 Satyaki Bhowmik
Satyaki Bhowmik Anwesha Bose
Anwesha Bose Amitava Sengupta
Amitava Sengupta- 1Stem Cell & Leukemia lab, CSIR-Indian Institute of Chemical Biology, IICB-Translational Research Unit of Excellence, Kolkata, West Bengal, India
- 2Academy of Scientific & Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh, India
- 3CSIR-IICB-Cancer Biology & Inflammatory Disorder Division, Kolkata, West Bengal, India
Age-related accumulation of somatic mutations in hematopoietic stem and progenitor cells (HSPCs), causing clonal hematopoiesis (CH), often precedes the development of hematologic malignancies. Chronic inflammation and aberrant cytokine expression that are common in aging, contribute to clonal expansion and genomic instability. Acute myeloid leukemia (AML) is an (epi)genetically and physiologically diverse malignancy, characterized by clonal proliferation and incomplete differentiation of HSPCs. The innate immune system, with pattern recognition receptors (PRRs), plays a pivotal role in maintaining hematopoietic homeostasis. Dysregulated signaling through PRRs disrupts hematopoiesis, fostering malignant cell proliferation. In addition, cytokines and interferons exert multifaceted effects on HSPCs, impacting their proliferation, differentiation, and survival. Therapeutic approaches targeting innate immune pathways, offer promising avenues for treating hematologic malignancies. Understanding the intricate crosstalk between innate immunity and hematopoiesis would provide insights into novel therapeutic strategies for combating hematologic malignancies, offering hope for improved patient outcomes and survival. In this review, we discuss about the malfunctioning innate immune-inflammatory axes in the context of abnormal hematopoiesis and the therapeutic approaches that are utilizing/targeting these pathways with efficacy. This review delves into the derangements of innate immune and inflammatory pathways implicated in the development of AML and myelodysplastic syndromes (MDS), shedding light on the therapeutic strategies targeting these pathways.
Introduction
The tightly regulated process of hematopoiesis ensures an uninterrupted production of blood cells throughout an individual’s lifespan. However, the hematopoietic system undergoes significant alterations with aging that predispose individuals to cytopenias, clonal hematopoiesis (CH) and hematologic malignancies. A major driver of these age related alterations is the gradual accumulation of mutations in hematopoietic stem and progenitor cells (HSPCs). A subset of these mutations gives a competitive advantage to the mutant clones allowing their disproportionate contribution to circulating blood cells. This condition, known as CH, has been connected to systemic inflammation and an elevated risk of myeloid malignancies. Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are the conditions that best illustrate the malignant manifestation of disturbed hematopoiesis. MDS often develops into secondary AML and is characterized by bone marrow failure, dysplastic hematopoiesis and recurrent somatic mutations. De novo AML, on the other hand, develops without a previous history of hematological disease but exhibits clonal dominance of genetically altered HSPCs with impaired differentiation.
Beyond genetic abnormalities, growing evidence highlights that chronic inflammation and innate immune dysregulation play a significant role in shaping the clonal landscape. Inflammatory signals can provide a selection pressure promoting the expansion of preleukemic clones. Thus, innate immune signaling emerges as a key regulator of normal hematopoiesis as well as leukemic transformation. The HSC niche is altered, aberrant myelopoiesis is promoted, clonal progression toward malignancy is accelerated when these pathways are dysregulated. In this review, we provide an overview of how innate immune signaling, aging, and CH interact with hematopoietic abnormalities. We highlight new treatment approaches that target these dysregulated networks and talk about how different immunological mechanisms contribute to the development of MDS and AML.
Hematopoietic abnormalities
Aging results in an accumulation of somatic mutations, that develop spontaneously in all the cells present in the body (1, 2). Base substitution mutations occur at an annual rate of 14.2 mutations, which indicate that the adult HSPCs acquire most of the mutations during the post-natal period (3). Some rare mutations grant an advantage to their host HSPCs and their progeny cells (clones) to grow successively and contribute a significant proportion to the adult mature blood cells pool, leading to CH, which is more prevalent with aging (4) (Figure 1). Since chronic inflammation is also linked to aging, one possible reason behind CH may be chronic inflammation (5). Most of the hematological malignancies harbor mutations that are common with those found in CH (6) (CH). However, the mutations found in the hematological malignancies may be present in healthy adult population as well (7) and to differentiate the non-malignant CH from the malignant one, a term CHIP (clonal hematopoiesis of indeterminate potential) was coined (8). According to Steensma, D. P. et al., CHIP comprises patients with cytopenia and cancer-associated mutations with no clinically detectable MDS as well as adults with normal peripheral blood counts. Though CHIP itself is not a disease, those who carry CHIP mutations, have been shown to develop MDS/AML at an annual rate of 0.5-1% with a greater probability of disease progression in patients with a higher VAF (9).
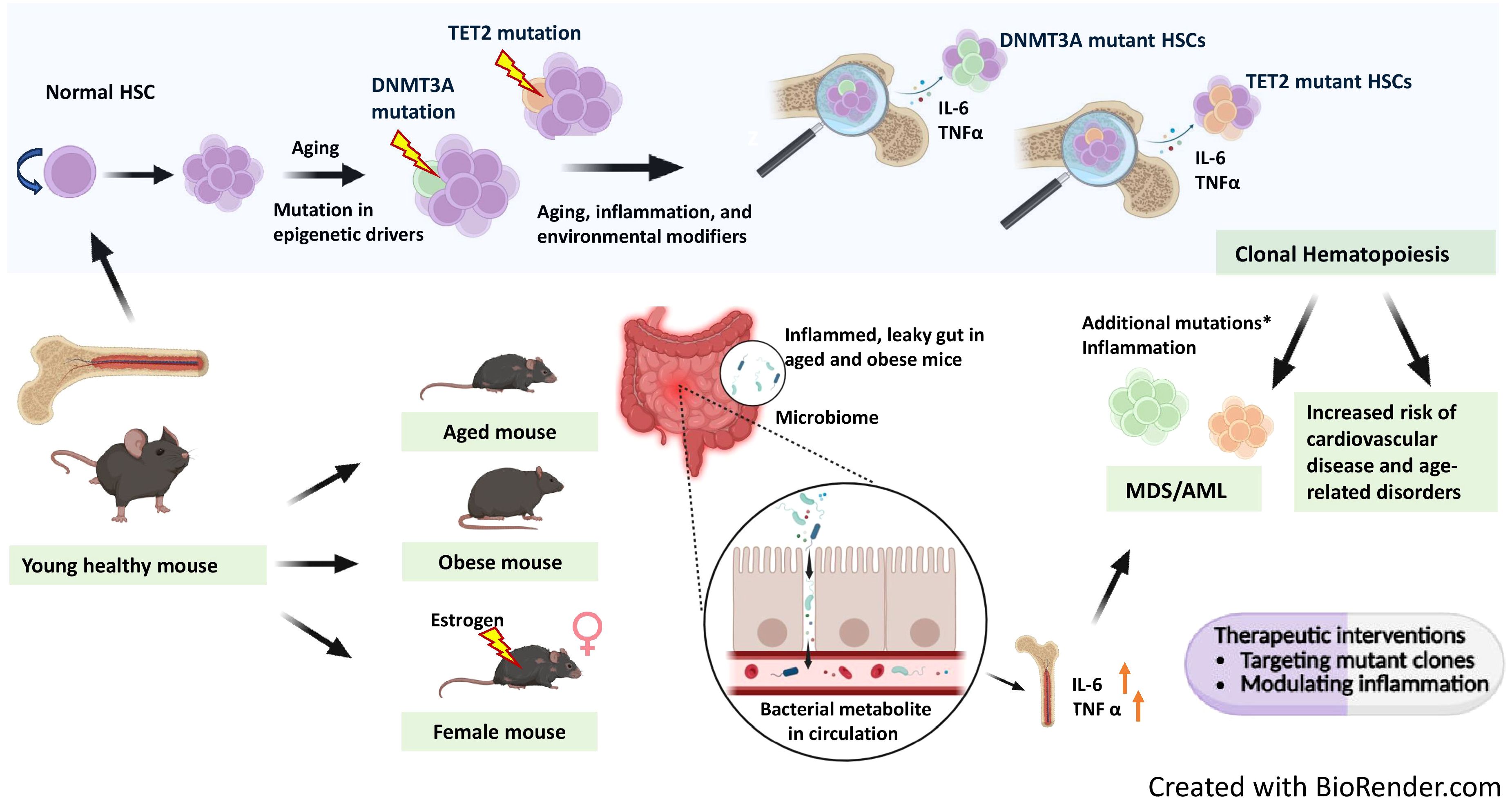
Figure 1. Interplay of age related clonal hematopoiesis with chronic inflammation in leukemia. Aging leads to the spontaneous accumulation of somatic mutations across all cells, including hematopoietic stem cells (HSCs). Many hematological malignancies harbor mutations overlapping with those found in clonal hematopoiesis (CH). CH can also arise as a consequence of chronic inflammation. With aging, mutations in epigenetic drivers like DNMT3A and TET2 accumulate, leading to expansion of mutant HSC clones. DNMT3A mutant clones expand further in female mice under the influence of estrogen. Mouse models of aging and obesity reveal gut inflammation and microbiome dysbiosis, resulting in a leaky gut that allows bacterial metabolites into circulation, further promoting systemic inflammation. Mutant HSCs produce elevated pro-inflammatory cytokines IL-6 and TNF-α, contributing to chronic inflammation. This chronic inflammatory environment, combined with additional mutations in genes involved in key pathways such as RNA splicing, epigenetic modification, cohesin complex function, transcription, DNA damage response, and signal transduction, can drive progression from myelodysplastic syndrome (MDS) to secondary AML. Ultimately, CH and associated inflammation increase the risk of cardiovascular disease and other age-related disorders. Therapeutic interventions targeting mutant hematopoietic clones and inflammatory pathways may help prevent progression from clonal hematopoiesis to myeloid malignancies. The figure is created with BioRender.com.
When a comprehensive diagnosis including a bone marrow examination is inconclusive in a patient appearing with one or more peripheral blood cytopenias, the term ‘idiopathic cytopenia of undetermined significance’ (ICUS) is used in general (10, 11). Peripheral blood cytopenias present a wide range of differential diagnoses, encompassing autoimmune diseases, inflammatory conditions, and various bone marrow disorders such as myelodysplastic syndromes (MDS), aplastic anemia, infiltrative processes, and toxic damage. Normal bone marrow activity can also be suppressed by some pharmaceuticals, as well as dietary inadequacies (12). Although a universally accepted definition of ICUS has not yet been established, the following diagnostic standards have been put forth: (I) at least four months of cytopenia, (II) failure to meet the basic diagnostic requirements for MDS, (III) exclusion of all other causes of cytopenia, and the absence of any genetic abnormalities in myeloid cells, such as mutations, karyotype abnormalities, or flow cytometry aberrations (13).
Patients with cytopenias who do not meet the diagnostic criteria for MDS (having < 10% dysplastic cells inside the bone marrow) or other hematologic illnesses and possess somatic mutations at a variant allele frequency (VAF) of ≥ 2% in genes linked to myeloid malignancies are referred to as having clonal cytopenia of unknown significance (CCUS) (14). The major differences between CHIP and CCUS are, clonal size is often greater in CCUS (VAF of 38% vs. 9% for CHIP), genes linked to a poor prognosis, such ASXL1, RUNX1, and TP53, are more frequently affected, and several genes are impacted in > 64% of CCUS patients compared to 8% of CHIP events (15–17). The comparison among these abnormalities has been discussed in Table 1.
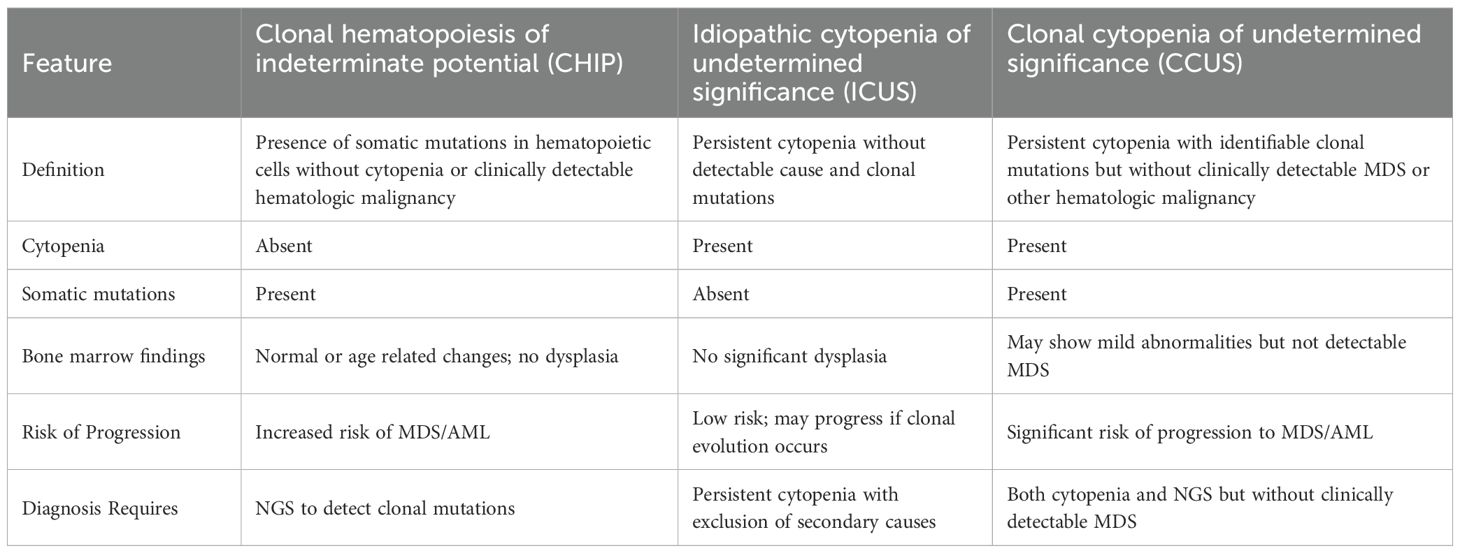
Table 1. Comparison of CHIP, ICUS and CCUS.
MDS is a collective term for describing the hematopoietic stem cell abnormalities in elderly patients. These disorders in common result in dysplasia of bone marrow precursors and peripheral cytopenia along with recurrent somatic gene mutations and chromosomal abnormalities (18). The most prevalent clinical symptom in MDS patients is moderate anaemia, although full myeloid bone marrow failure can also occur which frequently results in bleeding or infection-related mortality (19). Chronic inflammation encourages genomic instability through DNA mutations and epigenetic alterations, prevents tumour immune surveillance, and is predisposed to clonal evolution, all of which favour the development of cancer. As a result, the presence of MDS as well as ongoing chronic inflammation in the BM may eventually encourage the growth of leukemia. MDS patients may progress to secondary Acute myeloid leukemia (AML) that can be diagnosed by increased blast count in the bone-marrow and characterised by difference in proliferation and cell death (20). MDS and secondary AML cases share mutations in genes implicated in at least six key pathways (20).
However, in most of the cases reported, AML arises de-novo in healthy individuals with no previous report of hematological malignancies (21). AML can be characterized by uncontrolled clonal proliferation of immature hematopoietic precursors and incompletely differentiated myeloid blasts leading to impaired hematopoiesis in adults (22). The clonal proliferation and poor differentiation of mutant HSPCs define acute myeloid leukemia (AML), a genetically and physiologically diverse malignancy (23). Chimeric proteins are formed as a result of well-studied chromosomal translocations, which change the normal differentiation and maturation processes of myeloid precursor cells. However, in majority of AML patients, no large chromosomal abnormality is found (24), rather mutations are the key role players in those cases (25). Animal model studies have allowed to classify these mutations into different classes (26) like class I, which includes mutations in FLT3 (ITD and TKD), K/NRAS, TP53, c-KIT and class II, including NPM1 and CEBPA mutations (21). A third class of mutations include those in the epigenetic regulators like DNMT3A, TET2, IDH-1 and IDH-2 (21). Even before patients are diagnosed with AML, they may carry more mutations in some of these genes (DNMT3A, TET2, TP53, SRSF2, IDH2, SF3B1, JAK2, ASXL1), compared to control individuals and having any of these mutations at baseline assessment can be associated with a statistically significant increased risk of developing AML (27). Individuals, who have an autoimmune or chronic immune-stimulated condition, are more likely to acquire AML (28). This connection suggests that dysregulated cytokine expression, a common feature of auto-inflammatory disorders and chronic inflammation, may also encourage the growth of hematological malignancies. Although the role of genetic mutations and epigenetic modifications in the development of these malignancies have been well investigated, recent studies have found dysregulated immune and inflammatory axis to be one of the major players in the pathogenesis of AML.
Components of innate Immune system and their role in hematopoiesis
Germ line encoded Pattern Recognition Receptors (PRRs), present on the plasma membrane as well as within the cytoplasmic compartment of cellular components of the innate immune system for scanning the extra- and intra-cellular environments, have broad specificity to identify Pathogen Associated Molecular Patterns (PAMPs) and Damage Associated Molecular Patterns (DAMPs), which share a more or less similar structural pattern (29). This is evident from the recent studies that, innate immune signals and pathways that are specific to the activation of innate immune cells have a major role to play in normal, healthy HSPCs to regulate hematopoiesis.
Toll-like receptors (TLRs) are the first discovered PRRs, followed by the discovery of other PRR families and identification and characterization of their respective ligands that can initiate the immune signaling upon ligation. The TLRs play an important role in the innate immune system and can be present on the surface of immune as well as non-immune cells (endothelial and mesenchymal stem cells) as homodimers or heterodimers (30). TLR4 activation and induction of nuclear factor (NF)-κB has been shown to be important for subsequent activation of adaptive immune signaling (31). NF-κB plays a key role in inflammation and stress responses and is a key mediator of cell survival (32). Presence of NF-κB is evidently essential for proper functioning of HSCs including HSC proliferation, differentiation and self-renewal (33). Abnormal TLR signalling may lead to malfunctioning of haematopoiesis or haematopoietic malignancy (34, 35). On the contrary, absence of TLR signalling leads to lesser HSC repopulating defects (36).
An intracellular PAMP-sensing NOD-like receptor (NLR) family of receptors reside in the cytoplasm and is known to exert initial response against injury and stress (37). The members (NLRP1, NLRP3, NLRC4) are known to assemble canonical inflammasomes that are complemented by non-canonical pathways to activate different caspases in animal cells via well characterised mechanisms (37). Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) resembles the membrane-bound TLRs. NOD2 detects pathogen-associated motifs and triggers inflammatory signaling cascades that activate NF-κB (38). Ongoing studies have identified inflammasomes to be role players in the field of promoting human HSPC production. The Nlrp3 inflammasome can detect the influx of glucose and/or amino acids into cells, which causes adult human lymphocytic cells to proliferate and differentiate (39).
Interferons were primarily reported as soluble factors that possess antiviral and growth inhibitory activities (40). Interferons are mainly classified into two types: type I and type II. Type I interferons include IFN-α, IFN-β, IFN-ϵ, IFN-κ, IFN-ω, IFN-δ, and IFN-τ whereas, type II interferons include only IFN-γ (41). Following recognition of pathogen-associated molecular patterns (PAMPs), such as foreign or self-nucleic acids, type I IFNs are produced by the majority of cells (42). IFN signaling is initiated by the ligand-receptor interaction followed by the structural rearrangements of the ligand and dimerization of the receptor subunits and subsequent activation of the receptor associated JAKs leading to direct or indirect regulation of interferon stimulated genes (ISGs) and the downstream pathways (43). These ISGs can be engaged in a variety of activities, including the regulation of the immune system and cell death. IFN-α connects the innate and adaptive immune systems by promoting T cell proliferation and survival as well as NK cell cytotoxicity, in addition to having effects directly on tumour cells (44). IFN-α has been shown to possess the ability to remove the dormancy of HSCs and activate them in vivo (45). In contrast to this, chronic exposure to type I IFN takes the HSCs back to quiescence and saves the HSC pool from being exhausted (46).
The complement system was defined on the basis of its capacity to complement the immune responses mediated by cells and antibodies. Upon recognition of PAMPs and/or DAMPs, the complement effector molecules, which are in their precursor state and circulate in the serum and interstitial fluids, get quickly activated in a proteolytic and cascade-like way (47) The components of the complement system can also be produced and expressed locally by different immune and non-immune cell types (48–50). The observation that various cell types appear to contain a diverse range of complement components, receptors and regulators in a manner similar to the inflammasome, the term ‘complosome’ was coined to describe the intracellular complement system (51). The complosome meticulously regulates the production of intracellular reactive oxygen species (ROS) via both mitochondrial and cytosolic signaling pathways, thereby initiating stress-adaptive responses that enhance mitochondrial functionality and improve the overall fitness of HSPCs (52). These components have been extensively reviewed in several articles, thus, we are not citing them and their original references because of space constraints.
Malfunctioning of innate immune and inflammatory pathways in myeloid malignancies
Inflammasomes
The NLRP3 inflammasome is the subtype of inflammasome that has the greatest characterization and is implicated in autoimmune homeostasis as well as the pathophysiology of autoimmune and inflammatory disorders. The DAMP proteins S100A8 and S100A9, which are considerably elevated in MDS patient blood plasma, play a key function as activators of the Nlrp3 inflammasome in the pathogenesis of MDS (53). In response to elevated amounts of extracellular DAMPs, activation of the Nlrp3 inflammasome expands BM myeloid-derived suppressor cells, intensifies BM inflammation, damages hematopoietic cells, causes chromosomal abnormalities, eventually inducing pyroptosis (54). Nlrp3 inflammasome activated Caspase-1 is needed for pyroptosis, which subsequently causes cell enlargement, plasma membrane rupture, and the enormous release of intracellular DAMPs (extra cellular ATP, HMGB1, DNA, and ASC oligomers) as well as IL-1 and IL-18, which together recruit additional immune cells in the extracellular space and initiate the inflammatory cascade in the BM microenvironment (55). Nlrp3 inflammasome undoubtedly contributes to the development and progression of myeloproliferative neoplasm, but further in-depth research is required to understand how it affects this disease. Recent research indicates that, leukemic cells’ migratory and lethal dissemination are significantly influenced by the Nlrp3 inflammasome (56).
Cytokines
Cellular Proliferation, survival and therapy resistance in acute myeloid leukemia (AML) and other hematological cancers are significantly influenced by cytokines, their crucial functions and intricate interplay, in the setting of inflammation. The expression and levels of a number of cytokines (TNF-α, IFN-γ, TGF-β, IL-6 and IL-8) are found to be increased in case of MDS and signify abnormal inflammatory signaling and hematopoietic differentiation (57). Studies have found that individuals, who have chronic inflammation in their system, are more likely to acquire AML (28). An ex vivo cell viability screen in which primary AML samples were cultured in the presence of 94 distinct cytokines, to extensively analyse the impact of cytokines in modifying AML cell survival, identified IL-1β, exerting the most robust effects including increased cell growth and survival in almost 70% of 60 primary AML samples, irrespective of the mutational status (58). IL-1β treatment to AML cells increases the production of other cytokines, known to facilitate AML proliferation (59, 60). IL-6 plays important roles within the network of cytokines involved in the formation of leukemic blasts (61). In AML patients, plasma levels of IL-10 are notably elevated and are associated with elevated levels of IL-6 (62). By suppressing pro-leukemic cytokines at the transcriptional or post-transcriptional level, IL-10 may act to prevent the proliferation of AML cells (63). Jaiswal et al. have shown that macrophages from Tet2−/− bone marrow produce increased amounts of CXCL1, CXCL2 and CXCL3 (64). In individuals with Tet2 mutated CHIP, they have found increased IL-8 in serum. Tet2-deficient mice have increased IL-1β and inflammasome activation (65). Chronic inflammation and immunologic changes in bone marrow niche microenvironment are the effects of Tet2 deficiency (66). Yeaton et al. have demonstrated that disease development in aged animals is associated with an augmented inflammatory response and the establishment of an abnormal inflammatory monocytic cell population using single-cell transcriptome profiling of the bone marrow (67). In individuals with AML, the gene profile that characterises this inflammatory population is linked to a poor prognosis. S100A8 and S100A9 are damage associated molecular patterns (DAMPs), that are expressed in a variety of cells like endothelial cells, macrophages, osteoclasts, keratinocytes and dendritic cells upon inflammatory challenges (68) and can act as endogenous ligands for TLR4, resulting in dysregulated hematopoiesis in MDS by affecting HSPCs directly or indirectly (69). It has been shown that the S100A8 and S100A9 transcripts are overexpressed in the M4 and M5 AML compared to that in M0 and M1 (70). S100A8 and S100A9 appear to influence immunological check points to control leukemic proliferation (71).
Interferons
A study comparing the bone marrow-derived mesenchymal stem cells from 7 MDS patients found an upregulation of IFN-α/β signaling and ISG15 expression (72). MDS patients have an overexpression of IFN-γ in the bone marrow cells (73). In AML cells, chromosomal instability has been shown to result in micronuclei, subsequent DNA damage, and interferon signaling (74). During chronic infection, impairment of an IFN-γ mediated transcriptional network causes proliferation of Dnmt3a-loss of function HSC clones in a mouse model by decreasing stress induced apoptosis and inducing defective differentiation (75). IFN-γ secreted by T cells in the bone marrow microenvironment induces the expressions of PD1 on T cells and PDL1 on tumor cells resulting in the immune escape of the malignant cells (76). In the presence of recombinant interferons, colony forming ability of blast progenitors from peripheral blood isolated from AML patients was found to be suppressed relative to the control cultures (77). IFN-γ has been shown to have a suppressive effect on clonal proliferation of leukemic blasts and induce differentiation of the blasts isolated from patients with AML and CML (78). HSC function can be compromised upon chronic exposure to inflammation/inflammatory challenges (46, 79, 80). Several interferon-stimulated genes, including STAT1, IRF9, IFIT1, IFIT3, IFITM1 and IFI44L, were found to be upregulated in the CD34+ cells of MDS patients (81). Through interactions between the leukemic and mesenchymal stem cells, a subset of AML patients with high IFN-γ levels result in expansion of regulatory T cells (82).
Pattern recognition receptors
A number of TLRs like TLR2, TLR4 AND TLR6 have increased expression on the bone marrow CD34+ cells in MDS patients (83). Increased TLR2 and TLR4 levels in MDS patients are correlated with increased rates of apoptosis (84, 85). Despite increased expression with other TLRs in bone marrow cells during initial stage of MDS, TLR9 expression exhibits a significant downregulation when MDS gets transformed into AML (86). TLR2 and TLR4 act as receptors for HMGB1 (87) and knockdown of these receptors exhibited reversal of NLRP3 induction resulting from HMGB1 treatment (88). NF-κB activity was found to be increasing with the progression of MDS and was highest in the later stages of the disease (89), especially in elderly patients above the age of 75 years (90). Primary AML CD34+ cells exhibit easily observable NF-κB activity, in contrast to human CD34+ progenitor cells, which do not express NF-κB in the absence of stimulation (91). When NF-κB was inhibited in AML cell lines as well as in primary blasts isolated from AML and MDS patients, nutrient depletion driven apoptosis could be induced (92). During induction chemotherapy in AML patients, polymorphisms in TLR2 and TLR4 may result in infectious complications (93). CLL-1, also known as C‐type lectin domain family 12, member A (CLEC12A), has been identified as a marker for leukemic stem cells as it is present on the surface of CD34+CD38- cells in AML patients but can not be found on CD34+CD38 cells from healthy individuals (94, 95). The role of the cytokines and the PRRs have been discussed in Table 2.
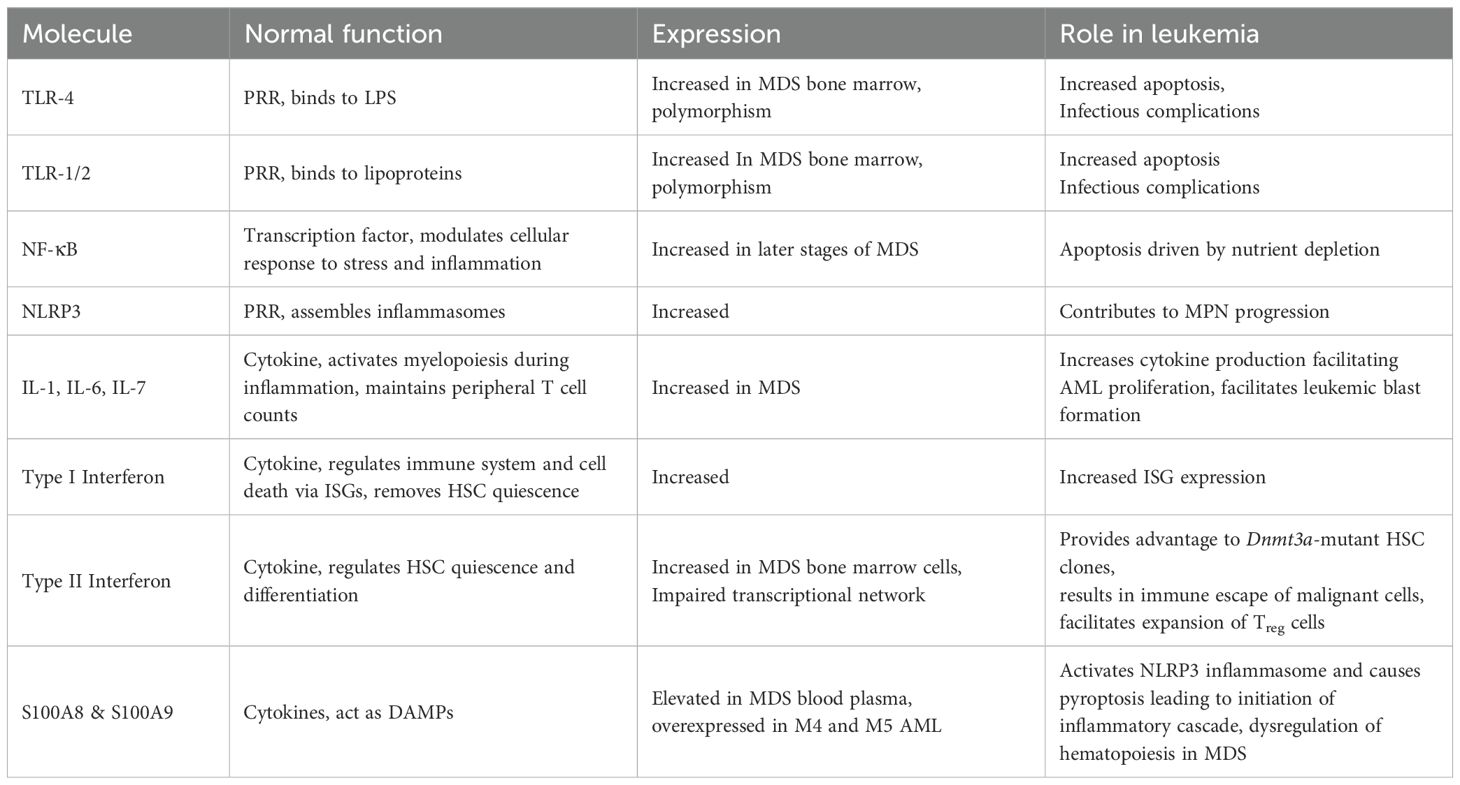
Table 2. Pathophysiological role of innate immune signaling components in leukemia.
Complosome
Various leukemic cell lines like HEL, K562, HL-60, HG-1a show mRNA expression for C3, C5, C3aR and C5aR1 (96). The potential function of the complosome in modulating the biology of these cells was found by deletion of the expression of C3 and C5 using CRISPR-Cas9, which hampered the migration of leukemic cells toward SDF-1 and eATP. C3 and C5 mRNA were also found to be significantly upregulated in AML blasts isolated from peripheral blood.
Immune signaling genes
Several cytokines, including interleukins and interferons have the ability to activate Janus Kinase 2 (JAK2), which regulates hematopoiesis. By fostering STAT phosphorylation, activated JAK2 primarily communicates with the STAT proteins. The transcription of downstream target genes is facilitated by subsequent dimerization and nuclear translocation of STAT. Numerous interleukins, such as IL-6, influence immune-inflammatory response, and immune cell development via signaling through the JAK-STAT pathway (97). JAK2 V617F is one of the most identified CH mutations, based on which different genetic murine models have been developed. Most of these mice exhibit MPN characteristics consistently (98, 99). Although it is not that clear how the same JAK2 mutation gives rise to different MPN subtypes, probably it is related to the involvement of different STATs in different subtypes (100).
Role of the bone marrow microenvironment/niche in CH progression
CH has been found to be strongly associated with infection, age-associated cytokine overproduction and other external inflammatory stimuli in the bone marrow microenvironment (101, 102). Pro-inflammatory cytokines released by immune cells to clear-off pathogens can stimulate the production of intracellular reactive oxygen and nitrogen species in adjacent cells within the microenvironment, setting a stage for somatic mutations in HSCs (103). These mutations may result in skewed differentiation and myeloid restricted progenitors. These devoted progenitors may have increased inflammatory signature that maintain a highly inflamed microenvironment for CH. As the inflammatory bone marrow ecosystem may include selection forces that promote the growth of CH clones, HSPCs with CH mutations may survive and be enriched throughout this process (104). For instance, Tet2-deficient HSPCs show increased expression of an anti-apoptotic long non-coding RNA as a result of IL-6-induced hyperactivation of the Shp2-Stat3 signaling cascade. Tet2 deficient HSCs are therefore resistant to apoptosis because of the inflammatory cytokine (105). Through impaired RIPK1-RIPK3-MLKL-mediated necroptosis signaling, Dnmt3a-mutated bone marrow cells also exhibit enhanced reconstitution potential in an aged bone marrow milieu in response to inflammatory insults (TNF-α) (106, 107). Although the underlying processes are unknown, the HSPCs with an Asxl1 mutation showed an anti-inflammatory gene expression profile (108). Regardless, one recent study has underscored telomere attrition as an instrument for clonal selection in aging hematopoiesis and leukemogenesis (109). Together, HSPCs with CH mutations gain clonal dominance over their normal counterparts by upregulating anti-inflammatory or anti-apoptotic genes, which increase their fitness in an inflammatory environment, and confer resistance to apoptosis.
Recent findings have shown that a few important components of the osteo-hematopoietic niche, such as mesenchymal stromal cells, endothelial cells, osteolineage cells, and the regulatory signals they transmit such as CXCL12, SCF, and osteopontin, gradually deteriorate with age. This degradation promotes the selection of mutant clones while also making the environment less favorable to normal hematopoietic stem cells (HSCs). Additionally, inflammatory cytokines (such as IL-1β, IL-6, and TNF-α) secreted by mutant HSPCs and their descendants actively remodel the bone marrow niche, reinforcing pro-inflammatory signaling loops and causing additional niche dysfunction (110) (Figure 1). Thus, a bidirectional interaction between mutant clones and their aging microenvironment shapes CH development, which is not just controlled by HSPC-intrinsic mutations. The influence of the aging microenvironment on clonal dynamics was illustrated in an elegant study employing DNMT3A R878H-mutant models (111). Transplantation studies performed into both young and elderly recipients demonstrated that DNMT3A-mutant HSCs acquire a competitive edge in aged bone marrow settings, which is mediated by increased TNF-α signaling. By influencing lineage bias toward myeloid vs lymphoid output, TNFR2 uncouples clone size from differentiation trajectory, whereas TNFR1 stimulates mutant clone proliferation. In addition to intrinsic signals from the stromal, endothelial and immune cells, microbial agents have been found to be important regulators of the bone marrow microenvironment in CH. Microbial components and PAMPs alter HSPC function and stimulate pre-leukemic myeloproliferation in Tet2-deficient models. For example, in Tet2-deficient mice, myeloproliferation can be induced by commensal microbiota-derived chronic inflammatory signaling, mainly through the IL-6 and IL-1β pathways (112). According to Yokomizo-Nakano et al., Tet2 loss after sequential exposure to microbial products enhances the signs of MDS, indicating that microbial signals could modify hematological niche for the growth of mutant clones in addition to their clonal expansion (113). Long-term IFN-γ exposure or chronic mycobacterial infection promotes the clonal proliferation of Dnmt3a-mutant HSCs through impaired differentiation and epigenetic reprogramming (106). Additionally, microbial translocation and dysbiosis offer a steady supply of PAMPs and metabolites that may affect HSPC activation and lineage output by altering systemic cytokine profiles (114). Overall, aging-associated intestinal alterations can cause systemic dissemination of microbial metabolites for pre-leukemic cell expansion. In this regard one recent elegant study has indicated that microbial metabolite [ADP-D-glycero-β-D-manno-heptose (ADP-heptose)], drives CH via ALPK1, suggesting that the ADP-heptose-ALPK1 axis is a promising therapeutic target to prevent progression of CH to overt leukemia and immune-related conditions (115). These results demonstrate that the bone marrow microenvironment actively influences the fitness and evolution of CH clones. Furthermore, they emphasize the idea that inflammatory remodeling of the hematopoietic niche with aging produces a milieu that is favorable for CH and may hasten the progression of AML/MDS.
Therapeutic approaches targeting innate immune pathways involved in CH
Poor progress has been achieved in prolonging the survival of high-risk people with AML. Although the use of targeted treatments in combination with chemotherapy has enhanced the chances of complete remission, recurrence rates have not changed much and remain the leading cause of treatment failure in the post-consolidation or post-transplantation setting. As a result, there is an increasing need to develop efficient and secure post-remission therapy strategies that may improve disease free survival and, as a result, overall survival.
IFN has the ability to stimulate apoptosis, inhibit cytokines that promote cell growth and proliferation, and increase the immunogenicity of AML cells (116). These immunomodulatory and anti-proliferative effects emphasise IFNs as a possible therapy for myeloid malignancies (117), especially in cases where the patients are tolerant to the conventional therapies. Several studies have shown that Interferon’s anti-leukemic efficacy is due to both direct action on leukemic blasts and indirect action through immune activation. IFN-α induces latent HSCs to enter the cell cycle (118). This suggests that administering IFN to AML patients can cause quiescent LSCs to multiply and become susceptible to conventional treatment. Minimal residual disease positive AML patients with t(8;21) mutations have reported a better 2-year overall survival upon IFN-α treatment after allogenic hematopoietic stem cell transplantation (119). It has been demonstrated that IFN- α maintenance therapy can lower the probability of relapse in AML patients with favourable risk (120). An ongoing phase I/II trial has shown that pegylated IFN-α did not significantly alter toxicity or acute graft-versus-host-disease (GVHD) risk when administered prophylactically after myeloablative conditioning in an AML cohort at high risk for relapse and resulted in relatively low rates of relapse suggesting a robust graft versus leukemia (GVL) response (121). Stimulating endogenous IFN synthesis, for example by turning on the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, is one method of exploiting interferon’s anti-cancer capabilities (122). cGAS/STING-dependent anti-leukemic action was demonstrated by the STING activator DMXAA in a mouse model of AML either by significantly prolonging the survival, or in some cases, curing the mice (123).
The innate immune system includes NK cells, which are able to detect the lack of certain proteins that may be downregulated on malignant cells, such as HLA proteins. They can also obliterate tumour cells right away. In vitro transformation of primary human NK cells into memory-like NK cells results in the production of IFN-γ and the manifestation of anti-leukemic characteristics (124). The first in human phase I study included three patients with relapsed/refractory AML received treatment with anti-CD33 CAR NK-92, but resulted in no durable response (125). Engineered CAR-NK cell treatments are becoming a popular new kind of therapy (126). TLR3,7,8, and 9 have been the focus of most of the clinical trials employing TLR agonists to treat hematological malignancies (127). A TLR8 ligand R848 has been shown to induce differentiation and stop proliferation of AML cells (128). It has been shown that two SNPs in the human TLR9 gene, T1486C and T1237C, downregulate the expression of TLR9 mRNA resulting in an improved outcome of graft transplant in AML patients (129). As the malfunctioning inflammatory pathways are one of the key responsible factors for maintaining the leukemic state of the immune cells, as a cell-intrinsic, self-directed immunotherapy these pathways may be applied. Ellegast et al. has shown using different genetic and protein degradation techniques in vitro and genetically in vivo to demonstrate AML cells’ reliance on IRF2BP2, which is shown to suppress IL-1β/TNFα signaling via NF-κB, and IRF2BP2 disruption causes an acute inflammatory state that culminates in AML cell death (130). In the mouse model of AML, therapy with an anti-S100A8 antibody led to a comparable effect on AML cell differentiation as treatment with recombinant S100A9 protein, both of which increased survival (70). S100A9 transgenic (S100A9Tg) mice and BM-MNC treated with S100A9 activate the programmed death 1 (PD-1) and programmed death-ligand 1 (PD-L1). MDS BM-MNC treated with recombinant PD-L1 experienced cell death, and in turn, PD-1/PD-L1 inhibition increased colony-forming ability in MDS patient BM-MNC and restored efficient hematopoiesis (71). Buteyn et al. have shown that a combination of synthetic NOD2 ligand and IFN- γ produced an inflammatory cytokine profile and activated NK cells in AML blasts which resulted in significantly increased mature CD27- CD11b+ NK cells in a mouse AML model, as well as significantly decreased disease burden and prolonged survival (131).
Even though CH is mostly linked to negative clinical outcomes, such as an elevated risk of systemic inflammation and hematologic malignancies, it is increasingly becoming evident that not all mutations linked to CH have harmful effects. The immune system may benefit functionally from certain mutations in some cases. CH mutations can increase the effectiveness of chimeric antigen receptor (CAR) T-cell treatment by improving the anticancer activity and T-cell persistence (132). Evidence from models of neurodegenerative diseases indicates that CH can stimulate positive immune responses in addition to cancer immunotherapy; CHIP carriers showed improved outcomes in Alzheimer’s disease and increased microglial activation, suggesting a possible protective role in neuroinflammation (133, 134). Spetzler et al. have identified mutations linked to CHIP in solid tumors that affect tumor-infiltrating lymphocytes (TILs) (135). This finding suggests that hematopoietic clones with CH mutations may directly impact the tumor-immune interface, possibly impacting immunotherapeutic response as well as immune surveillance. These results imply that CH should be considered a physiologically diverse state where the functional effects of mutations are extremely context-dependent, rather than just a pre-malignant or pathogenic condition.
Conclusion & future perspectives
Overall, the members of the innate immune system, like PRRs as well as cytokines and chemokines, have crucial roles in the regulation of the hematopoietic system. Malfunctioning of the components, as well as abnormal activation of the signaling pathways involved in the intricate interplay between these two systems may lead to genomic instability, compromised hematopoiesis and hematopoietic abnormality/malignancy like MDS and AML. The fact that CH can have context-dependent consequences, such as potentiating therapeutic responses or providing immunological benefits, further demonstrates that the biology of this process is more complex than previously appreciated. Current therapeutic approaches have shown exceptional efficacy and may prove themselves to be more effective in the treatment of AML in future. However, finding safe, well-tolerated post-remission techniques that can lower the relapse rate is a continuous problem in the treatment of AML. In cases of complete remission post stem cell transplantation, in specific subsets of AML patients maintenance therapy can be an important player (116). There is a dire need to sustain morphological and/or molecular complete remission in older patients, who have poor biological features at baseline.
Even with the significant advancements made in the field of CH, there are still many unanswered questions and unexplored areas. For example, early-life HSCs may be inherently less likely to acquire or spread somatic mutations, as shown by the low frequency of CH in children. Long-term effects on clonal evolution may arise from prenatal exposure to inflammation caused by environmental pollutants, which could still provide a mutagenic environment in utero. Early-life immune surveillance, in comparison to later stages of life, may provide better immunity against the outspread of mutant clones. However, as people age, the bone marrow microenvironment may get reprogrammed conducive to the proliferation and selection of pre-cancerous clones, which might lead to a higher frequency of CH among elderly individuals. Further influencing the process of CH development is the probable difference in the microbial components between childhood and adulthood that interact with the hematopoietic and immune systems. Future investigations addressing these aspects will improve our overall understanding of the processes in which immune competence, environmental exposure, stem cell physiology, ontogenty and microenvironmental signals interact to influence clonal development throughout life. Comprehensive systems biology understanding of immune signaling in the context of physiological aging associated clonal hematopoiesis, by leveraging genetic and meta-analysis coupled with cutting edge single-cell mutlimodal studies, will provide important insights in innate immune signaling and blood cell homeostasis.
Author contributions
SB: Writing – original draft, Writing – review & editing. AB: Writing – review & editing, Software, Visualization. AS: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study is supported by funding from Lady Tata Memorial Trust-Institutional Research Project Grant (LTMT-IRPG) (to A.S.), ‘S Ramachandran National Bioscience Award for Career Development’ from the Department of Biotechnology (DBT; 102/IFD/SAN/2749/2023-2024) (to A.S.), and Council for Scientific & Industrial Research, Govt. of India (CSIR; OLP-121/P07, RDS000003, MMP075204) (to A.S.). A.S. is an incumbent grantee of Collaborative Scientific Research Program (CSRP) supported by the Indo-French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA; 72T29-Au), and he was a recipient of Indian Council of Medical Research (ICMR)-DHR (Short-Term) International Fellowship (INDO/FRC/452/S-11/2019-20-lHD), and DBT Ramalingaswami Fellowship (BT/RLF/RE-ENTRY/06/2010). S.B. and A.B. acknowledge CSIR for supporting their doctoral Senior Research Fellowships.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Martincorena I and Campbell PJJS. Somatic mutation in cancer and normal cells. Sci. (2015) 349:1483–9. doi: 10.1126/science.aab4082
2. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Gene. (2015) 47:1402–7. doi: 10.1038/ng.3441
3. Osorio FG, Huber AR, Oka R, Verheul M, Patel SH, Hasaart K, et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. (2018) 25:2308–16. doi: 10.1016/j.celrep.2018.11.014
4. Jaiswal S and Ebert BLJS. Clonal hematopoiesis in human aging and disease. 366. (2019) 366(6465):eaan4673. doi: 10.1126/science.aan4673
5. Koschmieder S, Mughal T, Hasselbalch H, Barosi G, Valent P, Kiladjian J, et al. Myeloproliferative neoplasms and inflammation: whether to target the Malignant clone or the inflammatory process or both. Leuk. (2016) 30:1018–24. doi: 10.1038/leu.2016.12
6. Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lépine G, et al. The Journal of the American Society of Hematology, DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. (2017) 130:753–62. doi: 10.1182/blood-2017-04-777029
7. Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood. (2020) 136:1606–14. doi: 10.1182/blood.2019000989
8. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. The Journal of the American Society of Hematology, Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
9. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
10. Valent P, ICUS IDUS, and CHIP and CCUS. Diagnostic criteria, separation from MDS and clinical implications. Pathobiol: J Immunopathol Mol Cell Biol. (2019) 86:30–8. doi: 10.1159/000489042
11. Malcovati L and Cazzola M. The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematol Am Soc Hematol Educ Program. (2015) 2015:299–307. doi: 10.1182/asheducation-2015.1.299
12. Valent P. Low blood counts: immune mediated, idiopathic, or myelodysplasia. Hematol Am Soc Hematol Educ Program. (2012) 2012:485–91. doi: 10.1182/asheducation.V2012.1.485.3798522
13. Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. (2017) 8:73483–500. doi: 10.18632/oncotarget.19008
14. Steensma DP. Cytopenias + mutations - dysplasia = what? Blood. (2015) 126:2349–51. doi: 10.1182/blood-2015-10-672659
15. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
16. Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. (2015) 126:2355–61. doi: 10.1182/blood-2015-08-667063
17. Cargo CA, Rowbotham N, Evans PA, Barrans SL, Bowen DT, Crouch S, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. (2015) 126:2362–5. doi: 10.1182/blood-2015-08-663237
18. Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database. merge. (2012) 30:820. doi: 10.1200/JCO.2011.35.6394
19. Morel P, Hebbar M, Lai J, Duhamel A, Preudhomme C, Wattel E, et al. Cytogenetic analysis has strong independent prognostic value in de novo myelodysplastic syndromes and can be incorporated in a new scoring system: a report on 408 cases. Leuk. (1993) 7:1315–23.
20. Menssen AJ and Walter MJJB. Genetics of progression from MDS to secondary leukemia. Blood. (2020) 136:50–60. doi: 10.1182/blood.2019000942
21. De Kouchkovsky I and Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. (2016) 6:e441–1. doi: 10.1038/bcj.2016.50
22. Boila LD and Sengupta AJEH. Evolving insights on histone methylome regulation in human acute myeloid leukemia pathogenesis and targeted therapy. Exp Hematol. (2020) 92:19–31. doi: 10.1016/j.exphem.2020.09.189
23. Lowenberg B, Downing JR, and Burnett A. Acute myeloid leukemia. New England J Med. (1999) 341:1051–62. doi: 10.1056/NEJM199909303411407
24. C.G.A.R.N.J.N.E. Medicine, Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
25. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New England J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
26. Gilliland DG and Griffin JDJB. The Journal of the American Society of Hematology, The roles of FLT3 in hematopoiesis and leukemia. Blood. (2002) 100:1532–42. doi: 10.1182/blood-2002-02-0492
27. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. (2018) 24:1015–23. doi: 10.1038/s41591-018-0081-z
28. Kristinsson SY, Björkholm M, Hultcrantz M, Derolf ÅR, Landgren O, and Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol. (2011) 29:2897. doi: 10.1200/JCO.2011.34.8540
29. Medzhitov RJN. Recognition of microorganisms and activation of the immune response. Nat. (2007) 449:819–26. doi: 10.1038/nature06246
30. Chuenchor W, Jin T, Ravilious G, and Xiao TS. Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr Opin Immunol. (2014) 26:14–20. doi: 10.1016/j.coi.2013.10.009
31. Medzhitov R, Preston-Hurlburt P, and Janeway CAJN. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nat. (1997) 388:394–7. doi: 10.1038/41131
32. Baldwin AS Jr. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. (1996) 14:649–81. doi: 10.1146/annurev.immunol.14.1.649
33. Stein SJ and Baldwin ASJB. The Journal of the American Society of Hematology, Deletion of the NF-κB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. (2013) 121:5015–24. doi: 10.1182/blood-2013-02-486142
34. Hermouet S, Bigot-Corbel E, and Gardie B. Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediators Inflamm. (2015) 2015:145293. doi: 10.1155/2015/145293
35. Karthik L, Kumar G, Keswani T, Bhattacharyya A, Chandar SS, and Bhaskara Rao K. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS One. (2014) 9:e90972. doi: 10.1371/journal.pone.0090972
36. Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leuk. (2014) 28:1851–60. doi: 10.1038/leu.2014.68
37. Broz P and Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
38. Caruso R, Warner N, Inohara N, and Núñez GJI. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immun. (2014) 41:898–908. doi: 10.1016/j.immuni.2014.12.010
39. Frame JM, Long T, Schuster-Kubaczka C, Esain V, Lim SE, Daley GQ, et al. Inflammasome-mediated regulation of hematopoiesis in the vertebrate embryo. Nat. (2018) 132:330. doi: 10.1182/blood-2018-99-117076
40. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. (2003) 4:69–77. doi: 10.1038/ni875
41. Pestka S, Krause CD, and Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
42. Musella M, Manic G, De Maria R, Vitale I, and Sistigu AJO. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology. (2017) 6:e1314424. doi: 10.1080/2162402X.2017.1314424
43. Platanias LCJNRI. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
44. Fenton SE, Saleiro D, and Platanias LCJC. Type I and II interferons in the anti-tumor immune response. Cancers (Basel). (2021) 13:1037. doi: 10.3390/cancers13051037
45. Duchosal M and Trumpp AJN. IFNalpha activates dormant haematopoietic stem cells in vivo. Nat. (2009) 458:904908. doi: 10.1038/nature07815
46. Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. (2014) 211:245–62. doi: 10.1084/jem.20131043
47. Ricklin D, Hajishengallis G, Yang K, and Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
48. Heeger PS and Kemper CJI. Novel roles of complement in T effector cell regulation. Immunobiol. (2012) 217:216–24. doi: 10.1016/j.imbio.2011.06.004
49. Kolev M, Friec GL, and Kemper CJNRI. Complement—tapping into new sites and effector systems. Nat Rev Immunol. (2014) 14:811–20. doi: 10.1038/nri3761
50. Kolev M and Kemper C. Keeping it all going—Complement meets metabolism. Frontiers in Immunology. (2017) 8:1. doi: 10.3389/fimmu.2017.00001
51. West EE, Kolev M, and Kemper C. Complement and the regulation of T cell responses. Annual Rev Immunol. (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
52. Konopko A, Łukomska A, Ratajczak J, Kucia M, and Ratajczak MZJSCR. Reports, complosome regulates hematopoiesis at the mitochondria level. Stem Cell Rev Rep. (2025) 21:1–12. doi: 10.1007/s12015-025-10856-1
53. Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The Journal of the American Society of Hematology, The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. (2016) 128:2960–75. doi: 10.1182/blood-2016-07-730556
54. Croker BA, Silke J, and Gerlic M. Fight or flight-regulation of emergency hematopoiesis by pyroptosis and necroptosis. Curr Opin Hematol. (2015) 22:293. doi: 10.1097/MOH.0000000000000148
55. Shi J, Gao W, and Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biol Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
56. Ratajczak MZ, Adamiak M, Kucia M, Tse W, Ratajczak J, and Wiktor-Jedrzejczak W. The emerging link between the complement cascade and purinergic signaling in stress hematopoiesis. Front Immunol. (2018) 9:1295. doi: 10.3389/fimmu.2018.01295
57. Wang C, Yang Y, Gao S, Chen J, Yu J, Zhang H, et al. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit Rev Oncol Hematol. (2018) 122:123–32. doi: 10.1016/j.critrevonc.2017.12.013
58. Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC, et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. (2017) 18:3204–18. doi: 10.1016/j.celrep.2017.03.018
59. Beauchemin V, Villeneuve L, Rodriguez-Cimadevilla J, Rajotte D, Kenney J, Clark S, et al. Interleukin-6 production by the blast cells of acute myeloblastic leukemia: Regulation by endogenous interleukin-1 and biological implications. J Cell Physiol. (1991) 148:353–61. doi: 10.1002/jcp.1041480305
60. Cozzolino F, Torcia M, Bettoni S, Aldinucci D, Burgio VL, Petti MC, et al. Interleukin-1 and interleukin-2 control granulocyte-and granulocyte-macrophage colony-stimulating factor gene expression and cell proliferation in cultured acute myeloblastic leukemia. Int J Cancer. (1990) 46:902–7. doi: 10.1002/ijc.2910460525
61. Oster W, Cicco NA, Klein H, Hirano T, Kishimoto T, Lindemann A, et al. Participation of the cytokines interleukin 6, tumor necrosis factor-alpha, and interleukin 1-beta secreted by acute myelogenous leukemia blasts in autocrine and paracrine leukemia growth control. J Clinic Investig. (1989) 84:451–7. doi: 10.1172/JCI114186
62. Sanchez-Correa B, Bergua JM, Campos C, Gayoso I, Arcos MJ, Bañas H, et al. Cytokine profiles in acute myeloid leukemia patients at diagnosis: survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine. (2013) 61:885–91. doi: 10.1016/j.cyto.2012.12.023
63. Westermann F, Kube D, Haier B, Bohlen H, Engert A, Zuehlsdorf M, et al. Interleukin 10 inhibits cytokine production of human AML cells. Annals Oncol. (1996) 7:397–404. doi: 10.1093/oxfordjournals.annonc.a010607
64. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
65. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Sci. (2017) 355:842–7. doi: 10.1126/science.aag1381
66. Cull AH, Snetsinger B, Buckstein R, Wells RA, and Rauh J. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. (2017) 55:56–70. e13. doi: 10.1016/j.exphem.2017.08.001
67. Yeaton A, Cayanan G, Loghavi S, Dolgalev I, Leddin EM, Loo CE, et al. The impact of inflammation-induced tumor plasticity during myeloid transformation. Cancer Discov. (2022) 12:2392–413. doi: 10.1158/2159-8290.CD-21-1146
68. Hsu K, Champaiboon C, Guenther BD, Sorenson BS, Khammanivong A, Ross KF, et al. Chemistry, Anti-infective protective properties of S100 calgranulins. Antiinflamm Antiallergy Agents Med Chem. (2009) 8:290–305. doi: 10.2174/187152309789838975
69. Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell (2016) 19:613–27. doi: 10.1016/j.stem.2016.08.021
70. Laouedj M, Tardif MR, Gil L, Raquil M-A, Lachhab A, Pelletier M, et al. The Journal of the American Society of Hematology, S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood. (2017) 129:1980–90. doi: 10.1182/blood-2016-09-738005
71. Cheng P, Eksioglu EA, Chen X, Kandell W, Le Trinh T, Cen L, et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leuk. (2019) 33:2034–46. doi: 10.1038/s41375-019-0397-9
72. Kim M, Hwang S, Park K, Kim SY, Lee YK, and Lee D. Increased expression of interferon signaling genes in the bone marrow microenvironment of myelodysplastic syndromes. PLoS One. (2015) 10:e0120602. doi: 10.1371/journal.pone.0120602
73. Kitagawa M, Saito I, Kuwata T, Yoshida S, Yamaguchi S, Takahashi M, et al. Overexpression of tumor necrosis factor (TNF)-α and interferon (IFN)-γ by bone marrow cells from patients with myelodysplastic syndromes. Leuk. (1997) 11:2049–54. doi: 10.1038/sj.leu.2400844
74. Jin N, Lera RF, Yan RE, Guo F, Oxendine K, Horner VL, et al. Chromosomes, and Cancer, Chromosomal instability upregulates interferon in acute myeloid leukemia. Genes, Chromosomes and Cancer. (2020) 59:627–38. doi: 10.1002/gcc.22880
75. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell. (2021) 28:1428–42. doi: 10.1016/j.stem.2021.03.002
76. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng Q-R, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leuk. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
77. Takanashi M, Motoji T, Oshimi K, and Mizoguchi H. Effect of recombinant interferons on colony formation of blast progenitors in acute myeloblastic leukemia. Exp Hematol. (1987) 15:946–51.
78. Geissler K, Tricot G, Leemhuis T, Walker E, and Broxmeyer HE. Differentiation-inducing effect of recombinant human tumor necrosis factor α and γ-interferon in vitro on blast cells from patients with acute myeloid leukemia and myeloid blast crisis of chronic myeloid leukemia. Cancer Res. (1989) 49:3057–62.
79. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. (2011) 186:5367–75. doi: 10.4049/jimmunol.1003438
80. Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. (2016) 17:2584–95. doi: 10.1016/j.celrep.2016.11.031
81. Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta M, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leuk. (2010) 24:756–64. doi: 10.1038/leu.2010.31
82. Ferrell PB and Kordasti SJCCR. Hostile takeover: tregs expand in IFNγ-rich AML microenvironment. Clinical Cancer Res. (2022) 28:2986–8. doi: 10.1158/1078-0432.CCR-22-1030
83. Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, et al. Toll-like receptor alterations in myelodysplastic syndrome. Leuk. (2013) 27:1832–40. doi: 10.1038/leu.2013.180
84. Maratheftis CI, Andreakos E, Moutsopoulos HM, and Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clinical Cancer Res. (2007) 13:1154–60. doi: 10.1158/1078-0432.CCR-06-2108
85. Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, and Pagliuca AJB. The Journal of the American Society of Hematology, The role of apoptosis, proliferation, and the Bcl-2–related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood. (2000) 96:3932–8. doi: 10.1182/blood.V96.12.3932.h8003932_3932_3938
86. Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, et al. Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp Mol Pathol. (2010) 88:293–8. doi: 10.1016/j.yexmp.2010.01.009
87. Yuan S, Liu Z, Xu Z, Liu J, and . Zhang J. High mobility group box 1 (HMGB1): a pivotal regulator of hematopoietic Malignancies. J Hematol Oncol. (2020) 13:1–19. doi: 10.1186/s13045-020-00920-3
88. Liu N, Wu Y, Wen X, Li P, Lu F, and Shang H. Chronic stress promotes acute myeloid leukemia progression through HMGB1/NLRP3/IL-1β signaling pathway. J Mol Med (Berl). (2021) 99:403–14. doi: 10.1007/s00109-020-02011-9
89. Kerbauy DM, Lesnikov V, Abbasi N, Seal S, Scott B, and Deeg HJJB. NF-κB and FLIP in arsenic trioxide (ATO)-induced apoptosis in myelodysplastic syndromes (MDSs). Blood. (2005) 106:3917–25. doi: 10.1182/blood-2005-04-1424
90. de Matos AG, Ribeiro Junior HL, de Paula Borges D, Okubo BM, de Sousa JC, Barbosa MC, et al. Interleukin-8 and nuclear factor kappa B are increased and positively correlated in myelodysplastic syndrome. Med Oncol. (2017) 34:1–7. doi: 10.1007/s12032-017-1023-1
91. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. The Journal of the American Society of Hematology, Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. (2001) 98:2301–7. doi: 10.1182/blood.V98.8.2301
92. Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, et al. NF-κB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. (2007) 26:4071–83. doi: 10.1038/sj.onc.1210187
93. Schrenk KG, Schnetzke U, Stegemann K, von Lilienfeld-Toal M, Hochhaus A, and Scholl S. Efficacy of antifungal prophylaxis with oral suspension posaconazole during induction chemotherapy of acute myeloid leukemia. J Cancer Res Clin Oncol. (2015) 141:1661–8. doi: 10.1007/s00432-015-1962-x
94. Van Rhenen A, Van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The Journal of the American Society of Hematology, The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. (2007) 110:2659–66. doi: 10.1182/blood-2007-03-083048
95. Toft-Petersen M, Nederby L, Kjeldsen E, Kerndrup GB, Brown GD, Hokland P, et al. Unravelling the relevance of CLEC12A as a cancer stem cell marker in myelodysplastic syndrome. Br J Haematol. (2016) 175:393–401. doi: 10.1111/bjh.14270
96. Ratajczak MZ, Konopko A, Jarczak J, Kazek M, Ratajczak J, and Kucia M. Complosome as a new intracellular regulatory network in both normal and Malignant hematopoiesis. Leuk. (2025) 39:1571–7. doi: 10.1038/s41375-025-02613-7
97. Morris R, Kershaw NJ, and Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci: Publ Protein Soc. (2018) 27:1984–2009. doi: 10.1002/pro.3519
98. Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. (2010) 17:584–96. doi: 10.1016/j.ccr.2010.05.015
99. Akada H, Yan D, Zou H, Fiering S, Hutchison RE, and Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. (2010) 115:3589–97. doi: 10.1182/blood-2009-04-215848
100. Chen E, Beer PA, Godfrey AL, Ortmann CA, Li J, Costa-Pereira AP, et al. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell. (2010) 18:524–35. doi: 10.1016/j.ccr.2010.10.013
101. Trowbridge JJ and Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med. (2021) 218. doi: 10.1084/jem.20201544
102. King KY, Huang Y, Nakada D, and Goodell MA. Environmental influences on clonal hematopoiesis. Exp Hematol. (2020) 83:66–73. doi: 10.1016/j.exphem.2019.12.005
103. Kay J, Thadhani E, Samson L, Engelward B, and damage I-iDNA. mutations and cancer. DNA Repair. (2019) 83:102673. doi: 10.1016/j.dnarep.2019.102673
104. Loberg MA, Bell RK, Goodwin LO, Eudy E, Miles LA, SanMiguel JM, et al. Sequentially inducible mouse models reveal that Npm1 mutation causes Malignant transformation of Dnmt3a-mutant clonal hematopoiesis. Leuk. (2019) 33:1635–49. doi: 10.1038/s41375-018-0368-6
105. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–849 e5. doi: 10.1016/j.stem.2018.10.013
106. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. (2021) 28:1428–1442 e6. doi: 10.1016/j.stem.2021.03.002
107. Liao M, Chen R, Yang Y, He H, Xu L, Jiang Y, et al. Aging-elevated inflammation promotes DNMT3A R878H-driven clonal hematopoiesis. Acta Pharm Sinica B. (2022) 12:678–91. doi: 10.1016/j.apsb.2021.09.015
108. Avagyan S, Henninger JE, Mannherz WP, Mistry M, Yoon J, Yang S, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science. (2021) 374:768–72. doi: 10.1126/science.aba9304
109. McLoughlin MA, Cheloor Kovilakam S, Dunn WG, Gu M, Tobin J, Pershad Y, et al. Telomere attrition becomes an instrument for clonal selection in aging hematopoiesis and leukemogenesis. Nat Genet. (2025) 57(9):2215–25. doi: 10.1038/s41588-025-02296-x
110. Mistry JJ, Young KA, Colom Diaz PA, Maestre IF, Levine RL, and Trowbridge JJ. Mesenchymal stromal cell senescence induced by Dnmt3a -mutant hematopoietic cells is a targetable mechanism driving clonal hematopoiesis and initiation of hematologic Malignancy. bioRxiv: preprint server Biol. (2024). doi: 10.1101/2024.03.28.587254
111. SanMiguel JM, Eudy E, Loberg MA, Young KA, Mistry JJ, Mujica KD, et al. Distinct tumor necrosis factor alpha receptors dictate stem cell fitness versus lineage output in Dnmt3a-mutant clonal hematopoiesis. Cancer Discov. (2022) 12:2763–73. doi: 10.1158/2159-8290.CD-22-0086
112. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nat. (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z
113. Yokomizo-Nakano T, Hamashima A, Kubota S, Bai J, Sorin S, Sun Y, et al. Exposure to microbial products followed by loss of Tet2 promotes myelodysplastic syndrome via remodeling HSCs. J Exp Med. (2023) 220. doi: 10.1084/jem.20220962
114. Galloway-Pena JR and Jobin C. Microbiota influences on hematopoiesis and blood cancers: new horizons? Blood Cancer Discov. (2023) 4:267–75. doi: 10.1158/2643-3230.bcd-22-0172
115. Agarwal P, Sampson A, Hueneman K, Choi K, Jakobsen NA, Uible E, et al. Microbial metabolite drives ageing-related clonal haematopoiesis via ALPK1. Nat. (2025) 642:201–11. doi: 10.1038/s41586-025-08938-8
116. Molica M, Breccia M, Foa R, Jabbour E, and Kadia TM. Maintenance therapy in AML: the past, the present and the future. Am J Hematol. (2019) 94:1254–65. doi: 10.1002/ajh.25620
117. Alsufyani A, Alanazi R, Woolley JF, Dahal L.N.J.M.C.R., and Dog O. New trick: type I IFN–based treatment for acute myeloid leukemiaType-I IFN in AML. Mol Cancer Res. (2021) 19:753–6. doi: 10.1158/1541-7786.MCR-20-0871
118. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nat. (2009) 458:904–8. doi: 10.1038/nature07815
119. Mo XD, Wang Y, Zhang XH, Xu LP, Yan CH, Chen H, et al. Interferon-α Is effective for treatment of minimal residual disease in patients with t (8; 21) acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation: results of a prospective registry study. Oncologists. (2018) 23:1349–57. doi: 10.1634/theoncologist.2017-0692
120. Jiang H, Liu X-H, Kong J, Wang J, Jia J-S, Lu S-Y, et al. Interferon-α as maintenance therapy can significantly reduce relapse in patients with favorable-risk acute myeloid leukemia. Leuk Lymphoma. (2021) 62:2949–56. doi: 10.1080/10428194.2021.1948027
121. Magenau JM, Pawarode A, Riwes MM, Parkin B, Anand S, Ghosh M, et al. Transplantation, A Phase I/II clinical trial of type 1 interferon for reduction of relapse after HCT in high risk AML. Blood Advances. (2019) 25:S12–3. doi: 10.1016/j.bbmt.2018.12.078
122. Small S, Numan Y, and Platanias LCJB. Innate immune mechanisms and immunotherapy of myeloid Malignancies. Biomedicines. (2021) 9:1631. doi: 10.3390/biomedicines9111631
123. Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr., Duttagupta P, et al. STING pathway activation stimulates potent immunity against acute myeloid leukemia. Cell Rep. (2016) 15:2357–66. doi: 10.1016/j.celrep.2016.05.023
124. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8(357):. doi: 10.1126/scitranslmed.aaf2341
125. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. (2018) 8:1083.
126. Albinger N, Hartmann J, and Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Therapy. (2021) 28:513–27. doi: 10.1038/s41434-021-00246-w
127. Li W, Wang F, Guo R, Bian Z, and Song Y. Targeting macrophages in hematological Malignancies: recent advances and future directions. J Hematol Oncol. (2022) 15:110. doi: 10.1186/s13045-022-01328-x
128. Ignatz-Hoover JJ, Wang H, Moreton SA, Chakrabarti A, Agarwal MK, Sun K, et al. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia. (2015) 29:918–26. doi: 10.1038/leu.2014.293
129. Elmaagacli A, Steckel N, Ditschkowski M, Hegerfeldt Y, Ottinger H, Trenschel R, et al. Toll-like receptor 9, NOD2 and IL23R gene polymorphisms influenced outcome in AML patients transplanted from HLA-identical sibling donors. Bone Marrow Transplant. (2011) 46:702–8. doi: 10.1038/bmt.2010.166
130. Ellegast JM, Alexe G, Hamze A, Lin S, Pimkin M, Ross L, et al. Unleashing cell-intrinsic inflammation as a strategy to kill AML blasts. Cancer Discov. (2021) 138:3305. doi: 10.1182/blood-2021-151511
131. Buteyn NJ, Santhanam R, Merchand-Reyes G, Murugesan RA, Dettorre GM, Byrd JC, et al. Activation of the intracellular pattern recognition receptor NOD2 promotes acute myeloid leukemia (AML) cell apoptosis and provides a survival advantage in an animal model of AML. J Immunol. (2020) 204:1988–97. doi: 10.4049/jimmunol.1900885
132. Jain N, Zhao Z, Feucht J, Koche R, Iyer A, Dobrin A, et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nat. (2023) 615:315–22. doi: 10.1038/s41586-022-05692-z
133. Bouzid H, Belk JA, Jan M, Qi Y, Sarnowski C, Wirth S, et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nat Med. (2023) 29:1662–70. doi: 10.1038/s41591-023-02397-2
134. Matatall KA, Wathan TK, Nguyen M, Chen H, McDonald A, Qi G, et al. TET2-mutant myeloid cells mitigate Alzheimer’s disease progression via CNS infiltration and enhanced phagocytosis in mice. Cell stem cell. Cell Stem Cell. (2025) 32:1285–1298 e8. doi: 10.1016/j.stem.2025.06.006
Keywords: hematopoiesis, innate immunity, inflammation, acute myeloid leukemia, pattern recognition receptors, mutation, cytokines, interferon signaling
Citation: Bhowmik S, Bose A and Sengupta A (2025) Innate immune-inflammatory signaling milieu in myeloid leukemia and aging-associated clonal hematopoiesis pathologies. Front. Immunol. 16:1660709. doi: 10.3389/fimmu.2025.1660709
Received: 06 July 2025; Accepted: 30 September 2025;
Published: 16 October 2025.
Edited by:
Daniel Scott-Algara, Institut Pasteur, FranceReviewed by:
Jc Balandrán, New York University, United StatesMariusz Z Ratajczak, University of Louisville Physicians, United States
Copyright © 2025 Bhowmik, Bose and Sengupta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amitava Sengupta, YW1pdGF2YXNlbmd1cHRhLmlpY2JAY3Npci5yZXMuaW4=; YW1pdGF2YS5paWNiQGdtYWlsLmNvbQ==