 Yibo Zhang
Yibo Zhang Xuewen Li
Xuewen Li- Department of Cardiovascular Medicine, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, China
Transient receptor potential (TRP) channels are non-selective cation channels with diverse physiological functions, widely expressed across various cell types. These channels play crucial roles in maintaining homeostasis and contribute to the progression of cardiovascular diseases, particularly atherosclerosis. Atherosclerosis is a chronic vascular inflammatory condition marked by lipid accumulation and fibrous tissue proliferation in the arterial intima. TRP channels regulate intracellular ionic gradients and activate downstream signaling pathways, thereby influencing the function of vascular endothelial and smooth muscle cells. Therefore, they are increasingly implicated in the pathogenesis of cardiovascular and cerebrovascular diseases. Emerging evidence has demonstrated that pharmacological modulation (antagonism or activation) of TRP channels regulates programmed cell death mechanisms, positioning these channels as key modulators of atherosclerotic plaque dynamics. Specifically, TRP channels modulate various cell death modalities, including apoptosis, autophagy, and necrosis, while also influencing inflammatory responses and oxidative stress-related pathways that potentiate cellular death. These interconnected mechanisms significantly contribute to the development of atherosclerotic lesions. This review systematically examined the mechanistic roles of TRP channels in atherosclerosis via regulation of cell death pathways, aiming to provide a comprehensive understanding of their pathophysiological functions and to support the development of targeted molecular therapies.
1 Introduction
Atherosclerosis is a chronic inflammatory disease that primarily affects medium- to large-sized arteries, including the coronary, carotid, and aortic arteries, and is a major cause of cardiovascular morbidity and mortality (1). The development of atherosclerosis begins with damage to the arterial endothelium—the inner lining of blood vessels—which regulates vascular tone, inhibits thrombosis, and controls leukocyte migration (2). Factors such as chronic hypertension can mechanically damage endothelial cells (3). Once the endothelium is compromised, low-density lipoprotein (LDL) cholesterol can infiltrate the intima-media layer of the arterial wall, where it becomes oxidized and forms highly inflammatory oxidized LDL. Oxidized LDL draws immune cells, including macrophages and T-lymphocytes, to this site (4). These cells release inflammatory mediators such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and macrophage colony-stimulating factor (M-CSF), which further activate endothelial cells and promote the migration of more inflammatory cells (5) Macrophages and other immune cells also engulf oxidized LDL to form foam cells—a key component of atherosclerotic plaques—contributing to thickening and hardening of the arterial wall (6). In parallel, vascular smooth muscle cells (VSMCs) in the arterial wall, stimulated by inflammatory mediators, migrate from the media to the intima, where they proliferate and secrete extracellular matrix, further driving plaque formation and growth (7). Over time, the accumulation of lipids, inflammatory cells, and VSMCs leads to the formation of visible plaques within the arterial walls. These plaques gradually enlarge, causing arterial narrowing and restricting blood flow (8). Plaque ruptures—often due to weakening of the fibrous cap by inflammatory cells—exposes the lipid core, triggering platelet aggregation and thrombosis. This further obstructs the artery and may lead to acute cardiovascular events, such as myocardial infarction (9). Atherosclerosis remains the primary pathological basis of cardiovascular disease worldwide, underscoring the need for more effective strategies in prevention and treatment (10).
Emerging evidence have highlighted the pivotal regulatory role of transient receptor potential (TRP) channels in cardiovascular pathophysiology. These ion channels are widely expressed across vascular endothelia and smooth muscle cells, where they regulate key cellular processes. For example, TRPV4 plays a central role in vasodilation, and its dysfunction has been linked to cardiovascular disorders (11). Similarly, TRPC1 is abundantly expressed in cardiomyocytes, where its aberrant activity contributes to pathological cardiac remodeling processes such as hypertrophy and dysregulated angiogenesis (12). TRPM4 channels, widely expressed in mammalian cardiomyocytes, regulate cardiac electrophysiology by modulating ion flux; both gain- and loss-of-function mutations in TRPM4 disrupt cardiac conduction and repolarization, precipitating arrhythmogenic syndromes, including Brugada syndrome and congenital long QT syndrome (13). These functional attributes underscore the integral role of TRP channels in cardiovascular pathogenesis and position them as promising targets in cardiovascular research and therapy.
2 TRP channels
TRP channels are a family of non-selective cation channels embedded in the cell membrane. These polymodal channels exhibit exceptional capacity to detect diverse cellular and environmental stimuli, including thermal, mechanical, and chemical signals, thereby modulating transmembrane flux of monovalent (Na+, K+) and divalent (Ca2+, Mg2+) cations. Their ubiquitous expression across multiple tissue types and cellular systems underscores their fundamental roles in maintaining ionic homeostasis and signal transduction (14).
Structurally, functional TRP channels adopt tetrameric configurations comprising either homomeric or heteromeric subunit assemblies. This quaternary organization creates a central ion-conducting pore, with a specific subunit composition that determines the biophysical properties and regulatory mechanisms of the channel. The tetrameric architecture enables cooperative gating responses to integrated stimuli while maintaining ion selectivity profiles critical for physiological functions (15). Phylogenetic analysis based on structural homology and sequence conservation has classified mammalian TRP channels into seven evolutionarily distinct subfamilies: TRPV (vanilloid), TRPM (melastatin), TRPML (mucolipin), TRPC (canonical), TRPN (NOMPC-like), TRPP (polycystin), and TRPA (ankyrin) (16). This systematic classification reflects their functional diversification, with each subfamily displaying distinct activation mechanisms and tissue-specific roles. The following are the key physiological and pathological functions of each TRP subfamily:
● TRPC channel regulates intracellular calcium ion (Ca2+) homeostasis through a tight association with G protein-coupled receptor (GPCR) signaling cascades. GPCRs orchestrate diverse physiological and pathological processes ranging from muscle cell fusion and immunomodulation to vasoconstriction and tumor suppression (17).
● TRPV subfamily serves as multimodal sensors capable of integrating biochemical ligands, thermal gradients, pH fluctuations, and mechanical stimuli to modulate cellular responses (18).
● TRPM subfamily demonstrates functional pleiotropy, participating in temperature sensation, vascular morphogenesis, cancer biology, neuroinflammatory pathways, and metabolic disorders such as type 2 diabetes (19).
● TRPML subfamily governs lysosomal trafficking and autophagy. Dysregulation of TRPML channels precipitates neurodegenerative pathologies through impaired lysosomal proteostasis (20).
● TRPN channel, absent in mammals but present in organisms like Drosophila, mediate mechanosensory transduction through specialized force-gated ion channels critical for auditory and tactile perception (21).
● TRPP channel, considered the most evolutionarily ancient subfamily, function as cation-permeable channels at the plasma membrane and as Ca2+ release channels within endoplasmic reticulum membranes (22).
● TRPA subfamily operates as polymodal detectors of noxious stimuli, including reactive oxygen species (ROS), electrophilic compounds, cold temperatures, and endogenous damage-associated molecular patterns (DAMPs), thereby bridging extracellular stress signals to intracellular inflammatory cascades (23).
2.1 Relationship between atherosclerosis and TRP channels
Distinct TRP channel subtypes exert different regulatory functions across cellular compartments and pathological stages of atherosclerosis, synergistically modulating disease progression. Clinical evidence have shown that TRPC6 and TRPV1 mRNA are upregulated in atherosclerotic endothelial cells, contributing to pro-inflammatory cascades that drive plaque formation. These findings suggest TRPC6 and TRPV1 as promising therapeutic targets for atherosclerosis management (24). Mechanistically, TRPV1 activation modulates sirtuin-1 expression through epigenetic regulation, thereby attenuating nuclear factor kappa-B (NF-κB) signaling. This pathway suppresses ox-LDL-induced foam cell formation via vascular smooth muscle cell lipid uptake, ultimately mitigating the development of atherosclerotic lesions (25).
Emerging evidence has revealed a critical interplay between TRPV1 and endothelial nitric oxide synthase (eNOS) in the pathogenesis of atherosclerosis (26). TRPV1 activation in endothelial cells elevates intracellular Ca2+ concentration, thereby upregulating calmodulin kinase-II (CaMKII) expression. The resultant Ca2+–CaMKII complex directly phosphorylates eNOS at Ser1177, a prerequisite for enzymatic activation (27). Supporting this mechanism, ex vivo studies using human umbilical vein endothelial cells isolated from deoxycorticosterone-salt hypertensive mice demonstrated that TRPV1 agonism induces nitric oxide (NO) synthesis concurrent with Ser1177 phosphorylation (28). The natural compound sesamin exerts TRPV1-dependent cardioprotective effects via multilevel signaling modulation. Sesamin binding to TRPV1 triggers Ca2+ influx, which sequentially activates both CaMKII and Ca2+/calmodulin-dependent protein kinase kinase-beta. This dual kinase activation cascade potentiates eNOS activity by enhancing Ser1177 phosphorylation. Furthermore, sesamin-mediated TRPV1 stimulation initiates a parallel pathway involving soluble adenylate cyclase. Elevated cytosolic Ca2+ activates sAC, increasing cyclic adenosine monophosphate production and protein kinase A (PKA) activity. PKA-dependent phosphorylation synergistically amplifies eNOS activation, culminating in sustained NO release that mitigates endothelial dysfunction under atherosclerotic conditions (29).
TRPM7 is significantly upregulated in response to oxLDL stimulation during atherosclerotic plaque formation. Functionally, TRPM7 serves as a critical modulator of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signaling axis, orchestrating VSMC proliferation and migration under ox-LDL challenge. Mechanistically, TRPM7-mediated Ca2+/Mg2+ influx activated downstream ERK1/2 phosphorylation through sequential MEK1/2 activation, a process validated by attenuation of these effects upon TRPM7 suppression by siRNA or pharmacological inhibitors (2-APB). This signaling cascade potentiates VSMC phenotypic switching from contractile to synthetic states, a hallmark of neointimal hyperplasia in atherosclerosis (30).
TRPV4 channels modulate ox-LDL uptake by macrophages through calcium influx-dependent mechanisms. Genetic ablation of TRPV4 or pharmacological inhibition using selective antagonists effectively suppressed ox-LDL-induced foam cell formation by attenuating lipid internalization without altering CD36-mediated surface binding. The mechanoregulatory role of TRPV4 is further potentiated by pathophysiological matrix stiffness, which enhances TRPV4 plasma membrane localization and calcium permeability (31). Concurrently, biomechanical stimuli such as elevated matrix stiffness and shear stress induce endothelial dysfunction via TRPV4-mediated signaling cascades. Activation of the TRPV4/miR-6740/endothelin-1 axis disrupts endothelial barrier integrity, exacerbating vascular inflammation and atherosclerotic plaque progression (32).
TRPM2 channel mediate pro-inflammatory macrophage polarization through NLRP3 inflammasome activation. Mechanistically, TRPM2-dependent Ca2+ influx facilitates NLRP3 inflammasome assembly via mitochondrial ROS (mtROS)-mediated thioredoxin-interacting protein dissociation, thereby amplifying IL-1β secretion and foam cell formation. Genetic ablation of TRPM2 in Apoe-/- mice fed with high-fat diet showed decreased lesional CD68+ macrophage infiltration and increased collagen content (33).
These findings suggest that TRP channels are integral to key atherosclerotic mechanisms—endothelial dysfunction, inflammatory cascade response, foam cell formation, and plaque stability. Targeting these channels may offer novel therapeutic strategies for both the prevention and treatment of atherosclerosis.
3 Cell death in atherosclerosis
Cell death plays a critical role in the pathogenesis of atherosclerosis. The final stage of cellular life cycle is marked by the irreversible loss of cellular function and structural integrity, with far-reaching consequences for the pathological progression of atherosclerosis (34). Cell death is classified as Accidental Cell Death or Regulated Cell Death (RCD), depending on whether it is genetically controlled. RCD, synonymous with Programmed Cell Death (PCD), is a highly regulated and orderly process essential for maintaining tissue homeostasis and normal development (35). RCD involves several distinct processes including apoptosis, autophagy, necroptosis, pyroptosis, and ferroptosis. Each modality plays a unique role in the development of atherosclerosis (36).
RCD plays a non-negligible and vital role in atherosclerosis by influencing disease progression and plaque stability through various mechanisms. Iron-dependent regulation of cell death arises from an imbalance in iron, antioxidants, and lipid metabolism. Recent studies have highlighted its close association with atherosclerosis development (37). When ferroptosis occurs, it causes excessive macrophage death through the release of large quantities of inflammatory factors and oxidants. This intensifies inflammatory responses and oxidative stress, and promotes atherosclerotic plaque destabilization (38). Ferroptosis inhibitors like Ferrostatin-1 (Fer-1) effectively reduce iron-related death in macrophages. Ferroptosis inhibitors, such as Fer-1, can effectively reduce iron-mediated macrophage death. This action alleviates inflammatory responses and oxidative stress, thereby contributing to atherosclerotic plaque stabilization (39).
Parthanatos, an ROS-induced PCD, occurs in endothelial cells during atherosclerosis. It causes cell death via ROS - induced DNA damage and PARP -1 activation. Endothelial cells are vital for vascular homeostasis and are key components of the vascular lining. Parthanatos-induced endothelial dysfunction promotes inflammatory cell infiltration and adhesion, driving the progression of atherosclerosis (40, 41). Cellular pyroptosis is a necrotic PCD. VSMC pyroptosis is important in atherosclerosis. It involves NLRP3-associated caspase-1, activating gasdermin D, forming plasma membrane pores, and causing proinflammatory cell lysis (42). During atherosclerosis, VSMC pyroptosis releases many inflammatory factors like IL-1β and IL-18, further activating the inflammatory response and promoting vessel wall inflammation and tissue damage. In addition, excessive VSMC pyroptosis-induced cell death compromises vessel wall structure and function, contributing to plaque instability and rupture (43).
3.1 Relationship between TRP channels and cell death
TRP channels regulate numerous physiological and pathological processes, including various forms of cell death. TRPV1 antagonist capsazepine and calcium chelator BAPTA can alleviate mitochondrial dysfunction and cell apoptosis caused by abnormal increase in Ca2+, thereby improving anaplastic thyroid carcinoma (44). Beyond regulating the influx of intracellular Ca2+, TRP channels modulate cell death through autophagy regulation, signaling pathways, and apoptosis induction. For example, in gastric cancer cells, treatment with capsaicin increases TRPV6 expression, leading to elevated intracellular Ca2+ levels, Bax protein activation, increased mitochondrial permeability, and apoptosis promotion. TRPV6 overexpression enhances p53 activation via the JNK pathway. As an important tumor suppressor, activated p53 promotes cell cycle arrest and apoptosis, thereby inhibiting the growth of gastric cancer cells (45). When TRPV1 channel is activated, capsaicin-mediated Ca2+ influx raises intracellular Ca2+ levels and activates ROS production. Subsequently, ROS activate AMPK, inducing oxidative modification of the key autophagy protein Atg4C and upregulating Beclin-1 (46). Atg4C and Beclin-1 initiate autophagy by promoting autophagosome formation and regulate autophagosome-lysosome fusion, facilitating the degradation of autophagy substrates and recycling of cellular components (47). In atherosclerotic diseases, TRP channels modulate cell death through several mechanisms—namely Ca2+-dependent mitochondrial stress, ROS production, apoptotic signaling, and autophagic flux—contributing to vascular cell dysfunction, inflammation, and plaque instability.
4 TRP channels and apoptosis
Apoptosis is a form of PCD characterized by specific morphological changes and enzyme-dependent biochemical processes. These processes include nuclear condensation, cell membrane blebbing, and DNA fragmentation, culminating in orderly cell death without the release of cellular contents to minimize tissue damage. Often termed “cell suicide,” apoptosis is triggered by internal cell signals rather than external physical or chemical factors. It removes damaged or unnecessary cells and is crucial for multicellular organism development and the maintenance of tissue homeostasis (48).
TRP channels modulate apoptosis across a variety of cell types and pathophysiological conditions. For instance, in renal ischemia/reperfusion injury models, TRPM2 channel activation increases intracellular calcium ions, triggering oxidative stress, mitochondrial dysfunction, and apoptosis (49). Similarly, TRPV1 activation by heat or acidic pH causes calcium overload and neuronal apoptosis via downstream apoptotic signaling (23). In triple-negative breast cancer, TRPM7 inhibition enhances apoptosis induced by TNF-related apoptosis-inducing ligand, suggesting a protective, anti-apoptotic role for TRPM7 in cancer cells (50). TRPV6 inhibition disrupts intracellular calcium ion homeostasis and affects cell survival signaling pathways, leading to increased cardiomyocyte apoptosis. Conversely, the upregulation of TRPV6 can protect cardiomyocytes and enhance their anti-apoptotic capacity, suggesting that TRPV6 plays a protective role in pathological conditions such as myocardial ischemia-reperfusion injury (51).
Current research on TRP channel-regulated apoptosis in atherosclerosis predominantly focuses on the TRPC subfamily. oxLDL stimulation activates the TRPC1 channel, inducing sustained Ca2+ influx that disrupts intracellular Ca2+ homeostasis. This disturbance activates mitochondrial-dependent apoptotic pathways, ultimately leading to VSMC death. Caveolin-1 enhances VSMC sensitivity to oxLDL by maintaining TRPC1 expression and facilitating its translocation to the plasma membrane. Mechanistically, oxLDL accumulates in the caveolar membrane domains through its oxidized lipid components, triggering TRPC1 translocation from intracellular compartments to Caveolin-1-enriched plasma membrane regions via actin cytoskeleton reorganization. Pathologically, VSMC apoptosis promotes atherosclerotic plaque progression by expanding the necrotic core and thinning the fibrous cap, thereby increasing plaque vulnerability to rupture and subsequent thrombotic events (52).
TRPC3 is a critical regulator of macrophage survival signaling in the pathogenesis of atherosclerosis. Genetic ablation of TRPC3 enhances susceptibility to BAX/BAK-mediated intrinsic apoptosis. This pro-survival function is mechanistically linked to TRPC3-mediated constitutive calcium oscillations that sustain PI3K/Akt/NF-κB signaling through calcineurin-dependent pathways (53). The pathophysiological significance of TRPC3 extends to efferocytosis regulation—a crucial process maintaining plaque stability through apoptotic cell clearance. TRPC3-deficient macrophages exhibit compromised efferocytic capacity (54). Consequently, impaired efferocytosis in TRPC3-/- mice leads to elevation of apoptotic cell burden in advanced atherosclerotic plaques, accompanied by increased necrotic core formation and elevated IL-1α/IL-6 secretion. These findings establish TRPC3 as a dual modulator of macrophage survival and clearance, and its coordinated action prevents plaque necrosis and inflammatory exacerbations (55). Similarly, miR-26a suppresses TRPC3 expression. Overexpression of miR-26a can reduce TRPC3 protein levels, thereby inhibiting TRPC3-mediated cell apoptosis and alleviating the progression of atherosclerosis, whereas overexpression of TRPC3 can reverse the anti-apoptotic effects of miR-26a. TRPC3 activation can promote NF-κB activation, leading to increased production of inflammatory mediators and, consequently, aggravating endothelial cell inflammation and apoptosis (56).
Beyond its impact on TRPC3, miR-26a also directly targets the 3’ untranslated region of TRPC6 mRNA to suppress its protein expression. In both high-fat diet-fed ApoE-/- mice and ox-LDL-treated human aortic endothelial cells, ox-LDL significantly downregulated miR-26a through its receptor LOX1, leading to upregulated TRPC6 expression. This subsequently induces an intracellular calcium overload, activates the mitochondrial apoptotic pathway, and ultimately promotes endothelial apoptosis. Such mechanisms compromise vascular barrier integrity and accelerate lipid deposition and plaque formation. Diminished expression of miR-26a in hemodynamically disturbed regions correlates with weakened anti-apoptotic effects, highlighting its crucial role in maintaining endothelial homeostasis. Restoring miR-26a expression or targeted inhibition of TRPC6 may interrupt calcium signaling pathways (57).
In addition to the TRPC subfamily, other TRP channels play a role in regulating apoptosis. Studies have indicated that TRPV1 significantly modulates VSMC apoptosis. TRPV1 activation increases eNOS and NO production (27). NO reduces apoptosis via multiple mechanisms such as inhibiting mitochondrial cytochrome c release and decreasing caspase family activation (5, 58).
In atherosclerotic mouse and cell models, TRPV6 expression is significantly reduced, while pro-apoptotic factors such as Bax and cleaved caspase-3 are elevated. Conversely, TRPV6 overexpression in cell models lowers these pro-apoptotic factors and increases the anti-apoptotic Bcl-2, thus inhibiting apoptosis. Genetic knockdown of PKA partially abrogates TRPV6’s anti-apoptotic capacity. Subsequent pathway analysis confirmed TRPV6-mediated phosphorylation of uncoupling protein 2 (UCP2), establishing the PKA/UCP2 axis as the primary conduit for atheroprotective functions (58).
These findings suggest that targeting TRP channels can intervene in the core pathological process of apoptosis, offering a novel therapeutic strategy for the treatment of atherosclerosis.
5 TRP channels and autophagy
Autophagy is an evolutionarily conserved catabolic process that maintains cellular homeostasis by degrading damaged organelles and misfolded proteins. This process involves the formation of double-membrane autophagosomes, which fuse with lysosomes to degrade and recycle cytoplasmic contents (59). Autophagy is typically activated under metabolic stress, such as nutrient deprivation, and serves as a survival mechanism through controlled “self-digestion” (60). Based on distinct molecular mechanisms, autophagy manifests in three principal forms:
● Macroautophagy mediates bulk degradation through de novo formation of autophagosomes that engulf cytoplasmic regions.
● Microautophagy directly internalizes substrates via lysosomal membrane invagination to sequester specific organelles.
● Chaperone-mediated autophagy selectively translocates soluble proteins containing KFERQ-like motifs through LAMP-2A receptor complexes in lysosomal membranes.
These complementary pathways synergistically regulate proteostasis, organelle turnover, and metabolic adaptation to maintain cellular functional integrity (61).
TRP channels primarily modulate autophagy by regulating intracellular Ca2+ homeostasis. Different TRP subtypes affect autophagy via specific mechanisms. In TRPML1, released Ca2+ activates Ca2+/calmodulin-dependent phosphatase (CaN), which dephosphorylates TFEB, a transcription factor that facilitates its nuclear translocation. TFEB promotes lysosomal and autophagy-related gene expression, initiating autophagy-related gene transcription (62). In some studies, TRPC1 expression and intracellular Ca2+ levels increased when cells were exposed to hypoxia or starvation. Further research has shown that TRPC1-mediated Ca2+ influx is closely linked to autophagy induction. For example, after treating cells with DMOG or DFO, TRPC1 expression, Ca2+ influx, and expression of autophagy markers Beclin-1 and LC3A increased, suggesting that TRPC1-mediated Ca2+ influx plays a role in autophagy regulation (63). In HEK293 cells, LC3-II levels and basal autophagy increased significantly after Tet-induced TRPM7 channel expression, along with a higher pAMPK/AMPK ratio, indicating that TRPM7 channel activates basal autophagy via the AMPK signaling pathway. In SH-SY5Y cells, TRPM7-specific shRNA-mediated endogenous TRPM7 channel knockdown significantly reduced basal autophagy, confirming the importance of TRPM7 channels in maintaining basal autophagy (64). In addition to the classic autophagy pathways, alterations in lipid metabolism are considered key initiators of atherosclerosis. Autophagy degrades intracellular lipids and lipid droplets, regulates cellular lipid levels, and ameliorates atherosclerosis (10). Foam cell formation is a key step in the development of atherosclerosis. Foam cells are formed when macrophages or smooth muscle cells uptake excess lipids. Autophagy clears these lipids, reduces foam cell accumulation, and inhibits atherosclerotic progression (65). Autophagy also modulates reverse cholesterol transport, facilitating cholesterol efflux from arterial VSMCs to peripheral tissues, and reducing lipid deposition (66).
Studies on TRP channels and autophagy have primarily focused on the TRPV subfamily. The TRPV1 channel exerts atheroprotective effects through autophagy-mediated suppression of foam cell differentiation. Pharmacological activation of TRPV1 by capsaicin significantly attenuates ox-LDL-induced foam cell formation in VSMCs via enhanced autophagic flux. Mechanistically, TRPV1 activation initiates a coordinated AMPK/mTOR signaling cascade that drives autophagosome biogenesis and subsequent lysosomal fusion. This autolysosomal maturation enables efficient degradation of lipid droplets through lipophagy, as evidenced by reduced neutral lipid accumulation and intracellular cholesterol ester content. Critical functional validation comes from autophagy-deficient models: siRNA-mediated Atg7 silencing completely abrogates TRPV1’s ability to mitigate ox-LDL-induced lipid overload, while preserving cellular cholesterol efflux capacity. These experimental findings established that the basal autophagy machinery is an essential mediator of TRPV1’s cytoprotective effects against atherosclerotic foam cell transformation (67). The engineered CuS-TRPV1 nanocomposites demonstrate targeted therapeutic effects via photothermal channel activation. These nanoparticles exhibited dual functionality: specific TRPV1 receptor targeting through surface ligand interactions, and efficient near-infrared photothermal conversion. Upon NIR irradiation, localized hyperthermia induces TRPV1 conformational changes, triggering rapid calcium influx. This calcium surge activates a Ca2+/calmodulin-dependent kinase kinase β-mediated AMPK phosphorylation cascade, subsequently upregulating autophagic flux. The enhanced autophagy machinery promotes ATP-binding cassette transporter A1 (ABCA1) lysosomal trafficking and stabilization. This coordinated mechanism reduces intracellular cholesterol ester accumulation and suppresses foam cell differentiation (68).
Along with the TRPV family, the TRPA1 subfamily has attracted attention in atherosclerosis research. Studies have indicated that inhibiting or knocking out TRPA1 exacerbates lipid accumulation induced by ox-LDL and elevates intracellular cholesterol and triglyceride levels. Activating TRPA1 likely curbs intracellular lipid accumulation by enhancing ABC-dependent cholesterol efflux. Pre-treatment with the TRPA1 antagonist HC030031 does not affect ox-LDL binding but reduces cholesterol efflux dependent on apoAI or HDL. HC030031 treatment lowers the protein levels of ABCA1 and ABCG1, but does not significantly affect SR-BI protein levels, suggesting that TRPA1 activation primarily inhibit lipid accumulation by upregulating ABCA1- and ABCG1-mediated cholesterol efflux (69). However, recent studies have not directly confirmed whether TRPA1 suppresses lipid production by regulating autophagy. ABCA1 is a key regulator of cholesterol efflux, and its dysfunction can lead to the accumulation of intracellular cholesterol and other lipids (70). When autophagy is impaired, the p62/mTOR/LXRα signaling pathway inhibits the expression of ABCA1 and ABCG1. In contrast, the activation of autophagy can alleviate this inhibition. Therefore, we speculate that the mechanism by which TRPA1 inhibits lipid accumulation is closely related to autophagy (71).
6 TRP channels and necrosis
Cellular necrosis plays a critical role in the progression of atherosclerosis by promoting inflammatory responses and destabilizing atherosclerotic plaques. Damage-associated molecular patterns (DAMPs), such as high-mobility group protein B1, are released from necrotic cells. These DAMPs bind to receptors and trigger NF-κB dependent pro-inflammatory cytokine transcription, which in turn promotes atherosclerosis initiation and progression (72). The accumulation of necrotic cellular debris contributes to the formation of a necrotic core, a defining histopathological feature of mature atherosclerotic plaques. Expansion of this necrotic core compromises plaque stability by weakening the fibrous cap, increasing the likelihood of plaque rupture. Subsequent exposure to thrombogenic components such as tissue factors initiates coagulation cascades, significantly elevating the risk of acute cardiovascular complications, including myocardial infarction and cerebrovascular events (72).
The progression of atherosclerotic lesions demonstrates a critical association between TRPC3 channel expression dynamics and necrotic processes. Emerging evidence has revealed stage-specific modulation of plaque pathology through TRPC3 regulation in bone marrow-derived cells. During early atherogenesis, genetic deficiency of TRPC3 in bone marrow cells significantly attenuates plaque development, manifesting as reduced lesion area, diminished cellular components, and inflammatory cell infiltration. This regulatory mechanism undergoes functional adaptation in advanced disease stages. Late-stage atherosclerotic plaques with bone marrow-specific TRPC3 deficiency exhibit three hallmark stability features: reduction in necrotic core volume, enhanced collagen deposition, and significant fibrous cap thickening. Mechanistic analysis identified macrophage-specific TRPC3 signaling as the principal mediator of these effects. Targeted deletion of macrophages decreases necrotic core expansion by impairing proinflammatory cytokine production and enhancing efferocytosis. These findings establish TRPC3 as a pathophysiological regulator, with therapeutic implications for stage-specific intervention strategies (73, 74).
7 TRP channels and inflammation
The development of atherosclerosis is accompanied by inflammation, which plays a key role in all disease stages. Inflammation begins with early foam cell accumulation and lipid streak formation, continues through middle-stage fibrous plaque development, and persists until late-stage plaque rupture and thrombosis. Moreover, inflammation can directly destabilize coronary artery plaque structure (75).
TRPV1 activation significantly inhibits inflammatory responses in endothelial cells, evidenced by reduced pro-inflammatory cytokines production (such as TNF-α, IL-6, and MCP-1), decreased expression of adhesion molecules (such as ICAM-1 and VCAM-1), and diminished adhesion of monocytes to endothelial cells (76). Dietary capsaicin is considered an effective agonist of TRPV1 channel (77). In animal models of atherosclerosis, most in vivo studies on TRPV1 have used dietary capsaicin treatment to improve atherosclerotic plaques or reduce associated inflammation. Capsaicin significantly inhibit lipopolysaccharide (LPS)-induced upregulation of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) via enhancing nuclear factor IA and suppressing NF-κB expression (78). TRPV1 activation enhances Ser1177 phosphorylation of eNOS through the Ca2+/PI3K/Akt signaling pathway, thereby promoting NO production and subsequently inhibiting inflammatory responses in endothelial cells. In LPS-induced inflammatory models, TRPV1 agonist capsaicin significantly reduced the expression of inflammatory factors (TNF-α, IL-6, MCP-1) and adhesion molecules (ICAM-1, VCAM-1), while suppressing NF-κB activity to attenuate monocyte-endothelial cell adhesion. These effects were all dependent on activation of the TRPV1-Ca2+-eNOS/NO axis. This anti-inflammatory action was also validated in renal microvascular endothelial cells of salt-sensitive hypertensive mice, suggesting that TRPV1 may alleviate inflammatory damage under pathological conditions by modulating endothelial function (79).
TRPM2-/- mice show reduced production of ROS and blunted inflammatory responses. When fed a high cholesterol diet, TRPM2-/- mice show no difference in serum cholesterol levels compared with TRPM2+/+ mice, but they have significantly smaller aortic plaque areas, attenuating the progression of atherosclerosis. In the aortic root plaques of TRPM2-/- mice, decreased expression of CD68, α-SMA and PCNA suggests that TRPM2 may promote macrophage infiltration and smooth muscle cell migration to lesion sites. In addition, reduced expression of ICAM-1, MCP-1 and TNFα in TRPM2-/- mice suggests that TRPM2 may promote monocyte adhesion and vascular inflammation (80, 81).
8 TRP channels and oxidative stress
Oxeiptosis, first described in 2018, is a novel, caspase-independent form of cellular death (82). This apoptosis-like mechanism, propelled by ROS induction, is orchestrated through the coordinated interplay of ROS sensor KEAP1, phosphatase PGAM5, and pro-apoptotic factor AIFM1 (83). Oxeiptosis exhibits a pronounced association with ROS, a key pathological mediator of atherosclerosis (84). The aberrant accumulation of ROS within vascular endothelial cells serves as the central driver of coronary artery dysfunction and acts as a potent catalyst for plaque formation through redox signaling cascades. While emerging evidence implicates oxeiptosis in atherogenesis, its precise mechanistic underpinnings remain unclear. Therefore, this review specifically focused on delineating the regulatory role of TRP channels within the ROS metabolic network in atherosclerosis with the aim of paving the way for innovative therapeutic strategies (85).
The TRP channel superfamily, functioning as pivotal redox sentinels, dynamically modulate the redox equilibrium between ROS generation and scavenging through orchestrating intracellular Ca2+ homeostasis and signaling axes such as MAPK cascades and NF-κB pathways, thereby critically influencing cellular fate decisions (86). High-glucose-treated rat aortic smooth muscle cells exhibit markedly increased intracellular ROS generation concomitant with upregulated TRPM7 protein expression and potentiated whole-cell currents. TRPM7 knockdown and pharmacological ROS suppression via the free radical scavenger N-acetylcysteine effectively attenuate high glucose-induced phosphorylation of MEK1/2 and ERK1/2. These findings suggest that ROS-mediated modulation of TRPM7 expression serves as a crucial upstream event governing MEK/ERK signaling pathway activation (87).
In vitro studies show that capsaicin intake activates TRPV1, thereby upregulating UCP2 expression, reducing mitochondrial membrane potential, decreasing oxidative stress in mitochondrial respiratory chain complex I, and consequently inhibiting excessive ROS generation. Furthermore, TRPV1 activation enhanced UCP2 expression through phosphorylation of PKA. As a mitochondrial antioxidant defense factor, UCP2 restricts ROS production via mild uncoupling effect, demonstrating that TRPV1 activation improves coronary artery function by suppressing mitochondrial ROS generation via the upregulation of UCP2 and enhanced PKA activity (88).
TRPM2 is a nonselective cation channel that is primarily activated by ROS and adenosine diphosphate ribose (ADPR). Activation of TRPM2 channels increases calcium influx and exacerbates oxidative stress through enhanced ROS generation (89). In atherosclerosis, ROS produced at inflammatory sites can activate TRPM2 channels. Additionally, PARP-1 [poly(ADP-ribose) polymerase-1] becomes activated under oxidative stress, catalyzing NAD+ cleavage into ADPR, which subsequently binds to the NUDT9-H domain of TRPM2 to further open the channel. Excessive ROS perpetuates TRPM2 activation, establishing a positive feedback loop that intensifies intracellular oxidative stress (90).
9 Current landscape of TRP channel-targeted drug development
Current research on TRP channel-targeted pharmaceuticals focuses on modulating ion channel activity to intervene in disease pathogenesis. TRP channels hold significant pathological relevance in cardiovascular diseases. Among them, TRPV1 has emerged as a pivotal target for the development of anti-atherosclerotic drugs, owing to its central role in regulating vascular inflammation, oxidative stress, and programmed cell death.
Capsaicin serves, a dietary natural agonist of the TRPV1 channel (77), suppresses the expression of inflammatory cytokines and adhesion molecules in endothelial cells through TRPV1 activation. It also reduces monocyte-endothelial adhesion and enhances eNOS phosphorylation via the Ca2+/PI3K/Akt pathway to stimulate NO production, thereby improving endothelial function and vascular homeostasis. Furthermore, TRPV1 activation induces the AMPK/mTOR signaling axis, enhances autophagic flux, and promotes lipid droplet degradation, inhibiting ox-LDL-induced foam cell formation in VSMCs (67, 79) Capsaicin can also potentially activate TRPV6 channels (45), with recent studies demonstrating that TRPV6 overexpression suppresses VSMC apoptosis via the PKA/UCP2 signaling axis (58). Although no direct empirical evidence currently links capsaicin-induced TRPV6 activation to attenuated atherosclerosis progression, this mechanism represents a promising novel direction for investigating the anti-atherosclerotic properties of capsaicin. Beyond its well-characterized activation of TRP channels, capsaicin demonstrates additional anti-atherosclerotic properties. For example, it modulates serum lipid profiles and reduces the levels of pro-inflammatory cytokines and LPS by remodeling gut microbial composition and function, thereby ameliorating HFD-induced atherosclerosis (91). Furthermore, capsaicin alleviates HFD-induced hyperlipidemia and atherosclerotic progression in animal models by mitigating hypercholesterolemia. It achieves this effect by suppressing endoplasmic reticulum stress, reducing oxidative damage, and improving endothelial dysfunction (92, 93). Collectively, these multifaceted mechanisms highlight the significant therapeutic potential of capsaicin as an anti-atherosclerotic agent.
Beyond the well-established role of capsaicin-sensitive pathways, recent studies in 2025 have unveiled a novel immunotherapeutic strategy: a peptide vaccine (designated P1) targeting the E3 domain of TRPM2. This vaccine elicits active immunization in mice, prompting the endogenous production of anti-TRPM2 blocking antibodies that potently inhibit TRPM2 channel activity. This intervention attenuates TRPM2-mediated Ca2+ influx, mitigates vascular inflammation and macrophage infiltration, diminishes the accumulation of CD68+ macrophages and PCNA+ proliferating cells, and enhances atherosclerotic plaque stability. In an ApoE(-/-) murine model of atherosclerosis, administration of the P1 vaccine resulted in a significant reduction in the aortic plaque area and the necrotic core size within the aortic root (94). This immunotherapeutic approach delineates a promising pathway for the treatment of atherosclerosis through targeting TRP channels, underscoring the need for further investigation into the clinical translational potential of other TRP channel members.
10 Conclusion and future direction
TRP channels, widely expressed in cardiovascular cells—including cardiomyocytes, vascular endothelial cells, and VSMC—have emerged as key regulators of cell death in atherosclerosis. (Figure 1) They contribute to the progression of atherosclerosis by modulating multiple forms of cell death, including apoptosis, necrosis, autophagy, oxidative stress-induced death and inflammation-induced death. (Table 1) Understanding the specific contribution of TRP channel subtypes on disease progression is crucial. Targeting upstream or downstream signaling processes of TRP channels, rather than global activation or inhibition, may offer alternative therapeutic approaches.
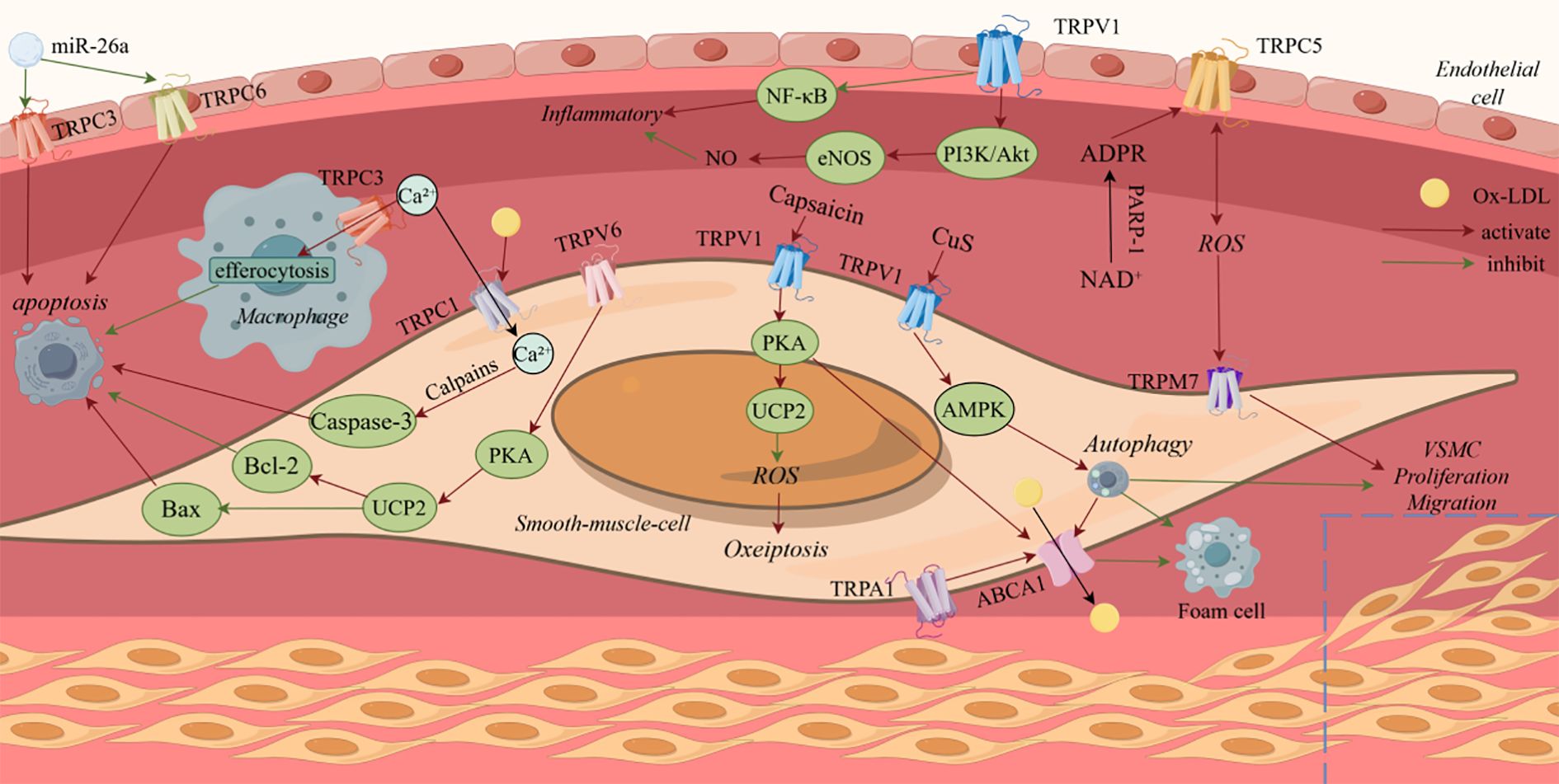
Figure 1. Summary of the signal transduction pathways transient receptor potential channels regulate cell death in atherosclerosis.
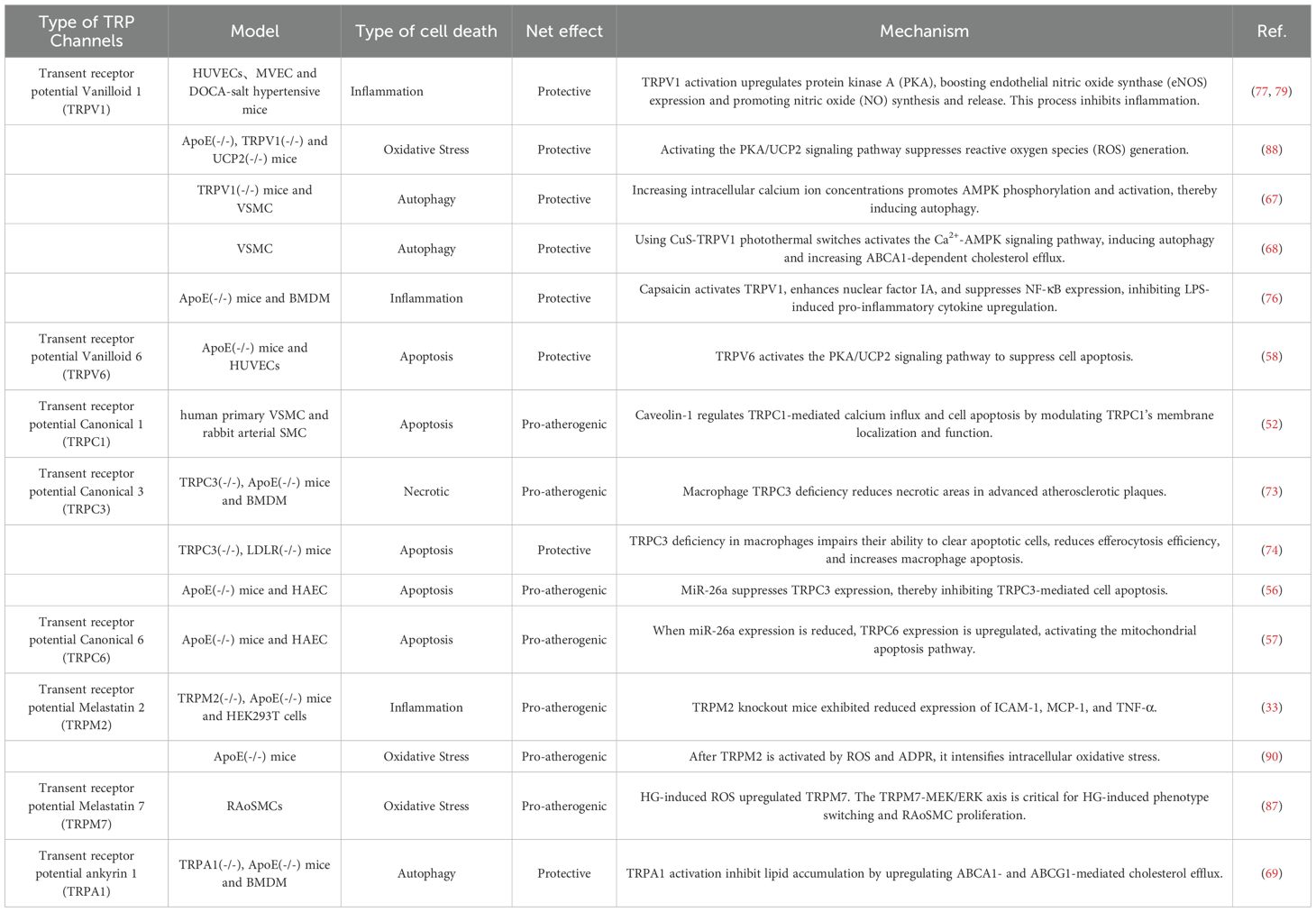
Table 1. Summary of the way transient receptor potential channels regulate cell death in atherosclerosis.
Current research on the role of TRP channels in atherosclerosis-related cell death has mainly focused on apoptosis. However, given the complexity and multi-pathway nature of cell death involving different forms, future studies may explore other cell death mechanisms. For example, ferroptosis, an emerging form of cell death, is gaining attention for its potential role in atherosclerosis (95).
TRP channels are also involved in ferroptosis regulation. For example, TRPV4 promotes calcium influx and activates the Ca2+/CaM/CaMKII signaling pathway, which facilitate vesicle formation and transport, leading to iron efflux and upregulation of LAMP2 and related vesicle transport proteins—ultimately triggering ferroptosis (96). In ozone-induced lung injury, increased TRPA1 expression activates the PI3K/Akt pathway, which reduces the expression of mitochondrial fusion protein OPA1, leading to mitochondrial dysfunction and ferroptosis (97). However, these findings are based on non-atherosclerotic systems, and their direct relevance to vascular cells remains unverified. Moreover, no study has confirmed TRPV4/TRPA1-mediated ferroptosis in human atherosclerotic plaques or animal models of atherosclerosis. This research gap highlights the need for targeted validation, such as cell-specific knockout models, to assess whether these mechanisms contribute to plaque instability or lipid accumulation in vivo.
Pyroptosis, a form of inflammatory PCD, plays an important role in cell death. TRPC6 modulates zinc influx and upregulates A20 expression, thereby inhibiting pyroptosis in renal tubular epithelial cells and ameliorating renal ischemia/reperfusion injury (98). However, this anti-pyroptotic role contrasts with the pro-apoptotic function of TRPC6 in atherosclerosis. No direct evidence links TRPC6 to pyroptosis in vascular cells, and its net effect on plaque inflammation remains speculative. While TRP channels hold promise in regulating novel death modalities such as ferroptosis and pyroptosis, their atherosclerosis-specific roles require rigorous validation to transcend speculative models.
Future studies should further investigate the roles of TRP channels in novel cell death modes. Such research would expand our understanding of the known functions of TRP channels in different cell types, such as endothelial and smooth muscle cells, and their specific regulatory mechanisms in various forms of cell death, such as apoptosis and autophagy. Such investigations may provide novel mechanistic insights and support the development of TRP channel–targeted therapies for more effective prevention and treatment of atherosclerosis.
Author contributions
YZ: Conceptualization, Writing – original draft, Writing – review & editing. WW: Visualization, Writing – original draft, Writing – review & editing. XML: Writing – original draft, Writing – review & editing. LZ: Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing. XWL: Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (No. 82170398) and the Fundamental Research Program of Shanxi Province (No. 202203021211078).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592:524–33. doi: 10.1038/s41586-021-03392-8, PMID: 33883728
2. Trimm E and Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2022) 20:197–210. doi: 10.1038/s41569-022-00770-1, PMID: 36198871
3. Gimbrone MA and García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/circresaha.115.306301, PMID: 26892962
4. Mineo C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc Res. (2020) 116:1254–74. doi: 10.1093/cvr/cvz338, PMID: 31834409
5. Cheng C, Zhang J, Li X, Xue F, Cao L, Meng L, et al. NPRC deletion mitigated atherosclerosis by inhibiting oxidative stress, inflammation and apoptosis in ApoE knockout mice. Signal Transduction Targeted Ther. (2023) 8:290. doi: 10.1038/s41392-023-01560-y, PMID: 37553374
6. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, and Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med. (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8, PMID: 28785870
7. Allahverdian S, Chaabane C, Boukais K, Francis GA, and Bochaton-Piallat M-L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. (2018) 114:540–50. doi: 10.1093/cvr/cvy022, PMID: 29385543
8. Stroope C, Nettersheim FS, Coon B, Finney AC, Schwartz MA, Ley K, et al. Dysregulated cellular metabolism in atherosclerosis: mediators and therapeutic opportunities. Nat Metab. (2024) 6:617–38. doi: 10.1038/s42255-024-01015-w, PMID: 38532071
9. Fuentes QE, Fuentes QF, Andrés V, Pello OM, de Mora JF, and Palomo GI. Role of platelets as mediators that link inflammation and thrombosis in atherosclerosis. Platelets. (2012) 24:255–62. doi: 10.3109/09537104.2012.690113, PMID: 22671308
10. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z, PMID: 31420554
11. Zhang DX and Gutterman DD. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. (2011) 57:133–9. doi: 10.1097/FJC.0b013e3181fd35d1, PMID: 20881603
12. Camacho Londoño JE, Tian Q, Hammer K, Schröder L, Camacho Londoño J, Reil JC, et al. A background Ca2+entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J. (2015) 36:2257–66. doi: 10.1093/eurheartj/ehv250, PMID: 26069213
13. Amarouch M-Y, El Hilaly J, and Yu H. Inherited cardiac arrhythmia syndromes: focus on molecular mechanisms underlying TRPM4 channelopathies. Cardiovasc Ther. (2020) 2020:1–10. doi: 10.1155/2020/6615038, PMID: 33381229
14. Koivisto A-P, Belvisi MG, Gaudet R, and Szallasi A. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat Rev Drug Discov. (2021) 21:41–59. doi: 10.1038/s41573-021-00268-4, PMID: 34526696
15. Zhao Y, McVeigh BM, and Moiseenkova-Bell VY. Structural pharmacology of TRP channels. J Mol Biol. (2021) 433:166914. doi: 10.1016/j.jmb.2021.166914, PMID: 33676926
16. Diver MM, Lin King JV, Julius D, and Cheng Y. Sensory TRP channels in three dimensions. Annu Rev Biochem. (2022) 91:629–49. doi: 10.1146/annurev-biochem-032620-105738, PMID: 35287474
17. Wang H, Cheng X, Tian J, Xiao Y, Tian T, Xu F, et al. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol Ther. (2020) 209:107497. doi: 10.1016/j.pharmthera.2020.107497, PMID: 32004513
18. Randhawa PK and Jaggi AS. TRPV1 channels in cardiovascular system: A double edged sword? Int J Cardiol. (2017) 228:103–13. doi: 10.1016/j.ijcard.2016.11.205, PMID: 27863349
19. Wu F, Bu S, and Wang H. Role of TRP channels in metabolism-related diseases. Int J Mol Sci. (2024) 25:692. doi: 10.3390/ijms25020692, PMID: 38255767
20. Rosato AS, Tang R, and Grimm C. Two-pore and TRPML cation channels: Regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol Ther. (2021) 220:107713. doi: 10.1016/j.pharmthera.2020.107713, PMID: 33141027
21. Gonzalez C, Lee J, Moon S, Cha Y, and Chung YD. Drosophila TRPN( = NOMPC) channel localizes to the distal end of mechanosensory cilia. PloS One. (2010) 5:e11012. doi: 10.1371/journal.pone.0011012, PMID: 20543979
22. Du J, Fu J, X-m X, and Shen B. The functions of TRPP2 in the vascular system. Acta Pharmacologica Sinica. (2016) 37:13–8. doi: 10.1038/aps.2015.126, PMID: 26725733
23. Zhang M, Ma Y, Ye X, Zhang N, Pan L, and Wang B. TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases. Signal Transduction Targeted Ther. (2023) 8:261. doi: 10.1038/s41392-023-01464-x, PMID: 37402746
24. Thilo F, Vorderwülbecke BJ, Marki A, Krueger K, Liu Y, Baumunk D, et al. Pulsatile atheroprone shear stress affects the expression of transient receptor potential channels in human endothelial cells. Hypertension. (2012) 59:1232–40. doi: 10.1161/hypertensionaha.111.183608, PMID: 22566504
25. Zhang M-J, Zhou Y, Chen L, Wang X, Pi Y, Long C-Y, et al. Impaired SIRT1 promotes the migration of vascular smooth muscle cell-derived foam cells. Histochem Cell Biol. (2016) 146:33–43. doi: 10.1007/s00418-016-1408-9, PMID: 26883442
26. Ching LC, Kou YR, Shyue SK, Su KH, Wei J, Cheng LC, et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc Res. (2011) 91:492–501. doi: 10.1093/cvr/cvr104, PMID: 21493704
27. Hollis M and Wang DH. Transient receptor potential vanilloid in blood pressure regulation. Curr Opin Nephrol Hypertension. (2013) 22:170–6. doi: 10.1097/MNH.0b013e32835c8d4c, PMID: 23274405
28. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, and Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. (1999) 399:601–5. doi: 10.1038/21224, PMID: 10376603
29. Pham TH, Jin SW, Lee GH, Park JS, Kim JY, Thai TN, et al. Sesamin induces endothelial nitric oxide synthase activation via transient receptor potential vanilloid type 1. J Agric Food Chem. (2020) 68:3474–84. doi: 10.1021/acs.jafc.9b07909, PMID: 32077699
30. Lin J, Zhou S, Zhao T, Ju T, Zhang L, and Imhof B. TRPM7 channel regulates ox-LDL-induced proliferation and migration of vascular smooth muscle cells via MEK-ERK pathways. FEBS Letters. (2016) 590:520–32. doi: 10.1002/1873-3468.12088, PMID: 26900082
31. Goswami R, Merth M, Sharma S, Alharbi MO, Aranda-Espinoza H, Zhu X, et al. TRPV4 calcium-permeable channel is a novel regulator of oxidized LDL-induced macrophage foam cell formation. Free Radical Biol Med. (2017) 110:142–50. doi: 10.1016/j.freeradbiomed.2017.06.004, PMID: 28602913
32. Song X, Sun Z, Chen G, Shang P, You G, Zhao J, et al. Matrix stiffening induces endothelial dysfunction via the TRPV4/microRNA-6740/endothelin-1 mechanotransduction pathway. Acta Biomaterialia. (2019) 100:52–60. doi: 10.1016/j.actbio.2019.10.013, PMID: 31606530
33. Zong P, Feng J, Yue Z, Yu AS, Vacher J, Jellison ER, et al. TRPM2 deficiency in mice protects against atherosclerosis by inhibiting TRPM2–CD36 inflammatory axis in macrophages. Nat Cardiovasc Res. (2022) 1:344–60. doi: 10.1038/s44161-022-00027-7, PMID: 35445217
34. Li M, Wang Z-W, Fang L-J, Cheng S-Q, Wang X, and Liu N-F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Disease. (2022) 13:467. doi: 10.1038/s41419-022-04923-5, PMID: 35585052
35. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduction Targeted Ther. (2022) 7:286. doi: 10.1038/s41392-022-01110-y, PMID: 35963853
36. Tang D, Kang R, Berghe TV, Vandenabeele P, and Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5, PMID: 30948788
37. Wang Y, Zhao Y, Ye T, Yang L, Shen Y, and Li H. Ferroptosis signaling and regulators in atherosclerosis. Front Cell Dev Biol. (2021) 9:809457. doi: 10.3389/fcell.2021.809457, PMID: 34977044
38. Liang D, Minikes AM, and Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. (2022) 82:2215–27. doi: 10.1016/j.molcel.2022.03.022
39. Ma J, Zhang H, Chen Y, Liu X, Tian J, and Shen W. The role of macrophage iron overload and ferroptosis in atherosclerosis. Biomolecules. (2022) 12:1702. doi: 10.3390/biom12111702, PMID: 36421722
40. Huang P, Chen G, Jin W, Mao K, Wan H, and He Y. Molecular mechanisms of parthanatos and its role in diverse diseases. Int J Mol Sci. (2022) 23:7292. doi: 10.3390/ijms23137292, PMID: 35806303
41. Zheng D, Liu J, Piao H, Zhu Z, Wei R, and Liu K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. (2022) 13:1039241. doi: 10.3389/fimmu.2022.1039241, PMID: 36389728
42. Zhaolin Z, Guohua L, Shiyuan W, and Zuo W. Role of pyroptosis in cardiovascular disease. Cell Proliferation. (2018) 52:e12563. doi: 10.1111/cpr.12563, PMID: 30525268
43. Lin L, Zhang M-X, Zhang L, Zhang D, Li C, and Li Y-l. Autophagy, Pyroptosis, and ferroptosis: new regulatory mechanisms for atherosclerosis. Front Cell Dev Biol. (2022) 9:809955. doi: 10.3389/fcell.2021.809955, PMID: 35096837
44. Xu S, Cheng X, Wu L, Zheng J, Wang X, Wu J, et al. Capsaicin induces mitochondrial dysfunction and apoptosis in anaplastic thyroid carcinoma cells via TRPV1-mediated mitochondrial calcium overload. Cell Signal. (2020) 75:109733. doi: 10.1016/j.cellsig.2020.109733, PMID: 32771398
45. Chow J, Norng M, Zhang J, and Chai J. TRPV6 mediates capsaicin-induced apoptosis in gastric cancer cells—Mechanisms behind a possible new “hot” cancer treatment. Biochim Biophys Acta (BBA) - Mol Cell Res. (2007) 1773:565–76. doi: 10.1016/j.bbamcr.2007.01.001, PMID: 17292493
46. Farfariello V, Amantini C, and Santoni G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J Leukocyte Biol. (2012) 92:421–31. doi: 10.1189/jlb.0312123, PMID: 22753949
47. Kongara S and Karantza V. The interplay between autophagy and ROS in tumorigenesis. Front Oncol. (2012) 2:171. doi: 10.3389/fonc.2012.00171, PMID: 23181220
48. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. (2019) 43:582–92. doi: 10.1002/cbin.11137, PMID: 30958602
49. Khanahmad H, Mirbod SM, Karimi F, Kharazinejad E, Owjfard M, Najaflu M, et al. Pathological mechanisms induced by TRPM2 ion channels activation in renal ischemia-reperfusion injury. Mol Biol Rep. (2022) 49:11071–9. doi: 10.1007/s11033-022-07836-w, PMID: 36104583
50. Shi R, Fu Y, Zhao D, Boczek T, Wang W, and Guo F. Cell death modulation by transient receptor potential melastatin channels TRPM2 and TRPM7 and their underlying molecular mechanisms. Biochem Pharmacol. (2021) 190:114664. doi: 10.1016/j.bcp.2021.114664, PMID: 34175300
51. Li Z, Meng Z, Lu J, Chen FM, Wong W-T, Tse G, et al. TRPV6 protects ER stress-induced apoptosis via ATF6α-TRPV6-JNK pathway in human embryonic stem cell-derived cardiomyocytes. J Mol Cell Cardiol. (2018) 120:1–11. doi: 10.1016/j.yjmcc.2018.05.008, PMID: 29758225
52. Ingueneau C, Huynh-Do U, Marcheix B, Athias A, Gambert P, Nègre-Salvayre A, et al. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J Cell Mol Med. (2008) 13:1620–31. doi: 10.1111/j.1582-4934.2008.00593.x, PMID: 20187291
53. Tano JY, Smedlund K, Lee R, Abramowitz J, Birnbaumer L, and Vazquez G. Impairment of survival signaling and efferocytosis in TRPC3-deficient macrophages. Biochem Biophys Res Commun. (2011) 410:643–7. doi: 10.1016/j.bbrc.2011.06.045, PMID: 21684255
54. Adkar SS and Leeper NJ. Efferocytosis in atherosclerosis. Nat Rev Cardiol. (2024) 21:762–79. doi: 10.1038/s41569-024-01037-7, PMID: 38750215
55. Tano J-YK, Lee RH, and Vazquez G. Macrophage function in atherosclerosis. Channels. (2014) 6:141–8. doi: 10.4161/chan.20292, PMID: 22909953
56. Feng M, Xu D, and Wang L. miR-26a inhibits atherosclerosis progression by targeting TRPC3. Cell Biosci. (2018) 8:4. doi: 10.1186/s13578-018-0203-9, PMID: 29387339
57. Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y, et al. MicroRNA-26a prevents endothelial cell apoptosis by directly targeting TRPC6 in the setting of atherosclerosis. Sci Rep. (2015) 5:9401. doi: 10.1038/srep09401, PMID: 25801675
58. Zheng L, Zhang H, Li X, and Pandey V. Overexpression of TRPV6 inhibits coronary atherosclerosis–related inflammatory response and cell apoptosis via the PKA/UCP2 pathway. Cardiovasc Ther. (2024) 2024:7053116. doi: 10.1155/2024/7053116, PMID: 39742020
59. Glick D, Barth S, and Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. (2010) 221:3–12. doi: 10.1002/path.2697, PMID: 20225336
60. Rabinovich-Nikitin I, Kirshenbaum E, and Kirshenbaum LA. Autophagy, clock genes, and cardiovascular disease. Can J Cardiol. (2023) 39:1772–80. doi: 10.1016/j.cjca.2023.08.022, PMID: 37652255
61. Yamamoto H and Matsui T. Molecular mechanisms of macroautophagy, microautophagy, and chaperone-mediated autophagy. J Nippon Med Sch. (2024) 91:2–9. doi: 10.1272/jnms.JNMS.2024_91-102, PMID: 37271546
62. Di Paola S, Scotto-Rosato A, and Medina DL. TRPML1: the ca((2+))retaker of the lysosome. Cell Calcium. (2018) 69:112–21. doi: 10.1016/j.ceca.2017.06.006, PMID: 28689729
63. Sukumaran P, Sun Y, Vyas M, and Singh BB. TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Disease. (2015) 6:e1674–e. doi: 10.1038/cddis.2015.7, PMID: 25741599
64. Oh HG, Chun YS, Park C-S, Kim T-W, Park MK, and Chung S. Regulation of basal autophagy by transient receptor potential melastatin 7 (TRPM7) channel. Biochem Biophys Res Commun. (2015) 463:7–12. doi: 10.1016/j.bbrc.2015.05.007, PMID: 25983327
65. Shao B-z, Han B-z, Zeng Y-x, Su D-f, and Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacologica Sinica. (2016) 37:150–6. doi: 10.1038/aps.2015.87, PMID: 26750103
66. Miao J, Zang X, Cui X, and Zhang J. Autophagy, hyperlipidemia, and atherosclerosis. Adv Exp Med Biol. (2020) 1207:237–64. doi: 10.1007/978-981-15-4272-5_18, PMID: 32671753
67. Li BH, Yin YW, Liu Y, Pi Y, Guo L, Cao XJ, et al. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Disease. (2014) 5:e1182–e. doi: 10.1038/cddis.2014.146, PMID: 24743737
68. Gao W, Sun Y, Cai M, Zhao Y, Cao W, Liu Z, et al. Copper sulfide nanoparticles as a photothermal switch for TRPV1 signaling to attenuate atherosclerosis. Nat Commun. (2018) 9:231. doi: 10.1038/s41467-017-02657-z, PMID: 29335450
69. Zhao J-F, Shyue S-K, Kou YR, Lu T-M, and Lee T-S. Transient receptor potential ankyrin 1 channel involved in atherosclerosis and macrophage-foam cell formation. Int J Biol Sci. (2016) 12:812–23. doi: 10.7150/ijbs.15229, PMID: 27313495
70. Matsuo M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J Pharmacol Sci. (2022) 148:197–203. doi: 10.1016/j.jphs.2021.11.005, PMID: 35063134
71. Liang X, Wang C, Sun Y, Song W, Lin J, Li J, et al. p62/mTOR/LXRα pathway inhibits cholesterol efflux mediated by ABCA1 and ABCG1 during autophagy blockage. Biochem Biophys Res Commun. (2019) 514:1093–100. doi: 10.1016/j.bbrc.2019.04.134, PMID: 31101336
72. Puylaert P, Zurek M, Rayner KJ, De Meyer GRY, and Martinet W. Regulated necrosis in atherosclerosis. Arteriosclerosis Thrombosis Vasc Biol. (2022) 42:1283–306. doi: 10.1161/atvbaha.122.318177, PMID: 36134566
73. Tano J-Y, Solanki S, Lee RH, Smedlund K, Birnbaumer L, and Vazquez G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovasc Res. (2014) 101:138–44. doi: 10.1093/cvr/cvt231, PMID: 24101197
74. Solanki S, Dube PR, Birnbaumer L, and Vazquez G. Reduced necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice with macrophage-specific loss of trpc3. Sci Rep. (2017) 7:42526. doi: 10.1038/srep42526, PMID: 28186192
75. Grebe A, Hoss F, and Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. (2018) 122:1722–40. doi: 10.1161/circresaha.118.311362, PMID: 29880500
76. Ávila DL, Fernandes-Braga W, Silva JL, Santos EA, Campos G, Leocádio PCL, et al. Capsaicin improves systemic inflammation, atherosclerosis, and macrophage-derived foam cells by stimulating PPAR gamma and TRPV1 receptors. Nutrients. (2024) 16:3167. doi: 10.3390/nu16183167, PMID: 39339767
77. Munjuluri S, Wilkerson DA, Sooch G, Chen X, White FA, and Obukhov AG. Capsaicin and TRPV1 channels in the cardiovascular system: the role of inflammation. Cells. (2021) 11:18. doi: 10.3390/cells11010018, PMID: 35011580
78. Zhao JJ, Hu YW, Huang C, Ma X, Kang CM, Zhang Y, et al. Dihydrocapsaicin suppresses proinflammatory cytokines expression by enhancing nuclear factor IA in a NF-κB-dependent manner. Arch Biochem Biophys. (2016) 604:27–35. doi: 10.1016/j.abb.2016.06.002, PMID: 27267730
79. Wang Y, Cui L, Xu H, Liu S, Zhu F, Yan F, et al. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis. (2017) 260:13–9. doi: 10.1016/j.atherosclerosis.2017.03.016, PMID: 28324760
80. Zhang Y, Ying F, Tian X, Lei Z, Li X, Lo C-Y, et al. TRPM2 promotes atherosclerotic progression in a mouse model of atherosclerosis. Cells. (2022) 11:1423. doi: 10.3390/cells11091423, PMID: 35563730
81. Zong P, Feng J, Yue Z, Yu AS, Vacher J, Jellison ER, et al. TRPM2 deficiency in mice protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. Nat Cardiovasc Res. (2022) 1:344–60. doi: 10.1038/s44161-022-00027-7, PMID: 35445217
82. Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. (2018) 19:130–40. doi: 10.1038/s41590-017-0013-y, PMID: 29255269
83. Chen KQ, Wang SZ, Lei HB, and Liu X. Mini-review: research and progress of oxeiptosis in diseases. Front Cell Dev Biol. (2024) 12:1428250. doi: 10.3389/fcell.2024.1428250, PMID: 38966429
84. Scaturro P and Pichlmair A. Oxeiptosis-a cell death pathway to mitigate damage caused by radicals. Cell Death Differ. (2018) 25:1191–3. doi: 10.1038/s41418-018-0134-3, PMID: 29844568
85. Batty M, Bennett MR, and Yu E. The role of oxidative stress in atherosclerosis. Cells. (2022) 11:3843. doi: 10.3390/cells11233843, PMID: 36497101
86. Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membrane Biol. (2006) 209:31–41. doi: 10.1007/s00232-005-0839-3, PMID: 16685599
87. Yang M, Fang J, Liu Q, Wang Y, and Zhang Z. Role of ROS-TRPM7-ERK1/2 axis in high concentration glucose-mediated proliferation and phenotype switching of rat aortic vascular smooth muscle cells. Biochem Biophys Res Commun. (2017) 494:526–33. doi: 10.1016/j.bbrc.2017.10.122, PMID: 29079194
88. Xiong S, Wang P, Ma L, Gao P, Gong L, Li L, et al. Ameliorating endothelial mitochondrial dysfunction restores coronary function via transient receptor potential vanilloid 1-mediated protein kinase A/uncoupling protein 2 pathway. Hypertension. (2016) 67:451–60. doi: 10.1161/hypertensionaha.115.06223, PMID: 26667415
89. Chen Z, Cheng Z, Ding C, Cao T, Chen L, Wang H, et al. ROS-activated TRPM2 channel: calcium homeostasis in cardiovascular/renal system and speculation in cardiorenal syndrome. Cardiovasc Drugs Ther. (2023) 39:615–31. doi: 10.1007/s10557-023-07531-3, PMID: 38108918
90. Li Y-S, Ren H-C, Li H, Xing M, and Cao J-H. From oxidative stress to metabolic dysfunction: The role of TRPM2. Int J Biol Macromolecules. (2025) 284:138081. doi: 10.1016/j.ijbiomac.2024.138081, PMID: 39603285
91. Dai Z, Li S, Meng Y, Zhao Q, Zhang Y, Suonan Z, et al. Capsaicin ameliorates high-fat diet-induced atherosclerosis in apoE(-/-) mice via remodeling gut microbiota. Nutrients. (2022) 14:4334. doi: 10.3390/nu14204334, PMID: 36297020
92. Kim HJ. Capsaicin supplementation prevents western diet-induced hyperleptinemia by reducing endoplasmic reticulum stress in apolipoprotein E-deficient mice. Food Nutr Res. (2023) 67:10.29219/fnr.v67.9610. doi: 10.29219/fnr.v67.9610, PMID: 38084147
93. Yang S, Liu L, Meng L, and Hu X. Capsaicin is beneficial to hyperlipidemia, oxidative stress, endothelial dysfunction, and atherosclerosis in Guinea pigs fed on a high-fat diet. Chem Biol Interact. (2019) 297:1–7. doi: 10.1016/j.cbi.2018.10.006, PMID: 30342015
94. Ying F, Zhang Y, Li X, Meng Z, Li J, Lo CY, et al. Active immunization using TRPM2 peptide vaccine attenuates atherosclerotic progression in a mouse model of atherosclerosis. Vaccines (Basel). (2025) 13:241. doi: 10.3390/vaccines13030241, PMID: 40266108
95. Xu X, Xu XD, Ma MQ, Liang Y, Cai YB, Zhu ZX, et al. The mechanisms of ferroptosis and its role in atherosclerosis. BioMed Pharmacother. (2024) 171:116112. doi: 10.1016/j.biopha.2023.116112, PMID: 38171246
96. Li M, Zheng J, Wu T, He Y, Guo J, Xu J, et al. Activation of TRPV4 induces exocytosis and ferroptosis in human melanoma cells. Int J Mol Sci. (2022) 23:4146. doi: 10.3390/ijms23084146, PMID: 35456964
97. Weng J, Liu Q, Li C, Feng Y, Chang Q, Xie M, et al. TRPA1-PI3K/Akt-OPA1-ferroptosis axis in ozone-induced bronchial epithelial cell and lung injury. The Science of the total environment. (2024) 918:170668. doi: 10.1016/j.scitotenv.2024.170668, PMID: 38320701
Keywords: atherosclerosis, cell death, TRP channels, autophagy, apoptosis
Citation: Zhang Y, Wang W, Li X, Li X and Zheng L (2025) Transient receptor potential channels as key regulators of cell death in atherosclerosis. Front. Immunol. 16:1661805. doi: 10.3389/fimmu.2025.1661805
Received: 08 July 2025; Accepted: 08 September 2025;
Published: 23 September 2025.
Edited by:
Xiaofeng Yang, Temple University, United StatesReviewed by:
Marcos Edgar Herkenhoff, Santa Catarina State University, BrazilLiang Hong, University of Illinois Chicago, United States
Copyright © 2025 Zhang, Wang, Li, Li and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuewen Li, eHVld2VubGkxMDEwQDEyNi5jb20=; Lei Zheng, OTQ0ODA3OTNAcXEuY29t