 Yunshen Li
Yunshen Li Jianan Zhao
Jianan Zhao Shicheng Guo
Shicheng Guo Dongyi He
Dongyi He- 1Department of Rheumatology, Shanghai Guanghua Hospital of Integrative Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
Osteoarthritis (OA) is a common joint disorder characterized by the degeneration of cartilage and inflammation, affecting millions worldwide. The disease’s complex pathogenesis involves various cell types, such as chondrocytes, synovial cells, osteoblasts, and immune cells, contributing to the intricate interplay of factors leading to tissue degradation and pain. RNA interference (RNAi) therapy, particularly through the use of small interfering RNA (siRNA), emerges as a promising avenue for OA treatment due to its capacity for specific gene silencing. siRNA molecules can modulate post-transcriptional gene expression, targeting key pathways involved in cellular proliferation, apoptosis, senescence, autophagy, biomolecule secretion, inflammation, and bone remodeling. This review delves into the mechanisms by which siRNA targets various cell populations within the OA milieu, offering a comprehensive overview of the potential therapeutic benefits and challenges in clinical application. By summarizing the current advancements in siRNA delivery systems and therapeutic targets, we provide a solid theoretical foundation for the future development of novel siRNA-based strategies for OA diagnosis and treatment, paving the way for innovative and more effective approaches to managing this debilitating disease.
1 Introduction
Osteoarthritis (OA) is a prevalent chronic joint disease characterized by the degeneration of cartilage and inflammation (1). The risk factors for OA encompass obesity, injury, genetic predisposition, among others. It is noteworthy that the incidence is higher among females compared to males, and age stands as the foremost risk factor (2, 3). The pathogenesis of osteoarthritis primarily involves the articular cartilage, subchondral bone, and synovium, yet its specific mechanisms remain not fully elucidated (4). Therapeutic interventions for OA mainly include non-pharmacologic management, pharmacologic management, and surgical interventions. Recommended non-pharmacological approaches include education, self-management, exercise, and weight loss (5). Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen are frontline pharmaceuticals in the treatment of OA, recommended for use in clinical guidelines. However, the cautious administration of NSAIDs is imperative to avert potential adverse effects. While acetaminophen’s efficacy falls short of that of NSAIDs, its safety profile renders it suitable for patients contraindicated for NSAIDs (6). Surgical interventions exhibit notable efficacy in long-term improvement of patients’ physiological function and alleviation of pain; nevertheless, they are not exempt from potential side effects (7). Moreover, an increasing body of evidence suggests a correlation between psychological factors and the onset of OA pain. Utilizing psychological approaches, such as coping skills training (CST) and emotional disclosure, has proven effective in alleviating OA pain (8). As the pathogenic mechanisms of OA are continually elucidated, an increasing number of pharmaceuticals targeting the pathophysiological mechanisms of the disease are under development to replace the current predominantly palliative treatments, such as chondroitin sulfate and hyaluronan, collectively known as disease-modifying osteoarthritis drugs (DMOADs). These drugs, targeting cartilage, inflammatory pathways, and subchondral bone, hold promising prospects (9).
RNA interference (RNAi) denotes the duplex RNA’s inhibitory effect on genes, constituting a natural mechanism within organisms to silence genes (10). Small interfering RNA (siRNA) represents a category of double-stranded RNA molecules, spanning 21–23 nucleotides, formed through enzymatic cleavage of double-stranded RNA (11). siRNA associates with various proteins to form the RNA-induced silencing complex (RISC). Subsequently, it pairs with target mRNA, cleaving the mRNA and ultimately impeding the transcriptional process (12). Given its selective ability to silence almost any gene, siRNA therapy holds promise as a clinical approach for targeting specific genes in the treatment of OA. The first siRNA therapy was approved in 2018, utilized for the treatment of transthyretin-mediated amyloidosis. Since then, siRNA-based therapies for various diseases have entered clinical trials, indicating the broad prospects of siRNA in treating diseases (13). Furthermore, diverse delivery systems based on siRNA have been developed to enhance the efficiency of siRNA reaching its target, encompassing polymers, lipids, antibodies, nanocarriers, and peptide segments (14). This review comprehensively summarizes the progress in utilizing siRNA for the treatment of OA from a cellular perspective, including targeting chondrocytes, fibroblast-like synoviocytes, and osteoblasts, offering a novel outlook on the application of siRNA in OA therapy.
2 Targeting chondrocytes
The pathological manifestations of OA are diverse, with the degeneration of articular cartilage being a primary characteristic. Articular cartilage primarily comprises water and organic extracellular components. Chondrocytes represent the exclusive cell type within the cartilage matrix, responsible for maintaining the synthesis and degradation equilibrium of the extracellular matrix (ECM) (15). Upon exposure to mechanical, inflammatory, and metabolic factors, anomalies arise in the function of chondrocytes. This results in a reduction in the generation of ECM molecules and an augmentation in the production of proteinases. The disruption of ECM homeostasis is thus initiated, consequently promoting the degeneration of articular cartilage (16).
2.1 Targeting the apoptosis of chondrocytes
The proliferation and apoptosis of chondrocytes undergo alterations in OA. Research indicates a diminished proliferation and an increased apoptosis of chondrocytes in OA, which is associated with the progression of OA (17–19). Targeting the proliferation and apoptosis of chondrocytes holds the potential to alleviate the progression of OA.
Downregulating the expression of DNA methyltransferase 3 alpha (DNMT3A) using siRNA can reduce apoptosis and induce proliferation of chondrocytes in knee OA rats (20). 15-Lipoxygenase-1 (15-LOX-1), a lipid metabolism enzyme, can facilitate apoptosis of chondrocytes induced by sodium nitroprusside (SNP) and inhibit chondrocyte proliferation. Inhibiting arachidonate 15-lipoxygenase (ALOX15) expression using siRNA significantly mitigates the destruction of articular surface and bone, as well as the formation of osteophytes, thereby considerably alleviating destabilization of the medial meniscus (DMM) induced OA in rats (21). The expression of KH RNA binding domain containing, signal transduction associated 1 (KHDRBS1) increases in chondrocytes of OA model rats stimulated by tumor necrosis factor α (TNF-α). KHDRBS1 siRNA inhibits the activation of the nuclear factor kappa B (NF-κB) signaling pathway and significantly reduces chondrocyte apoptosis by reducing caspase-3 expression (22). Secreted phosphoprotein 1 (SPP1) is an extracellular matrix molecule that induces apoptosis of OA chondrocytes by activating the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT) pathway and inhibits proliferation. Knocking down SPP1 using siRNA promotes chondrocyte proliferation and inhibits chondrocyte apoptosis through the reduction of caspase-3 and caspase-9 expression (23). Phosphatase and tensin homologue (PTEN) is a crucial tumor suppressor gene, and its expression is significantly upregulated in OA chondrocytes. PTEN affects cell proliferation by inhibiting the PI3K/AKT pathway. PTEN siRNA significantly inhibits apoptosis and promotes proliferation of OA chondrocytes (24). Downregulating ribosomal protein L38 (RPL38) using siRNA, leading to the upregulation of suppressor of cytokine signaling 2 (SOCS2) expression and activation of the janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) pathway, resulted in a reduction of chondrocyte apoptosis induced by interleukin 1β (IL-1β). This process also alleviated histological phenomena such as reduction of articular chondrocytes, cartilage degradation and erosion in OA mice, thereby delaying the progression of OA (25).
2.2 Targeting chondrocyte senescence
In OA, chondrocyte senescence increases, and it can propagate to adjacent healthy chondrocytes through cell communication, inducing senescence (26). The aggregation of senescent chondrocytes promotes the progression of OA by disrupting the ECM homeostasis (27). The senescence-associated secretory phenotype (SASP) expressed by senescent chondrocytes is one of the key contributors to ECM impairment, causing an imbalance between ECM synthesis and degradation through the secretion of various cytokines and proteases such as interleukin 6 (IL-6), matrix metalloproteinases 13 (MMP13), and a disintegrin and metalloprotease with thrombospondin motifs 5 (ADAMTS5) (28). Research targeting cellular senescence provides insights for siRNA therapy.
Senescent cells reduce apoptosis by producing apoptosis inhibitor proteins (IAPs). Knocking down genes encoding anti-apoptotic proteins baculoviral IAP repeat containing 2 (BIRC2), baculoviral IAP repeat containing 3 (BIRC3), and X-linked inhibitor of apoptosis (XIAP) induces the clearance of senescent cells. Furthermore, the IAP inhibitor (AT-406) further alleviated cartilage degeneration and tibial subchondral bone reconstruction in rats with post-traumatic osteoarthritis (PTOA), thereby decelerating the progression of OA (29). Asporin is an extracellular matrix protein that induces chondrocyte senescence by targeting transforming growth factor β1 (TGF-β1)–Smad family member 2 (SMAD2) pathway. Knocking down asporin using siRNA inhibits senescence in chondrocytes and alleviates cartilage destruction in DMM-induced OA mice through TGF-β1 pathway (30). Inhibiting excitatory amino acid transporter protein 1 (EAAT1) enhances the response of senescent chondrocytes to ferroptosis, inducing cell death, with no significant impact on normal cells. Moreover, the EAAT1 inhibitor (UCPH-101) induced the clearance of senescent chondrocytes and mitigated cartilage degeneration (31). Mitofusin 2 (MFN2) regulates mitochondrial fusion, which plays a role in cell metabolism and aging, while dysregulation of MFN2 can lead to cartilage destruction. The expression of MFN2 is elevated during OA and aging, while knocking down MFN2 using siRNA can reverse age-related metabolic changes in rat chondrocytes (32). Additionally, tribbles homolog 3 (TRB3) siRNA reduces senescence in OA chondrocytes by reducing p16 and p21 levels, presenting another target for addressing chondrocyte senescence (33).
2.3 Targeting chondrocyte autophagy
The relationship between chondrocyte autophagy and apoptosis is intricate. Chondrocyte autophagy eliminates aged organelles and proteins, thereby maintaining internal homeostasis and protecting cells from apoptosis. Under the influence of external pathological factors, cellular autophagy is impaired, leading to the progression of OA (34). Hence, utilizing siRNA to restore chondrocyte autophagic function might be a means to treat OA.
The mechanistic target of rapamycin (mTOR) pathway is a crucial cellular autophagy inhibition pathway, regulated by PI3K/AKT and AMP-activated protein kinase (AMPK) (35). MicroRNA 7 (miR-7) siRNA inhibits the phosphorylation of the PI3K/AKT/mTOR pathway by reducing interleukin 17A (IL-17A) expression, and promotes the conversion of microtubule associated protein light chain 3 (LC3) from LC3-I to LC3-II, enhances beclin 1 (BECN1) expression, and suppresses sequestosome 1 (SQSTM1) expression, thereby restoring autophagic dysfunction in IL-1β induced chondrocytes. This significantly reduces cartilage destruction and the progression of OA in model rats (36). The upregulation of MFN2 in OA model mice, through activating the NF-κB and p38 mitogen-activated protein kinase (MAPK) pathways and inhibiting the PI3K/AKT/mTOR pathway, promotes inflammation and leads to excessive autophagy in chondrocytes. Knocking down MFN2 can suppress inflammation and cartilage degeneration in OA rat chondrocytes, thereby slowing down the progression of OA (37). The upregulation of transient receptor potential cation channel subfamily V member 5 (TRPV5) expression in OA is mitigated by TRPV5 siRNA, which reduces intracellular Ca2+ influx and enhances autophagy in MIA-induced OA rat chondrocytes. Furthermore, ruthenium red (a TRPV5 inhibitor) delays OA progression by reducing cartilage destruction (38). TRB3 inhibits autophagy by suppressing the autophagic receptor p62 and is upregulated in TNF-α-induced OA chondrocytes. Knocking down TRB3 using siRNA promotes autophagy in chondrocytes, making it a potential target for targeting chondrocyte autophagic function (33).
2.4 Targeting chondrocyte secretion
In OA, the aberrant function of chondrocytes leads to the release of degradative enzymes, such as matrix metalloproteinases (MMPs) and a disintegrin and metalloprotease with thrombospondin motifs (ADAMTSs) (4). These enzymes further contribute to cartilage degradation. The degradation products of cartilage can serve as damage-associated molecular patterns (DAMPs), entering the synovium and inducing the production of inflammatory factors. This, in turn, further stimulates chondrocytes to produce degradative enzymes, forming a vicious cycle (39).
MMP13 is upregulated in PTOA and contributes to cartilage destruction by degrading type II collagen. Using a nano-platform to deliver MMP13 siRNA reduces cartilage degradation, synovial hyperplasia and osteophyte growth in PTOA mice model, slowing down the progression of PTOA with favorable long-term therapeutic effects (40). Intra-articular injection of MMP13 siRNA and/or ADAMTS5 siRNA inhibited cartilage degradation in early-stage OA mice model (41). The use of nanoparticle delivery for lysine demethylase 6B (KDM6B) siRNA lowered the expression of MMP13 in mice model, and significantly alleviated the progression of OA by reducing cartilage degradation (42). The level of β-catenin is increased in chondrocytes of OA mice. Catenin beta 1 (CTNNB1) encodes β-catenin, and knocking down CTNNB1 reduces the expression of matrix metalloproteinases 3 (MMP3), MMP13, and a disintegrin and metalloprotease with thrombospondin motifs 4 (ADAMTS4). This suggests that CTNNB1 siRNA may inhibit cartilage degradation (43). Jian Zhang et al. discovered a drug that co-delivers curcumin and endothelial PAS domain protein 1 (EPAS1) siRNA, which alleviates mice cartilage degradation and slows down the progression of OA by reducing the expression of MMP3, MMP13, ADAMTS5 in OA chondrocytes (44). The activation of the interleukin 1 (IL-1) signaling pathway can lead to the progression of OA. Matrix metalloproteinases 9 (MMP9) siRNA reduces the shedding of syndecan-4 (SDC4), thereby lowering the sensitivity of chondrocytes to the IL-1 signaling pathway (45). Furthermore, miR-7 siRNA inhibits the expression of MMP3, MMP13, and ADAMTS5 in IL-1β-induced chondrocytes, thereby promoting ECM homeostasis and delaying OA progression (36).
3 Targeting fibroblast-like synoviocytes
Synovial inflammation is another characteristic feature of OA. The aberrant function of chondrocytes leads to the secretion of proteolytic enzymes, causing the production of inflammatory and metabolic products that affect the adjacent synovium. The progression of synovial inflammation can further exacerbate cartilage damage (46).
The production of pro-inflammatory cytokines such as IL-1β, TNF-α, IL-6, can exacerbate cartilage degradation and is associated with hyperalgia in OA (47, 48). Silencing NLR family pyrin domain containing 1 (NLRP1) reduces the production of IL-1β induced by P2X4 purinoceptor (P2X4) in OA fibroblast-like synoviocytes (49). RELA proto-oncogene, NF-kB subunit (RELA) siRNA significantly inhibits the induction of IL-1β and TNF-α in the synovial fluid, thereby alleviating synovial inflammation and cartilage degradation in early-stage OA rats through inhibition of the NF-κB pathway (50). Utilizing nanoparticles to deliver cyclin dependent kinase inhibitor 2A (CDKN2A) siRNA results in a reduction of IL-1β, IL-6, and TNF-α expression in fibroblast-like synoviocytes of model rats, and reduces cartilage destruction and pain (51). Hyaluronan contributes to the composition of synovial fluid for joint lubrication. In OA, hyaluronan degradation occurs, promoting inflammation. The expression of cell migration inducing hyaluronidase 1 (CEMIP) increases in OA fibroblast-like synoviocytes, and knocking down CEMIP reduces the degradation of hyaluronan in OA fibroblast-like synoviocytes (52). Glutaminase (GLS) siRNA inhibits glutamine–glutamate metabolism, reducing the IL-6 inflammatory response in OA fibroblast-like synoviocytes (53).
Proliferation of fibroblast-like synoviocytes is one of the characteristics of synovial inflammation and can lead to hyperplasia of the synovial lining (54). Knocking down latent transforming growth factor beta binding protein 1 (LTBP-1) using siRNA reduces fibroblast-like synoviocytes proliferation by downregulating the transforming growth factor β (TGF-β) signaling pathway (55).
Methyltransferase like 3 (METTL3) mediated m6A modification inhibits autophagy in OA fibroblast-like synoviocytes by regulating autophagy related 7 (ATG7) RNA and promotes cell senescence. METTL3 siRNA is able to inhibit expression of SASP-related genes and alleviate senescence in fibroblast-like synoviocytes. Furthermore, intra-articular injection of METTL3 siRNA delayed the progression of DMM-induced OA in mice (56). Fibroblast-like synoviocytes are considered the primary cells involved in synovial fibrosis in OA, and chronic joint pain is closely associated with synovial fibrosis (57). Inhibiting the upregulated expression of hypoxia inducible factor 1 subunit alpha (HIF-1A) in knee OA model rats reduces pyroptosis in fibroblast-like synoviocytes and significantly decreases the expression of synovial fibrogenic markers (58).
4 Targeting osteoblasts
In OA, subchondral bone undergoes remodeling due to excessive loading and changes in mechanical environment (59). The abnormal remodeling of subchondral bone can further exacerbate cartilage degradation (60). Osteoblasts regulate bone formation, remodeling and mineralization (61). Osteoblasts undergo changes in phenotype in OA, such as elevated alkaline phosphatase activity and increased secretion of osteocalcin. Additionally, alterations in signaling pathways like wingless-type MMTV integration site family (WNT) and TGF-β contribute to the abnormal remodeling of subchondral bone (62, 63). Hence, targeting osteoblasts with siRNA represents a potential therapeutic approach.
In OA, there is an increased production of endogenous hepatocyte growth factor (HGF), which, by stimulating the production of TGF-β1, inhibits osteoblast responsiveness to bone morphogenetic protein 2 (BMP-2), leading to abnormal mineralization. HGF siRNA restores osteoblast responsiveness to BMP-2 and upregulates the WNT signaling pathway to nearly normal levels (64). Leptin expression is significantly increased in OA osteoblasts and can lead to elevated levels of alkaline phosphatase and osteocalcin, as well as increased osteoblast proliferation. Partially reducing the alkaline phosphatase activity and osteocalcin release is possible by inhibiting leptin or its receptor using siRNA, indicating the potential to inhibit bone remodeling (65). Silencing ALOX15 promotes AMPK phosphorylation, inhibits mechanistic target of rapamycin complex 1 (mTORC1) phosphorylation, thereby suppressing expression levels of TGF-β1. This, in turn, enhances osteoblast autophagy and ultimately alleviates the progression of OA (66). Activation of Toll-like receptor 4 (TLR4) and innate immune activation can exacerbate cartilage degradation in OA. The antidepressant amitriptyline can bind to TLR4, inhibiting TLR4, IL-1 receptor, and NLR family pyrin domain containing 3 (NLRP3) dependent innate immune responses in OA chondrocytes, synoviocytes, and osteoblasts. Similarly, silencing NLRP3 using siRNA has a comparable effect (67).
5 Targeting other cells
The pathological changes in OA involve the entire joint, and current research predominantly focuses on the abnormalities in cartilage. Research on other cells to treat OA is limited, such as macrophages or mesenchymal stem cells (68). Nevertheless, siRNA targeting these cells has shown promising prospects.
Xu Chen et al. engineered nanoparticles capable of releasing nitric oxide and notch receptor 1 (NOTCH1) siRNA. By inhibiting macrophage inflammatory responses, these nanoparticles reduced OA cartilage damage without significant side effects (69).
Mesenchymal stem cells derived from bone marrow are ideal for tissue repair due to their differentiation potential. Inhibiting BLACAT1 overlapping LEMD1 locus (BLACAT1) promotes the proliferation and osteogenic differentiation of bone marrow mesenchymal stem cells under inflammatory conditions, demonstrating potential for treating OA (70).
6 Discussion
OA is a chronic degenerative joint disease that, as it progresses, often leads to disability and pain, significantly impacting the quality of life (71). Currently, the treatment approach for OA primarily focuses on mitigating disease symptoms and arresting its progression. In recent years, with an increasingly profound understanding of OA, it is recognized as a comprehensive, multifactorial joint disorder, intimately connected to the interactions among articular cartilage, synovium, subchondral bone, and their constituent cells. Due to its inherent property of selectively silencing genes, siRNA stands as a potential candidate among DMOADs. Recent investigations have disclosed that employing siRNA to target chondrocytes, fibroblast-like synoviocytes, osteoblasts, osteoblasts, and other cells and molecules involved in the development of OA can effectively decelerate the progression of the ailment. Additionally, a substantial volume of research centered around siRNA delivery systems has enhanced the precision and duration of siRNA delivery to target tissues. While there is currently no siRNA therapy specifically targeting osteoarthritis (according to clinicaltrials.gov), an increasing number of siRNA therapies targeting various diseases are entering clinical trials or gaining approval. These include cancer, hypertension, hypercholesterolemia, as well as some rare genetic disorders such as hemophilia and primary hyperoxaluria (72). These studies collectively highlight the broad potential of employing siRNA in the treatment of OA.
However, the constraints of current siRNA therapies cannot be disregarded. The primary method of siRNA therapy involves intravenous administration using nano-carriers, including lipid carriers, polymer carriers, and inorganic carriers (12). Lipid carriers, due to their positively charged nature, may undergo aggregation with serum proteins (73). On the other hand, negatively charged siRNA can distribute through the bloodstream to the reticuloendothelial system (RES) and are more readily phagocytosed, compared to neutral or positively charged counterparts (11). Furthermore, siRNA permits a degree of mismatch with the target mRNA, resulting in the silencing of non-target genes, known as off-target effects (74). Competition with the endogenous RNAi pathway and off-target effects can lead to hepatotoxicity (75). Cationic lipids and polymers, among other delivery methods involving internalization, can also induce immune responses by activating Toll-like receptor 7 (TLR7) and Toll-like receptor 8 (TLR8) (76). To address these issues, numerous improvement strategies have been applied to optimize siRNA delivery, including chemical modifications, siRNA-ligand conjugation, siRNA-polymer conjugation, and others (77).
Beyond the inherent limitations of siRNA therapy, the selection of target genes represents one of the challenges in utilizing siRNA therapy for OA treatment. Given that OA is a disease affecting the entire joint, silencing target genes for OA may also potentially diminish therapeutic efficacy through the impact on physiological activities of other tissues, or even inadvertently accelerate disease progression. The activation of the PI3K/AKT/mTOR pathway can enhance skeletal muscle protein synthesis, whereas inhibition of the PI3K/AKT/mTOR pathway can increase protein breakdown, marking a signature of muscle atrophy. Moreover, muscular weakness constitutes a risk factor in the development of OA (78, 79). IL-6 is released during and after exercise to enhance muscle energy supply and plays a crucial role in the repair of acute muscle injuries by activating satellite cells (80, 81). Inhibiting IL-6 could have repercussions on muscle function, potentially exacerbating the progression of OA.
In this review, we summarized the advancements in utilizing siRNA to target various cells for the treatment of OA, including chondrocytes, fibroblast-like synoviocytes, osteoblasts, and others (Table 1 and Figure 1). These studies, encompassing both in vitro experiments and in vivo trials, consistently demonstrate the potential of siRNA therapy. However, siRNA therapy still confronts numerous challenges, such as the selection of therapeutic targets, more efficient carriers for siRNA delivery, and addressing the immunogenicity of siRNA. Hence, the therapeutic potential of siRNA remains vast, with significant room for further advancement.
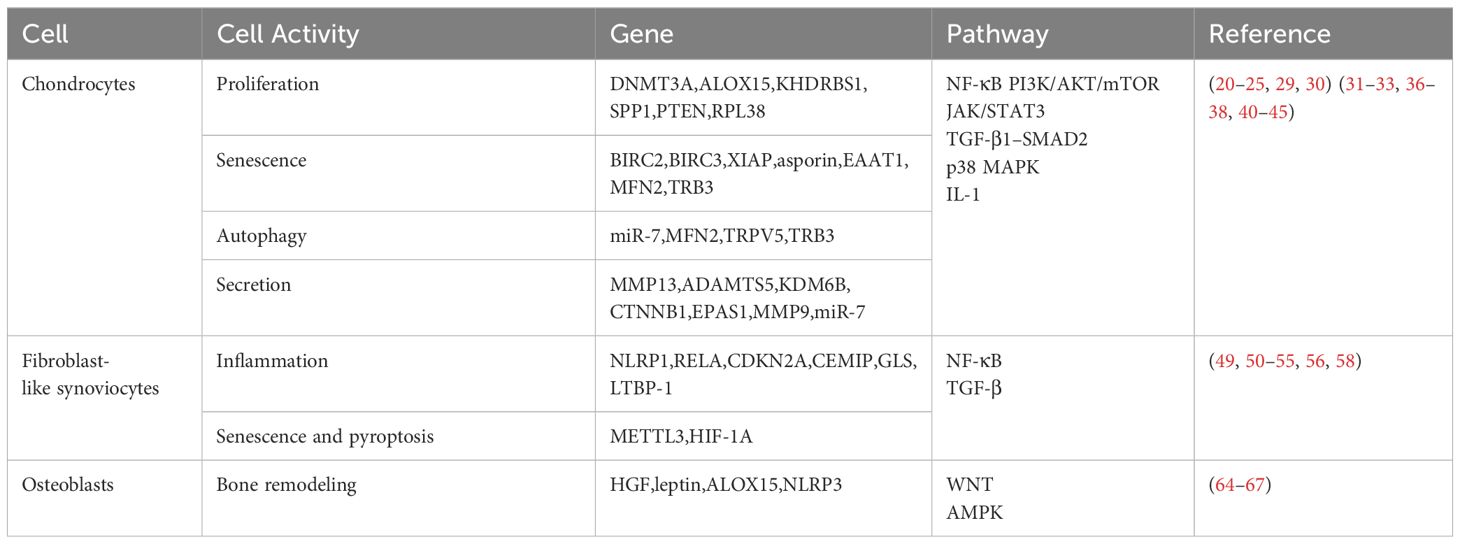
Table 1 Gene targets, cell activity and pathways table.
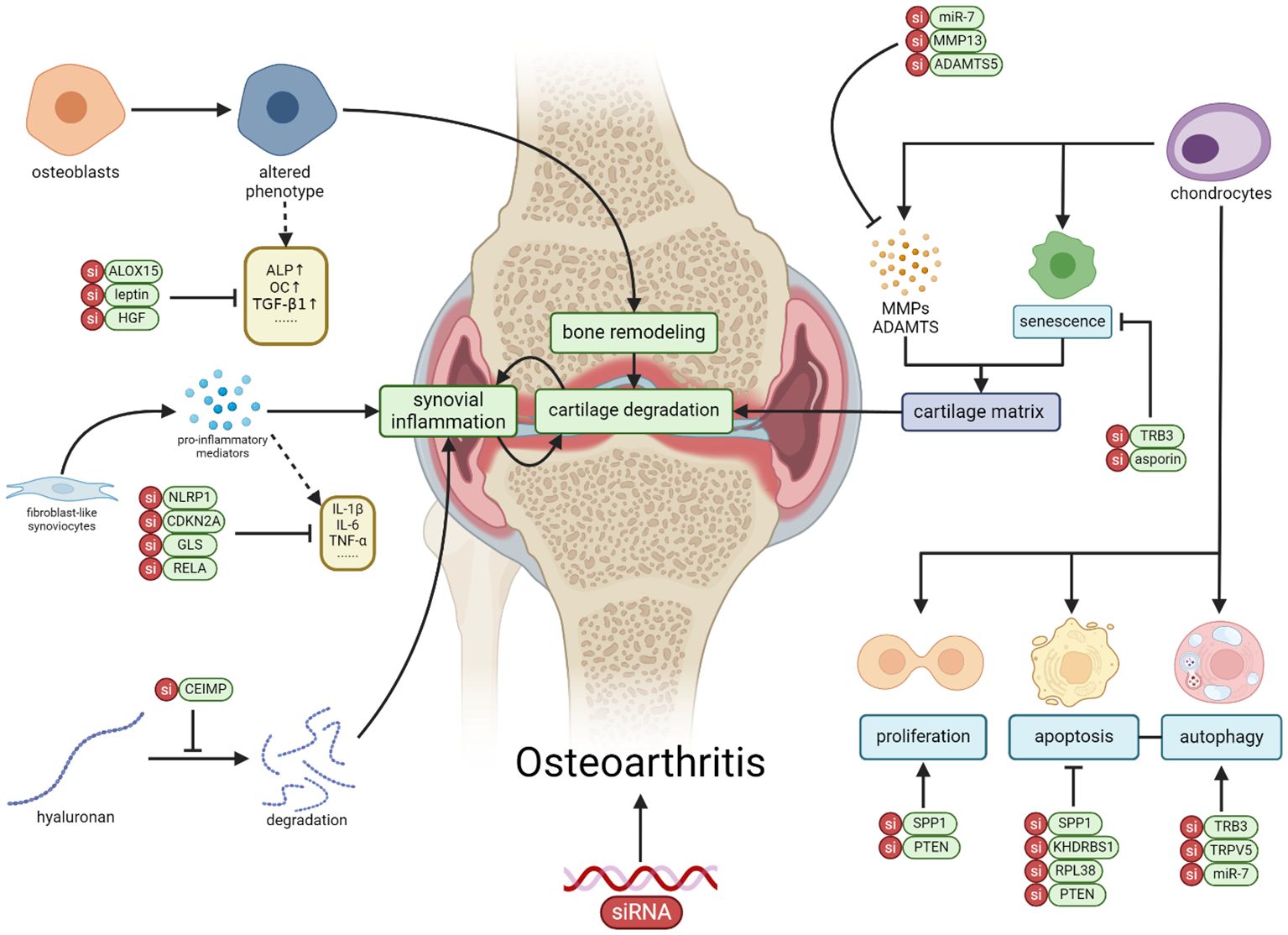
Figure 1 The application of siRNA therapy in OA involves targeting genes and signaling pathways in various cell types within OA, including chondrocytes, synovial fibroblasts, osteoblasts, macrophages, and mesenchymal stem cells. Through the correction of cellular activities such as proliferation, apoptosis, senescence, autophagy, and inflammation, siRNA exhibits the potential to alleviate the progression of OA (Created with BioRender.com).
Author contributions
YL: Writing – original draft, Writing – review & editing. JZ: Writing – original draft, Writing – review & editing. SG: Writing – review & editing. DH: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the National Natural Science Funds of China (82074234), Shanghai He Dongyi Famous Chinese Medicine Studio Construction Project (SHGZS-202220), Shanghai Municipal Health Commission (202340274), Traditional therapies and preparations for arthropathy transform the research and development of Doctor (RCJD2021B04).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Molnar V, Matišić V, Kodvanj I, Bjelica R, Jeleč Ž, Hudetz D, et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. Int J Mol Sci. (2021) 22:9208. doi: 10.3390/ijms22179208
2. Vina ER, Kwoh CK. Epidemiology of osteoarthritis: literature update. Curr Opin Rheumatol. (2018) 30:160–7. doi: 10.1097/BOR.0000000000000479
3. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. (2019) 393:1745–59. doi: 10.1016/S0140–6736(19)30417–9
4. Glyn-Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet. (2015) 386:376–87. doi: 10.1016/S0140–6736(14)60802–3
5. Nelson AE, Allen KD, Golightly YM, Goode AP, Jordan JM. A systematic review of recommendations and guidelines for the management of osteoarthritis: The Chronic Osteoarthritis Management Initiative of the U.S. Bone and Joint Initiative. Semin Arthritis Rheumatol. (2014) 43:701–12. doi: 10.1016/j.semarthrit.2013.11.012
6. Abramoff B, Caldera FE. Osteoarthritis: pathology, diagnosis, and treatment options. Med Clin North Am. (2020) 104:293–311. doi: 10.1016/j.mcna.2019.10.007
7. de l’Escalopier N, Anract P, Biau D. Surgical treatments for osteoarthritis. Ann Phys Rehabil Med. (2016) 59:227–33. doi: 10.1016/j.rehab.2016.04.003
8. Keefe FJ, Somers TJ. Psychological approaches to understanding and treating arthritis pain. Nat Rev Rheumatol. (2010) 6:210–6. doi: 10.1038/nrrheum.2010.22
9. Huang Z, Ding C, Li T, Yu SPC. Current status and future prospects for disease modification in osteoarthritis. Rheumatology. (2018) 57:iv108–23. doi: 10.1093/rheumatology/kex496
11. Alshaer W, Zureigat H, Al Karaki A, Al-Kadash A, Gharaibeh L, Hatmal MM, et al. siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur J Pharmacol. (2021) 905:174178. doi: 10.1016/j.ejphar.2021.174178
12. Friedrich M, Aigner A. Therapeutic siRNA: state-of-the-art and future perspectives. BioDrugs Clin Immunother Biopharm Gene Ther. (2022) 36:549–71. doi: 10.1007/s40259–022-00549–3
13. Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Delivery Rev. (2019) 144:133–47. doi: 10.1016/j.addr.2019.05.004
14. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. (2013) 12:967–77. doi: 10.1038/nmat3765
15. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primer. (2016) 2:16072. doi: 10.1038/nrdp.2016.72
16. Li MH, Xiao R, Li JB, Zhu Q. Regenerative approaches for cartilage repair in the treatment of osteoarthritis. Osteoarthritis Cartilage. (2017) 25:1577–87. doi: 10.1016/j.joca.2017.07.004
17. Zou L, Liu J, Lu H. Influence of protein kinase RIPK4 expression on the apoptosis and proliferation of chondrocytes in osteoarthritis. Mol Med Rep. (2018) 17:3078–84. doi: 10.3892/mmr.2017.8209
18. Zhu Z, Gao S, Chen C, Xu W, Xiao P, Chen Z, et al. The natural product salicin alleviates osteoarthritis progression by binding to IRE1α and inhibiting endoplasmic reticulum stress through the IRE1α-IκBα-p65 signaling pathway. Exp Mol Med. (2022) 54:1927–39. doi: 10.1038/s12276-022-00879-w
19. Tian F, Wang J, Zhang Z, Yang J. LncRNA SNHG7/miR-34a-5p/SYVN1 axis plays a vital role in proliferation, apoptosis and autophagy in osteoarthritis. Biol Res. (2020) 53:9. doi: 10.1186/s40659–020-00275–6
20. Gu W, Shi Z, Song G, Zhang H. MicroRNA-199–3p up-regulation enhances chondrocyte proliferation and inhibits apoptosis in knee osteoarthritis via DNMT3A repression. Inflammation Res. (2021) 70:171–82. doi: 10.1007/s00011–020-01430–1
21. Chen K, Yan Y, Li C, Yuan J, Wang F, Huang P, et al. Increased 15-lipoxygenase-1 expression in chondrocytes contributes to the pathogenesis of osteoarthritis. Cell Death Dis. (2017) 8:e3109. doi: 10.1038/cddis.2017.511
22. Xu L, Sun C, Zhang S, Xu X, Zhai L, Wang Y, et al. Sam68 promotes NF-κB activation and apoptosis signaling in articular chondrocytes during osteoarthritis. Inflammation Res. (2015) 64:895–902. doi: 10.1007/s00011–015-0872–3
23. Lin Z, Tian XY, Huang XX, He LL, Xu F. microRNA-186 inhibition of PI3K-AKT pathway via SPP1 inhibits chondrocyte apoptosis in mice with osteoarthritis. J Cell Physiol. (2019) 234:6042–53. doi: 10.1002/jcp.27225
24. Huang Z, Zhang N, Ma W, Dai X, Liu J. MiR-337–3p promotes chondrocytes proliferation and inhibits apoptosis by regulating PTEN/AKT axis in osteoarthritis. BioMed Pharmacother. (2017) 95:1194–200. doi: 10.1016/j.biopha.2017.09.016
25. Shi L, Hu H, Sun P, Li Z, Ji L, Liu S, et al. RPL38 knockdown inhibits the inflammation and apoptosis in chondrocytes through regulating METTL3-mediated SOCS2 m6A modification in osteoarthritis. Inflammation Res. (2022) 71:977–89. doi: 10.1007/s00011-022-01579-x
26. Xie J, Wang Y, Lu L, Liu L, Yu X, Pei F. Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res Rev. (2021) 70:101413. doi: 10.1016/j.arr.2021.101413
27. Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. (2009) 17:971–9. doi: 10.1016/j.joca.2009.03.002
28. Yagi M, Endo K, Komori K, Sekiya I. Comparison of the effects of oxidative and inflammatory stresses on rat chondrocyte senescence. Sci Rep. (2023) 13:7697. doi: 10.1038/s41598–023-34825–1
29. Peilin W, Songsong T, Chengyu Z, Zhi C, Chunhui M, Yinxian Y, et al. Directed elimination of senescent cells attenuates development of osteoarthritis by inhibition of c-IAP and XIAP. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2618–32. doi: 10.1016/j.bbadis.2019.05.017
30. Liu L, Zhao C, Zhang H, Lu Y, Luo B, Yao Z, et al. Asporin regulated by miR-26b-5p mediates chondrocyte senescence and exacerbates osteoarthritis progression via TGF-β1/Smad2 pathway. Rheumatol Oxf. (2022) 61:2631–43. doi: 10.1093/rheumatology/keab725
31. Wen Z, Xia G, Liang C, Wang X, Huang J, Zhang L, et al. Selective clearance of senescent chondrocytes in osteoarthritis by targeting excitatory amino acid transporter protein 1 to induce ferroptosis. Antioxid Redox Signal. (2023) 39:262–77. doi: 10.1089/ars.2022.0141
32. Xu L, Wu Z, He Y, Chen Z, Xu K, Yu W, et al. MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthritis Cartilage. (2020) 28:1079–91. doi: 10.1016/j.joca.2019.11.011
33. Gu Y, Yan R, Wang Y, Zeng Y, Yao Q. High TRB3 expression induces chondrocyte autophagy and senescence in osteoarthritis cartilage. Aging. (2022) 14:5366–75. doi: 10.18632/aging.204066
34. Xu K, He Y, Moqbel SAA, Zhou X, Wu L, Bao J. SIRT3 ameliorates osteoarthritis via regulating chondrocyte autophagy and apoptosis through the PI3K/Akt/mTOR pathway. Int J Biol Macromol. (2021) 175:351–60. doi: 10.1016/j.ijbiomac.2021.02.029
35. Dalle Pezze P, Ruf S, Sonntag AG, Langelaar-Makkinje M, Hall P. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat Commun. (2016) 7:13254. doi: 10.1038/ncomms13254
36. Zhou X, Li J, Zhou Y, Yang Z, Yang H, Li D, et al. Down-regulated ciRS-7/up-regulated miR-7 axis aggravated cartilage degradation and autophagy defection by PI3K/AKT/mTOR activation mediated by IL-17A in osteoarthritis. Aging. (2020) 12:20163–83. doi: 10.18632/aging.103731
37. Deng X, Xu H, Pan C, Hao X, Liu J, Shang X, et al. Moderate mechanical strain and exercise reduce inflammation and excessive autophagy in osteoarthritis by downregulating mitofusin 2. Life Sci. (2023) 332:122020. doi: 10.1016/j.lfs.2023.122020
38. Wei Y, Wang Y, Wang Y, Bai L. Transient receptor potential vanilloid 5 mediates ca2+ Influx and inhibits chondrocyte autophagy in a rat osteoarthritis model. Cell Physiol Biochem. (2017) 42:319–32. doi: 10.1159/000477387
39. Lambert C, Zappia J, Sanchez C, Florin A, Dubuc JE, Henrotin Y. The damage-associated molecular patterns (DAMPs) as potential targets to treat osteoarthritis: perspectives from a review of the literature. Front Med. (2020) 7:607186. doi: 10.3389/fmed.2020.607186
40. Bedingfield SK, Colazo JM, Di Francesco M, Yu F, Liu DD, Di Francesco V, et al. Top-Down Fabricated microPlates for Prolonged, Intra-articular Matrix Metalloproteinase 13 siRNA Nanocarrier Delivery to Reduce Post-traumatic Osteoarthritis. ACS Nano. (2021) 15:14475–91. doi: 10.1021/acsnano.1c04005
41. Hoshi H, Akagi R, Yamaguchi S, Muramatsu Y, Akatsu Y, Yamamoto Y, et al. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. (2017) 368:379–87. doi: 10.1007/s00441-016-2563-y
42. Jin Y, Liu Z, Li Z, Li H, Zhu C, Li R, et al. Histone demethylase JMJD3 downregulation protects against aberrant force-induced osteoarthritis through epigenetic control of NR4A1. Int J Oral Sci. (2022) 14:34. doi: 10.1038/s41368–022-00190–4
43. Hu S, Mao G, Zhang Z, Wu P, Wen X, Liao W, et al. MicroRNA-320c inhibits development of osteoarthritis through downregulation of canonical Wnt signaling pathway. Life Sci. (2019) 228:242–50. doi: 10.1016/j.lfs.2019.05.011
44. Zhang ZJ, Hou YK, Chen MW, Yu XZ, Chen SY, Yue YR, et al. A pH-responsive metal-organic framework for the co-delivery of HIF-2α siRNA and curcumin for enhanced therapy of osteoarthritis. J Nanobiotechnol. (2023) 21:18. doi: 10.1186/s12951–022-01758–2
45. Bollmann M, Pinno K, Ehnold LI, Märtens N, Märtson A, Pap T, et al. MMP-9 mediated Syndecan-4 shedding correlates with osteoarthritis severity. Osteoarthritis Cartilage. (2021) 29:280–9. doi: 10.1016/j.joca.2020.10.009
46. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis)! Osteoarthritis Cartilage. (2013) 21:16–21. doi: 10.1016/j.joca.2012.11.012
47. Liao Y, Ren Y, Luo X, Mirando AJ, Long JT, Leinroth A, et al. Interleukin-6 signaling mediates cartilage degradation and pain in posttraumatic osteoarthritis in a sex-specific manner. Sci Signal. (2022) 15:eabn7082. doi: 10.1126/scisignal.abn7082
48. Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. (2010) 6:625–35. doi: 10.1038/nrrheum.2010.159
49. Fan C, Zhao X, Guo X, Cao X, Cai J. P2X4 promotes interleukin−1β production in osteoarthritis via NLRP1. Mol Med Rep. (2014) 9:340–4. doi: 10.3892/mmr.2013.1748
50. Chen LX, Lin L, Wang HJ, Wei XL, Fu X, Zhang JY, et al. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector-mediated NF-kappaBp65-specific siRNA. Osteoarthritis Cartilage. (2008) 16:174–84. doi: 10.1016/j.joca.2007.06.006
51. Park H, Lee HR, Shin HJ, Park JA, Joo Y, Kim SM, et al. p16INK4a-siRNA nanoparticles attenuate cartilage degeneration in osteoarthritis by inhibiting inflammation in fibroblast-like synoviocytes. Biomater Sci. (2022) 10:3223–35. doi: 10.1039/D1BM01941D
52. Shiozawa J, de Vega S, Cilek MZ, Yoshinaga C, Nakamura T, Kasamatsu S, et al. Implication of HYBID (Hyaluronan-binding protein involved in hyaluronan depolymerization) in hyaluronan degradation by synovial fibroblasts in patients with knee osteoarthritis. Am J Pathol. (2020) 190:1046–58. doi: 10.1016/j.ajpath.2020.01.003
53. Farah H, Wijesinghe SN, Nicholson T, Alnajjar F, Certo M, Alghamdi A, et al. Differential metabotypes in synovial fibroblasts and synovial fluid in hip osteoarthritis patients support inflammatory responses. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23063266
54. Sanchez-Lopez E, Coras R, Torres A, Lane NE, Guma M. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. (2022) 18:258–75. doi: 10.1038/s41584–022-00749–9
55. Wang X, Dong C, Li N, Ma Q, Yun Z, Cai C, et al. Modulation of TGF−β activity by latent TGF−β−binding protein 1 in human osteoarthritis fibroblast−like synoviocytes. Mol Med Rep. (2018) 17:1893–900. doi: 10.3892/mmr.2017.8086
56. Chen X, Gong W, Shao X, Shi T, Zhang L, Dong J, et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis. (2022) 81:87–99. doi: 10.1136/annrheumdis-2021–221091
57. Maglaviceanu A, Wu B, Kapoor M. Fibroblast-like synoviocytes: Role in synovial fibrosis associated with osteoarthritis. Wound Repair Regen. (2021) 29:642–9. doi: 10.1111/wrr.12939
58. Zhang L, Zhang L, Huang Z, Xing R, Li X, Yin S, et al. Increased HIF-1α in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev. (2019) 2019:6326517. doi: 10.1155/2019/6326517
59. Goldring SR. Alterations in periarticular bone and cross talk between subchondral bone and articular cartilage in osteoarthritis. Ther Adv Musculoskelet Dis. (2012) 4:249–58. doi: 10.1177/1759720X12437353
60. Chen Y, Huang YC, Yan CH, Chiu KY, Wei Q, Zhao J, et al. Abnormal subchondral bone remodeling and its association with articular cartilage degradation in knees of type 2 diabetes patients. Bone Res. (2017) 5:17034. doi: 10.1038/boneres.2017.34
61. Maruotti N, Corrado A, Cantatore FP. Osteoblast role in osteoarthritis pathogenesis. J Cell Physiol. (2017) 232:2957–63. doi: 10.1002/jcp.25969
62. Hilal G, Martel-Pelletier J, Pelletier JP, Ranger P, Lajeunesse D. Osteoblast-like cells from human subchondral osteoarthritic bone demonstrate an altered phenotype in vitro: possible role in subchondral bone sclerosis. Arthritis Rheumatol. (1998) 41:891–9. doi: 10.1002/1529–0131(199805)41:5<891::AID-ART17>3.0.CO;2-X
63. Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res Ther. (2007) 9:R100. doi: 10.1186/ar2301
64. Abed E, Bouvard B, Martineau X, Jouzeau JY, Reboul P, Lajeunesse D. Elevated hepatocyte growth factor levels in osteoarthritis osteoblasts contribute to their altered response to bone morphogenetic protein-2 and reduced mineralization capacity. Bone. (2015) 75:111–9. doi: 10.1016/j.bone.2015.02.001
65. Mutabaruka MS, Aoulad Aissa M, Delalandre A, Lavigne M, Lajeunesse D. Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res Ther. (2010) 12:R20. doi: 10.1186/ar2925
66. Wan Y, Lv Y, Li L, Yin Z. 15-Lipoxygenase-1 in osteoblasts promotes TGF-β1 expression via inhibiting autophagy in human osteoarthritis. BioMed Pharmacother. (2020) 121:109548. doi: 10.1016/j.biopha.2019.109548
67. Franco-Trepat E, Alonso-Pérez A, Guillán-Fresco M, Jorge-Mora A, Crespo-Golmar A, López-Fagúndez M, et al. Amitriptyline blocks innate immune responses mediated by toll-like receptor 4 and IL-1 receptor: Preclinical and clinical evidence in osteoarthritis and gout. Br J Pharmacol. (2022) 179:270–86. doi: 10.1111/bph.15707
68. Knights AJ, Farrell EC, Ellis OM, Lammlin L, Junginger LM, Rzeczycki PM, et al. Synovial fibroblasts assume distinct functional identities and secrete R-spondin 2 in osteoarthritis. Ann Rheum Dis. (2023) 82:272–82. doi: 10.1136/ard-2022–222773
69. Chen X, Liu Y, Wen Y, Yu Q, Liu J, Zhao Y, et al. A photothermal-triggered nitric oxide nanogenerator combined with siRNA for precise therapy of osteoarthritis by suppressing macrophage inflammation. Nanoscale. (2019) 11:6693–709. doi: 10.1039/C8NR10013F
70. Ji Y, Fang QY, Wang SN, Zhang ZW, Hou ZJ, Li JN, et al. Lnc-RNA BLACAT1 regulates differentiation of bone marrow stromal stem cells by targeting miR-142–5p in osteoarthritis. Eur Rev Med Pharmacol Sci. (2020) 24:2893–901. doi: 10.26355/eurrev_202003_20653
71. Fu K, Robbins SR, McDougall JJ. Osteoarthritis: the genesis of pain. Rheumatol Oxf Engl. (2018) 57:iv43–50. doi: 10.1093/rheumatology/kex419
72. Ranasinghe P, Addison ML, Dear JW, Webb DJ. Small interfering RNA: Discovery, pharmacology and clinical development-An introductory review. Br J Pharmacol. (2023) 180:2697–720. doi: 10.1111/bph.15972
73. Bhavsar D, Subramanian K, Sethuraman S, Krishnan UM. Translational siRNA therapeutics using liposomal carriers: prospects & challenges. Curr Gene Ther. (2012) 12:315–32. doi: 10.2174/156652312802083611
74. Bartoszewski R, Sikorski AF. Editorial focus: understanding off-target effects as the key to successful RNAi therapy. Cell Mol Biol Lett. (2019) 24:69. doi: 10.1186/s11658–019-0196–3
75. Janas MM, Schlegel MK, Harbison CE, Yilmaz VO, Jiang Y, Parmar R, et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat Commun. (2018) 9:723. doi: 10.1038/s41467–018-02989–4
76. Raja MAG, Katas H, Amjad MW. Design, mechanism, delivery and therapeutics of canonical and Dicer-substrate siRNA. Asian J Pharm Sci. (2019) 14:497–510. doi: 10.1016/j.ajps.2018.12.005
77. Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. (2020) 5:101. doi: 10.1038/s41392-020-0207-x
78. Yoshida T, Delafontaine P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells. (2020) 9:1970. doi: 10.3390/cells9091970
79. Øiestad BE, Juhl CB, Culvenor AG, Berg B, Thorlund JB. Knee extensor muscle weakness is a risk factor for the development of knee osteoarthritis: an updated systematic review and meta-analysis including 46 819 men and women. Br J Sports Med. (2022) 56:349–55. doi: 10.1136/bjsports-2021–104861
80. Gomarasca M, Banfi G, Lombardi G. Myokines: The endocrine coupling of skeletal muscle and bone. Adv Clin Chem. (2020) 94:155–218. doi: 10.1016/bs.acc.2019.07.010
Keywords: osteoarthritis, siRNA, chondrocytes, fibroblast-like synoviocytes, osteoblasts
Citation: Li Y, Zhao J, Guo S and He D (2024) siRNA therapy in osteoarthritis: targeting cellular pathways for advanced treatment approaches. Front. Immunol. 15:1382689. doi: 10.3389/fimmu.2024.1382689
Received: 06 February 2024; Accepted: 17 May 2024;
Published: 04 June 2024.
Edited by:
Xin Zhang, Duke University, United StatesReviewed by:
Ha-Reum Lee, Chungnam National University, Republic of KoreaChristopher Vann, Duke University, United States
Zheng Liu, Virginia Commonwealth University, United States
Copyright © 2024 Li, Zhao, Guo and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=
†These authors have contributed equally to this work