 Gareth J. Morgan
Gareth J. Morgan Tatiana Prokaeva
Tatiana Prokaeva- 1Boston University Amyloidosis Center, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
- 2Section of Hematology and Medical Oncology, Department of Medicine, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
- 3Department of Pathology and Laboratory Medicine, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
Introduction: Systemic AL amyloidosis is caused by deposition of monoclonal antibody light chains (LC) as insoluble amyloid fibrils in multiple tissues, leading to irreversible and eventually fatal organ damage. Each patient has a unique LC sequence that appears to define its propensity to aggregate. The complexity and diversity of LC sequences has impeded efforts to understand why some LCs aggregate to cause disease while others do not.
Methods: We investigated residue changes, relative to the inferred precursor germline sequences, in monoclonal LCs associated with AL amyloidosis and multiple myeloma (MM), derived from the AL-Base resource. Consensus matrices, calculated using healthy polyclonal repertoire sequences from Observed Antibody Space (OAS), were used to determine the relative frequency of each residue in the monoclonal LC sequences.
Results: A subset of residues observed in AL-associated LCs was uncommon in the healthy repertoire, but these residues were highly diverse and were also observed in MM-associated LCs. We identified multiple positions that more frequently harbor uncommon residues in AL-associated LCs than OAS-derived LCs, including several positions that have previously been identified. However, each individual residue change occurs in only a small fraction of LCs, indicating that many types of residue change can contribute to disease. Furthermore, positions where residue changes occur most frequently were not enriched in amyloidosis-associated residues.
Discussion: These data provide a framework for future investigations into sequence determinants of amyloid propensity, supporting efforts towards earlier recognition and diagnosis of AL amyloidosis.
1 Introduction
Aggregation of amyloid fibrils derived from antibody light chain (LC) proteins leads to systemic AL amyloidosis (1). This incurable disease causes progressive organ failure due to displacement of healthy tissue and other toxic mechanisms (2, 3). Amyloid-forming LCs are secreted from monoclonal B cells, most commonly plasma cells that aberrantly proliferate in the bone marrow (2, 3). However, AL amyloidosis is rare and most individuals with an expanded population of such clonal plasma cells, known as monoclonal gammopathy or plasma cell dyscrasia (PCD), do not experience clinically significant amyloid deposition (4). In multiple myeloma (MM), a plasma cell cancer, circulating LC levels are high but amyloid deposition is relatively rare and generally does not cause symptoms (5). Transcriptomic analysis of plasma cells has not identified consistent differences between AL amyloidosis and MM, although both diseases involve similar genomic structural variants (6). Instead, the sequence and properties of the secreted monoclonal LC are hypothesized to determine risk of amyloidosis (7–10). LCs that can aggregate as amyloid fibrils are said to be “amyloidogenic”, but the sequence and structural determinants of amyloidogenicity are not well understood (11). Predicting which monoclonal LCs carry a risk of aggregation would allow earlier diagnosis of AL amyloidosis, potentially leading to improved patient outcomes. Therefore, it is important to understand how amyloidogenic LCs differ from those of the healthy immune repertoire and MM clones.
The major challenge to such a prediction is the diversity of LC sequences. LCs are components of antibodies that have undergone recombination and somatic hypermutation in response to antigen (8). A typical LC accumulates 5–15 amino acid residue changes, relative to the germline-encoded variable, joining and constant precursor genes (IGVL, IGJL and IGCL, respectively) from which it was derived (11). These mutations are primarily created by activation-induced cytidine deaminase, leading to a characteristic pattern of mutations that differs between precursor genes (12, 13). Healthy humans have an estimated repertoire of 106–107 unique LC sequences, several orders of magnitude less than the heavy chain (HC) repertoire, but far more complex than most other proteins (14).
All LCs must fold to their native state in order to be secreted from plasma cells (15). LCs form two structural domains with similar immunoglobulin folds, variable (VL) and constant (CL), corresponding to the IGVL-IGJL and IGCL gene sequences, respectively. Sequence variation is concentrated in the three complementarity determining regions (CDRs 1–3) of the VL domain, which form the antigen binding site. However, residue changes in framework regions (FRs 1–4), which account for the remainder of the VL domain sequence, also contribute to LC diversity. Peptides from the VL domain, in non-native conformations, form the core of most AL amyloid fibrils (9, 16–23). Residue changes in the CL-domain can also influence amyloidogenicity (23–26), but these are uncommon and less well characterized because most studies have focused on VL domains. CL domain sequences are often not determined or reported.
Several hundred monoclonal LC sequences associated with AL amyloidosis and other PCDs, primarily MM, have been determined (27–34). These sequences are collected in AL-Base, a public repository run by Boston University Amyloidosis Center (11, 35). A subset of precursor variable genes (IGKV and IGLV, encoding κ and λ LCs, respectively) is more common in AL amyloidosis than in MM or the polyclonal immune repertoire (11, 27, 28, 36). Notably, around 75% of AL clones secrete a λ LC, compared to 40% of MM clones (11). Among healthy individuals, approximately one third of circulating antibodies has a λ LC (37). One precursor gene, IGLV6-57, accounts for 18% of AL LCs, making it the most frequently used precursor, versus 1% of MM clones and 2% of the healthy repertoire (11). In contrast, IGKV1-33, IGLV1-44, IGLV2-14 and IGLV3-1 collectively account for 44% of AL clones but are also relatively common in MM (27% of clones) and the polyclonal repertoire (17% of sequences) (11). Therefore, specific residue changes, relative to the germline sequences of these genes, appear to predispose LCs towards or away from amyloid formation.
Two approaches to investigating the roles of LC sequence in AL amyloidosis are to compare multiple sequences (11, 36, 38–40) and to investigate the effects of specific residue changes on a single sequence (41–44). In both cases, a key challenge is selecting the most appropriate control sequences. Most investigators have used germline or MM LCs as supposedly “non-amyloidogenic” controls. Germline LCs represent an unmutated, wild-type-like reference, whereas MM LCs are, like AL LCs, produced from aberrant plasma cells after selection for antigen binding. However, both types of LC can be induced to aggregate in vitro, although this typically occurs less readily than for AL LCs (43–46). Neither control is ideal due to the number of residue differences between any two LCs, since the effect of each position depends on the rest of the sequence. A further problem is that the identity of the germline gene can only be unambiguously determined from the genome sequence, which is not generally available. Sequences derived from mRNA or peptides must be aligned to the germline gene with the highest homology. This is complicated by paralogs and multiple alleles of most germline genes (8, 47, 48). Therefore, identifying the effect of individual residue changes—and thereby predicting the properties of a new sequence—is not possible without extensive biochemical characterization (41–44).
Individual residue changes may destabilize the LC native state, enhance formation of amyloid fibrils or both (41–44). However, it is difficult to extrapolate beyond the specific LC that was studied. Only a few residue changes are known to contribute to amyloidogenicity in more than one LC. Examples include the R25G polymorphism in the IGLV6-57 gene, which alters the stability of the native state (46), and N-glycosylation of asparagine 86 in LCs derived from IGKV1-family genes (38, 49), although the mechanisms by which N-glycosylation promote amyloid formation are unknown (50). More recent attempts to identify amyloidogenic sequences have used machine learning algorithms to distinguish amyloid from non-amyloid LCs (51–54). However, these algorithms performed less well on a test cohort of several hundred sequences that we compiled following the recent revision of AL-Base (11).
Here, we analyzed the recently extended AL-Base data (11), as well as a larger set of polyclonal sequences from the Observed Antibody Space (OAS) resource (55, 56). We evaluated the frequency and positions of individual residue changes that are observed in AL amyloidosis, relative to MM and the polyclonal repertoire. By creating consensus matrices for each precursor gene from thousands of OAS sequences, we estimated the likelihood of observing each possible residue at each position in the LC sequence in the healthy polyclonal repertoire. This procedure allowed “uncommon” residues to be identified, which we hypothesize are important in determining the properties of the LC. We used this approach to prospectively identify positions that frequently harbor uncommon residues in AL-associated LCs. We also evaluated the frequency of individual residue changes that have previously been proposed to be amyloidogenic.
2 Methods
2.1 Terminology and nomenclature
Throughout this manuscript, we use the IMGT nomenclature and numbering system for LC genes, proteins and residues (47, 57). For each antibody, single germline precursor variable and joining genes (IGVL and IGJL, encompassing both κ and λ gene fragments from the IGK and IGL loci, respectively) are recombined to yield a variable region (IGKV-IGKJ for κ or IGLV-IGLJ for λ), encoding the ~110-residue LC VL-domain protein. The IMGT numbering system has 127 possible residue positions in LCs, including conserved gaps to accommodate insertions. This numbering system allows direct comparison between the different types of immunoglobulin domains, so several positions such as 58–64 are very rarely occupied by residues within human LCs. “Residue position” always refers to IMGT numbering.
2.2 Light chain sequences
This study focuses on the frequency of individual residue changes within LC VL-domains associated with AL amyloidosis, MM, or the polyclonal immune repertoire represented by OAS (56). We refer to these sequences as AL, MM and OAS LCs, respectively. The overall workflow is shown in Supplementary Figure 1. A “set” of sequences is defined as all the LCs from a specific germline gene and disease or repertoire of origin. Only complete IGVL-IGJL sequences with no ambiguous or missing residues were used for analysis. Sequences with CDR3 insertions longer than the available space in IMGT numbering were also excluded. We did not consider nucleotide sequences or CL domains, since these are not available for all LCs. We refer to “residue changes” rather than “mutations” since only protein sequences were analyzed and to avoid ambiguity between germline and somatic mutations.
In total, 746 AL, 969 MM and 8,047,747 OAS LC sequences were analyzed. AL LCs included monoclonal LC sequences from the AL-Base “AL-PCD” category, comprising all sequences from individuals with a diagnosis of AL amyloidosis, regardless of the underlying hematological malignancy. Note that this selection differed from that used in our previous studies (11, 50), which excluded LC sequences from the AL/MM subcategory. MM LCs included sequences from the “MM” and “SMM” (smoldering MM) subcategories, but excluded those with a diagnosis of light chain deposition disease. Unique OAS LCs were identified from 66 OAS samples derived from healthy adults as previously described (11).
Twenty IGVL genes (8 IGKV and 12 IGLV), for which at least five AL LCs were available in the AL-Base data, were analyzed: IGKV1-12, IGKV1-16, IGKV1-33, IGKV1-39, IGKV1-5, IGKV3-15, IGKV3-20, IGKV4-1; and IGLV1-36, IGLV1-40, IGLV1-44, IGLV1-47, IGLV1-51, IGLV2-8, IGLV2-14, IGLV2-23, IGLV3-1, IGLV3-19, IGLV3–21 and IGLV6-57. Results in the main body of the manuscript focus on LCs derived from IGKV1–33 and IGLV2-14, which are frequently observed in AL, MM and OAS LCs. Data for other germline genes is shown in Supplementary Data Sheet 1.
2.3 Alignment
LC nucleotide and protein sequences were assigned to germline precursor genes using the IMGT High-VQuest or DomainGapAlign tools, respectively (58). Paralogous IGKV genes from the proximal and distal loci (59) were counted as being from the proximal locus (e.g., LCs assigned to IGKV1-33 and IGKV1D-33 were both counted as being derived from IGKV1-33). For nucleotide sequences, the translation of the sequence provided by High-VQuest was used. OAS LCs are assigned to a precursor gene as part of the deposition process (56) so these assignments were not reanalyzed. Residues were aligned to the IMGT numbering system using the ANARCI tool (60), which more consistently assigned sequence gaps than other tools for these sequences. The analyses described below are sensitive to misaligned gaps in the IMGT numbering system, which may be misinterpreted as insertions or deletions (indels). There were 71 AL-Base sequences where alignment by ANARCI led to indels, relative to the assigned germline sequence. These were checked manually and 43 alignments were modified to favor residue replacements over indels, so as to maintain the positions of gaps that are present in germline sequences and minimize alignment biases.
2.4 Consensus matrices and uncommon residue frequency
For each IGVL gene, we separately aligned the AL, MM or OAS VL domain sequences using ANARCI (60). Following the approach of Sheng and coworkers (13), we constructed consensus matrices to define the distribution of residues at every position. An example matrix is shown in Supplementary Figure 2. The identity of the assigned IGVL allele and IGJL gene were not considered. The number and fraction of sequences with each residue at all 127 IMGT positions was calculated. For each group of sequences, this procedure yielded a 127 × 21 matrix, corresponding to the fraction of each residue, including gaps, at each position. We created a total of 59 matrices, corresponding to AL, MM and OAS LCs from each gene except IGLV1-36, for which no MM LCs were present in AL-Base. This approach avoided multiple sequence alignments, which are computationally expensive for large numbers of sequences and are prone to errors around the indels which are frequent in CDR3. The OAS matrices are provided in Supplementary Table 1.
To evaluate the residue-wise variability at each position within the matrices, we calculated the Gini coefficient (61), implemented in the R ineq package (62). This is a measure of the inequality of a distribution, which varies between 0 (corresponding to equal fractions of all possible residues) and 1 (a single residue observed in all sequences). To compare the residue-wise differences between sets of sequences, we calculated the Pearson correlation coefficient (ρ) between the columns of the relevant consensus matrices for each pair of residues (ρ = 1 indicates identical distributions of residues).
To compare pairs of consensus matrices, we calculated difference matrices by subtraction. For global comparison between groups of sequences, we calculated the pairwise Euclidian distance between each consensus matrix, which is the square root of the sum of squared differences between each of the positions in the matrices. These distances are equivalent to the total magnitude of the corresponding difference matrix. We subjected the results to hierarchical clustering analysis in R (63) to yield a phylogenetic tree (or dendrogram).
For all AL-Base sequences analyzed, the frequency of each residue within the appropriate OAS consensus matrix was calculated. Based on the distributions of these frequencies, we defined “common” residues as those appearing in ≥ 10% of OAS sequences derived from that germline gene and “uncommon” residues as those appearing in < 10% of OAS sequences. To visualize the frequency in OAS of each residue within a sequence of interest, we plotted the values from the corresponding consensus matrix as a “frequency profile”.
For each group of LCs, the number sequences harboring common and uncommon residues at each position was calculated. For prospective analysis and creation of frequency profile plots, all positions were considered. For previously identified positions, numbers of sequences were counted with both the specific residue change as published, and with any uncommon residue at that position. Residue changes described by Hurle and coworkers (41) were assigned to the germline gene of the LC in which they were originally described. Insertion of proline within CDR3 of an IGKV1-33-derived LC was identified as potentially amyloidogenic by Randles and coworkers (64). This is referred to as 95ProIns in the original publication, which uses the Kabat numbering system, and P115PP here using the IMGT numbering system. Identifying these insertions is difficult because of the variable length of CDR3, which can lead to ambiguous alignments. Sequences were identified where two consecutive proline residues occur around IMGT position 115 and at least one residue is inserted into the gap within CDR3, which corresponds to positions 110–113 of the germline IGKV1-33 sequence.
To compare the frequencies of residue changes within groups of LCs, we calculated the odds ratios (OR), 95% confidence intervals (CI) and associated p-values for each pairwise comparison. These parameters were calculated directly from counts, rather than estimated from a generalized linear model. To avoid division by zero, a correction factor of 0.1 was added to all counts before ORs were calculated. OR significance was calculated directly from the natural logarithms of ORs and their standard errors using the normal distribution, implemented in R (63). Positions with a positive OR for the AL vs. OAS comparison, but where only germline residues were observed in AL LCs, were excluded because these were determined to be artifacts caused by the large difference in sample sizes. P-values were corrected for multiple testing using the false discovery rate (FDR) method (65), where FDR < 0.05 was considered statistically significant.
2.5 Sequence logos
To visualize residue frequency at each position across the LC sequence, we generated customized sequence logo plots (shown in Supplementary Data Sheet 1) using the R package ggseqlogo (66) that differed from the original sequence logo concept (67). Because residue changes were distributed throughout the LC sequence, the information content at each position was dominated by the germline sequence. To highlight non-germline residues, we excluded the most frequent residue from the consensus matrix and created a logo where the height of each letter represented only its frequency in the alignment.
2.6 Structural analysis
Crystal structures of germline IGKV1-33, IGLV2-14 and IGLV6-57 VL-domains were available for analysis (PDB entries 2Q20 (43) 6SM1 (44) and 2W0K (68), respectively). In addition, we generated VL domain models for the 20 IGVL genes studied using the Alphafold 3 server https://alphafoldserver.com/ (69). Protein sequences corresponding to the *01 allele of each IGKV or IGLV gene and appropriate IGKJ1*01 or IGLJ1*01 gene were submitted using the default parameters. The resulting models had good agreement with the crystal structures (Cα root mean square deviations of 0.476, 0.435 and 0.679 Å for IGKV1-33, IGLV2-14 and IGLV6-57, respectively). Secondary structures and solvent-accessible surface areas were calculated using DSSP (70, 71). Residues with < 10% surface exposure, relative to reference peptides (72), were defined as buried and other residues were defined as solvent exposed. Data were mapped onto structures using ChimeraX (73).
2.7 Analysis software versions used
Most analysis was carried out using R v 4.2.2 (63) via the RStudio environment (74). The following packages were used: Biostrings v 2.66.0 (75), broom v 1.0.3 (76), cowplot v 1.1.1 (77), furrr v 0.3.1 (78), future v 1.33.0 (79), ggpubr v 0.6.0 (80), ggtree v 3.4.1 (81), ggseqlogo v 0.1 (66), ineq v 0.2-13 (62), msa v 1.30.1 (82), openxlsx v 4.2.8 (83), rstatix v 0.7.2 (84), and Tidyverse v 1.3.2 (85). Local installations of ANARCI (60) and DSSP v 2.1.0 (70, 71) were used on the Boston University Shared Computing Cluster. Alphafold 3 was accessed via the web interface, https://alphafoldserver.com/ (69). ANARCI can also be accessed via a web interface, https://opig.stats.ox.ac.uk/webapps/sabdab-sabpred/sabpred/anarci/.
3 Results
3.1 Sets of LCs derived from the same germline gene have similar patterns of residue changes
The frequency of mutations varies across LC sequences due to the biased mechanism of somatic hypermutation (13). Within each IGVL gene, alignments of OAS sequences provide a reference for this variability. We constructed consensus matrices from all OAS LCs for each IGVL gene, which define the frequency of each residue, including gaps, at all 127 IMGT positions. These matrices are visualized as heat maps in Figures 1A, B for IGKV1-33 and IGLV2-14, respectively. Results for other germline genes are shown in Supplementary Data Sheet 1. Frequency data for the OAS matrices are provided in Supplementary Table 1. The germline residue was present in > 90% of sequences at most positions. However, in positions that vary between alleles or are prone to mutation, the fraction of sequences with the most common residue was lower. These positions are frequent in CDRs but occur throughout the sequences. For the 20 IGVL genes where at least five AL LCs were available, we generated additional consensus matrices for AL and MM LCs and calculated differences between pairs of matrices. Data for IGKV1-33 and IGLV2-14 are shown in Figure 1, and data for other IGVL genes is shown in Supplementary Data Sheet 1.
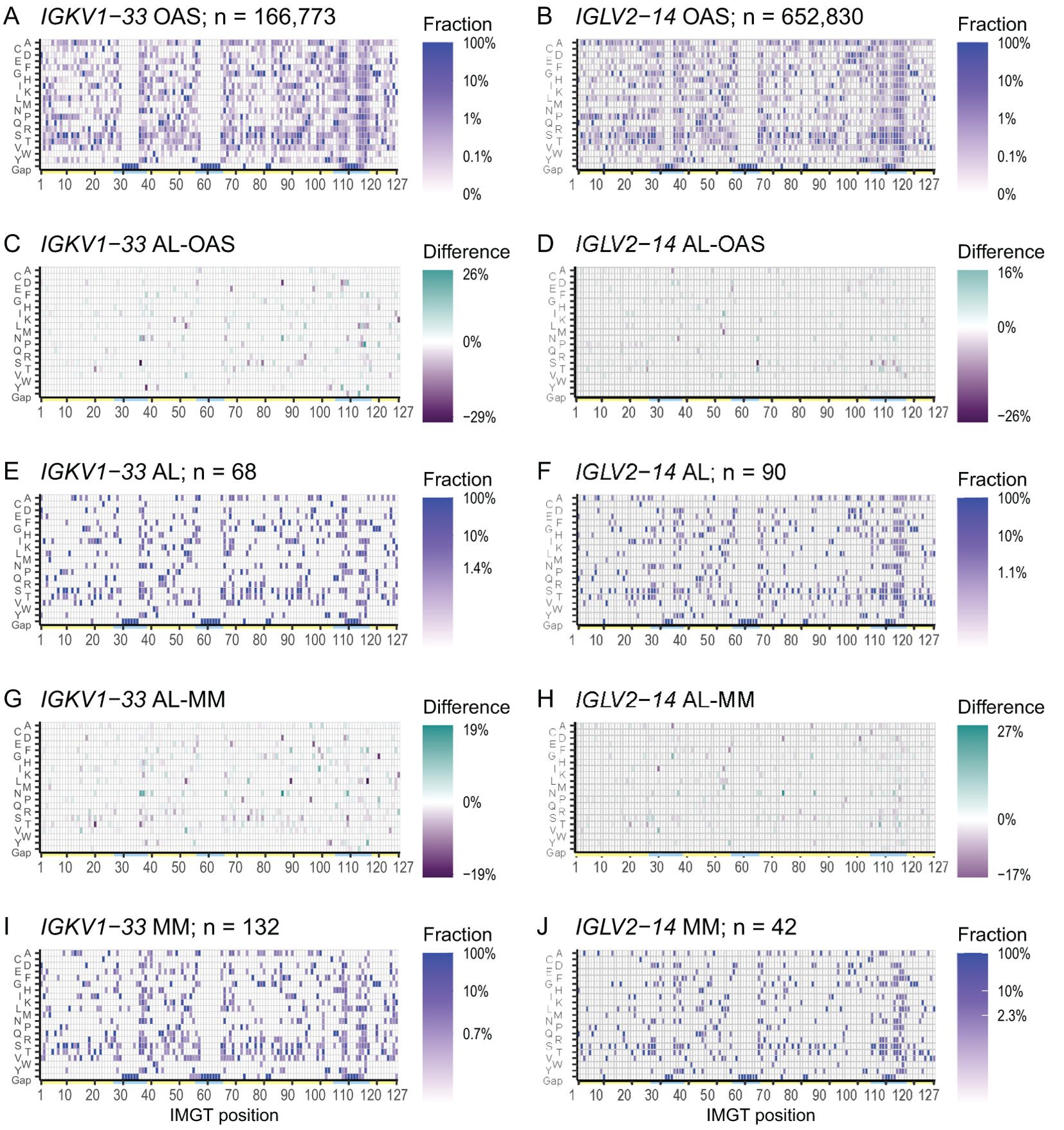
Figure 1. Patterns of residue use are similar between AL, MM and OAS LCs derived from the same IGVL gene. Heatmaps of the consensus matrices and difference matrices for alignments of AL, MM and OAS LCs derived from IGKV1-33 (left column) and IGLV2-14 (right column). Consensus matrix heatmaps (A, B, E, F, I, J) show the frequency of each residue, including gaps, in the corresponding multiple sequence alignment. The scale bars show the minimum frequency at which residues were observed. Yellow and cyan lines show the positions of FRs and CDRs, respectively. Difference heatmaps (C, D, G, H) are calculated by subtracting each position in the comparator matrix from that in the AL matrix. Residues which are more frequent in AL LCs are shown in green and residues which are less frequent in AL LCs are shown in purple. Heatmaps, sequence logo plots and statistics for 20 IGVL genes are shown in Supplementary Data Sheet 1.
To further evaluate the distribution of residue changes within sets of LCs, we generated sequence logos for AL, MM and OAS LCs, which show the frequency at which each residue was observed along the sequences (Supplementary Data Sheet 1). Quantitation of the variation within and between sets of LC sequences showed that the positions that differ between AL and MM LCs are also most variable positions within the sequences (Supplementary Data Sheet 1).
To investigate the relative contributions of within-gene and between-gene variability, we measured the pairwise distances between each of the consensus matrices and analyzed the resulting data using hierarchical clustering (Supplementary Figure 3). This analysis showed that AL LCs were consistently more closely related to MM and OAS LCs derived from the same gene than to AL LCs derived from other genes.
3.2 Uncommon residues are associated with amyloidogenicity in specific sequences
We calculated the frequency at which every residue in AL or MM LC sequences was observed in the OAS consensus matrices. Histograms of these frequencies are shown in Figure 2A. Both AL and MM sequences show a bimodal distribution of residue frequencies. Based on these distributions, we defined a residue as “uncommon” if it occurred in < 10% of sequences from the relevant OAS consensus matrix. Residues which occur in ≥ 10% or more of OAS sequences were defined as “common”. Similar proportions of residues were uncommon in both AL and MM LCs (Figure 2B) and uncommon residues were observed throughout the LC sequences in both groups (Figure 2). We examined the distribution of residues that occur in < 1% of OAS sequences (dark bars in Figure 2B), which also occurred throughout the LC sequence.
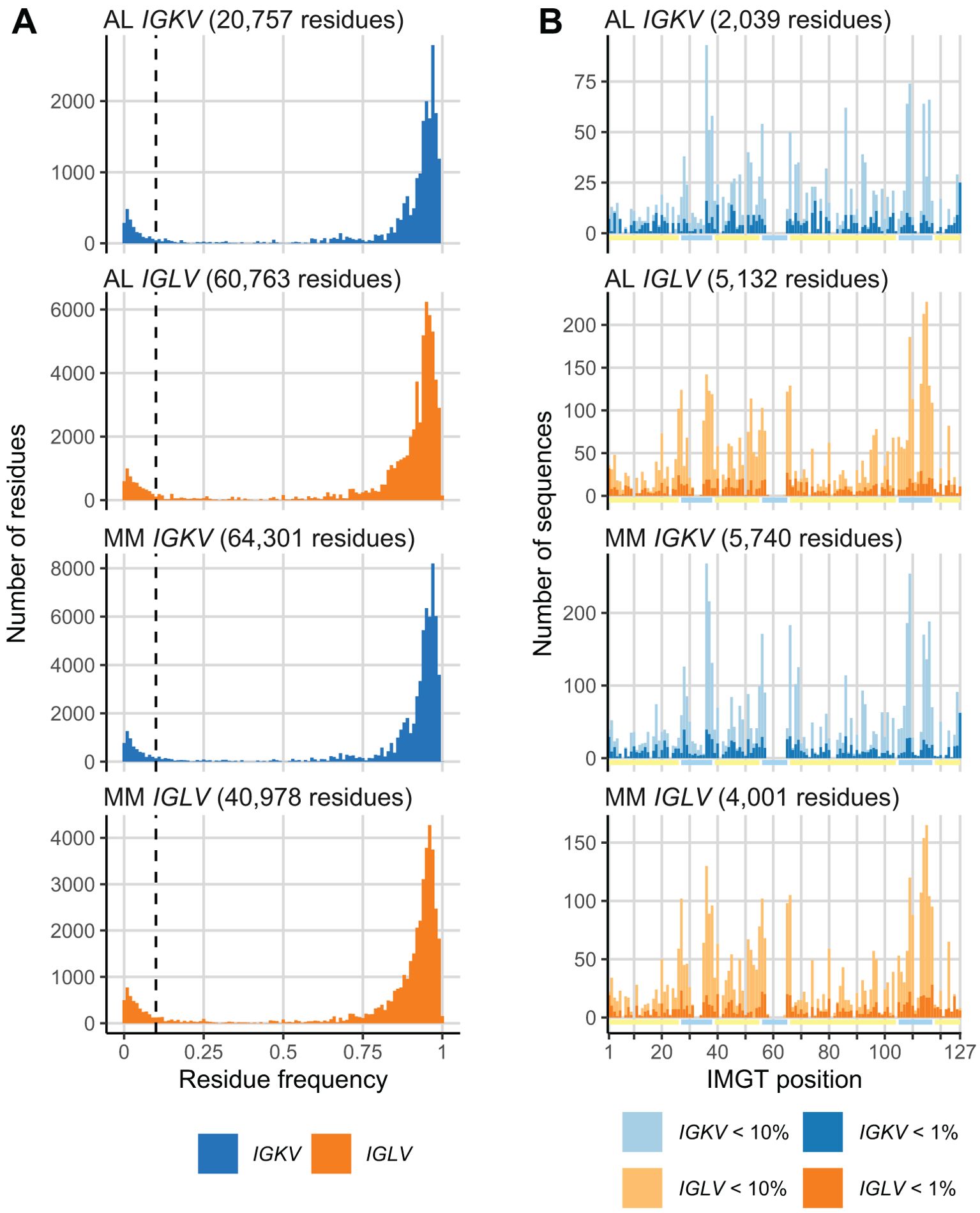
Figure 2. A subset of monoclonal LC residues is uncommon in the OAS repertoire. (A) Histograms showing the frequency at which each residue in AL and MM LCs is observed in the OAS consensus matrices. A 10% threshold, used to distinguish common and uncommon residues, is shown as a dashed line. The total number of residues in each set of sequences is shown. (B) Distribution of uncommon residues (present in < 10% of OAS sequences) across the sequences of LCs. Residues that occur in < 1% of OAS sequences are shown as darker bars. Yellow and cyan lines show the positions of FRs and CDRs, respectively. The total number of uncommon residues in each set of sequences is shown.
The distribution of residue frequencies was visualized graphically as a “frequency profile” for two amyloidogenic LCs, known as AL-09 and Pat-1, which are derived from the IGKV1-33 and IGLV2-14 genes, respectively (43, 44) (Figure 3). The frequency of each residue within the OAS repertoire alignments is shown. Baden and coworkers identified N34I and Y87H (N40I and Y103H in IMGT numbering) as destabilizing residue changes in AL-09 in vitro, relative to its germline sequence (43). These two residues were observed at lower frequencies within the OAS alignment for IGKV1-33 (0.81% and 0.77%, respectively) than the rest of the AL-09 sequence. Similarly, Kazman and coworkers identified L81V (IMGT L94V) as highly destabilizing to Pat-1 in vitro (44). This residue is the least common of all Pat-1 residues in the OAS alignment for IGLV2-14 (0.55%).
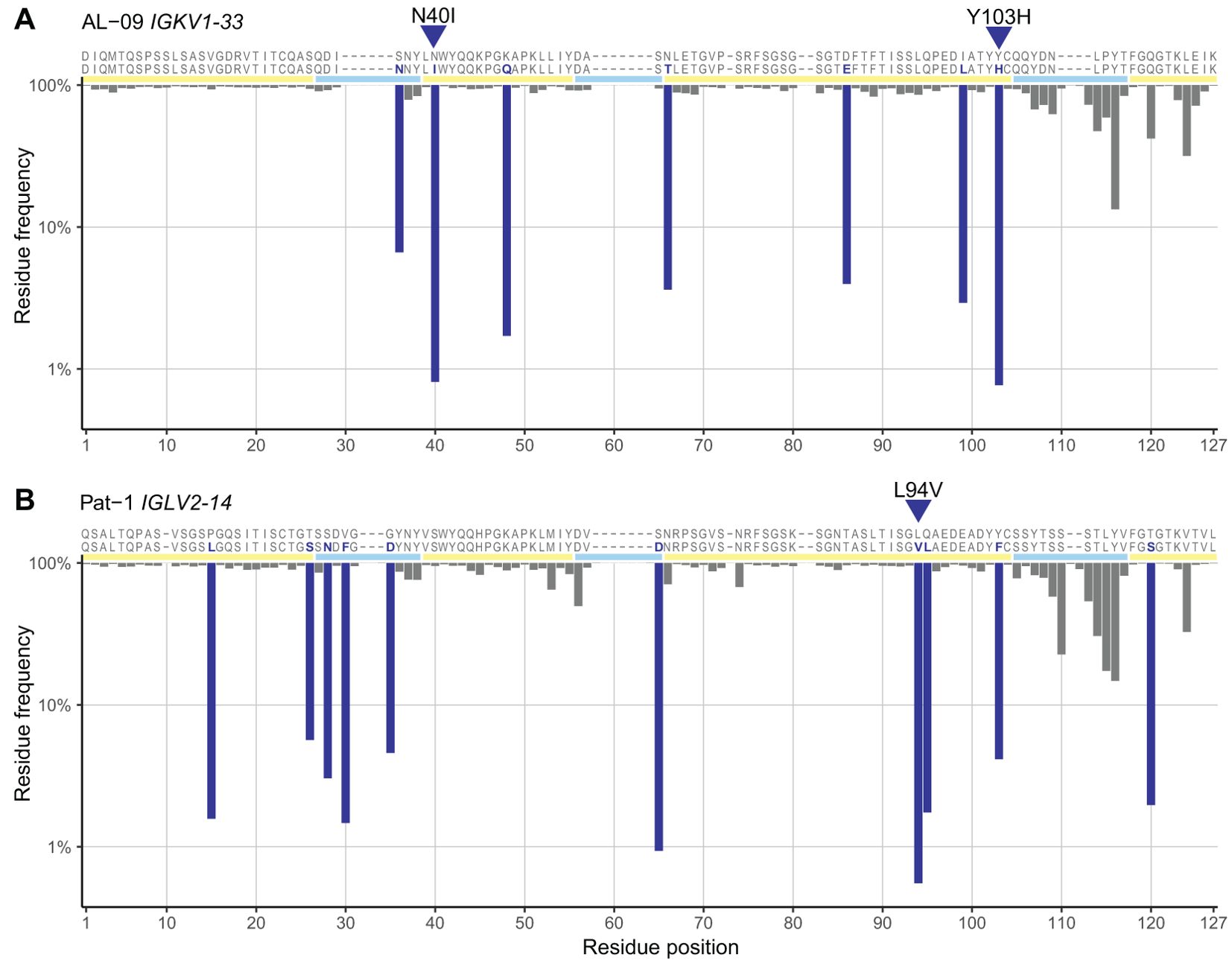
Figure 3. Residue frequency profiles of previously studied AL LCs. The sequences of two LCs, known as AL-09 (43) (A) and Pat-1 (44) (B) are shown with their assigned germline sequences, aligned to the IMGT numbering system. Residues that differ from the germline sequence are highlighted in dark blue and positions of FRs and CDRs are shown by yellow and cyan lines, respectively. The frequency of each residue and position within the OAS consensus matrices is shown as an inverted bar chart. Residue changes previously identified as destabilizing, which are the least common residues in each sequence, are indicated with blue triangles. The residue changes were originally reported with Kabat numbering (AL-09 N34I and Y87H) or sequential numbering (Pat-1 L81V).
3.3 Residue changes in many positions are associated with AL amyloidosis
To identify residue changes that are enriched or depleted in AL LCs compared with other LCs, we measured the proportion of sequences that harbor an uncommon residue at each position in all AL, MM and OAS LC sequences, segregated according to precursor gene. We calculated ORs and 95% confidence intervals for observing an uncommon residue at each position for AL vs. MM and AL vs. OAS LCs. Data at each position in LCs derived from IGKV1-33 and IGLV2-14 are shown for the AL vs. MM (Figure 4A) and AL vs. OAS (Figure 4B) comparisons. Equivalent results for other IGVL genes are shown in Supplementary Data Sheet 1, and details of all positions identified are reported in Supplementary Table 2. Without correction for multiple comparisons, six positions in IGKV1-33, but no positions in IGLV2-14, were significantly enriched for uncommon residues in AL vs. MM LCs (p < 0.05, orange symbols). Two positions in IGLV2-14 harbor uncommon residues less frequently in AL LCs than in MM LCs (p < 0.05, purple symbols). For the comparison between AL and OAS sequences, 26 positions among IGKV1-33 LCs and 7 positions among IGLV2-14 LCs were significantly enriched for uncommon residues (p < 0.05, orange symbols). Three positions in IGKV1-33 LCs and two positions in IGLV2-14 LCs were under-represented in uncommon residues for the AL vs. OAS comparison (p < 0.05, purple symbols).
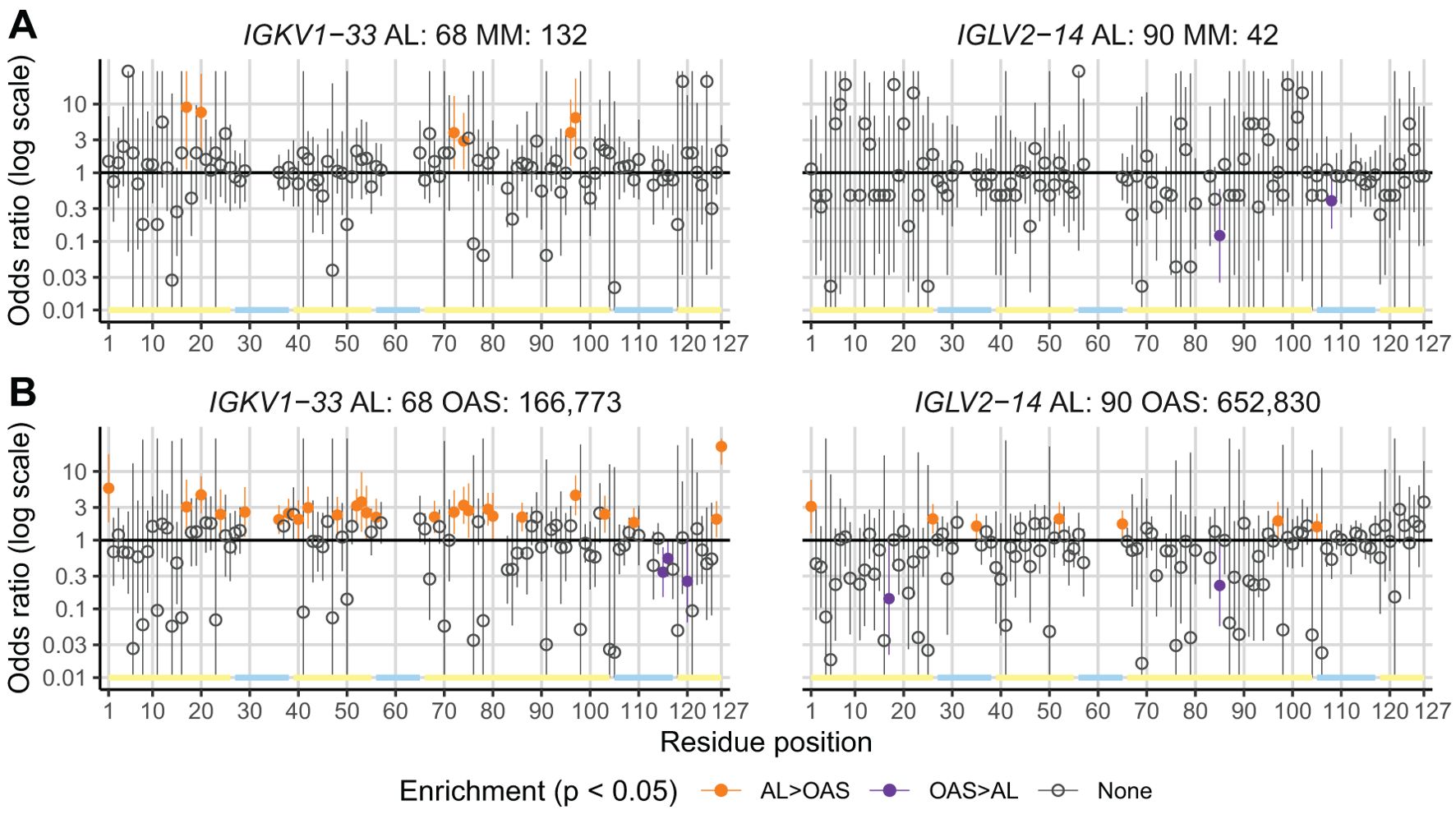
Figure 4. A subset of positions is enriched for uncommon residues in AL vs. MM or OAS LCs. Plots of ORs (points) and 95% confidence intervals (lines) for any uncommon residue at each position in LC sequences for the AL vs. MM (A) and AL vs. OAS (B) comparisons. Positions where AL LCs are enriched for uncommon residues (p < 0.05 before correction for multiple testing) are highlighted in orange. Positions where AL LCs were less likely to harbor uncommon residues are shown in purple. Positions corresponding to gaps in the germline sequence are not shown. The positions of FRs and CDRs are shown by yellow and cyan lines, respectively. Data for LCs derived from IGKV1–33 and IGLV2–14 are shown as examples; figures for other IGVL genes are shown in Supplementary Data Sheet 1 and additional data are provided in Supplementary Table 2.
Analysis of the 20 precursor genes where at least 5 AL sequences were available showed that the number and location of positions enriched for uncommon residues differ substantially between genes (Figure 5; Supplementary Table 2). Uncommon residues were over-represented in the AL vs. OAS comparison at 62 positions, or 125 gene/position combinations, encompassing all four FRs and three CDRs. Uncommon residues were under-represented in a further 20 positions, or 25 gene/position combinations. Notably, few of these positions were common between precursor genes, with 30 positions observed in only a single gene. The VL-domain N- and C-termini were the most frequently enriched positions (6 and 9 genes, respectively), although this may reflect systematic biases due to sequencing strategies. Other positions enriched in uncommon residues in multiple genes were positions 52 (6 genes), 85 (5 genes), 97 (6 genes) and 103 (5 genes). We correlated the variability at each position with its degree of enrichment in the AL vs. OAS comparison and observed that uncommon residues were most frequently enriched at positions that were conserved in OAS LCs (Supplementary Figure 4). Excluding the N- and C-termini, 85 of 110 positions that were enriched for uncommon residues in AL vs. OAS were within FRs (Figure 5; Supplementary Figure 4).
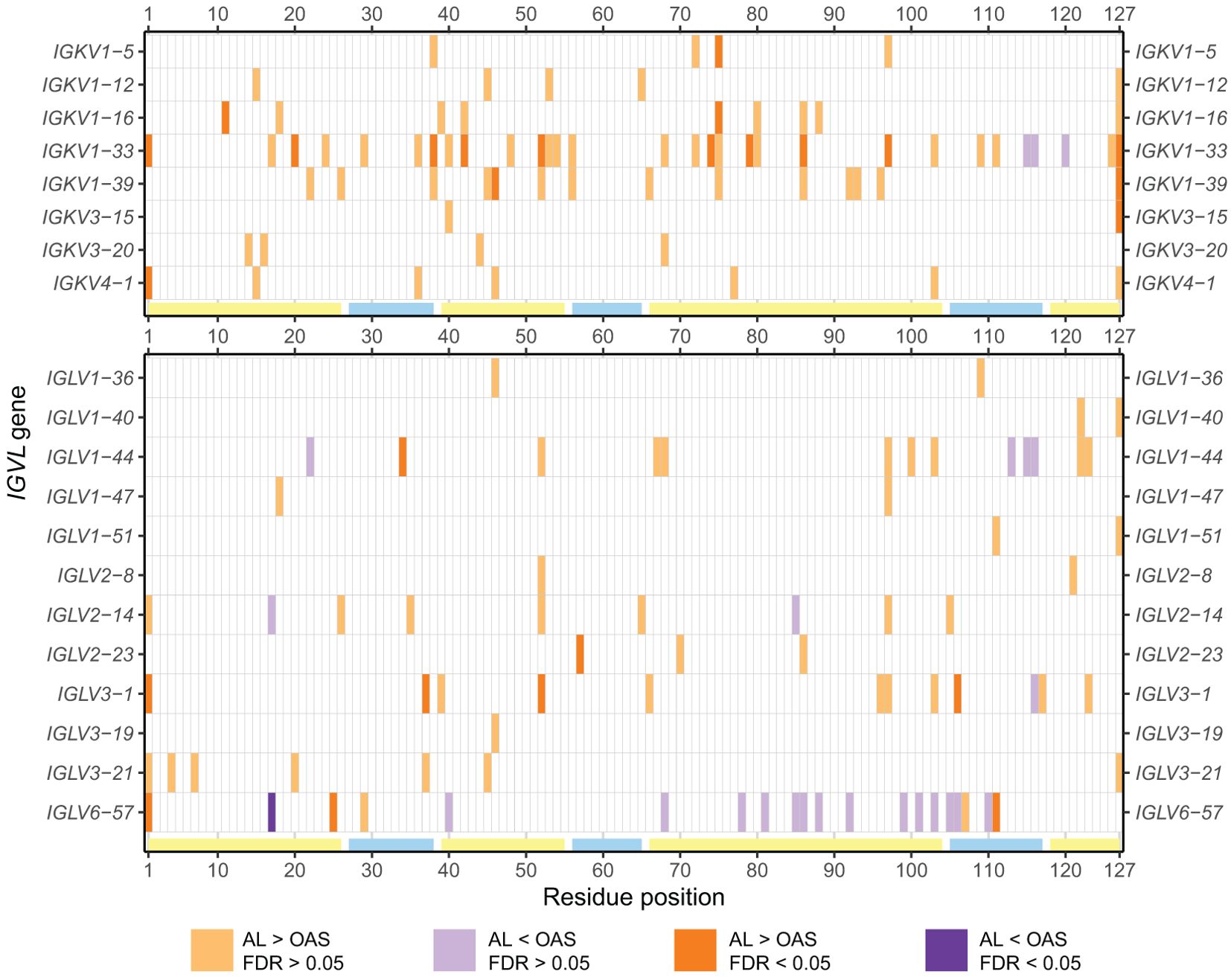
Figure 5. Positions enriched in uncommon residues vary between IGVL genes. Positions where uncommon residues are significantly more (orange) or less (purple) frequently observed in AL vs. OAS LCs are shown for all IGVL genes examined (p < 0.05). Positions for which significance was retained after correction for multiple testing (FDR < 0.05) are shown as darker shades. The positions of FRs and CDRs are shown by yellow and cyan lines, respectively. Additional information is provided in Supplementary Table 2.
We mapped these positions onto the available crystal structures of isolated VL-domains corresponding to IGKV1-33, IGLV2-14 and IGLV6-57 (43, 44, 68) (Figure 6). Structural mapping of these data for other IGVL genes, using computational models of the isolated VL domains, is shown in Supplementary Figure 5. Positions enriched for uncommon residues occur in a variety of structural contexts, including both solvent-exposed and buried residues; those in hydrogen-bonded secondary structures and unstructured loops; and residues that interact with the LC’s heavy chain partner. These details are provided in Supplementary Table 2. Again, there were no clear patterns of which positions were enriched for AL-associated residues
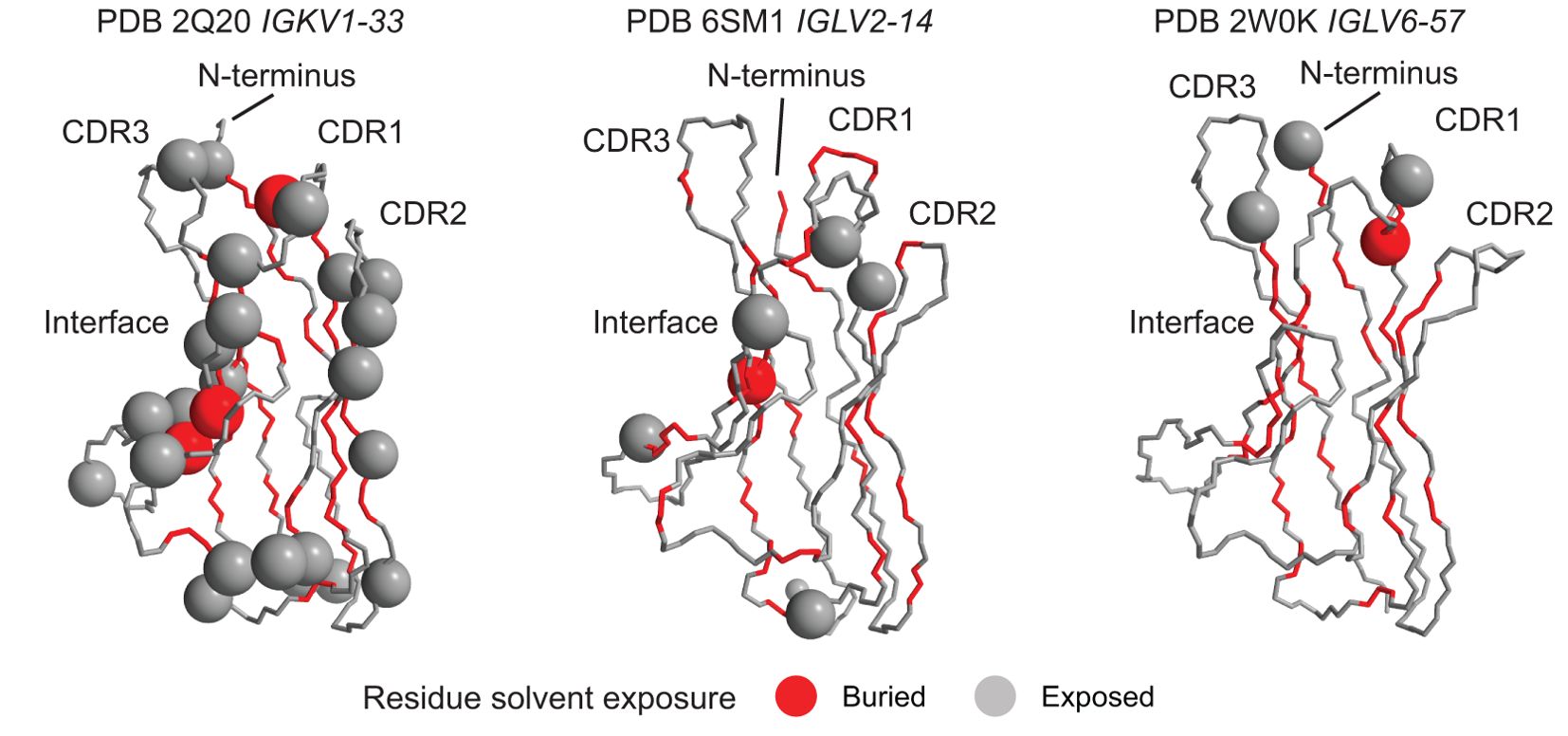
Figure 6. Diverse structural regions are more frequently mutated in AL than OAS LCs. Positions where uncommon residues are more frequent in AL vs. OAS LCs (orange panels in Figure 5) are shown as spheres on the available crystal structures of isolated VL-domains. Other residues are shown as a backbone trace. Residues whose sidechains are not accessible to the solvent are colored red and those with solvent-exposed sidechains are colored gray. The concave β-sheet that forms interface with the heavy chain in antibodies and with a partner LC in homodimers is indicated. Note that not all N-terminal residues are resolved in the structures. Data for other genes are shown in Supplementary Figure 5.
We next used this data to reexamine positions and residue changes that have previously been identified as associated with AL amyloidosis. A total of 48 positions were studied (Tables 1–3). Additional information for these positions is provided in Supplementary Table 3. We divided the residue changes into three groups, associated with all IGKV-derived LCs, specific IGKV genes and specific IGLV genes.
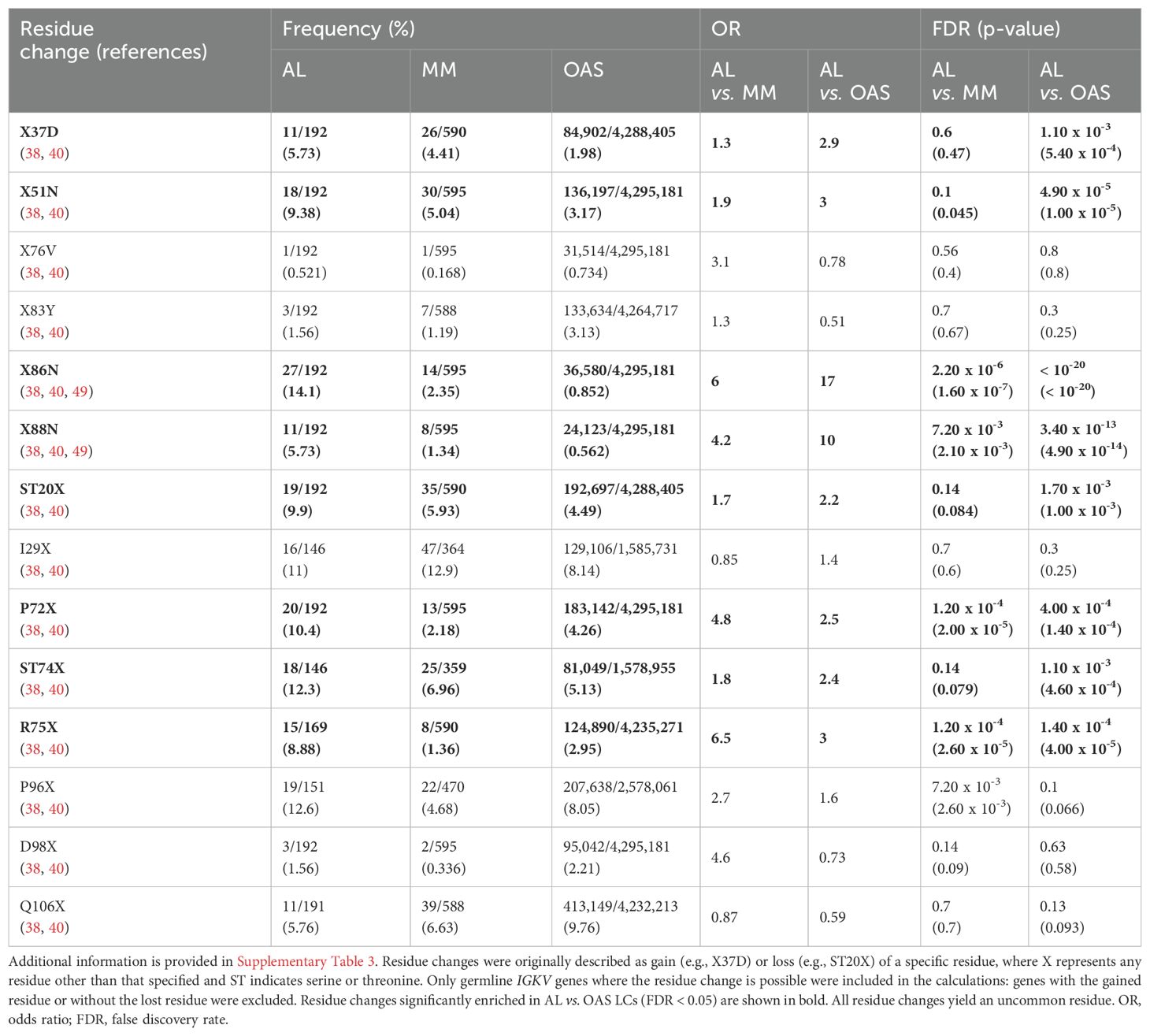
Table 1. Frequency in AL-Base and OAS LC sequences of residue changes previously reported as amyloidogenic among all IGKV LCs.
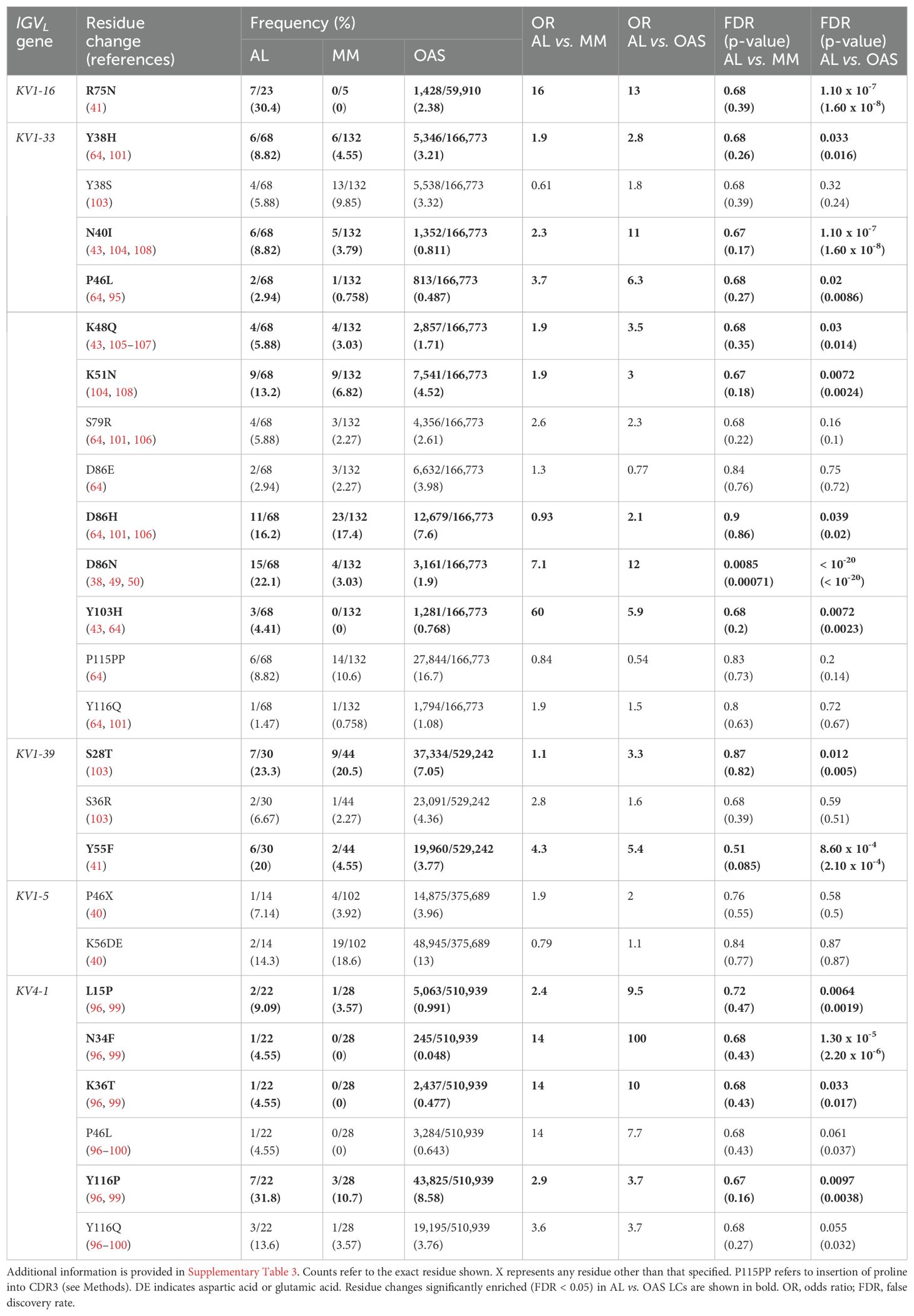
Table 2. Frequency within AL-Base and OAS LC sequences of residue changes previously reported as amyloidogenic for specific IGKV germline genes.
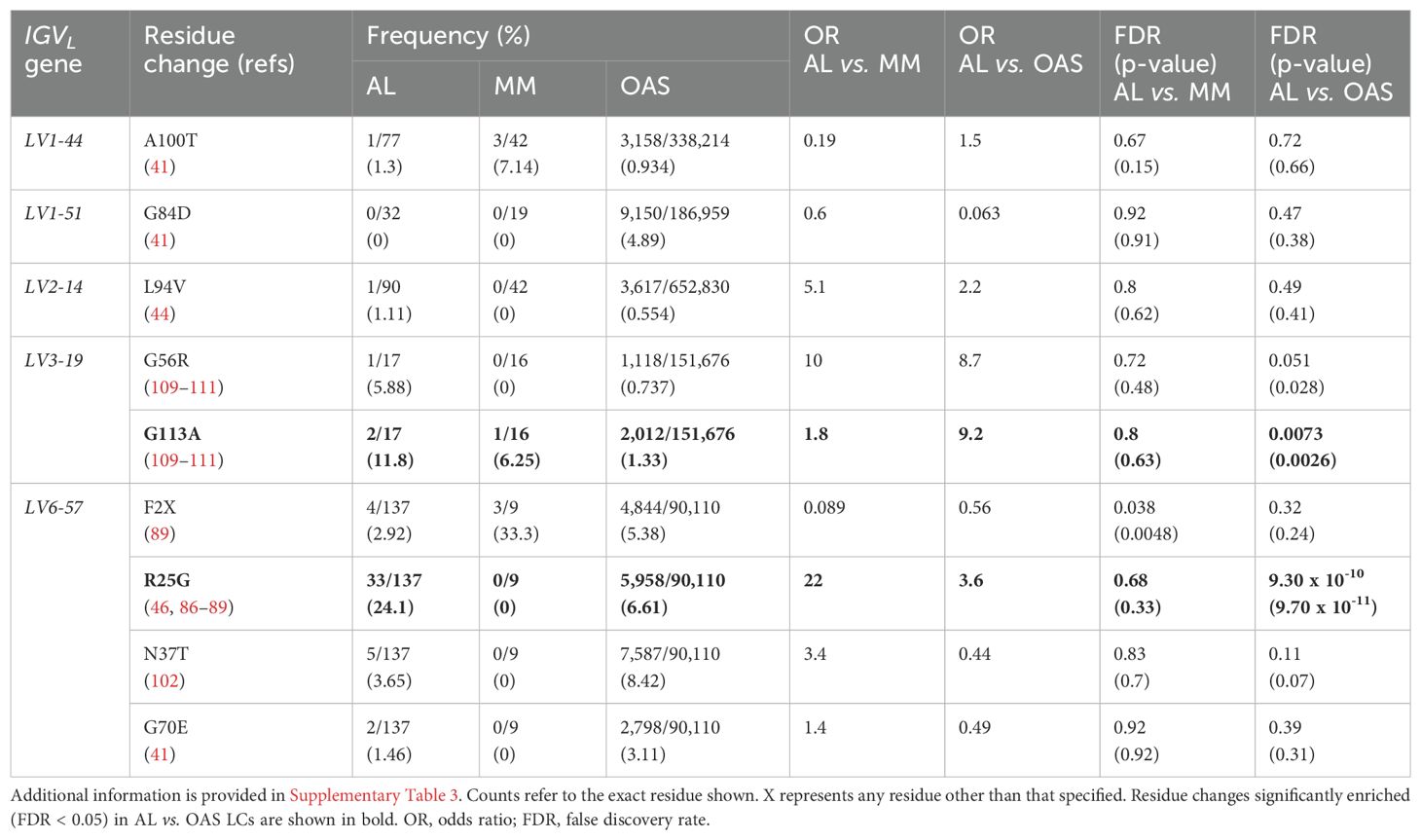
Table 3. Frequency within AL-Base and OAS LC sequences of residue changes previously reported as amyloidogenic for specific IGLV germline genes.
Table 1 and Figure 7 show residue changes proposed to be associated with amyloidosis in all IGKV-derived LCs. All changes yielded an uncommon residue at the specified position. Eight of 14 residue changes were significantly enriched in AL vs. OAS LCs. Five residue changes were significantly enriched in AL vs. MM LCs, of which four were also significantly enriched compared to OAS LCs. However, the frequency of each residue change in AL LCs was low, accounting for at most 12% of IGKV-derived AL LCs. The most significantly enriched residue change in this group was gain of an asparagine residue at position 86 (X86N), which was previously identified as a site for N-glycosylation (38, 49, 50).
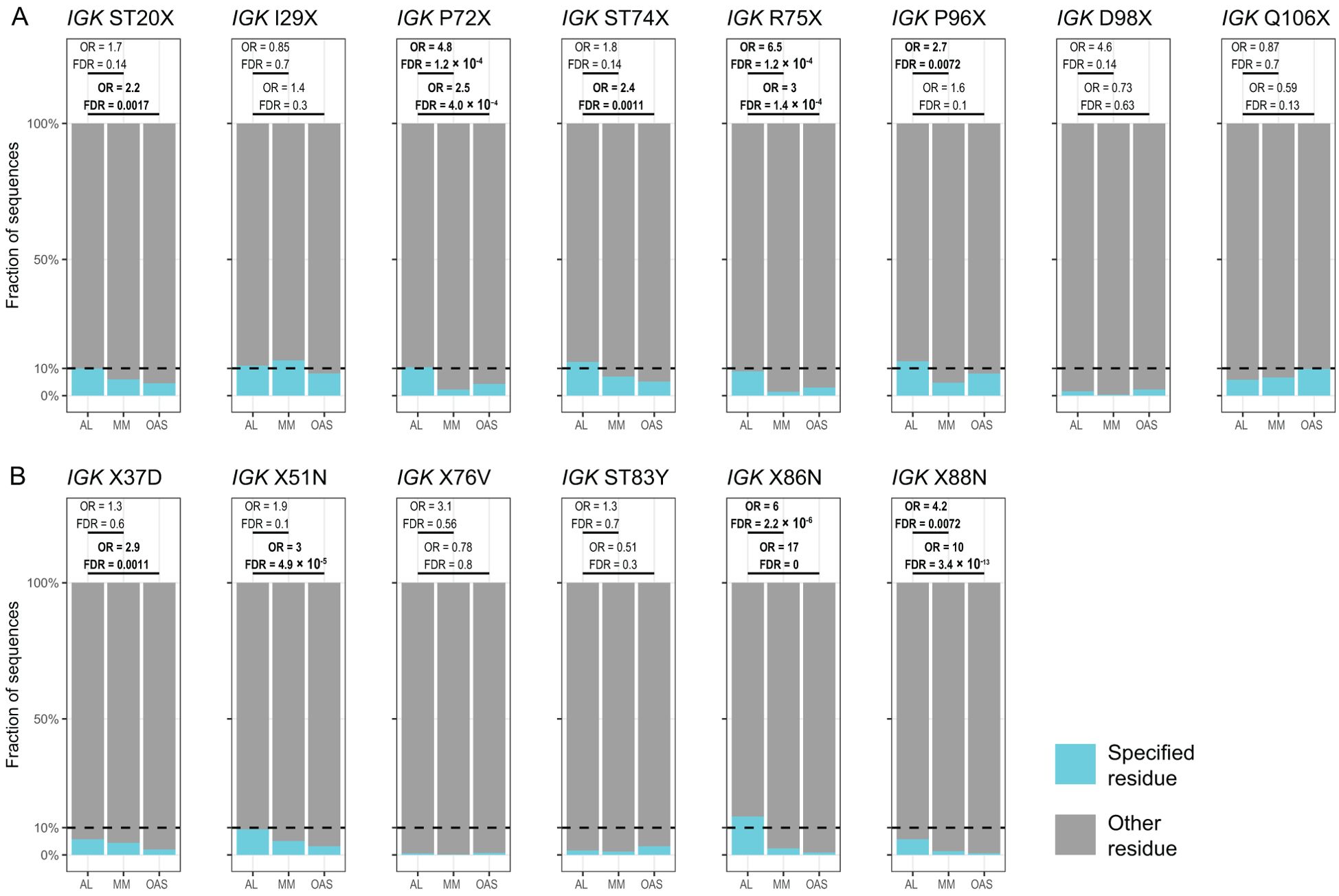
Figure 7. Enrichment of previously identified amyloidogenic residue changes in IGKV-derived LCs. Additional information is shown in Table 1 and Supplementary Table 3. The fraction of AL, MM and OAS LCs harboring each residue is shown in cyan. ST indicates serine or threonine; X indicates any residue other than the specified residue. ORs and significance are indicated for the AL vs. MM (upper line) and AL vs. OAS (lower line) comparisons. Significant comparisons (FDR < 0.05) are highlighted in bold. A 10% threshold, used to define uncommon residues, is shown as a dashed line. (A) Residue changes identified as replacement of the germline residue by any residue, calculated for all IGKV genes where the indicated residue is present. (B) Residue changes identified as acquisition of the specified residue from any germline residue.
Table 2 and Figure 8 show residue changes identified in specific IGKV genes, and Table 3 and Figure 9 show residue changes identified in specific IGLV genes. The tables show the frequency of the specified substitution and the figures show the frequencies of both the specified residue and all uncommon residues. Equivalent calculations for any uncommon residue at each position are provided in Supplementary File 1. Of the 34 residue changes, 32 yielded uncommon residues at that position. Seventeen of the 25 previously identified residue changes in IGKV genes are significantly enriched (OR > 1, FDR < 0.05) among AL vs. OAS LCs (Table 2). Any uncommon residue was significantly more frequent in AL than OAS LCs at 10 positions (Figure 8). Three residue changes in IGLV genes were significantly enriched among AL vs. OAS LCs (Table 2), but uncommon residues were only significantly enriched at position 25 of IGLV6-57 (Figure 9). No positions were enriched for specified or uncommon residues in the AL vs. MM comparison.
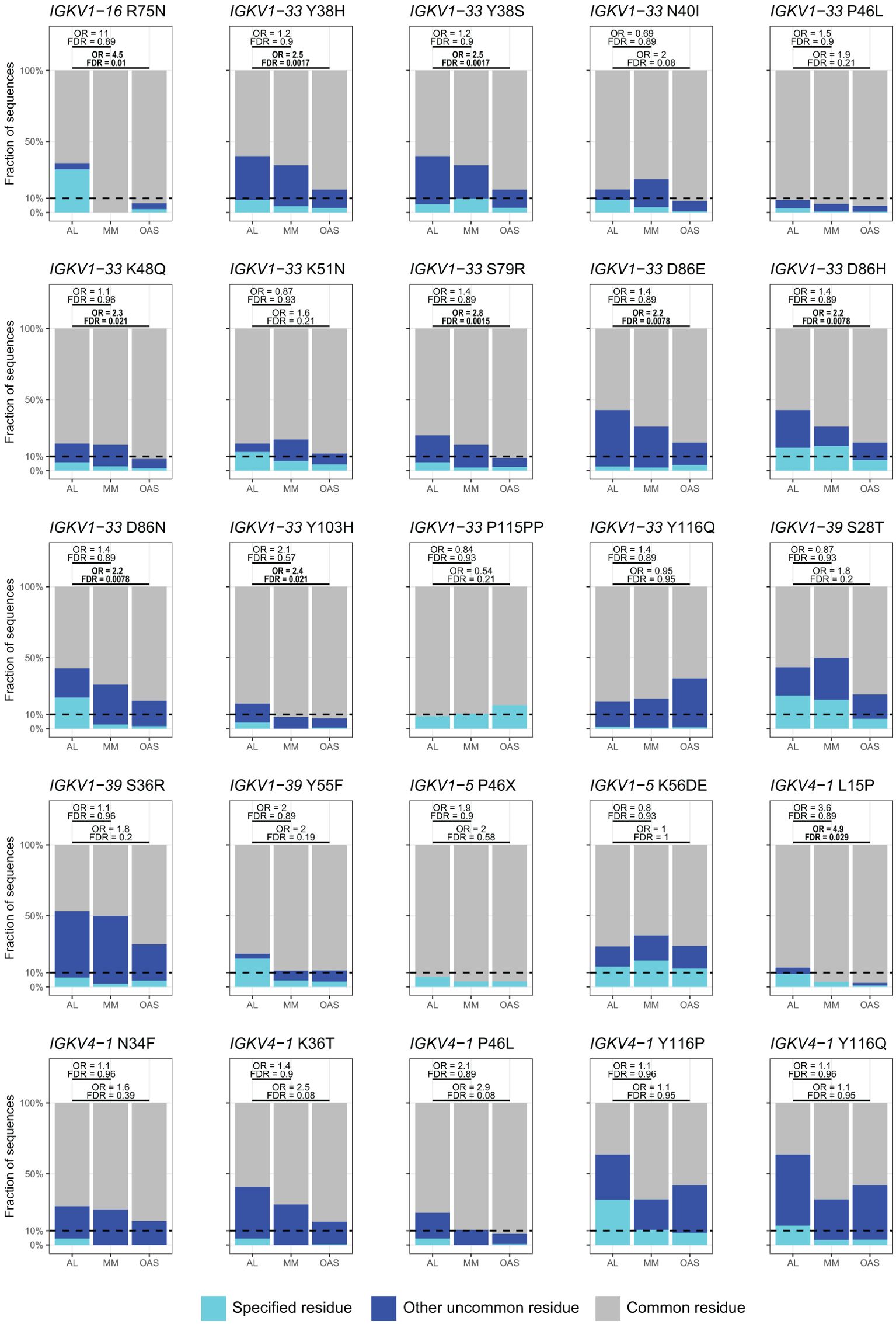
Figure 8. Enrichment of previously identified amyloidogenic residue changes in LCs derived from specific IGKV genes. Additional information is shown in Table 2 and Supplementary Table 3. The fraction of AL, MM and OAS LCs harboring the specified residue change is shown in cyan and the fraction harboring any other uncommon residue is shown in dark blue. P115PP indicates insertion of a second proline residue next to the germline-encoded proline within CDR3 of IGKV1-33. ORs and significance, calculated for all uncommon residues at each position (Supplementary File 1), are indicated for the AL vs. MM (upper line) and AL vs. OAS (lower line) comparisons. Significant comparisons (FDR < 0.05) are highlighted in bold. A 10% threshold, used to define uncommon residues, is shown as a dashed line.
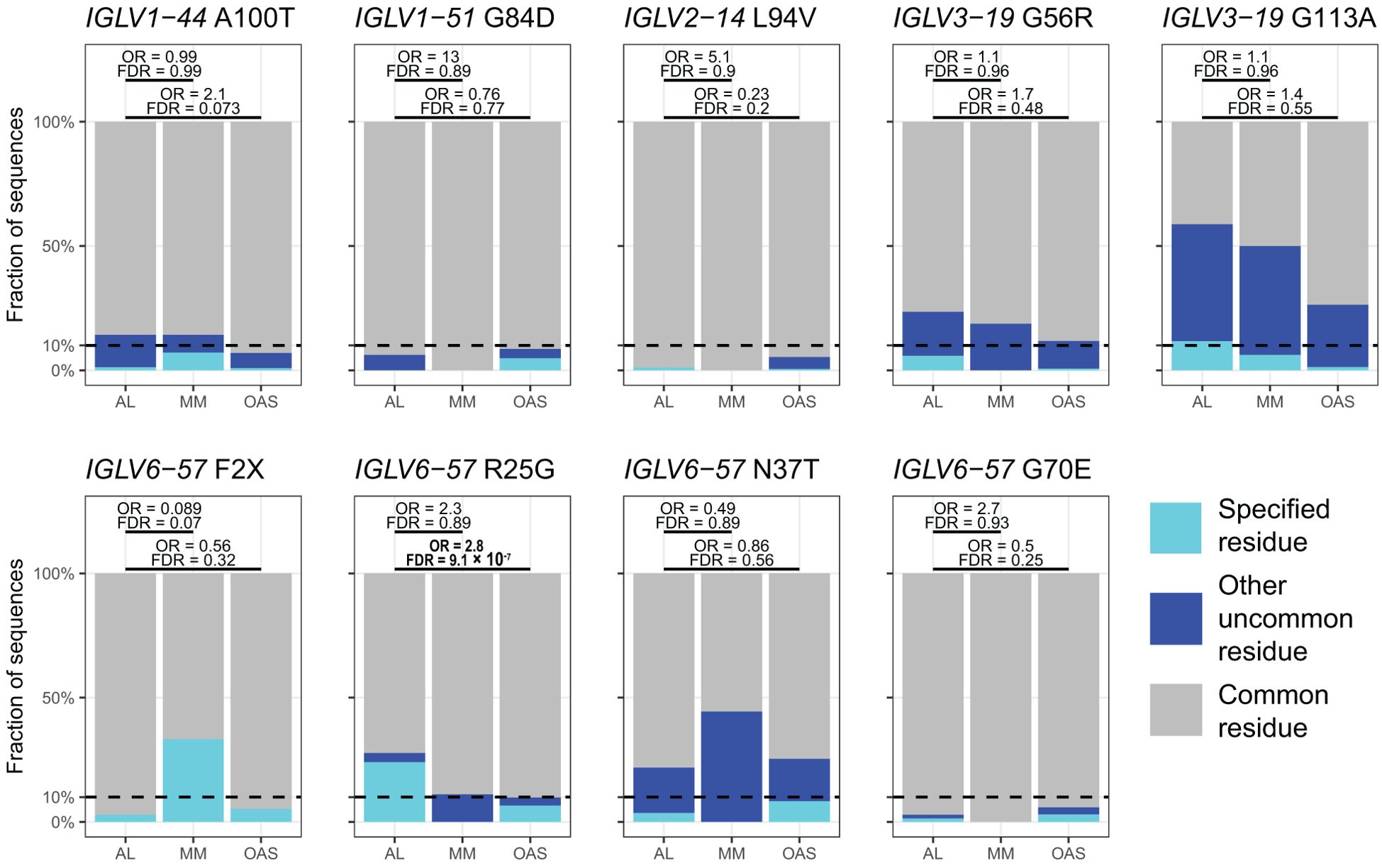
Figure 9. Enrichment of previously identified amyloidogenic residue changes in LCs derived from specific IGLV genes. Additional information is shown in Table 3 and Supplementary Table 3. The fraction of AL, MM and OAS LCs harboring the specified residue change is shown in cyan, while the fraction harboring any other uncommon residue is shown in dark blue. ORs and significance, calculated for all uncommon residues at each position (Supplementary Table 3), are indicated for the AL vs. MM (upper line) and AL vs. OAS (lower line) comparisons. Significant comparisons (FDR < 0.05) are highlighted in bold. A 10% threshold, used to define uncommon residues, is shown as a dashed line.
3.4 Glycine at position 25 of IGLV6–57 LCs is associated with AL amyloidosis
Our prospective analysis identified position 25 in IGLV6-57 as the site of an amyloid-associated residue change, in agreement with previous studies (46, 86–90). This residue change is associated with a germline-encoded polymorphism, rather than a somatic hypermutation: the IGLV6-57 *01, *03 and *04 alleles encode an arginine residue (R25), whereas the IGLV6-57*02 allele has a C>G nucleotide transversion that encodes a glycine residue at position 25 (G25). The presence of G25 has been proposed to be associated with AL amyloidosis (46, 86–90), which is supported by our analysis (Table 3; Figure 9). R25 forms a cation-π interaction that stabilizes the native VL domain, while G25 is destabilizing in the context of the VL-domain and the full-length LC (46, 91). We therefore investigated the distribution of residues at position 25 in AL, MM and OAS LC sequences derived from IGLV6-57 as a function of the assigned allele.
Among 137 AL LCs, 33 were G25 (24%), 99 were R25 (72%) and five had other residues at this position (Supplementary Figure 6). None of the 9 IGLV6-57-derived MM LCs, and 5,958 of 90,110 IGLV6-57-derived OAS LCs harbored G25 (6.6%). The OR for non-argenine residues between AL and MM was 2.32 (p = 0.42). The ORs between AL and OAS were 2.81 for non-argenine residues (p = 6.17 × 10-8) and 3.65 for glycine residues (p = 9.67 × 10-11). All AL LCs with G25 were assigned to the IGLV6-57*02 allele (Supplementary Figure 6). Of the 36 IGLV6-57*02 AL LC sequences, 33 harbored a glycine at position 25, two had alanine and one had a gap when aligned to IMGT numbering. The single MM LC assigned to IGLV6-57*02 had alanine at position 25. OAS LCs were assigned only to the *01 and *02 alleles and all sequences with G25 were derived from the *02 allele.
We repeated this analysis on 46 positions within 15 IGVL genes where there were residue differences between alleles (Supplementary Figure 7). The five other IGVL genes studied had either a single allele or no residue differences between the alleles. In many cases, there was substantial heterogeneity at these positions. However, none of these positions showed a significant difference between AL and MM, other than IGLV6-57 R25G, after correction for multiple testing.
4 Discussion
Using monoclonal LC sequences from AL-Base and polyclonal sequences from OAS, we have developed a new framework for analyzing potentially amyloidogenic LC sequences. Consensus matrices derived from the polyclonal antibody repertoire provide a germline gene-specific reference frequency for each LC residue at each position (Figure 1; Supplementary Data Sheet 1 and Supplementary Table 1). The consensus matrices allowed the variability at each position to be assessed (Figure 2), thereby identifying “uncommon” residues that may have important roles in amyloidogenicity (Figure 3). We calculated the relative likelihood of observing uncommon residues at each position in LCs derived from 20 IGVL genes (Figure 4 and Supplementary Data Sheet 1), observing that each gene has a unique set of residues that are more frequently mutated in AL than in MM or OAS LCs (Figure 5; Supplementary Table 2). These residues are distributed throughout the LC sequences and structures and do not generally correspond to regions with the most variable residues (Figures 5, 6; Supplementary Data). We observed that residue changes that have previously been identified as amyloidogenic were present in only a minority of AL LC sequences, even when enriched relative to MM or OAS LCs (Figures 7–9; Tables 1–3; Supplementary Table 3). These residue changes yielded an uncommon residue in 46 of 48 cases (95.8%), supporting our hypothesis that uncommon residues are associated with amyloidogenicity.
Our data provide new insights into the contributions to LC amyloidogenicity of precursor IGVL gene identity and somatic hypermutation. Identifying LCs that could deposit as amyloid in humans is an important goal, both to elucidate molecular mechanisms of disease and to aid diagnosis. Identifying and evaluating the sequence of a clonal LC associated with pre-symptomatic PCD could allow earlier intervention, which is critical for patients’ survival (2, 92). A diagnostic tool would need to distinguish between monoclonal LCs secreted by proliferative plasma cells, so MM LCs, which circulate at high levels but are not reported as forming amyloid are an appropriate control. Several computational approaches have been published (38, 51–54), but due to the rarity of amyloidosis these tools are insufficiently selective to be used clinically (11).
Although precursor gene use is an important contributor to amyloid propensity, most AL clones express LCs derived from a gene that is common in the healthy repertoire and MM (11). Therefore, the specific pattern of somatic hypermutations must play a role in pathology. Sequence conservation is associated with protein structure and stability (93, 94), and many studies have looked for residue changes that are associated with amyloidosis (10, 38–46, 49, 64, 68, 86–89, 95–111). However, because the likelihood of each residue change depends on the underlying DNA sequence of each germline gene (13), the distribution of residues at each position differs between genes (Figure 1) and the positions of amyloid-associated residue changes are distinct for each gene (Figure 5). Consistent with this, the distance between consensus matrices corresponding to AL and MM LCs from the same gene is less than that between AL LCs from closely related genes (Supplementary Figure 3). These factors hamper locus-wide comparisons between IGVL genes (38, 40, 112, 113). Therefore, any evaluation of residue changes needs to be within sequences derived from the same IGVL gene. Polymorphism between IGVL alleles further blurs the distinction between wild-type and variant sequences, so that even the word “mutation” is often used ambiguously in the context of antibodies. This problem is compounded by the limited annotation of human immunoglobulin alleles (48).
Our approach of defining uncommon residues avoids these problems and allows a focus on positions which are differentially mutated between sets of LCs, rather than highly variable in healthy LCs. For many amyloidogenic proteins, single residue changes are sufficient to cause disease and are readily identified (114), but this is not the case for LCs (11). We suggest that defining common and uncommon residues at each position relative to a consensus matrix, rather than wild-type and variant residues, is a more useful framework for considering LC sequence diversity in AL amyloidosis. Because the frequency at which a residue is observed in the OAS LC sequences may report on its compatibility within native antibodies (13, 93), the least common residues in a LC sequence may be particularly disruptive. The frequency profiles shown in Figure 3 may be a useful tool to simplify the mutational analysis of LCs, by allowing prioritization of residue changes for detailed study. Several studies have systematically mutated residues to identify changes that promote unfolding or aggregation, either from the inferred germline residue to the AL-associated residue, or vice versa (41, 43, 44). However, the time and effort required to exhaustively test each individual residue change has limited the number of LCs studied. For the two examples shown, we observed that the residue changes identified by biophysical analysis (43, 44) yield the most uncommon residues in the sequence. We hypothesize that such residue changes have an outsized effect on the behavior of the LC. However, uncommon residues are similarly frequent among AL and MM LCs, and distributed throughout the sequence (Figure 2). Therefore, the presence of an uncommon residue is neither necessary nor sufficient for amyloidogenesis and these potentially amyloidogenic residue changes still need to be investigated. Studying positions that are enriched in uncommon residues in multiple germline genes may reveal shared mechanisms of amyloidogenicity. For example, replacements of a surface-exposed leucine at position 52 are enriched in AL LCs derived from the IGKV1-33, IGKV1-39, IGLV1-44, IGLV2-14, IGLV2-8 and IGLV3-1 genes (Figure 5; Supplementary Data 3). Such replacements could alter the structure and interactions of both the native LC and its amyloid fibrils. The consensus matrices that we constructed for each IGVL gene capture the likelihood of every possible residue change, allowing an estimate of how well the protein can tolerate changes at that position. The large number of available sequences in OAS allow highly specific residue frequencies to be used to evaluate residue changes, rather than relying on generic substitution matrices such BLOSUM (115). There is incomplete overlap between the positions where specific residues are enriched in AL LCs and those where any uncommon residue is enriched, due to the differences in residue frequencies between the sets of LCs (Figures 8, 9). These differences likely reflect the distribution of residues at each position. Due to the large number of potential residue changes, we did not attempt to predict the effects of each change or classify residues by physicochemical properties. However, structural hypotheses about the potential effect of different classes of residue changes could be evaluated based on examination of the appropriate column of the consensus matrix.
Several studies have sought mutational “hotspots” or regions where AL LCs are frequently mutated to increase their amyloidogenicity (112, 113, 116, 117). Our data instead support a model where many individual residue changes can promote amyloidogenicity, but specific recurrent residue changes account for only a small fraction of amyloidogenic sequences (Figures 7–9). Importantly, the residues that are most frequently mutated in AL LCs (primarily CDR1 and CDR3, Figure 1) are not enriched in uncommon residues when compared to MM or OAS LCs (Figures 5, 6; Supplementary Figure 4). This counter-intuitive observation is due to the biased, gene-specific distribution of somatic hypermutations during affinity maturation (13). These results are consistent with previous observations that a limited number of residue changes have substantial effects on the properties of LCs, perhaps analogous to “driver” and “passenger” mutations associated with cancer (118). Examples include R25G in IGLV6-57 (46) and the X86N and X88N residue changes in IGKV1 LCs that create an N-glycosylation site (49). Such a model implies that the effects of each residue change must be determined to predict the risk of amyloidosis associated with a new LC sequence. Residue changes can alter the structure of the native VL-domain, the amyloid fibril and the intermediate states between these endpoints, as well as interactions of these states with other tissue factors (119). However, few LC proteins have been subject to the mutational analysis needed to determine the effects of an uncommon residue on the behavior of the protein. An advantage of sequence-based approaches to predicting amyloidogenicity is that they are agnostic to mechanism and can identify residue changes that modulate different processes.
This study is limited by the number and diversity of monoclonal AL and MM LCs available, which limits the statistical power of our analyses. In particular, the low numbers of monoclonal LCs derived from several genes limited our ability to detect positions enriched in uncommon residues. To compensate for this, we have reported positions where significance was lost after correction for multiple testing to highlight potentially relevant positions, acknowledging that this increases the risk of including false positives. We anticipate that additional monoclonal LC sequences will become available, allowing validation of our results on independent data. The residue frequencies for OAS LCs, particularly polymorphisms such as IGLV6-57 R25G, depend on the genotypes of the individuals whose immune repertoires were sequenced, which we did not attempt to infer. We did not consider the roles of the CL-domain or the LC’s heavy chain partner due to the lack of available sequences. Furthermore, it remains unclear whether MM and OAS LCs can truly be considered non-amyloidogenic, since these proteins can be induced to aggregate in vitro and their ability to form amyloid in patients is unknown. We assume that because amyloidosis is rare even in the context of a PCD, most normal repertoire LCs would not cause disease if overexpressed, but this is not known.
Amyloid formation by antibody LCs remains poorly understood. Our data clearly show that there are no individual residue changes that unambiguously lead to amyloidosis and provide an important resource for further investigation of LC amyloid propensity. By defining the mutational landscape of all LCs, we enable a more quantitative approach to both computational and laboratory studies of sequences.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://wwwapp.bumc.bu.edu/BEDAC_ALBase/ and https://opig.stats.ox.ac.uk/webapps/oas/.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
GM: Data curation, Visualization, Writing – review & editing, Project administration, Validation, Formal analysis, Methodology, Investigation, Writing – original draft, Resources, Software, Funding acquisition, Conceptualization. TP: Validation, Data curation, Conceptualization, Investigation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project was supported by NIH awards R01CA279808 and 1UL1TR001430; the Wildflower Foundation; the Karin Grunebaum Cancer Research Foundation; and the Boston University Amyloid Research Fund. AL-Base was originally funded by NIH award R01HL68705.
Acknowledgments
This work was made possible by the generosity of patients who allowed their samples and data to be used for research, and scientists who shared the LC sequences that are compiled in AL-Base. Analysis was carried out using the Boston University Shared Computing Cluster which is administered by Boston University’s Research Computing Services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1622207/full#supplementary-material
References
1. Buxbaum JN, Eisenberg DS, Fändrich M, McPhail ED, Merlini G, Saraiva MJM, et al. Amyloid nomenclature 2024: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. (2024) 31:249–56. doi: 10.1080/13506129.2024.2405948
2. Sanchorawala V. Systemic light chain amyloidosis. N Engl J Med. (2024) 390:2295–307. doi: 10.1056/NEJMra2304088
3. Merlini G, Dispenzieri A, Sanchorawala V, Schönland SO, Palladini G, Hawkins PN, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. (2018) 4:38. doi: 10.1038/s41572-018-0034-3
4. Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. (2018) 378:241–9. doi: 10.1056/NEJMoa1709974
5. Desikan KR, Dhodapkar MV, Hough A, Waldron T, Jagannath S, Siegel D, et al. Incidence and impact of light chain associated (AL) amyloidosis on the prognosis of patients with multiple myeloma treated with autologous transplantation. Leuk Lymphoma. (1997) 27:315–9. doi: 10.3109/10428199709059685
6. Alameda D, Goicoechea I, Vicari M, Arriazu E, Nevone A, Rodriguez S, et al. Tumor cells in light-chain amyloidosis and myeloma show distinct transcriptional rewiring of normal plasma cell development. Blood. (2021) 138:1583–9. doi: 10.1182/blood.2020009754
7. Blancas-Mejía LM and Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. (2013) 82:745–74. doi: 10.1146/annurev-biochem-072611-130030
8. Del Pozo-Yauner L, Herrera GA, Perez Carreon JI, Turbat-Herrera EA, Rodriguez-Alvarez FJ, and Ruiz Zamora RA. Role of the mechanisms for antibody repertoire diversification in monoclonal light chain deposition disorders: when a friend becomes foe. Front Immunol. (2023) 14:1203425. doi: 10.3389/fimmu.2023.1203425
9. Morgan GJ. Understanding and overcoming biochemical diversity in AL amyloidosis. Isr J Chem. (2023) 31:249–56. doi: 10.1002/ijch.202300128
10. Absmeier RM, Rottenaicher GJ, Svilenov HL, Kazman P, and Buchner J. Antibodies gone bad - the molecular mechanism of light chain amyloidosis. FEBS J. (2023) 290:1398–419. doi: 10.1111/febs.16390
11. Morgan GJ, Nau AN, Wong S, Spencer BH, Shen Y, Hua A, et al. An updated AL-base reveals ranked enrichment of immunoglobulin light chain variable genes in AL amyloidosis. Amyloid. (2025) 32:129–38. doi: 10.1080/13506129.2024.2434899
12. Methot SP and Di Noia JM. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol. (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
13. Sheng Z, Schramm CA, Kong R, NISC Comparative Sequencing Program, Mullikin JC, Mascola JR, et al. Gene-specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic hypermutation. Front Immunol. (2017) 8:537. doi: 10.3389/fimmu.2017.00537
14. Briney B, Inderbitzin A, Joyce C, and Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. (2019) 566:393–7. doi: 10.1038/s41586-019-0879-y
15. Feige MJ, Hendershot LM, and Buchner J. How antibodies fold. Trends Biochem Sci. (2010) 35:189–98. doi: 10.1016/j.tibs.2009.11.005
16. Radamaker L, Lin Y-H, Annamalai K, Huhn S, Hegenbart U, Schönland SO, et al. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat Commun. (2019) 10:1103. doi: 10.1038/s41467-019-09032-0
17. Swuec P, Lavatelli F, Tasaki M, Paissoni C, Rognoni P, Maritan M, et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat Commun. (2019) 10:1269. doi: 10.1038/s41467-019-09133-w
18. Radamaker L, Karimi-Farsijani S, Andreotti G, Baur J, Neumann M, Schreiner S, et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat Commun. (2021) 12:6434. doi: 10.1038/s41467-021-26553-9
19. Radamaker L, Baur J, Huhn S, Haupt C, Hegenbart U, Schönland S, et al. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat Commun. (2021) 12:875. doi: 10.1038/s41467-021-21126-2
20. Heerde T, Rennegarbe M, Biedermann A, Savran D, Pfeiffer PB, Hitzenberger M, et al. Cryo-EM demonstrates the in vitro proliferation of an ex vivo amyloid fibril morphology by seeding. Nat Commun. (2022) 13:85. doi: 10.1038/s41467-021-27688-5
21. Lavatelli F, Natalello A, Marchese L, Ami D, Corazza A, Raimondi S, et al. Truncation of the constant domain drives amyloid formation by immunoglobulin light chains. J Biol Chem. (2024) 300:107174. doi: 10.1016/j.jbc.2024.107174
22. Karimi-Farsijani S, Pfeiffer PB, Banerjee S, Baur J, Kuhn L, Kupfer N, et al. Light chain mutations contribute to defining the fibril morphology in systemic AL amyloidosis. Nat Commun. (2024) 15:5121. doi: 10.1038/s41467-024-49520-6
23. Schulte T, Chaves-Sanjuan A, Speranzini V, Sicking K, Milazzo M, Mazzini G, et al. Helical superstructures between amyloid and collagen in cardiac fibrils from a patient with AL amyloidosis. Nat Commun. (2024) 15:6359. doi: 10.1038/s41467-024-50686-2
24. Klimtchuk ES, Gursky O, Patel RS, Laporte KL, Connors LH, Skinner M, et al. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry. (2010) 49:9848–57. doi: 10.1021/bi101351c
25. Rottenaicher GJ, Absmeier RM, Meier L, Zacharias M, and Buchner J. A constant domain mutation in a patient-derived antibody light chain reveals principles of AL amyloidosis. Commun Biol. (2023) 6:209. doi: 10.1038/s42003-023-04574-y
26. Rennella E, Morgan GJ, Kelly JW, and Kay LE. Role of domain interactions in the aggregation of full-length immunoglobulin light chains. Proc Natl Acad Sci U.S.A. (2019) 116:854–63. doi: 10.1073/pnas.1817538116
27. Perfetti V, Casarini S, Palladini G, Vignarelli MC, Klersy C, Diegoli M, et al. Analysis of V(lambda)-J(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloid-associated germline gene segment. Blood. (2002) 100:948–53. doi: 10.1182/blood-2002-01-0114
28. Perfetti V, Palladini G, Casarini S, Navazza V, Rognoni P, Obici L, et al. The repertoire of λ light chains causing predominant amyloid heart involvement and identification of a preferentially involved germline gene, IGLV1-44. Blood. (2012) 119:144–50. doi: 10.1182/blood-2011-05-355784
29. Abraham RS, Geyer SM, Price-Troska TL, Allmer C, Kyle RA, Gertz MA, et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood. (2003) 101:3801–8. doi: 10.1182/blood-2002-09-2707
30. Prokaeva T, Spencer B, Kaut M, Ozonoff A, Doros G, Connors LH, et al. Soft tissue, joint, and bone manifestations of AL amyloidosis: clinical presentation, molecular features, and survival. Arthritis Rheum. (2007) 56:3858–68. doi: 10.1002/art.22959
31. Cascino P, Nevone A, Piscitelli M, Scopelliti C, Girelli M, Mazzini G, et al. Single-molecule real-time sequencing of the M protein: Toward personalized medicine in monoclonal gammopathies. Am J Hematol. (2022) 97:E389–92. doi: 10.1002/ajh.26684
32. Nau A, Shen Y, Sanchorawala V, Prokaeva T, and Morgan GJ. Complete variable domain sequences of monoclonal antibody light chains identified from untargeted RNA sequencing data. Front Immunol. (2023) 14:1167235. doi: 10.3389/fimmu.2023.1167235
33. Javaugue V, Pascal V, Bender S, Nasraddine S, Dargelos M, Alizadeh M, et al. RNA-based immunoglobulin repertoire sequencing is a new tool for the management of monoclonal gammopathy of renal (kidney) significance. Kidney Int. (2022) 101:331–7. doi: 10.1016/j.kint.2021.10.017
34. Comenzo RL, Wally J, Kica G, Murray J, Ericsson T, Skinner M, et al. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: association with dominant amyloid-related organ involvement and survival after stem cell transplantation. Br J Haematol. (1999) 106:744–51. doi: 10.1046/j.1365-2141.1999.01591.x
35. Bodi K, Prokaeva T, Spencer B, Eberhard M, Connors LH, and Seldin DC. AL-Base: a visual platform analysis tool for the study of amyloidogenic immunoglobulin light chain sequences. Amyloid. (2009) 16:1–8. doi: 10.1080/13506120802676781
36. Kourelis TV, Dasari S, Theis JD, Ramirez-Alvarado M, Kurtin PJ, Gertz MA, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood. (2017) 129:299–306. doi: 10.1182/blood-2016-10-743997
37. Fahey JL. Structural basis for the differences between type I and type II human γ-globulin molecules. J Immunol. (1963) 91:448–59. doi: 10.4049/jimmunol.91.4.448
38. Stevens FJ. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid. (2000) 7:200–11. doi: 10.3109/13506120009146835
39. Oberti L, Rognoni P, Barbiroli A, Lavatelli F, Russo R, Maritan M, et al. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci Rep. (2017) 7:16809. doi: 10.1038/s41598-017-16953-7
40. Stevens FJ, Weiss DT, and Solomon A. Structural bases of light chain-related pathology. In: The Antibodies. London: CRC Press (1999). p. 175–208.
41. Hurle MR, Helms LR, Li L, Chan W, and Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci U.S.A. (1994) 91:5446–50. doi: 10.1073/pnas.91.12.5446
42. Puri S, Gadda A, Polsinelli I, Barzago MM, Toto A, Sriramoju MK, et al. The critical role of the variable domain in driving proteotoxicity and aggregation in full-length light chains. J Mol Biol. (2025) 437:168958. doi: 10.1016/j.jmb.2025.168958
43. Baden EM, Randles EG, Aboagye AK, Thompson JR, and Ramirez-Alvarado M. Structural insights into the role of mutations in amyloidogenesis. J Biol Chem. (2008) 283:30950–6. doi: 10.1074/jbc.M804822200
44. Kazman P, Vielberg M-T, Pulido Cendales MD, Hunziger L, Weber B, Hegenbart U, et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. Elife. (2020) 9:e52300. doi: 10.7554/eLife.52300.sa2
45. Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, and Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry. (1999) 38:14101–8. doi: 10.1021/bi991131j
46. del Pozo Yauner L, Ortiz E, Sánchez R, Sánchez-López R, Güereca L, Murphy CL, et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins. (2008) 72:684–92. doi: 10.1002/prot.21934
47. Lefranc M-P and Lefranc G. Immunoglobulins or antibodies: IMGT® bridging genes, structures and functions. Biomedicines. (2020) 8:319. doi: 10.3390/biomedicines8090319
48. Mikocziova I, Greiff V, and Sollid LM. Immunoglobulin germline gene variation and its impact on human disease. Genes Immun. (2021) 22:205–17. doi: 10.1038/s41435-021-00145-5
49. Nevone A, Girelli M, Mangiacavalli S, Paiva B, Milani P, Cascino P, et al. An N-glycosylation hotspot in immunoglobulin κ light chains is associated with AL amyloidosis. Leukemia. (2022) 36:2076–85. doi: 10.1038/s41375-022-01599-w
50. Morgan GJ, Yung Z, Spencer BH, Sanchorawala V, and Prokaeva T. Predicting structural consequences of antibody light chain N-glycosylation in AL amyloidosis. Pharm (Basel). (2024) 17:1542. doi: 10.3390/ph17111542
51. Garofalo M, Piccoli L, Romeo M, Barzago MM, Ravasio S, Foglierini M, et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat Commun. (2021) 12:3532. doi: 10.1038/s41467-021-23880-9
52. Zhou Y, Liu W, Luo C, Huang Z, Samarappuli Mudiyanselage Savini G, Zhao L, et al. Ab-Amy 2.0: Predicting light chain amyloidogenic risk of therapeutic antibodies based on antibody language model. Methods. (2025) 233:11–8. doi: 10.1016/j.ymeth.2024.11.005
53. Zhou Y, Huang Z, Gou Y, Liu S, Yang W, Zhang H, et al. AB-Amy: machine learning aided amyloidogenic risk prediction of therapeutic antibody light chains. Antib Ther. (2023) 6:147–56. doi: 10.1093/abt/tbad007
54. Rawat P, Prabakaran R, Kumar S, and Gromiha MM. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci Rep. (2021) 11:13785. doi: 10.1038/s41598-021-93019-9
55. Olsen TH, Boyles F, and Deane CM. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. (2022) 31:141–6. doi: 10.1002/pro.4205
56. Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, and Krawczyk K. Observed antibody space: A resource for data mining next-generation sequencing of antibody repertoires. J Immunol. (2018) 201:2502–9. doi: 10.4049/jimmunol.1800708
57. Lefranc M-P, Pommié C, Kaas Q, Duprat E, Bosc N, Guiraudou D, et al. IMGT unique numbering for immunoglobulin and T cell receptor constant domains and Ig superfamily C-like domains. Dev Comp Immunol. (2005) 29:185–203. doi: 10.1016/j.dci.2004.07.003
58. Manso T, Folch G, Giudicelli V, Jabado-Michaloud J, Kushwaha A, Nguefack Ngoune V, et al. IMGT® databases, related tools and web resources through three main axes of research and development. Nucleic Acids Res. (2022) 50:D1262–72. doi: 10.1093/nar/gkab1136
59. Lefranc M-P. Nomenclature of the human immunoglobulin kappa (IGK) genes. Exp Clin Immunogenet. (2001) 18:161–74. doi: 10.1159/000049195
60. Dunbar J and Deane CM. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. (2016) 32:298–300. doi: 10.1093/bioinformatics/btv552
61. Dorfman R. A formula for the gini coefficient. Rev Econ Stat. (1979) 61:146. doi: 10.2307/1924845
62. Zeileis A. Measuring Inequality, Concentration, and Poverty. R package ineq version 0.2-13 (2014). Available online at: https://CRAN.R-project.org/package=ineq (accessed Mar 03, 2025).
63. R Core Team. R: A Language and Environment for Statistical Computing (2022). Available online at: https://www.R-project.org/ (Accessed March 3, 2025).
64. Randles EG, Thompson JR, Martin DJ, and Ramirez-Alvarado M. Structural alterations within native amyloidogenic immunoglobulin light chains. J Mol Biol. (2009) 389:199–210. doi: 10.1016/j.jmb.2009.04.010
65. Benjamini Y and Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc. (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
66. Wagih O. ggseqlogo: A ‘ggplot2’ extension for drawing publication-ready sequence logos (2017). Available online at: https://CRAN.R-project.org/package=ggseqlogo (Accessed March 3, 2025).
67. Schneider TD and Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. (1990) 18:6097–100. doi: 10.1093/nar/18.20.6097
68. Hernández-Santoyo A, del Pozo Yauner L, Fuentes-Silva D, Ortiz E, Rudiño-Piñera E, Sánchez-López R, et al. A single mutation at the sheet switch region results in conformational changes favoring lambda6 light-chain fibrillogenesis. J Mol Biol. (2010) 396:280–92. doi: 10.1016/j.jmb.2009.11.038
69. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
70. Kabsch W and Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. (1983) 22:2577–637. doi: 10.1002/bip.360221211
71. Touw WG, Baakman C, Black J, te Beek TAH, Krieger E, Joosten RP, et al. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. (2015) 43:D364–8. doi: 10.1093/nar/gku1028
72. Topham CM and Smith JC. Tri-peptide reference structures for the calculation of relative solvent accessible surface area in protein amino acid residues. Comput Biol Chem. (2015) 54:33–43. doi: 10.1016/j.compbiolchem.2014.11.007
73. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. (2023) 32:e4792. doi: 10.1002/pro.4792
74. RStudio Team. Posit. In: Posit (2022). Available online at: http://www.rstudio.com/ (accessed Mar 03, 2025).
75. Pagès H, Aboyoun P, Gentleman R, and DebRoy S. Biostrings: Efficient manipulation of biological strings (2022). Available online at: https://bioconductor.org/packages/Biostrings (Accessed March 3, 2025).
76. Robinson D, Hayes A, and Couch S. broom: Convert Statistical Objects into Tidy Tibbles (2023). Available online at: https://CRAN.R-project.org/package=broom (Accessed March 3, 2025).
77. Wilke CO. cowplot: Streamlined Plot Theme and Plot Annotations for `ggplot2’ (2020). Available online at: https://CRAN.R-project.org/package=cowplot (Accessed March 3, 2025).
78. Vaughan D and Dancho M. furrr: Apply Mapping Functions in Parallel using Futures (2022). Available online at: https://CRAN.R-project.org/package=furrr (Accessed March 3, 2025).
79. Bengtsson H. A unifying framework for parallel and distributed processing in R using futures. R J. (2021) 13:208. doi: 10.32614/RJ-2021-048
80. Kassambara A. ggpubr: `ggplot2’ Based Publication Ready Plots (2023). Available online at: https://CRAN.R-project.org/package=ggpubr (Accessed March 3, 2025).
81. Yu G, Smith DK, Zhu H, Guan Y, and Lam TT. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. (2017) 8:28–36. doi: 10.1111/2041-210X.12628
82. Bodenhofer U, Bonatesta E, Horejs-Kainrath C, and Hochreiter S. msa: an R package for multiple sequence alignment. Bioinformatics. (2015) 31:3997–9. doi: 10.1093/bioinformatics/btv494
83. Schauberger P, Walker A, Braglia L, Sturm J, Garbuszus JM, Barbone JM, et al. Read, Write and Edit xlsx Files. R package openxlsx version 4.2.8 (2025). Available online at: https://CRAN.R-project.org/package=openxlsx (accessed Mar 03, 2025).
84. Kassambara A. rstatix: Pipe-Friendly Framework for Basic Statistical Tests (2023). Available online at: https://CRAN.R-project.org/package=rstatix (Accessed March 3, 2025).
85. Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, et al. Welcome to the tidyverse. J Open Source Software. (2019) 4:1686. doi: 10.21105/joss.01686
86. Solomon A, Frangione B, and Franklin EC. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. (1982) 70:453–60. doi: 10.1172/JCI110635
87. Blancas-Mejia LM, Tellez LA, del Pozo-Yauner L, Becerril B, Sanchez-Ruiz JM, and Fernandez-Velasco DA. Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: the relation between unfolding and fibrillogenesis. J Mol Biol. (2009) 386:1153–66. doi: 10.1016/j.jmb.2008.12.069
88. Maya-Martinez R, French-Pacheco L, Valdés-García G, Pastor N, and Amero C. Different dynamics in 6aJL2 proteins associated with AL amyloidosis, a conformational disease. Int J Mol Sci. (2019) 20:4078. doi: 10.3390/ijms20174078
89. del Pozo-Yauner L, Wall JS, González Andrade M, Sánchez-López R, Rodríguez-Ambriz SL, Pérez Carreón JI, et al. The N-terminal strand modulates immunoglobulin light chain fibrillogenesis. Biochem Biophys Res Commun. (2014) 443:495–9. doi: 10.1016/j.bbrc.2013.11.123
90. González-Andrade M, Becerril-Luján B, Sánchez-López R, Ceceña-Álvarez H, Pérez-Carreón JI, Ortiz E, et al. Mutational and genetic determinants of λ6 light chain amyloidogenesis. FEBS J. (2013) 280:6173–83. doi: 10.1111/febs.12538
91. Morgan GJ and Kelly JW. The kinetic stability of a full-length antibody light chain dimer determines whether endoproteolysis can release amyloidogenic variable domains. J Mol Biol. (2016) 428:4280–97. doi: 10.1016/j.jmb.2016.08.021
92. Zhou P, Mansukhani MM, Yeh R, Lu J, Xia H, Koganti L, et al. Seeking amyloidosis very early: Free light chain differentials and IGLV gene use as screening variables for light-chain amyloidosis in λ monoclonal gammopathies. Br J Cancer Res. (2024) 7:681–8. doi: 10.31488/bjcr.193
93. Steipe B, Schiller B, Plückthun A, and Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J Mol Biol. (1994) 240:188–92. doi: 10.1006/jmbi.1994.1434
94. Sternke M, Tripp KW, and Barrick D. Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc Natl Acad Sci U.S.A. (2019) 116:11275–84. doi: 10.1073/pnas.1816707116
95. Blancas-Mejía LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, et al. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J Mol Biol. (2014) 426:347–61. doi: 10.1016/j.jmb.2013.10.016
96. Stevens PW, Raffen R, Hanson DK, Deng YL, Berrios-Hammond M, Westholm FA, et al. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Protein Sci. (1995) 4:421–32. doi: 10.1002/pro.5560040309
97. Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, and Millett I. Fink AL. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry. (2001) 40:3525–35. doi: 10.1021/bi001782b
98. Qin Z, Hu D, Zhu M, and Fink AL. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry. (2007) 46:3521–31. doi: 10.1021/bi061716v
99. Raffen R, Dieckman LJ, Szpunar M, Wunschl C, Pokkuluri PR, Dave P, et al. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci. (1999) 8:509–17. doi: 10.1110/ps.8.3.509
100. Khurana R, Souillac PO, Coats AC, Minert L, Ionescu-Zanetti C, Carter SA, et al. A model for amyloid fibril formation in immunoglobulin light chains based on comparison of amyloidogenic and benign proteins and specific antibody binding. Amyloid. (2003) 10:97–109. doi: 10.3109/13506120309041731
101. Marin-Argany M, Güell-Bosch J, Blancas-Mejía LM, Villegas S, and Ramirez-Alvarado M. Mutations can cause light chains to be too stable or too unstable to form amyloid fibrils. Protein Sci. (2015) 24:1829–40. doi: 10.1002/pro.2790
102. Peterle D, Klimtchuk ES, Wales TE, Georgescauld F, Connors LH, Engen JR, et al. A conservative point mutation in a dynamic antigen-binding loop of human immunoglobulin λ6 light chain promotes pathologic amyloid formation. J Mol Biol. (2021) 433:167310. doi: 10.1016/j.jmb.2021.167310
103. Klimtchuk ES, Peterle D, Bullitt E, Connors LH, Engen JR, and Gursky O. Role of complementarity-determining regions 1 and 3 in pathologic amyloid formation by human immunoglobulin κ1 light chains. Amyloid. (2023) 30:364–78. doi: 10.1080/13506129.2023.2212397
104. Schormann N, Murrell JR, Liepnieks JJ, and Benson MD. Tertiary structure of an amyloid immunoglobulin light chain protein: a proposed model for amyloid fibril formation. Proc Natl Acad Sci U.S.A. (1995) 92:9490–4. doi: 10.1073/pnas.92.21.9490
105. Blancas-Mejía LM, Horn TJ, Marin-Argany M, Auton M, Tischer A, and Ramirez-Alvarado M. Thermodynamic and fibril formation studies of full length immunoglobulin light chain AL-09 and its germline protein using scan rate dependent thermal unfolding. Biophys Chem. (2015) 207:13–20. doi: 10.1016/j.bpc.2015.07.005
106. Misra P, Blancas-Mejia LM, and Ramirez-Alvarado M. Mechanistic insights into the early events in the aggregation of immunoglobulin light chains. Biochemistry. (2019) 58:3155–68. doi: 10.1021/acs.biochem.9b00311
107. Peterson FC, Baden EM, Owen BAL, Volkman BF, and Ramirez-Alvarado M. A single mutation promotes amyloidogenicity through a highly promiscuous dimer interface. Structure. (2010) 18:563–70. doi: 10.1016/j.str.2010.02.012
108. Kobayashi Y, Tsutsumi H, Abe T, Ikeda K, Tashiro Y, Unzai S, et al. Decreased amyloidogenicity caused by mutational modulation of surface properties of the immunoglobulin light chain BRE variable domain. Biochemistry. (2014) 53:5162–73. doi: 10.1021/bi5007892
109. Rottenaicher GJ, Weber B, Rührnößl F, Kazman P, Absmeier RM, Hitzenberger M, et al. Molecular mechanism of amyloidogenic mutations in hypervariable regions of antibody light chains. J Biol Chem. (2021) 296:100334. doi: 10.1016/j.jbc.2021.100334
110. Pradhan T, Annamalai K, Sarkar R, Huhn S, Hegenbart U, Schönland S, et al. Seeded fibrils of the germline variant of human λ-III immunoglobulin light chain FOR005 have a similar core as patient fibrils with reduced stability. J Biol Chem. (2020) 295:18474–84. doi: 10.1074/jbc.RA120.016006
111. Annamalai K, Liberta F, Vielberg M-T, Close W, Lilie H, Gührs K-H, et al. Common fibril structures imply systemically conserved protein misfolding pathways. In Vivo. Angew Chem Int Ed Engl. (2017) 56:7510–4. doi: 10.1002/anie.201701761
112. Schreiner S, Berghaus N, Poos AM, Raab MS, Besemer B, Fenk R, et al. Sequence diversity of kappa light chains from patients with AL amyloidosis and multiple myeloma. Amyloid. (2024) 31:86–94. doi: 10.1080/13506129.2023.2295221
113. Poshusta TL, Sikkink LA, Leung N, Clark RJ, Dispenzieri A, and Ramirez-Alvarado M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One. (2009) 4:e5169. doi: 10.1371/journal.pone.0005169
114. Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. (2014) 35:E2403–12. doi: 10.1002/humu.22619
115. Henikoff S and Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U.S.A. (1992) 89:10915–9. doi: 10.1073/pnas.89.22.10915
116. Berghaus N, Schreiner S, Poos AM, Raab MS, Goldschmidt H, Mai EK, et al. Comparison of IGLV2–14 light chain sequences of patients with AL amyloidosis or multiple myeloma. FEBS J. (2023) 290:4256–67. doi: 10.1111/febs.16805
117. Ruiz-Zamora RA, Guillaumé S, Al-Hilaly YK, Al-Garawi Z, Rodríguez-Alvarez FJ, Zavala-Padilla G, et al. The CDR1 and other regions of immunoglobulin light chains are hot spots for amyloid aggregation. Sci Rep. (2019) 9:3123. doi: 10.1038/s41598-019-39781-3
118. Ostroverkhova D, Przytycka TM, and Panchenko AR. Cancer driver mutations: predictions and reality. Trends Mol Med. (2023) 29:554–66. doi: 10.1016/j.molmed.2023.03.007
Keywords: systemic AL amyloidosis, protein aggregation, amyloidogenesis, immunoglobulin light chains, somatic hypermutation, rare disease, plasma cells
Citation: Morgan GJ and Prokaeva T (2025) Clone-specific residue changes at multiple positions are associated with amyloid formation by antibody light chains. Front. Immunol. 16:1622207. doi: 10.3389/fimmu.2025.1622207
Received: 02 May 2025; Accepted: 30 June 2025;
Published: 01 August 2025.
Edited by:
Luis Del Pozo-Yauner, University of South Alabama, United StatesReviewed by:
Robert W. Maul, National Institute on Aging (NIH), United StatesLuis Del Pozo-Yauner, University of South Alabama, United States
Stefano Ricagno, University of Milan, Italy
Julio Isael Perez-Carreon, National Institute of Genomic Medicine (INMEGEN), Mexico
Copyright © 2025 Morgan and Prokaeva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gareth J. Morgan, Z2ptb3JnYW5AYnUuZWR1