 Zhenglei Shen1†Jingying Zhu2†
Zhenglei Shen1†Jingying Zhu2† Ni Luo1Lei Feng1
Ni Luo1Lei Feng1 Heng Yue1Liying Song1Kunmei Liu1Huaxian Li1
Heng Yue1Liying Song1Kunmei Liu1Huaxian Li1 Honghua Cao1Yeying Zhou1
Honghua Cao1Yeying Zhou1 Yasar Mehmood Yousafzai3Zia Asad4
Yasar Mehmood Yousafzai3Zia Asad4 Youyu Qiu5
Youyu Qiu5 Shiwen Zhang2*
Shiwen Zhang2*- 1Department of Hematology, the Third Affiliated Hospital of Kunming Medical University, Yunnan Cancer Hospital, Peking University Cancer Hospital Yunnan, Kunming, China
- 2Department of Head and Neck Thyroid Surgery Department, The Third Affiliated Hospital of Kunming Medical University, Yunnan Cancer Hospital, Peking University Cancer Hospital Yunnan, Kunming, China
- 3Institute of Pathology and Diagnostic Medicine Khyber Medical University, Peshawar, Pakistan
- 4Molecular Biologist Public Health Reference Laboratory, Peshawar, Pakistan
- 5Radiology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
Background: Acute myeloid leukemia (AML) is a hematological malignancy with a high incidence of febrile neutropenia during the first two treatment cycles. This study aims to develop a gene signature related to neutrophil extracellular traps (NETs) to enhance understanding of AML mechanisms and identify potential prognostic biomarkers.
Methods: A consistent cluster analysis was conducted on 151 AML patients from the TCGA dataset. A differential analysis was performed to identify the differentially expressed genes (DEGs) specific to different subtypes and the training cohort (normal vs tumour). The NETs-related differentially expressed genes (NR-DEGs) were obtained through the overlapping of the two sets of differentially expressed genes. Univariate Cox and Least absolute shrinkage and selection operator (LASSO) regression analysis were employed to construct a NETs-related AML prognostic signature. Furthermore, an immune feature estimation and functional enrichment analysis was conducted between the two risk subgroups.
Results: Two distinct AML subtypes were identified, exhibiting markedly disparate survival outcomes. A total of 1,700 and 1,941 DEGs were identified in the different subtypes and training cohort (normal vs. tumour), respectively. Thirteen NR-DEGs were identified. Subsequently, a NETs-related prognostic signature was constructed based on the 3 prognostic genes (MPO, CCL3, and TLR8). An independent prognostic analysis indicated that the risk score and age could be employed as independent prognostic factors. Our findings revealed the presence of five markedly differentially expressed immune cells between the two risk subgroups. Ultimately, it was determined that all three genes were associated with the ‘chemokine signalling pathway’.
Conclusion: The prognostic signature comprised of MPO, CCL3, and TLR8 based on NETs was established, which provided theoretical basis and reference value for the research of AML.
1 Introduction
Acute myeloid leukemia (AML) is a clonal hematopoietic stem cell disorder marked by the rapid proliferation of immature myeloid cells (1). The cornerstone of AML treatment has traditionally been chemotherapy; however, many patients experience drug resistance and high relapse rates, contributing to persistently poor overall survival outcomes (2). Recently, advancements in therapeutic strategies, including molecularly targeted therapies, specific antibodies, and immune checkpoint inhibitors, have aimed to address these resistance issues and enhance treatment efficacy (3). Furthermore, research has identified ALOX5AP as a promising prognostic marker in AML, indicating its potential role as a therapeutic target (4). Despite these advancements, the aggressive nature of AML and the scarcity of effective prognostic markers present significant challenges in improving patient survival rates. Thus, identifying new prognostic genes is crucial for monitoring patient outcomes and understanding the pathogenesis of AML.
The immune system’s role in AML has become a critical area of research, with immune dysregulation identified as a key factor in disease progression and treatment resistance (5). Neutrophils, the predominant immune cells in the bone marrow and peripheral blood, have a multifaceted role in cancer initiation, progression, and dissemination. In particular, neutrophil extracellular traps (NETs), which consist of decondensed chromatin enriched with antimicrobial proteins, have emerged as significant players in inflammation and cancer advancement (6). In the context of AML, NETs have been shown to modulate the immune response and may profoundly influence patient outcomes (7). However, the genes regulating NET formation and their prognostic implications in AML remain poorly understood. This knowledge gap has prompted our investigation into the potential of NETs-related genes as innovative prognostic biomarkers in AML (8).
In this study, we aimed to identify a NETs-related prognostic signature that could predict prognosis in AML patients By leveraging bioinformatics analyses on large-scale gene expression datasets, we sought to identify prognostic genes related to NETs in AML, establish a prognostic model and uncover the molecular underpinnings of these genes in AML and explore their potential as therapeutic targets. These findings contribute to offer a new perspective on the role of the immune system in AML prognosis.
2 Materials and methods
2.1 Data source
The TCGA database (https://www.cancer.gov/ccg/research/genome-sequencing/tcga) was utilized to obtain gene expression profiles from 151 blood samples of AML patients. Additionally, RNA sequencing data from 386 healthy peripheral blood samples were sourced from the GTEx database (https://commonfund.nih.gov/GTEx). The merged dataset from these sources was designated as the training cohort (batch effects were corrected for TCGA and GTEx data through the ComBat method). For validation of the risk model, gene expression data from the GSE71014 dataset (platform: GPL10558) were collected from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) , which included 104 bone marrow samples. A total of 136 neutrophil extracellular trap (NET)-related genes (NRGs) were derived from the existing literature (9).
2.2 Consensus clustering of the NRGs model
The ‘ConsensusClusterPlus’ package was employed to conduct consensus cluster analysis on AML samples (N=151), utilizing the expression levels of all genes available in the TCGA database. Additionally, the ‘survminer’ package (10) was utilized to analyze Kaplan-Meier (KM) survival curves, focusing on overall survival (OS) across different subtypes (p <0.05). Subsequently, the ‘estimate’ package was implemented to compare Immune, Stromal, and ESTIMATE scores among the various subtypes via the Wilcoxon rank-sum test (p <0.05) (11).
2.3 Differential analysis
The ‘DESeq2’ package (12) was utilized to identify differentially expressed genes (DEGs) across different subtypes and within the training cohort (normal vs. tumor). The criteria for significance were set as |log2FC| > 1 and adjusted p < 0.05. Both volcano plots and heat maps were generated to illustrate the identified DEGs.
2.4 Gene functional enrichment analysis
NETs-related differential genes (NR-DEGs) were derived by intersecting NRGs and DEGs from different subtypes and the training cohort. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the NR-DEGs were performed using the ‘clusterProfiler’ package (13). Adjusted p < 0.05 were considered statistically significant.
2.5 Risk score-based subgroup analysis of NR-reated AML patients
Univariate Cox regression analysis was performed on NRG-DEGs to obtain candidate prognostic genes (p < 0.05, Hazard Ratio (HR) ≠ 1). Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis was conducted based on candidate prognostic genes. Ten-fold cross-validation was adopted to screen prognostic genes based on the optimal lambda value. After screening out the prognostic genes, the risk scores of AML patients with available survival data were calculated based on the LASSO coefficient of each prognostic gene and the expression level of the genes. Then, AML patients with available survival data were categorized into two risk subgroups (low-risk vs. high-risk) based on the median risk score calculated per sample to construct a risk model. The prognostic reliability of the risk score and model was assessed by K-M and receiver operating characteristic (ROC) curve (1-, 3-, 5-years), respectively. Additionally, after cross-platform normalization of the GSE71014 dataset using the normalizeBetweenArrays function in the ‘limma’ package, external validation of the risk model was performed.
2.6 Clinical nomogram model
Univariate Cox regression analysis was performed for Risk score, gender, age, FAB category, and cytogenetics risk. Clinical factors with p < 0.05 and HR ≠ 1 were selected for proportional hazards (PH) test. Subsequently, multivariate Cox regression analysis was conducted on the clinical factors that passed the PH test. Clinical factors with p < 0.05 and HR ≠ 0 were selected as independent prognostic factors. The nomogram containing independent factors was drawn to predict 1-, 3-, and 5-year survival probability of AML patients. Evaluation of the predictive effect was done by the calibration curve. If the slope of the calibration curve was close to 1, it indicated that the prediction performance of the nomogram was good.
2.7 Gene set enrichment analysis
To explore the potential biological pathways involving prognostic genes, GSEA was performed in the training set. First, the correlation between each single prognostic gene and all genes in the training set was calculated, and then all genes were ranked from largest to smallest according to the correlation coefficients. Finally, GSEA was performed using the ‘clusterProfiler’ package (14). Adjusted p < 0.05 were deemed statistically significant.
2.8 Immune feature estimation and chemotherapy analysis
The relative abundance of 22 infiltrating immune cell types across different risk groups was assessed using the CIBERSORT algorithm (15). The Wilcoxon rank-sum test was used to compare the infiltration differences of 22 types of immune cells between the high-risk and low-risk groups, and the Benjamini Hochberg (BH) method was employed to correct the p-values (adjusted p < 0.05). Additionally, the expression levels of 20 immune checkpoint molecules were compared between the high- and low-risk groups (p < 0.05) (16). Standardized gene expression data were submitted to the tumor immune dysfunction and exclusion (TIDE) official website to obtain the TIDE score for AML, and the correlation between TIDE and risk score was analyzed.
2.9 Potential drug prediction analysis
Using the DGIdb database, we identified potential targeted therapies to explore prospective treatment options for the prognostic genes associated with AML.
2.10 Statistical analysis
In this study, the Wilcoxon rank-sum test was used to compare differences between the two groups, and the Log-rank test was employed to evaluate survival differences among different groups. A P-value less than 0.05 was regarded as statistically significant.
3 Results
3.1 The NRGs associated with prognosis in AML
Consensus clustering analysis identified two distinct subtypes among 151 AML patients: Cluster 1 (comprising 94 samples) and Cluster 2 (comprising 57 samples), based on the expression levels of 58,387 genes (Figures 1A, B). Uniform Manifold Approximation and Projection (UMAP) provided clear differentiation between these 2 clusters (Supplementary Figure 1). Kaplan-Meier (K-M) survival analysis indicated a significant difference in survival between Cluster 1 and Cluster 2, with patients in Cluster 2 exhibiting poorer OS probabilities (Figure 1C). Additionally, the distribution of various clinicopathological features was presented for each subtype (Table 1), revealing notable disparities in gender and age. Importantly, we observed that immune, stromal, and ESTIMATE scores were significantly elevated in Cluster 2 relative to Cluster 1 (Figure 1D).
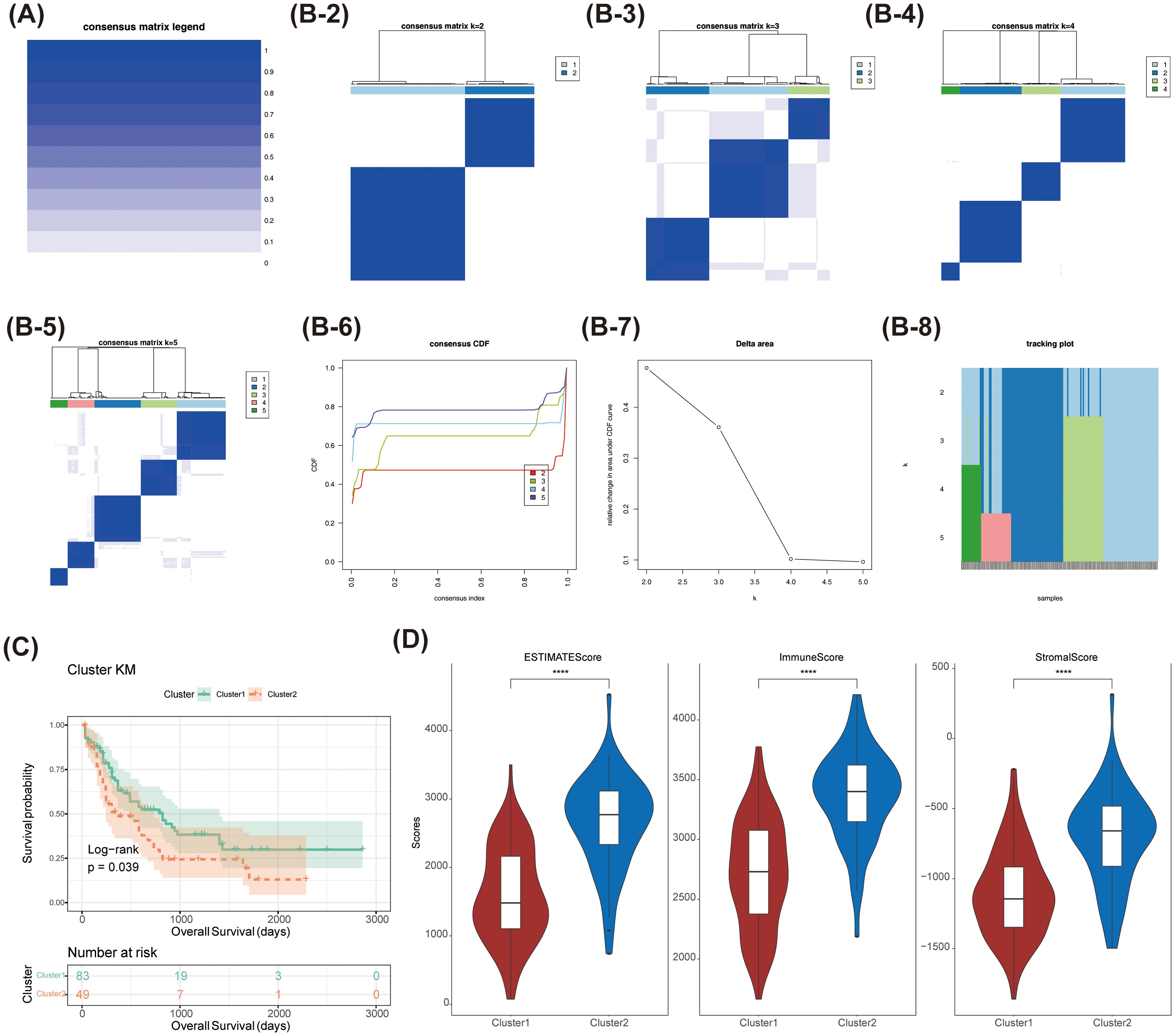
Figure 1. The consensus clustering analysis of myeloid leukaemia (AML) was conducted. (A-B-2-8)Comparison of clinical characteristics of patients with different subtypes of acute myeloid leukaemia (AML) (C) Survival analysis of patients with different subtypes of acute myeloid leukaemia (AML). The survival probability of Cluster 1 was higher than that of Cluster 2 (Log-rank p = 0.039). (D) Comparison of immune score and stromal score in patients with different subtypes of acute myeloid leukaemia (AML). **** stands for p < 0.0001.
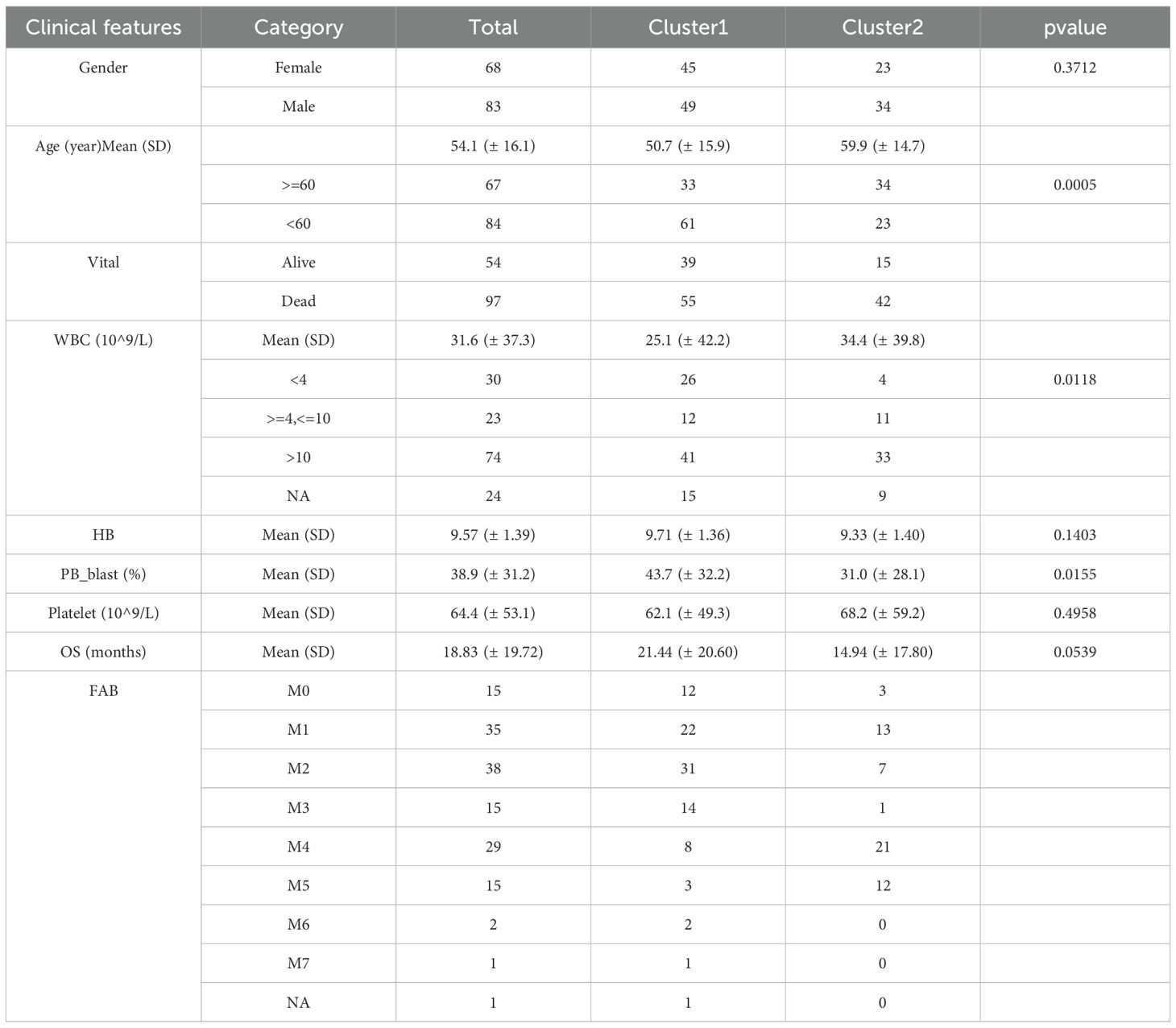
Table 1. Comparison of clinical characteristics among different subtypes of acute myeloid leukemia (AML) patients.
3.2 Identification of NR-DEGs in AML
A total of 1,700 DEGs were identified between the two clusters, comprising 760 upregulated and 940 downregulated genes (Figures 2A, B). In a parallel analysis, 1,941 DEGs (1,075 upregulated and 866 downregulated) were detected between normal and tumor groups (Figures 2C, D). Ultimately, we identified 13 NR-DEGs (Figure 2E). Functional enrichment analysis was performed to further elucidate the roles of these NR-DEGs in AML. The top 10 items for each classification were illustrated in a bubble diagram (Figure 2F; Supplementary Figures 2A, B). Our analysis revealed that these genes were primarily associated with the ‘positive regulation of inflammatory response’ and ‘positive regulation of cytokine production.’ Furthermore, these genes were implicated in the ‘Toll-like receptor signaling pathway’ and the formation of ‘Neutrophil extracellular traps’ (Figure 2G; Supplementary Figures 2C, D).
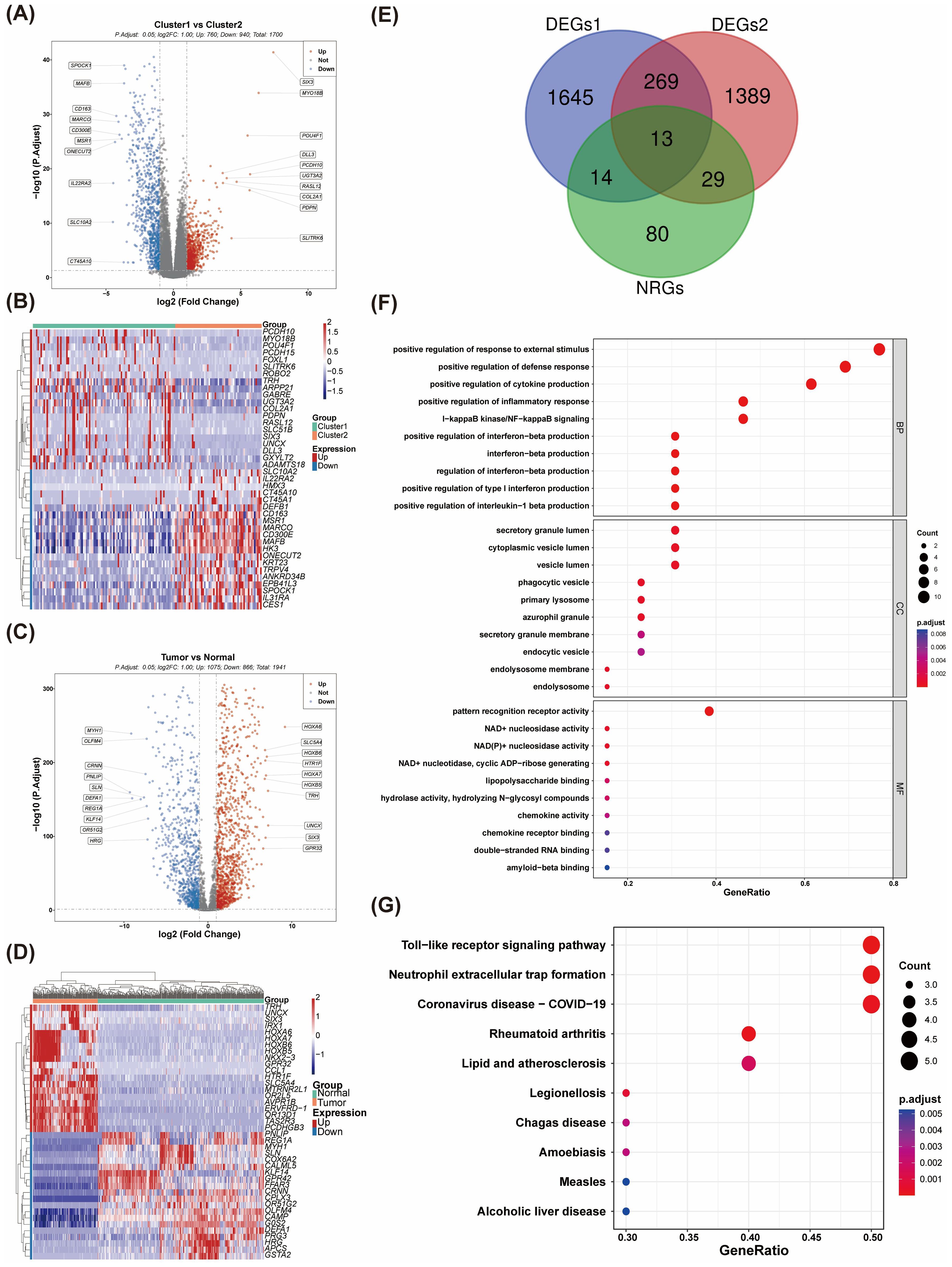
Figure 2. The identification and enrichment analysis of NR-DEGs. (A) The volcano plot of differentially expressed genes (DEGs) between Cluster 1 and Cluster 2. (B) The expression heatmap of DEGs between Cluster 1 and Cluster 2. (C) Volcano plots were generated to visualize the DEGs between AML and control samples. (D) Differential gene expression profiles in AML vs. normal tissues (E) A Venn diagram was constructed, revealing that a total of 13 NR-DEGs were identified. (F, G) Functional enrichment analyses were performed on the NR-DEGs through (GO) and Kyoto Encyclopedia of Genes and Genomes KEGG pathway analyses.
3.3 NR-DEGs-based gene signature for AML outcome prediction
Univariate regression analysis identified five significant prognostic genes (HR ≠ 1 & P < 0.05) within the training cohort (Figure 3A). To extract key prognostic indicators, we conducted LASSO regression on these five significant genes. Ultimately, three predictive genes were identified: MPO, CCL3, and TLR8 (Figures 3B, C). These genes were used to establish a NETs-related prognostic signature for AML. Patients were classified into two risk groups (high-risk and low-risk) based on the gene signature (Figures 3D, E). Strikingly, significant survival differences were observed between the 2 risk groups, with high-risk patients demonstrating poorer survival outcomes (Figure 3F). To assess the reliability of the model, the area under the curve (AUC) values for predicting 1-, 3-, and 5-year survival rates of AML patients were 0.68, 0.74, and 0.76, respectively, in the training set, indicating the model’s predictive capability regarding the survival status of AML patients (Figure 3G).
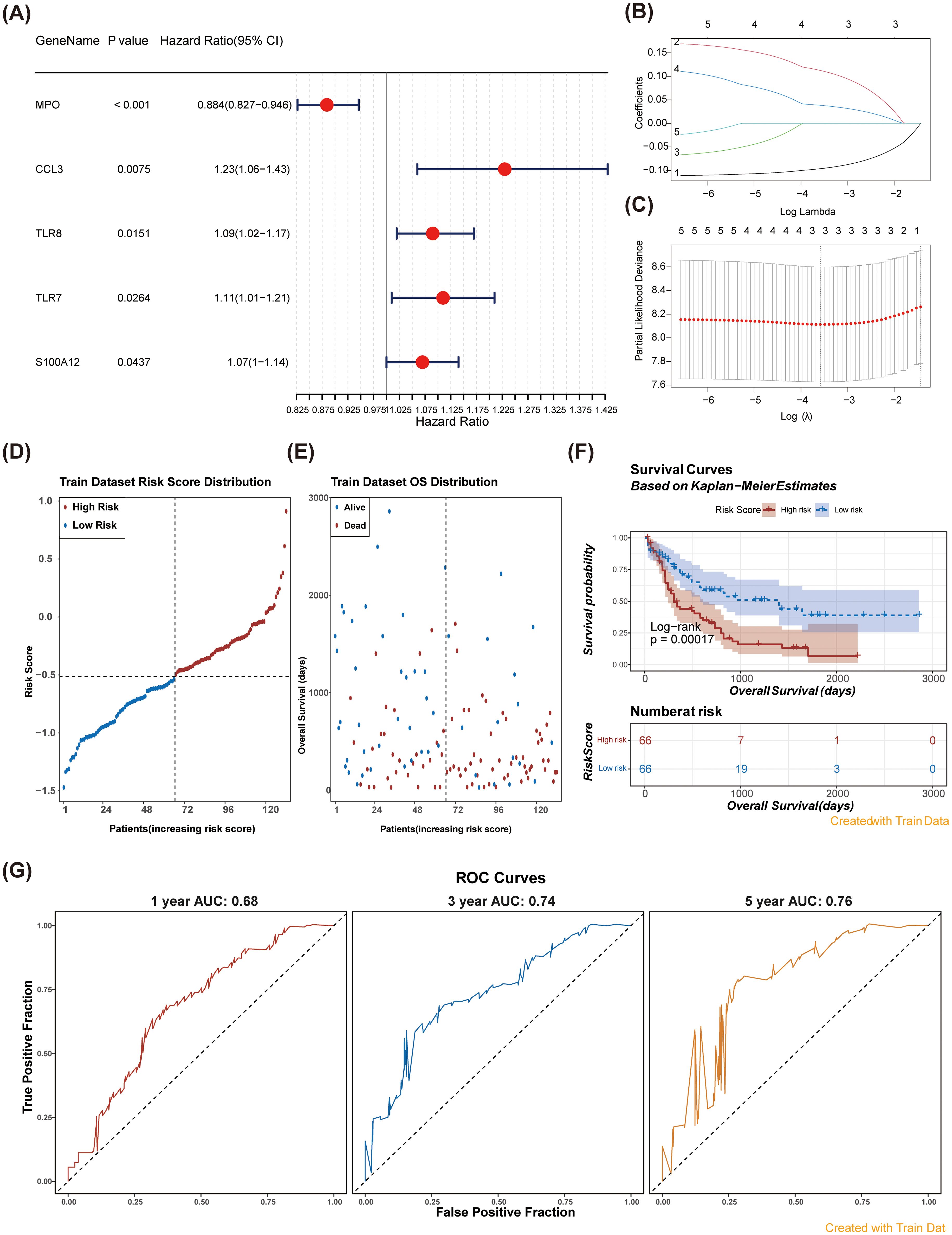
Figure 3. Identification of prognostic genes and construction of a risk model were performed. (A) Forest plots for univariate Cox regression analysis of NR-DEGs. Univariate Cox regression analysis identified five significant prognostic genes. (B, C) Least absolute shrinkage and selection operator (LASSO) analysis was performed on the five genes obtained from univariate Cox regression analysis. (D) Risk score distribution in patients: high vs. low risk groups over time. (E) Overall survival distribution of patients by risk score in the training dataset. (F) Kaplan-Meier survival curves for high and low risk patients based on risk scores. The survival probability of the low-risk group was higher than that of the high-risk group (Log-rank p = 0.00017). (G) ROC Curves for model performance at 1, 3, and 5 years using training data. The AUC values were all greater than 0.6.
Subsequently, we validated the risk model in an external dataset (GSE71014). Consistent with our training set findings (Figures 4A, B), patients in the high-risk group exhibited significantly poor overall survival (Figure 4C). The AUC values for the 1-, 3-, and 5-year survival predictions were consistently greater than 0.60 (Figure 4D), demonstrating that the NETs-related gene signature exhibited robust predictive performance in the validation cohort.
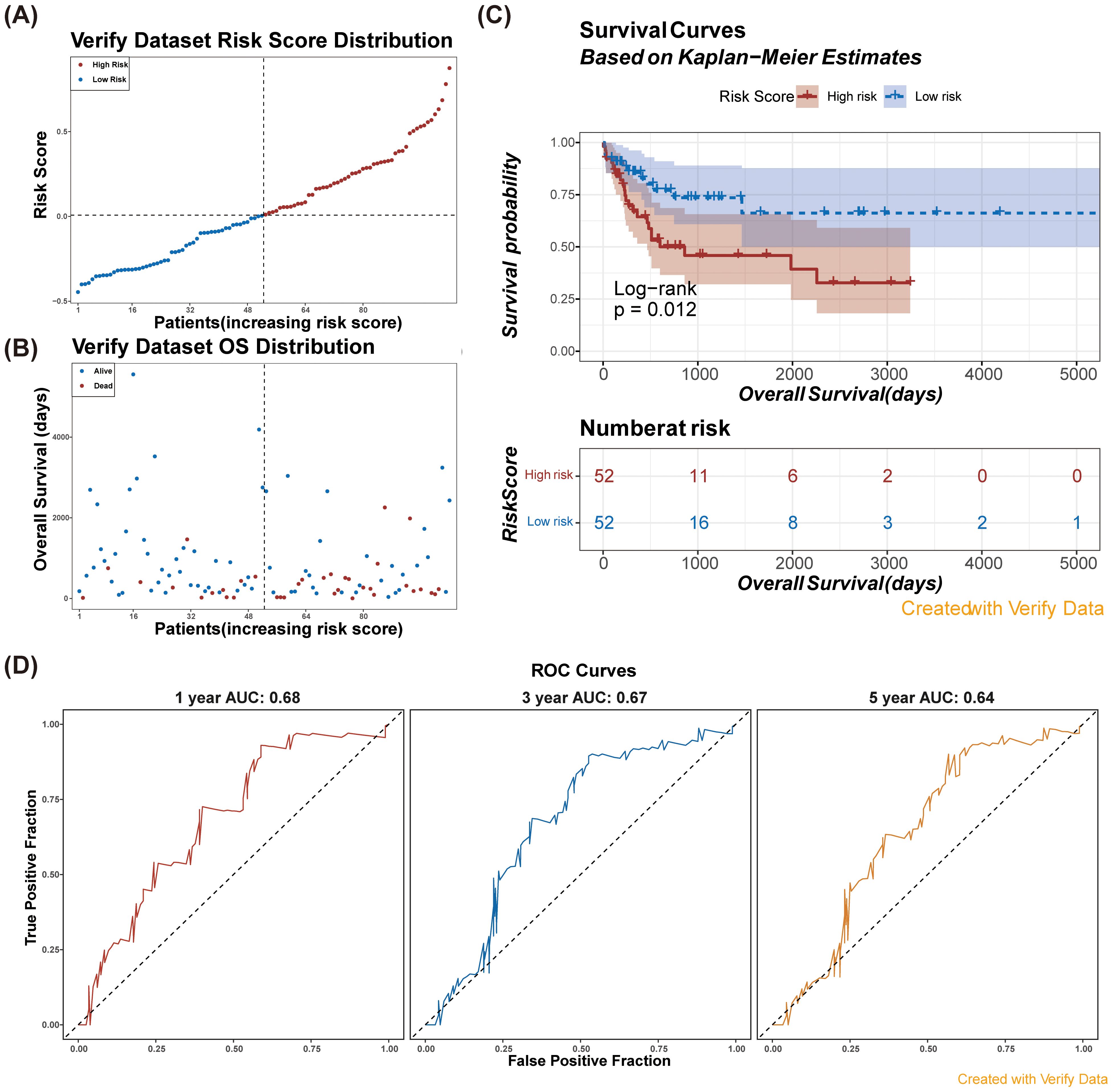
Figure 4. Verify the risk model in verification dataset (GSE71014). (A) Risk score distribution among patients in the verification dataset. (B) Overall survival distribution of patients in the verification dataset by risk score. (C) Kaplan-Meier survival curves for high and low risk patients based on risk scores in the verification dataset. Consistent with the results of the training set, the survival probability of the low-risk group was higher than that of the high-risk group.n (D) ROC curves for model performance at 1, 3, and 5 years using verification data.
3.4 Independent prognostic analysis for AML patients
Univariate Cox regression analyses indicated that risk score, cytogenetic risk, and age were significant prognostic factors (p < 0.05) (Figure 5A). The PH hypothesis test confirmed that risk score and age were appropriate for inclusion in the multivariate Cox regression model. These factors were validated as independent prognostic indicators for AML (Figure 5B). A nomogram depicting the predicted 1-, 3-, and 5-year survival rates was generated (Figure 5C), and the calibration curve demonstrated the accuracy and feasibility of the nomogram (Figure 5D).
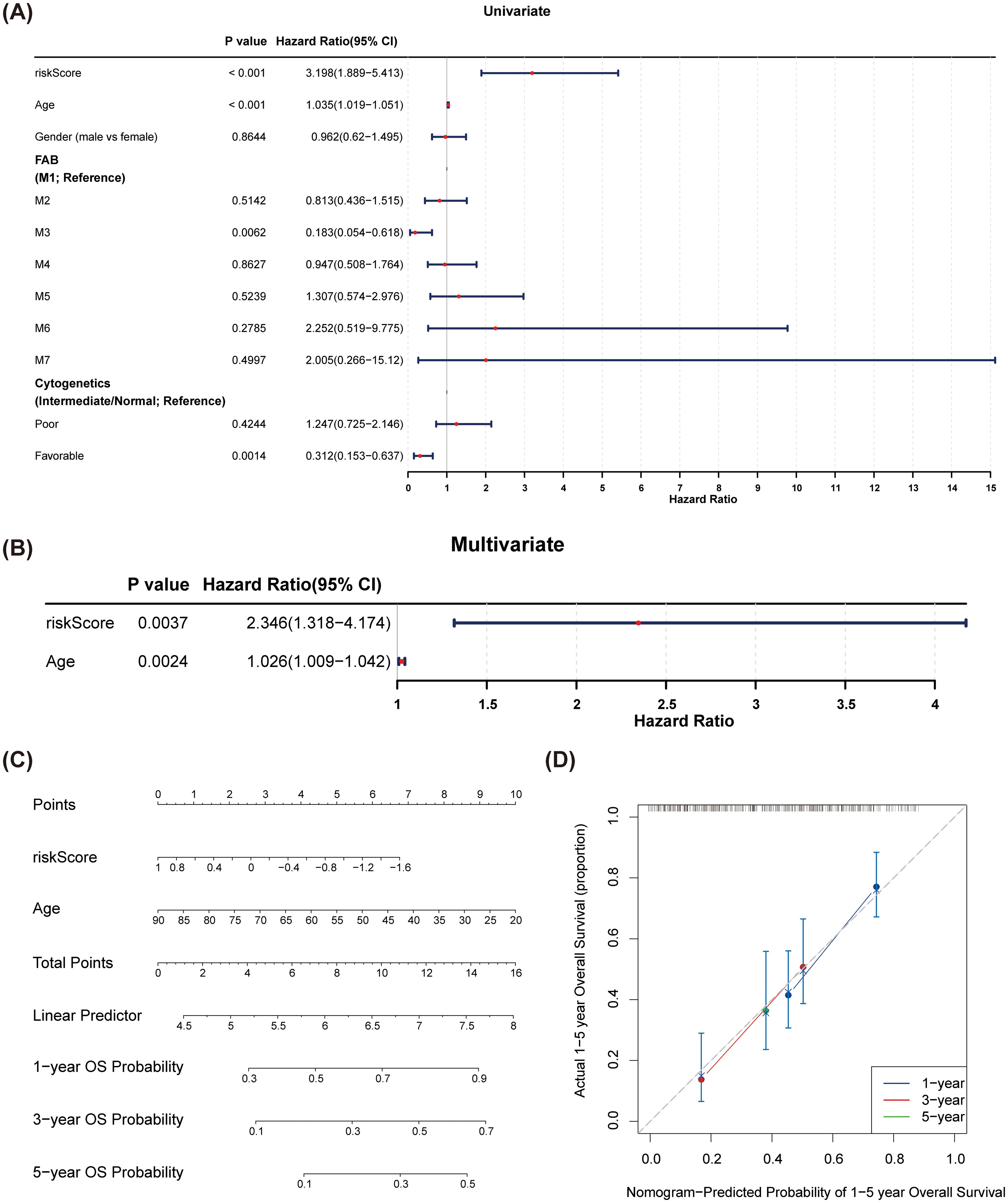
Figure 5. Construction and verification of the nomogram. (A) Forest plots of univariate Cox regression analyses for risk scores and clinical factors. Cytogenetics was divided into three categories: poor, intermediate, and favorable. The intermediate group was set as the reference group, and comparisons were made between poor vs. intermediate and favorable vs. intermediate. (B) Forest plots of multivariate Cox regression analyses for risk scores and clinical factors. Risk score and age were identified as independent prognostic factors. (C) A nomogram was constructed based on independent prognostic factors to predict the 1-, 3-, and 5-year overall survival probabilities of AML patients. (D) The calibration curves of the nomogram. The slopes of 1-, 3-, and 5-year survival were all close to 1, indicating high prediction accuracy of the nomogram.
3.5 The prognostic genes were related to chemokine relevant pathways
To explore the potential roles of the identified prognostic genes, we conducted GSEA on MPO, CCL3, and TLR8. Notably, we found that these three prognostic genes were significantly associated with the ‘chemokine signaling pathway’ (Figure 6). Furthermore, MPO (Figure 6A) and CCL3 (Figure 6B) were linked to the ‘cytokine-cytokine receptor interaction’ pathway, while TLR8 was enriched in the ‘Toll-like receptor signaling pathway’ (Figure 6C).
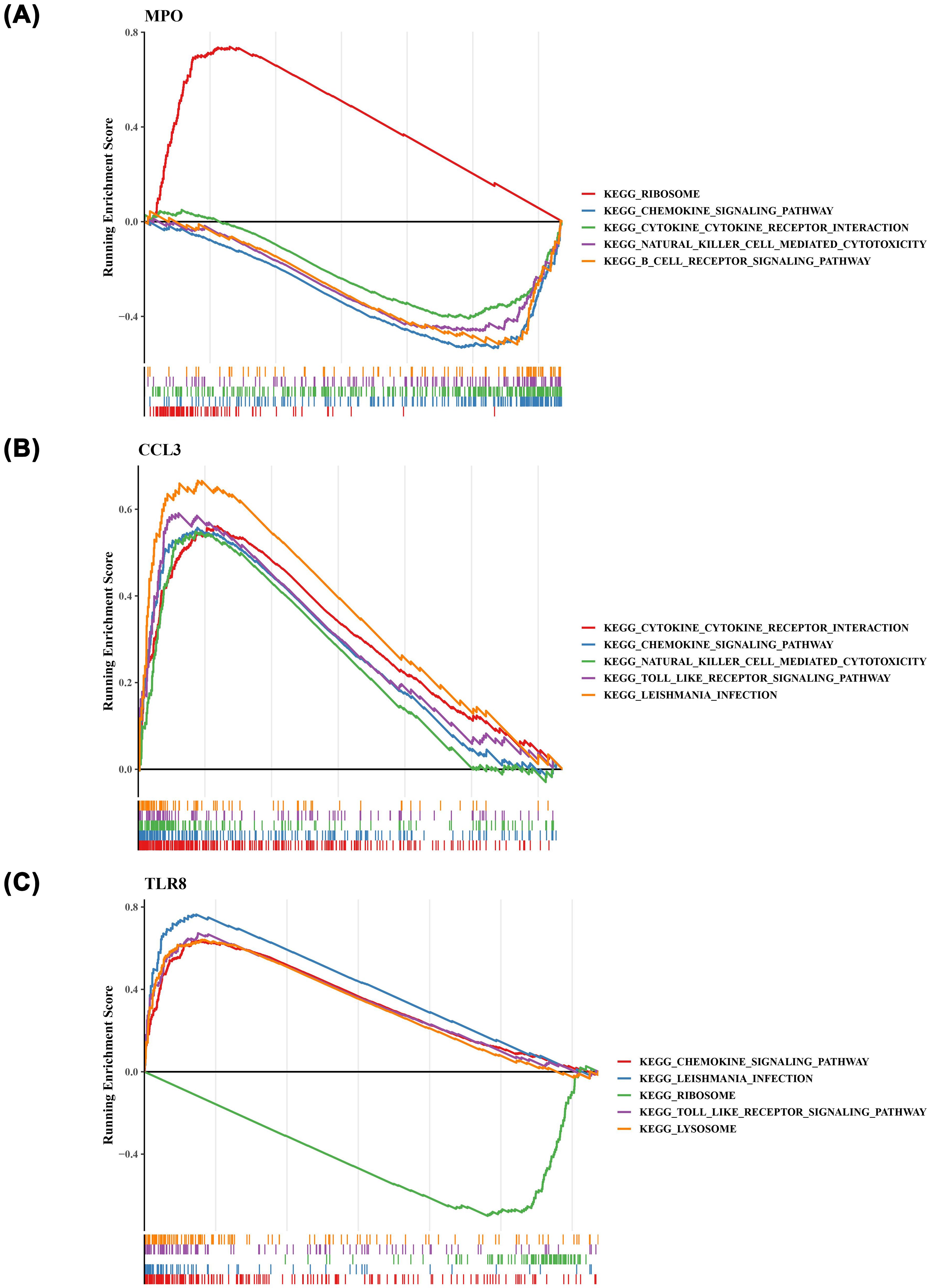
Figure 6. The gene set enrichment analysis (GSEA) result plots of the three prognostic genes. (A) MPO (B) CCL3 (C) TLR8. .
3.6 Immune infiltration analysis in risk subgroups
To investigate the immune microenvironment within AML, we analyzed the expression levels of 22 immune cell types across the 2 identified risk groups (Supplementary Figure 3). Our analysis revealed significant disparities in the abundances of five immune cell types, including resting mast cells, monocytes, follicular helper T cells, activated CD4 memory T cells, and resting CD4 memory T cells (Figure 7A). Additionally, all immune checkpoints exhibited differential expression between the high-risk and low-risk groups (Figure 7B). Importantly, we observed a strong positive correlation between the risk score and immune dysfunction, while the risk score showed a negative association with immune exclusion (Figure 7C).
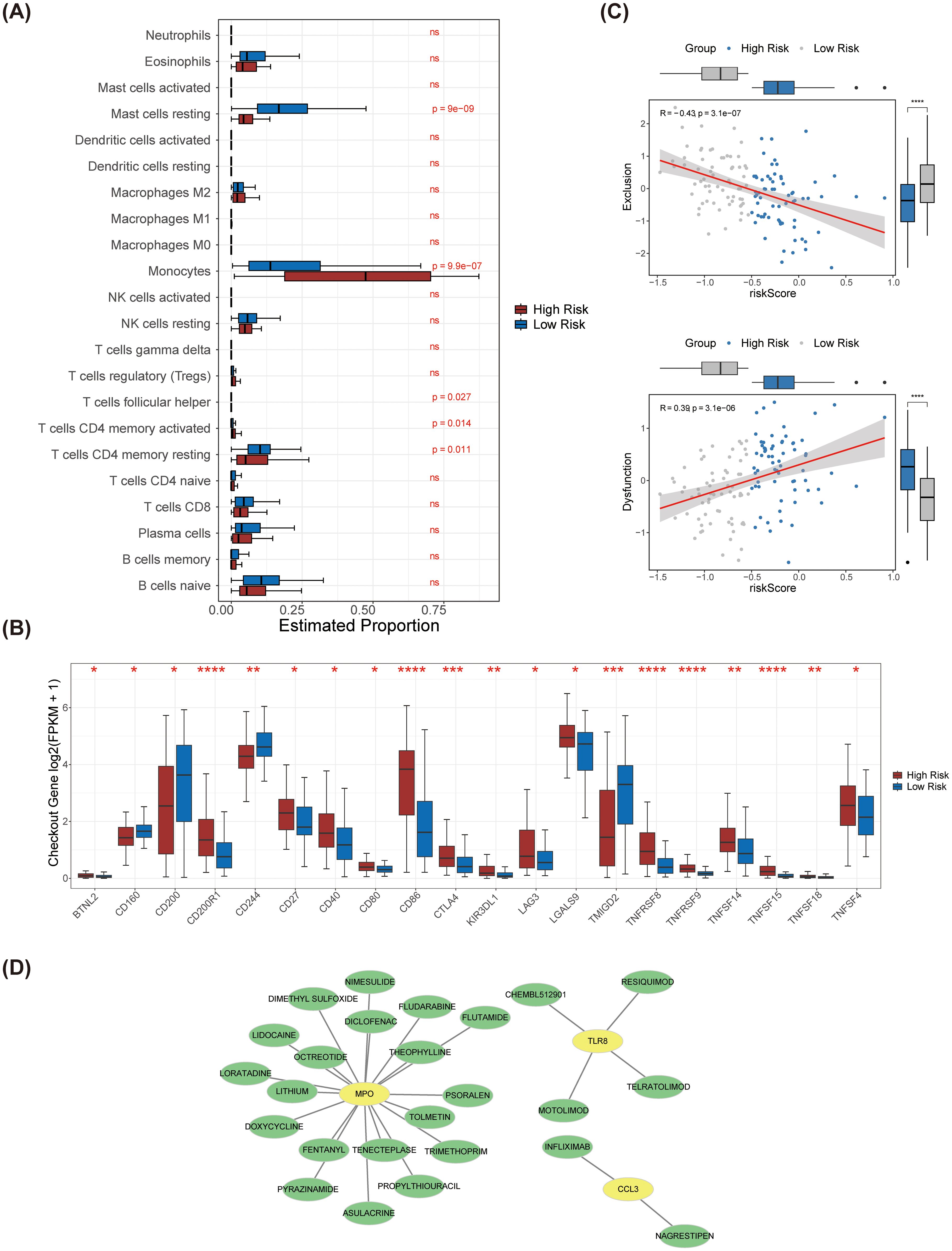
Figure 7. Immune infiltration analysis between high and low risk groups and drug prediction for prognostic genes. (A) The differences in the infiltration levels of 22 immune cells between the high and low-risk groups. (B) The differences in the expression of immune checkpoints between the high and low-risk groups. (C) Scatter plots of correlation analysis between exclusion, dysfunction and the risk score. Exclusion was significantly negatively correlated with the risk score, while dysfunction was significantly positively correlated with the risk score. (D) The prognostic gene-drug network diagram. Potential drugs were predicted for the three prognostic genes. **** represents p < 0.0001, *** p < 0.001, on behalf of ** p < 0.01, * on behalf of p < 0.05, ns on behalf of 0.05.
3.7 Prognostic genes-drug network
With the use of the database, we discovered 25 potential medications that target MPO, CCL3, and TLR8 (Figure 7D). The drugs that targeting MPO included tolmetin, asulacrine, and doxycycline etc. The drugs that targeting TLR8 included CHEMBL512901, resiquimod, telratolimod, and motolimod. The drugs that targeting CCL3 included infliximab and nagrestipen.
4 Discussion
This study presents novel insights into the prognostic gene expression patterns, biological functions, and potential clinical applications in AML through comprehensive bioinformatics analyses and extensive gene expression data. Our findings not only refine the molecular classification of AML but also reveal promising targets for future therapeutic strategies.
Utilizing consensus clustering analysis, we identified two distinct subtypes—Cluster 1 and Cluster 2—among a cohort of 151 AML patients based on the expression levels of 58,387 genes. This discovery represents a significant advancement in the molecular classification of AML, offering a novel approach to patient stratification driven by gene expression profiles. PCA further validated the clear segregation of these two clusters, corroborating existing literature on the inherent heterogeneity of AML. K-M survival analysis indicated a marked difference in survival outcomes between the two clusters, with patients in Cluster 2 demonstrating significantly poorer prognoses. These findings align with prior studies (3) linking specific AML subtypes to unfavorable outcomes. Notably, we observed significant variations in gender and age distributions between the two clusters, underscoring the potential impact of divergent biological characteristics and environmental factors on AML pathogenesis. Furthermore, Cluster 2 exhibited elevated immune, stromal, and ESTIMATE scores compared to Cluster 1, indicating a more active tumor microenvironment and a state of immune suppression. This observation echoes the work of Shaul and Fridlender (4), which highlights the role of tumor-associated neutrophils in shaping the tumor microenvironment and modulating the immune response.
In this study, we identified five significant genes through univariate regression analysis and further validated three key prognostic genes—Myeloperoxidase (MPO), Macrophage Inflammatory Protein-1α (CCL3), and Toll-Like Receptor 8 (TLR8)—using LASSO regression analysis. These genes were leveraged to construct a novel NETs-related prognostic signature for acute AML, marking a significant advancement in the molecular prognostic landscape of AML. Our prognostic model stratified AML patients into high-risk and low-risk groups, revealing significant survival disparities between these cohorts, with the high-risk group exhibiting markedly poorer outcomes. This finding is consistent with existing literature that underscores the relationship between specific gene expression patterns and AML prognosis (6). Our research not only delineates genes associated with AML prognosis but also formulates a robust risk model based on these markers. The model demonstrated commendable predictive performance, achieving AUC values of 0.68, 0.74, and 0.76 for 1-, 3-, and 5-year survival predictions, respectively. This is in line with the observations of Fu et al. (7), who classified AML through molecular subtypes that correlate with distinct prognoses. Our study enhances these findings by providing more precise prognostic insights through a dedicated gene signature. Validation of the risk model in the external dataset GSE71014 yielded results congruent with those from the training set, further affirming the robustness and reliability of our model.
MPO, a member of the myeloperoxidase protein family, is an enzyme predominantly expressed in neutrophils and is capable of generating reactive oxygen species (ROS) with potent oxidizing properties (8). Previous studies have shown that MPO and Fas/FasL expression can suppress CD4+ and CD8+ T cells, contributing to tumor progression (17).In the context of AML, MPO expression is closely associated with chemotherapy sensitivity, given its influence on ROS production within AML cells, which can consequently affect their responsiveness to chemotherapeutic agents. The findings of our study advocate for MPO as a prognostic marker in AML and provide a theoretical foundation for novel therapeutic strategies targeting ROS pathways. This aligns with previous research that emphasizes MPO’s significant role in AML prognosis. By incorporating MPO into our prognostic model, we further substantiate its critical involvement in treatment responses in AML.
CCL3, also referred to as Macrophage Inflammatory Protein-1α (MIP-1α), encodes a chemokine that directs the migration of immune cells, particularly leukocytes, to sites of inflammation or infection (9). In the realm of AML, the role of CCL3 is especially pertinent; it not only facilitates immune cell recruitment but may also influence disease progression and patient prognosis. Our study revealed that CCL3 expression in AML correlated significantly with patient risk scores and alterations in immune cell infiltration levels. This is supported by previous investigations demonstrating CCL3’s multifaceted role in various cancers, particularly regarding its regulation of the immune microenvironment in AML. Moreover, our study elucidates the direct association between CCL3 and AML prognosis, offering new insights for future therapeutic strategies aimed at this disease.
TLR8 is a pattern recognition receptor that plays a pivotal role in recognizing microbial components and activating the host’s innate immune response (10, 11). In the context of cancer, TLR8 is implicated in the activation of antitumor immune responses. As a member of the Toll-like receptors (TLRs) family, TLR8 interacts with damage-associated molecular patterns (DAMPs), modulating antigen presentation and cytokine release, thereby directly influencing T-cell activation and the clearance of tumor cells (18). Our study indicates that TLR8 may significantly influence the immune microenvironment of AML, potentially impacting disease progression and treatment strategies. This finding aligns with previous research demonstrating TLR8’s critical role in the immune response to various cancers. By highlighting the association between TLR8 and AML prognosis, our study offers a new theoretical foundation for the development of immunotherapies designed to leverage the innate immune system’s capabilities.
Univariate Cox regression analysis in this study identified risk score, cytogenetic risk, and age as significant prognostic factors in AML. These findings are consistent with the literature that emphasizes the importance of age and cytogenetic features in AML prognosis (12). For example, Nair et al. (13) noted in their review of AML treatment strategies that age is a key determinant of prognosis, often with older patients experiencing poorer outcomes. Furthermore, cytogenetic risk categories are a standard component of AML prognostic classification, closely correlated with treatment response and survival rates. Our study not only reaffirms the significance of these traditional prognostic factors but also establishes risk score and age as independent prognostic indicators in AML through PH hypothesis testing. This aligns with the findings of Kremer et al. (14, 19, 20), who reported that the regulation of the CXCR4 signaling pathway influences apoptosis in AML cells, potentially impacting prognosis. By providing a comprehensive risk score, our study refines prognostic assessments, enabling more accurate identification of high-risk patient populations. The generated nomogram forecasts 1-year, 3-year, and 5-year survival rates, while the calibration curve substantiates the nomogram’s effectiveness. The development of this prognostic tool is crucial, as it empowers clinicians and patients to better understand treatment outcome probabilities, aiding in informed decision-making. This aligns with research by Waugh and Wilson (15, 21), which discussed the role of the IL-8 pathway in cancer prognosis and treatment response. Our study extends these insights by offering a risk score-based prognostic model, providing a novel perspective for individualized treatment approaches in AML.
The GSEA of MPO, CCL3 and TLR8 revealed that these 3 prognostic genes were closely associated with the “chemokine signaling pathway, we conducted GSEA on MPO, CCL3, and TLR8, revealing that these 3 prognostic genes are closely linked to the “chemokine signaling pathway.” This discovery holds significant importance in AML research, as it elucidates potential mechanisms through which these genes may contribute to AML progression. Specifically, MPO and CCL3 are associated with “cytokine-cytokine receptor interaction,” while TLR8 is enriched in the “Toll-like receptor signaling pathway.” These results are in accordance with existing studies that emphasize the roles of chemokines and cytokines in shaping the immunological microenvironment and influencing disease progression in AML (16, 22).
In this study, we examined the immune microenvironment of AML patients by contrasting high-risk and low-risk subgroups, focusing on the expression levels of 22 immune cell types. Our analysis revealed significant differences in the abundances of five immune cell populations: resting mast cells, monocytes, follicular helper T cells, activated CD4 memory T cells, and resting CD4 memory T cells. These findings underscore the complexity of the immune landscape in AML and highlight the potential significance of various immune cell types in AML prognosis. Furthermore, we noted variations in the expression of immune checkpoints between the high-risk and low-risk groups, suggesting possible associations with immune escape mechanisms. Our study not only identifies the differential expression of immune cells but also correlates these findings with the risk scores of AML patients. This is in line with the work of Anderson et al. (23), who reported the expression of C-C chemokine receptor 1 in hematopoietic neoplasms, which may facilitate immune cell recruitment and influence tumor microenvironment regulation. Our research refines these observations by elucidating the relationship between specific immune cell subsets and AML risk scores, offering a fresh perspective for understanding the immune microenvironment in AML. Such insights can inform future treatment strategies; for instance, therapies targeting specific immune cell populations might effectively modulate the immune response, thereby enhancing the prognosis for AML patients. Our findings are corroborated by the research of Smits et al. (24), who demonstrated that the Toll-like receptor 7/8 agonist resiquimod boosts the immunostimulatory capacity of human AML cells, likely correlating with the differences in immune checkpoint expression identified in our study.
Through univariate regression analysis, five significant genes were identified, and 3 key prognostic genes were further confirmed—MPO, CCL3, and TLR8 —using LASSO regression analysis. These genes were utilized to construct a NETs-related prognostic signature for AML, an innovative finding in the molecular prognostic research of AML. This prognostic model categorized AML patients into high-risk and low-risk groups, revealing significant survival differences between the 2 with the high-risk group having poorer outcomes. This result is consistent with existing studies that emphasize the association between specific gene expression patterns and AML prognosis (6). Our study not only identified genes associated with AML prognosis but also developed a risk model based on these genes. The model demonstrated good predictive performance, with AUC values of 0.68, 0.74, and 0.76 for 1-, 3-, and 5-year survival predictions, respectively. This is in agreement with the findings of Fu et al. (7), who classified AML through molecular subtypes and found these subtypes to be associated with different prognoses. Our study further refined these findings by providing more precise prognostic information through a specific gene signature. Validation of the risk model in an external dataset, GSE71014, yielded results consistent with the training set, further confirming the robustness and reliability of our model.
The significance of this study lies in the identification of a novel gene signature associated with prognosis in AML, as well as insights into how these genes regulate the immune microenvironment and influence disease progression. This discovery provides new molecular targets for personalized treatment strategies in AML, thereby enhancing patient treatment responses and outcomes. However, our study has certain limitations, including its retrospective design and reliance on publicly available datasets, which may introduce biases that affect the generalizability of our findings. Therefore, future research should aim to incorporate prospective study designs and larger, more diverse patient cohorts to validate our prognostic models. Meanwhile, verification is carried out in a sample queue containing rich stroma. Additionally, experimental studies, including in vitro and in vivo assays, are necessary to confirm the roles of MPO, CCL3, and TLR8 in NETs and immune regulation in AML (25, 26). Clinical trials should also be considered to evaluate the practical utility of our prognostic model in guiding treatment decisions and improving patient outcomes (27, 28).
5 Conclusion
We identified 3 prognostic genes: MPO, CCL3, and TLR8. The expression of MPO is closely associated with chemotherapy sensitivity, potentially influencing the response of leukemia cells to chemotherapeutic agents by regulating the production of ROS. CCL3 expression is significantly correlated with patient risk scores and levels of immune cell infiltration, which may affect disease progression and patient prognosis by modulating immune cell infiltration in the tumor microenvironment, potentially playing a role in immune evasion and progression of AML. TLR8 may impact the tumor microenvironment by activating immune responses, thereby influencing disease progression and treatment strategies. The expression of TLR8 is closely related to the regulation of the immune microenvironment, which may provide a theoretical basis for developing new immunotherapeutic strategies. This study provides a novel prognostic signature for AML, offering insights into the molecular mechanisms underlying the disease and potential avenues for targeted therapy. Future research should focus on validating these findings in larger and more diverse cohorts, as well as exploring the functional roles of the identified genes in the progression of AML and the response to treatment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Ethics Committee of Yunnan Cancer Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
ZS: Funding acquisition, Writing – original draft, Writing – review & editing. JZ: Writing – original draft, Writing – review & editing. NL: Data curation, Writing – original draft, Writing – review & editing. LF: Data curation, Writing – original draft, Writing – review & editing. HY: Data curation, Writing – original draft, Writing – review & editing. LS: Investigation, Writing – original draft, Writing – review & editing. KL: Investigation, Writing – original draft, Writing – review & editing. HL: Investigation, Writing – original draft, Writing – review & editing. HC: Investigation, Writing – original draft, Writing – review & editing. YZ: Data curation, Writing – original draft, Writing – review & editing. YY: Methodology, Writing – original draft, Writing – review & editing. ZA: Methodology, Writing – original draft, Writing – review & editing YQ: Methodology, Writing – original draft, Writing – review & editing. SZ: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (NO. 81360089), the Applied Basic Research in Yunnan Province of China (NO. 202001AY070001-070), the High-level Talents Training support program in Yunnan Province (YNWR-MY-2020-015), Yunnan Province "ten thousand people plan" famous doctor special project (grant No. YNWR-MY-2019-050), Yunnan Provincial Health Commission - Medical Leading Talents Program (grant No. L-2019012), and the Science and Technology Department of Yunnan Province - Key project of Basic Research Program (grant No. 202301AY070001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1580750/full#supplementary-material
Supplementary Figure 1 | UMAP analysis plots of different subtypes.
Supplementary Figure 2 | The GO analysis results of NR-DEGs. (A) The GO enrichment string graph of DEGs between Cluster 1 and Cluster 2. (B) The GO enrichment string graph of DEGs between AML samples and control samples. (C) Analysis of toll-like receptor signaling pathway and neutrophil extracellular trap formation. (D) Gene expression analysis of toll-like receptor signaling pathway and neutrophil extracellular trap formation.
Supplementary Figure 3 | The proportion of 22 levels of immune cell infiltration in the samples.
References
1. Burnett AK, Wetzler M, and Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. (2011) 29:487–94. doi: 10.1200/JCO.2010.33.2928
2. Zou Y, Xie J, Zheng S, Liu W, Tang Y, and Tian W. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int J Surg. (2022) 72:106936. doi: 10.1016/j.ijsu.2022.106936
3. Nair R, Ramya N, and Baldauf HM. New strategies to treat AML: novel insights into AML survival pathways and combination therapies. Leukemia. (2021) 35:299–311. doi: 10.1038/s41375-020-01069-1
4. Bailey C, Pich O, Thol K, Watkins TBK, Luebeck J, and Rowan A. Origins and impact of extrachromosomal DNA. Nature. (2023) 613:193–200. doi: 10.1038/s41586-024-08107-3
5. Shaul ME and Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. (2019) 16:601–20. doi: 10.1038/s41571-019-0222-4
6. Lin J and Wilbur WJ. PubMed related articles: a probabilistic topic-based model for content similarity. BMC Bioinf. (2007) 8:423. doi: 10.1186/1471-2105-8-423
7. Wu J, Zhang F, Zheng X, Zhang J, Cao P, and Sun Z. Identification of renal ischemia reperfusion injury subtypes. Front Immunol. (2022) 13:1047367. doi: 10.3389/fimmu.2022.1047367
8. Wu T, Hu E, Xu S, Chen M, Guo P, and Dai Z. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). (2021) 2:100141. doi: 10.1016/j.xinn.2021.100141
9. Landry C, Peterson SD, and Arami A. A fusion approach to improve accuracy and estimate prediction interval for cuffless blood pressure monitoring. Sci Rep. (2022) 12:7948. doi: 10.1038/s41598-022-12087-7
10. Love MI, Huber W, and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
11. Yu G, Wang LG, Han Y, and He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. (2012) 16:284–7. doi: 10.1089/omi.2011.0118
12. Lu W, Li Y, Dai Y, and Chen K. Dominant myocardial fibrosis and complex immune microenvironment jointly shape the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Front Cardiovasc Med. (2022) 9:900810. doi: 10.3389/fcvm.2022.900810
13. Xu S, Tang L, and Liu Z. Hypoxia-related lncRNA correlates with prognosis and immune microenvironment in lower-grade glioma. Front Immunol. (2021) 12:731048. doi: 10.3389/fimmu.2021.731048
14. Fu D, Zhang B, Wu S, Feng J, and Jiang H. Molecular subtyping of acute myeloid leukemia through ferroptosis-derived signature. Front Cell Dev Biol. (2023) 11:1207642. doi: 10.3389/fcell.2023.1207642
15. Yamazaki Y, Ando K, Horio K, Horio K, Tsutsumi C, and Imanishi D. Expression of myeloperoxidase enhances the chemosensitivity of leukemia cells through the generation of reactive oxygen species and the nitration of protein. Leukemia. (2008) 22:1028–36. doi: 10.1038/leu.2008.8
16. Yazdani Z, Mousavi Z, Ghasemimehr N, Kalantary Khandany B, Nikbakht R, and Jafari E. Differential regulatory effects of chemotherapeutic protocol on CCL3, CCL4, CCL5, CCR5 axes in acute myeloid leukemia patients with monocytic lineage. Life Sci. (2020) 240:117071. doi: 10.1016/j.lfs.2019.117071
17. Tahimic CGT, Wang Y, and Bikle DD. Anabolic effects of IGF-1 signaling on the skeleton. Front Endocrinol. (2013) 4:6IF. doi: 10.3389/fendo.2013.00006IF
18. Sorenson PW and Caprio JC. Chemoreception. In: Evans DH, editor. The physiology of fishes. CRC Press, Boca Raton, FL (1998). p. 375–405.
19. Kremer KN, Peterson KL, and Schneider PA. CXCR4 chemokine receptor signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak. J Biol Chem. (2013) 288:22899–914. doi: 10.1074/jbc.M113.449926
20. Hu X, Mei S, Meng W, Xue S, Jiang L, and Yang Y. CXCR4-mediated signaling regulates autophagy and influences acute myeloid leukemia cell survival and drug resistance. Cancer Lett. (2018) 425:1–12. doi: 10.1016/j.canlet.2018.03.024
21. Waugh DJ and Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. (2008) 14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843
22. Wang Y, Gao A, Zhao H, Lu P, Cheng H, and Dong F. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1α/CCL3. Leukemia. (2016) 30:1897–908. doi: 10.1038/leu.2016.81
23. Khorramdelazad H, Mortazavi Y, Momeni M, Arababadi MK, Khandany BK, and Moogooei M. Lack of correlation between the CCR5-Δ32 mutation and acute myeloid leukemia in Iranian patients. Indian J Hematol Blood Transfus. (2015) 31:29–31. doi: 10.1007/s12288-014-0408-y
24. Anderson MW, Zhao S, and Ai WZ. C-C chemokine receptor 1 expression in human hematolymphoid neoplasia. Am J Clin Pathol. (2010) 133:473–83. doi: 10.1309/AJCP1TA3FLOQTMHF
25. van den Ancker W, van Luijn MM, Ruben JM, Westers TM, Bontkes HJ, and Ossenkoppele GJ. Targeting Toll-like receptor 7/8 enhances uptake of apoptotic leukemic cells by monocyte-derived dendritic cells but interferes with subsequent cytokine-induced maturation. Cancer Immunol Immunother. (2011) 60:37–47. doi: 10.1007/s00262-010-0917-y
26. Smits EL, Cools N, Lion E, Van Camp K, Ponsaerts P, and Berneman ZN. The Toll-like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol Immunother. (2010) 59:35–46. doi: 10.1007/s00262-009-0721-8
27. Goenka A, Khan F, Verma B, Sinha P, Dmello CC, and Jogalekar MP. Tumor microenvironment signaling and therapeutics in cancer progression. Cancer Commun (Lond). (2023) 43:525–61. doi: 10.1002/cac2.12416
Keywords: acute myeloid leukemia, neutrophil extracellular trap, prognostic signature, risk subgroups, tumor microenvironment
Citation: Shen Z, Zhu J, Luo N, Feng L, Yue H, Song L, Liu K, Li H, Cao H, Zhou Y, Yousafzai YM, Asad Z, Qiu Y and Zhang S (2025) Establishment of prognostic signature based on neutrophil extracellular traps-related genes in acute myeloid leukemia: a bioinformatics analysis. Front. Immunol. 16:1580750. doi: 10.3389/fimmu.2025.1580750
Received: 21 February 2025; Accepted: 13 August 2025;
Published: 24 October 2025.
Edited by:
Shenghui Zhang, First Affiliated Hospital of Wenzhou Medical University, ChinaReviewed by:
Aditya Yashwant Sarode, Columbia University, United StatesRashid Mehmood, St. Jude Children’s Research Hospital, United States
Copyright © 2025 Shen, Zhu, Luo, Feng, Yue, Song, Liu, Li, Cao, Zhou, Yousafzai, Asad, Qiu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiwen Zhang, enN3NDMwMTk3QHNpbmEuY29t
†These authors have contributed equally to this work