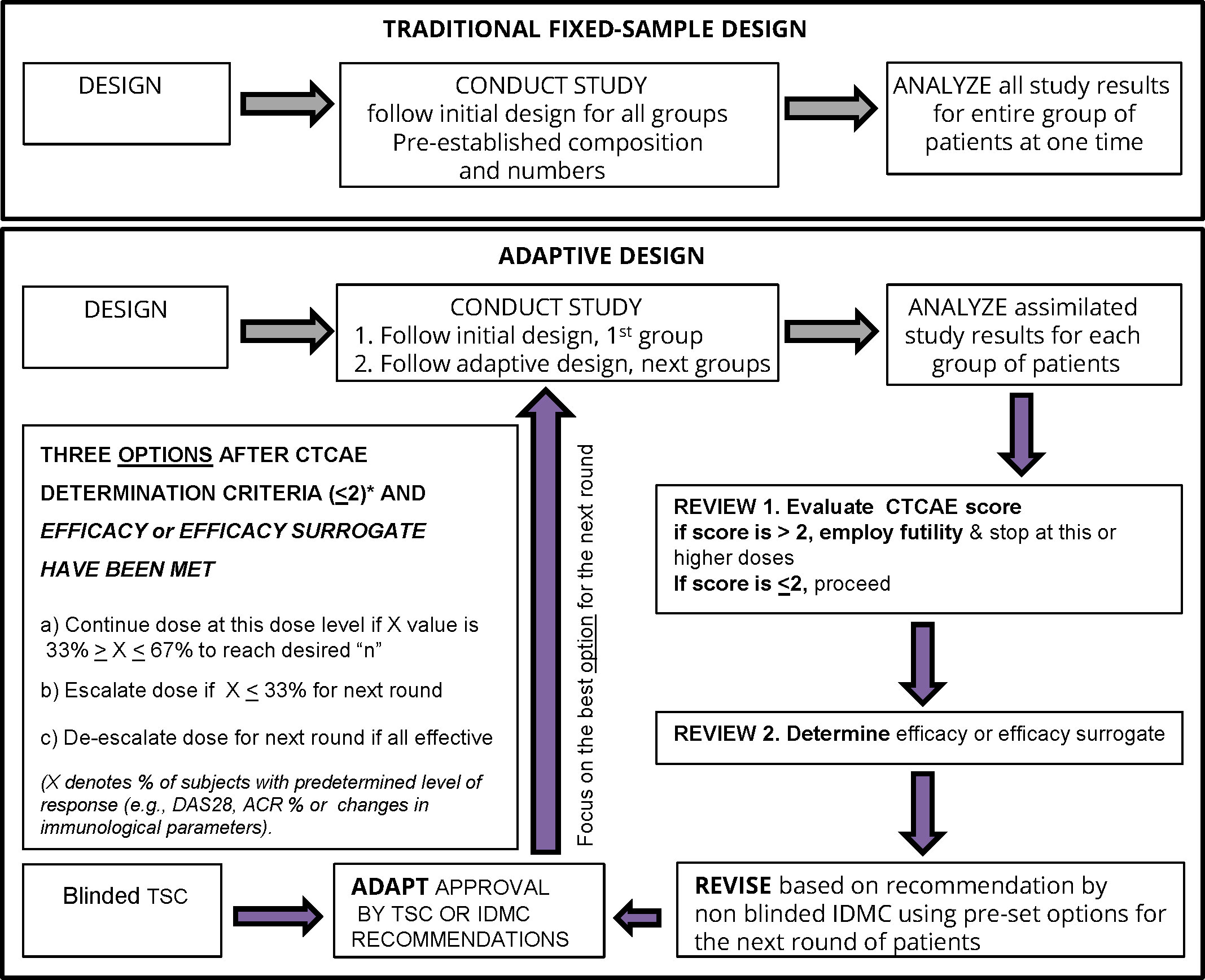
Figure 4 Bayesian optimal interval (BOIN) adaptive design study proposed for a clinical trial of therapeutic vaccines such as CEL-4000 for RA. The figure is adapted from Figure 1 in Pallman et al. (69). The decision steps for a conventional (traditional fixed-sample) and a BOIN study are compared. For the BOIN adaptive study, if the 1st step passes (for safety or toxicity) with CTCAE <2 then continuation proceeds to the 2nd step for determination of efficacy or a surrogate of efficacy. The criteria for the 4 options for continuing to the next round are listed in the largest box in the left side of the figure. The options are based on tests that determine the percent of responders (X) as indicated by pre-determined rules for evaluating disease activity (DAS28*or ACR* response %), toxicity, or treatment efficacy either by response rate with a pre-determined X value or favorable surrogate efficacy parameters (e.g. biomarker indicative of positive result such as decreased inflammatory (TNFα, IFNγ, IL-17) or increased anti-inflammatory (IL-4, IL-10, TGFβ) levels and/or favorable ratios and/or specific numbers of appropriate cell types (e.g., Th1, Th2, Th17, Treg) or other select biomarkers). Abbreviations: ACR, American College of Rheumatology; CTCAE, Common Criteria Terminology for Adverse Events (77); DAS28, Disease Activity Score for 28 joints; IDMC, Independent Data Monitoring Committee; TSC, Trial Safety Committee.
The adaptive design is best demonstrated and formalized in the Bayesian optimal interval (BOIN) studies (78–82). The advantage of the adaptive or Bayesian design approach is that it allows examination of both the safety/toxicity and efficacy parameters (lessening of the symptoms or surrogates of efficacy, as criteria) all at the same time and with pre-established and agreed-upon rules for discontinuation or redirection at specified points as the study progresses. The BOIN approach is recognized by the US FDA and European Medicines Evaluation Agency (EMEA) authorities as appropriate for certain FIH/FIM studies and has been used in oncology clinical trials for new peptide therapies including peptide vaccines (78–83). Bayesian design is based on a method of statistical inference in which Bayes’ theorem is used to update the probability (p value) for a hypothesis being tested as more evidence or information becomes available, e.g., after the start of the treatment of the subjects enrolled in the study. The FDA is encouraging the use of the adaptive design, such as the BOIN approach, because it is more ethical (providing therapy and demonstrating safety and non-toxicity in diseased instead of healthy individuals) and often requires fewer subjects by not continuing if prevalent futility elements are present.
For CEL-4000, the emphasis of the first trial on RA patients would be on the safety, and lack of toxicity, including immunotoxicity (e.g., an unexpected cytokine storm). If these criteria are met, the study would then proceed to the second step of efficacy, as indicated by reduction of symptoms or a surrogate indication of therapeutic success, such as (non-neutralizing) Ab production to CEL-4000 and changes in the production of cytokines (or ratios) or the types of T cells indicative of beneficial immunomodulation. Although the adjuvant (manufactured by Seppic and called ISA51vg or Montanide) used in our LEAPS vaccine formulations has proven safe in numerous human trials for cancer vaccines (including the ones using peptides similar to CEL-4000) for many years (see recent review by Melssen et al. (84)), it may also be necessary to evaluate the adjuvant’s safety separately.
It is proposed that subject selection will follow the most recent recommendations of the EULAR and the ACR for the management or RA using synthetic and biological DMARDs (13, 14) as approved by the study sponsor’s independent data monitoring committee (IDMC). Further, appropriate inclusion and exclusion criteria will be applied. The subject selection/inclusion criteria and the study design will have to be approved and performed at an appropriate clinical site.
The adaptive or Bayesian design with the BOIN approach seems to be the best for testing an immunomodulatory therapy, such as the CEL-4000 vaccine, in RA patients for several reasons (Figure 4). It would allow examination of numerous parameters at the same time, e.g., safety and toxicity, while also setting milestones for efficacy (beneficial changes in symptoms) or surrogates of efficacy (e.g., reduction of pro-inflammatory cytokines, and/or increase in anti-inflammatory/regulatory cytokines and Tregs). If a dose is judged as not safe, a futility designation would be made, and the study at that dose would be discontinued and possibly a lower dose would be tested. However, if a dose is judged as safe, then the study would proceed to examine efficacy or a surrogate of efficacy in the same subjects, and if appropriate, to consider an escalation or de-escalation of the dose in the next round of the study (77). The adaptive study design can also be applied to immunological parameters for determination of the best surrogate biomarkers indicative of treatment efficacy. These decisions can be made on the same day when the safety review is performed by the IDMC. The BOIN approach would be more economical, efficient, and compassionate for the participants than the traditional, fixed-sample study design (Figure 4).
Refinement of the CEL-4000 vaccine study would include various considerations, e.g., the criteria for patient inclusion or exclusion for the study, specific details regarding the futility designations, and potential comparisons with established RA treatments. Patient inclusion may potentially include screening for the presence of T cell cytokines that are promoting inflammation or other ex vivo parameters indicative of T cell and/or Ab reactivity to the vaccine if resources of such tests are available. Table 3 shows some of the risk factors and established laboratory findings related to RA for which the potential study population could be screened. In addition to age, gender, and smoking status, the patient’s HLA alleles, and RA-specific laboratory parameters (85–87) can be reviewed, keeping in mind that RA is a heterogeneous disease. Unfortunately, the field of RA-related biomarker tests has not advanced as fast as desired in order to allow identification of the best biomarkers to focus on at this stage of product development. For further insights, the reader is referred to the description of a RA activity biomarker test (MBNA Vectra®, now provided by LabCorp) (85–87) and a RA genetics review (88).
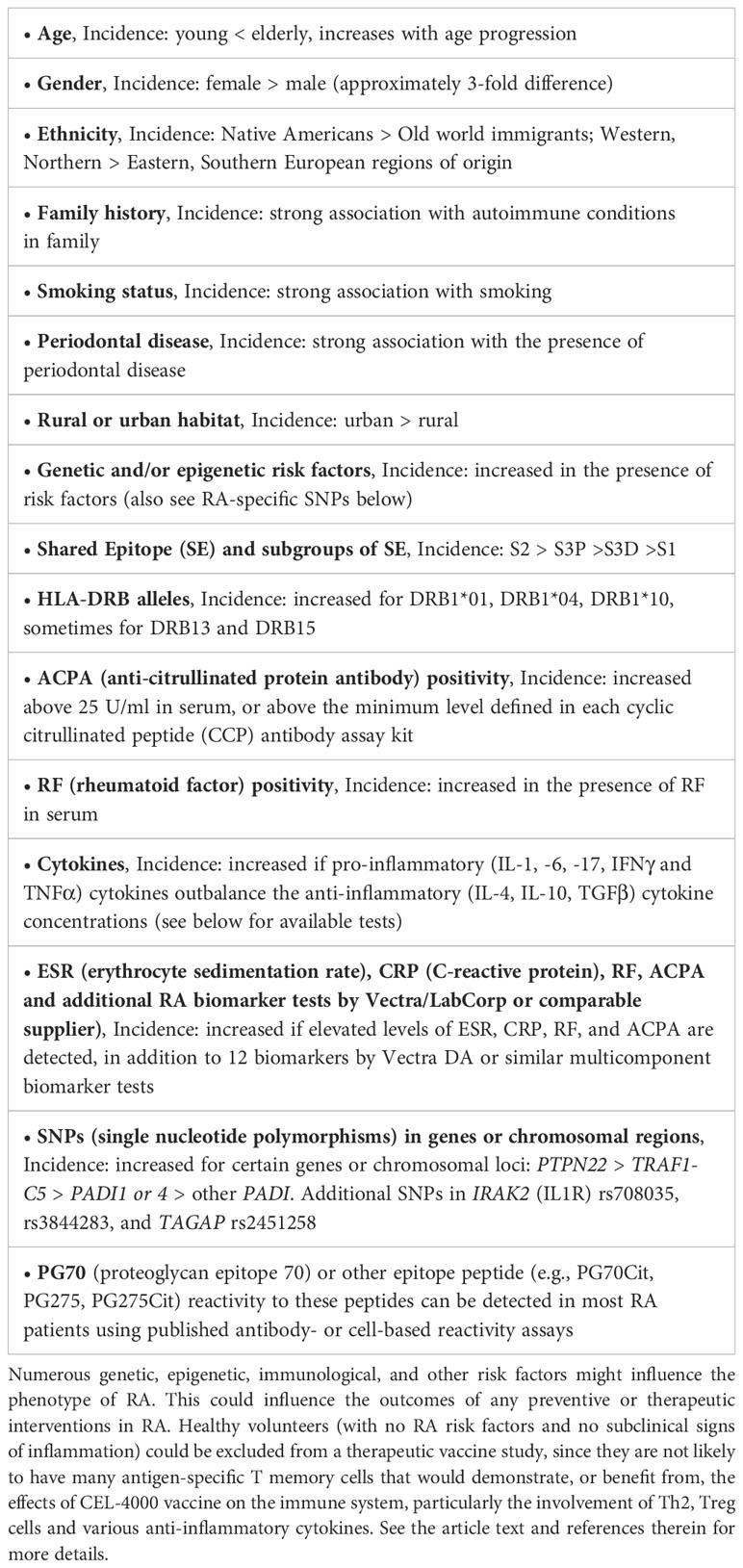
Table 3 Risk factors and laboratory parameters to consider in a BOIN study population for testing CEL-4000 in RA patients.
5 Conclusions
The current therapeutic arsenal for the treatment of RA mainly consists of immunosuppressive or ablative drugs, which carry the risk of facilitating recurrent or primary infectious diseases or cancer. A therapeutic vaccine that can rebalance inflammatory disease-promoting T cell immune responses in an antigen-specific manner is highly desirable. Evaluation of a treatment for an autoimmune inflammatory disease such as RA can only be performed in individuals who have the disease. Non-diseased but at-risk individuals (e.g., siblings of RA patients showing the presence of risk factors or laboratory-determined parameters indicative of subclinical RA) may also be appropriate study subjects. However, patient selection is not straightforward, as RA phenotypes show great heterogeneity and are under the influence of numerous environmental, genetic, and epigenetic factors. Treatment efficacy in an adaptive study would ultimately be demonstrated by delayed or reduced disease progression, but also by favorable changes in surrogate immune responses indicative of an antigen-specific decrease in pro-inflammatory and/or an increase in anti-inflammatory cytokines and corresponding increase in the number or function of Treg cells. Immunomodulation by CEL-4000 will likely occur in individuals already possessing disease promoting and antigen-specific Teff/Tm cells requiring testing in these individuals. Ultimately, an antigen-specific immunomodulating vaccine, such as CEL-4000, should deliver therapy for RA without weakening important immune defense mechanisms against microbial infections or cancer.
Author contributions
DZ: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. ZS: Formal analysis, Visualization, Writing – review & editing. AM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. KSR: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. RC: Data curation, Formal analysis, Investigation, Software, Writing – review & editing. KM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Visualization, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The CEL-4000 and DerG-PG275Cit (CEL-5000) vaccine-related research, reviewed in this article, was supported by the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) of USA under award number R44 AR063504-02A1 for DZ as well as by CEL-SCI Corporation, and was also supported under award numbers NIH/NIAMS R01 AR064206 and R01 AR062991 for KM and TT Glant at Rush University Medical Center. No additional funding was provided to write this review article. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
This paper is dedicated to the memory of Tibor T Glant, MD, PhD, DMSc (Department of Orthopedic Surgery, Rush University Medical Center, Chicago, IL) who provided the PGIA and GIA mouse models of RA for our preclinical vaccine studies and was an inspiration to us all. We would like to acknowledge and thank Dr. Glant for his contributions to the understanding of the immunopathology of RA and providing animal models of RA, allowing the development and testing of CEL-4000 and related vaccines. We thank Juan Schmukler, MD (Department of Internal Medicine, Division of Rheumatology, Rush University Medical Center, Chicago, IL), for his input on currently used FDA-approved therapeutics in RA.
Conflict of interest
Authors DZ and RC were employed by CEL-SCI Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, CEL-SCI Corporation, Rush University Medical Center, Augusta University/University of Georgia Medical Partnership, or the University of Debrecen, Faculty of Medicine.
References
1. Rosenthal KS, Mikecz K, Steiner HL, Glant TT, Finnegan A, Carambula RE, et al. Rheumatoid arthritis vaccine therapies: perspectives and lessons from therapeutic ligand epitope antigen presentation system vaccines for models of rheumatoid arthritis. Expert Rev Vaccines. (2015) 14:1–18. doi: 10.1586/14760584.2015.1026330
2. Markovics A, Rosenthal KS, Mikecz K, Carambula RE, Ciemielewski JC, Zimmerman DH. Restoring the balance between pro-inflammatory and anti-inflammatory cytokines in the treatment of rheumatoid arthritis: new insights from animal models. Biomedicines. (2021) 10:44. doi: 10.3390/biomedicines10010044
3. Shawky AM, Almalki FA, Abdalla AN, Abdelazeem AH, Gouda AM. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. (2022) 14(5):1001. doi: 10.3390/pharmaceutics14051001
4. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK–STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. (2017) 77:521–46. doi: 10.1007/s40265-017-0701-9
5. Caporali R, Germinario S, Kacsándi D, Choy E, Szekanecz Z. Start RA treatment – biologics or JAK-inhibitors? Autoimmun Rev. (2023) 10:3429. doi: 10.1016/j.autrev.2023.103429
6. Zimmerman DH, Mikecz K, Markovics A, Carambula RE, Ciemielewski JC, Toth DM, et al. Vaccination by two derG LEAPS conjugates incorporating distinct proteoglycan (PG, aggrecan) epitopes provides therapy by different immune mechanisms in a mouse model of rheumatoid arthritis. Vaccines. (2021) 9:448. doi: 10.3390/vaccines9050448
7. Mikecz K, Glant TT, Markovics A, Rosenthal KS, Kurko J, Carambula RE, et al. An epitope-specific DerG-PG70 LEAPS vaccine modulates T cell responses and suppresses arthritis progression in two related murine models of rheumatoid arthritis. Vaccine. (2017) 35:4048–56. doi: 10.1016/j.vaccine.2017.05.009
8. Silvagni E, Giollo A, Sakellariou G, Ughi N, D’Amico ME, Scirè CA, et al. One year in review 2020: novelties in the treatment of rheumatoid arthritis. Clin Exp Rheumatol. (2020) 38:181–94. doi: 10.55563/clinexprheumatol/n6zc67
9. Thomas J, Bansback N, Barber C, Wells G, Hazlewood G. Personalized medicine in rheumatoid arthritis: Combining biomarkers and patient preferences to guide therapeutic decisions. Best Pract Res Clin Rheumatol. (2022) 36:101812. doi: 10.1016/j.berh.2022.101812
10. Frazzei G, Musters A, de Vries N, Tas SW, van Vollenhoven RF. Prevention of rheumatoid arthritis: A systematic literature review of preventive strategies in at-risk individuals. Autoimmun Rev. (2023) 22:103217. doi: 10.1016/j.autrev.2022.103217
11. Pozsgay J, Szekanecz Z, Sármay G. Antigen-specific immunotherapies in rheumatic diseases. Nat Rev Rheumatol. (2017) 13:525–37. doi: 10.1038/nrrheum.2017.107
12. Kobezda T, Ghassemi-Nejad S, Mikecz K, Glant TT, Szekanecz Z. Of mice and men: how animal models advance our understanding of T-cell function in RA. Nat Rev Rheumatol. (2014) 10:160–70. doi: 10.1038/nrrheum.2013.205
13. Smolen JS, Landewé RBM, Bergstra SA, Kerschbaumer A, Sepriano A, Aletaha D, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. (2023) 82:3–18. doi: 10.1136/ard-2022-223356
14. Fraenkel L, Bathon JM, England BR, St.Clair EW, Arayssi T, Carandang K, et al. 2021 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. (2021) 73:1108–23. doi: 10.1002/art.41752
15. Santana E, Rosenthal K. Ablative treatment of inflammatory and other diseases with Monoclonal Antibodies (Mabs) or Janus Kinases (Nibs) increases risk for infection. Infect Dis Clin Pract. (2023) 31(6):e1317. doi: 10.1097/IPC.0000000000001317
16. Curtis JR, Yamaoka K, Chen Y-H, Bhatt DL, Gunay LM, Sugiyama N, et al. Malignancy risk with tofacitinib versus TNF inhibitors in rheumatoid arthritis: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheum Dis. (2023) 82:331–43. doi: 10.1136/ard-2022-222543
17. Molander V, Bower H, Frisell T, Delcoigne B, Di Giuseppe D, Askling J. Venous thromboembolism with JAK inhibitors and other immune-modulatory drugs: A Swedish comparative safety study among patients with rheumatoid arthritis. Ann Rheum Dis. (2022) 82:189–97. doi: 10.1136/ard-2022-223050
18. Corrigall VM, Panayi GS. Autoantigens and immune pathways in rheumatoid arthritis. Crit Rev Immunol. (2002) 22:281–93.
19. Bugatti S, Vitolo B, Caporali R, Montecucco C, Manzo A. B cells in rheumatoid arthritis: from pathogenic players to disease biomarkers. BioMed Res Int. (2014) 2014:681678. doi: 10.1155/2014/681678
20. Fert-Bober J, Darrah E, Andrade F. Insights into the study and origin of the citrullinome in rheumatoid arthritis. Immunol Rev. (2020) 294:133–47. doi: 10.1111/imr.12834
21. Nandakumar KS, Fang Q, Wingbro Ågren I, Bejmo ZF. Aberrant activation of immune and non-immune cells contributes to joint inflammation and bone degradation in rheumatoid arthritis. Int J Mol Sci. (2023) 24(21):15883. doi: 10.3390/ijms242115883
22. Martin E, Capini C, Duggan E, Lutzky VP, Stumbles P, Pettit AR, et al. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-κB. Arthritis Rheum. (2007) 56:2255–66. doi: 10.1002/art.22655
23. Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, et al. PD-L1– and calcitriol-dependent liposomal antigen-specific regulation of systemic inflammatory autoimmune disease. JCI Insight. (2019) 4(18):e126025. doi: 10.1172/jci.insight.126025
24. Shin TH, Kim HS, Kang TW, Lee BC, Lee HY, Kim YJ, et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. (2016) 7(12):e2524. doi: 10.1038/cddis.2016.442
25. van Herwijnen MJC, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci U.S.A. (2012) 109(35):14134–9. doi: 10.1073/pnas.1206803109
26. Zhang B, Wang Y, Yuan Y, Sun J, Liu L, Huang D, et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann Rheum Dis. (2021) 80:176–84. doi: 10.1136/annrheumdis-2020-217844
27. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:290ra87–290ra87. doi: 10.1126/scitranslmed.aaa9301
28. Sonigra A, Nel HJ, Wehr P, Ramnoruth N, Patel S, van Schie KA, et al. Randomized phase I trial of antigen-specific tolerizing immunotherapy with peptide/calcitriol liposomes in ACPA+ rheumatoid arthritis. JCI Insight. (2022) 7:1–17. doi: 10.1172/jci.insight.160964
29. Park EH, Lim H-S, Lee S, Roh K, Seo K-W, Kang K-S, et al. Intravenous infusion of umbilical cord blood-derived mesenchymal stem cells in rheumatoid arthritis: A phase ia clinical trial. Stem Cells Transl Med. (2018) 7:636–42. doi: 10.1002/sctm.18-0031
30. Nam Y, Jung SM, Rim YA, Jung H, Lee K, Park N, et al. Intraperitoneal infusion of mesenchymal stem cell attenuates severity of collagen antibody induced arthritis. PloS One. (2018) 13:e0198740. doi: 10.1371/journal.pone.0198740
31. Wang L, Wang L, Cong X, Liu G, Zhou J, Bai B, et al. Human umbilical cord mesenchymal stem cell therapy for patients with active rheumatoid arthritis: safety and efficacy. Stem Cells Dev. (2013) 22:3192–202. doi: 10.1089/scd.2013.0023
32. Yang Y, He X, Zhao R, Guo W, Zhu M, Xing W, et al. Serum IFN-γ levels predict the therapeutic effect of mesenchymal stem cell transplantation in active rheumatoid arthritis. J Transl Med. (2018) 16:165. doi: 10.1186/s12967-018-1541-4
33. Ghoryani M, Shariati-Sarabi Z, Tavakkol-Afshari J, Mohammadi M. The sufficient immunoregulatory effect of autologous bone marrow-derived mesenchymal stem cell transplantation on regulatory T cells in patients with refractory rheumatoid arthritis. J Immunol Res. (2020) 2020:3562753. doi: 10.1155/2020/3562753
34. Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol. (2014) 10(9):543–51. doi: 10.1038/nrrheum.2014.105
35. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. (2005) 52:2212–21. doi: 10.1002/art.21195
36. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. (2009) 10:1000–7. doi: 10.1038/ni.1774
37. Wright GP, Stauss HJ, Ehrenstein MR. Therapeutic potential of Tregs to treat rheumatoid arthritis. Semin Immunol. (2011) 23:195–201. doi: 10.1016/j.smim.2011.07.004
38. van Vollenhoven RF, Keystone EC, Strand V, Pacheco-Tena C, Vencovský J, Behrens F, et al. Efficacy and safety of tregalizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase IIb, randomised, placebo-controlled trial. Ann Rheum Dis. (2018) 77:495–9. doi: 10.1136/annrheumdis-2017-212478
39. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
40. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
41. Orvain C, Boulch M, Bousso P, Allanore Y, Avouac J. Is there a place for chimeric antigen receptor-T cells in the treatment of chronic autoimmune rheumatic diseases? Arthritis Rheumatol (Hoboken NJ). (2021) 73:1954–65. doi: 10.1002/art.41812
42. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. (2019) 25:e123–7. doi: 10.1016/j.bbmt.2018.12.756
43. Hafron JM, Wilfehrt HM, Ferro C, Harmon M, Flanders SC, McKay RR. Real-world effectiveness of sipuleucel-T on overall survival in men with advanced prostate cancer treated with androgen receptor-targeting agents. Adv Ther. (2022) 39:2515–32. doi: 10.1007/s12325-022-02085-6
44. Moorman CD, Sohn SJ, Phee H. Emerging therapeutics for immune tolerance: tolerogenic vaccines, T cell therapy, and IL-2 therapy. Front Immunol. (2021) 12:1–32. doi: 10.3389/fimmu.2021.657768
45. Van EW, Jansen MAA, Ludwig IS, Leufkens P, Goes C, VanLaar JM, et al. Heat shock proteins can be surrogate autoantigens for induction of antigen specific therpeutic tolerance in rheumatoid arthritis. Front Immunol. (2019) 10:1–6. doi: 10.3389/fimmu.2019.00279
46. Shuai Z, Zheng S, Wang K, Wang J, Leung PSCC, Xu B. Reestablish immune tolerance in rheumatoid arthritis. Front Immunol. (2022) 13:1–15. doi: 10.3389/fimmu.2022.1012868
47. Nagy NA, de Haas AM, Geijtenbeek TBH, van Ree R, Tas SW, van Kooyk Y, et al. Therapeutic liposomal vaccines for dendritic cell activation or tolerance. Front Immunol. (2021) 12:1–16. doi: 10.3389/fimmu.2021.674048
48. König R, Huang LY, Germain RNN. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature. (1992) 356:796–8. doi: 10.1038/356796a0
49. Glant TT, Radacs M, Nagyeri G, Olasz K, Laszlo A, Boldizsar F, et al. Proteoglycan-induced arthritis and recombinant human proteoglycan aggrecan G1 domain-induced arthritis in BALB/c mice resembling two subtypes of rheumatoid arthritis. Arthritis Rheum. (2011) 63:1312–21. doi: 10.1002/art.30261
50. Glant TT, Mikecz K. Proteoglycan aggrecan-induced arthritis - a murine autoimmune model of rheumatoid arthritis. Methods Mol Med. (2004) 102:313–38. doi: 10.1385/1-59259-805-6:313
51. Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. (2021) 371:145–53. doi: 10.1126/science.aay3638
52. Cappellano G, Abreu H, Casale C, Dianzani U, Chiocchetti A. Nano-microparticle platforms in developing next-generation vaccines. Vaccines. (2021) 9:1–18. doi: 10.3390/vaccines9060606
53. van Alem CMA, Metselaar JM, van Kooten C, Rotmans JI. Recent advances in liposomal-based anti-inflammatory therapy. Pharmaceutics (2021) 13(7):1004. doi: 10.3390/pharmaceutics13071004
54. Wenhart C, Holthoff H-P, Reimann A, Li Z, Faßbender J, Ungerer M. A fructosylated peptide derived from a collagen II T cell epitope for long-term treatment of arthritis (FIA-CIA) in mice. Sci Rep. (2021) 11:17345. doi: 10.1038/s41598-021-95193-2
55. Cappellano G, Comi C, Chiocchetti A, Dianzani U. Exploiting PLGA-based biocompatible nanoparticles for next-generation tolerogenic vaccines against autoimmune disease. Int J Mol Sci. (2019) 20:1–16. doi: 10.3390/ijms20010204
56. Klein SL, Morgan R. The impact of sex and gender on immunotherapy outcomes. Biol Sex Differ. (2020) 11:24. doi: 10.1161/CIRCRESAHA.121.319915
57. Li C, Han Y, Luo X, Qian C, Li Y, Su H, et al. Immunomodulatory nano-preparations for rheumatoid arthritis. Drug Delivery. (2023) 30:9–19. doi: 10.1080/10717544.2022.2152136
58. Zhao X, Long J, Liang F, Liu N, Sun Y, Xi Y. Dynamic profiles, biodistribution and integration evaluation after intramuscular/intravenous delivery of a novel therapeutic DNA vaccine encoding chicken type II collagen for rheumatoid arthritis in vaccinated normal rodent. J Nanobiotechnol. (2019) 17:1–10. doi: 10.1186/s12951-019-0528-5
59. Jansen MAAA, Spiering R, Broere F, van Laar JM, Isaacs JD, van Eden W, et al. Targeting of tolerogenic dendritic cells towards heat-shock proteins: a novel therapeutic strategy for autoimmune diseases? Immunology. (2018) 153:51–9. doi: 10.1111/imm.12811
60. Heydari R, Koohi F, Rasouli M, Rezaei K, Abbasgholinejad E, Bekeschus S, et al. Exosomes as rheumatoid arthritis diagnostic biomarkers and therapeutic agents. Vaccines. (2023) 11(3):687. doi: 10.3390/vaccines11030687
61. Zimmerman DH, Taylor P, Bendele A, Carambula R, Duzant Y, Lowe V, et al. CEL-2000: A therapeutic vaccine for rheumatoid arthritis arrests disease development and alters serum cytokine/chemokine patterns in the bovine collagen type II induced arthritis in the DBA mouse model. Int Immunopharmacol. (2010) 10:412–21. doi: 10.1016/j.intimp.2009.12.016
62. van de Stadt LA, de Koning MHMT, van de Stadt RJ, Wolbink G, Dijkmans BAC, Hamann D, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. (2011) 63:3226–33. doi: 10.1002/art.30537
63. Markovics A, Ocskó T, Katz RS, Buzás EI, Glant TT, Mikecz K. Immune recognition of citrullinated proteoglycan aggrecan epitopes in mice with proteoglycan-induced arthritis and in patients with rheumatoid arthritis. PloS One. (2016) 11:e0160284. doi: 10.1371/journal.pone.0160284
64. Immunogenicity testing of therapeutic protein products — developing and validating assays for anti-drug antibody detection. Guidance for Industry. Food and Drug Administration (2019). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating-assays-anti-drug.
65. Zimmerman DH, Morris S, Rouse D, Worthington K, Elliott D, Rosenthal K. Immunization with peptide heterconjugates primes a t helper cell type 1-associated antibody (IgG2a) response that recognizes the native epitope on the 38-kDA protein of Mycobacterium tuberculosis. Vaccine Res. (1996) 5:103–18.
66. Rosenthal KS, Mao H, Horne WI, Wright C, Zimmerman D. Immunization with a LEAPS heteroconjugate containing a CTL epitope and a peptide from beta-2-microglobulin elicits a protective and DTH response to herpes simplex virus type 1. Vaccine. (1999) 17:535–42. doi: 10.1016/S0264-410X(98)00231-X
67. Goel N, Rong Q, Zimmerman D, Rosenthal KS. A L.E.A.P.S. heteroconjugate vaccine containing a T cell epitope from HSV-1 glycoprotein D elicits Th1 responses and protection. Vaccine. (2003) 21:4410–20. doi: 10.1016/S0264-410X(03)00429-8
68. Zimmerman DH, Rosenthal KS. The L.E.A.P.S. approach to vaccine development. Front Biosci. (2005) 10:790–8. doi: 10.2741/1572
69. Pallmann P, Bedding AW, Choodari-Oskooei B, Dimairo M, Flight L, Hampson LV, et al. Adaptive designs in clinical trials: Why use them, and how to run and report them. BMC Med. (2018) 16:1–15. doi: 10.1186/s12916-018-1017-7
70. Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry. Food and Drug Administration (2019). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry.
71. Collins S, Zineh I. Determination Letter Fit for Purpose. FDA - Cent Drug Eval Res (2021) p. 1–3. Available at: https://www.fda.gov/media/155363/download.
72. Gao X, Fernandes LL, Klein M, Collins S. Statistical review and evaluation fit-for-purpose initiative BOIN(2020). Available online at: https://www.fda.gov/media/155364/download.
73. Phase II AutoImmune Hepatitis. ClinicalTrials.gov (2019). Available at: https://clinicaltrials.gov/ct2/show/NCT03593460.
74. An Adaptive Phase II Study to Evaluate the Efficacy, Pharmacodynamics, Safety and Tolerability of GSK2586184. ClinicalTrials.gov (2017). Available at: https://clinicaltrials.gov/ct2/show/NCT01777256.
75. Phase II Study Evaluating the Safety and Efficacy of GSK315234A in Patients With Rheumatoid Arthritis. ClinicalTrials.gov (2016). Available at: https://clinicaltrials.gov/ct2/show/NCT00674635.
76. Multiple endpoints in clinical trials: guidance for industry. Food and Drug Administration (2022). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/multiple-endpoints-clinical-trials-guidance-industry.
77. Common Terminology Criteria for Adverse Events (CTCAE).v.5.0. U.S. Department of Health and Human Services. Available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_50.
78. Liu S, Yuan Y. Bayesian optimal interval designs for phase I clinical trials. J R Stat Soc Ser C. (2015) 64:507–23. doi: 10.1111/rssc.12089
79. Liu S, Yuan Y. Erratum to: Bayesian optimal interval designs for phase I clinical trials (Journal of the Royal Statistical Society: Series C (Applied Statistics), (2015), 64, 3, (507-523), 10.1111/rssc.12089). J R Stat Soc Ser C Appl Stat. (2022) 71:491–2. doi: 10.1111/rssc.12541
80. Lin R, Zhou Y, Yan F, Li D, Yuan Y. BOIN12: Bayesian optimal interval phase I/II trial design for utility-based dose finding in immunotherapy and targeted therapies. JCO Precis Oncol. (2020) 4):1393–402. doi: 10.1200/PO.20.00257
81. Zhou Y, Lee JJ, Yuan Y. A utility-based Bayesian optimal interval (U-BOIN) phase I/II design to identify the optimal biological dose for targeted and immune therapies. Stat Med. (2019) 38:5299–316. doi: 10.1002/sim.8361
82. Takeda K, Taguri M, Morita S. BOIN-ET: Bayesian optimal interval design for dose finding based on both efficacy and toxicity outcomes. Pharm Stat. (2018) 17:383–95. doi: 10.1002/pst.1864
83. Trial of a Personalized and Adaptive Neoantigen Dose-Adjusted Vaccine Concurrently With Pembrolizumab (PANDA-VAC). ClinicalTrials.gov (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT04266730.
84. Melssen MM, Fisher CT, Slingluff CL, Melief CJM. Peptide emulsions in incomplete Freund’s adjuvant create effective nurseries promoting egress of systemic CD4 + and CD8 + T cells for immunotherapy of cancer. J Immunother Cancer. (2022) 10(9):e004709. doi: 10.1136/jitc-2022-004709
85. Meznerics FA, Kemény LV, Gunther E, Bakó E, Dembrovszky F, Szabó B, et al. Multibiomarker disease activity score: an objective tool for monitoring rheumatoid arthritis? A systematic review and meta-analysis. Rheumatology (Oxford). (2023) 62(6):2048–59. doi: 10.1093/rheumatology/keac715
86. Centola M, Cavet G, Shen Y, Ramanujan S, Knowlton N, Swan KA, et al. Development of a multi-biomarker disease activity test for rheumatoid arthritis. PloS One. (2013) 8:e60635. doi: 10.1371/journal.pone.0060635
87. Rech J, Hueber AJ, Finzel S, Englbrecht M, Haschka J, Manger B, et al. Prediction of disease relapses by multibiomarker disease activity and autoantibody status in patients with rheumatoid arthritis on tapering DMARD treatment. Ann Rheum Dis. (2016) 75:1637–44. doi: 10.1136/annrheumdis-2015-207900
Keywords: peptide vaccine, immunotherapy, rheumatoid arthritis, cytokines, proteoglycan (PG, aggrecan), PG-induced arthritis, collagen-induced arthritis
Citation: Zimmerman DH, Szekanecz Z, Markovics A, Rosenthal KS, Carambula RE and Mikecz K (2024) Current status of immunological therapies for rheumatoid arthritis with a focus on antigen-specific therapeutic vaccines. Front. Immunol. 15:1334281. doi: 10.3389/fimmu.2024.1334281
Received: 06 November 2023; Accepted: 08 February 2024;
Published: 06 March 2024.
Edited by:
Esther M. Lafuente, Complutense University of Madrid, SpainReviewed by:
Rong Xiu Li, Shanghai Jiao Tong University, ChinaAgnieszka Paradowska-Gorycka, National Institute of Geriatrics, Rheumatology and Rehabilitation, Poland
Copyright © 2024 Zimmerman, Szekanecz, Markovics, Rosenthal, Carambula and Mikecz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel H. Zimmerman, RFppbW1lcm1hbkBjZWwtc2NpLmNvbQ==