 Yanchun Cao
Yanchun Cao Ke Tang2†
Ke Tang2†- 1School of Traditional Chinese and Western Medicine, Gansu University of Chinese Medicine, Lanzhou, Gansu, China
- 2Department of Neurology, Affiliated Hospital of Gansu University of Chinese Medicine, Lanzhou, Gansu, China
- 3Department of Psychiatry, The No.2 people’s hospital of Lanzhou, Lanzhou, Gansu, China
- 4Department of Psychiatry, Xiaogan City social welfare and medical rehabilitation center, Xiaogan, Hubei, China
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by amyloid-β (Aβ) plaques, neurofibrillary tangles, and chronic neuroinflammation. While microglia and astrocytes dominate CNS immune responses, emerging evidence implicates peripheral innate immune cells (PIICs)—including neutrophils, monocytes, dendritic cells, NK cells, and myeloid-derived suppressor cells (MDSCs)—as critical modulators of AD pathogenesis. This review synthesizes recent advances linking PIIC-related genetic polymorphisms to AD susceptibility and progression. We highlight how PIICs traffic into the brain via chemokine signaling, where they exhibit stage-specific effects: early recruitment may limit Aβ deposition via phagocytosis, whereas chronic infiltration exacerbates neuroinflammation and neuronal death. Paradoxically, some PIICs exert immunosuppressive effects that could be harnessed therapeutically. We further discuss preclinical strategies to modulate PIIC function, such as CCR2 inhibition, neutrophil depletion, and MDSC adoptive transfer. By bridging peripheral and central immunity, this review unveils PIICs as promising targets for next-generation AD therapies, advocating for precision immunomodulation tailored to disease stages.
1 Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative condition, is pathologically defined by the accumulation of extracellular Aβ plaques alongside intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein (1). Although aging populations have intensified its global impact, current therapeutic strategies targeting conventional amyloid/tau paradigms show restricted efficacy (2). Emerging evidence indicates that AD pathogenesis encompasses diverse mechanisms, such as metabolic disturbances, impaired mitochondrial function, and particularly neuroinflammatory processes that initiate prior to observable pathology and propel disease advancement (3–5). Although transient neuroinflammatory responses serve protective functions, persistent inflammation worsens neuronal injury and hastens cognitive deterioration (6, 7), positioning it as a critical intervention point for halting the transition from mild cognitive impairment to AD (8).
Central nervous system (CNS) neuroinflammation is primarily regulated by microglia and astrocytes (9, 10), whereas PIICs contribute underrecognized yet vital functions. Diverging from traditional perceptions of innate immunity as nonspecific, PIICs—encompassing monocytes (PMCs), natural killer (NK) cells, and bone marrow progenitors—demonstrate adaptive characteristics through “trained immunity” enabling long-term immunological memory (11, 12). Their involvement manifests contrasting effects: beneficial immune monitoring during acute phases versus detrimental contributions in chronic AD progression (13). Contemporary findings reveal that PIICs (including polymorphonuclear neutrophils [PMNs], PMCs, dendritic cells [DCs], and NK cells) migrate into brain tissue, intensifying inflammatory cascades and neuronal damage (14–16). Chemotactic signals from peripheral Aβ recruit these cells, prompting secretion of proinflammatory factors and compromising blood-brain barrier integrity (17). While MDSCs have been extensively studied in oncology (18), their role in AD remains elusive. This review summarizes current knowledge on PIIC-associated genetic polymorphisms, quantitative and qualitative alterations in AD, and mechanistic insights into their CNS infiltration. Elucidating these processes may reveal innovative targets for pharmacological modulation in AD.
2 Peripheral innate immune in Alzheimer’s disease
CD33
Advances in GWAS have consistently identified genetic variants influencing AD susceptibility, with a significant proportion localized to myeloid cell-specific genes—over one-third of risk loci exhibit myeloid-selective expression (19, 20). Epigenetic analyses further indicate that AD-associated GWAS loci are disproportionately enriched in enhancer regions regulating innate immune activity (21). Understanding how polymorphisms in PIIC-related receptors, cytokines, and complement factors contribute to AD is thus essential for unraveling disease mechanisms and developing targeted therapies. Expressed mainly on microglia and myeloid cells, the transmembrane receptor CD33 modulates intercellular adhesion and innate immune signaling (22). An AD-linked risk allele of CD33 correlates with increased receptor expression in affected brains, impairing microglial phagocytosis and Aβ42 clearance (23, 24). Initial evidence implicating CD33 emerged from family-based GWAS, which detected the rs3826656 variant within the gene’s 3′ region (25). Subsequent work by Bradshaw et al. (26) revealed elevated rs3865444-associated CD33 transcription in AD patients, with expression levels inversely correlated with cognitive performance. Mendelian randomization studies further support a causal role for CD33-dependent immune dysregulation in AD (27).
2.2 TREM family receptors in immune regulation
Of the innate immune markers associated with AD, TREM2 has garnered the most attention due to its dual-phase impact on disease progression: early protective effects via Aβ clearance followed by later detrimental neuroinflammatory responses (28–30). This membrane-bound receptor enhances phagocytic activity and modulates microglial function (31). The R47H variant of TREM2 elevates AD risk by 2–3-fold (32). Adjacent to TREM2 on chromosome 6p21.1, TREM1 is predominantly active in monocytes, macrophages, and neutrophils during immune challenges (33). In Han Chinese populations, the TREM1 SNP rs2062323 exhibits a protective effect, with the T allele significantly reducing AD susceptibility.
2.3Cytokine and complement in Alzheimer’s disease
Cytokines serve as pivotal mediators of PIIC-driven immunity. Genetic studies have associated polymorphisms in interleukin genes. Notably, carriers of risk alleles in IL1β, IL6, IL10, and TNF-αdisplay heightened susceptibility (34–37). While IL1α (rs1800587) and IL33 (rs11792633) correlate with late-onset AD in Han Chinese cohorts, IL1β lacks such an association (37). Complement system genes (CLU, CR1, SERPINA3, CFH, C4) also harbor AD-linked SNPs (38, 39). CLU, a major risk locus for late-onset AD, contains protective variants (rs11136000, rs2279590, rs9331888) that reduce Aβ accumulation (39, 40). Patients homozygous for the rs11136000-C allele exhibit the highest Aβ burden, whereas TT genotypes show minimal deposition (41). In APOE4-positive individuals, the C allele significantly elevates CSF tau levels, with CC homozygotes exceeding CT heterozygotes (42). The rs9331888-GG genotype further correlates with diminished hippocampal volume (41). Beyond common variants, rare mutations in PLCG2, ABCA7, SORL1, and ECE2 also modulate AD risk (43, 44). Collectively, PIIC-related genetic variants intricately influence AD pathogenesis, offering mechanistic insights and potential therapeutic avenues. Future studies must consider ethnic, geographic, and demographic variables when assessing these polymorphisms.
3 Peripheral innate immune cells in Alzheimer’s disease
3.1 Polymorphonuclear neutrophils
As the predominant myeloid cell type in human peripheral blood, neutrophils (PMNs) play crucial roles in maintaining tissue homeostasis while also contributing to inflammatory damage during sterile inflammation (45, 46). Research using AD mouse models reveals their early involvement in disease pathogenesis, with cerebral accumulation preceding clinical symptoms and subsequent release of pro-inflammatory factors (47, 48). A meta-analysis by Huang etal. demonstrated significantly elevated peripheral PMN counts in patients with mild cognitive impairment and AD compared with healthy controls, implicating oxidative stress, immune dysregulation and neuroinflammation in driving this expansion (49). Through enhanced antigen presentation, these cells stimulate T lymphocyte activation, creating a feedback loop that amplifies TNF-α production (50).
Chronic TNF-α modulation in 3×Tg AD mice demonstrated dual effects: promoting PMN infiltration (CD45Hi/CD11b+/GR1+/1A8+) alongside Aβ/tau accumulation, yet paradoxically enhancing memory function (51). The neurotoxic potential of activated PMNs stems from myeloperoxidase (MPO), ROS, and NET generation - all capable of compromising blood-brain barrier integrity (52, 53), with circulating MPO levels predicting cognitive deterioration (54). Experimental PMN depletion during early-stage AD yields lasting cognitive improvements in aged models, highlighting their disease-modifying capacity (47, 55). Clinical studies consistently associate elevated peripheral PNLR values with AD progression markers, including CSF Aβ reduction, tau elevation, and hippocampal volume loss (15, 50). This ratio may signify dysregulated immune responses correlating with amyloid accumulation and progressive cognitive impairment (15, 56). Mechanistic studies employing microfluidic AD models reveal Aβ-stimulated microglia secrete IL-6, IL-8, and CCL2, with cytokine neutralization preventing PMN CNS migration (57). Aβ42 further potentiates neuroinvasion by modulating LFA-1 affinity states, increasing endothelial ICAM-1 binding (47). Pharmacological LFA-1 inhibition attenuates PMN recruitment, ameliorates neuropathology, and restores cognitive function in AD mice (47, 58). Intracerebral PMNs localize to amyloid deposits, where MPO, NETs, and IL-17 perpetuate neuroinflammation (47). Their interactions with microglia induce MIF and IL-2 secretion (57), while the latter reduces plaque load and enhances synaptic function (59, 60). Thus, PMNs demonstrate pleiotropic neuroprotective and neurotoxic effects in AD pathogenesis (Figure 1).
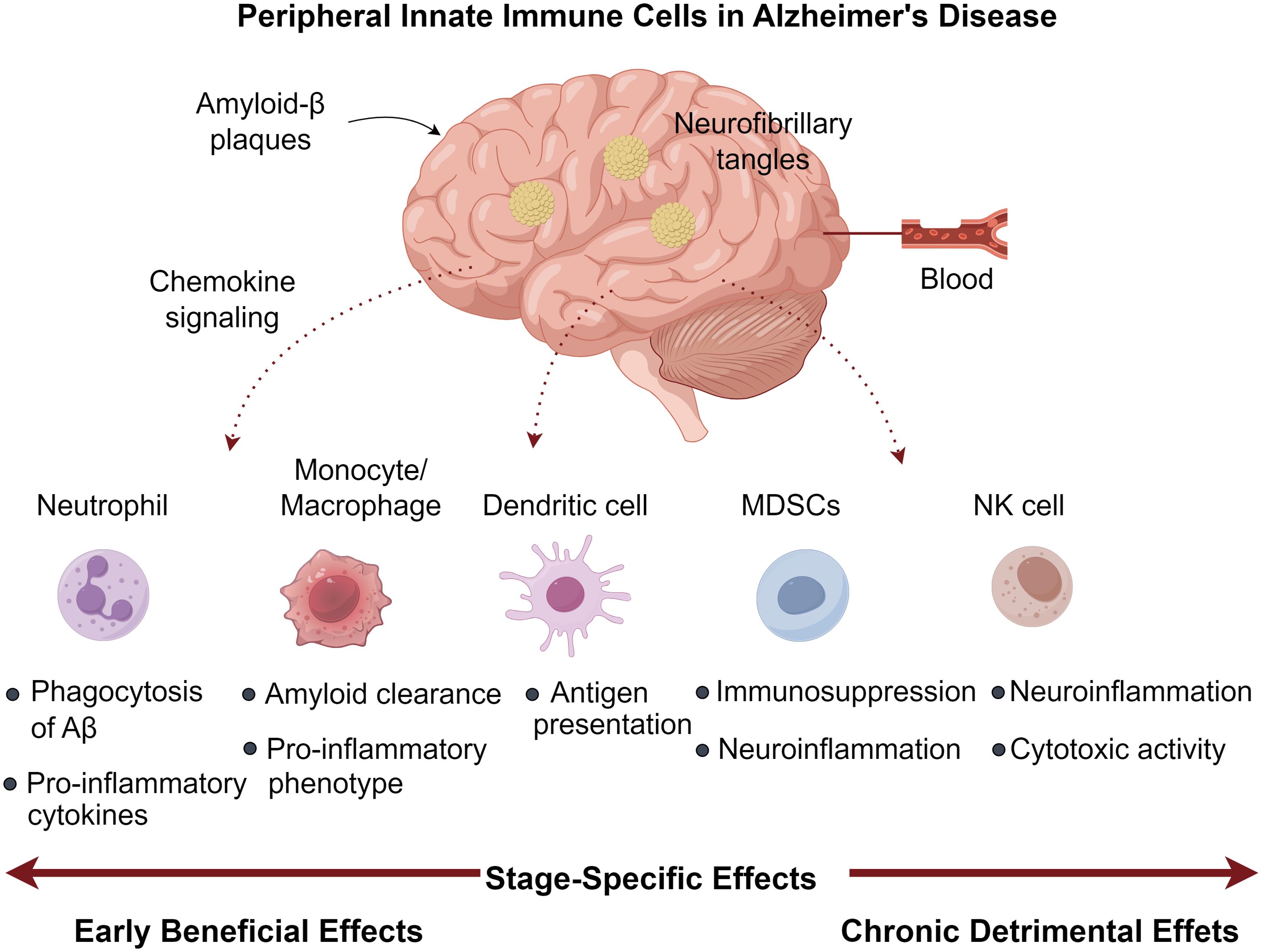
Figure 1. Peripheral innate immune cells in Alzheimer’s disease.
3.2 Peripheral monocytes/macrophages
Mononuclear phagocytes (PMCs) exhibit remarkable functional plasticity, dynamically responding to pathological stimuli through subset-specific adaptations. Human monocyte populations are classified based on surface markers into three distinct subsets: classical (CD14+CD16+), intermediate (CD14+CD16+), and non-classical (CD14dimCD16+). In Alzheimer’s disease (AD), shifts in monocyte distribution occur, characterized by increased non-classical and intermediate subsets alongside decreased classical monocytes (61). During neuroinflammatory conditions, classical monocytes preferentially infiltrate the CNS, where microglial activation by Aβ upregulates CCR2 chemokines, facilitating monocyte recruitment, macrophage differentiation, and amyloid clearance (62). Genetic ablation of CCR2 in AD models reduces cerebral PMC numbers (63), whereas pharmacological CCR2 blockade exacerbates amyloid accumulation and cognitive decline (64). Clinical studies reveal diminished monocytic CCR2 expression but elevated circulating CCL2 in AD patients, indicating impaired CCR2-CCL2 signaling and defective migration (65).
The mitochondrial translocator protein (TSPO), highly expressed in AD-associated microglia (66), has recently been identified as a regulator of PMC chemotaxis, with TSPO inhibitors suppressing Aβ-induced monocyte migration (67). Despite entering the CNS, PMCs demonstrate impaired phagocytic activity, leading to neuronal damage (68). Age-dependent reductions in Aβ42 uptake across all monocyte subsets are more pronounced in AD, as reported by Chen et al. (69), potentially due to decreased Toll-like receptor 2 (TLR2) expression, which compromises Aβ recognition. Furthermore, PMCs adopt a pro-inflammatory state, releasing elevated levels of cytokines (IL-6, IL-1β, TNF) and inflammasome components (NLRP3, IL-18), alongside increased HLA-DRA expression, thereby exacerbating neurodegeneration (61). Notably, PMC transcriptional profiles shift during disease progression, with anti-inflammatory IL-10 predominating in early-stage AD before transitioning to a sustained pro-inflammatory signature (61), suggesting temporally distinct roles—neuroprotective in prodromal phases but detrimental in later stages. Overexpression of ACE in CD115+ PMCs has been shown to mitigate neuropathology and improve cognition in APPRE/PS1 transgenic mice (70). Emerging therapeutic strategies may involve bone marrow-derived progenitor cell transplantation to restore functional PMC populations (32) (Table 1).
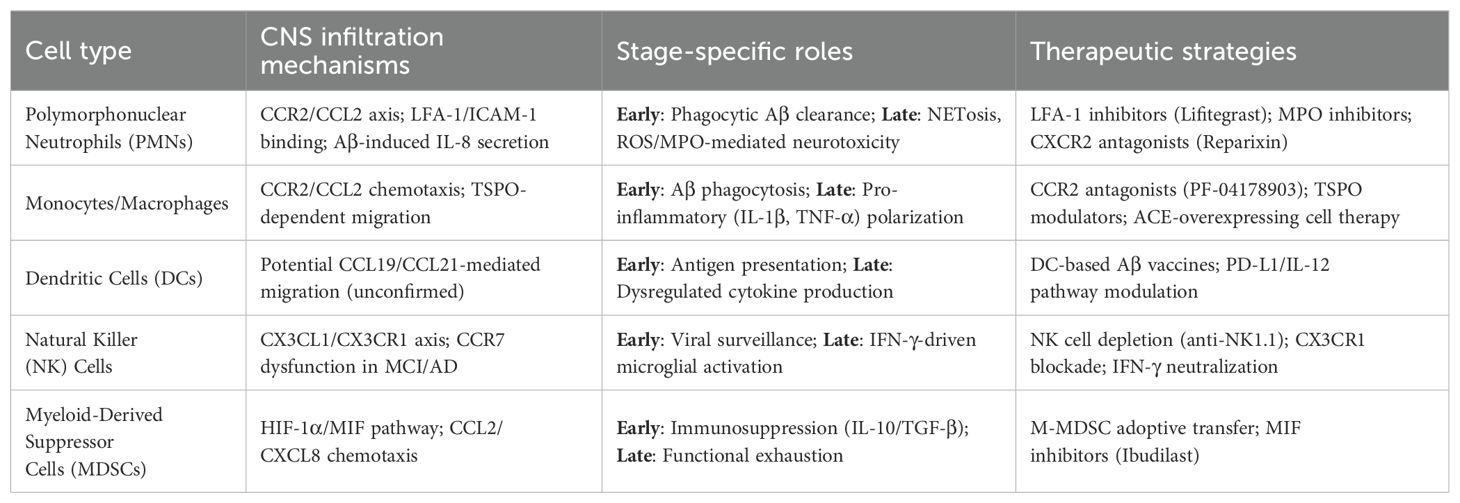
Table 1. Peripheral Innate Immune Cells (PIICs) in Alzheimer’s disease: mechanisms and therapeutic implications.
3.3 Dendritic cells
Dendritic cells (DCs) serve as critical intermediaries between innate and adaptive immune responses. As these antigen-presenting cells undergo maturation, their migration from circulation to peripheral tissues results in decreased blood DC populations (71). While definitive evidence of peripheral DC infiltration into the AD brain remains elusive, studies in APP/PS1 transgenic mice demonstrate that systemic DC depletion leads to increased amyloid plaque burden, suggesting their potential role in cerebral amyloid clearance (72). This hypothesis is supported by clinical observations of decreased circulating DC precursors (73) and myeloid DC subsets (74) in AD patients, consistent with possible CNS migration. The causal relationship between DC reduction and neurodegenerative processes requires further clarification.
Research by Ciaramella et al. (74) identified an association between lower myeloid DC counts and both disease advancement and depressive symptoms in AD. In contrast, 5×FAD transgenic models exhibit altered DC function characterized by increased IL-12 and MIP-1 production in mesenteric lymph node myeloid DCs, coupled with decreased PD-L1 expression, indicating potential DC dysfunction in AD (75) (Table 1). Current understanding of DC involvement in AD remains limited, with particularly scarce data regarding DC-based immunization strategies. One study demonstrated cognitive improvement in APPswe/PSEN1ΔE9 mice following co-immunization with Aβ4-pulsed DCs and splenocytes (76). DC-based immunization approaches may offer advantages over conventional protein vaccines by simultaneously engaging both innate and adaptive immunity to generate targeted antibody responses and enhance pathogen clearance. However, clinical translation requires thorough investigation of multiple parameters including DC development, phenotypic characteristics, proliferative potential, antigen recognition profiles, post-activation behavior, and safety considerations (71).
3.4 Natural killer cells
Natural killer (NK) cells, a specialized group of innate immune lymphocytes, are increasingly recognized as key modulators of neuroinflammatory processes (77). These cytotoxic lymphocytes are primarily categorized into two functionally distinct populations based on CD56 expression levels: the relatively scarce CD56bright subset that specializes in cytokine production, and the more prevalent CD56dim population that mediates cell-killing activity (77). Current understanding of NK cell characteristics in Alzheimer’s pathology reveals conflicting observations. While some investigations report no significant differences in NK cell numbers or functional capacity between AD patients and healthy individuals (78), other studies demonstrate decreased peripheral NK cell counts with concurrent upregulation of immune-related genes (79). Research by Qi et al. (77) revealed a reduction in both NK cell numbers and cytotoxic potential in AD patients, along with identification of an expanded CX3CR1+TBX21+ NK subpopulation showing an inverse correlation with cognitive scores. Conversely, Solerte et al. (80) reported enhanced cytotoxic activity and increased TNF-α/IFN-γ production in AD-derived NK cells compared to age-matched controls, similarly associating with poorer cognitive outcomes. Experimental NK cell elimination in 3×Tg AD mice was shown to reduce neuroinflammation and cognitive deficits while preserving neural progenitor populations, though without affecting amyloid deposition (81).
The functional state of NK cells is determined by the integration of signals from both stimulatory and inhibitory surface receptors. Analysis of receptor expression patterns revealed preserved levels of CD57, NKG2D and CD94 in mild cognitive impairment (MCI) and early AD patients relative to healthy elderly subjects, while NKG2A showed selective reduction in MCI cases, a change that may promote NK cell activation (82). These partially contradictory findings collectively suggest that while NK cell alterations occur in AD, their exact role in disease mechanisms remains unclear. Comprehensive investigations employing single-cell transcriptomics coupled with advanced immunophenotyping are needed to fully characterize NK cell diversity across different AD stages. Although NK cell presence has been confirmed in AD animal models, their involvement in human disease requires further validation (83). Glial cells (microglia and astrocytes) contribute to NK cell recruitment and sustained neuroimmune activation through cytokine and chemokine secretion (84). NK cell-derived IFN-γ can polarize microglia toward a pro-inflammatory state, resulting in suppressed hippocampal neurogenesis and the development of cognitive deficits and depressive-like symptoms (85). Distinct NK cell subsets exhibit differential chemokine receptor expression patterns: while CX3CR1 facilitates CD56dimCD16+ NK cell brain infiltration in multiple sclerosis (86), its expression remains unaltered on NK cells from MCI and AD patients (82). Interestingly, CCR7 (the receptor for CCL19/CCL21) shows increased expression in MCI-derived NK cells, yet their migratory response to CCL19 is impaired in both MCI and AD cases (82) (Table 1).
3.5 Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) represent a heterogeneous population of immature myeloid precursors that emerge in response to inflammatory signals. These immunoregulatory cells are broadly classified into monocytic (M-MDSCs) and granulocytic (PMN-MDSCs) subtypes, both capable of suppressing effector immune cell functions through potent inhibition of mature myeloid and lymphocyte populations (87). Under chronic inflammatory conditions, MDSCs migrate to affected tissues where they help regulate immune responses and prevent excessive activation. In Alzheimer’s disease, available evidence suggests a dynamic pattern of MDSC involvement, with expansion during initial disease phases followed by progressive decline (88). Clinical studies reveal distinct alterations in MDSC populations across disease stages. Le Page et al. (89) observed increased circulating CD33+HLA-DR- M-MDSCs and CD33+HLA-DR-CD11b+CD15+ MDSCs in mild cognitive impairment (MCI) patients compared to both AD patients and healthy controls. Similarly, Thome et al. (61) reported enhanced MDSC frequency and immunosuppressive activity in early AD, associated with reduced pro-inflammatory gene expression in peripheral blood mononuclear cells - effects that diminished with disease progression (Table 1).
The neuroprotective potential of MDSCs stems from their ability to secrete immunomodulatory cytokines that promote microglial polarization toward an anti-inflammatory M2 phenotype. While this shift may help mitigate neuroinflammation and neuronal damage, it could simultaneously impair clearance of pathological protein aggregates (90). The temporal pattern of MDSC dynamics suggests an endogenous regulatory mechanism that becomes inadequate as AD advances (88). This observation has spurred interest in therapeutic strategies aimed at sustaining MDSC-mediated immunosuppression, as demonstrated by studies showing that M-MDSC transplantation can counteract immune dysregulation and cognitive deficits in AD mouse models (91). Despite these findings, direct evidence of MDSC infiltration in the AD brain remains elusive. Potential mechanisms for CNS recruitment may involve the HIF-1α/MIF pathway, known to mediate MDSC trafficking in cancer (92), given the elevated cerebrospinal fluid MIF levels observed in AD patients (93). Additionally, various chemokines upregulated in AD may facilitate MDSC migration toward affected brain regions (90). Further research is needed to clarify the spatial and temporal distribution of MDSCs in AD pathogenesis and their potential as therapeutic targets (Figure 1).
4 Therapeutic landscape
Despite decades of research, therapeutic options for AD remain limited, with most treatments offering only symptomatic relief rather than disease modification (94). Recently, however, the therapeutic landscape has evolved to include several disease-modifying treatments (DMTs), particularly monoclonal antibodies that target Aβ (95). For example, aducanumab and lecanemab, two anti-Aβ monoclonal antibodies aim to reduce Aβ plaque burden through immunotherapeutic mechanisms (96). Aducanumab selectively binds to aggregated Aβ and facilitates its clearance, while lecanemab preferentially targets soluble Aβ protofibrils, mitigating their synaptotoxic effects (97). Other experimental approaches include tau-targeting immunotherapies, such as semorinemab, that aim to neutralize pathological tau species and interrupt their propagation between neurons (98). However, these therapies have shown limited benefits in clinical trials, and cause adverse effects such as amyloid-related imaging abnormalities-edema (99, 100). Compared to these approaches, immunomodulatory strategies targeting peripheral innate immune cells represent a novel paradigm. These include DC-based immunization approaches and CCR2 regulator to facilitate monocyte infiltration and macrophage differentiation to restore immune homeostasis. While still in preclinical stages, these therapies offer the advantage of modulating upstream inflammatory signals that orchestrate central pathology. Thus, contextualizing these immunotherapies within the current AD brain immune landscape not only clarifies their unique positioning but also highlights the need for stage-specific, precision immunomodulation that integrates both central and peripheral immune mechanisms.
5 Conclusion
Recent advances have revealed that PIICs are not mere bystanders but active participants in Alzheimer’s disease (AD) progression. This review highlights the stage-specific and context-dependent roles of PIIC subsets, such as neutrophils, monocytes/macrophages, dendritic cells, NK cells, and MDSCs, in shaping the neuroinflammatory landscape of AD. Early recruitment of PIICs may aid in Aβ clearance and immunoregulation, while persistent activation exacerbates neurotoxicity via cytokine storms, oxidative stress, and blood–brain barrier disruption. Genetic polymorphisms in PIIC-associated genes, such as CD33 and TREM2, further underscore their mechanistic relevance and suggest population-specific susceptibilities.
Targeting innate immune pathways offers a promising yet complex therapeutic avenue. Modulation of chemokine receptors, cytokine secretion, and macrophage function has shown efficacy in preclinical models. However, challenges remain, including the heterogeneity of immune responses across AD stages, potential off-target effects, and limited understanding of long-term immunomodulation. Future drug development should prioritize precision strategies that harness the protective capacity of PIICs while minimizing chronic inflammation. Integrating multi-omics profiling, advanced immunophenotyping, and longitudinal clinical data will be essential to refine these approaches. Ultimately, a deeper understanding of innate immunity may unlock novel interventions capable of altering AD trajectory in its earliest and most modifiable phases.
Author contributions
YC: Writing – original draft. KT: Writing – original draft. PM: Writing – original draft. RZ: Writing – original draft. YY: Writing – original draft. TL: Writing – original draft. YZ: Writing – original draft. XP: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Project of Gansu Provincial Administration of Chinese Medicine (GZKG-2024-59), Natural Science Foundation of Gansu Province (24JRRA1039). Gansu Province science and technology plan project (22JR11RA127).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Si ZZ, Zou CJ, Mei X, Li XF, Luo H, Shen Y, et al. Targeting neuroinflammation in Alzheimer’s disease: from mechanisms to clinical applications. Neural Regener Res. (2023) 18:708–15. doi: 10.4103/1673-5374.353484
2. Cai Y, Liu J, Wang B, Sun M, and Yang H. Microglia in the neuroinflammatory pathogenesis of alzheimer’s disease and related therapeutic targets. Front Immunol. (2022) 13:856376. doi: 10.3389/fimmu.2022.856376
3. Princiotta Cariddi L, Mauri M, Cosentino M, Versino M, and Marino F. Alzheimer’s disease: from immune homeostasis to neuroinflammatory condition. Int J Mol Sci. (2022) 23:13008. doi: 10.3390/ijms232113008
4. Liu Y and Aguzzi A. Immunotherapy for neurodegeneration? Science. (2019) 364:130–1. doi: 10.1126/science.aaw0685
5. Liu Y, Tan Y, Cheng G, Ni Y, Xie A, Zhu X, et al. Customized intranasal hydrogel delivering methylene blue ameliorates cognitive dysfunction against alzheimer’s disease. Adv Mater. (2024) 36:e2307081. doi: 10.1002/adma.202307081
6. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, and Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflamm. (2018) 15:276. doi: 10.1186/s12974-018-1313-3
7. Yang C and Xu P. The role of transforming growth factor β1/Smad pathway in Alzheimer’s disease inflammation pathology. Mol Biol Rep. (2023) 50:777–88. doi: 10.1007/s11033-022-07951-8
8. Schmidt-Morgenroth I, Michaud P, Gasparini F, and Avrameas A. Central and peripheral inflammation in mild cognitive impairment in the context of alzheimer’s disease. Int J Mol Sci. (2023) 24:10523. doi: 10.3390/ijms241310523
9. Boboc IKS, Cojocaru A, Nedelea G, Catalin B, Bogdan M, and Calina D. Chronic administration of ion channel blockers impact microglia morphology and function in a murine model of alzheimer’s disease. Int J Mol Sci. (2023) 24:14474. doi: 10.3390/ijms241914474
10. Deng Q, Wu C, Parker E, Liu TC, Duan R, and Yang L. Microglia and astrocytes in alzheimer’s disease: significance and summary of recent advances. Aging Dis. (2024) 15:1537–64. doi: 10.14336/AD.2023.0907
11. Netea MG, Latz E, Mills KH, and O’Neill LA. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol. (2015) 16:675–9. doi: 10.1038/ni.3178
12. Netea MG. Training innate immunity: the changing concept of immunological memory in innate host defence. Eur J Clin Invest. (2013) 43:881–4. doi: 10.1111/eci.2013.43.issue-8
13. Huang B, Zhenxin Y, Chen S, Tan Z, Zong Z, Zhang H, et al. The innate and adaptive immune cells in alzheimer’s and parkinson’s diseases. Oxid Med Cell Longev. (2022) 2022:1315248. doi: 10.1155/2022/1315248
14. Shi M, Chu F, Tian X, Aerqin Q, Zhu F, and Zhu J. Role of adaptive immune and impacts of risk factors on adaptive immune in alzheimer’s disease: are immunotherapies effective or off-target? Neuroscientist. (2022) 28:254–70. doi: 10.1177/1073858420987224
15. Hou JH, Ou YN, Xu W, Zhang PF, Tan L, and Yu JT. Association of peripheral immunity with cognition, neuroimaging, and Alzheimer’s pathology. Alzheimers Res Ther. (2022) 14:29. doi: 10.1186/s13195-022-00968-y
16. Bettcher BM, Tansey MG, Dorothée G, and Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol. (2021) 17:689–701. doi: 10.1038/s41582-021-00549-x
17. Shi M, Chu F, Zhu F, and Zhu J. Peripheral blood amyloid-β involved in the pathogenesis of Alzheimer’s disease via impacting on peripheral innate immune cells. J Neuroinflamm. (2024) 21:5. doi: 10.1186/s12974-023-03003-5
18. Joshi S and Sharabi A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther. (2022) 235:108114. doi: 10.1016/j.pharmthera.2022.108114
19. Haage V and De Jager PL. Neuroimmune contributions to Alzheimer’s disease: a focus on human data. Mol Psychiatry. (2022) 27:3164–81. doi: 10.1038/s41380-022-01637-0
20. McGeer PL and McGeer EG. Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets. (2015) 19:497–506. doi: 10.1517/14728222.2014.988707
21. Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. (2015) 518:365–9. doi: 10.1038/nature14252
22. Griciuc A, Federico AN, Natasan J, Forte AM, McGinty D, Nguyen H, et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum Mol Genet. (2020) 29:2920–35. doi: 10.1093/hmg/ddaa179
23. Eskandari-Sedighi G, Jung J, and Macauley MS. CD33 isoforms in microglia and Alzheimer’s disease: Friend and foe. Mol Aspects Med. (2023) 90:101111. doi: 10.1016/j.mam.2022.101111
24. Haure-Mirande JV, Audrain M, Ehrlich ME, and Gandy S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol Neurodegener. (2022) 17:55. doi: 10.1186/s13024-022-00552-w
25. Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. (2008) 83:623–32. doi: 10.1016/j.ajhg.2008.10.008
26. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. (2013) 16:848–50. doi: 10.1038/nn.3435
27. Tang C, Lei X, Ding Y, Yang S, Ma Y, and He D. Causal relationship between immune cells and neurodegenerative diseases: a two-sample Mendelian randomisation study. Front Immunol. (2024) 15:1339649. doi: 10.3389/fimmu.2024.1339649
28. Fassler M, Rappaport MS, Cuño CB, and George J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J Neuroinflamm. (2021) 18:19. doi: 10.1186/s12974-020-01980-5
29. Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. (2019) 22:191–204. doi: 10.1038/s41593-018-0296-9
30. Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, et al. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology. (2016) 105:196–206. doi: 10.1016/j.neuropharm.2016.01.028
31. Niso-Santano M, Fuentes JM, and Galluzzi L. Immunological aspects of central neurodegeneration. Cell Discov. (2024) 10:41. doi: 10.1038/s41421-024-00666-z
32. Dujardin P, Vandenbroucke RE, and Van Hoecke L. Fighting fire with fire: The immune system might be key in our fight against Alzheimer’s disease. Drug Discov Today. (2022) 27:1261–83. doi: 10.1016/j.drudis.2022.01.004
33. Wang ZT, Fu Y, Chen SD, Huang YY, Ma YH, Wang YJ, et al. Association of rs2062323 in the TREM1 gene with Alzheimer’s disease and cerebrospinal fluid-soluble TREM2. CNS Neurosci Ther. (2023) 29:1657–66. doi: 10.1111/cns.14129
34. Lin E, Kuo PH, Liu YL, Yang AC, and Tsai SJ. Association and interaction effects of interleukin-12 related genes and physical activity on cognitive aging in old adults in the Taiwanese population. Front Neurol. (2019) 10:1065. doi: 10.3389/fneur.2019.01065
35. Babić Leko M, Nikolac Perković M, Klepac N, Štrac D, Borovečki F, Pivac N, et al. IL-1β, IL-6, IL-10, and TNFα Single nucleotide polymorphisms in human influence the susceptibility to alzheimer’s disease pathology. J Alzheimers Dis. (2020) 75:1029–47. doi: 10.3233/JAD-200056
36. Yoo TJ. Anti-inflammatory gene therapy improves spatial memory performance in a mouse model of alzheimer’s disease. J Alzheimers Dis. (2022) 85:1001–8. doi: 10.3233/JAD-215270
37. Tian M, Deng YY, Hou DR, Li W, Feng XL, and Yu ZL. Association of IL-1, IL-18, and IL-33 gene polymorphisms with late-onset Alzheimer's disease in a Hunan Han Chinese population. Brain Res. (2015) 1596:136–45. doi: 10.1016/j.brainres.2014.11.019
38. Ma Z, Yang F, Fan J, Li X, Liu Y, Chen W, et al. Identification and immune characteristics of molecular subtypes related to protein glycosylation in Alzheimer’s disease. Front Aging Neurosci. (2022) 14:968190. doi: 10.3389/fnagi.2022.968190
39. Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. (2022) 54:412–36. doi: 10.1038/s41588-022-01024-z
40. Zorzetto M, Datturi F, Divizia L, Pistono C, Campo I, De Silvestri A, et al. Complement C4A and C4B gene copy number study in alzheimer’s disease patients. Curr Alzheimer Res. (2017) 14:303–8. doi: 10.2174/1567205013666161013091934
41. Tan L, Wang HF, Tan MS, Tan CC, Zhu XC, Miao D, et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci Rep. (2016) 6:26027. doi: 10.1038/srep26027
42. Zhou Y, Hayashi I, Wong J, Tugusheva K, Renger JJ, and Zerbinatti C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PloS One. (2014) 9:e103187. doi: 10.1371/journal.pone.0103187
43. Wang H, Dombroski BA, Cheng PL, Tucci A, Si YQ, Farrell JJ, et al. Structural variation detection and association analysis of whole-genome-sequence data from 16,905 alzheimer’s diseases sequencing project subjects. medRxiv. (2023) 21:e70277. doi: 10.1101/2023.09.13.23295505
44. Liao X, Cai F, Sun Z, Zhang Y, Wang J, Jiao B, et al. et al: Identification of Alzheimer’s disease-associated rare coding variants in the ECE2 gene. JCI Insight. (2020) 5:e135119. doi: 10.1172/jci.insight.135119
45. Kolaczkowska E and Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
46. Ng LG, Ostuni R, and Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
47. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
48. Zenaro E, Piacentino G, and Constantin G. The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis. (2017) 107:41–56. doi: 10.1016/j.nbd.2016.07.007
49. Huang LT, Zhang CP, Wang YB, and Wang JH. Association of peripheral blood cell profile with alzheimer’s disease: A meta-analysis. Front Aging Neurosci. (2022) 14:888946. doi: 10.3389/fnagi.2022.888946
50. Dong X, Nao J, Shi J, and Zheng D. Predictive value of routine peripheral blood biomarkers in alzheimer’s disease. Front Aging Neurosci. (2019) 11:332. doi: 10.3389/fnagi.2019.00332
51. Gabbita SP, Johnson MF, Kobritz N, Eslami P, Poteshkina A, Varadarajan S, et al. Oral TNFα Modulation alters neutrophil infiltration, improves cognition and diminishes tau and amyloid pathology in the 3xTgAD mouse model. PloS One. (2015) 10:e0137305. doi: 10.1371/journal.pone.0137305
52. Rossi B, Constantin G, and Zenaro E. The emerging role of neutrophils in neurodegeneration. Immunobiology. (2020) 225:151865. doi: 10.1016/j.imbio.2019.10.014
53. Pietronigro EC, Della Bianca V, Zenaro E, and Constantin G. NETosis in alzheimer’s disease. Front Immunol. (2017) 8:211. doi: 10.3389/fimmu.2017.00211
54. Bawa KK, Krance SH, Herrmann N, Cogo-Moreira H, Ouk M, Yu D, et al. A peripheral neutrophil-related inflammatory factor predicts a decline in executive function in mild Alzheimer’s disease. J Neuroinflamm. (2020) 17:84. doi: 10.1186/s12974-020-01750-3
55. Aries ML and Hensley-McBain T. Neutrophils as a potential therapeutic target in Alzheimer’s disease. Front Immunol. (2023) 14:1123149. doi: 10.3389/fimmu.2023.1123149
56. Mehta NH, Zhou L, Li Y, McIntire LB, Nordvig A, Butler T, et al. Peripheral immune cell imbalance is associated with cortical beta-amyloid deposition and longitudinal cognitive decline. Sci Rep. (2023) 13:8847. doi: 10.1038/s41598-023-34012-2
57. Park J, Baik SH, Mook-Jung I, Irimia D, and Cho H. Mimicry of central-peripheral immunity in alzheimer’s disease and discovery of neurodegenerative roles in neutrophil. Front Immunol. (2019) 10:2231. doi: 10.3389/fimmu.2019.02231
58. Pietronigro E, Zenaro E, Bianca VD, Dusi S, Terrabuio E, Iannoto G, et al. Blockade of α4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci Rep. (2019) 9:12055. doi: 10.1038/s41598-019-48538-x
59. Li SQ, Yu Y, Han JZ, Wang D, Liu J, Qian F, et al. Deficiency of macrophage migration inhibitory factor attenuates tau hyperphosphorylation in mouse models of Alzheimer’s disease. J Neuroinflamm. (2015) 12:177. doi: 10.1186/s12974-015-0396-3
60. Alves S, Churlaud G, Audrain M, Michaelsen-Preusse K, Fol R, Souchet B, et al. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain. (2017) 140:826–42. doi: 10.1093/brain/aww330
61. Thome AD, Faridar A, Beers DR, Thonhoff JR, Zhao W, Wen S, et al. Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol Neurodegener. (2018) 13:61. doi: 10.1186/s13024-018-0293-1
62. Monoranu CM, Hartmann T, Strobel S, Heinsen H, Riederer P, Distel L, et al. Is there any evidence of monocytes involvement in alzheimer’s disease? A pilot study on human postmortem brain. J Alzheimers Dis Rep. (2021) 5:887–97. doi: 10.3233/ADR-210052
63. Naert G and Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. (2011) 31:6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011
64. Rossi B, Santos-Lima B, Terrabuio E, Zenaro E, and Constantin G. Common peripheral immunity mechanisms in multiple sclerosis and alzheimer’s disease. Front Immunol. (2021) 12:639369. doi: 10.3389/fimmu.2021.639369
65. Lee WJ, Liao YC, Wang YF, Lin IF, Wang SJ, and Fuh JL. Plasma MCP-1 and cognitive decline in patients with alzheimer’s disease and mild cognitive impairment: A two-year follow-up study. Sci Rep. (2018) 8:1280. doi: 10.1038/s41598-018-19807-y
66. Zhang L, Hu K, Shao T, Hou L, Zhang S, Ye W, et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta Pharm Sin B. (2021) 11:373–93. doi: 10.1016/j.apsb.2020.08.006
67. Conti E, Grana D, Angiulli F, Karantzoulis A, Villa C, Combi R, et al. TSPO modulates oligomeric amyloid-β-induced monocyte chemotaxis: relevance for neuroinflammation in alzheimer’s disease. J Alzheimers Dis. (2023) 95:549–59. doi: 10.3233/JAD-230239
68. Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. (2005) 7:221–32. doi: 10.3233/JAD-2005-7304
69. Chen SH, Tian DY, Shen YY, Cheng Y, Fan DY, Sun HL, et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl Psychiatry. (2020) 10:423. doi: 10.1038/s41398-020-01113-9
70. Danziger R, Fuchs DT, Koronyo Y, Rentsendorj A, Sheyn J, Hayden EY, et al. The effects of enhancing angiotensin converting enzyme in myelomonocytes on ameliorating Alzheimer’s-related disease and preserving cognition. Front Physiol. (2023) 14:1179315. doi: 10.3389/fphys.2023.1179315
71. Brezovakova V, Valachova B, Hanes J, Novak M, and Jadhav S. Dendritic cells as an alternate approach for treatment of neurodegenerative disorders. Cell Mol Neurobiol. (2018) 38:1207–14. doi: 10.1007/s10571-018-0598-1
72. Butovsky O, Kunis G, Koronyo-Hamaoui M, and Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur J Neurosci. (2007) 26:413–6. doi: 10.1111/j.1460-9568.2007.05652.x
73. Ciaramella A, Salani F, Bizzoni F, Pontieri FE, Stefani A, Pierantozzi M, et al. Blood dendritic cell frequency declines in idiopathic Parkinson’s disease and is associated with motor symptom severity. PloS One. (2013) 8:e65352. doi: 10.1371/journal.pone.0065352
74. Ciaramella A, Salani F, Bizzoni F, Orfei MD, Caltagirone C, Spalletta G, et al. Myeloid dendritic cells are decreased in peripheral blood of Alzheimer’s disease patients in association with disease progression and severity of depressive symptoms. J Neuroinflamm. (2016) 13:18. doi: 10.1186/s12974-016-0483-0
75. Ano Y, Ikado K, Uchida K, and Nakayama H. Amyloid β-induced mesenteric inflammation in an alzheimer’s disease transgenic mouse model. Curr Alzheimer Res. (2020) 17:52–9. doi: 10.2174/1567205017666200212160343
76. Wang F, Liu H, Shen X, Ao H, Moore N, Gao L, et al. Combined treatment of amyloid-β1-42 stimulated bone marrow-derived dendritic cells plus splenocytes from young mice prevents the development of Alzheimer’s disease in APPswe/PSENldE9 mice. Neurobiol Aging. (2015) 36:111–22. doi: 10.1016/j.neurobiolaging.2014.06.029
77. Qi C, Liu F, Zhang W, Han Y, Zhang N, Liu Q, et al. Alzheimer’s disease alters the transcriptomic profile of natural killer cells at single-cell resolution. Front Immunol. (2022) 13:1004885. doi: 10.3389/fimmu.2022.1004885
78. Busse M, Michler E, von Hoff F, Dobrowolny H, Hartig R, Frodl T, et al. Alterations in the peripheral immune system in dementia. J Alzheimers Dis. (2017) 58:1303–13. doi: 10.3233/JAD-161304
79. Lu Y, Li K, Hu Y, and Wang X. Expression of immune related genes and possible regulatory mechanisms in alzheimer’s disease. Front Immunol. (2021) 12:768966. doi: 10.3389/fimmu.2021.768966
80. Solerte SB, Cravello L, Ferrari E, and Fioravanti M. Overproduction of IFN-gamma and TNF-alpha from natural killer (NK) cells is associated with abnormal NK reactivity and cognitive derangement in Alzheimer’s disease. Ann N Y Acad Sci. (2000) 917:331–40. doi: 10.1111/j.1749-6632.2000.tb05399.x
81. Zhang Y, Fung ITH, Sankar P, Chen X, Robison LS, Ye L, et al. Depletion of NK cells improves cognitive function in the alzheimer disease mouse model. J Immunol. (2020) 205:502–10. doi: 10.4049/jimmunol.2000037
82. Le Page A, Bourgade K, Lamoureux J, Frost E, Pawelec G, Larbi A, et al. NK Cells are Activated in Amnestic Mild Cognitive Impairment but not in Mild Alzheimer’s Disease Patients. J Alzheimers Dis. (2015) 46:93–107. doi: 10.3233/JAD-143054
83. Netzahualcoyotzi C, Santillán-Cigales JJ, Adalid-Peralta LV, and Velasco I. Infiltration of immune cells to the brain and its relation to the pathogenesis of Alzheimer’s and Parkinson’s diseases. J Neurochem. (2024) 168:2316–34. doi: 10.1111/jnc.v168.9
84. Solana C, Tarazona R, and Solana R. Immunosenescence of natural killer cells, inflammation, and alzheimer’s disease. Int J Alzheimers Dis. (2018) 2018:3128758. doi: 10.1155/2018/3128758
85. Zhang J, He H, Qiao Y, Zhou T, He H, Yi S, et al. Priming of microglia with IFN-γ impairs adult hippocampal neurogenesis and leads to depression-like behaviors and cognitive defects. Glia. (2020) 68:2674–92. doi: 10.1002/glia.v68.12
86. Infante-Duarte C, Weber A, Krätzschmar J, Prozorovski T, Pikol S, Hamann I, et al. Frequency of blood CX3CR1-positive natural killer cells correlates with disease activity in multiple sclerosis patients. FASEB J. (2005) 19:1902–4. doi: 10.1096/fj.05-3832fje
87. Salminen A. Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (Berl). (2021) 99:1553–69. doi: 10.1007/s00109-021-02123-w
88. Tamberi L, Belloni A, Pugnaloni A, Rippo MR, Olivieri F, Procopio AD, et al. The influence of myeloid-derived suppressor cell expansion in neuroinflammation and neurodegenerative diseases. Cells. (2024) 13:643. doi: 10.3390/cells13070643
89. Le Page A, Garneau H, Dupuis G, Frost EH, Larbi A, Witkowski JM, et al. Differential phenotypes of myeloid-derived suppressor and T regulatory cells and cytokine levels in amnestic mild cognitive impairment subjects compared to mild alzheimer diseased patients. Front Immunol. (2017) 8:783. doi: 10.3389/fimmu.2017.00783
90. Salminen A, Kaarniranta K, and Kauppinen A. The potential importance of myeloid-derived suppressor cells (MDSCs) in the pathogenesis of Alzheimer’s disease. Cell Mol Life Sci. (2018) 75:3099–120. doi: 10.1007/s00018-018-2844-6
91. Cheng X, Chi L, Lin T, Liang F, Pei Z, Sun J, et al. Exogenous monocyte myeloid-derived suppressor cells ameliorate immune imbalance, neuroinflammation and cognitive impairment in 5xFAD mice infected with Porphyromonas gingivalis. J Neuroinflamm. (2023) 20:55. doi: 10.1186/s12974-023-02743-8
92. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
93. Popp J, Bacher M, Kölsch H, Noelker C, Deuster O, Dodel R, et al. Macrophage migration inhibitory factor in mild cognitive impairment and Alzheimer’s disease. J Psychiatr Res. (2009) 43:749–53. doi: 10.1016/j.jpsychires.2008.10.006
94. Tatulian SA. Challenges and hopes for Alzheimer’s disease. Drug Discov Today. (2022) 27:1027–43. doi: 10.1016/j.drudis.2022.01.016
95. Jin Y, Du Q, Song M, Kang R, Zhou J, Zhang H, et al. Amyloid-β-targeting immunotherapies for Alzheimer’s disease. J Control Release. (2024) 375:346–65. doi: 10.1016/j.jconrel.2024.09.012
96. Jucker M and Walker LC. Alzheimer’s disease: From immunotherapy to immunoprevention. Cell. (2023) 186:4260–70. doi: 10.1016/j.cell.2023.08.021
97. Shi M, Chu F, Zhu F, and Zhu J. Impact of anti-amyloid-β Monoclonal antibodies on the pathology and clinical profile of alzheimer’s disease: A focus on aducanumab and lecanemab. Front Aging Neurosci. (2022) 14:870517. doi: 10.3389/fnagi.2022.870517
98. Ayalon G, Lee SH, Adolfsson O, Foo-Atkins C, Atwal JK, Blendstrup M, et al. Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer’s disease. Sci Transl Med. (2021) 13:eabb2639. doi: 10.1126/scitranslmed.abb2639
99. Slomski A. Anti-tau antibody semorinemab fails to slow alzheimer disease. Jama. (2022) 328:415. doi: 10.1001/jama.2022.12727
Keywords: Alzheimer’s disease, peripheral innate immunity, neuroinflammation, trained immunity, genetic polymorphisms, myeloid-derived suppressor cells (MDSCs)
Citation: Cao Y, Tang K, Ma P, Zhang R, Yang Y, Li T, Zhang Y and Peng X (2025) The role of peripheral innate immune cells in Alzheimer’s disease progression. Front. Immunol. 16:1616939. doi: 10.3389/fimmu.2025.1616939
Received: 24 April 2025; Accepted: 01 July 2025;
Published: 16 July 2025.
Edited by:
Claudia Gonzalez-Espinosa, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Weronika Bielska, Medical University of Lodz, PolandCopyright © 2025 Cao, Tang, Ma, Zhang, Yang, Li, Zhang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoming Peng, NjIzMTA0NzcxQHFxLmNvbQ==
†These authors have contributed equally to this work