 Wanqiu Xia
Wanqiu Xia Xianghan Zhang2†
Xianghan Zhang2† Lei Fang
Lei Fang- 1Department of Gynaecology, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
- 2Capital Medical University, Beijing, China
- 3Department of General Surgery, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, China
- 4Key Laboratory of Preservation of Human Genetic Resources and Disease Control in China, Harbin Medical University, Ministry of Education, Harbin, China
Tumor-associated macrophages (TAMs) are central to tumor progression, immune suppression, and resistance to therapy, making them promising targets in cancer immunotherapy. TAMs exhibit functional heterogeneity, polarizing into pro-tumor (M2-like) and anti-tumor (M1-like) phenotypes under different microenvironmental cues. M2-like TAMs promote immune evasion, angiogenesis, and metastasis, while M1-like TAMs enhance antitumor immunity. Combining TAM-targeted therapies with immune checkpoint inhibitors is also emerging as a potential strategy to enhance immunotherapy efficacy. This review outlines TAM-mediated immunosuppression mechanisms, including the secretion of VEGF, TGF-β, and immune checkpoint molecules like PD-L1. We also summarize the current strategies targeting TAMs, such as blocking the CD47/SIRPα axis, using CD40 agonists, and PI3Kγ inhibitors, which have shown promise in preclinical studies. Overall, this review underscores TAMs as pivotal therapeutic targets and proposes future directions to optimize combinatorial immunotherapy for enhanced clinical outcomes.
1 Introduction
Recent advancements in cancer immunotherapy have led to significant breakthroughs across various malignancies (1, 2). The core of these therapies involves reactivating innate and adaptive immune responses to induce robust antitumor immunity. Among these strategies, immune checkpoint inhibitors (ICIs), particularly monoclonal antibodies targeting programmed cell death-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), have shown notable therapeutic efficacy in multiple solid tumors (3–5). Despite their success, ICI monotherapy benefits only a subset of patients, with substantial intertumoral and interindividual variability in treatment outcomes (6). Moreover, many initially responsive patients develop acquired resistance to ICIs during treatment (7), underscoring the need for deeper insights into tumor progression mechanisms and novel therapeutic targets to improve immunotherapy efficacy.
The tumor microenvironment (TME) is a complex, immunosuppressive landscape comprised of diverse tumor cells, infiltrating immune cells, and stromal components (8–10). A growing body of evidence highlights that the immunosuppressive characteristics of the TME present a major obstacle to the success of immunotherapeutic strategies (11, 12). The TME is enriched with various immunosuppressive cell subsets, such as tumor-associated macrophages (TAMs) and regulatory T (Treg) cells. Notably, TAMs are the most abundant immune cell population within the TME (13). Emerging evidence underscores TAMs as central players in tumor angiogenesis, metastasis, immune evasion, and therapeutic resistance (14, 15). Importantly, targeting TAMs therapeutically has been shown to reduce resistance to ICIs (16). Both preclinical and clinical studies have demonstrated that combining TAM-targeted therapies with immune checkpoint blockade enhances antitumor efficacy (17). These findings position TAMs as a promising target for cancer immunotherapy. This review will discuss recent developments in TAM-targeted strategies and the current limitations of this approach.
2 Heterogeneity and plasticity of TAMs
Macrophages demonstrate remarkable plasticity and functional heterogeneity, assuming divergent roles within the TME based on external cues (18–21). Traditionally, they are dichotomized into M1 and M2 phenotypes: M1 macrophages, activated by IFN-γ, LPS, or TNF-α, exhibit tumoricidal and pro-inflammatory activity via secretion of IL-1β, IL-12, IL-23, and reactive nitrogen species; M2 macrophages, induced by IL-4, IL-10, or glucocorticoids, secrete IL-10 and TGF-β and express Arg1 and CD206, facilitating tissue repair, angiogenesis, and tumor progression (22). However, this binary model is now considered overly reductive, as macrophages often co-express M1 and M2 markers along a transcriptional continuum (18). IL−4/STAT6 signaling is a central driver of M2-like phenotypes: IL−4 binding to its receptor activates Janus kinases (JAKs), which phosphorylate STAT6, enabling STAT6 to translocate into the nucleus and induce transcription of anti-inflammatory and pro-tumor genes such as Arg1, MRC1 (CD206), and CCL18. Conversely, the NF−κB pathway, activated by stimuli such as LPS or TNF-α, is a hallmark of M1 polarization. Nuclear translocation of NF−κB subunits (p65/p50) induces transcription of pro-inflammatory cytokines (IL−1β, IL−12, TNF-α) that sustain anti-tumor immunity. Additionally, in the hypoxic tumor microenvironment, HIF-1α and HIF-2α stabilize and interact with co-activators to preferentially drive the expression of VEGF, CXCL12, and other genes that skew macrophages toward an immunosuppressive M2-like state. These signaling axes dynamically shape the transcriptional landscape of TAMs and contribute to their phenotypic plasticity within tumors.
Macrophage polarization is regulated by key transcription factors. M1 macrophages are driven by IRF5 and STAT1 signaling, while M2 macrophages are regulated by IRF3, IRF4, and STAT6, which promote anti-inflammatory gene transcription including Arg1 and IL-10 (23). Thus, reprogramming macrophage polarization holds therapeutic potential. In tumors, TAMs exhibit dynamic polarization. M1-like TAMs mediate anti-tumor effects through phagocytosis, ADCC, production of ROS and NO, and secretion of IFN-γ and IL-12 to enhance NK and CTL activity (24–26). Conversely, M2-like TAMs promote tumorigenesis by supporting angiogenesis, EMT, ECM remodeling, and immune suppression. They secrete VEGF, PDGF, EGF, FGF, TGF-β, MMPs, and cathepsins, and inhibit immunity via PD-L1, CD47/SIRPα, and cytokines like IL-10 and CCL2 (25–28). TAM heterogeneity contributes to variable clinical outcomes. Elevated TAM density is associated with poor prognosis in over 80% of cancers (11), particularly in lung and breast cancers, where M2-like TAMs associate with reduced survival (29). In breast cancer, stromal TAMs are more predictive of poor outcomes than intratumoral TAMs (30). Similarly, high TAM infiltration correlates with aggressive phenotypes in gastric, bladder, and skin cancers, multiple myeloma, and Hodgkin lymphoma (29). However, in colorectal cancer, TAMs may exert anti-tumor effects, correlating with CD8+ T cell infiltration and fewer metastases. Still, CD163+ TAMs in colorectal tumors predict poor outcomes (31). Collectively, the evidence supports the use of TAM infiltration levels as a predictive biomarker for patient prognosis in many solid tumors. Evaluating specific TAM subsets may offer more accurate prognostic insights in clinical oncology.
3 Pro-tumorigenic mechanisms of TAMs
3.1 Hypoxic microenvironment induces M2 polarization of TAMs
Owing to the rapid proliferation and expansion of tumor tissues, the TME often becomes hypoxic (32). Upon recruitment to the tumor site, macrophages are subjected to this hypoxic milieu, which activates multiple intracellular signaling pathways, including the hypoxia-inducible factor (HIF) pathway, the VEGF pathway, and the NF-κB pathway (33–35). These signaling cascades promote the accumulation of cytokines such as VEGF and eotaxin within tumor tissues, subsequently driving the polarization of macrophages toward the immunosuppressive M2 phenotype (36). M2-TAMs further secrete chemotactic factors including CCL2, CCL5, and macrophage colony-stimulating factor-1 (CSF-1), thereby contributing to immunosuppression and establishing a supportive niche for tumor angiogenesis, metastasis, and invasion (37, 38). M2-polarized macrophages actively reshape the TME by releasing IL−10 and TGF−β (39). IL−10 suppresses antigen presentation by dendritic cells and macrophages, downregulates MHC-II and costimulatory molecules, and inhibits cytotoxic T lymphocyte (CTL) activity, leading to an immune-permissive environment (40). TGF−β exerts pleiotropic effects by inducing epithelial–mesenchymal transition (EMT) in tumor cells, activating cancer-associated fibroblasts, and stimulating extracellular matrix deposition, all of which promote local fibrosis, tumor invasion, and immune exclusion (41). Importantly, the hypoxic TME also drives upregulation of biomarkers such as PD-L1 on circulating tumor cells (CTCs), which represents a mechanism of adaptive immune resistance; PD-L1-positive CTCs interact with PD-1 on T cells to dampen immune surveillance and are now being explored as predictors of ICI response (42, 43).
3.2 TAM-mediated angiogenesis
To accommodate the increased metabolic and oxygen demands associated with the high proliferation rate of tumor cells, TAMs undergo functional adaptation to promote angiogenesis and support tumor growth (44). The imbalance between pro-angiogenic and anti-angiogenic factors in the hypoxic tumor milieu leads to aberrant neovascularization, resulting in vasculature that is typically abnormal, immature, and highly permeable compared to normal blood vessels (45). Angiogenesis within tumors is a coordinated process involving both malignant and stromal cells and requires degradation of the basement membrane along with endothelial cell proliferation and migration (46). TAMs actively participate in this process by secreting matrix metalloproteinases (MMPs) and cathepsins that degrade extracellular matrix components (47). Furthermore, they produce key pro-angiogenic factors such as VEGF, platelet-derived growth factor (PDGF), basic fibroblast growth factor (b-FGF), and chemokines including CCL2 and CXCL8, which collectively facilitate the formation of a vascular network essential for sustained tumor expansion and dissemination (48).
VEGF plays a central mechanistic role: it binds VEGFR2 on endothelial cells, stimulating their proliferation, migration, and survival, while also increasing vascular permeability (49, 50). VEGF also indirectly suppresses anti-tumor immunity by impairing dendritic cell maturation and promoting anergic or exhausted T cell phenotypes, thereby coupling angiogenesis with immune evasion (51). Additionally, TGF−β released by M2 macrophages augments angiogenesis through induction of extracellular matrix remodeling, fibroblast activation, and production of angiogenic ligands, while IL−10 reduces inflammatory cues that might otherwise restrain angiogenesis (52, 53). Together, these factors foster a structurally and functionally aberrant vascular network that facilitates tumor perfusion and dissemination. Notably, factors such as VEGF-A and CCL2 can recruit circulating monocytes, and their expression levels positively correlate with TAM accumulation and vascular density in certain tumor types (44, 54). Of particular interest is a monocyte subpopulation characterized by the expression of the tyrosine kinase receptor Tie2 which has gained increasing attention for its role in promoting angiogenesis (55, 56). Biel et al. demonstrated that angiopoietin-1 (Ang-1), the ligand of Tie2, is expressed by endothelial cells and promotes perivascular alignment of TEMs. These cells subsequently secrete Wnt-7b, which targets endothelial cells and induces VEGF production, thus enhancing angiogenesis (57–60). Collectively, TAMs act in concert with tumor-derived angiogenic factors to facilitate neovascularization, laying the groundwork for tumor proliferation and progression.
3.3 TAMs and tumor metastasis
Tumor invasion and metastasis represent the leading cause of cancer-related mortality (61, 62). Substantial evidence supports a functional association between TAM recruitment and tumor cell dissemination. TAMs participate in the formation of the pre-metastatic niche, thereby fostering colonization at distant sites. Moreover, within the metastatic microenvironment, TAMs facilitate tumor cell extravasation and survival by mediating immune evasion (63). Epithelial-to-mesenchymal transition (EMT) is a critical step in the metastatic cascade, whereby epithelial tumor cells acquire migratory and invasive properties, enabling survival and dissemination via hematogenous or lymphatic routes. TAMs have been implicated in the regulation of this process (64). For instance, TGF-β secreted by TAMs can induce EMT in tumor cells. In teratomas, TAM accumulation is associated with elevated TGF-β expression, which in turn triggers EMT and promotes metastasis (65). Additionally, TAMs secrete proteolytic enzymes such as cathepsins, MMPs, and serine proteases, which degrade basement membranes and extracellular matrix, facilitating tumor cell escape from the primary site. Furthermore, tumor-derived CSF-1 interacts with epidermal growth factor (EGF) signaling in macrophages, promoting their perivascular accumulation and enabling immune evasion by tumor cells (54). These findings suggest that TAM accumulation significantly enhances tumor cell invasiveness and metastatic potential. Therefore, targeting TAMs may offer a promising strategy to counteract metastasis and enhance the efficacy of cancer immunotherapy (Figure 1).
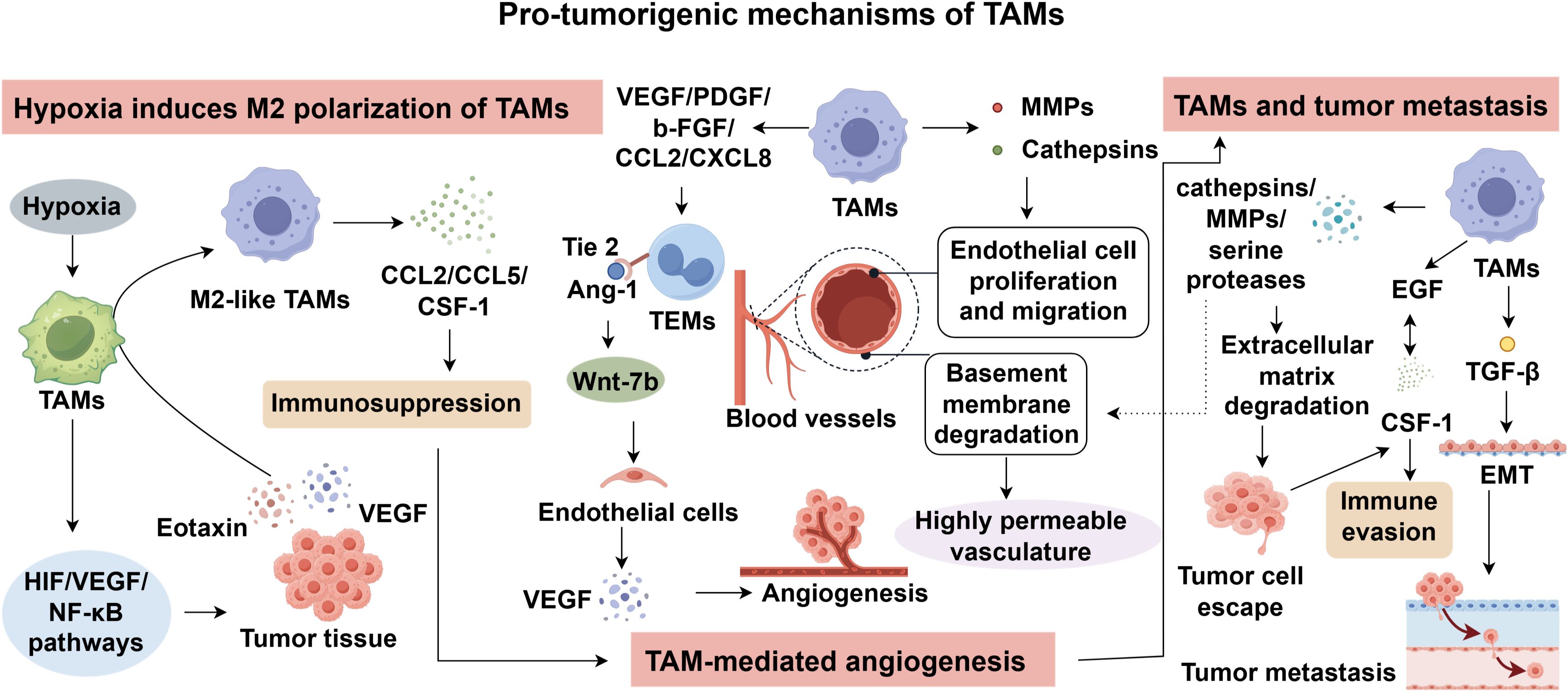
Figure 1. Pro-tumorigenic mechanisms of TAMs.
4 Advances in targeting TAMs for cancer therapy
4.1 Inhibition of pro-tumor TAMs
4.1.1 Blocking monocyte/macrophage recruitment
Inhibiting monocyte/macrophage recruitment to the TME limits pro-tumor TAM accumulation. CCL2–CCR2 and CXCL12–CXCR4 axes are key mediators of this process (66). CCL2, secreted by tumor and stromal cells, recruits CCR2+ monocytes, promoting TAM differentiation; anti-CCL2 antibodies reduce TAM infiltration and tumor progression (67). However, CCL2 blockade withdrawal accelerated metastasis in breast cancer models, likely due to reactive monocytosis (68). Phase I trials of CCL2/CCR2 inhibitors showed modest benefit, while Phase II lacked efficacy, possibly due to compensatory CCL2 upregulation (69). Notably, CCR2 antagonists plus chemotherapy showed improved outcomes in pancreatic cancer compared to chemotherapy alone (70). CXCL12 recruits immunosuppressive TAMs and impairs T cell activation; CXCR4 blockade reduces TAM chemotaxis and delays tumor growth in preclinical models (71, 72). Similar effects were seen in ovarian and prostate cancers (25). Additionally, the CX3CL1/CX3CR1 axis promotes TAM-driven skin carcinogenesis (73) (Table 1).
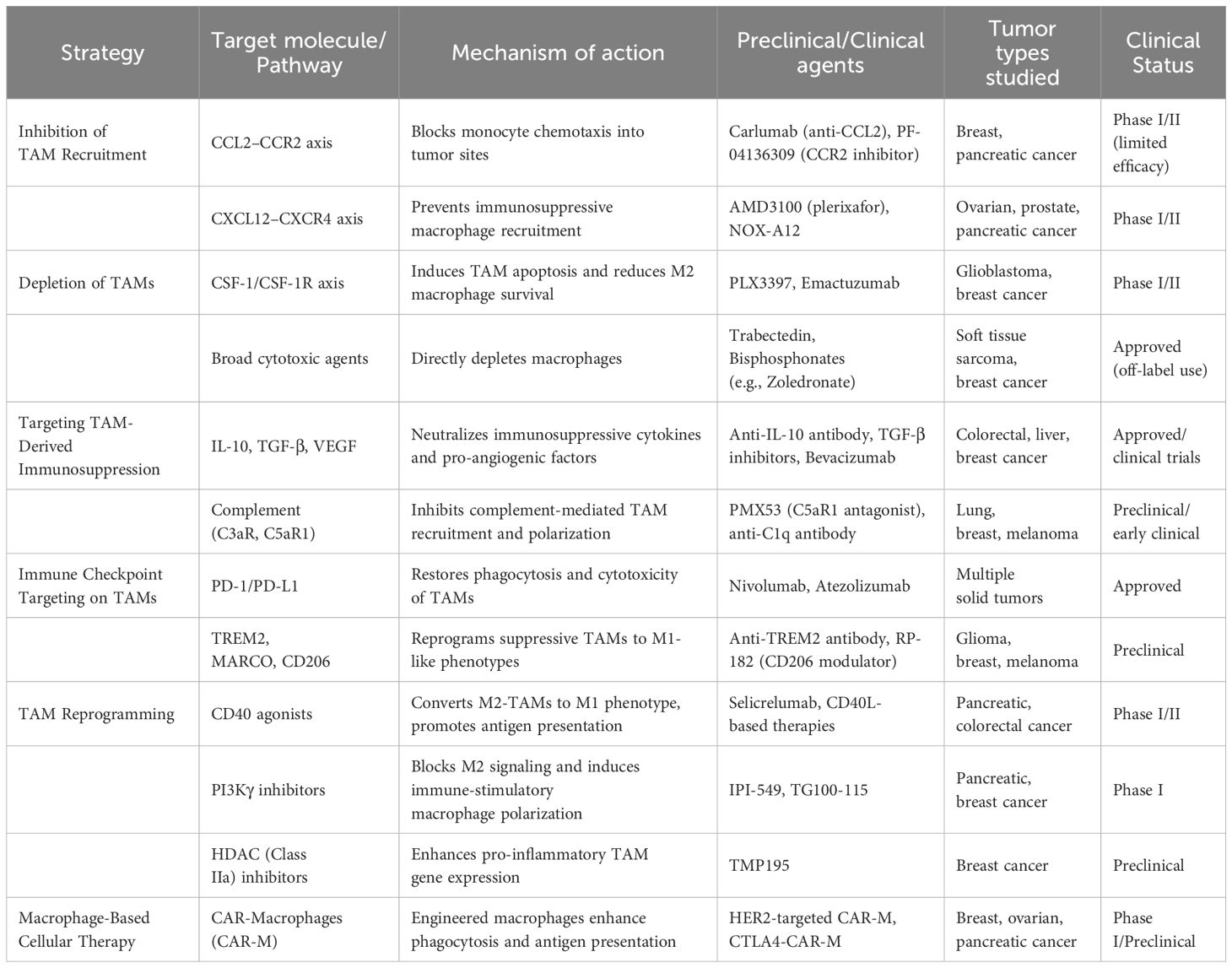
Table 1. Therapeutic strategies targeting tumor-associated macrophages (TAMs) in cancer immunotherapy.
4.1.2 Targeting pro-tumor complement components
Beyond blocking recruitment, inducing TAM apoptosis offers another avenue to deplete these cells. The CSF-1/CSF-1R axis is critical for monocyte/macrophage differentiation, maturation, and survival. Inhibition of this pathway induces TAM death and attenuates their pro-tumor functions (14, 74). Agents such as trabectedin and bisphosphonates have been shown to eliminate macrophages through apoptosis. Trabectedin induces DNA damage and G2/M arrest in tumor cells, and exhibits anti-proliferative activity in melanoma (75). Bisphosphonates enhance immunosurveillance, inhibit tumor invasiveness, and reduce angiogenesis, while also synergizing with other anticancer agents (76). Both preclinical and clinical studies in breast cancer have validated the anti-tumor potential of bisphosphonates targeting TAMs (77). Recent evidence underscores the tumor-promoting role of complement in human and murine cancers. Activation via classical (C1q), alternative, or lectin pathways generates C3a/C5a and the membrane attack complex. C5a recruits MDSCs, enhancing immunosuppression, while C3a/C3aR signaling drives TAM recruitment and immune evasion. In squamous carcinoma, urokinase-positive macrophages mediate C5a release, promoting pro-tumor TAMs and suppressing T cell cytotoxicity (78). Prognostic complement markers include C4d, C5a/C5aR1, and C1s/C4d (79, 80). High expression of complement genes correlates with poor prognosis in melanoma, glioma, and ccRCC (79). C1q+ TAMs induce immunosuppression via PD-1, LAG-3, and PD-L2 (80). Complement inhibition synergizes with ICB; dual C5a or C3aR and PD-1 blockade enhances CD8+ T cells, reduces MDSCs, and improves survival (78).
4.1.3 Immune checkpoints on TAMs
TAM subsets defined by surface markers, such as CD163+ and CD206+ macrophages, have distinct functional roles and are associated with different prognostic outcomes across multiple cancer types. Scavenger receptors on TAMs are promising targets for macrophage reprogramming. CD163, an M2 marker, is strongly linked to immunosuppressive functions and poor prognosis in cancers such as pancreatic cancer and melanoma (25) Depletion or functional blockade of CD163+ TAMs has been shown to enhance T-cell–mediated immunity and improve responses to PD−1 blockade in preclinical studies (81). Similarly, CD206 defines another immunosuppressive TAM subset that secretes high levels of IL−10 and promotes tumor immune evasion. RP-182, a synthetic peptide targeting CD206, eliminates CD206+ cells and reprograms TAMs into M1-like macrophages, enhancing phagocytosis and antitumor activity, with synergistic effects in immunotherapy models (25). Given the distinct prognostic implications of CD163+ and CD206+ subsets, specifically targeting these populations has emerged as a precision strategy to improve clinical outcomes. MARCO, enriched in glioblastoma TAMs, also supports immunosuppression; its blockade reprograms TAMs (14). TAMs expressing PD-1 show reduced phagocytosis, and tumor PD-L1 impairs both T cell and macrophage functions; PD-1/PD-L1 blockade restores immunity (82). TREM2, upregulated in TAMs across cancers, limits PD-1 blockade efficacy; anti-TREM2 antibodies are under clinical evaluation (25, 83). These findings highlight that focusing on specific immunosuppressive TAM subsets, such as CD163+, CD206+, MARCO+, or TREM2+ macrophages, may enable more precise interventions and better therapeutic efficacy in combination with immune checkpoint inhibitors.
4.2 Activation of anti-tumoral TAMs
While depletion of TAMs has demonstrated antitumor potential, the tumoricidal capacity of macrophages also merits attention. Besides the protumoral, immunosuppressive TAM phenotype dominant in the TME, antitumoral macrophages exist. As complete depletion may lead to chronic inflammation or infection, reprogramming TAMs into antitumor phenotypes is a promising alternative (14, 84). Myeloid cells, including macrophages, express SIRPα, which binds CD47 to inhibit phagocytosis (25) Thus, the CD47/SIRPα axis represents a target for immunotherapy. Blocking this axis restores macrophage phagocytosis and elicits antitumor immunity (85). Anti-CD47 antibodies suppress tumor growth, enhance antitumoral macrophage recruitment, and activate CD8+ T cells in multiple tumor models, including glioblastoma, where CD47 blockade reprogrammed TAMs (25, 85). Anti-CD47 therapy also synergizes with immune checkpoint blockade, amplifying efficacy and reducing metastasis (85). Other TAM-modulating agents include CD40 agonists, PI3Kγ inhibitors, and class IIa histone deacetylase (HDAC) inhibitors. CD40, a TNF receptor family member expressed on tumor cells and antigen-presenting cells, including macrophages, induces proinflammatory cytokine release and upregulation of CD80 and CD86 upon activation, sustaining T cell responses (29, 86). In pancreatic ductal adenocarcinoma, CD40 activation converted immunosuppressive TAMs to immunostimulatory ones, restoring immune surveillance (29). In murine colon cancer, CD40 agonists combined with CSF-1R inhibitors reduced immunosuppressive cells and increased antitumoral TAMs (33), and also showed synergy with checkpoint blockade and chemotherapy (86). Thus, CD40 activation facilitates macrophage repolarization and enhances immunotherapy response (29).
PI3Kγ is highly expressed in myeloid cells and promotes immunosuppression via NF-κB inhibition (86, 87). PI3Kγ blockade reactivates T cell responses and inhibits tumor progression (87). In breast and pancreatic cancer models, PI3Kγ deletion or inhibition reprogrammed TAMs, alleviated immunosuppression, and reduced tumor invasion and metastasis (86). Moreover, PI3Kγ inhibitors synergized with checkpoint blockade to improve tumor control and prognosis in vivo (86). HDACs regulate gene expression by removing acetyl groups from histone and non-histone proteins (88). TMP195, a selective class IIa HDAC inhibitor, promoted recruitment and differentiation of phagocytic, proinflammatory macrophages in the TME, reprogramming TAMs and reducing tumor burden and metastasis. In murine breast cancer models, TMP195 also enhanced tumor killing when combined with chemotherapy or checkpoint inhibitors (89). Overall, reprogramming TAMs into tumoricidal phenotypes offers substantial therapeutic promise and a novel avenue for cancer immunotherapy.
4.3 Macrophage-based therapy
In recent years, chimeric antigen receptor T cell (CAR-T) therapy has achieved remarkable success in the treatment of hematologic malignancies. However, its efficacy in solid tumors remains limited, primarily due to tumor heterogeneity and the profoundly immunosuppressive tumor microenvironment (90). As an alternative, chimeric antigen receptor macrophages (CAR-M) have emerged as a novel form of cell-based immunotherapy designed to harness the innate phagocytosis, cytokine release, activation of the tumor microenvironment, and antigen-presenting capacity of macrophages. Unlike CAR-T cells, CAR-M can infiltrate solid tumors more effectively and remodel the immunosuppressive milieu through secretion of proinflammatory cytokines, enhanced phagocytosis, and cross-priming of tumor-specific T cells (91). Currently, several clinical trials are underway or in development to evaluate the therapeutic potential of CAR-M across different malignancies. Phase 1 clinical trial of CT-0508 has shown that HER2-targeted CAR-M therapy achieves significant antitumor responses in murine tumor models (NCT04660929) (92). This approach not only mediates direct tumor cell killing but also promotes the phenotypic shift from M2- to M1-type macrophages, thereby amplifying T cell-mediated antitumor responses. However, several limitations remain to be addressed. Unlike T cells, macrophages have limited proliferative and expansion capacity, which may significantly constrain the overall therapeutic efficacy. Additionally, excessive macrophage activation may lead to overproduction of proinflammatory cytokines, resulting in potential cytotoxicity (91). Despite these challenges, CAR-M therapy represents an exciting frontier in solid tumor immunotherapy, and continued investigation and optimization are warranted.
5 Conclusion
TAMs play pivotal roles in tumor progression, immune suppression, and therapeutic resistance, orchestrating various aspects of tumor biology, including angiogenesis, metastasis, and immune evasion. Their functional heterogeneity and plasticity enable TAMs to adopt pro-tumor (M2-like) or anti-tumor (M1-like) phenotypes, contributing to the complex TME. While M1-like TAMs are associated with anti-tumor immunity, M2-like TAMs support tumor progression by promoting angiogenesis, immune suppression, and metastasis. These findings underscore the potential of targeting TAMs in cancer immunotherapy, with strategies like macrophage reprogramming, recruitment inhibition, and immune checkpoint blockade showing promise in preclinical and early-phase clinical trials.
However, several challenges remain in TAM-targeted therapies. The dynamic heterogeneity of TAM subsets necessitates the identification of precise biomarkers to guide treatment selection. Additionally, compensatory mechanisms and the impact of standard therapies on TAM function require further investigation. Future research should focus on understanding TAM diversity through spatial multi-omics, optimizing combination regimens with immune checkpoint inhibitors (ICIs), and exploring novel targets such as complement cascades (C3aR/C5aR) and scavenger receptors (TREM2). By addressing these gaps, TAM-targeted strategies may significantly enhance immunotherapy outcomes and expand their clinical applicability across various cancers.
Author contributions
WX: Writing – original draft. XZ: Writing – original draft. YW: Writing – original draft. ZH: Writing – original draft. XG: Writing – original draft, Writing – review & editing. LF: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the following source. Excellent youth project of the fourth Affiliated Hospital of Harbin Medical University (HYDSYYXQN202401). The Open Project Program of Key Laboratory of Preservation of Human Genetic Resources and Disease Control in China (Harbin Medical University), Ministry of Education (No. LPHGRD2024-001). Foundation of the Fourth Affiliated Hospital of Harbin Medical University (KYJB2024-02). Postdoctoral Fellowship Program of CPSF under Grant Number GZC20251365. Heilongjiang province exchange medical research institute (070500020261).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang M, Liu C, Tu J, Tang M, Ashrafizadeh M, Nabavi N, et al. Advances in cancer immunotherapy: historical perspectives, current developments, and future directions. Mol Cancer. (2025) 24:136. doi: 10.1186/s12943-025-02305-x
2. Zeiser R. Immunotherapy in cancer. Semin Arthritis Rheum. (2025) 72s:152666. doi: 10.1016/j.semarthrit.2025.152666
3. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, et al. The next decade of immune checkpoint therapy. Cancer Discov. (2021) 11:838–57. doi: 10.1158/2159-8290.CD-20-1680
4. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/Anti-CTLA-4 combined therapy. Cancer Cell. (2019) 35:238–55.e236. doi: 10.1016/j.ccell.2019.01.003
5. Guo X, Cui T, Sun L, Fu Y, Cheng C, Wu C, et al. A STT3A-dependent PD-L1 glycosylation modification mediated by GMPS drives tumor immune evasion in hepatocellular carcinoma. Cell Death Differ. (2025) 32:944–58. doi: 10.1038/s41418-024-01432-0
6. Havel JJ, Chowell D, and Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
7. Vesely MD, Zhang T, and Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol. (2022) 40:45–74. doi: 10.1146/annurev-immunol-070621-030155
8. Xie H, Xi X, Lei T, Liu H, and Xia Z. CD8(+) T cell exhaustion in the tumor microenvironment of breast cancer. Front Immunol. (2024) 15:1507283. doi: 10.3389/fimmu.2024.1507283
9. Deng Y, Shi M, Yi L, Naveed Khan M, Xia Z, and Li X. Eliminating a barrier: Aiming at VISTA, reversing MDSC-mediated T cell suppression in the tumor microenvironment. Heliyon. (2024) 10:e37060. doi: 10.1016/j.heliyon.2024.e37060
10. Zhang Y, Qin N, Wang X, Liang R, Liu Q, Geng R, et al. Glycogen metabolism-mediated intercellular communication in the tumor microenvironment influences liver cancer prognosis. Oncol Res. (2024) 32:563–76. doi: 10.32604/or.2023.029697
11. Komohara Y, Fujiwara Y, Ohnishi K, and Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Delivery Rev. (2016) 99:180–5. doi: 10.1016/j.addr.2015.11.009
12. Yan J, Ye G, Jin Y, Miao M, Li Q, and Zhou H. Identification of novel prognostic circRNA biomarkers in circRNA-miRNA-mRNA regulatory network in gastric cancer and immune infiltration analysis. BMC Genomics. (2023) 24:323. doi: 10.1186/s12864-023-09421-2
13. Pathria P, Louis TL, and Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
14. DeNardo DG and Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
15. Li X, Poire A, Jeong KJ, Zhang D, Ozmen TY, Chen G, et al. C5aR1 inhibition reprograms tumor associated macrophages and reverses PARP inhibitor resistance in breast cancer. Nat Commun. (2024) 15:4485. doi: 10.1038/s41467-024-48637-y
16. Chamseddine AN, Assi T, Mir O, and Chouaib S. Modulating tumor-associated macrophages to enhance the efficacy of immune checkpoint inhibitors: A TAM-pting approach. Pharmacol Ther. (2022) 231:107986. doi: 10.1016/j.pharmthera.2021.107986
17. Shi T, Zhang Y, Wang Y, Song X, Wang H, Zhou X, et al. DKK1 promotes tumor immune evasion and impedes anti-PD-1 treatment by inducing immunosuppressive macrophages in gastric cancer. Cancer Immunol Res. (2022) 10:1506–24. doi: 10.1158/2326-6066.CIR-22-0218
18. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
19. Zhang Y, Tang S, Gao Y, Lu Z, Yang Y, Chen J, et al. Application of exosomal miRNA mediated macrophage polarization in colorectal cancer: Current progress and challenges. Oncol Res. (2023) 32:61–71. doi: 10.32604/or.2023.043481
20. Zhai X, Zhang H, Xia Z, Liu M, Du G, Jiang Z, et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. (2024) 6:101032. doi: 10.1016/j.jhepr.2024.101032
21. Zhao Z, Luo Q, Liu Y, Jiang K, Zhou L, Dai R, et al. Multi-level integrative analysis of the roles of lncRNAs and differential mRNAs in the progression of chronic pancreatitis to pancreatic ductal adenocarcinoma. BMC Genomics. (2023) 24:101. doi: 10.1186/s12864-023-09209-4
22. Arora S, Dev K, Agarwal B, Das P, and Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. (2018) 223:383–96. doi: 10.1016/j.imbio.2017.11.001
23. Davuluri GVN and Chan CH. Regulation of intrinsic and extrinsic metabolic pathways in tumour-associated macrophages. FEBS J. (2023) 290:3040–58. doi: 10.1111/febs.16465
24. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S, et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. (2023) 14:1295257. doi: 10.3389/fimmu.2023.1295257
25. Mantovani A, Allavena P, Marchesi F, and Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
26. Bied M, Ho WW, Ginhoux F, and Blériot C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cell Mol Immunol. (2023) 20:983–92. doi: 10.1038/s41423-023-01061-6
27. Mitsudomi T and Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. (2010) 277:301–8. doi: 10.1111/j.1742-4658.2009.07448.x
28. Salmaninejad A, Valilou SF, Soltani A, Ahmadi S, Abarghan YJ, Rosengren RJ, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (Dordr). (2019) 42:591–608. doi: 10.1007/s13402-019-00453-z
29. Guerriero JL. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med. (2018) 24:472–89. doi: 10.1016/j.molmed.2018.03.006
30. Shen J, Ma H, Chen Y, and Shen J. ScRNA-seq reveals the correlation between M2 phenotype of tumor-associated macrophages and lymph node metastasis of breast cancer. Oncol Res. (2023) 31:955–66. doi: 10.32604/or.2023.029638
31. Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. (2019) 18:64. doi: 10.1186/s12943-019-0976-4
32. Cai Q, Zhang H, Huang Z, and Tian Q. Near-infrared II nanomaterials for hypoxic TME diagnosis and therapy. Nanomedicine (Lond). (2025) 20:1745–58. doi: 10.1080/17435889.2025.2525057
33. Bai R, Li Y, Jian L, Yang Y, Zhao L, and Wei M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: mechanisms and clinical treatment strategies. Mol Cancer. (2022) 21:177. doi: 10.1186/s12943-022-01645-2
34. de la Calle-Fabregat C, Calafell-Segura J, Gardet M, Dunsmore G, Mulder K, Ciudad L, et al. NF-κB and TET2 promote macrophage reprogramming in hypoxia that overrides the immunosuppressive effects of the tumor microenvironment. Sci Adv. (2024) 10:eadq5226. doi: 10.1126/sciadv.adq5226
35. Lu F, Ye M, Shen Y, Xu Y, Hu C, Chen J, et al. Hypoxic tumor-derived exosomal miR-4488 induces macrophage M2 polarization to promote liver metastasis of pancreatic neuroendocrine neoplasm through RTN3/FABP5 mediated fatty acid oxidation. Int J Biol Sci. (2024) 20:3201–18. doi: 10.7150/ijbs.96831
36. Lu Y, Han G, Zhang Y, Zhang L, Li Z, Wang Q, et al. M2 macrophage-secreted exosomes promote metastasis and increase vascular permeability in hepatocellular carcinoma. Cell Commun Signal. (2023) 21:299. doi: 10.1186/s12964-022-00872-w
37. Mitrofanova I, Zavyalova M, Riabov V, Cherdyntseva N, and Kzhyshkowska J. The effect of neoadjuvant chemotherapy on the correlation of tumor-associated macrophages with CD31 and LYVE-1. Immunobiology. (2018) 223:449–59. doi: 10.1016/j.imbio.2017.10.050
38. Henze AT and Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. (2016) 126:3672–9. doi: 10.1172/JCI84427
39. Sun Y, Yang H, Mei X, Xia J, Feng L, Gao J, et al. Cinobufagin inhibits invasion and migration of non-small cell lung cancer via regulating glucose metabolism reprogramming in tumor-associated macrophages. Drug Des Devel Ther. (2025) 19:6647–64. doi: 10.2147/DDDT.S531190
40. Yang Y, Sun Z, Li J, Song Y, and Xu W. Neutrophil-derived IL-10 increases CVB3-induced acute pancreatitis pathology via suppressing CD8(+)T cell activation while increasing macrophage STAT3-IL-6 cascade. Cytokine. (2024) 184:156784. doi: 10.1016/j.cyto.2024.156784
41. Zhang H, Nie J, Bao Z, Shi Y, Gong J, and Li H. FOXC1 promotes EMT and colorectal cancer progression by attracting M2 macrophages via the TGF-β/Smad2/3/snail pathway. Cell Signal. (2025) 130:111680. doi: 10.1016/j.cellsig.2025.111680
42. Wang J, Zhu Y, Chen Y, Huang Y, Guo Q, Wang Y, et al. Three-in-One oncolytic adenovirus system initiates a synergetic photodynamic immunotherapy in immune-Suppressive cholangiocarcinoma. Small. (2023) 19:e2207668. doi: 10.1002/smll.202207668
43. Strati A, Economopoulou P, Lianidou E, and Psyrri A. Clinical significance of PD-L1 status in circulating tumor cells for cancer management during immunotherapy. Biomedicines. (2023) 11:1768. doi: 10.3390/biomedicines11061768
44. Prenen H and Mazzone M. Tumor-associated macrophages: a short compendium. Cell Mol Life Sci. (2019) 76:1447–58. doi: 10.1007/s00018-018-2997-3
45. Netea-Maier RT, Smit JWA, and Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
46. Zeng J, Yang Y, Jiang C, Wu B, and Chen M. An HRL-SC/HIF-1α positive feedback loop enhances cell proliferation, migration and angiogenesis in dental pulp stem cells via PI3K/AKT signalling pathway. Int Endod J. (2025) 58:1006–24. doi: 10.1111/iej.14229
47. Dong S, Li X, Chen Z, Shi H, Wang Z, and Zhou W. MMP28 recruits M2-type tumor-associated macrophages through MAPK/JNK signaling pathway-dependent cytokine secretion to promote the Malignant progression of pancreatic cancer. J Exp Clin Cancer Res. (2025) 44:60. doi: 10.1186/s13046-025-03321-x
48. Cassuto J, Folestad A, Göthlin J, Malchau H, and Kärrholm J. VEGF-A, -C, -D, VEGFR1, -2, -3, PDGF-BB and FGF-2 join forces to induce vascular and lymphatic angiogenesis during bone healing of hip implants. Bone Rep. (2025) 26:101856. doi: 10.1016/j.bonr.2025.101856
49. Benmebarek MR, Oguz C, Seifert M, Ruf B, Myojin Y, Bauer KC, et al. Anti-vascular endothelial growth factor treatment potentiates immune checkpoint blockade through a BAFF- and IL-12-dependent reprogramming of the TME. Immunity. (2025) 58:926–45.e910. doi: 10.1016/j.immuni.2025.02.017
50. Biswas N, Mori T, Ragava Chetty Nagaraj NK, Xin H, Diemer T, Li P, et al. Adenosine diphosphate stimulates VEGF-independent choroidal endothelial cell proliferation: A potential escape from anti-VEGF therapy. Proc Natl Acad Sci U.S.A. (2025) 122:e2418752122. doi: 10.1073/pnas.2418752122
51. Mahaki H, Nobari S, Tanzadehpanah H, Babaeizad A, Kazemzadeh G, Mehrabzadeh M, et al. Targeting VEGF signaling for tumor microenvironment remodeling and metastasis inhibition: Therapeutic strategies and insights. BioMed Pharmacother. (2025) 186:118023. doi: 10.1016/j.biopha.2025.118023
52. Deng Y, Jia X, Liu L, He Q, and Liu L. The role of intestinal macrophage polarization in colitis-associated colon cancer. Front Immunol. (2025) 16:1537631. doi: 10.3389/fimmu.2025.1537631
53. Dong M, Zhang X, Peng P, Chen Z, Zhang Y, Wan L, et al. Hypoxia-induced TREM1 promotes mesenchymal-like states of glioma stem cells via alternatively activating tumor-associated macrophages. Cancer Lett. (2024) 590:216801. doi: 10.1016/j.canlet.2024.216801
54. Petty AJ and Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. (2017) 9:289–302. doi: 10.2217/imt-2016-0135
55. Zhang Y and Brekken RA. Are TEMs canceled? Questioning the functional relevance of tie2-expressing macrophages. Cancer Res. (2022) 82:1172–3. doi: 10.1158/0008-5472.CAN-22-0330
56. Jakab M, Rostalski T, Lee KH, Mogler C, and Augustin HG. Tie2 receptor in tumor-infiltrating macrophages is dispensable for tumor angiogenesis and tumor relapse after chemotherapy. Cancer Res. (2022) 82:1353–64. doi: 10.1158/0008-5472.CAN-21-3181
57. Biel NM and Siemann DW. Targeting the Angiopoietin-2/Tie-2 axis in conjunction with VEGF signal interference. Cancer Lett. (2016) 380:525–33. doi: 10.1016/j.canlet.2014.09.035
58. Turrini R, Pabois A, Xenarios I, Coukos G, Delaloye JF, and Doucey MA. TIE-2 expressing monocytes in human cancers. Oncoimmunology. (2017) 6:e1303585. doi: 10.1080/2162402X.2017.1303585
59. Eklund L, Kangas J, and Saharinen P. Angiopoietin-Tie signalling in the cardiovascular and lymphatic systems. Clin Sci (Lond). (2017) 131:87–103. doi: 10.1042/CS20160129
60. Sidibe A, Ropraz P, Jemelin S, Emre Y, Poittevin M, Pocard M, et al. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun. (2018) 9:355. doi: 10.1038/s41467-017-02610-0
61. Li Z, Zhou H, Xia Z, Xia T, Du G, Franziska SD, et al. HMGA1 augments palbociclib efficacy via PI3K/mTOR signaling in intrahepatic cholangiocarcinoma. biomark Res. (2023) 11:33. doi: 10.1186/s40364-023-00473-w
62. Zhang Y, Zhu X, Chen L, Gao T, Chen G, Zhu J, et al. β-Catenin mediated TAM phenotype promotes pancreatic cancer metastasis via the OSM/STAT3/LOXL2 axis. Neoplasia. (2025) 60:101096. doi: 10.1016/j.neo.2024.101096
63. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. (2017) 214:2695–713. doi: 10.1084/jem.20160392
64. Li L and Lu Y. Inhibition of hypoxia-induced cell motility by p16 in MDA-MB-231 breast cancer cells. J Cancer. (2010) 1:126–35. doi: 10.7150/jca.1.126
65. Quail DF and Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
66. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. (2019) 18:177. doi: 10.1186/s12943-019-1102-3
67. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
68. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
69. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. (2013) 31:760–8. doi: 10.1007/s10637-012-9869-8
70. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
71. Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. (2019) 378:131–8. doi: 10.1016/j.yexcr.2019.03.013
72. Mota JM, Leite CA, Souza LE, Melo PH, Nascimento DC, de-Deus-Wagatsuma VM, et al. Post-Sepsis state induces tumor-Associated macrophage accumulation through CXCR4/CXCL12 and favors tumor progression in mice. Cancer Immunol Res. (2016) 4:312–22. doi: 10.1158/2326-6066.CIR-15-0170
73. Conroy MJ and Lysaght J. CX3CL1 signaling in the tumor microenvironment. Adv Exp Med Biol. (2020) 1231:1–12. doi: 10.1158/2326-6066.CIR-15-0170
74. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
75. Carminati L, Pinessi D, Borsotti P, Minoli L, Giavazzi R, D’Incalci M, et al. Antimetastatic and antiangiogenic activity of trabectedin in cutaneous melanoma. Carcinogenesis. (2019) 40:303–12. doi: 10.1093/carcin/bgy177
76. Van Acker HH, Anguille S, Willemen Y, Smits EL, and Van Tendeloo VF. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol Ther. (2016) 158:24–40. doi: 10.1016/j.pharmthera.2015.11.008
77. Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. (2015) 5:35–42. doi: 10.1158/2159-8290.CD-14-0621
78. Pio R, Ajona D, Ortiz-Espinosa S, Mantovani A, and Lambris JD. Complementing the cancer-immunity cycle. Front Immunol. (2019) 10:774. doi: 10.3389/fimmu.2019.00774
79. Daugan MV, Revel M, Thouenon R, Dragon-Durey MA, Robe-Rybkine T, Torset C, et al. Intracellular factor H drives tumor progression independently of the complement cascade. Cancer Immunol Res. (2021) 9:909–25. doi: 10.1158/2326-6066.CIR-20-0787
80. Roumenina LT, Daugan MV, Noé R, Petitprez F, Vano YA, Sanchez-Salas R, et al. Tumor cells hijack macrophage-Produced complement C1q to promote tumor growth. Cancer Immunol Res. (2019) 7:1091–105. doi: 10.1158/2326-6066.CIR-18-0891
81. Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E, et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J Exp Med. (2019) 216:2394–411. doi: 10.1084/jem.20182124
82. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
83. Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. (2020) 182:872–85.e819. doi: 10.1016/j.cell.2020.06.032
84. Lu J, Dong B, Chen A, He F, Peng B, Wu Z, et al. Escherichia coli promotes DSS−induced murine colitis recovery through activation of the TLR4/NF−κB signaling pathway. Mol Med Rep. (2019) 19:2021–8. doi: 10.3892/mmr.2019.9848
85. Mantovani A, Marchesi F, Malesci A, Laghi L, and Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
86. Wan Y, Tong W, Zhou R, Li J, Yuan J, Wang F, et al. Habitual animal fat consumption in shaping gut microbiota and microbial metabolites. Food Funct. (2019) 10:7973–82. doi: 10.1039/C9FO01490J
87. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
88. Wright LH and Menick DR. A class of their own: exploring the nondeacetylase roles of class IIa HDACs in cardiovascular disease. Am J Physiol Heart Circ Physiol. (2016) 311:H199–206. doi: 10.1152/ajpheart.00271.2016
89. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, SChad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
90. Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: A new immunotherapy candidate against solid tumors. BioMed Pharmacother. (2021) 139:111605. doi: 10.1016/j.biopha.2021.111605
91. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
Keywords: Tumor-associated macrophages, tumor microenvironment, immunosuppression, immune checkpoints, treatment resistance, immunotherapy
Citation: Xia W, Zhang X, Wang Y, Huang Z, Guo X and Fang L (2025) Progress in targeting tumor-associated macrophages in cancer immunotherapy. Front. Immunol. 16:1658795. doi: 10.3389/fimmu.2025.1658795
Received: 03 July 2025; Accepted: 13 August 2025;
Published: 27 August 2025.
Edited by:
Jin Bin, Shandong University, ChinaReviewed by:
Min Miao, Ningbo University, ChinaCopyright © 2025 Xia, Zhang, Wang, Huang, Guo and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinyu Guo, Z3VveGlueXVfaG11QDE2My5jb20=; Lei Fang, ZmFuZ2xlaTgxOUAxNjMuY29t
†These authors have contributed equally to this work