 Zun-yue Zhang1,2†
Zun-yue Zhang1,2† Xin-feng Zhang
Xin-feng Zhang Cong-hui Xu
Cong-hui Xu Fang Huang
Fang Huang- 1School of Medicine, Yunnan University, Kunming, China
- 2Yunnan Technological Innovation Centre of Drug Addiction Medicine, Yunnan University, Kunming, China
- 3Department of Gastrointestinal and Hernia Surgery, The First Affiliated Hospital of Kunming Medical University, Kunming, China
Targeting tumor-initiating cells (TICs) in digestive system tumors is a feasible strategy to boost the effectiveness of cancer immunotherapy. Because of their stem cell-like properties, TICs can cause tumor heterogeneity, recurrence, and resistance to conventional medicines, which can seriously impair treatment outcomes. This review discusses the unique features of TICs inside various digestive system tumors, such as colorectal, pancreatic, liver, and gastric cancers. We look at the mechanisms that TICs evade immune recognition, including altered tumor microenvironment, decreased immunogenicity, and immune checkpoint molecule expression. Furthermore, we highlight potential strategies for TICs, such as differentiation therapies, inhibiting certain signaling pathways, and enhancing immune recognition through advanced immunotherapeutic approaches. The analysis also examines the potential for combination therapy, which include adoptive cell therapies, TIC-targeted strategies, and immune checkpoint inhibitors. Lastly, we address the challenges presented by TIC heterogeneity and immune escape mechanisms, emphasizing the need for more clinical research to back up these innovative tactics. All things considered, TIC targeting is a significant method to improve immunotherapy’s efficacy in treating digestive system cancers, which will ultimately help patients.
Introduction
Along with the great majority of non-tumorigenic cells, some tumors have a little number of cells that have the ability to self-renew and start new tumors. Tumor-initiating cells (TICs), also called cancer stem cells (CSCs) or CSC-like cells, are a group of cells that can produce diverse cell populations that closely mimic the original tumor’s makeup (1). TICs, a subset of cells within malignancies, may arise from alterations in progenitor or normal stem cells or from genetic defects. Because of their similar ability to differentiate into diverse lineages, TICs were formerly thought to be produced from normal stem cells. Transduction of the MLL-AF9 fusion protein into myeloid progenitor cells can generate leukemia in vivo, indicating that progenitor cells can develop into leukemic stem cells (2). Subsequent research has demonstrated that TICs can be created by genetically altering tumor endothelial cells and astrocytes to dedifferentiate (3, 4). This suggests that TICs may originate from a variety of sources, which could be a major factor in their differentiation from healthy stem cells. TICs differ from normal stem cells in their phenotypes and capabilities. TICs and normal stem cells are two distinct cell types with different characteristics and roles. Despite their similarities, they nevertheless differ significantly, particularly in terms of behavior, regulation, and their effects on health and illness (5). Normal stem cells help replace damaged or dying cells and maintain tissue homeostasis. TICs are believed to be responsible for tumor genesis, growth, and recurrence due to their stem-like properties, which leads to intratumor heterogeneity of cancer cells (6). While TICs can self-renew and manufacture more TICs and differentiated cancer cells, normal stem cells can divide and produce identical stem cells and differentiated cells. Normal stem cell division is tightly regulated by internal mechanisms to prevent excessive proliferation and maintain tissue integrity in response to specific stimuli from the milieu (the stem cell niche). However, if left untreated, TICs multiply, leading to the growth and advancement of malignancies (7). Normal stem cells frequently maintain genomic stability, have low rates of genetic mutation, and can efficiently repair DNA damage, whereas TICs directly contribute to intratumor heterogeneity (8, 9).
Whether TICs represent a distinct group of cancer cells or a functional state of some cancer cells is still up for dispute. TICs are now widely recognized as being crucial to therapeutic progress due to their ability to self-renew, resist chemotherapy, and respond to immune checkpoint inhibitors (10–12). The TIC concept has led to a reexamination of therapeutic alternatives for cancer cure. The issues of cancer recurrence and metastatic spread still require attention, even though many malignancies have been adequately remitted by a number of popular anti-cancer medicines. Most conventional medications are cytotoxic and highly non-selective because their aim is to destroy all rapidly proliferating cells. Sometimes, this approach can lead to a difference between a good clinical response (a significant reduction in tumor size) and an inadequate survival response. This could be due to TIC-driven recurrence after a significant number of cancer cells are killed without totally removing the TICs (13, 14). Digestive system tumors offer a fantastic chance to increase the efficacy of cancer immunotherapy. TIC is a cell population of extremely drug-resistant, asymmetrically dividing, tumor-initiating cells that arises after early success of tumor radiotherapy or chemotherapy. It has a strong correlation with tumor heterogeneity and is essential for clinical phenomena such as treatment resistance, tumor metastasis, tumor dormancy, and recurrence (12, 15). Digestive system tumors, such as those of the stomach, liver, pancreas, and colon, present a challenging issue since they usually exhibit resistance to standard therapies. Resistance (16–18), recurrence, and metastasis (3, 19) are believed to be influenced by the unique features of TICs, including their ability to self-renew, differentiate, and evade the immune system. Therefore, if the characteristics of TICs are well defined, the reasons for their variability and immune evasion mechanisms are investigated, and potential indicators are identified, targeting TICs may help cure digestive system cancers.
TICs in digestive system tumors
There is a subgroup of tumor cells called TICs that share traits with stem cells. Because they may self-renew and create the heterogeneity of tumor cells, they are important in tumor spread and recurrence. The TIC markers and their functions in gastrointestinal malignancies, including colorectal, stomach, liver, and pancreatic cancers, are listed in Table 1. TICs are often resistant to chemotherapy and radiation due to their quiescent nature, enhanced DNA repair processes, and ability to evade the immune system. They are therefore a crucial target for the efficacy of long-term treatment.
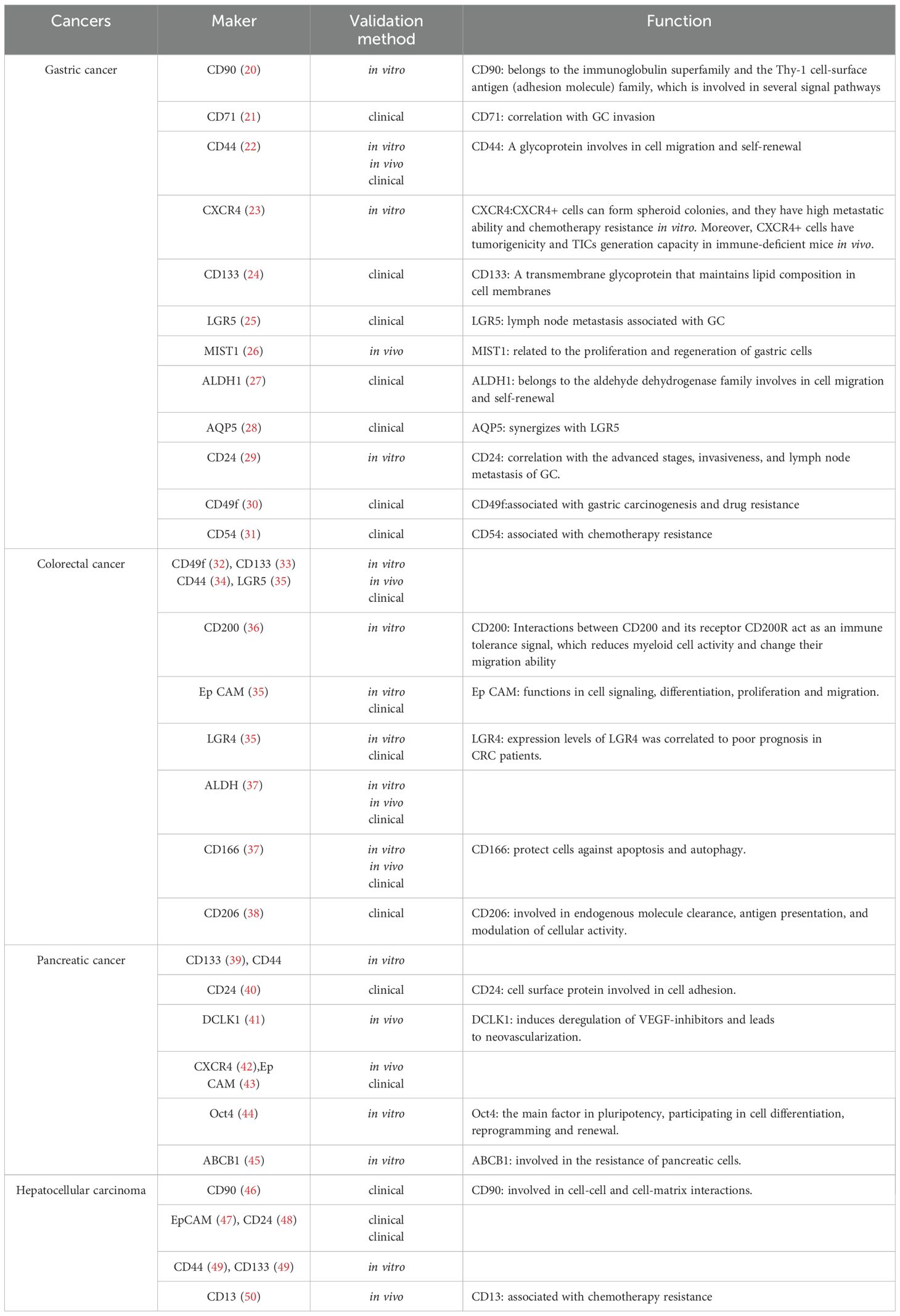
Table 1. Stem cell biomarkers and functions identified in gastrointestinal tumors.
Mechanisms of immune evasion by TICs
Immunosuppressive microenvironment
The tumor microenvironment (TME) is the primary site of conflict between tumor cells and the host immune system. Numerous studies have examined the relationship between immune modulation and cancer, specifically cancer dormancy (51). Non-cellular and cellular elements, including immune cells, cancer-associated fibroblasts, and other stromal cells, as well as secretomes and exosomes generated from these cells, comprise the TME. Together, these elements provide an environment that suppresses the immune system and promotes tumor growth. The TME is essentially a cellular environment based on tumors or TICs that significantly encourages the unchecked proliferation of tumor cells or TICs, which in turn influences the host system’s capacity to develop cancer (52). Immunotherapy stimulates the immune system to attack cancer cells in the TME. It aims to boost the activity of natural killer (NK) cells and cytotoxic CD8+ T lymphocytes (CTLs), as opposed to immunosuppressive cells such as mesenchymal-derived suppressor cells (MDSCs), regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and cancer-associated fibroblasts (CAFs) (53). The failure of cancer immunotherapy is often due to the fact that TICs can overcome anti-tumor immunity (54). TICs can evade tumor immunity by changing the TME, and these environmental changes can result in TIC phenotypic changes that facilitate tumor immune evasion (55, 56). For instance, regulatory myeloid cells, such as Tregs and MDSCs, are drawn to the TME by TIC-secreted CXCL12 (57).MDSCs, TAMs, and Tregs are the three most prevalent immunosuppressive cells in the TME. In their symbiotic connections with TICs, they interact and suppress tumor immunity. In the next sections, we shall discuss these cells’ interactions with TICs in greater detail. This article outlines the possible paths of TIC function, their upstream transcription factors, and the interaction between TICs and immunosuppressive cells in Figure 1.
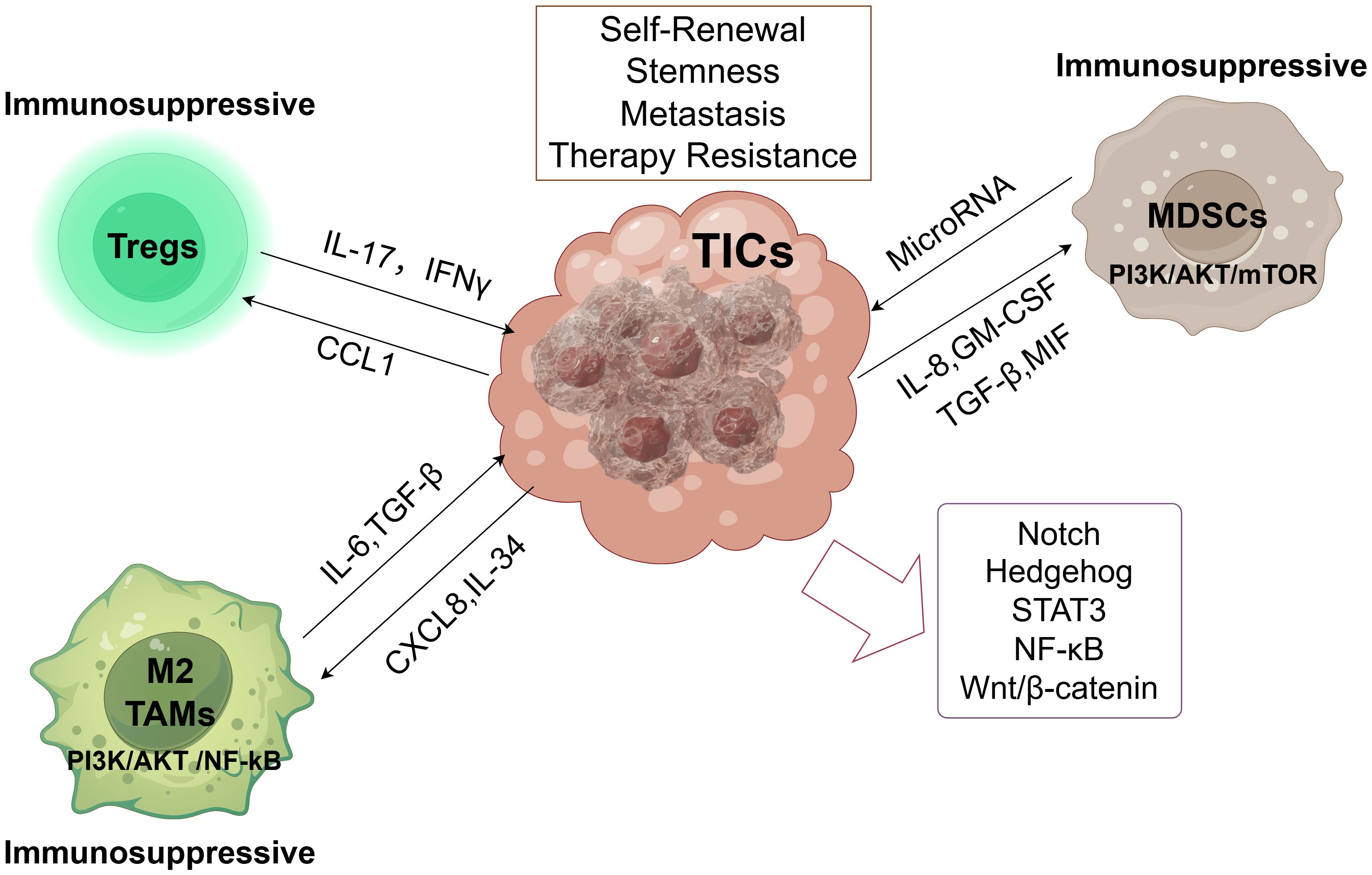
Figure 1. Tumor-initiating cells (TICs) orchestrate an immunosuppressive microenvironment. By releasing several soluble factors, TICs create an immunosuppressive environment that attracts MDSCs, TAMs, and Tregs to the tumor microenvironment. By encouraging tumor angiogenesis, chemoresistance, and tumor spreading, these immunosuppressive cells create an immunosuppressive environment. More significantly, they preserve the stemness and functionality of TICs. These immunosuppressive cells promote TIC self-renewal, stemness, metastasis, and therapy resistance, primarily through PI3K/AKT/mTOR signaling (MDSCs) and PI3K/AKT/NF-κB signaling (M2 TAMs). Key signaling pathways (Notch, Hedgehog, STAT3, NF-κB, Wnt/β-catenin) active within TICs regulate their core properties.
TICs and MDSCs
MDSCs are one of the most discussed biological entities in immunology. Despite differences in their classification and history, MDSCs are most commonly used to describe cells produced during chronic inflammation, particularly in late malignancy, and they contain T-cell immunosuppressive capabilities (58). MDSCs are composed of a heterogeneous population of myeloid cells, among which granulocytic or polymorphonuclear cells (PMN-MDSCs) and mononuclear cells (M-MDSCs) constitute subpopulations (59). PMN-MDSCs comprise over 80% of all MDSCs, whereas M-MDSCs can differentiate into TAMs (60). MDSCs are the primary organizers of inflammation associated with cancer because of their dynamic expression of several polarized inflammatory programs that promote tumor growth, including tumor angiogenesis, immunosuppression, tissue remodeling, and the maintenance of TIC stemness (61). The development of cancer is aided by MDSCs’ promotion of angiogenesis, invasion, metastasis, and cancer cell survival (58, 62). According to research, MDSCs regulate the phenotypic transition in TICs. By upregulating the expression of microRNAs (miRNAs), which are short (~22 nt) non-coding endogenous RNAs that post-transcriptionally influence gene expression in cancer cells (63), MDSCs contribute to the TIC phenotype (64). In particular, MDSCs promote TIC-associated gene expression, spheroid formation, and cancer spread while suppressing T-cell activation. MDSCs cause cancer cells to express miRNA-101. The corepressor gene C-terminal binding protein-2 (CtBP2), which specifically targets stemness core genes, is then suppressed by miRNA-101. higher tumorigenicity, higher metastatic potential, and increased stemness of cancer cells are the results of this suppression. Interestingly, poor survival is independently predicted by both increased tumor miRNA-101 expression and greater MDSC infiltration. These components function independently of one another, and a lower prognosis is also associated with decreased CtBP2 expression in malignancies (64). MDSCs can stimulate mesenchymal development and TIC proliferation (65). The method through which MDSCs promote TIC stemness depends on STAT3. Reprogramming brought on by STAT3 phosphorylation can give monocytes a pro-tumor immunosuppressive phenotype. By blocking STAT3, MDSC’s ability to produce TICs in vitro can be completely eradicated (65). Additionally, MDSCs can promote stemness in TICs and upregulate PD-L1 by activating the PI3K/AKT/mTOR pathway (66). On the other hand, TICs draw in and encourage MDSC infiltration, development, and activation via secreting soluble chemicals and exosomes specific to different cancer types. These consist of granulocyte-macrophage colony-stimulating factor (GM-CSF), TGF-β, IL-8, and macrophage migration inhibitory factor (MIF) (67–69). Additionally, interaction between MDSCs and macrophages has been shown in other investigations to polarize macrophages toward an M2 phenotype (70).
TICs and TAMs
TAMs are incredibly flexible cells that undergo many forms of functional activation in response to a range of stimuli. Infiltrating macrophages, whose activity is influenced by inflammatory and stress signals within the TME, can mediate tumor immune evasion (71). TAMs suppress anti-tumor immune responses by preventing CD8+ T lymphocytes from entering tumor sites or by decreasing their cytotoxic activity (72). The two primary populations of TAMs are M1 macrophages, which repress the tumor, and M2 macrophages, which promote growth (73). By secreting CXCL8, which in turn activates NF-κB and PI3K/AKT signaling, TICs may maintain their survival, proliferation, and self-renewal through cell-intrinsic pathways. Simultaneously, CXCL8 induces activation in TAMs via the CXCR2-JAK2/STAT3 axis, promoting an M2-like TAM phenotype through paracrine, cell-extrinsic pathways (74). TICs in liver cancer generate IL-34, a gene that p53 transcriptionally suppresses, as a result of p53 depletion. By increasing CD36-mediated fatty acid oxidation metabolism, IL-34 promotes M2-like polarization of TAMs. These IL-34-coordinated TAMs promote immunological evasion by inhibiting CD8+T cell-mediated anti-tumor immunity (75). Using soluble mediators including IL-6, TGF-β, WNT ligands, and pleiotrophic trophic proteins, or juxtacrine signaling discovered in co-culture experiments, TAMs can, on the one hand, cause phenotypic alterations and maintain stemness in TICs (76–80). Through the activation of nuclear factor-κB (NF-κB), AKT, and signal transducer and activator of transcription 3 (STAT3), TAMs promote self-renewal by signaling to TICs via ephrin type-A receptor 4 (EPHA4) and receptor-type tyrosine-protein phosphatase ζ (PTPRZ1) (78, 79). Pancreatic tumors respond better to chemotherapy and have fewer TICs when TAMs are targeted (81). In conclusion, factors, exosomes, or metabolites produced by TICs attract and polarize TAMs (82, 83). However, the promotion of TICs stemness and phenotypic alterations depends on TAM-derived paracrine substances or the physical interactions between TAMs and TICs (84, 85).
TICs and T cell
IL-17, which is released by T-helper 17 (Th17) cells, promotes TIC self-renewal and stemness maintenance. Both IL-17 stimulation and IL-17 introduction significantly enhance the growth of cancer and the ability to form spheroid in a dose-dependent manner. Furthermore, nude mice with higher levels of IL-17 gene expression are more carcinogenic (86). An important component of the TME, regulatory T cells (Tregs) can enhance carcinogenesis, metastasis, and chemotherapeutic drug resistance in cancer cells, as well as stimulate tumor angiogenesis and reduce anti-tumor immunity (87). There may be a connection between Tregs and TICs since, whereas TICs can draw Tregs into the TME, regs also support TIC stemness (87). Higher Th1 CD4+ T cell and/or CTL densities are associated with longer overall survival (88). These T-cell populations produce high quantities of the proinflammatory cytokine interferon-gamma (IFNγ), which is crucial for both local and systemic immunity (89). With its cytotoxic impact, immunostimulatory properties, and cell proliferation inhibitory effects, IFNγ is one of the primary mediators of anti-tumor immunity (90). However, studies have demonstrated that T-cell-derived IFNγ induces TICs to display certain functional traits, including as spheroid formation and resistance to chemotherapy or radiation, in addition to increasing stem cell markers (91). On the other hand, by upregulating programmed death-1 (PD-1) on CD8+ T-cells, TIC-produced PD-L1 can encourage CD8+ T-cell exhaustion (92).
Checkpoint molecule expression
TIC-secreted exosomes trigger the STAT3 pathway, which raises PD-L1 in TAMs (83). MDSCs can promote the stemness of TICs and raise PD-L1 by activating the PI3K/AKT/mTOR pathway (66).By stimulating the ITGB4/SNAI1/SIRT3 signaling pathway, PD-L1 can encourage tumor growth and metastasis, suggesting yet another potential mechanism for these pathways to work in concert (93). Prior studies have shown that the TIC-like population of malignancies, including colorectal cancer, has higher levels of PD-L1 (94). Through a STT3-dependent mechanism, PD-L1 preferentially expresses on TICs and aids in their immune evasion (95). This finding suggests a possible way that TICs evade immune surveillance. These results demonstrate the therapeutic potential of employing immune checkpoint inhibitors to target TICs. One intriguing approach to creating novel therapies that cause cancer patients’ tumors to partially or completely recede is immune checkpoint inhibition (96). Research suggests that checkpoint blockade therapy is highly effective in treating immunogenic malignancies with high levels of CTL infiltration (97). In particular, T-cell activation and proliferation potential can be restored by anti-PD-1 or anti-PD-L1 antibodies, which will allow immune cells to detect and eradicate cancerous cells more efficiently (98). Patients with PD-L1-positive tumors have much greater response rates to PD-1/PD-L1 blocking therapy than patients without PD-L1 expression, according to clinical evidence (99). Importantly, preclinical studies demonstrate that PD-1/PD-L1 blockade synergistically enhances the antitumor efficacy of TIC-targeted vaccines in murine cancer models (100).
Low immunogenicity
TICs surviving persistent immune pressure must circumvent genomic alterations that might induce novel innate and adaptive immune responses, ultimately manifesting an immunogenically attenuated phenotype (101). Comprehensive characterization of TIC-associated immune profiles represents a fundamental prerequisite for developing successful TIC-targeted immunotherapies. These immunological attributes - encompassing antigen processing/presentation machinery (including MHC complexes encoded by HLA genes), costimulatory/inhibitory signaling molecules, tumor-associated antigens (TAAs), and cytokine networks - represent critical determinants for effective immunotherapy development. The diminished MHC I expression observed in TICs may promote their survival through impaired T-cell recognition (102). TICs displaying attenuated MHC I presentation or deficient natural killer cell-activating ligand expression demonstrate impaired immune recognition, potentially conferring dual resistance to chemotherapy and immune-mediated elimination (103). Moreover, TICs exhibit systemic downregulation of antigen processing components including transporter associated with antigen processing (TAP), low molecular weight proteasome subunits (LMP), and β2-microglobulin (104). Immune profiling studies have identified distinct expression patterns of costimulatory (e.g., CD80/CD86) and coinhibitory (e.g., CTLA-4, PD-1/PD-L1, B7-H2/H3) molecules in TICs, revealing a predominant inhibitory signature with costimulatory molecule deficiency (105). This immunological tolerance is compounded by the weak immunogenicity of TAAs derived from TICs, which exhibit heterogeneous expression patterns within tumor masses due to immune selection pressures (106). Consequently, identifying novel tumor-specific neoantigens that stably associate with malignant progression and evade host immune editing remains a central challenge in immunotherapy development. Notably, soluble mediators in the TIC secretome warrant particular attention. Cytokines including CCL-2, IL-6, IL-8, IL-10, IL-13, and TNF-α show tumor-specific secretion profiles, with experimental evidence demonstrating TIC-derived cytokines mediate immune evasion through recruitment and activation of MDSCs and TAMs (107).
Strategies to target TICs
Inhibition of TIC-specific pathways
Numerous pluripotency-associated transcription factors, such as OCT4, SOX2, NANOG, KLF4, and MYC, regulate TICs. Experimental evidence indicates that TIC-derived cytokines mediate immune evasion through recruitment and activation of MDSCs and TAMs. Cytokines, such as CCL-2, IL-6, IL-8, IL-10, IL-13, and TNF-α, exhibit tumor-specific secretion profiles (108). Important signaling pathways, including Notch, Hedgehog (Hh), Wnt/β-catenin, FGF/FGFR, EGF/EGFR, NF-κB, MAPK, PTEN/PI3K, HER2, JAK/STAT, PI3K/AKT/mTOR, TGF/SMAD, and PPAR pathways, are also commonly dysregulated in TICs (108–111). Activation of the STAT3 and Notch3/mTOR pathways promotes higher PD-L1 expression in TICs than in non-TICs, especially in colorectal cancer, gastric cancer, and other gastrointestinal cancers. While STAT3 suppression has been demonstrated to restore T-cell function (112), PD-L1 overexpression increases TIC stemness through a self-sustaining positive feedback loop (113, 114). By successfully removing lung cancer TICs, lowering metastasis, and extending patient survival, napabacusin, a STAT3 inhibitor, has shown strong efficacy in clinical trials (115). One important mediator of TIC-TME interactions is NF-κB, whose suppression significantly lowers the expression of cancer stem cell markers (116). The transcription factor SOX2 orchestrates transcriptional networks that sustain cellular stemness (117) and confer anti-apoptotic capabilities (118). and it plays essential roles in maintaining both embryonic stem cell features and TIC characteristics (119–121). Numerous cancer types have been shown to have SOX2 as an oncogenic driver that is enhanced during carcinogenesis, metastasis, and recurrence (122, 123). In vivo, tumor growth and chemoresistance are greatly reduced when SOX2 is suppressed (123). By working in tandem with SOX2, the Wnt/β-catenin pathway plays a vital role in maintaining cancer stemness. SOX2 improves β-catenin’s nuclear localization and transcriptional activity by directly interacting with it (123, 124). An almost universal characteristic of sporadic colorectal cancers is aberrant Wnt/β-catenin signaling, which is mostly caused by APC mutations. Nuclear β-catenin levels rise as a result of pathway activation, which promotes the development of the T-cell factor/lymphoid enhancer factor complex and the subsequent upregulation of target genes such as AXIN2, SOX2, TCF7, c-MYC, and MMP7 (125, 126). By inducing TIC apoptosis, therapeutic blocking of this mechanism significantly lowers tumor recurrence (127–129). Numerous cancers have been shown to have deregulation of JAK/STAT signaling in TICs (130–132). By inhibiting STAT1 activation, clinical intervention with FDA-approved JAK1/2 inhibitor ruxolitinib or anti-IFNγ antibodies successfully stops IFNγ-mediated TIC production (91). Together, these results highlight the therapeutic potential of focusing on TICs’ aberrant signaling pathways to enhance clinical results.
Differentiation therapy
TICs exhibit characteristic stem cell characteristics, such as the ability to self-renew and the potential for multi-lineage differentiation, as a unique subpopulation within tumor heterogeneity. These cells promote the development of primary tumors, mediate resistance to treatment, and aid in distant metastases and tumor recurrence (133). According to new data, TICs are malleable cells that can switch between quiescent and proliferative states in both directions (134). By using pharmaceutical intervention to reroute tumor reprogramming toward terminal differentiation or death while reducing proliferative potential, differentiation treatment takes advantage of the reversible differentiation abnormalities seen in malignant cells (135, 136). The goal of the TIC differentiation therapeutic paradigm is to either cause TICs to mature terminally or transform them into therapy-sensitive non-mesenchymal equivalents. This approach seeks to reduce invasive potential and malignant development by converting aggressive, undifferentiated tumors into differentiated cell populations with reduced tumorigenicity. In the end, these differentiation-based strategies might eliminate tumors and exhaust the TIC reservoir (137). Genetically defined models provide mechanistic insights, but translational hurdles in solid tumors continue because of sample heterogeneity and constraints in in vitro culture. Targeted inhibition of this oncogenic chimeric protein with functional antibodies causes TIC differentiation and functional ablation in PTPRK-RSPO3 fusion-driven colon cancers, which is a noteworthy example (138).This proof-of-concept illustrates how differentiation treatment may be used to treat specific solid gastrointestinal cancers with distinct driver changes.
Enhancing immune recognition
TIC-specific antigens or surface markers, including as CD44, CD133, and EpCAM, can be targeted by vaccines and adoptive cell treatments (such CAR-T cells). Using embryonic stem cells as universal preventive cancer vaccines is a unique treatment approach made possible by the antigenic similarities between TICs and these cells. Anti-embryonic antigens can induce anti-tumor immune responses through cross-reactive immunity (139), as prior studies have confirmed that tumor-embryonic antigens (such as carcinoembryonic antigen) are co-expressed in both TICs and embryonic stem cells (140). In addition to being a rich source of TIC-specific antigens, embryonic stem cells have the ability to imitate embryonic niche conditions in their conditioned medium, which can aid in differentiation-based cancer therapy. As preventive measures against lung cancer (141),CRC (142) and ovarian cancer (143), inactivated embryonic stem cell-derived vaccines have been created with success and have shown effectiveness in inhibiting the growth and spread of tumors in animal models. In addition to inducing strong TIC-specific CTL responses, therapeutic cancer vaccines that target TICs have shown strong antitumor activity against SCC7 squamous cell carcinoma and D5 melanoma in mouse models (144). Targeting self-renewing TICs is one area in which chimeric antigen receptor (CAR)-T cell therapy, a novel immunotherapy strategy that involves genetically modifying T cells to express cancer-specific CARs (145), shows promise (146). In both syngeneic and xenograft murine models, preclinical research has shown that CAR-T cell therapy can completely eradicate established solid tumors and metastatic lesions in a variety of cancer types, including colon, breast, melanoma, and pancreatic cancer (147). Prior research has shown that Claudin18.2-targeted CAR-T cell therapy is remarkably effective in treating gastroesophageal junction cancer. With a median overall survival of 8.8 months and a disease control rate of 91.8%, the results of a phase II clinical trial (NCT04581473) revealed that some patients even experienced long-term remission (148). With a 6-month overall survival rate of 81.2%, Claudin18.2-targeted CAR-T cell treatment achieved an objective response rate of 57.1% and a disease control rate of 75.0% in patients with gastric cancer. According to these early findings, CAR-T cell treatment shows encouraging efficacy and tolerable safety in cancer patients (149).
Overcoming immunosuppression
Even though tumors have an immunosuppressive environment where cancer cells can inhibit the activation of immune cells through a variety of mechanisms, including the recruitment of TAMs, MDSCs, and Tregs, the attenuation of MHC class I expression, and the use of the PD-1/PD-L1 axis, the immune response against TICs can be strengthened by modulating the TME by using inhibitors of immune checkpoint molecules or reducing the recruitment of immunosuppressive cells. Many drugs that target immune checkpoint receptors, such as CTLA-4, PD-1 (cemiplimab (150)) and PD-L1(avelumab (151), durvalumab (152)). The use of the antibody ipilimumab, which targets cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), was authorized in 2011. Additionally, tremelimumab (CP-675206), a fully human monoclonal antibody, binds to the CTLA-4 molecule expressed on the surface of activated T lymphocytes and T regulatory cells. Tremelimumab prevents CTLA4 from binding to its target ligands (B7–1 and B7-2) by inhibiting the negative regulatory signal that CTLA4 sends on T cell priming (153). 2014 also saw the approval of pembrolizumab and nivolumab, two antibodies that block programmed cell death protein 1 (anti-PD-1) and its ligand 1 (anti-PD-L1) (154). Blocking CTLA4, PD-1, and PD-L1 inhibitory effects can promote and enable effective immune responses against tumor cells. Tregs, a vital part of the immune system, are important targets for treatment and can be used to predict the course and prognosis of cancer (155). Tregs have been shown to directly kill NK cells via β-galactoside-binding protein, which promotes lung metastasis (156). To stop cancer cells from spreading to the lungs, it is enough to target Treg cells (157). MDSCs, recognized as one of the main cellular components in the tumor microenvironment, promote tumor growth by carrying out immunosuppressive functions. They are now significant modulators of the cancer immune response and targets for cancer therapy (158). Modifying the TAM response may enhance immunotherapy. Many strategies to reduce TAMs have been studied in laboratory settings and are thought to be effective therapeutic interventions at this time (159). By inhibiting M-MDSC and TAM survival, some anti-CSF1R drugs that are being studied in cancer patients have demonstrated promising anticancer potential (160). Immunoevasion linked to TICs may be caused by a variety of extrinsic stimuli in addition to immunosuppressive cell activity. Because of their resistance to degradation, persistent organic pollutants like perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS), which are widely used in industrial applications like firefighting foams and water-reactive materials, have become common environmental contaminants, raising concerns about their possible health effects. Per- and polyfluoroalkyl substances (PFAS) may facilitate immune evasion by altering tumor-associated gene expression, controlling immune cell function, and improving TIC properties, according to earlier research using network toxicology, single-cell sequencing, spatial transcriptomics, and molecular simulation technologies (161, 162).
Combination therapies
Checkpoint inhibitors + TIC targeting
Checkpoint inhibitor monotherapy has demonstrated the most significant activity in tumor types with high PD-L1 expression and/or high microsatellite instability or mismatch repair deficiency, though this is limited to no more than one-third of cancer patients authorized for checkpoint inhibitor treatment (163). Because CTLA-4 and PD-1 antagonists are more effective when used jointly than when used alone, a combined immunotherapy regimen was recently approved (164). While PD-1 inhibitors target peripheral T cell activation, especially in the tumor context, CTLA-4 antagonists affect T cell priming (165). The robust immune responses shown in in vivo studies when both targets are blocked have strengthened the theoretical foundation for combined ICB (166). In conclusion, if CD8+ T cells are absent from the tumor microenvironment, blocking the PD-1/PD-L1 pathway will not work. Combining this tactic with CTLA-4 blockage may increase the quantity of activated CD8+ T cells (167). However, combining two ICBs will unavoidably cause more side effects, and their clinical application might be challenging (168). For tumors with low immunogenicity or stromal fibrosis, where the effect of checkpoint inhibition as a monotherapy is minimal or nonexistent, combining checkpoint inhibition with other treatments may result in a synergistic response (169). Some researchers suggest combining TIC targeting with immune checkpoint blocking to improve treatment outcomes. As was previously known, TICs are commonly accompanied by aberrant route changes. By combining ICB with targeting these pathways, treatment resistance can be reduced and patient outcomes can be greatly enhanced. Using methods like organoids, transcriptomics, genomic sequencing, and immunohistochemistry, suitable individuals should be identified and monitored both before and during treatment (170). Research has demonstrated that blocking oncogenic Myc signaling using epigenetic strategies (like JQ1) significantly reduces PD-L1 expression, which is why anti-PD-1 antibodies and JQ1 produce a synergistic immune response (171). These results suggest that combining immune checkpoint blockade with TIC targeting can significantly improve the efficacy of immunotherapy for malignancies.
Chemotherapy + immunotherapy/targeted therapy
Given the immunomodulatory and adjuvant effects of classical chemotherapy and its widespread clinical usage, combining immunotherapy with chemotherapy presents an enticing option to boost immunotherapy’s efficacy across a larger patient population (172). In particular, TIC-targeted chemotherapy seeks to either use surface receptors to transport chemotherapeutic drugs directly into TICs for efficient removal or disrupt basic intracellular pathways in TICs. The immunosuppressive TME is essential for preserving TICs’ stem-like characteristics. As a result, some immunotherapies that can rewire the TME have also shown promise in eliminating TICs (173). The use of combined treatment techniques is encouraged by the possibility for chemotherapy and immunotherapy medications to complement each other mechanistically, even though only a small number of patients benefit from either medication alone. Combining immunomodulators, like immune checkpoint inhibitors, can improve the antitumor immune response triggered by chemotherapeutic drugs while suppressing or even eliminating tumor growth and metastasis. Consequently, immunotherapy in combination with chemotherapy has recently been identified as a highly promising approach to improve these medications’ efficacy (172, 174). Pancreatic cancer (175), gastric cancer (176), CRC (177), and hepatocellular carcinoma (178) are among the gastrointestinal malignancies for which standard chemotherapy and immunotherapy have been shown to have beneficial therapeutic effects (179, 180). Intracellular proteins that are essential for the growth, development, or metabolism of TICs can be inhibited by chemotherapy drugs. For example, it has been demonstrated that ALDH1 inhibitors cause apoptosis in TICs and block the production of markers linked to stemness (181). Additionally, CD44-positive breast cancer TICs can be precisely targeted by gemcitabine-conjugated nanoparticles functionalized with CD44 antibodies, improving overall therapeutic efficacy (182). Glioblastoma TICs have been successfully eradicated by NK cell therapy in the field of immunotherapy (183). Furthermore, surface receptors on TICs have been used for targeted delivery of immunomodulators, much like chemotherapeutic medications and radioisotopes (184). There are intrinsic disadvantages to immunotherapy and chemotherapy, despite their notable clinical benefits. Chemotherapy drugs have the ability to directly destroy or inactivate dendritic cells and other immune cells (185). High-dose chemotherapy often results in a decrease in B cells, T cells, and NK cells (186). These cells possess every functional characteristic needed to expose effector T cells to antigens linked to tumors, hence stopping the formation of tumors in the host (187). The immunosuppression caused by the death of these cells may be a significant factor in decreased efficacy and tumor recurrence when immunotherapy and chemotherapy are used in combination.
Clinical trials
Clinical studies of medications that target surface indicators linked to CSCs Monoclonal antibodies (mAbs) that target surface indicators unique to CSCs have become a cutting-edge cancer treatment tool. Future and ongoing clinical trials that combine immunotherapy with TIC-targeted medicines for malignancies of the digestive system will yield vital information to improve these strategies. Targeted treatments have shown considerable promise in treating TICs, particularly solid tumors, as seen by recent developments in clinical research. To overcome tumor heterogeneity, improve safety even more, and investigate earlier clinical applications, more research is required. Targeted treatments for TICs have advanced from proof-of-concept to clinical translation, notwithstanding the lingering obstacles. In order to get from “prolonged survival” to “cure of recurrence,” future research must optimize current therapies through interdisciplinary collaboration (immunology, AI, materials science) and investigate early interventions. In Table 2, we provide an overview of some of the current clinical research on GI cancers.
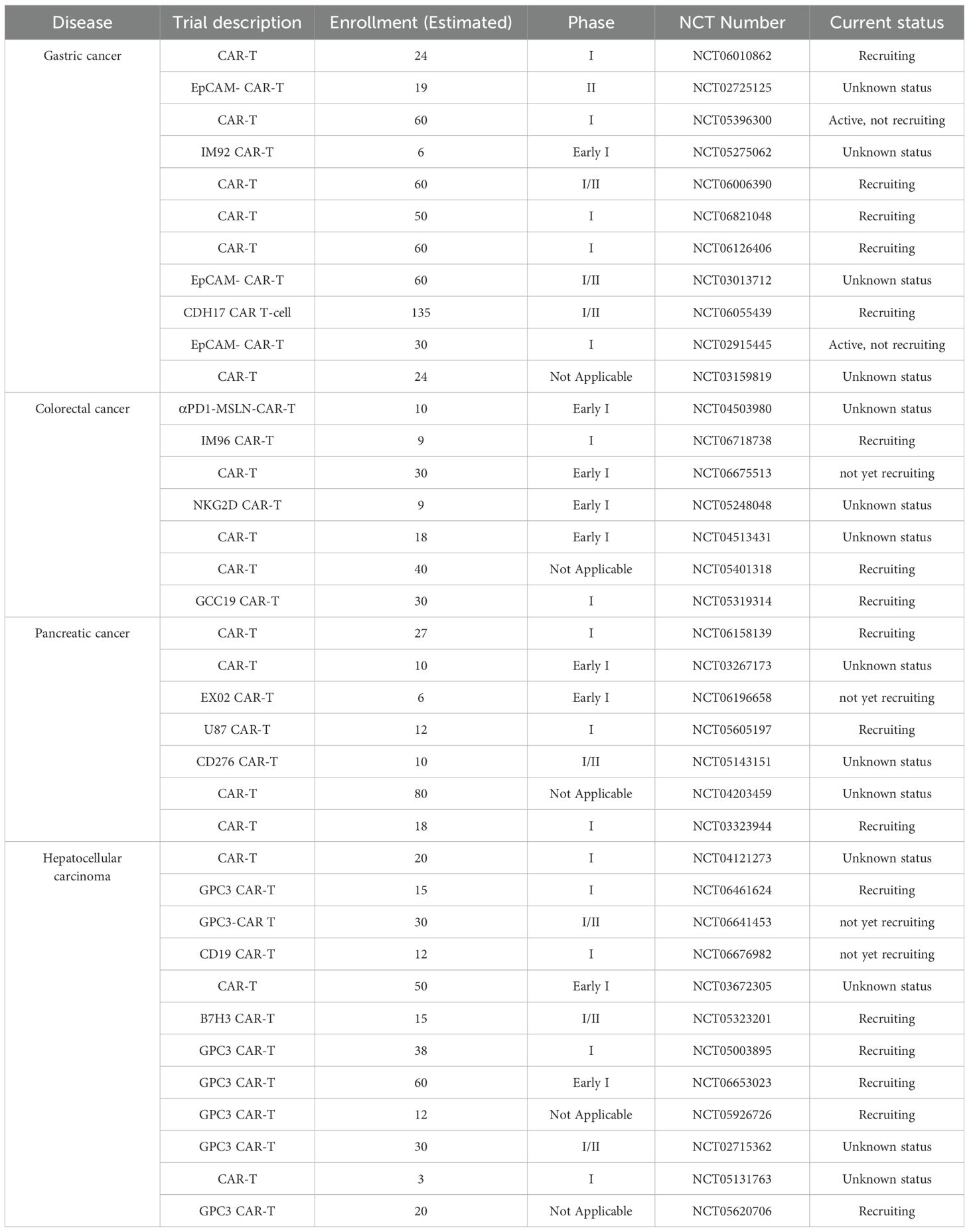
Table 2. TICs-directed immunotherapy in ongoing clinical trials.
Challenges and future directions
Many patients continue to get ineffective therapy despite advancements in cancer treatment, which results in disease progression, recurrence, and a worse overall survival rate. Limitations in basic research and obstacles in clinical trials are the two primary categories of current challenges. While clinical trials challenges primarily include unclear therapeutic targets, low clinical trial success rates, and a lack of specific biomarkers, basic research challenges primarily include a lack of knowledge about TIC heterogeneity, immune evasion mechanisms, and the limitations of current models.
One of the most crucial things to keep in mind is tumor heterogeneity. The identification of several clones with different DNA sequences within the same tumor has led to the widespread recognition of cancer as a heterogeneous illness (188). Intra-tumoral heterogeneity is increasingly understood to play a role in treatment failure and the advancement of disease (189). Such heterogeneity is regarded as a significant barrier to precision cancer therapy (190)and also results in reduced immune responses against cancer (191). Both genetic determinants (primarily involving developmental pathways through gene mutations and tumor microenvironment alterations) and non-genetic factors, such as epigenetic modifications like DNA methylation, histone modifications, chromatin accessibility, microRNAs, and other non-coding RNAs, are strongly implicated in functional heterogeneity (192–194). Furthermore, TIC-mediated immune evasion continues to be a major contributing factor to immunotherapy failure. TICs may generate more immune escape mechanisms as they mature. To comprehend these mechanisms and learn how to combat them, more research is required. Finally, few molecular targets have been effectively converted into clinical care, and clinical trials continue to have high failure rates, despite a great deal of attention being paid to identifying key molecular targets as possible avenues for therapeutic intervention against TIC activities.
Reliable biomarkers are still lacking in clinical research. Despite the development of certain TIC-associated indicators, their specificity is typically lacking. It is obvious that in order to target this cell population efficiently, a thorough understanding of TIC indicators is necessary. Further research is still required to assess and validate TIC markers. Furthermore, it might be necessary to create treatments that can eradicate TICs with a variety of genetic alterations and phenotypic traits (195).Immunotherapy failure may be caused by unclear treatment targets, and clinical translation is hampered by poor clinical trial success rates taken together.
In order to improve our knowledge of TIC-associated immune microenvironments and particular indicators to direct future study, future studies should concentrate on using single-cell and spatial transcriptomic technologies to map the immune microenvironment of TICs (196, 197). To overcome drug resistance and increase the effectiveness of immunotherapy for tumors of the digestive system like colorectal cancer, Hong et al., for example, proposed an integrative genomics and single-cell analysis framework to identify immune-related and potential drug targets in TIC-enriched populations (198). In order to define liver cancer heterogeneity and create risk stratification models, Yang et al. used multi-region sequencing in conjunction with spatial transcriptomics, offering mechanistic insights into clinical difficulties driven by TICs (199).Yang’s group recently created a computational pipeline that combines five statistical inference techniques and is named SiLi (Statistical Inference-based Synthetic Lethality Identification). Through SiLi analysis of large-scale sequencing datasets, they methodically discovered synthetic lethal interactions in liver cancer, providing possible approaches for creating analogous algorithms to create TIC-targeted synthetic lethal combination treatments that address metastasis and recurrence. Last but not least, researchers can trace the origins of tumor-initiating cells and pinpoint important regulatory pathways to guide future targeted therapies by fusing lineage tracing with CRISPR gene editing technology (200). Integrating multi-omics data to forecast treatment responses or immune evasion patterns across several tumors from intricate databases has become possible with the development of machine learning (201).
Conclusion
Targeting tumor-initiating cells in tumors of the digestive system is a promising way to improve cancer immunotherapy. By addressing the challenges posed by TICs, such as immune evasion and treatment resistance, therapies can improve patient outcomes and reduce tumor recurrence. Combination techniques, especially those that combine immune control and direct TIC targeting, are likely to determine the future of cancer treatment.
Author contributions
Z-yZ: Writing – original draft, Conceptualization, Data curation, Supervision. X-fZ: Conceptualization, Data curation, Formal analysis, Writing – original draft. C-hX: Conceptualization, Data curation, Writing – original draft. K-hW: Funding acquisition, Resources, Writing – review & editing. FH: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Yunnan Technological Innovation Centre of Drug Addiction Medicine (Grant Number: 202305AK340001), Yunnan Fundamental Research projects (Grant Number: 202301AU070099).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Taniguchi S, Elhance A, Van Duzer A, Kumar S, Leitenberger JJ, and Oshimori N. Tumor-initiating cells establish an IL-33-TGF-beta niche signaling loop to promote cancer progression. Science. (2020) 369:269-+. doi: 10.1126/science.aay1813
2. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. (2006) 442:818–22. doi: 10.1038/nature04980
3. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
4. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. (2012) 338:1080–4. doi: 10.1126/science.1226929
5. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauss A, et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
6. Huang Z, Wu T, Liu AY, and Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. (2015) 6:39550–63. doi: 10.18632/oncotarget.6098
7. Verga Falzacappa MV, Ronchini C, Reavie LB, and Pelicci PG. Regulation of self-renewal in normal and cancer stem cells. FEBS J. (2012) 279:3559–72. doi: 10.1111/j.1742-4658.2012.08727.x
8. Lagasse E. Cancer stem cells with genetic instability: the best vehicle with the best engine for cancer. Gene Ther. (2008) 15:136–42. doi: 10.1038/sj.gt.3303068
9. Wyles SP, Brandt EB, and Nelson TJ. Stem cells: the pursuit of genomic stability. Int J Mol Sci. (2014) 15:20948–67. doi: 10.3390/ijms151120948
10. Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, et al. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. (2019) 177:1172–1186 e1114. doi: 10.1016/j.cell.2019.03.025
11. Lu H, Xie Y, Tran L, Lan J, Yang Y, Murugan NL, et al. Chemotherapy-induced S100A10 recruits KDM6A to facilitate OCT4-mediated breast cancer stemness. J Clin Invest. (2020) 130:4607–23. doi: 10.1172/JCI138577
12. Batlle E and Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
13. Clayton S and Mousa SA. Therapeutics formulated to target cancer stem cells: Is it in our future? Cancer Cell Int. (2011) 11:7. doi: 10.1186/1475-2867-11-7
14. Junttila MR and de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. (2013) 501:346–54. doi: 10.1038/nature12626
15. Nassar D and Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. (2016) 11:47–76. doi: 10.1146/annurev-pathol-012615-044438
16. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
17. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. (2009) 458:780–3. doi: 10.1038/nature07733
18. Oravecz-Wilson KI, Philips ST, Yilmaz OH, Ames HM, Li L, Crawford BD, et al. Persistence of leukemia-initiating cells in a conditional knockin model of an imatinib-responsive myeloproliferative disorder. Cancer Cell. (2009) 16:137–48. doi: 10.1016/j.ccr.2009.06.007
19. Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. (2006) 12:5615–21. doi: 10.1158/1078-0432.CCR-06-0169
20. Jiang Y, He Y, Li H, Li HN, Zhang L, Hu W, et al. Expressions of putative cancer stem cell markers ABCB1, ABCG2, and CD133 are correlated with the degree of differentiation of gastric cancer. Gastric Cancer. (2012) 15:440–50. doi: 10.1007/s10120-012-0140-y
21. Ohkuma M, Haraguchi N, Ishii H, Mimori K, Tanaka F, Kim HM, et al. Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann Surg Oncol. (2012) 19:1357–64. doi: 10.1245/s10434-011-1739-7
22. Fu Y, Du P, Zhao J, Hu C, Qin Y, and Huang G. Gastric cancer stem cells: mechanisms and therapeutic approaches. Yonsei Med J. (2018) 59:1150–8. doi: 10.3349/ymj.2018.59.10.1150
23. Fujita T, Chiwaki F, Takahashi RU, Aoyagi K, Yanagihara K, Nishimura T, et al. Identification and Characterization of CXCR4-Positive Gastric Cancer Stem Cells. PloS One. (2015) 10:e0130808. doi: 10.1371/journal.pone.0130808
24. Rocco A, Liguori E, Pirozzi G, Tirino V, Compare D, Franco R, et al. CD133 and CD44 cell surface markers do not identify cancer stem cells in primary human gastric tumors. J Cell Physiol. (2012) 227:2686–93. doi: 10.1002/jcp.23013
25. Simon E, Petke D, Boger C, Behrens HM, Warneke V, Ebert M, et al. The spatial distribution of LGR5+ cells correlates with gastric cancer progression. PloS One. (2012) 7:e35486. doi: 10.1371/journal.pone.0035486
26. Nienhuser H, Kim W, Malagola E, Ruan T, Valenti G, Middelhoff M, et al. Mist1+ gastric isthmus stem cells are regulated by Wnt5a and expand in response to injury and inflammation in mice. Gut. (2021) 70:654–65. doi: 10.1136/gutjnl-2020-320742
27. Katsuno Y, Ehata S, Yashiro M, Yanagihara K, Hirakawa K, and Miyazono K. Coordinated expression of REG4 and aldehyde dehydrogenase 1 regulating tumourigenic capacity of diffuse-type gastric carcinoma-initiating cells is inhibited by TGF-beta. J Pathol. (2012) 228:391–404. doi: 10.1002/path.4020
28. Zhao R, He B, Bie Q, Cao J, Lu H, Zhang Z, et al. AQP5 complements LGR5 to determine the fates of gastric cancer stem cells through regulating ULK1 ubiquitination. J Exp Clin Cancer Res. (2022) 41:322. doi: 10.1186/s13046-022-02532-w
29. Fujikuni N, Yamamoto H, Tanabe K, Naito Y, Sakamoto N, Tanaka Y, et al. Hypoxia-mediated CD24 expression is correlated with gastric cancer aggressiveness by promoting cell migration and invasion. Cancer Sci. (2014) 105:1411–20. doi: 10.1111/cas.12522
30. Fukamachi H, Seol HS, Shimada S, Funasaka C, Baba K, Kim JH, et al. CD49f(high) cells retain sphere-forming and tumor-initiating activities in human gastric tumors. PloS One. (2013) 8:e72438. doi: 10.1371/journal.pone.0072438
31. Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. (2012) 22:248–58. doi: 10.1038/cr.2011.109
32. Haraguchi N, Ishii H, Mimori K, Ohta K, Uemura M, Nishimura J, et al. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int J Oncol. (2013) 43:425–30. doi: 10.3892/ijo.2013.1955
33. Sipos F and Muzes G. Interconnection of CD133 stem cell marker with autophagy and apoptosis in colorectal cancer. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252011201
34. Joosten SPJ, Spaargaren M, Clevers H, and Pals ST. Hepatocyte growth factor/MET and CD44 in colorectal cancer: partners in tumorigenesis and therapy resistance. Biochim Biophys Acta Rev Cancer. (2020) 1874:188437. doi: 10.1016/j.bbcan.2020.188437
35. AbdelMageed M, Ismail HTH, Olsson L, Lindmark G, Hammarstrom ML, Hammarstrom S, et al. Clinical significance of stem cell biomarkers epCAM, LGR5 and LGR4 mRNA levels in lymph nodes of colon cancer patients. Int J Mol Sci. (2021) 23. doi: 10.3390/ijms23010403
36. Zhang SS, Huang ZW, Li LX, Fu JJ, and Xiao B. Identification of CD200+ colorectal cancer stem cells and their gene expression profile. Oncol Rep. (2016) 36:2252–60. doi: 10.3892/or.2016.5039
37. Soleimani A, Saeedi N, Al-Asady AM, Nazari E, Hanaie R, Khazaei M, et al. Colorectal Cancer Stem Cell Biomarkers: Biological Traits and Prognostic Insights. Curr Pharm Des. (2024) 30:1386–97. doi: 10.2174/0113816128291321240329050945
38. Fan W, Yang X, Huang F, Tong X, Zhu L, and Wang S. Identification of CD206 as a potential biomarker of cancer stem-like cells and therapeutic agent in liver cancer. Oncol Lett. (2019) 18:3218–26. doi: 10.3892/ol.2019.10673
39. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. (2009) 28:3949–59. doi: 10.1038/onc.2009.252
40. Jacob J, Bellach J, Grutzmann R, Alldinger I, Pilarsky C, Dietel M, et al. Expression of CD24 in adenocarcinomas of the pancreas correlates with higher tumor grades. Pancreatology. (2004) 4:454–60. doi: 10.1159/000079824
41. Qiu W, Remotti HE, Tang SM, Wang E, Dobberteen L, Lee Youssof A, et al. Pancreatic DCLK1(+) cells originate distinctly from PDX1(+) progenitors and contribute to the initiation of intraductal papillary mucinous neoplasm in mice. Cancer Lett. (2018) 423:71–9. doi: 10.1016/j.canlet.2018.03.009
42. Kayali AG, Van Gunst K, Campbell IL, Stotland A, Kritzik M, Liu G, et al. The stromal cell-derived factor-1alpha/CXCR4 ligand-receptor axis is critical for progenitor survival and migration in the pancreas. J Cell Biol. (2003) 163:859–69. doi: 10.1083/jcb.200304153
43. Fong D, Steurer M, Obrist P, Barbieri V, Margreiter R, Amberger A, et al. Ep-CAM expression in pancreatic and ampullary carcinomas: frequency and prognostic relevance. J Clin Pathol. (2008) 61:31–5. doi: 10.1136/jcp.2006.037333
44. Lu Y, Zhu H, Shan H, Lu J, Chang X, Li X, et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. (2013) 340:113–23. doi: 10.1016/j.canlet.2013.07.009
45. Harpstrite SE, Gu H, Natarajan R, and Sharma V. Interrogation of multidrug resistance (MDR1) P-glycoprotein (ABCB1) expression in human pancreatic carcinoma cells: correlation of 99mTc-Sestamibi uptake with western blot analysis. Nucl Med Commun. (2014) 35:1067–70. doi: 10.1097/MNM.0000000000000158
46. Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. (2008) 13:153–66. doi: 10.1016/j.ccr.2008.01.013
47. Park DJ, Sung PS, Kim JH, Lee GW, Jang JW, Jung ES, et al. EpCAM-high liver cancer stem cells resist natural killer cell-mediated cytotoxicity by upregulating CEACAM1. J Immunother Cancer. (2020) 8. doi: 10.1136/jitc-2019-000301
48. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, and Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. (2011) 9:50–63. doi: 10.1016/j.stem.2011.06.005
49. Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. (2010) 126:2067–78. doi: 10.1002/ijc.24868
50. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. (2010) 120:3326–39. doi: 10.1172/JCI42550
51. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482:400–4. doi: 10.1038/nature10755
52. Nayak A, Warrier NM, and Kumar P. Cancer stem cells and the tumor microenvironment: targeting the critical crosstalk through nanocarrier systems. Stem Cell Rev Rep. (2022) 18:2209–33. doi: 10.1007/s12015-022-10426-9
53. Mu Q and Najafi M. Resveratrol for targeting the tumor microenvironment and its interactions with cancer cells. Int Immunopharmacol. (2021) 98:107895. doi: 10.1016/j.intimp.2021.107895
54. Yang C, Geng H, Yang X, Ji S, Liu Z, Feng H, et al. Targeting the immune privilege of tumor-initiating cells to enhance cancer immunotherapy. Cancer Cell. (2024) 42:2064–2081 e2019. doi: 10.1016/j.ccell.2024.10.008
55. Wu Y, Zhang J, Zhang X, Zhou H, Liu G, and Li Q. Cancer stem cells: A potential breakthrough in HCC-targeted therapy. Front Pharmacol. (2020) 11:198. doi: 10.3389/fphar.2020.00198
56. He X, Smith SE, Chen S, Li H, Wu D, Meneses-Giles PI, et al. Tumor-initiating stem cell shapes its microenvironment into an immunosuppressive barrier and pro-tumorigenic niche. Cell Rep. (2021) 36:109674. doi: 10.1016/j.celrep.2021.109674
57. Maccalli C, Rasul KI, Elawad M, and Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. (2018) 53:189–200. doi: 10.1016/j.semcancer.2018.09.006
58. Hegde S, Leader AM, and Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. (2021) 54:875–84. doi: 10.1016/j.immuni.2021.04.004
59. Gabrilovich DI and Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
60. Kumar V, Patel S, Tcyganov E, and Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
61. Sica A, Porta C, Amadori A, and Pasto A. Tumor-associated myeloid cells as guiding forces of cancer cell stemness. Cancer Immunol Immunother. (2017) 66:1025–36. doi: 10.1007/s00262-017-1997-8
62. Veglia F, Sanseviero E, and Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
63. Lu TX and Rothenberg ME. MicroRNA. J Allergy Clin Immunol. (2018) 141:1202–7. doi: 10.1016/j.jaci.2017.08.034
64. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. (2013) 39:611–21. doi: 10.1016/j.immuni.2013.08.025
65. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. (2014) 63:513–28. doi: 10.1007/s00262-014-1527-x
66. Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. (2020) 69:2477–99. doi: 10.1007/s00262-020-02628-2
67. Chikamatsu K, Takahashi G, Sakakura K, Ferrone S, and Masuyama K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck. (2011) 33:208–15. doi: 10.1002/hed.21420
68. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res. (2017) 77:2607–19. doi: 10.1158/0008-5472.CAN-16-2534
69. Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD, et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells. (2016) 34:2026–39. doi: 10.1002/stem.2393
70. Sinha P, Clements VK, Bunt SK, Albelda SM, and Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
71. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. (2019) 35:588–602 e510. doi: 10.1016/j.ccell.2019.02.009
72. Mantovani A, Allavena P, Marchesi F, and Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
73. Chanmee T, Ontong P, Konno K, and Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
74. Yuan W, Zhang Q, Gu D, Lu C, Dixit D, Gimple RC, et al. Dual Role of CXCL8 in Maintaining the Mesenchymal State of Glioblastoma Stem Cells and M2-Like Tumor-Associated Macrophages. Clin Cancer Res. (2023) 29:3779–92. doi: 10.1158/1078-0432.CCR-22-3273
75. Nian Z, Dou Y, Shen Y, Liu J, Du X, Jiang Y, et al. Interleukin-34-orchestrated tumor-associated macrophage reprogramming is required for tumor immune escape driven by p53 inactivation. Immunity. (2024) 57:2344–2361 e2347. doi: 10.1016/j.immuni.2024.08.015
76. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U.S.A. (2011) 108:12425–30. doi: 10.1073/pnas.1106645108
77. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. (2014) 352:160–8. doi: 10.1016/j.canlet.2014.05.008
78. Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun. (2017) 8:15080. doi: 10.1038/ncomms15080
79. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. (2014) 147:1393–404. doi: 10.1053/j.gastro.2014.08.039
80. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol. (2012) 189:444–53. doi: 10.4049/jimmunol.1103248
81. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. (2013) 73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731
82. Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. (2020) 10:371–81. doi: 10.1158/2159-8290.CD-19-0400
83. Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. (2018) 7:e1412909. doi: 10.1080/2162402X.2017.1412909
84. Chen P, Hsu WH, Han J, Xia Y, and DePinho RA. Cancer stemness meets immunity: from mechanism to therapy. Cell Rep. (2021) 34:108597. doi: 10.1016/j.celrep.2020.108597
85. Liu D, Lu Q, Wang X, Wang J, Lu N, Jiang Z, et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res. (2019) 29:365–78. doi: 10.1038/s41422-019-0155-6
86. Xiang T, Long H, He L, Han X, Lin K, Liang Z, et al. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene. (2015) 34:165–76. doi: 10.1038/onc.2013.537
87. Xu Y, Dong X, Qi P, Ye Y, Shen W, Leng L, et al. Sox2 communicates with tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cells. (2017) 35:2351–65. doi: 10.1002/stem.2720
88. Fridman WH, Pages F, Sautes-Fridman C, and Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
89. Kennedy R and Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. (2008) 222:129–44. doi: 10.1111/j.1600-065X.2008.00616.x
90. Kursunel MA and Esendagli G. The untold story of IFN-gamma in cancer biology. Cytokine Growth Factor Rev. (2016) 31:73–81. doi: 10.1016/j.cytogfr.2016.07.005
91. Beziaud L, Young CM, Alonso AM, Norkin M, Minafra AR, and Huelsken J. IFNgamma-induced stem-like state of cancer cells as a driver of metastatic progression following immunotherapy. Cell Stem Cell. (2023) 30:818–831 e816. doi: 10.1016/j.stem.2023.05.007
92. Gholami A. Cancer stem cell-derived exosomes in CD8(+) T cell exhaustion. Int Immunopharmacol. (2024) 137:112509. doi: 10.1016/j.intimp.2024.112509
93. Wang S, Li J, Xie J, Liu F, Duan Y, Wu Y, et al. Programmed death ligand 1 promotes lymph node metastasis and glucose metabolism in cervical cancer by activating integrin beta4/SNAI1/SIRT3 signaling pathway. Oncogene. (2018) 37:4164–80. doi: 10.1038/s41388-018-0252-x
94. Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X, et al. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PloS One. (2015) 10:e0135528. doi: 10.1371/journal.pone.0135528
95. Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. (2018) 9:1908. doi: 10.1038/s41467-018-04313-6
96. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, et al. The next decade of immune checkpoint therapy. Cancer Discov. (2021) 11:838–57. doi: 10.1158/2159-8290.CD-20-1680
97. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. (2017) 545:60–5. doi: 10.1038/nature22079
98. Wolchok JD and Chan TA. Cancer: Antitumour immunity gets a boost. Nature. (2014) 515:496–8. doi: 10.1038/515496a
99. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
100. Shi X, Zhang X, Li J, Mo L, Zhao H, Zhu Y, et al. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int J Cancer. (2018) 142:2106–17. doi: 10.1002/ijc.31219
101. Dunn GP, Old LJ, and Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
102. Schatton T, Schutte U, Frank NY, Zhan Q, Hoerning A, Robles SC, et al. Modulation of T-cell activation by Malignant melanoma initiating cells. Cancer Res. (2010) 70:697–708. doi: 10.1158/0008-5472.CAN-09-1592
103. Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA, et al. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol. (2007) 83:121–31. doi: 10.1007/s11060-006-9265-3
104. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. (2010) 16:800–13. doi: 10.1158/1078-0432.CCR-09-2730
105. Maccalli C, Volonte A, Cimminiello C, and Parmiani G. Immunology of cancer stem cells in solid tumours. A review Eur J Cancer. (2014) 50:649–55. doi: 10.1016/j.ejca.2013.11.014
106. Anichini A, Fossati G, and Parmiani G. Heterogeneity of clones from a human metastatic melanoma detected by autologous cytotoxic T lymphocyte clones. J Exp Med. (1986) 163:215–20. doi: 10.1084/jem.163.1.215
107. Schatton T and Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci. (2009) 1176:154–69. doi: 10.1111/j.1749-6632.2009.04568.x
108. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. (2020) 5:8. doi: 10.1038/s41392-020-0110-5
109. Visvader JE and Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. (2012) 10:717–28. doi: 10.1016/j.stem.2012.05.007
110. Regenbrecht CR, Lehrach H, and Adjaye J. Stemming cancer: functional genomics of cancer stem cells in solid tumors. Stem Cell Rev. (2008) 4:319–28. doi: 10.1007/s12015-008-9034-0
111. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. (2010) 143:313–24. doi: 10.1016/j.cell.2010.09.010
112. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. (2010) 9:67–78. doi: 10.1158/1535-7163.MCT-09-0734
113. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ Cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. (2016) 22:3571–81. doi: 10.1158/1078-0432.CCR-15-2665
114. Mansour FA, Al-Mazrou A, Al-Mohanna F, Al-Alwan M, and Ghebeh H. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology. (2020) 9:1729299. doi: 10.1080/2162402X.2020.1729299
115. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U.S.A. (2015) 112:1839–44. doi: 10.1073/pnas.1424171112
116. Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. (2010) 70:10464–73. doi: 10.1158/0008-5472.CAN-10-0732
117. Chen S, Xu Y, Chen Y, Li X, Mou W, Wang L, et al. SOX2 gene regulates the transcriptional network of oncogenes and affects tumorigenesis of human lung cancer cells. PloS One. (2012) 7:e36326. doi: 10.1371/journal.pone.0036326
118. Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y, et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J Mol Cell Biol. (2011) 3:230–8. doi: 10.1093/jmcb/mjr002
119. Takahashi K and Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
120. Hoshino H, Nagano H, Haraguchi N, Nishikawa S, Tomokuni A, Kano Y, et al. Hypoxia and TP53 deficiency for induced pluripotent stem cell-like properties in gastrointestinal cancer. Int J Oncol. (2012) 40:1423–30. doi: 10.3892/ijo.2012.1346
121. Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. (2007) 9:625–35. doi: 10.1038/ncb1589
122. Hutz K, Mejias-Luque R, Farsakova K, Ogris M, Krebs S, Anton M, et al. The stem cell factor SOX2 regulates the tumorigenic potential in human gastric cancer cells. Carcinogenesis. (2014) 35:942–50. doi: 10.1093/carcin/bgt410
123. Zhu Y, Huang S, Chen S, Chen J, Wang Z, Wang Y, et al. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by beta-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. (2021) 12:449. doi: 10.1038/s41419-021-03733-5
124. Tang Q, Chen J, Di Z, Yuan W, Zhou Z, Liu Z, et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/beta-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. (2020) 39:232. doi: 10.1186/s13046-020-01690-z
126. Yue Y, Xue Q, Yang J, Li X, Mi Z, Zhao G, et al. Wnt-activated olfactory ensheathing cells stimulate neural stem cell proliferation and neuronal differentiation. Brain Res. (2020) 1735:146726. doi: 10.1016/j.brainres.2020.146726
127. Kim JY, Lee HY, Park KK, Choi YK, Nam JS, and Hong IS. CWP232228 targets liver cancer stem cells through Wnt/beta-catenin signaling: a novel therapeutic approach for liver cancer treatment. Oncotarget. (2016) 7:20395–409. doi: 10.18632/oncotarget.7954
128. Yin X, Xiang T, Li L, Su X, Shu X, Luo X, et al. DACT1, an antagonist to Wnt/beta-catenin signaling, suppresses tumor cell growth and is frequently silenced in breast cancer. Breast Cancer Res. (2013) 15:R23. doi: 10.1186/bcr3399
129. Shi L, Fei X, Wang Z, and You Y. PI3K inhibitor combined with miR-125b inhibitor sensitize TMZ-induced anti-glioma stem cancer effects through inactivation of Wnt/beta-catenin signaling pathway. In Vitro Cell Dev Biol Anim. (2015) 51:1047–55. doi: 10.1007/s11626-015-9931-x
130. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U.S.A. (2007) 104:16158–63. doi: 10.1073/pnas.0702596104
131. Yang L, Dong Y, Li Y, Wang D, Liu S, Wang D, et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-kappaB/Notch1 pathway in non-small cell lung cancer. Int J Cancer. (2019) 145:1099–110. doi: 10.1002/ijc.32151
132. Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. (2013) 25:961–9. doi: 10.1016/j.cellsig.2013.01.007
133. Shackleton M, Quintana E, Fearon ER, and Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. (2009) 138:822–9. doi: 10.1016/j.cell.2009.08.017
134. da Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, and Munoz P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol. (2018) 53:48–58. doi: 10.1016/j.semcancer.2018.08.009
135. Leszczyniecka M, Roberts T, Dent P, Grant S, and Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. (2001) 90:105–56. doi: 10.1016/S0163-7258(01)00132-2
136. Sachs L. Control of normal cell differentiation and the phenotypic reversion of Malignancy in myeloid leukaemia. Nature. (1978) 274:535–9. doi: 10.1038/274535a0
137. Arima Y, Nobusue H, and Saya H. Targeting of cancer stem cells by differentiation therapy. Cancer Sci. (2020) 111:2689–95. doi: 10.1111/cas.14504
138. Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature. (2016) 529:97–100. doi: 10.1038/nature16466
139. Brewer BG, Mitchell RA, Harandi A, and Eaton JW. Embryonic vaccines against cancer: an early history. Exp Mol Pathol. (2009) 86:192–7. doi: 10.1016/j.yexmp.2008.12.002
140. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. (2000) 21:585–91. doi: 10.1093/carcin/21.4.585
141. Yaddanapudi K, Mitchell RA, Putty K, Willer S, Sharma RK, Yan J, et al. Vaccination with embryonic stem cells protects against lung cancer: is a broad-spectrum prophylactic vaccine against cancer possible? PloS One. (2012) 7:e42289. doi: 10.1371/journal.pone.0042289
142. Li Y, Zeng H, Xu RH, Liu B, and Li Z. Vaccination with human pluripotent stem cells generates a broad spectrum of immunological and clinical responses against colon cancer. Stem Cells. (2009) 27:3103–11. doi: 10.1002/stem.234
143. Zhang Z, Chen X, Chang X, Ye X, Li Y, and Cui H. Vaccination with embryonic stem cells generates effective antitumor immunity against ovarian cancer. Int J Mol Med. (2013) 31:147–53. doi: 10.3892/ijmm.2012.1195
144. Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. (2012) 72:1853–64. doi: 10.1158/0008-5472.CAN-11-1400
145. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
146. Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. (2021) 11:1192–211. doi: 10.1158/2159-8290.CD-20-1243
147. Zhao Y, Chen J, Andreatta M, Feng B, Xie YQ, Wenes M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. (2024) 42:1693–704. doi: 10.1038/s41587-023-02060-8
148. Qi C, Liu C, Gong J, Liu D, Wang X, Zhang P, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial final results. Nat Med. (2024) 30:2224–34. doi: 10.1038/s41591-024-03037-z
149. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. (2022) 28:1189–98. doi: 10.1038/s41591-022-01800-8
150. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. (2018) 379:341–51. doi: 10.1056/NEJMoa1805131
151. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. (2019) 380:1103–15. doi: 10.1056/NEJMoa1816047
152. Fujiwara Y, Iguchi H, Yamamoto N, Hayama M, Nii M, Ueda S, et al. Tolerability and efficacy of durvalumab in Japanese patients with advanced solid tumors. Cancer Sci. (2019) 110:1715–23. doi: 10.1111/cas.14003
153. Leach DR, Krummel MF, and Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
154. Sharma P and Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
155. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. (2006) 24:5373–80. doi: 10.1200/JCO.2006.05.9584
156. Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. (2009) 69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619
157. Biragyn A, Bodogai M, Olkhanud PB, Denny-Brown SR, Puri N, Ayukawa K, et al. Inhibition of lung metastasis by chemokine CCL17-mediated in vivo silencing of genes in CCR4+ Tregs. J Immunother. (2013) 36:258–67. doi: 10.1097/CJI.0b013e318294357c
158. Yin Z, Li C, Wang J, and Xue L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int J Cancer. (2019) 144:933–46. doi: 10.1002/ijc.31744
159. Anfray C, Ummarino A, Andon FT, and Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. (2019) 9. doi: 10.3390/cells9010046
160. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, and Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
161. Hong Y, Wang D, Liu Z, Chen Y, Wang Y, and Li J. Decoding per- and polyfluoroalkyl substances (PFAS) in hepatocellular carcinoma: a multi-omics and computational toxicology approach. J Transl Med. (2025) 23:504. doi: 10.1186/s12967-025-06517-z
162. Hong Y, Li J, Qiu Y, Wang Y, Du Z, Liu Z, et al. Integrative in silico analysis of per- and polyfluorinated alkyl substances (PFAS)-associated molecular alterations in human cancers: a multi-cancer framework for predicting toxicogenomic disruption. Int J Surg. (2025). doi: 10.1097/JS9.0000000000002632
163. Masucci GV, Cesano A, Hawtin R, Janetzki S, Zhang J, Kirsch I, et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I - pre-analytical and analytical validation. J Immunother Cancer. (2016) 4:76. doi: 10.1186/s40425-016-0178-1
164. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
165. Mellman I, Coukos G, and Dranoff G. Cancer immunotherapy comes of age. Nature. (2011) 480:480–9. doi: 10.1038/nature10673
166. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes. vivo J Immunol. (2015) 194:950–9. doi: 10.4049/jimmunol.1401686
167. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology. (2017) 153:1107–1119 e1110. doi: 10.1053/j.gastro.2017.06.017
168. Weber JS, Yang JC, Atkins MB, and Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. (2015) 33:2092–9. doi: 10.1200/JCO.2014.60.0379
169. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
170. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. (2017) 51:1357–69. doi: 10.3892/ijo.2017.4129
171. Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. (2017) 18:2162–74. doi: 10.1016/j.celrep.2017.02.011
172. Kim J, Manspeaker MP, and Thomas SN. Augmenting the synergies of chemotherapy and immunotherapy through drug delivery. Acta Biomater. (2019) 88:1–14. doi: 10.1016/j.actbio.2019.02.012
173. Liu Y and Wang H. Biomarkers and targeted therapy for cancer stem cells. Trends Pharmacol Sci. (2024) 45:56–66. doi: 10.1016/j.tips.2023.11.006
174. Gomez GG, Hutchison RB, and Kruse CA. Chemo-immunotherapy and chemo-adoptive immunotherapy of cancer. Cancer Treat Rev. (2001) 27:375–402. doi: 10.1053/ctrv.2001.0222
175. Kamath SD, Kalyan A, Kircher S, Nimeiri H, Fought AJ, Benson A 3rd, et al. Ipilimumab and gemcitabine for advanced pancreatic cancer: A phase ib study. Oncologist. (2020) 25:e808–15. doi: 10.1634/theoncologist.2019-0473
176. Li S, Yu W, Xie F, Luo H, Liu Z, Lv W, et al. Neoadjuvant therapy with immune checkpoint blockade, antiangiogenesis, and chemotherapy for locally advanced gastric cancer. Nat Commun. (2023) 14:8. doi: 10.1038/s41467-022-35431-x
177. Veen T, Kanani A, Lea D, and Soreide K. Clinical trials of neoadjuvant immune checkpoint inhibitors for early-stage operable colon and rectal cancer. Cancer Immunol Immunother. (2023) 72:3135–47. doi: 10.1007/s00262-023-03480-w
178. Liu X and Qin S. Immune checkpoint inhibitors in hepatocellular carcinoma: opportunities and challenges. Oncologist. (2019) 24:S3–S10. doi: 10.1634/theoncologist.2019-IO-S1-s01
179. Weiss GJ, Blaydorn L, Beck J, Bornemann-Kolatzki K, Urnovitz H, Schutz E, et al. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Invest New Drugs. (2018) 36:96–102. doi: 10.1007/s10637-017-0525-1
180. Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH Jr., Bagala C, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. (2014) 25:1750–5. doi: 10.1093/annonc/mdu205
181. Liu C, Qiang J, Deng Q, Xia J, Deng L, Zhou L, et al. ALDH1A1 activity in tumor-initiating cells remodels myeloid-derived suppressor cells to promote breast cancer progression. Cancer Res. (2021) 81:5919–34. doi: 10.1158/0008-5472.CAN-21-1337
182. Fan Y, Wang Q, Lin G, Shi Y, Gu Z, and Ding T. Combination of using prodrug-modified cationic liposome nanocomplexes and a potentiating strategy via targeted co-delivery of gemcitabine and docetaxel for CD44-overexpressed triple negative breast cancer therapy. Acta Biomater. (2017) 62:257–72. doi: 10.1016/j.actbio.2017.08.034
183. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. (2009) 182:3530–9. doi: 10.4049/jimmunol.0802845
184. Badrinath N and Yoo SY. Recent advances in cancer stem cell-targeted immunotherapy. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11030310
185. Tanaka H, Matsushima H, Mizumoto N, and Takashima A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. (2009) 69:6978–86. doi: 10.1158/0008-5472.CAN-09-1101
186. Steele TA. Chemotherapy-induced immunosuppression and reconstitution of immune function. Leuk Res. (2002) 26:411–4. doi: 10.1016/S0145-2126(01)00138-2
187. Dhodapkar MV, Dhodapkar KM, and Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. (2008) 15:39–50. doi: 10.1038/sj.cdd.4402247
188. Wersto RP, Liblit RL, Deitch D, and Koss LG. Variability in DNA measurements in multiple tumor samples of human colonic carcinoma. Cancer. (1991) 67:106–15. doi: 10.1002/1097-0142(19910101)67:1<106::AID-CNCR2820670120>3.0.CO;2-I
189. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
190. Tang M, Zhao Y, Zhao J, Wei S, Liu M, Zheng N, et al. Liver cancer heterogeneity modeled by in situ genome editing of hepatocytes. Sci Adv. (2022) 8:eabn5683. doi: 10.1126/sciadv.abn5683
191. McDonald KA, Kawaguchi T, Qi Q, Peng X, Asaoka M, Young J, et al. Tumor heterogeneity correlates with less immune response and worse survival in breast cancer patients. Ann Surg Oncol. (2019) 26:2191–9. doi: 10.1245/s10434-019-07338-3
192. Dick JE. Stem cell concepts renew cancer research. Blood. (2008) 112:4793–807. doi: 10.1182/blood-2008-08-077941
193. Meacham CE and Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. (2013) 501:328–37. doi: 10.1038/nature12624
194. Kreso A and Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
195. Wiesner SM, Freese A, and Ohlfest JR. Emerging concepts in glioma biology: implications for clinical protocols and rational treatment strategies. Neurosurg Focus. (2005) 19:E3. doi: 10.3171/foc.2005.19.4.4
196. Li J, Xu N, Hu L, Xu J, Huang Y, Wang D, et al. Chaperonin containing TCP1 subunit 5 as a novel pan-cancer prognostic biomarker for tumor stemness and immunotherapy response: insights from multi-omics data, integrated machine learning, and experimental validation. Cancer Immunol Immunother. (2025) 74:224. doi: 10.1007/s00262-025-04071-7
197. Chen S, Huang C, Li K, Cheng M, Zhang C, Xiong J, et al. Tumor-initiating cells escape tumor immunity via CCL8 from tumor-associated macrophages in mice. J Clin Invest. (2025) 135. doi: 10.1172/JCI180893
198. Hong Y, Li J, Xu N, Shu W, Chen F, Mi Y, et al. Integrative genomic and single-cell framework identifies druggable targets for colorectal cancer precision therapy. Front Immunol. (2025) 16:1604154. doi: 10.3389/fimmu.2025.1604154
199. Yang C, Zhang S, Cheng Z, Liu Z, Zhang L, Jiang K, et al. Multi-region sequencing with spatial information enables accurate heterogeneity estimation and risk stratification in liver cancer. Genome Med. (2022) 14:142. doi: 10.1186/s13073-022-01143-6
200. Islam M, Yang Y, Simmons AJ, Shah VM, Musale KP, Xu Y, et al. Temporal recording of mammalian development and precancer. Nature. (2024) 634:1187–95. doi: 10.1038/s41586-024-07954-4
Keywords: tumor-initiating cells (TICs), digestive system tumors, immunotherapy, tumor microenvironment, immune checkpoints, combination therapy, differentiation therapy
Citation: Zhang Z-y, Zhang X-f, Xu C-h, Wang K-h and Huang F (2025) The immunomodulatory role of tumor-initiating cells in digestive system tumors: from mechanisms to therapy. Front. Immunol. 16:1621464. doi: 10.3389/fimmu.2025.1621464
Received: 01 May 2025; Accepted: 08 July 2025;
Published: 24 July 2025.
Edited by:
Chen Yang, Southern Medical University, ChinaCopyright © 2025 Zhang, Zhang, Xu, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun-hua Wang, d2FuZ2t1bmh1YTE5NjRAMTI2LmNvbQ==; Fang Huang, aGZhbmdAeW51LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship