 Qingchen Li1*
Qingchen Li1* Xinyun Chen2
Xinyun Chen2- 1Department of Neurology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Neurology, the Third People’s Hospital of Zhengzhou, Zhengzhou, China
Background: Glial fibrillary acidic protein–immunoglobulin G (GFAP-IgG) can coexist with aquaporin-4–IgG (AQP4-IgG) or myelin oligodendrocyte glycoprotein–IgG (MOG-IgG). We aimed to investigate the clinical characteristics of patients with GFAP-IgG coexisting with AQP4-IgG or MOG-IgG.
Methods: We retrospectively collected data from 81 GFAP-IgG-positive patients and described and compared the clinical characteristics of those with GFAP-IgG coexisting with AQP4-IgG or MOG-IgG.
Results: (1) Among the 81 GFAP-IgG-positive patients, nine (11.1%) were positive for AQP4-IgG and seven (8.6%) were positive for MOG-IgG. The clinical manifestations of overlapping syndromes were diverse; all patients met the clinical phenotype of autoimmune GFAP astrocytopathy (A-GFAP-A) and also fulfilled the diagnostic criteria for neuromyelitis optica spectrum disorders or MOG antibody-associated disorders. Compared with the GFAP-AQP4 overlapping syndrome, the GFAP-MOG overlapping syndrome had a higher frequency of seizures (57.1% vs. 0, p = 0.019). (2) Compared with the nonoverlapping syndrome group, the overlapping syndrome group had more women (68.6% vs. 32.3%, p = 0.008), a higher incidence of optic neuritis (ON) (43.8% vs. 4.6%, p < 0.001), lower CSF white blood cell counts (median: 30 cells/mm3 vs. 94 cells/mm3, p = 0.001) and protein levels (median: 0.375 g/L vs. 0.78 g/L, p < 0.001), and a higher proportion of patients receiving long-term immunotherapy (68.8% vs.13.8%, p < 0.001).
Conclusions: Among patients with A-GFAP-A, 20% had concurrent AQP4-IgG or MOG-IgG, exhibiting distinct clinical features that suggest a different disease phenotype driven by overlapping autoimmune mechanisms.
1 Introduction
Autoimmune glial fibrillary acidic protein astrocytopathy (A-GFAP-A) is an autoimmune inflammatory disorder of the central nervous system (CNS) that affects the brain, meninges, spinal cord, and optic nerve. The primary clinical manifestations include corticosteroid-responsive encephalitis or meningoencephalitis, with or without myelitis. An immunoglobulin G (IgG) autoantibody that selectively targets GFAP in astrocytes has been identified as a highly specific diagnostic biomarker for this disease (1, 2). Neuromyelitis optica spectrum disorders (NMOSD) and myelin oligodendrocyte glycoprotein (MOG) antibody-associated disorders (MOGAD) are two immune-mediated inflammatory demyelinating diseases of the CNS. Aquaporin-4 (AQP4) antibody and MOG antibody serve as specific biomarkers for the diagnosis of these two diseases, respectively (3, 4).
The clinical manifestations of A-GFAP-A, NMOSD, and MOGAD exhibit significant overlap, and in some cases, GFAP-IgG may co-occur with AQP4-IgG or MOG-IgG (5–11). However, most previous studies were case reports, and no study has systematically compared the frequency, clinical features, and auxiliary examinations of these overlapping syndromes. Here, we analyzed the clinical characteristics of patients with GFAP-IgG coexisting with either AQP4-IgG or MOG-IgG, aiming to enhance our understanding of this phenomenon.
2 Materials and methods
2.1 Patients
The study was approved by the ethics committee of the First Affiliated Hospital of Zhengzhou University (2024-KY-2298).
In this study, we identified 81 GFAP-IgG-positive patients who were admitted to the First Affiliated Hospital of Zhengzhou University, China, from February 2020 to October 2024. The inclusion criteria included the following: (1) CSF and/or serum testing positive for GFAP-IgG by cell-based assays (CBA); (2) presentation with one or more clinical syndromes, including encephalitis, meningitis, myelitis, and optic neuritis (ON); (3) testing for AQP4-IgG and MOG-IgG by CBA; and (4) availability of clinical data. The exclusion criteria included other diseases, such as brain tumors, traumatic brain injury, Alzheimer’s disease, and toxic or metabolic CNS disorders.
2.2 Study design
We retrospectively analyzed the clinical data of these patients. Information was collected through medical records, telephone interviews, and outpatient follow-ups. The modified Rankin Scale (mRS) was used to assess disease severity and clinical outcomes at the last follow-up; an mRS score < 3 was considered a good outcome. Follow-up duration was defined as the time from the initial evaluation at disease onset to the last follow-up. Relapses were defined as the development of new neurological symptoms lasting at least 24 h, occurring 1 month after clinical improvement or stabilization.
All CSF and serum samples were collected during the early active disease stage. GFAP antibodies were detected using an indirect immunofluorescence CBA method, employing human embryonic kidney (HEK) 293 cells transfected with GFAP expression plasmids (Shanghai Genechem Co.,Ltd, Shanghai, China). Antibodies to AQP4 and MOG were detected using the fixed CBA. Among the 81 GFAP-IgG-positive patients, nine were positive for AQP4-IgG and seven were positive for MOG-IgG. We characterized the clinical features of these overlapping syndromes and compared them with those of patients without coexisting AQP4-IgG or MOG-IgG.
2.3 Statistical analysis
Statistical analyses were performed using SPSS version 26.0. Continuous variables were expressed as mean ± standard deviation (SD) or median (range), and categorical variables as counts and proportions. Continuous variables were compared using the Mann–Whitney U test or t-test, and categorical variables using the Chi-square test or Fisher’s exact test. A p-value < 0.05 was considered statistically significant.
3 Results
3.1 Case series study
3.1.1 Demographics and clinical manifestations
Among the 81 GFAP-IgG-positive patients, nine (11.1%) were positive for AQP4-IgG (patients 1–9; one man, eight women), and seven (8.6%) were positive for MOG-IgG (patients 10–16; four men, three women). The overlapping syndromes were defined as follows: the coexistence of GFAP-IgG and AQP4-IgG was categorized as the GFAP-AQP4 group, while the coexistence of GFAP-IgG and MOG-IgG was categorized as the GFAP-MOG group. The clinical data of patients with these two overlapping syndromes are summarized in Tables 1, 2.
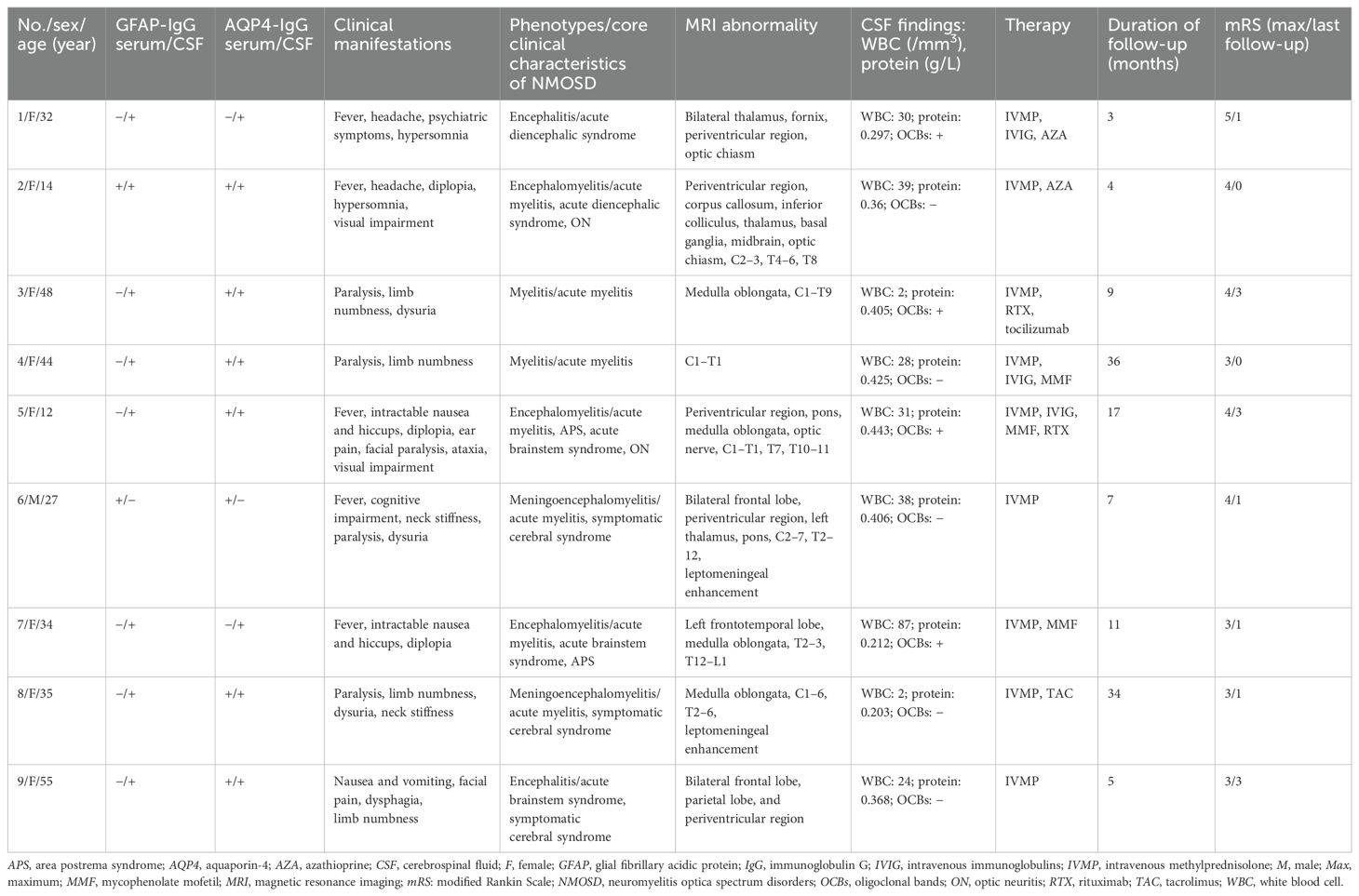
Table 1. Clinical data of patients with the GFAP-AQP4 overlapping syndrome.
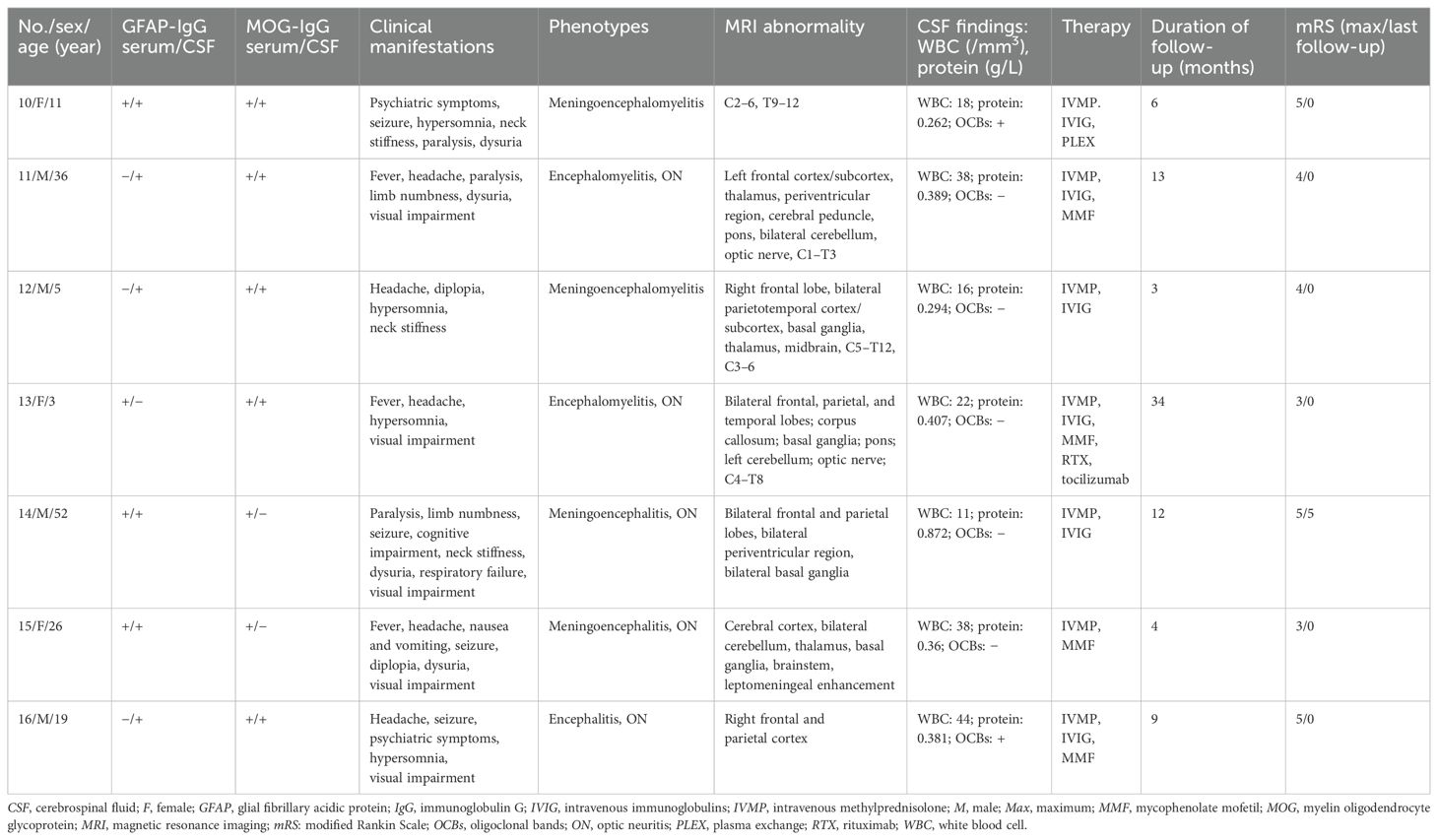
Table 2. Clinical data of GFAP-MOG overlapping syndrome.
The median age at onset was 34 years (range: 12–55 years) in the GFAP-AQP4 group and 19 years (range: 3–52 years) in the GFAP-MOG group. Patient 5 developed area postrema syndrome (APS) 40 days earlier, and patient 11 developed myelitis and ON 16 months earlier. However, antibodies against GFAP, AQP4, and MOG were not detected at that time. Upon experiencing new neurological events, both patients underwent antibody testing and were subsequently diagnosed. Tumor screening was performed in all patients, and elevated tumor markers were detected in three cases (CA72–4 in patients 5 and 16; CA19–9 in patient 15). However, no concurrent tumors were identified in the overlapping patients.
The clinical phenotypes in the GFAP-AQP4 group included encephalomyelitis (n = 3), meningoencephalomyelitis (n = 2), encephalitis (n = 2), and myelitis (n = 2), with two patients also presenting with ON. These syndromes fulfilled the core clinical characteristics of NMOSD, including acute myelitis (n = 7), acute brainstem syndrome (n = 3), symptomatic cerebral syndrome (n = 3), APS (n = 2), ON (n = 2), and acute diencephalic syndrome (n = 2). The clinical phenotypes in the GFAP-MOG group included meningoencephalomyelitis (n = 2), encephalomyelitis (n = 2), meningoencephalitis (n = 2), and encephalitis (n = 1); notably, five patients with ON. Compared with the patients in the GFAP-AQP4 group, those in the GFAP-MOG group had more seizures (57.1% vs. 0%, p = 0.019) (Table 3).
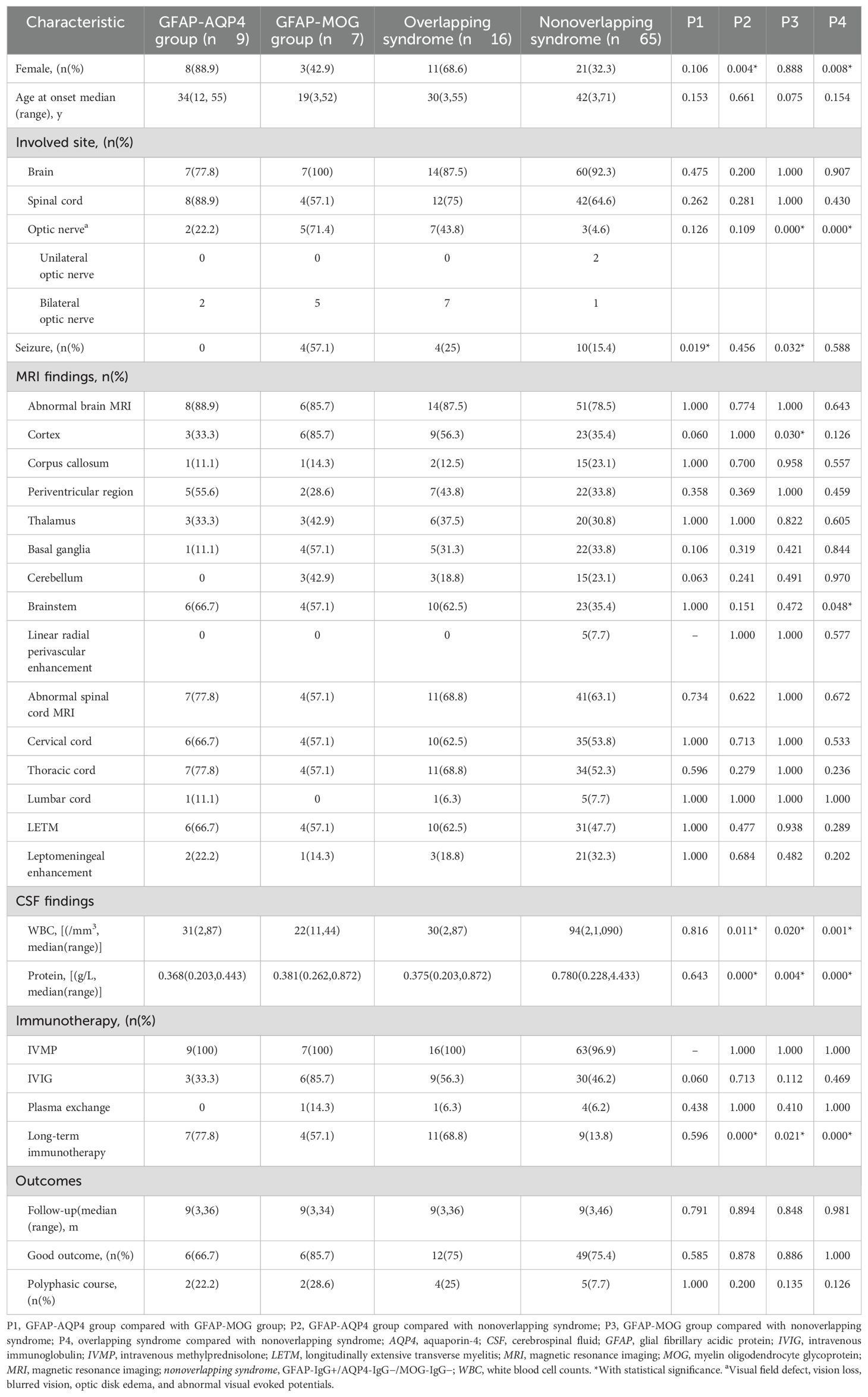
Table 3. Comparison of clinical characteristics among all groups.
3.1.2 MRI and CSF findings
Brain and spinal cord MRIs were performed in all patients. Brain lesions were observed in eight patients in the GFAP-AQP4 group and six patients in the GFAP-MOG group. In the GFAP-AQP4 group, common lesions were observed in the brainstem (n = 6), periventricular region (n = 5), cortex (n = 3), and thalamus (n = 3). In contrast, the GFAP-MOG group exhibited common lesions in the cortex (n = 6), basal ganglia (n = 4), brainstem (n = 4), thalamus (n = 3), and cerebellum (n = 3). Leptomeningeal enhancement was observed in two patients (patient 8 in the brainstem meninges and patient 15 in the meninges of the cerebellar and cerebral hemispheres). However, no patients exhibited periventricular radial linear enhancement. Spinal MRI revealed that six patients in the GFAP-AQP4 group and four patients in the GFAP-MOG group had longitudinally extensive spinal cord lesions, primarily located in the cervicothoracic cord. Patient 7 had spinal lesions involving no more than three vertebral segments, while patient 4 had spinal meningeal enhancement. MRI characteristics are summarized in Tables 1, 2 and Figures 1, 2.
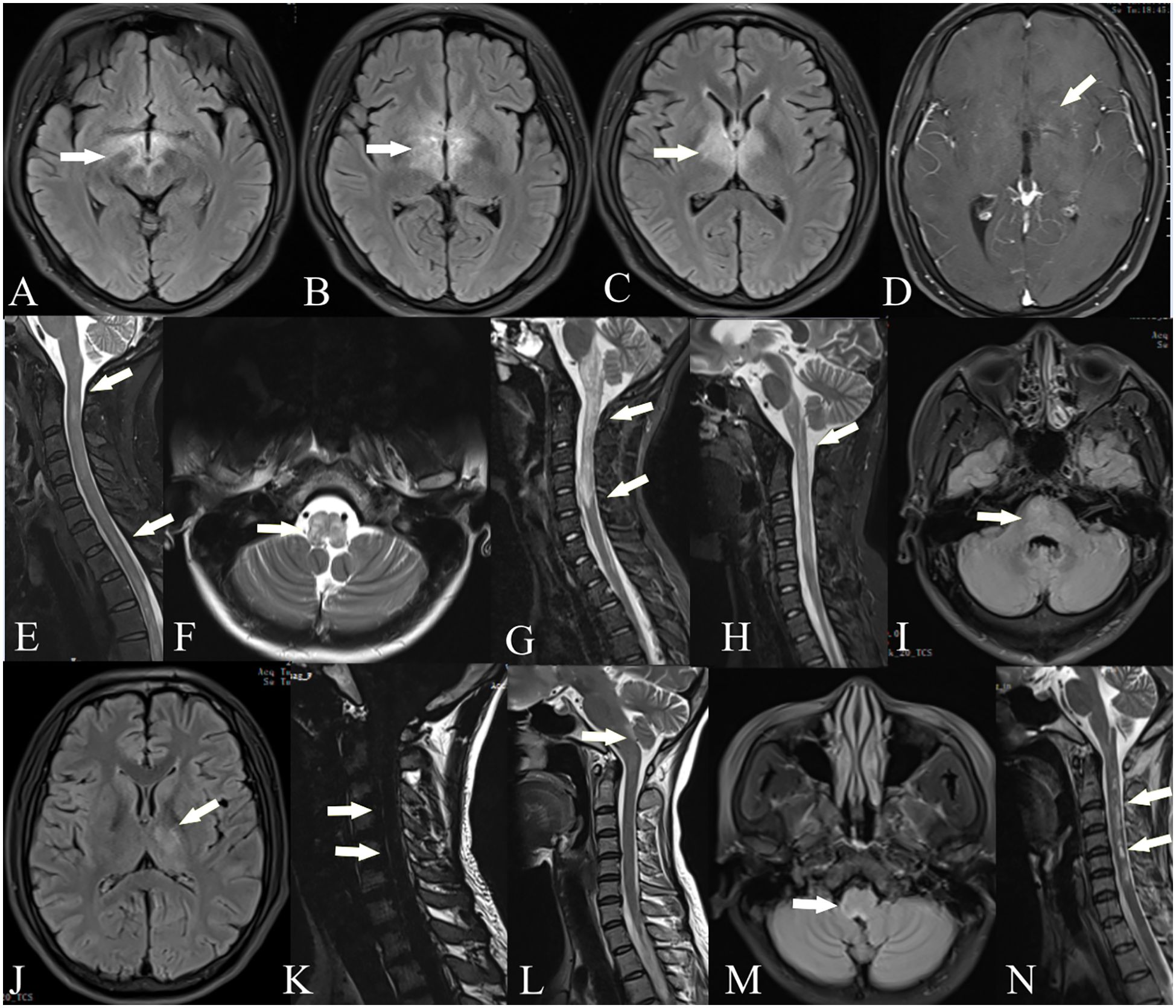
Figure 1. Brain and spinal cord MRI of GFAP-AQP4 overlapping syndrome. Patient 1: T2-FLAIR imaging showed lesions in the thalamus, fornix, optic chiasm, and around the third ventricle (A–C). Patient 2: Contrast-enhanced T1-weighted imaging showed spotty enhancement in the corpus callosum and around the third ventricle (D). Patient 3: T2-weighted imaging showed lesions in the spinal cord (E). Patient 5: T2-weighted imaging showed lesions in the medulla oblongata (F) and spinal cord (G); after treatment, the lesions were significantly reduced (H). Patient 6: T2-FLAIR imaging showed lesions in the paraventricular region, thalamus, and pons (I, J), with leptomeningeal enhancement in the cervical cord (K). Patient 7: T2-FLAIR imaging showed lesions in the area postrema (L, M). Patient 8: T2-weighted imaging showed lesions in the spinal cord (N).
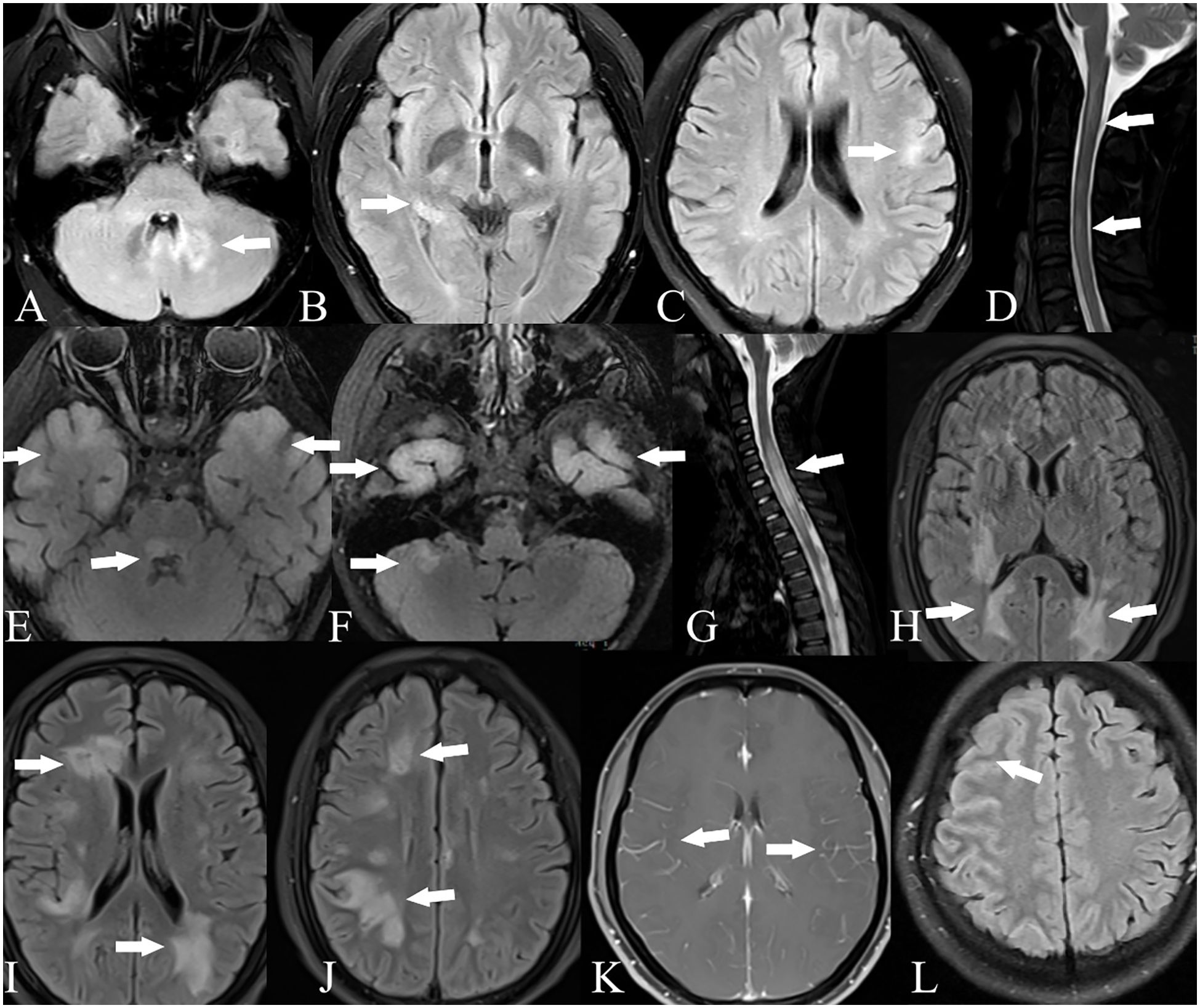
Figure 2. Brain and spinal cord MRI of GFAP-MOG overlapping syndrome. Patient 11: T2-FLAIR imaging showed lesions in the cerebellum (A), thalamus (B), and cortex (C); T2-weighted imaging showed lesions in the spinal cord (D). Patient 13: T2-FLAIR imaging showed lesions in the bilateral cortex, pons, and cerebellum (E, F); T2-weighted imaging showed lesions in the spinal cord (G). Patient 14: T2-FLAIR imaging showed lesions in the bilateral cortex, periventricular region, and basal ganglia (H–J). Patient 15: Contrast-enhanced T1-weighted imaging showed leptomeningeal enhancement (K). Patient 16: T2-FLAIR imaging showed lesions in the right frontal and parietal cortex (L).
CSF analysis revealed pleocytosis (> 5 cells/mm3) in seven patients from both the GFAP-AQP4 and GFAP-MOG groups. Increased protein levels (> 0.45 g/L) were found in only one patient (patient 14). CSF oligoclonal bands were positive in four patients in the GFAP-AQP4 group and two patients in the GFAP-MOG group. Next-generation sequencing (NGS) detected herpes simplex virus in patient 15. In addition, one patient (patient 1) in the GFAP-AQP4 group tested positive for anti-N-methyl-d-aspartate receptor (NMDAR) antibody, antiglutamic acid decarboxylase 65 (GAD65) antibody, and antiglycine receptor (GlyR) antibody in the CSF. Two patients (patients 10 and 16) in the GFAP-MOG group tested positive for anti-NMDAR antibody in both CSF and serum. No differences in MRI lesions or CSF findings were observed between the two overlapping syndromes (Table 3).
3.1.3 Treatment and outcome
All patients received intravenous methylprednisolone (IVMP; 0.5–1 g/day for 5 days in 14 patients, 0.3 g/day in patient 12, and 0.2 g/day in patient 13) during the acute stage, followed by a tapering course of oral methylprednisolone (decreased by 4 mg/day every 1–2 weeks). Nine patients received intravenous immunoglobulin (IVIG; 0.4 g/kg/day for 5 days), while one patient underwent plasma exchange. Additionally, seven patients in the GFAP-AQP4 group and three patients in the GFAP-MOG group received long-term immunotherapy. Most patients showed good outcomes after treatment.
During follow-up, three patients (patients 1, 4, and 8) in the GFAP-AQP4 group became negative for both GFAP-IgG and AQP4-IgG, with clinical improvement. Patient 3 experienced a recurrence of myelitis 7 months later, accompanied by elevated AQP4 antibody titers. In the GFAP-MOG group, three patients (patients 10, 13, and 15) became negative for both GFAP-IgG and MOG-IgG. Patient 13 experienced a recurrence of myelitis and ON 5 months later, with MOG-IgG reverting to positive. No differences in treatment and outcome were observed between the two overlapping syndromes (Table 3).
3.2 Comparison of clinical features among different groups
Of the 81 GFAP-IgG-positive patients, 16 patients with AQP4-IgG or MOG-IgG were classified into the overlapping syndrome group, while 65 patients without AQP4-IgG or MOG-IgG were classified into the nonoverlapping syndrome group.
3.2.1 Overlapping syndrome and nonoverlapping syndrome groups
Compared with the nonoverlapping syndrome group, the overlapping syndrome group had a higher proportion of women (68.6% vs. 32.3%, p = 0.008), more cases of ON (43.8% vs. 4.6%, p < 0.001), lower CSF white blood cell counts (median: 30 cells/mm3 vs. 94 cells/mm3, p = 0.001) and protein levels (median: 0.375 g/L vs. 0.78 g/L, p < 0.001). Furthermore, a greater proportion of patients in the overlapping syndrome group received long-term immunotherapy (68.8% vs. 13.8%, p < 0.001) (Table 3).
3.2.2 GFAP-AQP4 and nonoverlapping syndrome groups
Compared with the nonoverlapping syndrome group, the GFAP-AQP4 group had a higher proportion of women (88.9% vs. 32.3%, p = 0.004), lower CSF white blood cell counts (median: 31 cells/mm3 vs. 94 cells/mm3, p = 0.011) and protein levels (median: 0.368 g/L vs. 0.78 g/L, p < 0.001), and a greater proportion of patients receiving long-term immunotherapy (77.8% vs. 13.8%, p < 0.001) (Table 3).
3.2.3 GFAP-MOG and nonoverlapping syndrome groups
Compared with the nonoverlapping syndrome group, the GFAP-MOG group exhibited a higher frequency of ON (71.4% vs. 4.6%, p < 0.001) and seizures (57.1% vs. 15.4%, p = 0.032), more cortical lesions (85.7% vs. 35.4%, p = 0.003), lower CSF white blood cell counts (median: 22 cells/mm3 vs. 94 cells/mm3, p = 0.020) and protein levels (median: 0.381 g/L vs. 0.78 g/L, p = 0.004), and a greater proportion of patients receiving long-term immunotherapy (57.1% vs. 13.8%, p = 0.021) (Table 3).
4 Discussion
In recent years, with the expansion of the spectrum of antineuronal antibodies and advances in detection techniques, an increasing number of CNS cases with overlapping antibodies have been reported. Some patients may test positive for multiple neuronal antibodies or present with overlapping clinical phenotypes. Previous studies have shown that GFAP-IgG can coexist with other neuronal antibodies, most commonly NMDAR-IgG, AQP4-IgG, and MOG-IgG. However, the prevalence of coexisting antibodies varies considerably across different studies (1, 6, 11–13). In a report from the Mayo Clinic, Flanagan et al. found that among 102 A-GFAP-A patients, 41 (40%) had one or more coexisting antibodies. The most common was NMDAR-IgG, followed by AQP4-IgG (1). Yang et al. analyzed 30 GFAP-IgG-positive patients, of whom 10 (33.3%) were diagnosed with overlapping syndromes. AQP4-IgG was the most common coexisting antibody (16.6%), followed by NMDAR-IgG (6). However, some scholars have suggested that MOG-IgG is the most frequently coexisting antibody. A Chinese study of 35 children with A-GFAP-A showed that 11 tested positive for other neuronal antibodies, including MOG-IgG in five patients (14.3%) (11). Fang et al. also reported that MOG antibody coexistence was the most common type of A-GFAP-A overlapping syndrome, with an occurrence rate of 10.4% (12).
In our study, the clinical characteristics of these two overlapping syndromes can be summarized as follows: (1) In our cohort, the prevalence of GFAP-IgG coexisting with AQP4-IgG and MOG-IgG was comparable. (2) Symptoms of the overlapping syndromes were diverse, exhibiting features consistent with both A-GFAP-A and the diagnostic criteria for NMOSD or MOGAD. (3) Patients with GFAP-IgG coexisting with AQP4-IgG or MOG-IgG were more likely to be women, exhibited a higher prevalence of ON, and had milder CSF inflammatory changes. (4) Antibody detection is crucial for disease diagnosis; however, antibody levels may fluctuate during disease progression and in response to treatment.
Our study found a higher proportion of women in the GFAP-AQP4 group, consistent with the predominance of women observed in NMOSD. This may be due to the greater likelihood of AQP4 antibodies affecting women. Yang et al. reported that patients with overlapping syndromes tended to be younger (6); however, this age-related difference was not observed in our study. Although the clinical manifestations of overlapping syndromes are diverse and difficult to distinguish from those of nonoverlapping syndromes, several distinguishing features can still be identified. In our study, patients with overlapping syndromes exhibited a significantly higher prevalence of ON. Particularly in the GFAP-MOG group, ON was the most common phenotype. Patients with overlapping syndromes may present with bilateral ON or recurrent ON, with relapses predominantly occurring during steroid tapering. In addition, MRI revealed T2-hyperintensities in the optic nerves or optic chiasm, consistent with ON features observed in demyelinating diseases (3, 4, 9). In contrast, visual impairment in A-GFAP-A is heterogeneous, and optic disk edema is more likely attributable to venous inflammation rather than ON (12, 14, 15). NMOSD usually affects the optic nerves and spinal cord, with less involvement of the brain (16). However, in our cohort, 77.8% of patients in the GFAP-AQP4 group exhibited symptoms of encephalopathy. This discrepancy may be attributed to the coexistence of GFAP-IgG. APS is a characteristic manifestation in AQP4-IgG-positive NMOSD, with a reported prevalence of 16% to 43% (3). It can also occur in A-GFAP-A, albeit at a lower frequency (4%–11%) (17, 18). In our GFAP-AQP4 group, two patients experienced APS, similar to previous studies (19). Patients with MOGAD were more likely to experience seizures than those with NMOSD or A-GFAP-A (5, 20). Similarly, we observed a higher incidence of seizures in the GFAP-MOG group, with MRI revealing more cortical involvement. Therefore, seizures may serve as a potential indicator of MOG antibody positivity. These findings suggest that overlapping phenotypes may be driven by overlapping autoimmune mechanisms.
Intracranial lesions in A-GFAP-A predominantly involve the cerebral white matter, basal ganglia, hypothalamus, cerebellum, and brainstem. Periventricular radial linear enhancement and leptomeningeal enhancement are characteristic imaging features of the disease (1, 2, 5, 9). In our cohort, three patients in the overlapping syndrome group exhibited leptomeningeal enhancement, and five patients presented with clinical signs of meningeal irritation. These findings suggest a possible role for GFAP-IgG in these conditions. Moreover, periventricular lesions commonly seen in NMOSD and large space-occupying lesions typically associated with MOGAD can also be present in patients with overlapping syndromes. Patients with NMOSD and MOGAD often present with mild CSF pleocytosis and elevated protein levels, whereas those with A-GFAP-A exhibit a more severe inflammatory response (5, 16). In our study, the CSF of patients with overlapping syndromes exhibited mild inflammatory changes, closely resembling those observed in demyelinating diseases. These findings may reflect the pathogenic effects of AQP4-IgG or MOG-IgG.
Infection is a common immune trigger of antibody-mediated autoimmune disorders (21, 22). These pathogens may directly damage neuronal and glial cells, leading to antibody production and secondary autoimmune responses. In addition, certain components of these pathogens may share structural similarities with host antigens, increasing the risk of generating multiple antibodies. Unlike AQP4 or MOG, which target cell surface antigens and exert direct pathogenic effects, GFAP is an intracellular antigen. Rather than acting as a direct pathogenic target, GFAP serves as a biomarker of CD8+ T-cell-mediated inflammatory processes (1, 2). Autopsy findings from a GFAP-IgG-positive patient with meningoencephalomyelitis were nonspecific and showed no astrocyte involvement or demyelination (23), suggesting that GFAP-IgG may not be the directly pathogenic antibody responsible for astrocyte inflammation. AQP4-IgG or MOG-IgG from the systemic circulation may enter the CNS through a disrupted blood–brain barrier, potentially initiating primary inflammatory events and damaging astrocytes, while GFAP autoimmunity may occur as a secondary phenomenon. Elevated levels of GFAP have been shown to correlate with disease severity in AQP4-IgG-positive NMOSD and MOGAD, particularly during acute attacks (24, 25). Moreover, the presence of multiple antibody positivities may also be related to immune reconstitution (26). During the reduction or cessation of immunotherapy, the immune system recovers from immunosuppression and rebuilds itself, leading immune cells to attack autoantigens and generate new immune responses (27, 28). However, the precise mechanisms underlying overlapping syndromes remain poorly understood, and further research is warranted.
In addition to AQP4-IgG and MOG-IgG, GFAP-IgG can coexist with other antibodies, most commonly NMDAR-IgG (1). Interestingly, three patients with overlapping syndromes tested positive for NMDAR-IgG, including two in the GFAP-AQP4 group and one in the GFAP-MOG group. MOG and functional NMDARs may coexist on the surface of oligodendrocytes. During autoimmune processes, immune cells may mistakenly target MOG and NMDAR autoantigens, leading to the production of MOG-IgG and NMDAR-IgG (26, 29). The co-occurrence of GFAP-IgG, AQP4-IgG, and NMDAR-IgG is a strong predictor of ovarian teratoma. Flanagan et al. reported that among seven GFAP-IgG-positive patients who were concurrently positive for NMDAR-IgG and AQP4-IgG, five developed ovarian teratoma (1). GFAP autoimmunity may represent a paraneoplastic immune response triggered by tumors expressing neuronal and glial elements. However, no tumors were observed in our study of overlapping syndromes, possibly due to the limited sample size or short follow-up period. Additionally, some scholars have suggested that the incidence of tumors may be associated with ethnic specificity (30).
The diagnosis of overlapping syndrome primarily relies on antibody detection. For A-GFAP-A, CSF antibody testing demonstrates high specificity and sensitivity. However, the phenotypes of patients with positive serology are heterogeneous (1, 31–34). Therefore, patients with isolated serum GFAP-IgG positivity require additional clinical and radiological assessments. In our study, two patients with overlapping syndromes tested positive for GFAP-IgG only in the serum. Nevertheless, after a rigorous exclusion of other neurological disorders, both cases fulfilled the diagnostic criteria for A-GFAP-A. It should be noted that antibody testing alone is insufficient to establish a definitive diagnosis, as false-positive results may occur. Therefore, we recommend repeating antibody testing, particularly during disease relapses.
Although antibody overlap can complicate clinical diagnosis, the management of overlapping and nonoverlapping syndromes during the acute phase follows similar therapeutic principles. Patients with A-GFAP-A overlapping syndromes typically respond poorly to acute-phase therapy and exhibit higher relapse rates. Consequently, aggressive immunotherapy is recommended for these patients (6, 9, 11).
Here, we provide the following recommendations: (1) For patients who test positive for AQP4 or MOG antibodies, testing for GFAP antibodies is not recommended, as the presence of AQP4 or MOG antibodies is sufficient to guide subsequent treatment decisions. (2) For patients who test positive for GFAP antibodies, it is recommended to simultaneously test for AQP4 and MOG antibodies. Given the recurrent and disabling nature of NMOSD and MOGAD, long-term immunotherapy is of great significance. Detection of AQP4 and MOG antibodies may aid in informing subsequent treatment strategies. (3) If patients with encephalitis or myelitis develop ON either simultaneously or sequentially, it is recommended to test for AQP4 or MOG antibodies first, rather than GFAP antibodies.
This study has several limitations. First, as a retrospective study, it limits the collection of detailed clinical data, such as antibody titers, the possible presence of other unknown pathogenic antibodies, and comprehensive ophthalmological assessments. Second, the use of a fixed CBA for antibody detection may result in false-negative results, particularly in low-titer cases. Some patients did not undergo antibody testing during the recovery phase. Additionally, due to the small sample size and multiple statistical comparisons, there is a risk of type I error. Given the retrospective and exploratory nature of the study, p-values should not be interpreted with caution and should not be considered confirmatory.
In conclusion, A-GFAP-A can coexist with demyelinating diseases. When patients with A-GFAP-A present with atypical symptoms such as ON, or when patients with demyelinating diseases have meningeal involvement, the possibility of an overlapping syndrome should be considered. Accurate diagnosis requires both clinical assessment and antibody testing. Future research should aim to elucidate the association between antibodies and clinical phenotypes, identify pathogenic antibodies, and avoid relying solely on antibody presence for diagnosis.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
QL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft. XC: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors thank all the subjects who participated in this study, as well as the staff of the Department of Neuroimmunology Laboratory and Imaging of the First Affiliated Hospital of Zhengzhou University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
2. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. (2017) 81:298–309. doi: 10.1002/ana.24881
3. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
4. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD Panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
5. Xiao J, Zhang S, Chen X, Tang Y, Chen M, Shang K, et al. Comparison of clinical and radiological characteristics in autoimmune GFAP astrocytopathy, MOGAD and AQP4-IgG+ NMOSD mimicking intracranial infection as the initial manifestation. Mult Scler Relat Disord. (2022) 66:104057. doi: 10.1016/j.msard.2022.104057
6. Yang X, Xu H, Ding M, Huang Q, Chen B, Yang H, et al.Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodiesFront Neurol. (2018) 9:251. doi: 10.3389/fneur.2018.00251
7. Martin AJ, Strathdee J, and Wolfe N. Coexistent anti-GFAP and anti-MOG antibodies presenting with isolated meningitis and papillitis: more support for overlapping pathophysiology. BMJ Neurol Open. (2022) 4:e000236. doi: 10.1136/bmjno-2021-000236
8. Zhao C, Li A, Liu L, Wang J, and Fan D. Acute myelitis, recurrent optic neuritis, and seizures over 17 years. Front Neurol. (2020) 11:541146. doi: 10.3389/fneur.2020.541146
9. Ding J, Ren K, Wu J, Li H, Sun T, Yan Y, et al. Overlapping syndrome of MOG-IgG-associated disease and autoimmune GFAP astrocytopathy. J Neurol. (2020) 267:2589–93. doi: 10.1007/s00415-020-09869-2
10. Ji S, Liu C, Bi Z, Gao H, Sun J, and Bu B. Overlapping syndrome mimicking infectious meningoencephalitis in a patient with MOG and GFAP IgG. BMC Neurol. (2021) 21:348. doi: 10.1186/s12883-021-02381-8
11. Fang H, Hu W, Jiang Z, Yang H, Liao H, Yang L, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in children: a retrospective analysis of 35 cases. Front Immunol. (2021) 12:761354. doi: 10.3389/fimmu.2021.761354
12. Fang T, Wu W, He X, Liang Y, Lin Q, Dai K, et al. Clinical characteristics of overlapping syndrome in patients with GFAP-IgG and MOG-IgG: a case series of 8 patients and literature review. J Neurol. (2024) 271:6811–21. doi: 10.1007/s00415-024-12633-5
13. Liu L, Fang B, Qiao Z, Di X, Ma Q, Zhang J, et al. Clinical manifestation, auxiliary examination features, and prognosis of GFAP autoimmunity: a Chinese cohort study. Brain Sci. (2022) 12:1662. doi: 10.3390/brainsci12121662
14. Chen JJ, Aksamit AJ, McKeon A, Pittock SJ, Weinshenker BG, Leavitt JA, et al. Optic disc edema in glial fibrillary acidic protein autoantibody-positive meningoencephalitis. J Neuroophthalmol. (2018) 38:276–81. doi: 10.1097/WNO.0000000000000593
15. Greco G, Masciocchi S, Diamanti L, Bini P, Vegezzi E, Marchioni E, et al. Visual system involvement in glial fibrillary acidic protein astrocytopathy: two case reports and a systematic literature review. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200146. doi: 10.1212/NXI.0000000000200146
16. Lana-Peixoto MA and Talim N. Neuromyelitis optica spectrum disorder and anti-MOG syndromes. Biomedicines. (2019) 7:42. doi: 10.3390/biomedicines7020042
17. Deng B, Wang J, Yu H, Jin L, Qiu Y, Liu X, et al. Area postrema syndrome in autoimmune glial fibrillary acidic protein astrocytopathy: a case series and literature review. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e200029. doi: 10.1212/NXI.0000000000200029
18. Long Y, Liang J, Xu H, Huang J, Yang J, Gao C, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neuol. (2018) 25:477–83. doi: 10.1111/ene.13531
19. Lin H, Huang Y, Zeng H, Wang M, Guan S, Chen G, et al. Overlapping clinical syndromes in patients with glial fibrillary acidic protein IgG. Neuroimmunomodulation. (2020) 27:69–74. doi: 10.1159/000505730
20. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol. (2018) 75(1):65–71. doi: 10.1001/jamaneurol.2017.3196
21. Koga M, Takahashi T, Kawai M, Fujihara K, and Kanda T. A serological analysis of viral and bacterial infections associated with neuromyelitis optica. J Neurol Sci. (2011)300(1-2):19–22. doi: 10.1016/j.jns.2010.10.013
22. Frau J, Coghe G, Lorefice L, Fenu G, and Cocco E. The Role of microorganisms in the etiopathogenesis of demyelinating diseases. Life (Basel). (2023) 13:1309. doi: 10.3390/life13061309
23. Yamakawa M, Hogan KO, Leever J, and Jassam YN. Autopsy case of meningoencephalomyelitis associated with glial fibrillary acidic protein antibody. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1081. doi: 10.1212/NXI.0000000000001081
24. Chang X, Huang W, Wang L, Zhang J, Zhou L, Lu C, et al. Serum neurofilament light and GFAP are associated with disease severity in inflammatory disorders with aquaporin-4 or myelin oligodendrocyte glycoprotein antibodies. Front Immunol. (2021) 12:647618. doi: 10.3389/fimmu.2021.647618
25. Schindler P, Aktas O, Ringelstein M, Wildemann B, Jarius S, Paul F, et al. Glial fibrillary acidic protein as a biomarker in neuromyelitis optica spectrum disorder: a current review. Expert Rev Clin Immunol. (2023) 19:71–91. doi: 10.1080/1744666X.2023.2148657
26. Ren Y, Chen X, He Q, Wang R, and Lu W. Co-occurrence of anti-N-methyl-D-aspartate receptor encephalitis and anti-myelin oligodendrocyte glycoprotein inflammatory demyelinating diseases: a clinical phenomenon to be taken seriously. Front Neurol. (2019) 10:1271. doi: 10.3389/fneur.2019.01271
27. Van Obberghen EK, Cohen M, Rocher F, and Lebrun-Frenay C. Multiple immune disorders after natalizumab discontinuation: after the CIRIS, the SIRIS? Rev Neurol (Paris). (2017) 173:222–4. doi: 10.1016/j.neurol.2017.03.008
28. Giedraitienė N, Kizlaitienė R, and Kaubrys G. New autoimmune disorder development after immune reconstitution therapy for multiple sclerosis. Sci Rep. (2024) 14:30991. doi: 10.1038/s41598-024-82196-y
29. Fan S, Xu Y, Ren H, Guan H, Feng F, Gao X, et al. Comparison of myelin oligodendrocyte glycoprotein (MOG)-antibody disease and AQP4-IgG-positive neuromyelitis optica spectrum disorder (NMOSD) when they co-exist with anti-NMDA (N-methyl-D-aspartate) receptor encephalitis. Mult Scler Relat Disord. (2018) 20:144–52. doi: 10.1016/j.msard.2018.01.007
30. Zhu B, Sun M, Yang T, Yu H, and Wang L. Clinical, imaging features and outcomes of patients with anti-GFAP antibodies: a retrospective study. Front Immunol. (2023) 14:1106490. doi: 10.3389/fimmu.2023.1106490
31. Zhang Z, Zoltewicz JS, Mondello S, Newsom KJ, Yang Z, Yang B, et al.Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS One. (2014)9, e92698. doi: 10.1371/journal.pone.0092698
32. Wang KKW, Yang Z, Yue JK, Zhang Z, Winkler EA, Puccio AM, et al. Plasma anti-glial fibrillary acidic protein autoantibody levels during the acute and chronic phases of traumatic brain injury: a transforming research and clinical knowledge in traumatic brain injury pilot study. J Neurotrauma (2016) 33(13):1270–7. doi: 10.1089/neu.2015.3881
33. Wei P, Zhang W, Yang L, Zhang H, Xu X, Jiang Y, et al. Serum GFAP autoantibody as an ELISA-detectable glioma marker. Tumour Biol. (2013) 34:2283–92. doi: 10.1007/s13277-013-0770-7
Keywords: glial fibrillary acidic protein, aquaporin-4, myelin oligodendrocyte glycoprotein, autoimmunity, overlapping
Citation: Li Q and Chen X (2025) Clinical characteristics of patients with GFAP-IgG coexisting with AQP4-IgG or MOG-IgG. Front. Immunol. 16:1610486. doi: 10.3389/fimmu.2025.1610486
Received: 12 April 2025; Accepted: 03 July 2025;
Published: 24 July 2025.
Edited by:
Francesco Patti, University of Catania, ItalyReviewed by:
Eun-Jae Lee, University of Ulsan, Republic of KoreaEslam Shosha, McMaster University, Canada
Copyright © 2025 Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingchen Li, cWluZ2NoZW4wMDlAMTYzLmNvbQ==