 Manlio Vinciguerra*
Manlio Vinciguerra* Desislava K. Tsoneva
Desislava K. Tsoneva- Department of Translational Stem Cell Biology, Research Institute, Medical University Varna, Varna, Bulgaria
Histone variants are specialized isoforms of histone proteins that play crucial roles in regulating chromatin structure and function, influencing transcription, DNA repair, and cell cycle progression. Their dynamic incorporation into nucleosomes impacts gene expression and cellular identity, particularly in the context of inflammation during cell dedifferentiation and transdifferentiation. This mini-review provides a comprehensive overview of the role of histone variants in these processes, highlighting their significance in modulating inflammatory responses and cellular plasticity. We discuss mechanisms by which histone variants influence chromatin architecture and gene regulation, the interplay between histone variants and inflammatory pathways, and the specific roles of key histone variants such as H3.3, H2A.Z, and MacroH2A in dedifferentiation and transdifferentiation. Additionally, we explore the potential therapeutic implications of targeting histone variants to modulate inflammation and cellular plasticity in diseases like cancer and chronic inflammatory conditions. By summarizing existing knowledge and identifying gaps in understanding, this review underscores the importance of histone variants in inflammation-related cell plasticity and suggests future research directions further elucidating their roles and therapeutic potential.
Introduction
Histone variants are isoforms of the linker and core histone proteins that differ from canonical histones in their amino acid sequences and post-translational modifications (PTMs), leading to unique genomic locations and functions (1, 2). The dynamic incorporation of histone variants into nucleosomes significantly impacts nucleosome stability and creates functionally distinct chromatin domains essential for various cellular processes (2–4). In inflammation, histone variants modulate the inflammatory response by altering chromatin structure and gene expression. For example, accumulation of the histone variant H3.3 at specific loci is associated with increased transcriptional activity, contributing to inflammatory processes (5). In senescent cells, the loss of canonical histones H3 and H4 and the accumulation of selected histone variants lead to the secretion of pro-inflammatory factors, establishing a pro-inflammatory environment that drives chronic inflammation and tissue dysfunction (5). Cell dedifferentiation, the process by which specialized cells revert to a more primitive state, is closely linked to chromatin dynamics and histone variant incorporation. This involves the downregulation of key genes and the upregulation of genes typically suppressed in differentiated cells (6). Histone variants such as H3.3 and H2A.Z are associated with transcriptional regulation at active genes and can reset epigenetic states critical for dedifferentiation (7). Transdifferentiation, where one differentiated cell type converts into another, also involves histone variants. For instance, manipulating α cell-specific transcription factor Arx or ablation of Pdx1 in β cells can induce transdifferentiation of β cells into α cells and vice versa (8–10). The dynamic behavior of linker histones, such as H1 variants, influences chromatin compaction and accessibility, impacting both dedifferentiation and transdifferentiation (7). Inflammation-induced changes in histone variants and their PTMs are crucial in regulating gene expression during inflammation and cell dedifferentiation. These modifications can activate or repress gene expression, modulating inflammatory responses and contributing to the pathogenesis of inflammatory diseases (5, 11).
This mini-review explores the role of histone variants in inflammation during cell dedifferentiation and transdifferentiation. We discuss mechanisms by which histone variants influence these processes, the interplay between histone variants and inflammatory pathways, and the potential therapeutic implications of targeting histone variants in inflammation-related cellular plasticity (Figure 1). Understanding these mechanisms is vital for developing targeted therapies for diseases characterized by aberrant inflammation and cellular plasticity.
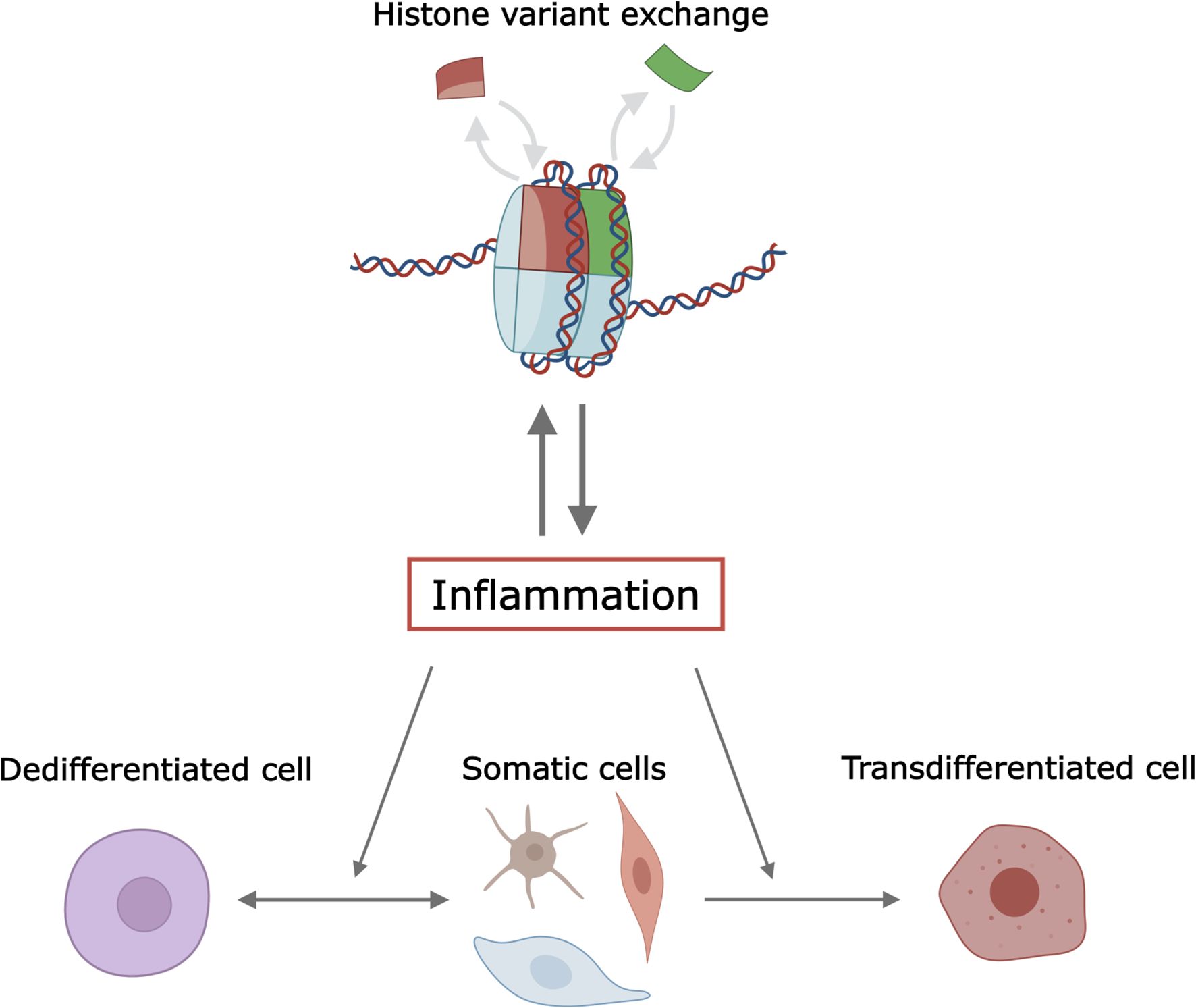
Figure 1. Illustration of the interplay between histone variants, inflammation, and cellular plasticity. The figure is created in Inkscape and partially with Biorender. 219.
Histone variants and inflammation in epigenetic remodeling
Histone variants, first described in 1977 (12), are critical chromatin components, differing from canonical histones (H1, H2A, H2B, H3, H4) in their amino acid sequences and PTMs. All canonical histones have variants, which contribute to structural and physical diversities to the nucleosome core particle (13). Since 2017, histone variants have followed a unified phylogeny-based nomenclature (14). In contrast to canonical histones being encoded by multiple genes, histone variants are usually encoded by one or a few genes, and some are produced in specific tissues. The differences from canonical histones result in unique histone variant genomic localization and functions, enhancing the complexity of chromatin architecture and enabling specialized roles in gene regulation, DNA repair, and cell cycle progression (3, 4, 15). Unlike the canonical histone mRNAs, the histone variant mRNAs are poly-adenylated, and the proteins are produced in cell cycle- and replication-independent manners. Therefore, while canonical histones are synthesized primarily during DNA replication, histone variants are expressed throughout the cell cycle and can replace canonical histones during various biological processes (16). Among the core histones, the H2A family has the greatest number of known variants, exhibiting the highest sequence divergence (17), with the “short H2A variants”, which lack a C-terminal tail, being the most divergent (5). The variants of H2A, primarily H2A.X, H2A.Z, and macroH2A, are well-established participants in the genome integrity protection (18). H3 variants, including H3.3, centromeric H3 variant (cenH3, also called CENPA in humans) play significant roles in active transcription and regulatory regions (19). Histone H2B and H4 are among the slowest evolving proteins with functional variants often tissue-restricted, such as the testis, or species-restricted, such as Trypanosoma for H2B and H4 variants, respectively (20, 21). Although less studied, histone H1 variants also contribute to chromatin structure and gene regulation (22).
Inflammation is a first responder against injury and infection and is also critical for the regeneration and repair of tissue after injury. Immune and non-immune cells directly respond to the local inflammatory cues and can undergo phenotypic and differentiation potential modifications (i.e. cell differentiation) or even cell lineage transitions (i.e. transdifferentiation) in physiological and pathological settings (see next section). In this respect, histone variants create unique chromatin states that influence transcriptional regulation (23) and therefore, cellular processes involving gene expression switch such as embryonic development, stem cell lineage commitment, somatic cell reprogramming, inflammatory signaling, and aging (24–26).
Specific examples include the histone variants H2A.J, H2AX, and macroH2A. H2A.J was shown to accumulate in the chromatin of senescent cells with persistent DNA damage caused by replication stress, phosphorylated H2AX (γ-H2AX)-marked double-strand DNA breaks, or RAS oncogene (27). Downregulation of H2A.J impedes inflammatory gene expression, including the expression of senescent-associated secretory proteins (SASP), while overexpression induces the expression of those genes (27). γ-H2AX is a well-established marker of double-strand DNA breaks, connecting DNA damage and chronic inflammation. Region-unspecific H2AX phosphorylation was observed following viral DNA replication onset, but not following nonreplicating virus infection (28). MacroH2A is a unique histone variant of H2A with its nonhistone region (NHR) contributing to promoter-specific repression of gene expression due to its large size (29). Such macroH2A-mediated gene expression inhibition is reflected in inflammatory signaling modulation. MacroH2A histones hinder the chromatin remodeling capacity of the SWI/SNF complex on macroH2A-containing nucleosomes and block specific NF-κB binding to nucleosomes (29), causing deregulated inflammatory responses by repressing expression of several pro-inflammatory genes and activation of inflammatory T cells (30). Conversely, NAP1 chaperone-mediated incorporation of H2A.Z in the nucleosome elicits a proinflammatory response by nucleosome remodeling at the TNFα promoter (31).
Cell differentiation and transdifferentiation: role for histone variants
Cell dedifferentiation involves the loss of specialized cellular identity and regression to a less differentiated state. It is a transient process by which cells become less specialized and return to an earlier cell state within the same lineage. Dedifferentiation implies an increase in cell potency, meaning that, following dedifferentiation, a cell may possess the ability to re-differentiate into more cell types than it did before dedifferentiation. On the other hand, transdifferentiation, also known as lineage reprogramming, is an uncommon process in which one mature somatic cell is transformed into another mature somatic cell without undergoing an intermediate pluripotent state or progenitor cell type (32, 33). Transdifferentiation is a type of metaplasia, which includes all cell fate switches, including the interconversion of stem cells. While several examples of transdifferentiation in invertebrates and in amphibians have been reported (34), the best example in humans and mice is the spontaneous fate switch of pancreatic α-cells into β-cells, which has been demonstrated for both healthy and diabetic pancreatic islets (35).
The dedifferentiation process is characterized by several mechanisms: 1. reduction in expression of lineage-specific genes, including essential transcription factors (TFs) and metabolic genes. In this respect, Hikichi et al. have shown that, of the large number of TFs expressed in a neural-lineage cell line, only a subset of TFs, when overexpressed, strongly interfered with the dedifferentiation triggered by the procedure to generate human induced pluripotent stem cells (iPSCs) (36). Among these TFs, ZBTB12 is a prominent molecular barrier to dedifferentiation in human iPSCs (37). Another example is shown by the dedifferentiation induction of β cells dedifferentiation following the deletion of the TF IRE1α, which results in the prevention of type 1 diabetes (38); 2. Activation of genes suppressed in normal differentiated cells and progenitor cell genes: almost any differentiated cell, in health and disease, can be returned to its pluripotent state by activating specific signaling pathways or by expressing the appropriate transcription factors (39). This can occur, for instance, through somatic reprogramming using Yamanaka factors (40), or by activation of the Wnt/β-catenin signaling pathway in epidermal cells (41) and endothelial cells (42) for regeneration; 3. Inflammation, oxidative stress, ER stress, and hypoxia contribute to dedifferentiation. IL-1β, IL-6, and TNFα have been shown to promote β-cell dedifferentiation in cultured human and mouse islets, with IL-1β being the most potent one of them (43, 44). Inflammation can also induce cancer dedifferentiation, as in metastatic melanoma (45). Oxidative and ER stresses reduce the functionality of endocrine cells by stimulating their de-/trans-differentiation through the loss of transcription factors critical for cell development, maturity, and regeneration, as observed in β-cells and thyrocytes (46, 47). As for hypoxia, a long-term hypoxic state maintains dedifferentiation in fetal cardiovascular progenitor cells through the Wnt/β-catenin signaling pathway (48), and favors transdifferentiation in a variety of cell types, including epithelial-to-mesenchymal (49), renal tubular cells into myofibroblasts (50), and pulmonary arterial endothelial cells into smooth muscle cells (51).
Histone variants significantly influence dedifferentiation and transdifferentiation by altering chromatin dynamics and gene expression. Incorporation of variants like H3.3 and H2A.Z at active genes is associated with transcriptional activation and resetting of epigenetic states vital for dedifferentiation and transdifferentiation (52–54). The histone variant H2A.Z is noteworthy for marking the 5’ ends of both active and inactive genes in euchromatin, influencing gene expression patterns during inflammation and cellular reprogramming (55) by enhancing the access of transcription factors to genes important for pluripotency (56). In response to vascular injury such as a myocardial infarction, vascular smooth muscle cells (VSMC) in the neointima undergo phenotypic switching from a differentiated to a dedifferentiated state, characterized by a significant reduction in contractile gene expression (57). H2A.Z occupies genomic regions near VSMC marker genes, and its occupancy is decreased in VSMCs undergoing dedifferentiation (58); its in vivo overexpression rescues injury-induced loss of VSMC and phenotypic switching from a dedifferentiated to a differentiated state, characterized by a significant reduction in contractile gene expression (57). H2A.Z occupies genomic regions near VSMC marker genes, and its occupancy is decreased in VSMCs undergoing dedifferentiation (58); its in vivo overexpression rescues injury-induced loss of VSMC and neointima formation (58). Histone variant H3.3B has also been implicated in the phenotypic transition of VSMCs, as well as in vascular inflammation in aortic dissection (59). Knock-down of H3.3 at the early stage of transdifferentiation - induced by forced expression of Scl, Lmo2, Runx1, and Bmi1 - gave rise to greater induced hematopoietic progenitor cells derived from mouse embryonic fibroblasts (54). The levels of histone variant H1.0 correlate with tumor cell differentiation and patient survival. Silencing H1.0 favors self-renewal, indicating that H1.0 helps maintain the differentiated state and may contribute to dedifferentiation (60). Linker histone B4, a variant of H1, is enriched at promoters of reactivated pluripotency genes and is required for pluripotency gene reactivation in oocytes (61), and transdifferentiation of pigmented epithelial cells in vivo (62).
MacroH2A histone variants, dedifferentiation and inflammation
MacroH2A histone variants represent a useful proof-of-concept to illustrate the role of histone variants at the crossroad between dedifferentiation and inflammation. Its above-mentioned NHR domain protrudes from the compact structure of the nucleosome, likely affecting the function and organization of the surrounding chromatin (63). Variants of the macroH2A family, like macroH2A1 (coded by H2AFY gene) and macroH2A2 (coded by H2AFY2 gene) regulate gene expression important for differentiation, stem cell reprogramming and tumor suppression. They can inhibit reprogramming by maintaining repressive chromatin states (60, 64–66). In fact, macroH2A’s role as a barrier to cellular plasticity might predict a possible role in preventing cancer, which can be considered a state of dedifferentiation (67–70). The levels of both macroH2A1 (and its exon splicing variants macroH2A1.1 and macroH2A1.2) and macroH2A.2 are strongly predictive of survival from numerous cancer types (71–81). MacroH2A-depleted low-malignancy melanoma exhibited enhanced tumor growth, cell motility, and metastasis, whereas its overexpression in highly malignant cells had the opposite effects (72). Moreover, in macroH2A-depleted melanoma, single-cell and spatial transcriptomics identified increased dedifferentiation in the tumor compartment, accumulation of cancer-associated fibroblasts and immunosuppressive monocytes, and depletion of functional cytotoxic T cells (82), resulting in tumor-infiltrating immune cell dysfunction and accelerated tumor growth and invasiveness.
Histone variants, inflammation, and cell plasticity
It has been established that inflammation, an integral part of the regenerative response that entails cytokine production by innate and adaptive immune cells, can trigger cell dedifferentiation or transdifferentiation (83–86). In turn, inflammatory cytokines such as TNF-α and IL-6 can modulate the expression and incorporation of histone variants, altering chromatin accessibility and transcriptional activity (60). MacroH2A histones mediate gene expression in response to pro-inflammatory signals (87–90). In the macroH2A-depleted melanoma model, macroH2A-deficient cancer-associated fibroblasts display increased myeloid chemoattractant activity as a consequence of hyperinducible expression of inflammatory genes, which was enforced by increased chromatin looping of their promoters to enhancers that gain H3K27ac (82). In high-fat diet-induced obesity models, macroH2A1 isoforms regulate metabolic health and tissue inflammation (91, 92). At the genomic level, macroH2A1.2 and macroH2A2 regulate the inflammatory-induced transcriptional response of cancer cells by affecting enhancer-promoter contacts, therefore, modulating enhancer activity and sensitivity to inflammatory cytokines (93). Thanos and coworkers’ earlier work has shown that silencing macroH2A regulates inflammatory cytokine IL-8 transcription (87). Other histone variants-related mechanisms interact too with the inflammatory process: for instance, H2A.Z nucleosomes at type I interferon (IFN)-stimulated gene (ISG) promoters modulate the biological response to IFN (94); H3.3 phosphorylation amplifies inflammatory stimulation-induced transcription (95) and induces proatherogenic gene expression (96); loss of the H3.3 chaperone DAXX in hematopoietic precursors leads to inflammation (97). The mRNA expression levels of both H2A.Z and H3.3 were found elevated in peripheral blood mononuclear (PBMC) cells of patients with rheumatoid arthritis (98). H3.3 has been identified as a crucial epigenetic regulator of the innate immune system. Following inflammatory stimuli, H3.3 is phosphorylated at its S31 site, promoting gene transcription at activated genes (95, 99). H3.3 has also been shown to exhibit a silencing function on viral genomes through H3.3-dependent chromatinization (100–102), therefore, modulating anti-viral immunity. Deletion of H3.3 in hematopoietic stem progenitor cells (HSPCs) results in a loss of adult homeostatic hematopoiesis, myeloid lineage bias, and premature HSC exhaustion (103). The complex interplay between histone variants and inflammation affects cellular plasticity, contributing to disease progression. Moreover, the relationship with inflammation appears to be bidirectional. It is well established that very high circulating levels of histones, including histone variants, induce systemic inflammation, sepsis-like symptoms, and multi-organ injury in animal models and in patients (104–107). Intracellularly, histone variants regulate inflammatory responses by influencing pro-inflammatory gene expression, while inflammation alters histone variant expression and incorporation, impacting chromatin dynamics. Understanding this interplay is crucial for elucidating the mechanisms underlying dedifferentiation and transdifferentiation in inflammatory contexts.
Discussion
Histone variants play a pivotal role in modulating inflammation during cell dedifferentiation and transdifferentiation by influencing chromatin structure and gene expression (Figure 1). Key variants such as H3.3, H2A.Z, and macroH2A are crucial in altering chromatin accessibility and transcriptional regulation, thereby modulating cellular plasticity. These insights present potential therapeutic avenues, including targeting specific histone variants to control inflammation and cellular reprogramming in diseases like cancer and diabetes. Despite these advancements, challenges persist in understanding the complex interplay between histone variants and other chromatin modifiers, as well as their context-dependent effects. Future research should aim to elucidate the precise mechanisms by which histone variants influence inflammation and cellular plasticity, utilizing advanced techniques like single-cell sequencing and CRISPR-based epigenome editing. Non-coding RNAs, such as lncRNAs and miRNA, are key players in the inflammatory response (108) and in cell reprogramming (109), respectively. The functional interactions between histone variants and non-coding RNAs remain mostly unexplored. Examples of functional interactions between non-coding RNA and histone variants include the conditional deletion of lncRNA Xist that disrupts histone macroH2A localization during X chromosome inactivation (110), and the lncRNA LHX1-DT-dependent regulation of cardiomyocyte differentiation through H2A.Z (111). Addressing systematically these epigenetic cross-talks and regulatory mechanisms will enhance our ability to develop targeted therapies that leverage the regulatory potential of histone variants in inflammation-related cellular plasticity processes.
Author contributions
MV: Writing – review & editing, Writing – original draft, Conceptualization. DT: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors acknowledge support from the Project 856871-TRANSTEM/European Commission Horizon 2020 Framework Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnaudo AM, Molden RC, and Garcia BA. Revealing histone variant induced changes via quantitative proteomics. Crit Rev Biochem Mol Biol. (2011) 46:284–94. doi: 10.3109/10409238.2011.577052
2. Buschbeck M and Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. (2017) 18:299–314. doi: 10.1038/nrm.2016.166
3. Biterge B and Schneider R. Histone variants: key players of chromatin. Cell Tissue Res. (2014) 356:457–66. doi: 10.1007/s00441-014-1862-4
4. Yin X, Zeng D, Liao Y, Tang C, and Li Y. The function of H2A histone variants and their roles in diseases. Biomolecules. (2024) 14:993. doi: 10.3390/biom14080993
5. Tan SYX, Zhang J, and Tee WW. Epigenetic regulation of inflammatory signaling and inflammation-induced cancer. Front Cell Dev Biol. (2022) 10:931493. doi: 10.3389/fcell.2022.931493
6. Bensellam M, Jonas JC, and Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol. (2018) 236:R109–43. doi: 10.1530/JOE-17-0516
7. Henikoff S and Smith MM. Histone variants and epigenetics. Cold Spring Harb Perspect Biol. (2015) 7:a019364. doi: 10.1101/cshperspect.a019364
8. Matsuoka Ta, Kawashima S, Miyatsuka T, Sasaki S, Shimo N, Katakami N, et al. Mafa enables Pdx1 to effectively convert pancreatic islet progenitors and committed islet α-cells into β-cells in vivo. Diabetes. (2017) 66:1293–300. doi: 10.2337/db16-0887
9. Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, et al. Converting adult pancreatic islet α Cells into β Cells by targeting both Dnmt1 and Arx. Cell Metab. (2017) 25:622–34. doi: 10.1016/j.cmet.2017.01.009
10. Moin ASM and Butler AE. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr Diabetes Rep. (2019) 19:83. doi: 10.1007/s11892-019-1194-6
11. Lin Y, Qiu T, Wei G, Que Y, Wang W, Kong Y, et al. Role of histone post-translational modifications in inflammatory diseases. Front Immunol. (2022) 13:852272. doi: 10.3389/fimmu.2022.852272
12. Franklin SG and Zweidler A. Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature. (1977) 266:273–5. doi: 10.1038/266273a0
13. Kurumizaka H, Kujirai T, and Takizawa Y. Contributions of histone variants in nucleosome structure and function. J Mol Biol. (2021) 433:166678. doi: 10.1016/j.jmb.2020.10.012
14. El Kennani S, Adrait A, Shaytan AK, Khochbin S, Bruley C, Panchenko AR, et al. MS_HistoneDB, a manually curated resource for proteomic analysis of human and mouse histones. Epigenet Chromatin. (2017) 10:2. doi: 10.1186/s13072-016-0109-x
15. Ragusa D and Vagnarelli P. Contribution of histone variants to aneuploidy: a cancer perspective. Front Genet. (2023) 14:1290903. doi: 10.3389/fgene.2023.1290903
16. Foroozani M, Holder DH, and Deal RB. Histone variants in the specialization of plant chromatin. Annu Rev Plant Biol. (2022) 73:149–72. doi: 10.1146/annurev-arplant-070221-050044
17. Jiang X, Soboleva TA, and Tremethick DJ. Short Histone H2A Variants: Small in Stature but not in Function. Cells. (2020) 9:867. doi: 10.3390/cells9040867
18. Oberdoerffer P and Miller KM. Histone H2A variants: Diversifying chromatin to ensure genome integrity. Semin Cell Dev Biol. (2023) 135:59–72. doi: 10.1016/j.semcdb.2022.03.011
19. Shi L, Wen H, and Shi X. The histone variant H3.3 in transcriptional regulation and human disease. J Mol Biol. (2017) 429:1934–45. doi: 10.1016/j.jmb.2016.11.019
20. Talbert PB, Ahmad K, Almouzni G, Ausió J, Berger F, Bhalla PL, et al. A unified phylogeny-based nomenclature for histone variants. Epigenet Chromatin. (2012) 5:7. doi: 10.1186/1756-8935-5-7
21. Draizen EJ, Shaytan AK, Mariño-Ramírez L, Talbert PB, Landsman D, and Panchenko AR. HistoneDB 2.0: a histone database with variants—an integrated resource to explore histones and their variants. Database. (2016) 2016:baw014. doi: 10.1093/database/baw014
22. Cheema M and Ausió J. The structural determinants behind the epigenetic role of histone variants. Genes. (2015) 6:685–713. doi: 10.3390/genes6030685
23. Weber CM and Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. (2014) 28:672–82. doi: 10.1101/gad.238873.114
24. Lo Re O and Vinciguerra M. Histone macroH2A1: A chromatin point of intersection between fasting, senescence and cellular regeneration. Genes. (2017) 8:367. doi: 10.3390/genes8120367
25. Zheng ZH, Sam TW, Zeng Y, Chu JJH, and Loh YH. Chromatin regulation in development: current understanding and approaches. Stem Cells Int. (2021) 2021:1–12. doi: 10.1155/2021/8817581
26. Cohen LRZ and Meshorer E. The many faces of H3.3 in regulating chromatin in embryonic stem cells and beyond. Trends Cell Biol. (2024) 34:1044–55. doi: 10.1016/j.tcb.2024.03.003
27. Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux PF, Bischof O, et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun. (2017) 8:14995. doi: 10.1038/ncomms14995
28. Nichols GJ, Schaack J, and Ornelles DA. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J Virol. (2009) 83:5987–98. doi: 10.1128/JVI.00091-09
29. Angelov D, Molla A, Perche PY, Hans F, Côté J, Khochbin S, et al. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol Cell. (2003) 11:1033–41. doi: 10.1016/S1097-2765(03)00100-X
30. Liu T, Zhang L, Joo D, and Sun SC. NF-κB signaling in inflammation. Sig Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
31. El Gazzar M, Liu T, Yoza BK, and McCall CE. Dynamic and selective nucleosome repositioning during endotoxin tolerance. J Biol Chem. (2010) 285:1259–71. doi: 10.1074/jbc.M109.067330
32. Orkin SH and Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. (2008) 132:631–44. doi: 10.1016/j.cell.2008.01.025
33. Graf T and Enver T. Forcing cells to change lineages. Nature. (2009) 462:587–94. doi: 10.1038/nature08533
34. Shen CN, Burke ZD, and Tosh D. Transdifferentiation, metaplasia and tissue regeneration. Organogenesis. (2004) 1:36–44. doi: 10.4161/org.1.2.1409
35. Van Der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dólleman S, et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. (2017) 25:911–26.e6. doi: 10.1016/j.cmet.2017.03.017
36. Hikichi T, Matoba R, Ikeda T, Watanabe A, Yamamoto T, Yoshitake S, et al. Transcription factors interfering with dedifferentiation induce cell type-specific transcriptional profiles. Proc Natl Acad Sci USA. (2013) 110:6412–7. doi: 10.1073/pnas.1220200110
37. Han D, Liu G, Oh Y, Oh S, Yang S, Mandjikian L, et al. ZBTB12 is a molecular barrier to dedifferentiation in human pluripotent stem cells. Nat Commun. (2023) 14:632. doi: 10.1038/s41467-023-36178-9
38. Lee H, Lee YS, Harenda Q, Pietrzak S, Oktay HZ, Schreiber S, et al. Beta cell dedifferentiation induced by IRE1α Deletion prevents type 1 diabetes. Cell Metab. (2020) 31:822–36.e5. doi: 10.1016/j.cmet.2020.03.002
39. Yao Y and Wang C. Dedifferentiation: inspiration for devising engineering strategies for regenerative medicine. NPJ Regener Med. (2020) 5:14. doi: 10.1038/s41536-020-00099-8
40. Yamanaka S. Pluripotent stem cell-based cell therapy—Promise and challenges. Cell Stem Cell. (2020) 27:523–31. doi: 10.1016/j.stem.2020.09.014
41. Zhang C, Chen P, Fei Y, Liu B, Ma K, Fu X, et al. Wnt/β-catenin signaling is critical for dedifferentiation of aged epidermal cells in vivo and in vitro. Aging Cell. (2012) 11:14–23. doi: 10.1111/j.1474-9726.2011.00753.x
42. Kohler EE, Baruah J, Urao N, Ushio-Fukai M, Fukai T, Chatterjeeet I, et al. Low-dose 6-bromoindirubin-3′-oxime induces partial dedifferentiation of endothelial cells to promote increased neovascularization. Stem Cells. (2014) 32:1538–52. doi: 10.1002/stem.1658
43. Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, Barbieux C, et al. The role of inflammation in β-cell dedifferentiation. Sci Rep. (2017) 7:6285. doi: 10.1038/s41598-017-06731-w
44. Liang R, Liu N, Wang G, Sun P, Liu Y, Zou J, et al. Cytohistologic analyses of β cell dedifferentiation induced by inflammation in human islets. Eur J Inflamm. (2021) 19:20587392211014416. doi: 10.1177/20587392211014416
45. Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B, et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. (2018) 8:935–43. doi: 10.1158/2159-8290.CD-17-1178
46. Ulianich L, Mirra P, Garbi C, Calì G, Conza D, Treglia A S, et al. The pervasive effects of ER stress on a typical endocrine cell: dedifferentiation, mesenchymal shift and antioxidant response in the thyrocyte. Front Endocrinol. (2020) 11:588685. doi: 10.3389/fendo.2020.588685
47. Dinić S, Arambašić Jovanović J, Uskoković A, Uskoković A, Mihailović M, Grdović N, et al. Oxidative stress-mediated beta cell death and dysfunction as a target for diabetes management. Front Endocrinol. (2022) 13:1006376. doi: 10.3389/fendo.2022.1006376
48. Knox C, Camberos V, Ceja L, Monteon A, Hughes L, Longo L, et al. Long-term hypoxia maintains a state of dedifferentiation and enhanced stemness in fetal cardiovascular progenitor cells. IJMS. (2021) 22:9382. doi: 10.3390/ijms22179382
49. Rana MK, Srivastava J, Yang M, Chen CS, and Barber DL. Hypoxia increases extracellular fibronectin abundance but not assembly during epithelial cell transdifferentiation. J Cell Sci. (2015) 128(6):1083–9. doi: 10.1242/jcs.155036
50. Manotham K, Tanaka T, Matsumoto M, et al. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. (2004) 65:871–80. doi: 10.1111/j.1523-1755.2004.00461.x
51. Wang J, Yan G, Guo H, Zhu Y, Shui X, He Y, et al. ITE promotes hypoxia-induced transdifferentiation of human pulmonary arterial endothelial cells possibly by activating transforming growth factor-β/Smads and MAPK/ERK pathways. J Cell Biochem. (2019) 120:19567–77. doi: 10.1002/jcb.29264
52. Arenas-Mena C. Indirect development, transdifferentiation and the macroregulatory evolution of metazoans. Phil Trans R Soc B. (2010) 365:653–69. doi: 10.1098/rstb.2009.0253
53. Santoro SW and Dulac C. Histone variants and cellular plasticity. Trends Genet. (2015) 31:516–27. doi: 10.1016/j.tig.2015.07.005
54. Fang HT, El Farran CA, Xing QR, Zhang LF, Li H, Lim B, et al. Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat Commun. (2018) 9:1537. doi: 10.1038/s41467-018-03904-7
55. Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, et al. Histone variant H2A.Z marks the 5′ Ends of both active and inactive genes in euchromatin. Cell. (2005) 123:233–48. doi: 10.1016/j.cell.2005.10.002
56. Dong F, Song Z, Yu J, Zhang B, Jiang BC, Shen Y, et al. Dynamic changes in occupancy of histone variant H2A.Z during induced somatic cell reprogramming. Stem Cells Int. (2016) 2016:3162363. doi: 10.1155/2016/3162363
57. Bennett MR, Sinha S, and Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
58. Yao F, Yu P, Li Y, Yuan X, Li Z, Zhang T, et al. Histone variant H2A.Z is required for the maintenance of smooth muscle cell identity as revealed by single-cell transcriptomics. Circulation. (2018) 138:2274–88. doi: 10.1161/CIRCULATIONAHA.117.033114
59. Zhang X, Che Y, Mao L, Li D, Deng J, Guo Y, et al. H3.3B controls aortic dissection progression by regulating vascular smooth muscle cells phenotypic transition and vascular inflammation. Genomics. (2023) 115:110685. doi: 10.1016/j.ygeno.2023.110685
60. Talbert PB and Henikoff S. Histone variants at a glance. J Cell Sci. (2021) 134:jcs244749. doi: 10.1242/jcs.244749
61. Jullien J, Åstrand C, Halley-Stott RP, Garrett N, and Gurdon JB. Characterization of somatic cell nuclear reprogramming by oocytes in which a linker histone is required for pluripotency gene reactivation. Proc Natl Acad Sci U S A. (2010) 107:5483–8. doi: 10.1073/pnas.1000599107
62. Maki N, Suetsugu-Maki R, Sano S, Nakamura K, Nishimura O, Tarui H, et al. Oocyte-type linker histone B4 is required for transdifferentiation of somatic cells in vivo. FASEB J. (2010) 24:3462–7. doi: 10.1096/fj.10-159285
63. Rivera-Casas C, Gonzalez-Romero R, Cheema MS, Ausió J, and Eirín-López JM. The characterization of macroH2A beyond vertebrates supports an ancestral origin and conserved role for histone variants in chromatin. Epigenetics. (2016) 11:415–25. doi: 10.1080/15592294.2016.1172161
64. Barrero MJ, Sese B, Martí M, and Izpisua Belmonte JC. Macro histone variants are critical for the differentiation of human pluripotent cells. J Biol Chem. (2013) 288:16110–6. doi: 10.1074/jbc.M113.466144
65. Gaspar-Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Leu NA, Leroy G, et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat Commun. (2013) 4:1565. doi: 10.1038/ncomms2582
66. Barrero MJ, Sese B, Kuebler B, Bilic J, Boue S, Martí M, et al. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. (2013) 3:1005–11. doi: 10.1016/j.celrep.2013.02.029
67. Friedmann-Morvinski D and Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. (2014) 15:244–53. doi: 10.1002/embr.201338254
68. Giallongo S, Řeháková D, Biagini T, Lo Re O, Raina P, Lochmanová G, et al. Histone Variant macroH2A1.1 Enhances Nonhomologous End Joining-dependent DNA Double-strand-break Repair and Reprogramming Efficiency of Human iPSCs. Stem Cells. (2022) 40:35–48. doi: 10.1093/stmcls/sxab004
69. Lo Re O, Mazza T, Giallongo S, Sanna P, Rappa F, Vinh Luong T, et al. Loss of histone macroH2A1 in hepatocellular carcinoma cells promotes paracrine-mediated chemoresistance and CD4+ CD25+ FoxP3+ regulatory T cells activation. Theranostics. (2020) 10:910–24. doi: 10.7150/thno.35045
70. Liorni N, Napoli A, Castellana S, Giallongo S, Řeháková D, Lo Re O, et al. Integrative CUT&Tag-RNA-seq analysis of histone variant macroH2A1-dependent orchestration of human induced pluripotent stem cell reprogramming. Epigenomics. (2023) 15:863–77. doi: 10.2217/epi-2023-0267
71. Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. (2009) 28:3423–8. doi: 10.1038/onc.2009.26
72. Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. (2010) 468:1105–9. doi: 10.1038/nature09590
73. Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, and Gamble MJ. QKI-mediated alternative splicing of the histone variant macroH2A1 regulates cancer cell proliferation. Mol Cell Biol. (2011) 31:4244–55. doi: 10.1128/MCB.05244-11
74. Rappa F, Greco A, Podrini C, Cappello F, Foti M, Bourgoin L, et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS One. (2013) 8:e54458. doi: 10.1371/journal.pone.0054458
75. Borghesan M, Fusilli C, Rappa F, Panebianco C, Rizzo G, Oben JA, et al. DNA hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. (2016) 76:594–606. doi: 10.1158/0008-5472.CAN-15-1336
76. Lo Re O, Fusilli C, Rappa F, Van Haele M, Douet J, Pindjakova J, et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology. (2018) 67:636–50. doi: 10.1002/hep.29519
77. Lo Re O, Douet J, Buschbeck M, Fusilli C, Pazienza V, Panebianco C, et al. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics. (2018) 13:829–45. doi: 10.1080/15592294.2018.1514239
78. Giallongo S, Lo Re O, and Vinciguerra M. Macro histone variants: emerging rheostats of gastrointestinal cancers. Cancers. (2019) 11:676. doi: 10.3390/cancers11050676
79. Bereshchenko O, Lo Re O, Nikulenkov F, Flamini S, Kotaskova J, Mazza T, et al. Deficiency and haploinsufficiency of histone macroH2A1.1 in mice recapitulate hematopoietic defects of human myelodysplastic syndrome. Clin Epigenet. (2019) 11:121. doi: 10.1186/s13148-019-0724-z
80. Giallongo S, Di Rosa M, Caltabiano R, Longhitano L, Reibaldi M, Distefano A, et al. Loss of macroH2A1 decreases mitochondrial metabolism and reduces the aggressiveness of uveal melanoma cells. Aging. (2020) 12:9745–60. doi: 10.18632/aging.103241
81. Buzova D, Petrilli LL, Frohlich J, Tsoneva DK, Bianco SD, Braghini MR, et al. Extracellular histones profiles of pediatric H3K27-altered diffuse midline glioma. Mol Diagn Ther. (2025) 29:129–41. doi: 10.1007/s40291-024-00754-6
82. Filipescu D, Carcamo S, Agarwal A, Tung N, Humblin E, Goldberg MS, et al. MacroH2A restricts inflammatory gene expression in melanoma cancer-associated fibroblasts by coordinating chromatin looping. Nat Cell Biol. (2023) 25:1332–45. doi: 10.1038/s41556-023-01208-7
83. Mills JC and Sansom OJ. Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal. (2015) 8:re8. doi: 10.1126/scisignal.aaa7540
84. Karin M and Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. (2016) 529:307–15. doi: 10.1038/nature17039
85. Cabillic F and Corlu A. Regulation of transdifferentiation and retrodifferentiation by inflammatory cytokines in hepatocellular carcinoma. Gastroenterology. (2016) 151:607–15. doi: 10.1053/j.gastro.2016.06.052
86. Zhang Y, Kang Z, Liu M, Wang L, and Liu F. Single-cell omics identifies inflammatory signaling as a trans-differentiation trigger in mouse embryos. Dev Cell. (2024) 59:961–78.e7. doi: 10.1016/j.devcel.2024.02.010
87. Agelopoulos M and Thanos D. Epigenetic determination of a cell-specific gene expression program by ATF-2 and the histone variant macroH2A. EMBO J. (2006) 25:4843–53. doi: 10.1038/sj.emboj.7601364
88. Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, and Kraus WL. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. (2010) 24:21–32. doi: 10.1101/gad.1876110
89. Gamble MJ and Kraus WL. Multiple facets of the unique histone variant macroH2A: From genomics to cell biology. Cell Cycle. (2010) 9:2568–74. doi: 10.4161/cc.9.13.12144
90. Hussey KM, Chen H, Yang C, Park E, Hah N, Erdjument-Bromage H, et al. The histone variant macroH2A1 regulates target gene expression in part by recruiting the transcriptional coregulator PELP1. Mol Cell Biol. (2014) 34:2437–49. doi: 10.1128/MCB.01315-13
91. Pazienza V, Panebianco C, Rappa F, Memoli D, Borghesan M, Cannito S, et al. Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenet Chromatin. (2016) 9:45. doi: 10.1186/s13072-016-0098-9
92. Chiodi V, Rappa F, Lo Re O, Chaldakov GN, Lelouvier B, Micale V, et al. Deficiency of histone variant macroH2A1.1 is associated with sexually dimorphic obesity in mice. Sci Rep. (2023) 13:19123. doi: 10.1038/s41598-023-46304-8
93. Corujo D, Malinverni R, Carrillo-Reixach J, Meers O, Garcia-Jaraquemada A, Le Pannérer MM, et al. MacroH2As regulate enhancer-promoter contacts affecting enhancer activity and sensitivity to inflammatory cytokines. Cell Rep. (2022) 39:110988. doi: 10.1016/j.celrep.2022.110988
94. Au-Yeung N and Horvath CM. Histone H2A.Z suppression of interferon-stimulated transcription and antiviral immunity is modulated by GCN5 and BRD2. iScience. (2018) 6:68–82. doi: 10.1016/j.isci.2018.07.013
95. Armache A, Yang S, Martínez De Paz A, Robbins LE, Durmaz C, Cheong JQ, et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature. (2020) 583:852–7. doi: 10.1038/s41586-020-2533-0
96. Jin YJ, Liang G, Li R, Alnouri MW, Bentsen M, Kuenne C, et al. Phosphorylation of endothelial histone H3.3 serine 31 by PKN1 links flow-induced signaling to proatherogenic gene expression. Nat Cardiovasc Res. (2025) 4:180–96. doi: 10.1038/s44161-024-00593-y
97. Gerber JP, Russ J, Chandrasekar V, Offermann N, Lee HM, Spear S, et al. Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation. Nat Cell Biol. (2021) 23:1224–39. doi: 10.1038/s41556-021-00774-y
98. Asadipour M, Hassan-Zadeh V, Aryaeian N, Shahram F, and Mahmoudi M. Histone variants expression in peripheral blood mononuclear cells of patients with rheumatoid arthritis. Int J Rheum Dis. (2018) 21:1831–7. doi: 10.1111/1756-185X.13126
99. Thorne JL, Ouboussad L, and Lefevre PF. Heterochromatin protein 1 gamma and IκB kinase alpha interdependence during tumour necrosis factor gene transcription elongation in activated macrophages. Nucleic Acids Res. (2012) 40:7676–89. doi: 10.1093/nar/gks509
100. Tsai K, Chan L, Gibeault R, Conn K, Dheekollu J, Domsic J, et al. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J Virol. (2014) 88:14350–63. doi: 10.1128/JVI.01895-14
101. Rai TS, Glass M, Cole JJ, Rather MI, Marsden M, Neilson M, et al. Histone chaperone HIRA deposits histone H3.3 onto foreign viral DNA and contributes to anti-viral intrinsic immunity. Nucleic Acids Res. (2017) 45:11673–83. doi: 10.1093/nar/gkx771
102. Cohen C, Corpet A, Roubille S, Maroui MA, Poccardi N, Rousseau A, et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog. (2018) 14:e1007313. doi: 10.1371/journal.ppat.1007313
103. Guo P, Liu Y, Geng F, Daman AW, Liu X, Zhong L, et al. Histone variant H3.3 maintains adult haematopoietic stem cell homeostasis by enforcing chromatin adaptability. Nat Cell Biol. (2022) 24:99–111. doi: 10.1038/s41556-021-00795-7
104. Kawai C, Kotani H, Miyao M, Ishida T, Jemail L, Abiruet H, et al. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am J Pathol. (2016) 186:829–43. doi: 10.1016/j.ajpath.2015.11.025
105. Rico MC, Perez-Leal O, Barbe MF, Amin M, Colussi DJ, Florez ML, et al. Extracellular acetylated histone 3.3 induces inflammation and lung tissue damage. Biomolecules. (2023) 13:1334. doi: 10.3390/biom13091334
106. Su F, Moreau A, Savi M, Salvagno M, Annoni F, Zhao L, et al. Circulating nucleosomes as a novel biomarker for sepsis: A scoping review. Biomedicines. (2024) 12:1385. doi: 10.3390/biomedicines12071385
107. Dutta S, Dutta S, Somanath PR, Narayanan SP, Wang X, and Zhang D. Circulating nucleosomes and histones in the development of lung injury and sepsis. CIMB. (2025) 47:133. doi: 10.3390/cimb47020133
108. Zhang Y, Liu H, Niu M, Wang Y, Xu R, Guo Y, et al. Roles of long noncoding RNAs in human inflammatory diseases. Cell Death Discov. (2024) 10:235. doi: 10.1038/s41420-024-02002-6
109. Luginbühl J, Sivaraman DM, and Shin JW. The essentiality of non-coding RNAs in cell reprogramming. Non-coding RNA Res. (2017) 2:74–82. doi: 10.1016/j.ncrna.2017.04.002
110. Csankovszki G, Panning B, Bates B, Pehrson JR, and Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet. (1999) 22:323–4. doi: 10.1038/11887
Keywords: histone variants, inflammation, differentiation, stem cells, epigenetics
Citation: Vinciguerra M and Tsoneva DK (2025) Histone variants: key regulators of inflammation in cell dedifferentiation and transdifferentiation. Front. Immunol. 16:1619100. doi: 10.3389/fimmu.2025.1619100
Received: 27 April 2025; Accepted: 16 June 2025;
Published: 27 June 2025.
Edited by:
Toyotaka Ishibashi, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaReviewed by:
Neha Mishra, University Medical Center Schleswig-Holstein, GermanyCopyright © 2025 Vinciguerra and Tsoneva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manlio Vinciguerra, bWFubGlvLnZpbmNpZ3VlcnJhQG11LXZhcm5hLmJn