 Bo Dou1†
Bo Dou1† Chentao Liu
Chentao Liu- 1Central South University, Xiangya Hospital, Pediatric Department, Changsha, Hunan, China
- 2Hunan Normal University, Hunan Provincial People's Hospital, Department of Respiratory Medicine, Changsha, Hunan, China
OTULIN deficiency is a complex disease characterized by a wide range of clinical manifestations, including skin rash, joint welling, lipodystrophy to pulmonary abscess, and sepsis shock. This disease is mechanistically linked to mutations in the OTULIN gene, resulting in an immune disorder that compromises the body’s ability to effectively combat pathogens and foreign stimuli. The OTULIN gene is responsible for encoding a deubiquitinating enzyme crucial for hydrolyzing Met1-poly Ub chains, and its dysfunction leads to dysregulated immune responses. Patients with OTULIN deficiency often exhibit an increase in monocytes, including neutrophils and macrophages, along with inflammatory clinical features. The onset of symptoms typically occurs at an early age. However, individuals with OTULIN haploinsufficiency are particularly susceptible to life-threatening staphylococcal infections. Currently, the most effective treatment for patients with OTULIN biallelic mutations involves the use of TNF-blocking agents, which target the dysregulated immune response. In conclusion, OTULIN deficiency presents a complex clinical picture with diverse manifestations, attributed to mutations in the OTULIN gene. Understanding the underlying mechanisms is crucial for developing targeted therapeutic interventions to address this challenging condition. Further research into the pathophysiology of OTULIN deficiency is essential for improving clinical management and outcomes for affected individuals.
1 Introduction
Immunological response, which is a body’s critical defense mechanism against pathogens and foreign substances, is tightly controlled at multiple levels including reversible modifications of various signaling molecules. Ubiquitination is an important post-translational modification that covalently attaches ubiquitin chains to target molecular proteins and either marks substrate proteins for proteasomal degradation or allows them to function as a scaffold to recruit other proteins and mediate downstream signaling (1–3). Linear ubiquitin chains are a unique type of ubiquitin chain, in which the ubiquitin molecules are linked together in a linear fashion through their N-terminal methionine and C-terminal glycine residues, and are generated by a multi-subunit E3 ligase complex called the linear ubiquitin chain assembly complex (LUBAC), which compromises three subunits: HOIL-1L (heme-oxidized iron-regulatory protein 2 ubiquitin ligase-1 like, also known as RBCK1), HOIP (HOIL-1L-interacting protein, also known as RNF31), and SHARPIN (SHANK-associated RH domain-interacting protein) (4, 5). Numerous publications reported functions of linear ubiquitin chains including regulating immune responses and inflammation, via NF-κB signaling in response to various stimuli, and preventing TNF-induced cell death (6–13). That disruption of LUBAC components leads to immunodeficient disorders and autoinflammatory disorders in humans (14), and embryonic lethality or inflammatory phenotype in mice (11, 12, 15), highlights the crucial role of LUBAC-mediated Met1 signaling in maintaining normal mammalian physiology. Ubiquitination is reversed by the deubiquitinating enzyme (DUB) via cleaving the ubiquitin molecules from their substrate (2). To date, almost 100 DUBs are identified in humans (2), but mainly A20, cylindromatosis (CYLD), and OTULIN have been characterized as key DUBs involved in the negative regulation of NF-κB activation and cell death (16). OTULIN, also known as Fam105B or Gumby, was recently identified in human and mice (17, 18), with exclusive specificity to hydrolyze Met1-poly Ub chains (4, 17–19). Herein, we briefly review the genetic and clinical features of OTULIN deficiency, focusing on immunological phenotypes. Finally, we summarize the state-of-the-art management options, shedding light on the current main research gaps.
2 OTULIN: structure, function, and mutations
The 352-amino acid protein OTULIN was initially discovered by Keusekotten et al. through a bioinformatics approach (17, 18, 20). This important deubiquitinating enzyme comprises a catalytic ovarian tumor (OTU) protease domain, an N-terminal interaction motif (PIM) involving peptide:N-glycanase (PNGase)/UBA-containing or UBX-containing proteins (PUB), and a C-terminal PSD95-Dlg1-ZO-1(PDZ) domain-binding motif (17, 18, 21, 22). OTU domain spans 80–345 amino acids, shares features of the papain-like family of cysteine peptidases, and employs a Cys129/His339/Asn341 catalytic triad (17, 20) (Figure 1). A striking function of OTULIN is its exclusive hydrolyzation to Met1-linked poly-Ub chains, without affecting other linkage types of poly-Ub chain (17, 18). This feature function originates from a highly conserved Ub binding site located in the OTU domain via a mechanism known as substrate-associated catalysis (17). Upon binding to Met1-linked poly Ub chains, the S1’ binding site in OTULIN orientates the Met1 residue of the proximal Ub toward the catalytic center, while the inter-Ub Lys residues remain remote from the catalytic center (17). However, binding of Ub chains with different linkages, but structurally similar Lys63-linked chains, would rotate the proximal Ub for several degrees and restrict proper binding to the S1’ binding site (17, 20). Ub substrates have also been demonstrated to directly contribute to OTULIN catalytic activity (17). In the absence of Ub, OTULIN Cys129, His399, and Asn341 adopt an inhibited catalytic triad conformation, and this auto-inhibition is released upon binding to Met1-linked proximal Ub (17). In addition, OTULIN selectively interacts with the LUBAC HOIP PUB domain via the N-terminal PIM, which covers Asp54–Met55–Tyr56–Arg57–Ala58 residues (21, 22) and serves as the sole site of interaction between OTULIN and HOIP, whereas the OTU domain’s presence is unnecessary for binding to HOIP (21, 22). Mutations in the OTULIN catalytic Cys129 do not affect the association between OTULIN and HOIP (15, 21).
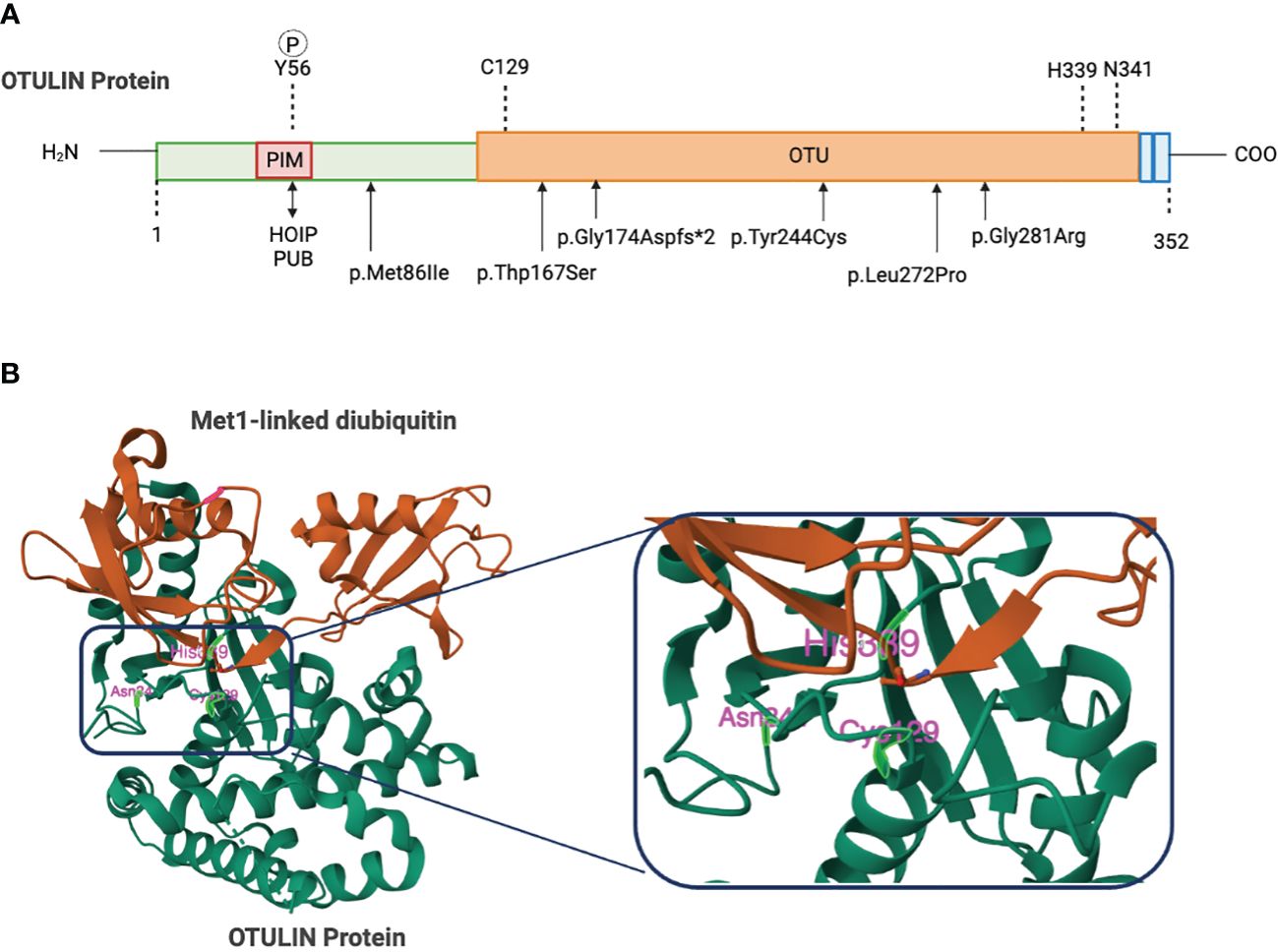
Figure 1 Domain organization of OTU deubiquitinates. (A) Schematic figure of the domain structures and important residues. The N-terminus of OTULIN contains the PUB-interaction motif (PIM) that mediates the interaction with the HOIP subunit of LUBAC. The OTU domain contains a catalytic triad consisting of Cys129, His339, and Asn342. The C-terminal OTULIN domain contains a PD2 binding motif (PDZbm) responsible for binding to the snx27. Gly174, Tyr244, Leu272, or Gly281 mutations in OTULIN catalytically inactivate OTULIN or reduce protein stability causing the severe inflammatory phenotype in ORAS patients. (B) Structure of OTULIN (green) bound to the Met1-linked Di-ubiquitin activating probe (brown) with a close-up view of the catalytic triad (Cys129, His339, and Asn341). Image created with Biorender.com.
Pathogens, tissue damage, and stress provoke host immune response and activate the downstream inflammatory signaling pathway. While immune receptors, such as TNFR1, NOD-like receptors (NLRs), CD40, toll-like receptors (TLRs), and the interleukin-1 receptor (IL-1R) instigate the assembly of receptor signaling complexes (RSCs), LUBAC recruitment via a Lys63 ubiquitin-dependent approach is a pivotal step in the course of these complex processes (5, 23–26). LUBAC recruitment facilitates Met1-linked ubiquitination of TNFR1, receptor-associated proteins, or pre-existing Lys63 polyubiquitin chains, and initiates downstream MAPK and NF-κB signaling by orchestrating the recruitment of the TAK1 and IKK complexes (24, 25, 27, 28). Numerous studies have demonstrated OTULIN’s role in counteracting LUBAC-mediated Met1 ubiquitination and subsequent NF-κB activation (17, 18, 29–33). However, deficiencies in OTULIN, overexpression of catalytic inactive OTULIN mutants, and OTULIN knockdown result in a remarkable increase in cellular Met1 Ub levels, leading to the accumulation of Met1-linked Ub on several pivotal proteins within inflammatory pathways, including TNFR1, receptor-interacting serine/threonine-protein kinase-1 (RIPK1), receptor-interacting serine/threonine-protein kinase-2 (RIPK2), NF-κB essential modulator (NEMO), and LUBAC (17, 23, 31–35). By contrast, overexpression of OTULIN leads to a reduction in TNFα-induced NF-κB activation and gene transcription (17). It is worth noting that unlike CYLD and A20, which exert their influence on NF-κB signaling through negative feedback mechanisms, OTULIN is not subject to transcriptional regulation by NF-κB (17, 18, 32, 36, 37).
The OTULIN gene is located on chromosome 5p15.2. To date, approximately 99 pathogenic or likely pathogenic germline variants are identified, affecting nearly 50 individuals (29–31, 38–40). Patients with variants in OTULIN gene and size of variants larger than 50 bps mainly present 5p- syndrome and are responsible for 72% of cases. Single genetic variants in OTULIN only account for 20%–30% of cases. Among these cases with single genetic mutation in OTULIN, missense mutations in OTULIN clustered within the OTU region underlie roughly 62.5% of cases, whereas null mutations (frameshift, nonsense, and splice site), located prior to or within the OTU region, represent approximately 37.5% of mutations (29–31, 39, 40). The impact of mutations in the OTULIN gene is quite variable, resulting in a range of clinical phenotypes. Loss-of-function mutations in the OTULIN gene often led to reduced levels of the OTULIN protein, which, in turn, contributes to the observed variability in clinical presentation. However, some mutations, such as OTULIN (L272P), are associated with only minimal degradation of the OTULIN protein (30, 31). Others, like OTULIN (G281R), result in almost complete loss of function (29). These findings underscore the complex and diverse effects of OTULIN gene mutations on protein expression and function, highlighting the need for further research to fully understanding the underlying mechanisms and potential therapeutic implication.
3 Clinical features: autoinflammation and immunodeficiency
3.1 Biallelic homozygous variants in OTULIN result in an early-onset autoinflammatory disease: OTULIN-related autoinflammatory syndrome
In 2016, two groups respectively identified a surplus of linear ubiquitin linkages in humans due to homozygous variants in OTULIN that result in an autoinflammatory disease: OTULIN‐related autoinflammatory syndrome (ORAS, also known as otulipenia) (30, 31). According to the 2022 Update of the IUIS Phenotypical Classification, OTULIN deficiency is classified among auto-immunodeficiency disorders of sterile inflammation, predominant at skin and joint (41). To date, 10 patients with ORAS from seven families have been identified (29–31, 40, 42, 43). The clinical landscapes of humans with biallelic hypomorphic mutations of OTULIN deficiency are complex and multifaceted (Figure 2) and dominated by premature birth at gestation from 28 + 6 weeks to 36 weeks, recurrent episodes of neonatal-onset fever, and rashes. Rashes showed pustules, scarring, or erythema with painful skin nodules. Skin biopsy revealed a predominantly septal panniculitis and neutrophilic dermatosis with vasculitis of small and medium-sized blood vessels. Other characteristic manifestations included protracted diarrhea, progressive lipodystrophy, joint swelling, arthralgia, failure to thrive, and spontaneous and progressive steatotic liver disease (30, 31, 44, 45). During the repeated episodes of systemic inflammation, patients present with elevated white blood cell count, serum C-reactive protein level, immunoglobulin levels, and serum autoantibodies without clear evidence for infections. Intriguingly, the raised white blood cell count comprises predominantly neutrophilia but normal lymphocyte subsets (30, 31). Consistent with this result, loss of OTULIN in T cells and B cells generated healthy mice with no overt inflammatory phenotypes, as assessed by blood cell, cytokine, and serum IgG analysis (30), whereas deletion of OTULIN in myeloid cells resulted in a strong inflammatory phenotype (30).
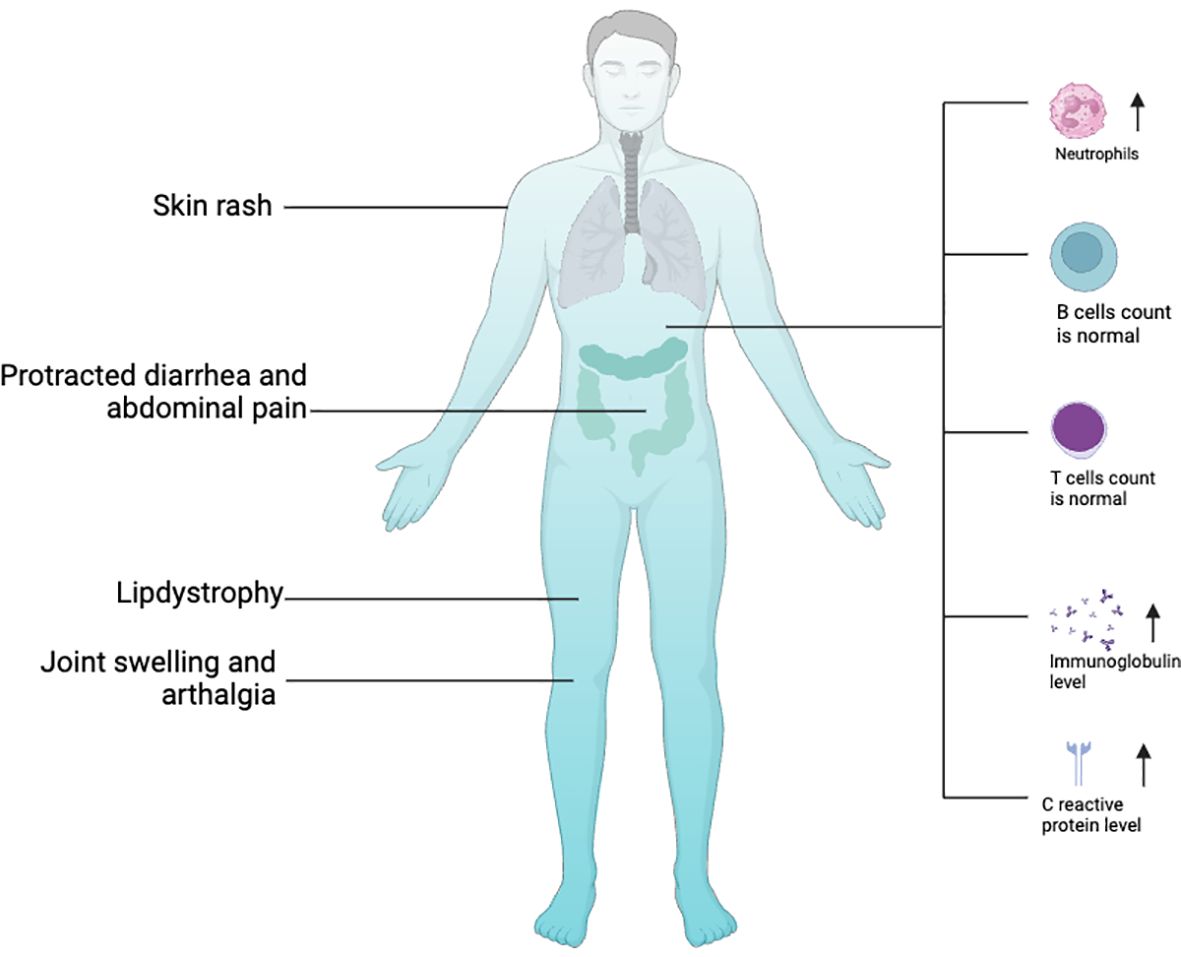
Figure 2 Clinical features of OTULIN with biallelic hypomorphic mutations. Image created with Biorender.com.
Using genetic testing, Mediterranean fever, chronic infantile neurological cutaneous articular syndrome, tumor necrosis factor-receptor associated periodic syndrome, and hyper IgD syndrome are excluded as the causes of the symptoms (Experimental Models and Subject Details). Four of the identified homozygous variants in the OTULIN gene, namely, p.Leu272Pro, p.Tyr244Cys, p.Gly174Aspfs*2, and p.Gly281Arg, are located in the OTU domain and confer predisposition of OTULIN stability, catalytic activity, and/or capability of binding to M1 ubiquitin (29–31), whereas the homozygous variant of c.864 + 2 T > C in OTULIN alters a donor splicing site and thus generates a truncated and unstable protein (40) (Figure 1). Numerous literatures had reported that disruption of LUBAC components lead to immunodeficient disorders and auto-inflammatory disorders. Loss-of-function mutations in HOIP or HOIL-1 are known to cause a severe multiorgan autoinflammatory disease in humans (14, 46) and lead to embryonic lethality in mice (11, 12). In line with the role of OTULIN in counteracting LUBAC, OTULIN-deficient skin fibroblasts from patients with ORAS showed reduced TNF-induced NF-κB and MAPK activation but enhanced sensitization to TNF-induced cell death relative to healthy controls (29). Intriguingly, the functional consequences of OTULIN deficiency in ORAS are cell type-specific (29, 31, 39). OTULIN deficiency in myeloid cells has been shown to result in increased NF-κB signaling and MAPK activation and cell death responses in vitro (29, 47). Patient-derived fibroblasts with OTULIN deficiency show a diminished response to TNF (29). Currently, there is limited evidence regarding OTULIN’s role in T cells. This may disrupt the homeostasis of T cells, leading to autoimmune diseases. However, OTULIN deficiency in epithelial cells disrupts intestinal barrier integrity and exacerbates intestinal inflammation, contributing to the pathogenesis of inflammatory bowel disease in ORAS (48). Nevertheless, the molecular mechanism of these cell type-dependent differences remains elusive.
Patients with biallelic hypomorphic variants in the OTULIN gene mainly present with an early-onset ORAS, whereas one Greek patient with two compound heterozygous variants in OTULIN was associated with fulminate atypical late-onset ORAS (29–31, 39). This has led to an expansion of the clinical spectrum of ORAS patients (39). This Greek patient, a 7-year-old boy, demonstrated age-appropriate psychomotor development and obesity features. He experienced a severe autoinflammatory episode characterized by multiorgan sterile abscess formation. Initially presenting with abdominal pain, subfebrile fever, leukocytosis, neutrophilia, and significantly elevated levels of C-reactive protein, the patient subsequently developed spiking fevers, inflammatory lesions on the wrist and ankle, and abscesses in the left lower pulmonary lobe, left axilla, and spleen. Inflammatory parameters continued to rise during the course of his illness. However, treatment with broad-spectrum antibiotics had not improved the patient’s condition, biopsies and smear remained sterile, and histopathological analysis of the skin revealed massive inflammatory infiltrates, predominantly consisting of granulocytes, monocytes, and macrophages. Lung and spleen showed signs of inflammation and necrosis. All blood culture, stool culture, and tracheal fluids remained sterile. On day 14 of in-patient stay, an autoinflammatory syndrome was suspected, and treatment with corticosteroids was started, which resulted in a decline of body temperature and CRP levels and in marked improvement of the patient’s condition. Two months following admission, the patient was discharged in good condition. Using a combination of whole exome sequencing and targeted Sanger sequencing, a compound heterozygous missense variant in exon 3 (c.258G>A; p.M86I) and exon 5 (c.500G>C; p.W167S) of the OTULIN gene was revealed (39). The patient inherited the p.M86I variant from his father, whereas his mother is a heterozygous carrier of the p.W167S mutation. The patient is the second child born to non-consanguineous parents. Both his parents and siblings are clinically well. In-depth biochemical analysis revealed that binding of OTULIN to linear ubiquitin is compromised by both variants; however, protein stability and catalytic activity are most affected by the OTULIN variant p.W167S (39).
Another possible clinical feature of OTULIN deficiency is embryonic lethal. Mice carrying loss-of-function homozygous variants W96R and D336E in Otulin resulted in abnormal cranial vasculature and die embryonically after embryonic day E12.5 (18). Genetic loss of Otulin in mice or expression of a knock-in C129A variant leads to embryonic lethality at E10.5 (30, 35), with the C129A-expression mice showing extensive cell death in the yolk sac and placenta (35). Tamoxifen-induced adult CreERT2-OtulinLacZ/flox mice become moribund within a day (30). Deficiency in functional OTULIN protein by these mutations suggest a potential role of the LUBAC/OTULIN axis in developmental Wnt signaling (18, 49), though exactly molecular mechanisms are unclear.
3.2 OTULIN haploinsufficiency underlies an immunodeficient disorder conferring predisposition of life-threatening staphylococcal disease
Autosomal recessive OTULIN mutations cause ORAS, manifesting early in life with autoinflammation without clear evidence of infections, due to the defective downregulation of NF-κB-dependent inflammatory signaling. Recently, a group of patients carrying heterozygous variants of OTULIN were identified (named immunodeficiency 107 by MedGen) (38, 43, 50). Clinical manifestations presented, to some extent, with phenotypic heterogeneity. Six carriers were apparently healthy, one-third of patients expressed relatively milder respiratory infections, and the other one-third expressed the severe phenotypes including necrotizing pneumonia, necrotizing cellulitis, recurrent furunculosis, abdominal sepsis, spontaneous abscess, and sepsis shock, thus indicating that rare heterozygous mutations of OTULIN confer predisposition to severe necrosis, typically, but not exclusively, after infection with Staphylococcus aureus. It should be mentioned that skin and/or lung are consistently affected in all cases. The age of clinical onset is variable, and the earliest age of clinical onset in this cohort is 1.5 years (38, 43). The penetrance is estimated at 90%.
In comparison with biallelic OTULIN deficiency, OTULIN haploinsufficiency causes an accumulation of linear ubiquitin in dermal fibroblasts, but TNF-receptor NF-κB signaling remains intact. Routine immunological tests including assessments of leukocyte differentiation and oxidative burst capacity revealed no explanatory defects in patients with OTULIN haploinsufficiency. Subtle immunological testing for peripheral blood mononuclear cells (PBMCs) by mass cytometry and RNA sequencing showed no differences in the abundance of leukocyte subsets or in the expression of lineage markers. The development of myeloid and lymphoid subsets in patients with OTULIN haploinsufficiency was normal. Both at baseline and after stimulation, the PBMCs of the patients had a capacity to secrete various cytokines, including TNF and IL-1β, similar to that of PBMCs from healthy controls (38). These observations indicate that human OTULIN haploinsufficiency conferring susceptibility to infections is driven by a defect of cell-intrinsic immunity in fibroblasts.
4 Current management and future perspectives
Given the broad spectrum of clinical manifestations, the diagnosis of OTULIN deficiency can be challenging. A strong clinical suspicion must arise when clinical features, such as neonatal onset of recurrent fever, erythematous rash with painful nodules, and painful joints, are presented. Because of the auto-inflammatory nature of ORAS, anti-inflammatory drugs currently represent an effective treatment. However, treatment of affected patients with prednisolone (general cortico-steroidal immunosuppressant), azathioprine, methotrexate (anti-proliferative immunosuppressive drugs), and Anakinra (recombinant IL-1R-antatonist) had little or no effect on clinical symptoms (30, 31). By contrast, TNF-blocking agents infliximab or etanercept drastically reduced the symptoms, and serum levels of CRP as well as WBC and neutrophil counts in blood returned to normal ranges, suggesting a TNF-mediated pathogenesis in the patients (30, 31). Hematopoietic stem cell transplantation (HSCT) has been treated in one patient who developed severe inflammatory symptoms after birth, including fever and panniculitis (29). At 7 months of age, this patient had fever, diarrhea, panniculitis, cachexia, and cataracts. She underwent an HSCT at 17 months of age, and her symptoms resolved for 9 months, at which point she relapsed with fever and panniculitis. It was found that she had a decrease in bone marrow chimerism and regrowth of native hematopoietic cells. At 29 months of age, she was treated with Etanercept and had resolution of symptoms (29).
Further research needs to explore effective treatments other than anti-inflammatory drugs. Gene editing technology, such as the CRISPR/Cas9 system, represents one of the most curative approaches, in the wake of a successful outcome in the treatment of other hereditary diseases. In addition, the use of homology-direct repair and somatic genetic rescue as a correction mechanism may be attempted.
5 Conclusions
Germline mutations of OTULIN cause various complex clinical phenotypes, including autoinflammatory disorders and immunodeficient disorders. Despite the emergent findings, the exact pathogenetic details underlying these genetic variants are elusive. Uncovering these subtle molecular mechanisms will help develop more efficient and targeted treatments for patients.
Author contributions
CL: Funding acquisition, Supervision, Visualization, Writing – review & editing, Writing – original draft. BD: Investigation, Writing – original draft, Data curation. WP: Writing – original draft. GJ: Investigation, Data curation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the Natural Science Foundation of Hunan Province (2023JJ30914) and the Hunan Provincial Health Commission scientific research project (C2019179).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iwai K, Fujita H, Sasaki Y. Linear ubiquitin chains: NF-kappaB signalling, cell death and beyond. Nat Rev Mol Cell Biol. (2014) 15:503–8. doi: 10.1038/nrm3836
2. Clague MJ, Urbe S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. (2019) 20:338–52. doi: 10.1038/s41580-019-0099-1
3. Swatek KN, Komander D. Ubiquitin modifications. Cell Res. (2016) 26:399–422. doi: 10.1038/cr.2016.39
4. Fiil BK, Gyrd-Hansen M. The Met1-linked ubiquitin machinery in inflammation and infection. Cell Death Differ. (2021) 28:557–69. doi: 10.1038/s41418-020-00702-x
5. Hrdinka M, Gyrd-Hansen M. The met1-linked ubiquitin machinery: emerging themes of (De)regulation. Mol Cell. (2017) 68:265–80. doi: 10.1016/j.molcel.2017.09.001
6. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
7. Zak DE, Schmitz F, Gold ES, Diercks AH, Peschon JJ, Valvo JS, et al. Systems analysis identifies an essential role for SHANK-associated RH domain-interacting protein (SHARPIN) in macrophage Toll-like receptor 2 (TLR2) responses. Proc Natl Acad Sci U.S.A. (2011) 108:11536–41. doi: 10.1073/pnas.1107577108
8. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. (2012) 46:746–58. doi: 10.1016/j.molcel.2012.04.014
9. Kumari S, Redouane Y, Lopez-Mosqueda J, Shiraishi R, Romanowska M, Lutzmayer S, et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife. (2014) 3. doi: 10.7554/eLife.03422
10. Rickard JA, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. (2014) 3. doi: 10.7554/eLife.03464
11. Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. (2014) 9:153–65. doi: 10.1016/j.celrep.2014.08.066
12. Taraborrelli L, Peltzer N, Montinaro A, Kupka S, Rieser E, Hartwig T, et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat Commun. (2018) 9:3910. doi: 10.1038/s41467-018-06155-8
13. Jahan AS, Elbaek CR, Damgaard RB. Met1-linked ubiquitin signalling in health and disease: inflammation, immunity, cancer, and beyond. Cell Death Differ. (2021) 28:473–92. doi: 10.1038/s41418-020-00676-w
14. Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. (2012) 13:1178–86. doi: 10.1038/ni.2457
15. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. (2011) 471:591–6. doi: 10.1038/nature09816
16. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Differ. (2017) 24:1172–83. doi: 10.1038/cdd.2017.46
17. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. (2013) 153:1312–26. doi: 10.1016/j.cell.2013.05.014
18. Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, MacLean TA, Srikumar T, et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature. (2013) 498:318–24. doi: 10.1038/nature12296
19. Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. (2013) 154:169–84. doi: 10.1016/j.cell.2013.05.046
20. Elliott PR. Molecular basis for specificity of the Met1-linked polyubiquitin signal. Biochem Soc Trans. (2016) 44:1581–602. doi: 10.1042/BST20160227
21. Schaeffer V, Akutsu M, Olma MH, Gomes LC, Kawasaki M, Dikic I. Binding of OTULIN to the PUB domain of HOIP controls NF-kappaB signaling. Mol Cell. (2014) 54:349–61. doi: 10.1016/j.molcel.2014.03.016
22. Elliott PR, Nielsen SV, Marco-Casanova P, Fiil BK, Keusekotten K, Mailand N, et al. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol Cell. (2014) 54:335–48. doi: 10.1016/j.molcel.2014.03.018
23. Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. (2015) 13:2258–72. doi: 10.1016/j.celrep.2015.11.009
24. Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Biophys Res Commun. (2016) 474:452–61. doi: 10.1016/j.bbrc.2016.04.141
25. Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U.S.A. (2013) 110:15247–52. doi: 10.1073/pnas.1314715110
26. Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature. (2015) 528:370–5. doi: 10.1038/nature16165
27. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. (2009) 136:1098–109. doi: 10.1016/j.cell.2009.03.007
28. Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
29. Damgaard RB, Elliott PR, Swatek KN, Maher ER, Stepensky P, Elpeleg O, et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol Med. (2019) 11. doi: 10.15252/emmm.201809324
30. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell. (2016) 166:1215–1230.e20. doi: 10.1016/j.cell.2016.07.019
31. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci U.S.A. (2016) 113:10127–32. doi: 10.1073/pnas.1612594113
32. Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker-Jensen S, et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol Cell. (2013) 50:818–30. doi: 10.1016/j.molcel.2013.06.004
33. Hrdinka M, Fiil BK, Zucca M, Leske D, Bagola K, Yabal M, et al. CYLD limits lys63- and met1-linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep. (2016) 14:2846–58. doi: 10.1016/j.celrep.2016.02.062
34. Douglas T, Saleh M. Post-translational modification of OTULIN regulates ubiquitin dynamics and cell death. Cell Rep. (2019) 29:3652–3663 e5. doi: 10.1016/j.celrep.2019.11.014
35. Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature. (2018) 559:120–4. doi: 10.1038/s41586-018-0256-2
36. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. (2000) 289:2350–4. doi: 10.1126/science.289.5488.2350
37. Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, et al. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. (2004) 279:36171–4. doi: 10.1074/jbc.M406638200
38. Spaan AN, Neehus AL, Laplantine E, Staels F, Ogishi M, Seeleuthner Y, et al. Human OTULIN haploinsufficiency impairs cell-intrinsic immunity to staphylococcal alpha-toxin. Science. (2022) 376:eabm6380. doi: 10.1126/science.abm6380
39. Zinngrebe J, Moepps B, Monecke T, Gierschik P, Schlichtig F, Barth TFE, et al. Compound heterozygous variants in OTULIN are associated with fulminant atypical late-onset ORAS. EMBO Mol Med. (2022) 14:e14901. doi: 10.15252/emmm.202114901
40. Nabavi M, Shahrooei M, Rokni-Zadeh H, Vrancken J, Changi-Ashtiani M, Darabi K, et al. Auto-inflammation in a patient with a novel homozygous OTULIN mutation. J Clin Immunol. (2019) 39:138–41. doi: 10.1007/s10875-019-00599-3
41. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. (2022) 42:1508–20. doi: 10.1007/s10875-022-01352-z
42. Raza R, Jimenez-Heredia R, Anwar MZ, Ullah A, Zia A, Rashid S, et al. Whole Exome Sequencing Revealed a Novel Sequence Variant in The OTULIN Underlying Auto-Inflammatory Syndrome. Research Square Platform LLC (2021). doi: 10.21203/rs.3.rs-164929/v1
43. Arts RJW, van der Linden TJ, van der Made CI, Hendriks MMC, van der Heijden WA, de Mast Q, et al. OTULIN haploinsufficiency-related fasciitis and skin necrosis treated by TNF inhibition. J Clin Immunol. (2023) 44:10. doi: 10.1007/s10875-023-01630-4
44. Damgaard RB, Jolin HE, Allison MED, Davies SE, Titheradge HL, McKenzie ANJ, et al. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. (2020) 27:1457–74. doi: 10.1038/s41418-020-0532-1
45. Verboom L, Martens A, Priem D, Hoste E, Sze M, Vikkula H, et al. OTULIN prevents liver inflammation and hepatocellular carcinoma by inhibiting FADD- and RIPK1 kinase-mediated hepatocyte apoptosis. Cell Rep. (2020) 30:2237–2247 e6. doi: 10.1016/j.celrep.2020.01.028
46. Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. (2015) 212:939–51. doi: 10.1084/jem.20141130
47. Doglio MG, Verboom L, Ruilova Sosoranga E, Frising UC, Asaoka T, Gansemans Y, et al. Myeloid OTULIN deficiency couples RIPK3-dependent cell death to Nlrp3 inflammasome activation and IL-1beta secretion. Sci Immunol. (2023) 8:eadf4404. doi: 10.1126/sciimmunol.adf4404
48. Verboom L, Anderson CJ, Jans M, Petta I, Blancke G, Martens A, et al. OTULIN protects the intestinal epithelium from apoptosis during inflammation and infection. Cell Death Dis. (2023) 14:534. doi: 10.1038/s41419-023-06058-7
49. Wang W, Li M, Ponnusamy S, Chi Y, Xue J, Fahmy B, et al. ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nat Commun. (2020) 11:3965. doi: 10.1038/s41467-020-17770-9
Keywords: OTULIN, deubiquitinase, genetic deficiency, autoinflammatory disorder, immunodeficient disorder
Citation: Dou B, Jiang G, Peng W and Liu C (2024) OTULIN deficiency: focus on innate immune system impairment. Front. Immunol. 15:1371564. doi: 10.3389/fimmu.2024.1371564
Received: 16 January 2024; Accepted: 04 April 2024;
Published: 07 May 2024.
Edited by:
Fuminori Tokunaga, Osaka Metropolitan University, JapanReviewed by:
Rune Damgaard, Technical University of Denmark, DenmarkCopyright © 2024 Dou, Jiang, Peng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chentao Liu, bGl1Y2hlbnRhbzIwMTdAb3V0bG9vay5jb20=
†These authors have contributed equally to this work