 Wanli Xu
Wanli Xu Zhilin Guo3
Zhilin Guo3 Tingyun Xu
Tingyun Xu Leyi Chen
Leyi Chen Wenan Xu
Wenan Xu- 1Shenzhen Clinical College of Stomatology, School of Stomatology, Southern Medical University, Shenzhen, Guangdong, China
- 2Shenzhen Stomatology Hospital (Pingshan) of Southern Medical University, Shenzhen, Guangdong, China
- 3School of Stomatology, Southern Medical University, Guangzhou, Guangdong, China
Chronic inflammatory diseases are widespread and often accompanied by comorbidities, making treatment challenging. Current immunosuppressive and anti-inflammatory therapies have limited efficacy and significant side effects, and are insufficient to address the complexity of coexisting conditions. This review explores recent advances in innate immune memory, also known as trained immunity, and its potential role in inflammatory diseases. We hypothesize that targeting the regulatory mechanisms of trained immunity may lead to novel therapeutic strategies that more effectively control inflammation and improve disease outcomes. Finally, we highlight that the interplay between trained immunity and inflammatory diseases remains incompletely understood, and further research is needed to elucidate its mechanisms and clinical translational potential.
1 Introduction
With the shifting landscape of modern disease burden, chronic inflammation-related disorders have emerged as a major health concern. Inflammatory diseases are central to a wide spectrum of conditions, including infections, autoimmune disorders, and metabolic syndromes, and are frequently accompanied by chronic comorbidities, forming complex pathological networks. Current therapeutic strategies, such as immunosuppressants, nonsteroidal anti-inflammatory drugs, and biologics, offer partial symptom relief but are often limited by suboptimal efficacy, considerable side effects, and risks of immune suppression-related complications. Particularly under the coexistence of Multiple chronic inflammatory diseases, treating a single disease entity often fails to achieve durable outcomes, highlighting the urgent need for novel therapeutic perspectives.
Recent advances in immunology have challenged the conventional view that immunological memory is exclusive to the adaptive immune system. It is now recognized that the innate immune system also possesses a form of memory-like response, termed trained immunity (1). This phenomenon was first identified in plants and invertebrates, which lack adaptive immunity, and is characterized by enhanced protection upon secondary challenge following a primary encounter. Trained immunity has since been observed in vertebrates as well, suggesting that it represents an evolutionarily conserved defense strategy. Mechanistically, trained immunity is mediated through epigenetic reprogramming, enabling innate immune cells to mount faster and more robust responses upon restimulation.
However, trained immunity presents a double-edged sword. While it confers beneficial effects in host defense against pathogens, its excessive activation may exacerbate pathological inflammation, thereby contributing to the progression of inflammatory diseases. Emerging evidence indicates that features of aberrant trained immunity are present in patients with inflammatory disorders and chronic comorbidities, providing a theoretical framework for reinterpreting disease mechanisms from an innate immune memory perspective. This review aims to provide a comprehensive overview of the concept and evolutionary origins of trained immunity, examine its roles in inflammatory diseases, and propose novel therapeutic approaches targeting trained immunity. By precisely modulating the activation state of trained immunity, it may be possible to suppress pathological inflammation while preserving essential immune defenses, offering promising new avenues for the treatment of inflammation-related diseases and chronic complications.
2 Another memory of immune system: trained immunity
The human immune system is categorized into innate and adaptive immunity. During inflammation, pathogen invasion is initially countered by physical barriers such as the skin and mucosal surfaces. Once pathogens breach these barriers, innate immune cells and molecules are rapidly activated, triggering the innate immune response. This leads to the immediate release of cytokines and chemokines, which recruit and activate immune cells such as neutrophils and macrophages to eliminate pathogens (2, 3). Subsequently, dendritic cells and macrophages present pathogen-derived antigens to T cells, initiating adaptive immune responses. Activated B cells produce specific antibodies, and immunological memory is established to enhance protection against future infections with the same pathogen (4). Historically, research has predominantly focused on adaptive immune memory. However, emerging evidence indicates that, beyond T and B lymphocytes, innate immune cells and even non-immune cells exhibit a memory-like response. Upon encountering microbial components or their products, these cells mount a faster and more robust reaction upon secondary exposure. This phenomenon suggests the existence of memory within the innate immune system. In 2011, Mihai Netea defined this biological process as “trained immunity” (5), while in 2017, Elaine Fuchs referred to it as “inflammatory memory” (6). Unlike the lifelong persistence of adaptive immune memory, trained immunity is transient, typically lasting from at least 3 months to up to 1 year (7). Notably, heterologous protection induced by live vaccines may persist for as long as 5 years (8). Recent studies also reveal that this immunological phenotype can be transmitted across generations (9). In contrast to the recombination-driven mechanisms of adaptive memory, trained immunity is mediated through reversible epigenetic modifications, including chromatin remodeling, long non-coding RNA (lncRNA) transcription, DNA methylation, and metabolic reprogramming. These alterations partly persist after the cessation of stimulation, thereby enabling a form of functional memory. Furthermore, while adaptive memory enhances responses to specific antigens, trained immunity provides broad-spectrum protection against diverse pathogens in a non-specific manner (10). In summary, both innate and adaptive immunological memory collaboratively maintain host defense and homeostasis.
3 The origin of trained immunity
This phenomenon of non-specific immune memory was first observed in plants and invertebrates, both of which, as evolutionarily ancient organisms, lack adaptive immune systems and rely solely on innate immune mechanisms to combat infections (11), does this imply that they possess immune memory? Upon exposure to attenuated microbes, plants can develop long-lasting, broad-spectrum resistance to various pathogens, including viruses, bacteria, fungi, and oomycetes (12). Systemic acquired resistance (SAR) is induced following pathogenic infection and spreads from the infection site to the entire plant. This process is mediated by signaling molecules (such as salicylic acid and jasmonic acid), as well as regulatory factors (such as NPR1 and SNI1), thereby establishing a systemic defense response (12). SAR represents a “memory-like” defensive strategy in plants, offering long-term protection across the whole organism, analogous to immunological memory in animals (13). These findings suggest that plants can also acquire memory capacity through innate immune pathways. Similarly, Drosophila exhibits protection against reinfection with the same pathogen after prior exposure to Streptococcus pneumoniae or Beauveria bassiana (14). Mosquitoes infected with plasmodium falciparum show partial protection upon re-infection (15). Moreover, water fleas (Daphnia) can transfer pathogen-specific immunity against pasteuria ramosa to their offspring via maternal transmission (16). Other invertebrates such as sponges, corals, small crustaceans, and shrimp also display features of immune memory, evidenced by stronger responses upon secondary challenge with the same donor (17, 18). These examples demonstrate that, despite lacking adaptive immune cells like T and B lymphocytes, invertebrates can still mount memory-like responses through their innate immune systems, offering protection upon re-exposure to identical or different pathogens. Such findings challenge the traditional view that innate immune responses are entirely non-adaptive and devoid of memory. The first vertebrate evidence of innate immune memory dates back to 1986, when researchers injected an avirulent, non-germinating strain of Candida albicans (PCA-2) into Rag1−/− mice lacking mature T and B cells, these mice exhibited protection against subsequent lethal systemic C. albicans infection and also showed cross-protection against Staphylococcus aureus, this protection was independent of T-cell-mediated mechanisms and instead relied on innate immune components, particularly macrophages and cytokine production (19). Further, the non-specific protective effects of Bacillus Calmette–Guérin (BCG) vaccination have significantly advanced the study of trained immunity. BCG, a live attenuated vaccine derived from Mycobacterium bovis, is widely used to prevent tuberculosis (20). In murine models, BCG vaccination has been shown to confer protection against secondary infection with Candida albicans (21). Beyond experimental models, epidemiological studies have reported that BCG vaccination in children not only protects against tuberculosis but also reduces overall morbidity and mortality from unrelated infections (22). In regions with high tuberculosis prevalence, widespread BCG vaccination not only helps curb tuberculosis transmission but may also offer collateral protection against other infectious diseases.
With the in-depth exploration of the non-specific protective effects of the BCG vaccine, it has become increasingly evident that the innate immune system is not devoid of memory capabilities, as traditionally believed. Instead, it possesses a certain degree of immune memory. This discovery has sparked widespread interest in trained immunity research in vertebrates. Researchers have begun investigating the forms, mechanisms, and potential applications of trained immunity in vertebrates from various perspectives and using multiple approaches (Figure 1). For example, the skin of mice previously exposed to inflammatory stimuli heals significantly faster upon subsequent injury compared to that of naïve mice (6). Offspring of pregnant mice that experienced infection exhibit stronger resistance to intestinal infections but are also more susceptible to intestinal inflammation (9). Lipopolysaccharide (LPS) induced trained immunity in lung-resident cells has been shown to confer robust protection against Streptococcus pneumoniae infection in mice (23). Subsequent studies have further explored the extensive cellular-level functions of trained immunity. Monocytes, macrophages, dendritic cells (DCs), and natural killer (NK) cells, all key components of the innate immune system, have recently been found to not only serve basic immune defense roles but also to exhibit memory-like characteristics. For instance, trained monocytes and macrophages produce higher levels of reactive oxygen species (ROS) and pro-inflammatory cytokines upon re-stimulation, enhancing their ability to initiate and sustain inflammatory responses and eliminate pathogens more effectively (24). NK cells expressing Ly49H expand rapidly following murine cytomegalovirus (MCMV) infection and demonstrate enhanced production of interferon-γ (IFN-γ) and stronger degranulation upon re-encounter with MCMV (25). Trained DCs have also been shown to process and present antigens more efficiently to T lymphocytes, thereby facilitating adaptive immune activation (26). Thus, innate and adaptive immunity act synergistically to protect the host. Interestingly, trained immunity is not limited to immune cells. Recent research shows that non-immune cells can also exhibit trained immunity. Inflammation induced by skin injury prompts epidermal stem cells (EpSCs) to retain long-term memory that accelerates wound healing upon subsequent injury (6). Hair follicle stem cells are capable of differentiating into epidermal cells while still maintaining their ability to produce hair after epidermal regeneration (27). The trained immunity-like behavior of EpSCs is particularly significant, as tissue stem cells are central to maintaining homeostasis and regeneration through sensing environmental cues and adjusting their behavior. Moreover, S. pneumoniae infection induces memory in respiratory epithelial cells, which enhances bacterial adhesion and facilitates infection upon reinfection (28). Additional studies have reported the presence of trained immunity in muscle stem cells, intestinal epithelial cells, and nasal mucosal epithelial stem cells (9, 29, 30). These findings collectively support the notion that the human immune system harbors a more primitive, innate form of immune memory.
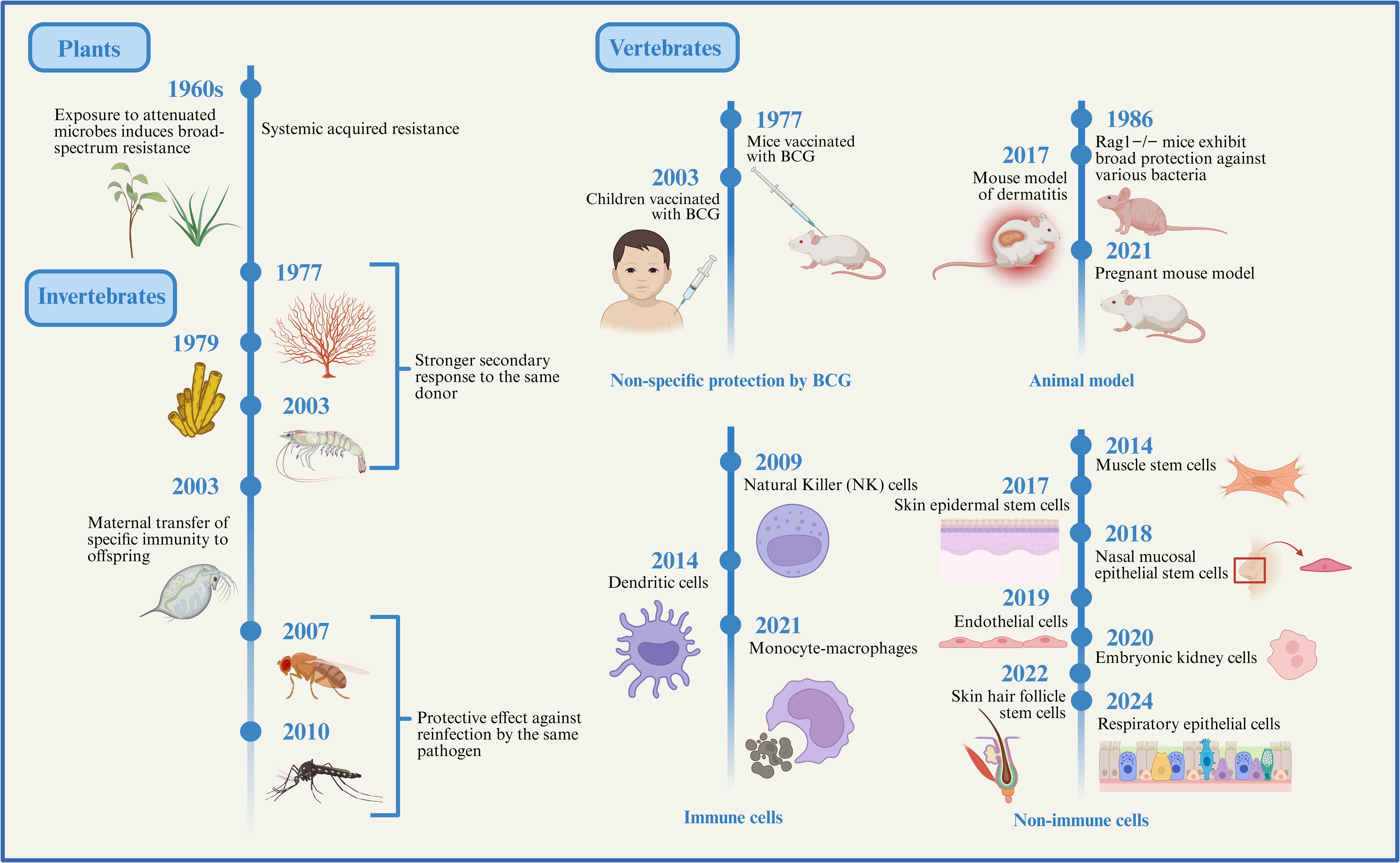
Figure 1. The evolution of trained immunity. Here, we outline the evolutionary background of innate immune memory. Although plants and invertebrates lack adaptive immune systems, their innate immune systems can still generate memory effects against prior infections, providing a certain degree of protection upon subsequent encounters with the same or different pathogens. Subsequently, studies on vertebrate animal models, immune cells, and non-immune cells have gradually revealed the existence and mechanisms of trained immunity, indicating that this form of immune memory has progressively developed and matured throughout evolution. Created with BioRender.com.
4 The dual role of trained immunity
Trained immunity may represent an evolutionarily primitive form of immune memory. It is a remarkable capacity of the body to recall prior adverse encounters and respond more swiftly upon re-exposure, thereby enhancing host defense and survival. For instance, pancreatic epithelial cells that recover from acute inflammation can rapidly reinitiate acinar-to-ductal metaplasia (ADM) during subsequent inflammatory events, effectively limiting tissue damage by promptly reducing zymogen production (31). Similarly, maternal exposure to inflammatory insults during pregnancy leads to elevated IL-6 levels in the systemic circulation, which induces IL-6R expression in fetal intestinal epithelial stem cells, this inflammatory imprint persists into adulthood, conferring increased resistance to intestinal infections in the offspring (9). In addition to epithelial barriers in the digestive tract, other barrier tissues benefit from trained immunity through enhanced tissue repair and antimicrobial capacity. Acute exposure to LPS induces long-lasting changes in airway macrophages, resulting in an innate immune memory phenotype that protects against bacterial pneumonia (23). Likewise, brief cutaneous exposure to imiquimod, an acute inflammatory trigger that activates IL-17-type psoriasis-like responses, enhances wound healing capacity thereafter (6). Protective effects of trained immunity have also been reported in various in vivo models. Pre-injection of β-glucan in mice significantly improves survival against S.aureus and reduces renal necrosis associated with systemic infection (32). In leukemic mice, β-glucan not only prolongs survival but also extends lifespan during experimental S. aureus sepsis (33). Intraperitoneal administration of cytosine–guanine (CpG) oligodeoxynucleotides confers protection against Escherichia coli-induced meningitis in neutropenic mice (34). Interestingly, compared with sedentary controls, bone marrow-derived macrophages (BMDMs) from exercise-trained mice show reduced LPS-induced NF-κB activation and proinflammatory gene expression, along with increased expression of M2-related genes, these changes are associated with improved mitochondrial quality, increased reliance on oxidative phosphorylation, and reduced ROS production (35, 36). These findings suggest that moderate, regular exercise may activate trained immunity, strengthening host defense while reducing chronic inflammation and disease incidence.
In contrast, chronic excessive inflammation or tissue injury represents another potential consequence of trained immunity, contributing to the pathogenesis of diseases such as atherosclerosis, rheumatoid arthritis, psoriasis, and inflammatory bowel disease. In these contexts, the maladaptive effects of trained immunity may exacerbate disease pathology. During tissue repair, skin stem cells retain epigenetic memory associated with migration and inflammation, endowing them with enhanced proliferative capacity, increased migratory ability, and heightened environmental sensitivity, these traits facilitate more efficient regeneration following recurrent injuries (37). However, it is noteworthy that such regenerative features significantly overlap with malignant characteristics of tumor cells, including uncontrolled proliferation and invasive migration (38), thereby potentially promoting tumor initiation and metastasis. Moreover, the persistent accumulation of inflammatory memory may lead to stem cell dysfunction, resulting in exaggerated immune responses and increased susceptibility to autoimmune diseases (e.g., psoriasis) and chronic inflammatory conditions. Over time, the abnormal buildup of epigenetic memory may become a key contributor to chronic inflammation and tumorigenesis. Similarly, although maternal inflammatory training of intestinal stem cells offers infection resistance to offspring, the concurrently heightened Th17 response may disrupt host–microbiota homeostasis, triggering immune overactivation and chronic inflammation, and thereby elevating the risk of intestinal inflammatory disorders in the progeny (9) (Figure 2). These findings suggest that while acute and transient injury can potentiate repair mechanisms, repeated insults may drive chronic inflammation and impair healing. Thus, trained immunity is a double-edged sword, its net effect depends on whether it enhances or aggravates the tissue response.
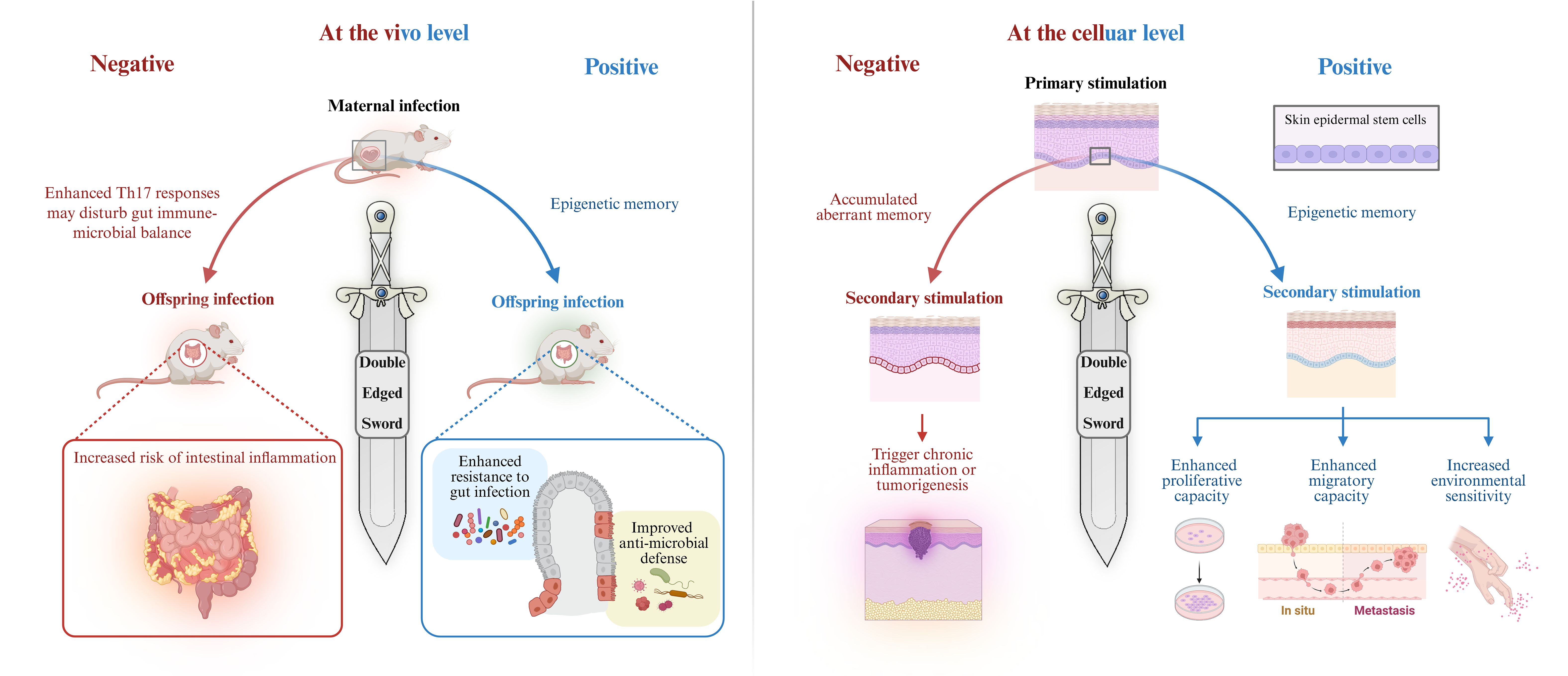
Figure 2. Trained immunity is a double-edged sword. Trained immunity plays a dual role in inflammation regulation and immune defense. At the organismal level, maternal inflammation during pregnancy elevates IL-6 levels, activating IL-6R expression in fetal intestinal epithelial stem cells and establishing lasting inflammatory memory. This memory enhances offspring resistance to intestinal infections. On the other hand, enhanced Th17 responses may disrupt the balance between the gut microbiota and the immune system, causing immune overactivation and chronic inflammation, thereby increasing the risk of intestinal inflammatory diseases in the offspring (9). At the cellular level, transient inflammatory stimulation by imiquimod endows EpSCs with memory capacity, promoting proliferation and migration, enhancing wound healing, and strengthening resistance to future pathogens, thereby providing protection. However, prolonged excessive stimulation of stem cells may lead to “overtrained,” accumulate abnormal memories, excessive proliferation and migration, and ultimately triggering chronic inflammation or tumorigenesis (6). Created with BioRender.com.
5 Trained immunity of inflammatory diseases
5.1 Autoimmune diseases
5.1.1 Sarcoidosis
Sarcoidosis is a non-caseating epithelioid granulomatous inflammatory disease, the formation of these granulomas disrupts tissue homeostasis, primarily affecting the lung parenchyma and leading to clinical symptoms such as cough and dyspnea, Multiple organ involvement (39). Despite the identification of various environmental, occupational, infectious, and genetic risk factors, the precise etiology of sarcoidosis remains unclear. It is thought to involve aberrant immune responses but is not classified as a typical autoimmune disorder. Glucocorticoids remain the first-line therapy, but their use carries a dose-dependent risk of severe adverse events (40). Anti-TNF-α biologics are effective in steroid-refractory or organ-threatening cases. For severe and progressive disease, third-line treatment may include biologics or advanced immunomodulators (40). Sarcoid granulomas consist of both immune and structural cells. Their formation begins when macrophages and dendritic cells recognize antigens via pattern recognition receptors (PRRs), triggering the secretion of cytokines (e.g., TNF-α, IL-1β, IL-6, IL-12) and chemokines (e.g., CXCL9, CXCL10, CCL2), these mediators promote macrophage recruitment and aggregation to form the granuloma core (41). Th17.1 and Th1 cells contribute to granuloma development and maintenance by secreting IL-17 and IFN-γ, thereby regulating immune cell recruitment and granuloma architecture (42). The granuloma center typically comprises CD68+ macrophages and multinucleated giant cells with M1-like characteristics, while the periphery is enriched in Th1 and Th17.1 cells (43). Together, these components sustain granulomatous inflammation and perpetuate the disease process.
Currently, limited evidence supports a direct role for trained immunity in sarcoidosis, However, some studies suggest that monocytes in sarcoidosis patients exhibit pathological features overlapping with the detrimental effects observed in trained immunity. Pathogen exposure may trigger trained immunity in sarcoidosis, peripheral blood mononuclear cells (PBMCs) from patients produce higher TNF and IL-6 levels upon bacterial or fungal stimulation than those from healthy controls and show abnormal expression of pathogen-sensing PRRs (44). metabolic alterations in monocytes have been identified as another hallmark, PBMCs from sarcoidosis patients show dysregulation of metabolic and oxidative phosphorylation pathways, particularly upregulation of the mechanistic target of rapamycin (mTOR) signaling pathway (45). Hyperactivation of mTOR promotes macrophage proliferation, inhibits apoptosis, and drives metabolic reprogramming, thereby supporting macrophage survival and granuloma persistence (46, 47). mTORC1 maintains granuloma structure, while JAK/STAT enhances cytokine production and immune cell activation, they suppress macrophage autophagy and promote formation of multinucleated giant cells, a hallmark of sarcoid granulomas (48). Downstream of mTOR, hypoxia-inducible factor-1α (HIF-1α) is excessively activated in sarcoid macrophages, exacerbating inflammation and fibrosis (49). Dysregulated mTOR and HIF-1α signaling disrupt glycolysis and impair the tricarboxylic acid (TCA) cycle, a metabolic cascade implicated in protective β-glucan-induced trained immunity (50). Metabolomic analysis of sarcoidosis patient serum reveals perturbations in glycolysis and TCA cycle activity, contributing to macrophage hypertrophy and multinucleated giant cell formation—the key components of granulomas. Activated pentose phosphate pathway (PPP) and elevated metabolic states in monocytes further correlate with granulomatous inflammation (43).
The precise epigenetic mechanisms in sarcoidosis remain poorly defined. However, bronchoalveolar lavage cells from sarcoidosis patients show aberrant DNA methylation in genes linked to immune responses, such as HLA-DPB2, CXCL7, and CCL16, suggesting dysregulated gene expression due to DNA methylation and chromatin remodeling (51, 52). These metabolic and potential epigenetic alterations may amplify monocyte dysfunction in sarcoidosis.
Although current studies on trained immunity in sarcoidosis are limited, its pathological features and underlying principles suggest opportunities for early intervention against adverse progression. When monocytes are in a “pre-trained” or “pre-stimulated” state, secondary stimulation may trigger persistent hyperactivation, leading to enhanced inflammation, multinucleated macrophage formation, and granuloma maintenance. This process may further promote tissue fibrosis, resulting in damage and functional impairment. Thus, while trained immunity may enhance host defense, it can also drive chronic inflammation. Further studies are needed to clarify the potential detrimental effects of trained immunity in sarcoidosis and to provide mechanistic insights for therapeutic strategies.
5.1.2 Multiple sclerosis
Multiple sclerosis (MS) is a neurodegenerative disorder caused by inflammatory damage to the myelin sheath in the brain and spinal cord. This disrupts neural signal transmission, leading to neurological symptoms, reduced quality of life, and disability. Common symptoms include fatigue, blurred vision, optic neuritis, limb weakness or sensory disturbances, dizziness, balance problems, cognitive impairment, and bladder dysfunction (53). In MS, B lymphocytes exhibit a proinflammatory profile that drives cortical pathology and contributes to neurological dysfunction (54). CD20, a specific surface marker of B cells, is the target of anti-CD20 monoclonal antibodies (e.g., ocrelizumab, ofatumumab, rituximab, ublituximab). These agents selectively deplete B cells through antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity without impairing antibody production by plasma cells (55, 56). Although anti-CD20 therapy effectively suppresses disease activity, it may trigger reactivation of latent infections (e.g., tuberculosis, hepatitis) and infusion-related reactions (e.g., fever, headache) (57). However, the role of innate immunity remains poorly understood. Comparing peripheral blood from MS patients and healthy controls showed that granulocytes significantly decreased in MS, while monocyte representation remained unchanged (58). CD64+ and PD-L1+ granulocytes showed no significant decline, whereas other subsets defined by distinct membrane markers displayed marked differences (58). These findings indicate that research on MS pathogenesis and therapy should also focus on elements of the innate immune response. Macrophages in MS patients tend to exhibit a proinflammatory phenotype. Even in the absence of inflammatory stimuli, these cells display increased expression of glycolytic genes, a metabolic feature resembling trained immunity. Additional alterations include downregulation of electron transport chain genes, TCA cycle disruption, and abnormal fatty acid metabolism. These changes impair oxidative metabolism, which is essential for anti-inflammatory macrophage function, and may contribute to or result from the proinflammatory state, perivascular macrophages with elevated glycolytic capacity show enhanced transmigration in MS animal models and are associated with immune cell infiltration (59). This intrinsic pro-inflammatory metabolic program, independent of external signals, suggests that macrophages may be more prone to activation and may even spontaneously generate low-grade inflammation. Such activity can sustain neuroinflammation even in the absence of clear external triggers. These intrinsic defects align with the pathological features of MS and suggest an important role for macrophages. In treated MS patients, levels of key metabolites were similar to those in healthy controls (59). This indicates that therapy has beneficial effects on the energy defects of innate immune cells and shows that macrophage metabolism is closely linked to disease state, possibly influenced by innate immune memory. These findings highlight the importance of macrophages in MS and their potential as therapeutic targets. Normalization of metabolite levels further demonstrates that reversal of this “trained” state is feasible and supports the strategy of targeting metabolic pathways to eliminate maladaptive training.
Epigenetic modifications in immune and glial cells also contribute to MS pathogenesis and progression. Differentially methylated regions and sites have been identified in immune cells and oligodendrocytes from MS patients compared to healthy controls (60). The promoter region of PADI2, encoding peptidyl arginine deiminase 2, an enzyme catalyzing myelin basic protein (MBP) citrullination, is hypomethylated in MS. This hypomethylation suppresses MBP production, compromising myelin stability, similar PADI2 hypomethylation has been observed in PBMCs from MS patients (61). Moreover, hypomethylation of the IL-17A promoter in T cells increases IL-17 expression, promoting central nervous system inflammation (62). In PBMCs, citrullination of histone H3 at H3Cit8 prevents heterochromatin protein 1 binding to H3K9me3, thereby suppressing TNF-α and IL-8 expression (63). Histone deacetylation is more prominent in chronic MS lesions than in early-stage lesions, and the efficiency of deacetylation declines as the disease progresses (64). Although current evidence does not establish a direct link between trained immunity and MS, immune cells in MS exhibit epigenetic reprogramming and metabolic changes. These trained-like features may enhance immune activation or induce tolerance, ultimately influencing clinical manifestations. Future studies should further investigate the role of trained immunity in MS, particularly across different disease stages.
5.1.3 Rheumatoid arthritis
Rheumatoid arthritis (RA) is the most common inflammatory arthritis and represents a systemic inflammatory disease that mainly affects the joints (65). In RA, synovial macrophages and T cells excessively secrete TNF-α, driving synovial hyperplasia and inducing IL-6 and IL-1 release, which forms an “inflammatory amplification loop” and leads to cartilage and bone destruction (66). Anti-TNF antibodies such as adalimumab block TNF-α binding to its receptors, reduce inflammatory mediator release, and alleviate inflammation and tissue damage (67). In patients with poor response to TNF inhibitors, anti-CD20 therapy is widely used, improving synovitis, slowing joint destruction, and delaying disease onset in high-risk individuals (68). This raises the question of whether trained immunity could be applied to intervene early in macrophage and T-cell overactivation to block the amplification loop. Increasing evidence indicates that innate immunity plays a critical role in disease initiation and persistence. Circulating monocytes from RA patients produce elevated levels of IL-1β and IL-6 upon in vitro stimulation (69), IL-6 contributes to joint destruction (70), while IL-1β promotes osteoclastogenesis, induces matrix metalloproteinase (MMP) expression in chondrocytes, and initiates synoviocyte proliferation (71). Together, these cytokines drive bone degradation. RA monocytes show activation of PI3K/mTOR and MAPK pathways, inhibition of mTOR reduces synovial osteoclast formation and protects against local bone and cartilage damage (72), highlighting the central role of cytokine-driven signaling in RA pathophysiology.
Epigenetic mechanisms also contribute to RA, involving histone acetylation, methylation, and DNA methylation. Increased expression of H3.3 in PBMCs from RA patients has been associated with histone acetylation markers (73). Hypomethylated and hypermethylated genomic regions have been identified in PBMCs, RA synovial fibroblasts, and RA synovial tissues (74–76). LPS stimulation of THP-1 cells from RA patients results in enhanced histone H3 and H4 acetylation at the CCL2 promoter (77). mTORC1 promotes the expression of lactate dehydrogenase A (LDHA) and Pyruvate kinase isozyme type M2 (PKM2) by activating HIF-1α and catalyzes the reductive carboxylation of glutamine to generate lipid precursors, thereby aggravating synovial inflammation (78, 79). The hypoxic synovial microenvironment further enhances angiogenesis and glycolysis through HIF-1α, creating a vicious cycle of inflammation and hypoxia (80). Dysregulated lipid metabolism in RA is characterized by elevated low-density lipoprotein Cholesterol (LDL-C), impaired high-density lipoprotein cholesterol (HDL-C) function, and abnormal proprotein convertase subtilisin/kexin Type 9 (PCSK9) accumulation, which directly drive inflammation and immune dysregulation. Serum PCSK9 levels correlate positively with Disease Activity Score in 28 Joints and Rheumatoid Factor and accelerate RA progression through dual mechanisms (81). These findings indicate that innate immune cells in RA also undergo metabolic changes resembling trained immunity, sustaining pro-inflammatory responses. In addition, under the context of studying central trained immunity as a basis for inflammatory comorbidities, experimental periodontitis-induced central trained immunity could be transferred to healthy mice via bone marrow transplantation, leading to more severe arthritis in a collagen antibody, induced arthritis model (82). Central trained immunity acted in a maladaptive manner, thereby exacerbating inflammation and increasing the risk of inflammatory comorbidities.
Future research should further investigate the specific mechanisms of trained immunity in RA, particularly how epigenetic reprogramming and metabolic shifts coordinate to regulate monocytes, macrophages, and possibly non-immune cells. Integrating trained immunity–targeted interventions with immunosuppressive therapies may offer more precise and effective treatment strategies for RA patients.
5.1.4 Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a chronic, multisystem inflammatory disease caused by abnormal immune activation that attacks self-tissues, representing a diffuse connective tissue disorder. It is an autoimmune disease targeting antigens derived from apoptotic microparticles (MPs) and neutrophil extracellular traps (NETs), and is typically B cell/antibody–driven (83). Conventional treatment relies on glucocorticoids and immunosuppressants, which have substantial side effects. Recently, belimumab and anifrolumab have shown clinical benefits by neutralizing the B cell activating factor to reduce abnormal B cell survival, and by blocking the type I interferon receptor to inhibit interferon-driven inflammation, improving disease activity, reducing flares, and lowering steroid use (84). Monocytes and macrophages are increasingly recognized as key contributors to disease pathogenesis (85). Macrophages from SLE patients exhibit impaired phagocytosis of apoptotic cells and reduced clearance of immune complexes (86). In lupus nephritis, macrophage infiltration serves as a predictor of disease progression (87). Emerging evidence also suggests a possible link between SLE and trained immunity. Circulating monocytes from SLE patients produce higher levels of proinflammatory cytokines in response to Toll-like receptor (TLR) agonists, indicating a trained phenotype. This response is associated with dysregulation of histone H3 lysine 4 trimethylation (H3K4me3) and increased expression of genes involved in metabolism and inflammation (88). However, excessive production of these cytokines may also promote adaptive immune activation, including autoreactive T and B cell responses and antinuclear antibody formation, potentially perpetuating inflammatory tissue damage. BMDMs from lupus-prone mice display hallmarks of trained immunity, such as enhanced mycobacterial killing and increased cytokine production. These functional changes are mechanistically linked to elevated glycolytic metabolism (89). Intraperitoneal injection of β-glucan into 12-week-old NZB/W F1 mice (SLE-prone) exacerbated extramedullary hematopoiesis in the spleen, promoted myeloid skewing, and worsened lupus nephritis, inducing maladaptive trained immunity (90). In SLE mouse models, hematopoietic stem and progenitor cells (HSPCs) exhibited transcriptional reprogramming and myeloid bias. The autoimmune inflammatory environment in SLE may train HSPCs to produce hyperresponsive myeloid cells, further promoting autoantibody generation and organ damage, and potentially driving maladaptive progression (91). Histone modifications have been extensively studied in both human and murine models of SLE (92). For example, elevated histone H3 acetylation and increased H3K4me2 levels at the CD70 gene promoter are positively correlated with disease activity (93). Aberrant patterns of histone acetylation and methylation have been observed in monocytes and T lymphocytes from SLE patients (94). Therapeutic interventions targeting metabolic and epigenetic processes have demonstrated benefit in SLE, suggesting that inhibition of trained immunity may contribute to these protective effects.
5.1.5 Others
In addition to the autoimmune diseases mentioned above, several skin inflammation related autoimmune or autoinflammatory disorders have also been associated with trained immunity. Such as psoriasis, an inflammatory skin disease, is characterized by erythematous plaques resulting from keratinocyte hyperproliferation (95), although the epigenetic elements of trained immunity in human psoriasis have not been fully defined, studies using imiquimod (IMQ)-induced murine psoriasis models have demonstrated that EpSCs retain innate immune memory by maintaining chromatin accessibility following inflammation. These accessible chromatin regions allow rapid activation of relevant genes upon secondary insult (6). Hyperimmunoglobulin D syndrome (HIDS), a monogenic autoinflammatory disorder, features erythematous rashes and recurrent sterile inflammation. Aberrant trained immunity in HIDS arises from mevalonate kinase pathway dysfunction, leading to metabolite accumulation that activates the AKT–mTOR–HIF-1α axis. This shifts immune cells from oxidative phosphorylation to glycolysis, giving circulating monocytes a persistently activated trained immunity profile (96). This process may further activate the NLRP3 inflammasome, resulting in excessive release of inflammatory cytokines such as IL-1β and amplifying systemic inflammation (97).
5.2 Degenerative diseases
5.2.1 Atherosclerosis
Atherosclerotic cardiovascular disease (ASCVD) is a chronic degenerative vascular disease characterized by low-grade, persistent inflammation of the vascular wall, in which monocytes and macrophages play a central role (98). Factors that exacerbate this chronic inflammatory state, including dyslipidemia, obesity, diabetes, hypertension, aging, and smoking, as well as nontraditional risks such as infections and chronic inflammatory diseases, can promote ASCVD progression (99). Trained immunity, through metabolic and epigenetic reprogramming of innate immune cells, enhances inflammatory responses upon secondary stimulation. Given its amplified proinflammatory characteristics, trained immunity may play a key role in infection-related ASCVD. In murine models, short-term exposure to low-dose LPS induces long-lasting monocyte polarization toward a proinflammatory phenotype, aggravating atherosclerotic lesion development (100). Endogenous ASCVD-associated stimuli, such as oxidized low-density lipoprotein (oxLDL), lipoprotein (a), aldosterone, and S100A4 protein, have also been shown to induce trained immunity in monocytes and macrophages. Trained monocytes exhibit enhanced adhesion to vascular endothelium, increased migratory capacity, and elevated inflammatory responses to TLR agonists (101). In trained macrophages, foam cell formation and upregulation of matrix metalloproteinases accelerate ASCVD progression (102).
HSPCs in the bone marrow contribute to trained immunity. Upon primary stimulation, such as infection, vaccination, or inflammation, HSPCs undergo transcriptional and epigenetic reprogramming, generating monocytes and macrophages with enhanced responsiveness. In mouse models of ASCVD (e.g., LDLR-/-), a high-fat diet expands bone marrow progenitors and promotes myelopoiesis, accompanied by long-term epigenetic changes linked to NLRP3 inflammasome activation and increased IL-1β secretion. Resulting monocytes adopt a primed state with heightened TLR responses for at least four weeks. Transplanting bone marrow from these trained mice into normal-diet recipients enlarges atherosclerotic lesions (103, 104). Monocytes and macrophages from trained HSPCs produce more proinflammatory cytokines (TNF-α, IL-6, MCP-1) and show enhanced vascular migration, promoting vascular inflammation and ASCVD progression (105). Hypercholesterolemia also drives HSPC proliferation and myeloid skewing, producing proinflammatory, pro-atherogenic myeloid cells (105).
Clinical observations support these findings. Using 18F-fluorodeoxyglucose positron emission tomography, researchers have detected significantly elevated glucose metabolism in human atherosclerotic plaques, particularly within lipid-rich necrotic cores and regions densely infiltrated by immune cell (106, 107), high-risk plaques, in particular, display increased glycolytic activity (108), and as a metabolic hallmark of trained immunity (109, 110). Although the direct involvement of trained immunity remains to be confirmed, high-risk plaques produce higher levels of proinflammatory cytokines and chemokines than low-risk, stable plaques (108). Furthermore, circulating monocytes isolated from symptomatic ASCVD patients exhibit a more proinflammatory phenotype compared to those from healthy individuals (111).
In addition to immune cells, vascular non-immune cells, such as endothelial cells and smooth muscle cells (SMCs), also participate in trained immunity during atherosclerosis. Vascular SMCs isolated from diabetic mouse models exhibit enhanced migratory capacity, upregulated expression of inflammatory genes, and increased adhesion to monocytes. These trained-like features persist over time and remain detectable even in vitro (112). Similarly, stimulation with oxLDL induces sustained proinflammatory “priming effects” in cultured coronary artery SMCs (113). These findings indicate that SMCs, like innate immune cells, can undergo trained immunity and maintain a long-term activated state. Endothelial cells also exhibit similar behavior. The pro-atherogenic lipid molecule lysophosphatidylcholine reprograms aortic endothelial cells, inducing sustained inflammatory activation (114). High glucose levels drive endothelial cells into a prolonged proinflammatory state, characterized by NF-κB upregulation and increased expression of pro-atherogenic genes such as MCP-1 and VCAM-1 (115). These “trained” vascular non-immune cells may play critical roles in the progression of atherosclerosis, particularly by promoting vascular inflammation and accelerating ASCVD development.
Treatment of atherosclerosis focuses on lowering blood lipids, controlling blood pressure, improving glucose metabolism, and using antiplatelet agents to slow disease progression (116). Anti-inflammatory approaches, such as the IL-1β inhibitor canakinumab in the CANTOS trial, have also been used to reduce cardiovascular events (117). Although these strategies lower event rates, they do not address chronic low-grade vascular inflammation and fail to fully halt atherosclerosis progression. Given these limitations, targeting trained immunity provides a novel therapeutic perspective. Interventions aimed at modulating trained immune responses in both immune and non-immune vascular cells may offer more effective approaches for the treatment of atherosclerosis.
5.2.2 Alzheimer’s disease
Alzheimer’s disease (AD) is the most common form of dementia, characterized by progressive cognitive decline and behavioral impairment, with risk increasing with age. It is an inflammatory and neurodegenerative disorder driven by extracellular accumulation of amyloid-β (Aβ) and intracellular aggregation of Tau protein (118). Aβ deposition activates microglia, triggering local inflammation and further promoting plaque formation (119). Current therapies include anti-Aβ monoclonal antibodies, anti-Tau treatments, and TREM2 agonists. Lecanemab, a humanized monoclonal antibody targeting the N-terminus of Aβ, reduces neurotoxicity by promoting Aβ clearance (120). Anti-Tau therapies target Tau’s N-terminus to inhibit abnormal aggregation and phosphorylation, slowing neurodegeneration. TREM2 agonists enhance microglial phagocytosis, promoting Aβ and Tau clearance and improving the neural microenvironment (121). However, microglia, the resident immune cells of the brain, normally play a protective role. In early AD, they secrete neurotoxic cytokines, sustaining chronic inflammation (122). In APP23 transgenic mice, a single intraperitoneal LPS injection (1×LPS) at 3 months increases Aβ plaque burden and total Aβ while reducing brain IL-10. Gene co-expression analysis shows upregulation of HIF-1 signaling and glycolysis-related genes. Microglia display higher mitochondrial membrane potential and lactate release, reflecting HIF-1α activation and a metabolic shift toward glycolysis. In contrast, repeated LPS injections (4×LPS) produce opposite effects, indicating that trained immunity exacerbates AD pathology, whereas immune tolerance mitigates it (123). In another model expressing mutant human APP, presenilin, and Tau, repeated LPS increases Tau phosphorylation without significantly changing Aβ deposition (124). These findings suggest that trained immunity contributes to neuroinflammation and neurodegeneration in AD. In early or preclinical stages, it is marked by heightened inflammatory responses and increased Aβ production, potentially causing neuronal damage. In later stages, chronic Aβ exposure or inflammation may induce tolerance, reducing cytokine release and promoting repair, though this response can be maladaptive or insufficient. Notably, microglia from 1×LPS- and 4×LPS-treated mice exhibit distinct levels of H3K4me1 and H3K27ac, suggesting that epigenetic remodeling plays a key role in trained immunity–associated microglial plasticity (123). Other myeloid cells, including DCs and neutrophils, may also participate in AD-related neuroinflammation and degeneration. These cells may even infiltrate the central nervous system across the blood–brain barrier and contribute to neuronal injury and cognitive decline (125–128). Collectively, AD progression may be driven by complex interactions among multiple immune cell types, with trained immunity and its epigenetic imprinting contributing to disease dynamics across different stages.
Epigenetic regulation plays a critical role in the progression of AD. Demethylation at the promoter region of the APP gene has been implicated in Aβ accumulation in the aging brain (129). Methylation changes in the Tau, particularly at CpG dinucleotide sites, have been shown to alter microtubule function, promoting aberrant Tau aggregation and the formation of neurofibrillary tangles (130, 131). In addition, hyperphosphorylated Tau can suppress gene expression by recruiting histone deacetylases (HDACs) to condense chromatin structure (129). Among HDAC family members, SIRT1 catalyzes the deacetylation of lysine 28 on Tau, inhibiting its normal function and facilitating aggregation (132). Decreased SIRT1 expression in the cortex is closely associated with the accumulation of both Aβ and Tau in AD patients (133), aberrant nuclear–cytoplasmic localization of H3K4me3 has also been reported in early-stage AD (94), though its functional significance remains unclear (134). These findings suggest that early epigenetic alterations may contribute to AD pathology.
Immune cells in AD may undergo increased aerobic glycolysis, a key metabolic driver of trained immunity. Under LPS and IFN-γ stimulation, microglial metabolism shifts toward glycolysis and the PPP (135). Metabolic dysregulation in microglia impairs Aβ clearance and sustains proinflammatory cytokine release (136). In AD animal models, mTOR signaling is activated early, whereas TREM2-deficient mice show mTOR pathway defects, leading to abnormal ATP levels and biosynthetic pathways (137). Although direct evidence of trained immunity in clinical AD is still limited, transcriptional, epigenetic, and metabolic alterations observed in immune cells, including microglia, monocytes, and dendritic cells, from both patients and animal models are consistent with mechanisms underlying innate immune memory. These insights support the hypothesis that trained immunity–like processes may contribute to the immunopathogenesis of AD.
5.3 Pneumonia
Pneumonia is a lung infection caused by bacteria, viruses, or other microorganisms, primarily affecting the alveoli. Among these, bacterial and viral pneumonia are the most common. Recent studies have linked both forms to trained immunity. S. pneumoniae is the leading cause of community-acquired pneumonia and remains a life-threatening pathogen (138). Infection with S. pneumoniae induces histone H3K4me2 modifications in respiratory epithelial cells, which persist for at least nine days after bacterial clearance by antibiotics, these epigenetic changes result in altered cellular metabolism and lysosomal transport, enhancing bacterial adhesion and promoting secondary infections (28). Similarly, LPS exposure induces long-term phenotypic changes and trained immunity in lung-resident macrophages, this response provides significant protection against S. pneumoniae but offers limited defense against SARS-CoV-2 (23). In the context of viral infections, influenza virus has been shown to induce trained immunity in alveolar macrophages, conferring long-lasting anti-tumor immunity. This effect persists for at least 30, 60 and even 120 days after infection, significantly inhibiting pulmonary tumor growth. The protective mechanism depends on IFN-γ, NK cells, and epigenetic and metabolic reprogramming (139). SARS-CoV-2 infection–induced pneumonia may also be associated with trained immunity. SARS-CoV-2, the causative agent of COVID-19, is transmitted primarily through respiratory droplets, similar to influenza. Clinical manifestations range from asymptomatic infection and mild upper respiratory symptoms to severe pneumonia with respiratory failure and death (140). By enhancing innate immune responses via epigenetic and metabolic reprogramming, trained immunity may provide early antiviral protection in COVID-19, reducing viral replication and inflammation. Interestingly, S.aureus skin infections can induce eosinophil-mediated innate immune memory via IL-33 and C5a, leading to bone marrow reprogramming. This alters mature myeloid cell function and exacerbates lung inflammation in systemic allergy models (141). Future research should explore the role of trained immunity in pneumonia of different etiologies. Understanding how epigenetic and metabolic reprogramming modulate immune responses may offer new therapeutic strategies for the prevention and treatment of pneumonia.
5.4 Gastrointestinal inflammation
Studies on trained immunity in gastrointestinal inflammation primarily focus on intestinal inflammation and pancreatitis. During maternal infection, elevated IL-6 crosses the placenta and acts on fetal intestinal epithelial cells. This proinflammatory signal induces epigenetic changes, promoting Th17 differentiation and expansion, enhancing resistance to intestinal infection but increasing risk of intestinal inflammation. These modifications include lasting changes in chromatin accessibility and transcriptional programs in intestinal epithelial stem cells, which may persist into adulthood (9). The integrity of the intestinal barrier is essential for maintaining host health. When this barrier is disrupted, commensal bacteria can translocate systemically and trigger inflammatory responses. One study showed that barrier damage allows Enterococcus faecalis, a member of the gut microbiota, to migrate to the bone marrow, where it induces trained immunity in myeloid progenitors via the Mincle receptor. This response may be either protective or detrimental, depending on context. Mice treated with heat-killed E. faecalis exhibited less weight loss and higher survival rates following infection with Candida albicans or influenza virus. However, in inflammatory models, trained immunity may aggravate disease, for instance, in dextran sulfate sodium-induced colitis, Mincle-deficient mice displayed milder pathological symptoms, indicating that trained immunity contributes to the pathology of inflammatory bowel disease (142). These findings offer novel insights into the complex interplay among the gut microbiota, immune system, and inflammation, which may inform new immunotherapies and improve understanding of diseases related to increased gut permeability. Moreover, inflammation is a major risk factor for pancreatic ductal adenocarcinoma. During pancreatitis, oncogenic kirsten rat sarcoma viral oncogene homolog (KRAS) mutations accelerate tumor progression. Notably, even transient inflammatory episodes can sensitize pancreatic epithelial cells to subsequent KRAS-driven transformation. This adaptive response involves persistent transcriptional and epigenetic reprogramming, enabling rapid activation of ADM during recurrent inflammation, thereby limiting tissue damage (31).
5.5 Allergic diseases
Allergic diseases have emerged as a global public health challenge, with rising prevalence imposing a significant socioeconomic burden. Chronic conditions such as allergic asthma, allergic rhinitis, food allergy, atopic dermatitis, and anaphylaxis impair quality of life and contribute to substantial healthcare expenditures (143). The treatment of allergic diseases has shifted from symptomatic control to a strategy combining immune modulation and targeted therapy. Allergen immunotherapy and biologics/monoclonal antibodies are emerging as key options, offering not only symptom relief but also the potential to induce tolerance and modify disease progression (144). Traditionally, allergic responses were thought to be driven primarily by the adaptive immune system, particularly IgE-mediated reactions. However, recent studies suggest that aberrant activation of the innate immune system also plays a critical role in the onset and progression of allergic diseases. Notably, trained immunity has been proposed as a potential mechanism. Early-life innate immune hyperactivation has been observed in allergic children, while asthmatic children display immune dysregulation characterized by reduced IFN-γ production and ILC2 expansion in response to rhinovirus or LPS stimulation (145). These phenomena may result from allergen- or virus-induced trained immunity, leading to long-lasting immune dysfunction and increased susceptibility to allergic inflammation. In allergic mouse models or patients with house dust mite–induced asthma, macrophages exhibit exaggerated production of TNF-α, CCL17, leukotrienes, PGE2, and IL-6 upon stimulation. This process depends on the TNF signaling pathway and involves 2-hydroxyglutarate accumulation and KDM1A-mediated demethylation, identifying potential therapeutic targets for allergic asthma (146). In a study using single-cell RNA sequencing, Li et al. (2022) identified an inflammatory neutrophil subpopulation in allergic asthma with molecular features of innate immune memory. Asthma may reprogram neutrophil populations, leading to expansion of G-CSFR+FcγRIIb+ neutrophils, which may represent memory-like neutrophils and serve as novel targets in neutrophil-dominant asthma (147). Two experimental studies further indicate that infection-induced reprogramming of innate immune cells may prevent asthma development. In one model, murine gammaherpesvirus 4 lung infection suppressed HDM-induced experimental asthma (HDM-EAA) through two mechanisms (1): downregulating costimulatory molecules CD80/CD86 on migratory dendritic cells, impairing Th2 activation; and (2) inducing embryonically derived alveolar macrophage apoptosis, while recruiting bone marrow–derived monocytes that differentiated into anti-inflammatory macrophages with sustained IL-10 production. These monocytes persisted in lung parenchyma for up to 28 days, suggesting the formation of long-term trained immunity via epigenetic reprogramming (148). In another study, neonatal peritoneal inoculation of EV-A71 virus induced glycolysis-dependent proinflammatory training in macrophages, these trained cells displayed prolonged (≥21 days) secretion of IL-6, TNF-α, and CCL17 and aggravated HDM-EAA. Adoptive transfer of EV-A71–trained BMDMs into naïve mice enhanced Th2 inflammation and allergen-specific IgE levels. Further analysis revealed temporal polarization of these trained macrophages: an early Th2-skewed phase, followed by a classical proinflammatory phenotype. This process was abrogated by the glycolysis inhibitor 2-deoxy-D-glucose (2-DG), confirming the central role of metabolic reprogramming in immune training (148). In addition to immune cells, other cell types such as airway epithelial cells may also contribute to trained immunity in allergic disease. Airway epithelial cells play key roles in asthma pathogenesis and respond to microbial compounds, allergens, and pollutants. Single-cell transcriptomic profiling in chronic rhinosinusitis revealed basal epithelial cells with Th2 cytokine memory characteristics, suggesting that epithelial cells may act as allergen memory reservoirs. Notably, microbial imprinting in the respiratory epithelium may dynamically regulate future immune responses by modulating cytokine output such as IL-1b and IL-8 (149), resembling the trained immunity seen in macrophages. Beyond asthma, food allergy has gained attention due to rising prevalence in Western countries. Trained immunity may influence food allergy development by reshaping the DCs–T cell interaction network and promoting Th2-skewed responses. In peanut-allergic infants, PBMCs exhibit excessive TNF-α production upon nonspecific stimulation. This hyperresponsive immune phenotype persists in allergic adolescents, marked by increased circulating dendritic/monocyte populations and elevated secretion of IL-6, IL-1β, and TNF-α upon LPS challenge (150). In summary, trained immunity provides a novel conceptual framework for understanding allergic diseases. It offers promising avenues for early intervention and the development of targeted therapies based on epigenetic and metabolic reprogramming of immune and non-immune cells.
Trained immunity exerts long-lasting effects on cell function through epigenetic regulation and metabolic reprogramming (Table 1) and shows bidirectional roles in immune regulation: disease-induced trained immunity can, in turn, exacerbate pathology, creating a self-reinforcing vicious cycle. Accordingly, new prevention and treatment strategies should aim to enhance its beneficial effects and intervene early in harmful reprogramming. These findings offer new perspectives for targeted therapies, though underlying mechanisms and clinical applications require further investigation.
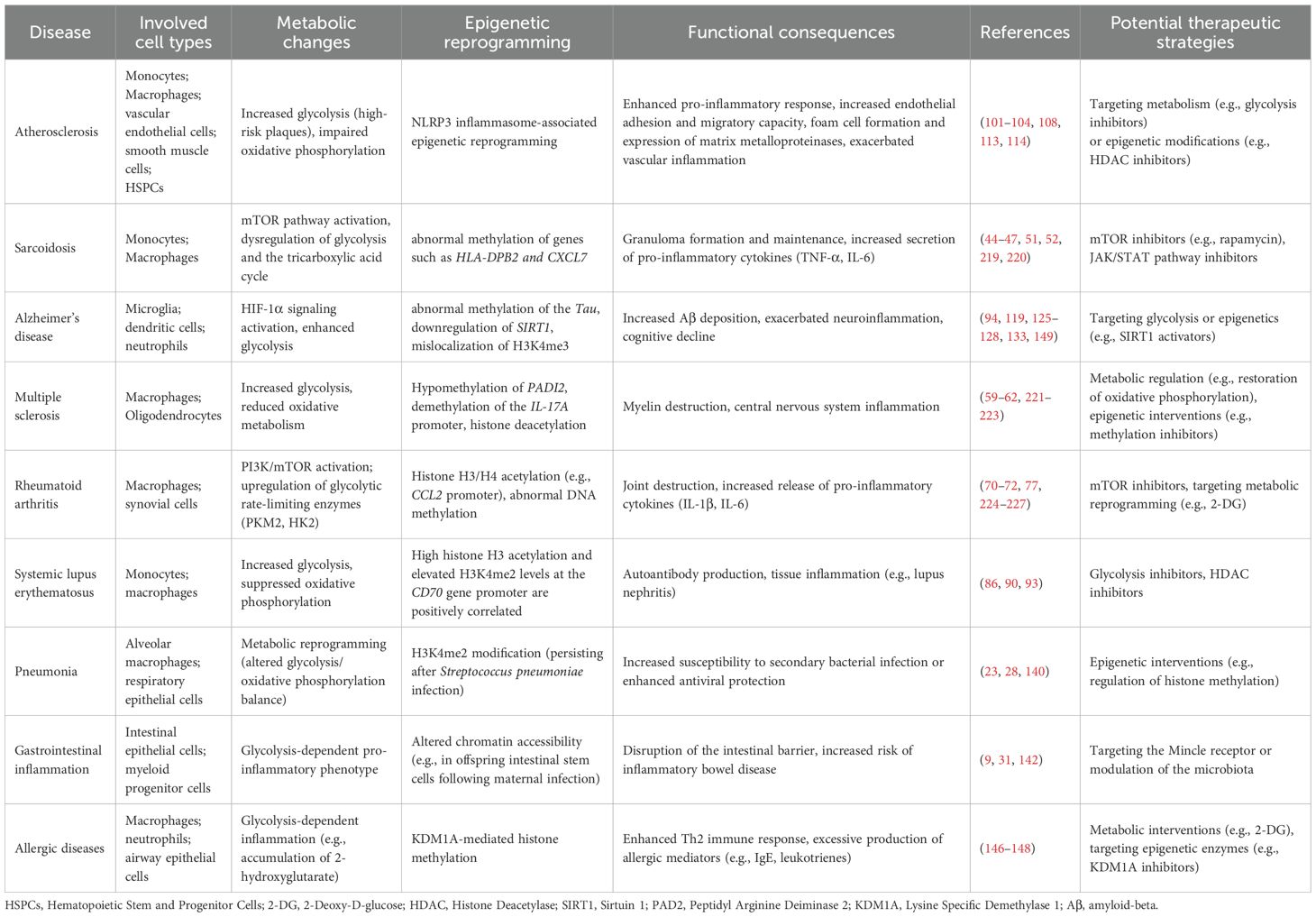
Table 1. Trained immunity in inflammatory diseases: from metabolic dysregulation to epigenetic reprogramming.
6 Treatment of inflammatory diseases based on training immune mechanisms
For inflammatory diseases, most current therapies target the adaptive immune system or provide symptomatic relief by suppressing inflammatory mediators. However, these approaches often fail to prevent disease recurrence or chronic progression. The discovery of trained immunity opens new therapeutic avenues by enabling interventions that target excessive innate immune activation. By modulating epigenetic remodeling and metabolic reprogramming within trained immunity pathways, it is possible to develop targeted therapies that reprogram inflammatory memory in myeloid cells at its source. In addition, regulatory vaccines and smart nanomaterials designed to modulate trained immunity, particularly those targeting bone marrow or inflamed tissues, offer novel, precision-based strategies for inflammatory disease management. These approaches hold promise for achieving long-term immune tolerance and sustained inflammation resolution (Figure 3).
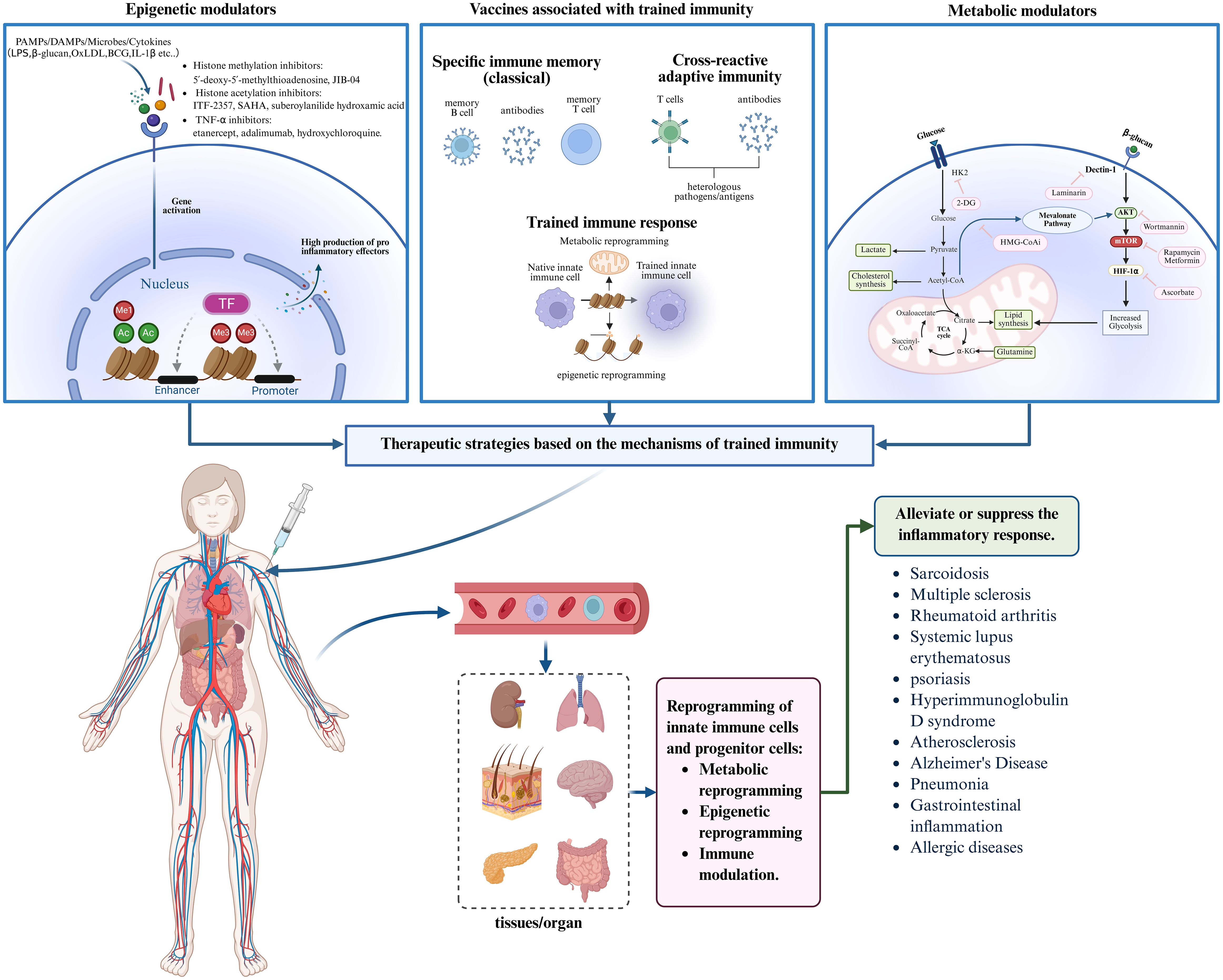
Figure 3. Trained immunity-based therapeutic strategies for inflammatory diseases. Trained immunity offers novel intervention strategies for inflammatory diseases. By developing epigenetic modulators, metabolic regulators, and trained immunity-related vaccines, these agents can be administered systemically and delivered via the bloodstream to target organs. This approach aims to modulate trained immunity through epigenetic remodeling, metabolic reprogramming, and immune activation, thereby effectively alleviating or suppressing inflammatory responses and improving disease outcomes. Created with BioRender.com.
6.1 Based on epigenetic reprogramming
Unlike classical adaptive immune memory, which relies on antigen receptor gene rearrangement, trained immunity operates through epigenetic modifications that increase chromatin accessibility at gene loci following primary stimulation. Upon secondary exposure, this enables rapid transcriptional activation of genes involved in inflammation, antimicrobial defense, and stress responses, without altering the underlying DNA sequence. In eukaryotic cells, genomic DNA is compacted into nucleosomes by structural histone proteins, rendering most gene regions inaccessible to transcription factors. Transcription initiation requires nucleosome displacement at promoter regions to expose regulatory elements, allowing transcription factor binding and recruitment of RNA polymerase (74, 151). Chromatin accessibility is regulated by epigenetic modifications, including histone methylation, histone acetylation, and DNA methylation, among which histone modifications are well-established drivers of inflammatory memory.
For instance, IFN-γ stimulation of HeLa cells increases H3K4me2 at the DRA promoter, which persists after withdrawal and across multiple cell cycles (152). Similarly, Primary S. pneumoniae infection induces H3K4me2 enrichment in respiratory epithelial cells (28). In murine EpSCs, inflammatory signals activate STAT3, which translocates to the nucleus and binds H3K4me1- and H3K27ac-marked chromatin, establishing an epigenetically poised state (6). Histone modifications also mediate trained immunity in vitro: in β-glucan–trained monocytes, promoters and enhancers of inflammation- and immunity-related genes gain H3K4me1, H3K4me3, and H3K27ac marks, maintaining transcriptionally active chromatin and enabling enhanced cytokine production upon restimulation (153). In contrast, LPS-induced tolerance is marked by attenuated transcriptional responses upon secondary LPS challenge. Tolerized macrophages fail to deposit active histone marks (e.g., H3K4me1, H3K27ac) at the promoters of tolerant genes, resembling the immune paralysis seen in sepsis, interestingly, β-glucan injection can reverse LPS-induced tolerance by restoring H3K27ac deposition and reactivating macrophage function. The BET family small-molecule inhibitor I-BET151 (GSK1210151A) is primarily used to suppress inflammation and for anti-tumor purposes. Evidence supporting its role as an mTOR inhibitor remains largely preclinical (154). Its limitation is the inability to reverse effects once tolerance is established, making it potentially ineffective in monocytes that have already experienced inflammatory activation. Nonetheless, β-Glucan and I-BET151 offer a theoretical basis and potential targets for developing immunomodulatory strategies against tolerance-related diseases such as sepsis (155).Similarly, BCG-induced trained immunity involves the remodeling of H3K9me3, H3K4me3, and H3K27ac histone marks (156, 157). Genome-wide H3K27ac ChIP-seq analyses revealed enrichment of this active histone modification at genes encoding the oxLDL receptor, a marker of atherosclerosis, and other inflammation-related genes in monocytes following BCG vaccination, suggesting transcriptional activation of these pro-atherogenic genes (157). The trained phenotype and gene-specific H3K4me3 enrichment persist for at least three months and up to one year post-vaccination (156). In atherosclerosis, oxLDL-induced trained immunity in monocyte-derived macrophages also promotes H3K4me3-mediated activation of genes involved in inflammation, chemotaxis, and foam cell formation (102). The histone methyltransferase inhibitor 5′-deoxy-5′-methylthioadenosine (MTA) reverses histone methylation and prevents chromatin remodeling, effectively abrogating the trained immunity phenotype induced by oxLDL. This finding offers a potential strategy for modulating inflammation in atherosclerosis (102, 158, 159). Histone methylation is regulated by a complex network of enzymes, including histone methyltransferases and demethylases, which modulate gene expression by adding or removing methyl groups at specific histone residues (160). The broad-spectrum Jumonji histone demethylase inhibitor JIB-04 reduces trained immunity by modulating the repressive H3K9 mark (161). To date, evidence for its therapeutic effects as an mTOR inhibitor is limited to pharmacological and animal studies, with no established clinical trials (162). Similarly, histone acetylation, catalyzed by histone acetyltransferases (HATs) and HDACs, plays a regulatory role in trained immunity (163). Class I/II HDAC inhibitors used in cancer therapy, including vorinostat (suberoylanilide hydroxamic acid) and others such as ITF-2357 and SAHA, have shown anti-inflammatory effects in inflammatory diseases (164). Vorinostat is not a classical mTOR antagonist but has been used clinically in combination with mTOR inhibitors or to modulate mTOR-related pathways (165). ITF-2357 has advanced in multiple clinical trials, with recent progress in some indications; for example, a phase II study (NCT00928707) showed that SAHA combined with hydroxyurea was well tolerated (166). In trained monocytes, elevated NAD+/NADH ratios and lactate accumulation modulate HDAC activity and influence downstream gene expression (167, 168). In THP1 cells from rheumatoid arthritis patients, LPS stimulation increases H3 and H4 acetylation at the CCL2 promoter, which can be reversed by TNF-α inhibitors such as etanercept and adalimumab (77). Besides. In SLE, hydroxychloroquine suppresses trained immunity by inhibiting H3K27ac and H3K4me3 modifications at inflammation-related genes (169).
Although less studied, DNA methylation also contributes to trained immunity. In human embryonic kidney cells, persistent TNF-α stimulation activates TET enzymes, which demethylate the CALCB enhancer and IL32 promoter, establishing a trained-like transcriptional state (170). In trained macrophages, long-term changes in gene expression are associated with altered DNA methylation patterns, particularly 5-methylcytosine (171). Whether modulating DNA demethylation can similarly correct inflammatory dysregulation remains an open question. Certain metabolites, including fumarate, α-ketoglutarate (α-KG), and succinate, regulate TET family demethylase activity, although direct DNA methylation changes have not been consistently observed in trained immunity models (50, 172). In conclusion, both histone modifications and DNA methylation serve as central regulatory mechanisms in trained immunity. Targeting these epigenetic pathways may offer a precise strategy to control excessive trained immune responses and alleviate inflammation-related diseases.
6.2 Based on metabolic reprogramming
Trained cells not only enhance gene expression via epigenetic remodeling but also reprogram metabolic pathways to meet elevated energy demands. In quiescent states, immune cells primarily rely on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) for energy. Upon activation, monocytes and macrophages shift toward glycolysis, lipid metabolism, and amino acid metabolism to support proinflammatory functions.
Glycolysis is intrinsically upregulated in the trained immunity process, independent of external stimuli (173). β-glucan-trained monocytes exhibit increased glucose uptake and lactate production, reflecting enhanced glycolytic flux (110). Similarly, BCG-trained monocytes show marked increases in glycolysis and glutaminolysis. Pharmacological or genetic inhibition of key glycolytic enzymes impairs trained immunity (109). The Akt/mTOR/HIF1α axis orchestrates this metabolic reprogramming by modulating HIF1α activity and glycolytic enzyme expression (174, 175). Similarly, inhibition of Akt/mTOR/HIF1α signaling suppresses β-glucan and BCG-induced trained immunity (109, 110). 2-DG, and metformin are established mTOR inhibitors. Studies using rapamycin-loaded high-density lipoprotein nanoparticles (mTORi-HDL) showed that they prevent post-transplant monocytes from developing trained macrophages upon exposure to alloantigens and HMGB1. mTORi-HDL accumulates in the graft and is taken up by myeloid cells; three intravenous injections reduce inflammatory cell infiltration, prolong graft survival, and suppress macrophage proinflammatory responses (176). A phase I trial determined that 2-DG combined with docetaxel at 63 mg/kg/day is clinically tolerable, showing manageable toxicity and modest antitumor activity (177). Experimental studies indicate that metformin activates Adenosine 5’-monophosphate (AMP)-activated protein kinase and/or inhibits mTORC1 via pathways including regulated in development and DNA damage responses 1 (178). Besides, mTOR activation occurs on lysosomal surfaces (179), where trained immunity is marked by lysosomal gene activation (155). Lysosomotropic agents like chloroquine and hydroxychloroquine impair lysosomal function and robustly inhibit trained immunity (169). The mTOR metabolic signaling pathway regulates HIF-1α activity. In HIF-1α knockout mice, the absence of HIF-1α signaling results in defective epigenetic programming and impaired production of proinflammatory cytokines (180). Succinate, a glycolytic intermediate, stabilizes HIF-1α by inhibiting its degradation, thereby sustaining chromatin accessibility. As an epigenetic modulator, succinate also suppresses histone and DNA methylation, supporting prolonged expression of glycolysis-related genes (109, 181). Collectively, the Akt/mTOR/HIF-1α axis coordinates metabolic and epigenetic reprogramming to drive and maintain trained immunity, representing a potential target for therapeutic intervention. In β-glucan–trained cells, the PPP is also upregulated. However, PPP inhibition has minimal impact on trained immunity, suggesting a limited role in this process (50). Several metabolism-targeting drugs are used not only in immune-related conditions such as cancer but also in inflammation research. 2-DG and dichloroacetic acid inhibit glycolysis and effectively block trained immunity induction in vitro (182). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a key glycolytic enzyme, also exerts immunomodulatory effects. Through metabolic reprogramming, GAPDH regulates trained macrophages and reduces allergen-induced airway inflammation in murine models of allergic asthma. Mechanistically, GAPDH promotes the transition of M2 macrophages to a proinflammatory M1 phenotype, thereby suppressing Th2 cell activation and exerting anti-inflammatory effects (183).
In addition to glycolysis, enhanced glutamine metabolism is a critical metabolic feature of trained immunity. Inhibition of glutaminase significantly suppresses β-glucan and BCG-induced trained immunity (50, 109). Both stimuli promote glutaminolysis through HIF-1α activation, increasing intracellular α-KG levels. α-KG enters the TCA cycle, supplying energy to support cell function (184). Supplementation with glutamine, arginine, and tryptophan further enhances immune responses in trained cells. These amino acids contribute to DNA synthesis, redox homeostasis, protein translation, and nitric oxide production, thereby improving innate immune cell function (185, 186).
Lipid metabolism also plays a vital role in trained immunity. β-glucan-induced training depends on mevalonate, a key intermediate in the cholesterol biosynthesis pathway. Monocytes from patients with HIDS, characterized by mevalonate accumulation, exhibit a trained immunity, like phenotype. Statins, which inhibit mevalonate production, effectively block β-glucan-induced training (50). Additionally, statins and liver X receptor antagonists suppress cholesterol biosynthesis and reduce mevalonate availability, thereby preventing mTOR-mediated trained immunity (96, 187). In addition, fluvastatin, an inhibitor of the rate-limiting enzyme HMG-CoA reductase, also attenuates β-glucan–induced training in vitro (184). In contrast, aldosterone promotes trained immunity by enhancing fatty acid synthesis, an effect that can be blocked by aldosterone receptor antagonists (188). Lipid signaling also regulates PPARs, SREBPs, mTOR, and various lipid mediators, modulating the metabolic state, inflammatory response, and functional output of trained immune cells (189–192). In summary, metabolic reprogramming in trained immunity involves the coordinated regulation of glucose, amino acid, and lipid metabolism. These pathways not only meet the energetic demands of immune activation and inflammation but also shape long-term gene expression through epigenetic mechanisms. Regulation of key signaling pathways such as HIF-1α, mTOR, and Akt, along with control over amino acid and lipid metabolism, provides a promising framework for developing therapeutic strategies against trained immunity–associated hyperinflammatory diseases.
6.3 Vaccines about trained immunity
Conventional vaccines primarily rely on the stimulation of antigen-specific T and B cells to elicit adaptive immune responses against specific pathogens. With growing insights into trained immunity, several classical vaccines have been shown to induce long-lasting functional reprogramming of innate immune cells, resulting in enhanced nonspecific protection upon subsequent infections. MV130, a sublingually administered inactivated polybacterial vaccine composed of 90% Gram-positive and 10% Gram-negative bacteria (193), rapidly induces DCs to secrete proinflammatory cytokines such as IL-6, TNF-α, and IL-1β. It also promotes Th1 and Th17 responses against both intracellular and extracellular pathogens, followed by IL-10 production to limit excessive inflammation (194). In a virus-induced asthma model, MV130 demonstrated nonspecific protection by enhancing neutrophil-mediated immunomodulation, thereby preventing asthma exacerbation during rhinovirus infection (195). A phase III randomized placebo-controlled trial involving 120 wheeze-prone children showed that daily sublingual administration of MV130 for six months significantly reduced wheezing episodes, symptoms, and medication use, with protective effects lasting another six months (196). In addition, allergen–mannan conjugate vaccines have been shown to reprogram monocytes into tolerogenic DCs via epigenetic and metabolic remodeling, supporting the induction of trained tolerance in allergen immunotherapy (197).
Vaccines typically consist of antigens and adjuvants. Due to the weak immunogenicity of antigens alone, adjuvants are essential to amplify and sustain immune responses (198). Many live-attenuated or inactivated vaccines act as self-adjuvants by stimulating PRRs. BCG is a classic example. BCG induces trained immunity via chromatin remodeling and metabolic rewiring, enhancing the functionality of monocytes and neutrophils. Upon secondary exposure to unrelated pathogens, trained cells exhibit increased cytokine production and antimicrobial capacity (199–201). A phase III clinical trial (NCT03296423) demonstrated that BCG vaccination enhances inflammatory cytokine secretion via epigenetic modulation, improving resistance to respiratory viral infections in the elderly. BCG also boosts influenza-specific antibody responses and modulates cytokine production in adults (202). Vaccination with AS03-adjuvanted influenza vaccine increases chromatin accessibility in innate immune cells and enhances interferon signaling, conferring broad antiviral resistance in vitro against dengue and Zika viruses. These findings suggest that AS03 functions not only as an adaptive immunity enhancer but also as a trained immunity inducer, offering broad-spectrum antiviral potential (203).
At the beginning of the COVID-19 pandemic in 2019, the absence of specific immunity and vaccines drew attention to BCG vaccination. It was considered a potential source of heterologous protection through cross-reactivity with viral antigens, nonspecific activation of lymphocytes, and enhancement of innate immunity via trained immunity. Animal studies have suggested that BCG reduces viral load and mitigated immunopathology in SARS-CoV-2 infection models (204). Unreviewed observational studies also reported lower COVID-19 mortality in countries with universal BCG vaccination, such as South Korea and Japan, compared with countries without such practice, including Italy and the United States (205). However, these associations are indirect and may be confounded by genetic, environmental, vaccine strain, and policy factors. Large-scale studies have generally shown that BCG does not prevent COVID-19 infection or severe disease, and some even indicated an increased risk of symptomatic COVID-19 among vaccine recipients (206–208). The role of BCG in COVID-19 protection remains uncertain, with several unresolved issues. For example, studies in children showed stronger effects when vaccination occurred before 9 months of age, but the influence of age at vaccination on COVID-19 protection is unknown (209). Trained monocytes gradually lose their enhanced cytokine responses, and the duration of heterologous immunity remains unclear (210). It is also unknown whether patients receiving intravesical BCG for bladder cancer gain protection against COVID-19 (211). Overall, the use of BCG in COVID-19 highlights the potential of trained immunity in broad antiviral defense, but faces challenges of inconsistent clinical evidence, uncertain durability, and limited vaccine resources. Other vaccines have also exhibited potential cross-protective effects. Influenza vaccination, by inducing trained immunity, was associated with reduced SARS-CoV-2 infection risk, particularly among healthcare workers and elderly populations (212). Additionally, varicella-zoster virus vaccines may also offer partial protection against COVID-19 (213).
Although trained immunity-based vaccines have not received the same level of global attention as other pandemic countermeasures, multiple clinical trials are ongoing to assess their potential benefits in the context of COVID-19. If proven effective, these vaccines could serve as a proof-of-concept for their use during future pandemics, especially as bridging strategies when pathogen-specific vaccines are unavailable or under development. Given their ability to induce trained immunity, vaccines and vaccine adjuvants should not be viewed solely as passive immunogenic agents. Instead, they represent potential tools for targeted activation of innate immune memory. Through rational antigen design and adjuvant optimization, it may be possible to strategically induce trained immune states to support infection control, immune modulation, and even chronic disease intervention.
6.4 Others
Beyond epigenetic and metabolic regulation, advances in nanomedicine have opened new avenues for modulating trained immunity. Nanomaterials not only enable efficient delivery of immunoregulatory agents but can also be engineered to target the bone marrow and myeloid precursors, thereby inducing or suppressing trained immune responses. For example, mTORi-HDL suppress trained immunity in myeloid cells and promote the expansion of anti-inflammatory Ly6C- macrophages, alleviating graft rejection and prolonging graft survival (214). Combined delivery of CD40 co-stimulation inhibitors using TRAF6i-HDL nanoparticles further disrupts both trained immunity and co-stimulatory pathways in myeloid cells, significantly improving transplant acceptance and inducing long-term immune tolerance (176). In addition, bone marrow–targeted nanomaterials engineered for spleen or inflammation site accumulation (e.g., tumors or atherosclerotic plaques) have demonstrated therapeutic potential. These particles either induce monocyte training in the spleen or reprogram myeloid cells at disease sites toward anti-inflammatory phenotypes (215). Although hepatic accumulation of certain nanomaterials may cause adverse effects, targeted delivery to the spleen and inflammatory foci offers promising immunomodulatory and anti-inflammatory benefits (216). In summary, chemical functionalization of nanomaterials allows the modulation of epigenetic and metabolic programs in myeloid progenitors, enabling the induction or inhibition of trained immunity. This strategy holds therapeutic potential for enhancing antimicrobial defense or suppressing maladaptive immune responses in diseases such as atherosclerosis and rheumatoid arthritis. Apart from nanotechnology, molecular targeting therapies are also being explored. IL-1β and GM-CSF are key mediators of trained immunity (180). Monoclonal antibodies against IL-1β have shown efficacy in inflammation-related cardiovascular diseases, reducing thrombotic events and attenuating trained immune responses following myocardial infarction (117). Anti–GM-CSF antibodies are currently under clinical investigation for the treatment of rheumatoid arthritis and related conditions (217). Moreover, RNA interference offers a novel approach to block trained immunity, associated signaling pathways. However, its clinical translation remains in early stages (218).
7 Conclusion
Trained immunity, as a novel mechanism of immune regulation based on epigenetic memory in innate immune cells, offers a new framework for understanding the pathophysiology of infectious responses and immune-mediated diseases. Although numerous preclinical and animal studies have provided evidence linking trained immunity to inflammatory diseases, several critical issues remain unresolved. First, more animal models and clinical data are needed to clarify the actual occurrence and mechanistic roles of trained immunity in various inflammatory disorders. Second, strategies to modulate trained immunity must be developed to prevent pathological inflammation caused by excessive stimulation, requiring intervention at the earliest stages of external exposure. Third, the reversible epigenetic modifications and short-lived memory effects of trained immunity offer promising intervention points; however, how to balance therapeutic utilization with the potential risks remains an important research focus. Lastly, identifying ways to integrate innate immune memory with adaptive immune memory may enable the development of more precise and effective therapeutic strategies by harnessing their complementary strengths. To date, the role of trained immunity has been increasingly recognized in SLE, allergic diseases, ASCVD, and MS. However, further studies are needed to elucidate the underlying mechanisms by which trained immunity contributes to disease activity and to determine whether inhibition of trained immunity can prevent disease exacerbation. In contrast, understanding of trained immunity in RA, AD, and sarcoidosis remains limited, although dysregulated metabolic and epigenetic pathways suggest a potential role. Other inflammatory diseases also warrant deeper investigation in this context.
In the future, integrating conventional treatment approaches with emerging immunomodulatory strategies may open new therapeutic avenues for managing complex inflammatory diseases.
Author contributions
WLX: Conceptualization, Data curation, Investigation, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. ZG: Conceptualization, Investigation, Visualization, Writing – review & editing. TX: Investigation, Writing – review & editing. JC: Investigation, Visualization, Writing – review & editing. LC: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. W-AX: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Shenzhen Medical Research Fund (A2302043) and Shenzhen Science and Technology Program (JCYJ20240813170459001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ochando J, Mulder WJM, Madsen JC, Netea MG, and Duivenvoorden R. Trained immunity - basic concepts and contributions to immunopathology. Nat Rev Nephrol. (2023) 19:23–37. doi: 10.1038/s41581-022-00633-5
2. Pradeu T, Thomma B, Girardin SE, and Lemaitre B. The conceptual foundations of innate immunity: Taking stock 30 years later. Immunity. (2024) 57:613–31. doi: 10.1016/j.immuni.2024.03.007
3. Carpenter S and O’Neill LAJ. From periphery to center stage: 50 years of advancements in innate immunity. Cell. (2024) 187:2030–51. doi: 10.1016/j.cell.2024.03.036
4. Chi H, Pepper M, and Thomas PG. Principles and therapeutic applications of adaptive immunity. Cell. (2024) 187:2052–78. doi: 10.1016/j.cell.2024.03.037
5. Netea MG, Quintin J, and van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
6. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. (2017) 550:475–80. doi: 10.1038/nature24271
7. Vuscan P, Kischkel B, Joosten LAB, and Netea MG. Trained immunity: General and emerging concepts. Immunol Rev. (2024) 323:164–85. doi: 10.1111/imr.13326
8. Nankabirwa V, Tumwine JK, Mugaba PM, Tylleskär T, and Sommerfelt H. Child survival and BCG vaccination: a community based prospective cohort study in Uganda. BMC Public Health. (2015) 15:175. doi: 10.1186/s12889-015-1497-8
9. Lim AI, McFadden T, Link VM, Han SJ, Karlsson RM, Stacy A, et al. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science. (2021) 373:eabf3002. doi: 10.1126/science.abf3002
10. Dominguez-Andres J and Netea MG. Long-term reprogramming of the innate immune system. J Leukoc Biol. (2019) 105:329–38. doi: 10.1002/jlb.Mr0318-104r
11. Kurtz J and Franz K. Innate defence: evidence for memory in invertebrate immunity. Nature. (2003) 425:37–8. doi: 10.1038/425037a
12. Durrant WE and Dong X. Systemic acquired resistance. Annu Rev Phytopathol. (2004) 42:185–209. doi: 10.1146/annurev.phyto.42.040803.140421
13. Lemaitre B, Reichhart JM, and Hoffmann JA. Drosophila host defense: differential induction of antimicrobial peptide genes after infection by various classes of microorganisms. Proc Natl Acad Sci U S A. (1997) 94:14614–9. doi: 10.1073/pnas.94.26.14614
14. Pham LN, Dionne MS, Shirasu-Hiza M, and Schneider DS. A specific primed immune response in Drosophila is dependent on phagocytes. PloS Pathog. (2007) 3:e26. doi: 10.1371/journal.ppat.0030026
15. Rodrigues J, Brayner FA, Alves LC, Dixit R, and Barillas-Mury C. Hemocyte differentiation mediates innate immune memory in Anopheles Gambiae mosquitoes. Science. (2010) 329:1353–5. doi: 10.1126/science.1190689
16. Little TJ, O’Connor B, Colegrave N, Watt K, and Read AF. Maternal transfer of strain-specific immunity in an invertebrate. Curr Biol. (2003) 13:489–92. doi: 10.1016/s0960-9822(03)00163-5
17. Hildemann WH, Raison RL, Cheung G, Hull CJ, Akaka L, and Okamoto J. Immunological specificity and memory in a scleractinian coral. Nature. (1977) 270:219–23. doi: 10.1038/270219a0
18. Moret Y and Siva-Jothy MT. Adaptive innate immunity? Responsive-mode prophylaxis in the mealworm beetle, Tenebrio molitor. Proc Biol Sci. (2003) 270:2475–80. doi: 10.1098/rspb.2003.2511
19. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
20. Tran V, Liu J, and Behr MA. BCG vaccines. Microbiol Spectr. (2014) 2:Mgm2–0028-2013. doi: 10.1128/microbiolspec.MGM2-0028-2013
21. Blanden RV, Lefford MJ, and Mackaness GB. The host response to Calmette-Guérin bacillus infection in mice. J Exp Med. (1969) 129:1079–107. doi: 10.1084/jem.129.5.1079
22. Aaby P and Benn CS. Saving lives by training innate immunity with bacille Calmette-Guerin vaccine. Proc Natl Acad Sci U S A. (2012) 109:17317–8. doi: 10.1073/pnas.1215761109
23. Kang A, Ye G, Afkhami S, Aleithan F, Singh K, Dvorkin-Gheva A, et al. LPS-induced lung tissue-resident trained innate immunity provides differential protection against pneumococci and SARS-CoV-2. Cell Rep. (2024) 43:114849. doi: 10.1016/j.celrep.2024.114849
24. Ferreira AV, Koeken V, Matzaraki V, Kostidis S, Alarcon-Barrera JC, de Bree LCJ, et al. Glutathione metabolism contributes to the induction of trained immunity. Cells. (2021) 10:971. doi: 10.3390/cells10050971
25. Sun JC, Beilke JN, and Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
26. Murphy DM, Mills KHG, and Basdeo SA. The effects of trained innate immunity on T cell responses; clinical implications and knowledge gaps for future research. Front Immunol. (2021) 12:706583. doi: 10.3389/fimmu.2021.706583
27. Larsen SB, Cowley CJ, Sajjath SM, Barrows D, Yang Y, Carroll TS, et al. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell. (2021) 28:1758–74.e8. doi: 10.1016/j.stem.2021.07.001
28. Chevalier C, Chica C, Matheau J, Pain A, Connor MG, and Hamon MA. Epithelial cells maintain memory of prior infection with Streptococcus pneumoniae through di-methylation of histone H3. Nat Commun. (2024) 15:5545. doi: 10.1038/s41467-024-49347-1
29. Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. (2018) 560:649–54. doi: 10.1038/s41586-018-0449-8
30. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. (2014) 510:393–6. doi: 10.1038/nature13255
31. Del Poggetto E, Ho IL, Balestrieri C, Yen EY, Zhang S, Citron F, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science. (2021) 373:eabj0486. doi: 10.1126/science.abj0486
32. Marakalala MJ, Williams DL, Hoving JC, Engstad R, Netea MG, and Brown GD. Dectin-1 plays a redundant role in the immunomodulatory activities of β-glucan-rich ligands in vivo. Microbes Infect. (2013) 15:511–5. doi: 10.1016/j.micinf.2013.03.002
33. Di Luzio NR and Williams DL. Protective effect of glucan against systemic Staphylococcus aureus septicemia in normal and leukemic mice. Infect Immun. (1978) 20:804–10. doi: 10.1128/iai.20.3.804-810.1978
34. Ribes S, Meister T, Ott M, Redlich S, Janova H, Hanisch UK, et al. Intraperitoneal prophylaxis with CpG oligodeoxynucleotides protects neutropenic mice against intracerebral Escherichia coli K1 infection. J Neuroinflamm. (2014) 11:14. doi: 10.1186/1742-2094-11-14
35. Zhang H, Chen T, Ren J, Xia Y, Onuma A, Wang Y, et al. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nat Metab. (2021) 3:843–58. doi: 10.1038/s42255-021-00402-x
36. Murugathasan M, Jafari A, Amandeep A, Hassan SA, Chihata M, and Abdul-Sater AA. Moderate exercise induces trained immunity in macrophages. Am J Physiol Cell Physiol. (2023) 325:C429–c42. doi: 10.1152/ajpcell.00130.2023
37. Gonzales KAU, Polak L, Matos I, Tierney MT, Gola A, Wong E, et al. Stem cells expand potency and alter tissue fitness by accumulating diverse epigenetic memories. Sci (New York NY). (2021) 374:eabh2444. doi: 10.1126/science.abh2444
38. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
39. Fu Y, Li Y, Xu L, Liu S, Wang M, Xiao L, et al. Immunology repertoire study of pulmonary sarcoidosis T cells in CD4+, CD8+ PBMC and tissue. Oncotarget. (2017) 8:89515–26. doi: 10.18632/oncotarget.20085
40. Drent M, Crouser ED, and Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med. (2021) 385:1018–32. doi: 10.1056/NEJMra2101555
41. Bonham CA. Biomarkers in sarcoidosis: can microRNAs fill the gap? Am J Respir Cell Mol Biol. (2018) 58:1–2. doi: 10.1165/rcmb.2017-0344ED
42. Krausgruber T, Redl A, Barreca D, Doberer K, Romanovskaia D, Dobnikar L, et al. Single-cell and spatial transcriptomics reveal aberrant lymphoid developmental programs driving granuloma formation. Immunity. (2023) 56:289–306.e7. doi: 10.1016/j.immuni.2023.01.014
43. Nakamizo S, Sugiura Y, Ishida Y, Ueki Y, Yonekura S, Tanizaki H, et al. Activation of the pentose phosphate pathway in macrophages is crucial for granuloma formation in sarcoidosis. J Clin Invest. (2023) 133:e171088. doi: 10.1172/jci171088
44. Terčelj M, Stopinšek S, Ihan A, Salobir B, Simčič S, Wraber B, et al. In vitro and in vivo reactivity to fungal cell wall agents in sarcoidosis. Clin Exp Immunol. (2011) 166:87–93. doi: 10.1111/j.1365-2249.2011.04456.x
45. Garman L, Pelikan RC, Rasmussen A, Lareau CA, Savoy KA, Deshmukh US, et al. Single cell transcriptomics implicate novel monocyte and T cell immune dysregulation in sarcoidosis. Front Immunol. (2020) 11:567342. doi: 10.3389/fimmu.2020.567342
46. Bueno-Beti C, Lim CX, Protonotarios A, Szabo PL, Westaby J, Mazic M, et al. An mTORC1-dependent mouse model for cardiac sarcoidosis. J Am Heart Assoc. (2023) 12:e030478. doi: 10.1161/jaha.123.030478
47. Linke M, Pham HT, Katholnig K, Schnöller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. (2017) 18:293–302. doi: 10.1038/ni.3655
48. Pacheco Y, Lim CX, Weichhart T, Valeyre D, Bentaher A, and Calender A. Sarcoidosis and the mTOR, rac1, and autophagy triad. Trends Immunol. (2020) 41:286–99. doi: 10.1016/j.it.2020.01.007
49. Jeny F, Bernaudin JF, Valeyre D, Kambouchner M, Pretolani M, Nunes H, et al. Hypoxia promotes a mixed inflammatory-fibrotic macrophages phenotype in active sarcoidosis. Front Immunol. (2021) 12:719009. doi: 10.3389/fimmu.2021.719009
50. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
51. Kan RL, Chen J, and Sallam T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. (2022) 38:182–93. doi: 10.1016/j.tig.2021.06.014
52. Yang IV, Konigsberg I, MacPhail K, Li L, Davidson EJ, Mroz PM, et al. DNA methylation changes in lung immune cells are associated with granulomatous lung disease. Am J Respir Cell Mol Biol. (2019) 60:96–105. doi: 10.1165/rcmb.2018-0177OC
54. Maggi P, Bulcke CV, Pedrini E, Bugli C, Sellimi A, Wynen M, et al. B cell depletion therapy does not resolve chronic active multiple sclerosis lesions. EBioMedicine. (2023) 94:104701. doi: 10.1016/j.ebiom.2023.104701
55. de Sèze J, Maillart E, Gueguen A, Laplaud DA, Michel L, Thouvenot E, et al. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front Immunol. (2023) 14:1004795. doi: 10.3389/fimmu.2023.1004795
56. Carlson AK, Amin M, and Cohen JA. Drugs targeting CD20 in multiple sclerosis: pharmacology, efficacy, safety, and tolerability. Drugs. (2024) 84:285–304. doi: 10.1007/s40265-024-02011-w
57. Chisari CG, Sgarlata E, Arena S, Toscano S, Luca M, and Patti F. Rituximab for the treatment of multiple sclerosis: a review. J Neurol. (2022) 269:159–83. doi: 10.1007/s00415-020-10362-z
58. Pavelek Z, Angelucci F, Souček O, Krejsek J, Sobíšek L, Klímová B, et al. Innate immune system and multiple sclerosis. Granulocyte numbers are reduced in patients affected by relapsing-remitting multiple sclerosis during the remission phase. J Clin Med. (2020) 9:1468. doi: 10.3390/jcm9051468
59. Fransson J, Bachelin C, Ichou F, Guillot-Noël L, Ponnaiah M, Gloaguen A, et al. Multiple sclerosis patient macrophages impaired metabolism leads to an altered response to activation stimuli. Neurol Neuroimmunol Neuroinflamm. (2024) 11:e200312. doi: 10.1212/nxi.0000000000200312
60. Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat Neurosci. (2014) 17:121–30. doi: 10.1038/nn.3588
61. Calabrese R, Zampieri M, Mechelli R, Annibali V, Guastafierro T, Ciccarone F, et al. Methylation-dependent PAD2 upregulation in multiple sclerosis peripheral blood. Mult Scler. (2012) 18:299–304. doi: 10.1177/1352458511421055
62. Iridoy Zulet M, Pulido Fontes L, Ayuso Blanco T, Lacruz Bescos F, and Mendioroz Iriarte M. Epigenetic changes in neurology: DNA methylation in multiple sclerosis. Neurologia. (2017) 32:463–8. doi: 10.1016/j.nrl.2015.03.011
63. Sharma P, Azebi S, England P, Christensen T, Møller-Larsen A, Petersen T, et al. Citrullination of histone H3 interferes with HP1-mediated transcriptional repression. PloS Genet. (2012) 8:e1002934. doi: 10.1371/journal.pgen.1002934
64. Pedre X, Mastronardi F, Bruck W, López-Rodas G, Kuhlmann T, and Casaccia P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J Neurosci. (2011) 31:3435–45. doi: 10.1523/jneurosci.4507-10.2011
65. Misra DP. Clinical manifestations of rheumatoid arthritis, including comorbidities, complications, and long-term follow-up. Best Pract Res Clin Rheumatol. (2025) 39:102020. doi: 10.1016/j.berh.2024.102020
66. Yuan Y, Mu N, Li Y, Gu J, Chu C, Yu X, et al. TNF-α Promotes synovial inflammation and cartilage bone destruction in rheumatoid arthritis via NF-κB/YY1/miR-103a-3p axis. FASEB J. (2025) 39:e70876. doi: 10.1096/fj.202501452R
67. Xia X, He C, Xue Z, Wang Y, Qin Y, Ren Z, et al. Single cell immunoprofile of synovial fluid in rheumatoid arthritis with TNF/JAK inhibitor treatment. Nat Commun. (2025) 16:2152. doi: 10.1038/s41467-025-57361-0
68. Gerlag DM, Safy M, Maijer KI, Tang MW, Tas SW, Starmans-Kool MJF, et al. Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: the PRAIRI study. Ann Rheum Dis. (2019) 78:179–85. doi: 10.1136/annrheumdis-2017-212763
69. Lioté F, Boval-Boizard B, Weill D, Kuntz D, and Wautier JL. Blood monocyte activation in rheumatoid arthritis: increased monocyte adhesiveness, integrin expression, and cytokine release. Clin Exp Immunol. (1996) 106:13–9. doi: 10.1046/j.1365-2249.1996.d01-820.x
70. Yokota K, Sato K, Miyazaki T, Aizaki Y, Tanaka S, Sekikawa M, et al. Characterization and function of tumor necrosis factor and interleukin-6-induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol. (2021) 73:1145–54. doi: 10.1002/art.41666
71. Shiratori T, Kyumoto-Nakamura Y, Kukita A, Uehara N, Zhang J, Koda K, et al. IL-1β Induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: A possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of kindlin-3 and talin-1. J Immunol. (2018) 200:218–28. doi: 10.4049/jimmunol.1602035
72. Lu Y, Parker N, Kleindl PJ, Cross VA, Wollak K, Westrick E, et al. Antiinflammatory activity of a novel folic acid targeted conjugate of the mTOR inhibitor everolimus. Mol Med. (2015) 21:584–96. doi: 10.2119/molmed.2015.00040
73. Asadipour M, Hassan-Zadeh V, Aryaeian N, Shahram F, and Mahmoudi M. Histone variants expression in peripheral blood mononuclear cells of patients with rheumatoid arthritis. Int J Rheum Dis. (2018) 21:1831–7. doi: 10.1111/1756-185x.13126
74. Calo E and Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. (2013) 49:825–37. doi: 10.1016/j.molcel.2013.01.038
75. Liu CC, Fang TJ, Ou TT, Wu CC, Li RN, Lin YC, et al. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol Lett. (2011) 135:96–9. doi: 10.1016/j.imlet.2010.10.003
76. Nakano K, Whitaker JW, Boyle DL, Wang W, and Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. (2013) 72:110–7. doi: 10.1136/annrheumdis-2012-201526
77. Lin YC, Lin YC, Huang MY, Kuo PL, Wu CC, Lee MS, et al. Tumor necrosis factor-alpha inhibitors suppress CCL2 chemokine in monocytes via epigenetic modification. Mol Immunol. (2017) 83:82–91. doi: 10.1016/j.molimm.2017.01.009
78. Wang X, Pan L, Niu D, Zhou J, Shen M, Zeng Z, et al. Jingfang Granules alleviates the lipid peroxidation induced ferroptosis in rheumatoid arthritis rats by regulating gut microbiota and metabolism of short chain fatty acids. J Ethnopharmacol. (2025) 339:119160. doi: 10.1016/j.jep.2024.119160
79. Huang Y, Yue S, Qiao J, Dong Y, Liu Y, Zhang M, et al. Identification of diagnostic genes and drug prediction in metabolic syndrome-associated rheumatoid arthritis by integrated bioinformatics analysis, machine learning, and molecular docking. Front Immunol. (2024) 15:1431452. doi: 10.3389/fimmu.2024.1431452
80. Gong X, Su L, Huang J, Liu J, Wang Q, Luo X, et al. An overview of multi-omics technologies in rheumatoid arthritis: applications in biomarker and pathway discovery. Front Immunol. (2024) 15:1381272. doi: 10.3389/fimmu.2024.1381272
81. Mayboroda OA, Lageveen-Kammeijer GSM, Wuhrer M, and Dolhain R. An integrated glycosylation signature of rheumatoid arthritis. Biomolecules. (2023) 13:1106. doi: 10.3390/biom13071106
82. Li X, Wang H, Yu X, Saha G, Kalafati L, Ioannidis C, et al. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell. (2022) 185:1709–27.e18. doi: 10.1016/j.cell.2022.03.043
83. Nandakumar KS and Nündel K. Editorial: Systemic lupus erythematosus - predisposition factors, pathogenesis, diagnosis, treatment and disease models. Front Immunol. (2022) 13:1118180. doi: 10.3389/fimmu.2022.1118180
84. Parodis I, Lindblom J, Levy RA, Zen M, Cetrez N, Gomez A, et al. Attainment of remission and low disease activity after treatment with belimumab in patients with systemic lupus erythematosus: a post-hoc analysis of pooled data from five randomised clinical trials. Lancet Rheumatol. (2024) 6:e751–e61. doi: 10.1016/s2665-9913(24)00162-0
85. Arnaud L, Chasset F, and Martin T. Immunopathogenesis of systemic lupus erythematosus: An update. Autoimmun Rev. (2024) 23:103648. doi: 10.1016/j.autrev.2024.103648
86. Frank MM, Hamburger MI, Lawley TJ, Kimberly RP, and Plotz PH. Defective reticuloendothelial system Fc-receptor function in systemic lupus erythematosus. N Engl J Med. (1979) 300:518–23. doi: 10.1056/nejm197903083001002
87. Hill GS, Delahousse M, Nochy D, Rémy P, Mignon F, Méry JP, et al. Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int. (2001) 59:304–16. doi: 10.1046/j.1523-1755.2001.00492.x
88. Yanginlar C, Rother N, Post T, Jacobs M, Jonkman I, Brouns M, et al. Trained innate immunity in response to nuclear antigens in systemic lupus erythematosus. J Autoimmun. (2024) 149:103335. doi: 10.1016/j.jaut.2024.103335
89. Mills TS, Kain B, Burchill MA, Danis E, Lucas ED, Culp-Hill R, et al. A distinct metabolic and epigenetic state drives trained immunity in HSC-derived macrophages from autoimmune mice. Cell Stem Cell. (2024) 31:1630–49.e8. doi: 10.1016/j.stem.2024.09.010
90. Zervopoulou E, Grigoriou M, Doumas SA, Yiannakou D, Pavlidis P, Gasparoni G, et al. Enhanced medullary and extramedullary granulopoiesis sustain the inflammatory response in lupus nephritis. Lupus Sci Med. (2024) 11:1106. doi: 10.1136/lupus-2023-001110
91. Grigoriou M, Banos A, Filia A, Pavlidis P, Giannouli S, Karali V, et al. Transcriptome reprogramming and myeloid skewing in haematopoietic stem and progenitor cells in systemic lupus erythematosus. Ann Rheum Dis. (2020) 79:242–53. doi: 10.1136/annrheumdis-2019-215782
92. Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, and van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol. (2015) 179:68–74. doi: 10.1111/cei.12359
93. Zhou Y, Qiu X, Luo Y, Yuan J, Li Y, Zhong Q, et al. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus. (2011) 20:1365–71. doi: 10.1177/0961203311413412
94. Zhan Y, Guo Y, and Lu Q. Aberrant epigenetic regulation in the pathogenesis of systemic lupus erythematosus and its implication in precision medicine. Cytogenet Genome Res. (2016) 149:141–55. doi: 10.1159/000448793
95. Armstrong AW and Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. Jama. (2020) 323:1945–60. doi: 10.1001/jama.2020.4006
96. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46.e9. doi: 10.1016/j.cell.2017.11.025
97. Silva R. Hyper-igD syndrome: caused by deficiency on ras prenylation and trained immunity? J Clin Immunol. (2023) 43:1740–2. doi: 10.1007/s10875-023-01548-x
98. Moore KJ, Sheedy FJ, and Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
99. Christ A, Bekkering S, Latz E, and Riksen NP. Long-term activation of the innate immune system in atherosclerosis. Semin Immunol. (2016) 28:384–93. doi: 10.1016/j.smim.2016.04.004
100. Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun. (2016) 7:13436. doi: 10.1038/ncomms13436
101. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/circulationaha.116.020838
102. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, and Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/atvbaha.114.303887
103. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
104. van Kampen E, Jaminon A, van Berkel TJ, and Van Eck M. Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis. J Leukoc Biol. (2014) 96:833–41. doi: 10.1189/jlb.1A0114-017R
105. Seijkens T, Hoeksema MA, Beckers L, Smeets E, Meiler S, Levels J, et al. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. (2014) 28:2202–13. doi: 10.1096/fj.13-243105
106. Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. (2006) 48:1818–24. doi: 10.1016/j.jacc.2006.05.076
107. Liu J, Kerwin WS, Caldwell JH, Ferguson MS, Hippe DS, Alessio AM, et al. High resolution FDG-microPET of carotid atherosclerosis: plaque components underlying enhanced FDG uptake. Int J Cardiovasc Imaging. (2016) 32:145–52. doi: 10.1007/s10554-015-0739-2
108. Tomas L, Edsfeldt A, Mollet IG, Perisic Matic L, Prehn C, Adamski J, et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur Heart J. (2018) 39:2301–10. doi: 10.1093/eurheartj/ehy124
109. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
110. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
111. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
112. Li SL, Reddy MA, Cai Q, Meng L, Yuan H, Lanting L, et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes. (2006) 55:2611–9. doi: 10.2337/db06-0164
113. Schnack L, Sohrabi Y, Lagache SMM, Kahles F, Bruemmer D, Waltenberger J, et al. Mechanisms of trained innate immunity in oxLDL primed human coronary smooth muscle cells. Front Immunol. (2019) 10:13. doi: 10.3389/fimmu.2019.00013
114. Li X, Wang L, Fang P, Sun Y, Jiang X, Wang H, et al. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J Biol Chem. (2018) 293:11033–45. doi: 10.1074/jbc.RA118.002752
115. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. (2008) 205:2409–17. doi: 10.1084/jem.20081188
116. Pedro-Botet J, Climent E, and Benaiges D. Atherosclerosis and inflammation. New therapeutic approaches. Med Clin (Barc). (2020) 155:256–62. doi: 10.1016/j.medcli.2020.04.024
117. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
118. Rostagno AA. Pathogenesis of alzheimer’s disease. Int J Mol Sci. (2022) 24:107. doi: 10.3390/ijms24010107
119. Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. (2016) 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
120. Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in early symptomatic alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. Jama. (2023) 330:512–27. doi: 10.1001/jama.2023.13239
121. Self WK and Holtzman DM. Emerging diagnostics and therapeutics for Alzheimer disease. Nat Med. (2023) 29:2187–99. doi: 10.1038/s41591-023-02505-2
122. Sarlus H and Heneka MT. Microglia in alzheimer’s disease. J Clin Invest. (2017) 127:3240–9. doi: 10.1172/jci90606
123. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
124. Kitazawa M, Oddo S, Yamasaki TR, Green KN, and LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. (2005) 25:8843–53. doi: 10.1523/jneurosci.2868-05.2005
125. Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, et al. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. (1998) 4:480–9. doi: 10.1007/BF03401753
126. Bossù P, Spalletta G, Caltagirone C, and Ciaramella A. Myeloid dendritic cells are potential players in human neurodegenerative diseases. Front Immunol. (2015) 6:632. doi: 10.3389/fimmu.2015.00632
127. Ciaramella A, Salani F, Bizzoni F, Orfei MD, Caltagirone C, Spalletta G, et al. Myeloid dendritic cells are decreased in peripheral blood of Alzheimer’s disease patients in association with disease progression and severity of depressive symptoms. J Neuroinflamm. (2016) 13:18. doi: 10.1186/s12974-016-0483-0
128. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
129. Liu X, Jiao B, and Shen L. The epigenetics of alzheimer’s disease: factors and therapeutic implications. Front Genet. (2018) 9:579. doi: 10.3389/fgene.2018.00579
130. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. (2010) 464:1201–4. doi: 10.1038/nature08890
131. Iovino M, Agathou S, González-Rueda A, Del Castillo Velasco-Herrera M, Borroni B, Alberici A, et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain. (2015) 138:3345–59. doi: 10.1093/brain/awv222
132. Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. (2011) 2:252. doi: 10.1038/ncomms1255
133. Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr., Bennett DA, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. (2009) 68:48–58. doi: 10.1097/NEN.0b013e3181922348
134. Mastroeni D, Delvaux E, Nolz J, Tan Y, Grover A, Oddo S, et al. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol Aging. (2015) 36:3121–9. doi: 10.1016/j.neurobiolaging.2015.08.017
135. Gimeno-Bayón J, López-López A, Rodríguez MJ, and Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res. (2014) 92:723–31. doi: 10.1002/jnr.23356
136. Yang F, Zhao D, Cheng M, Liu Y, Chen Z, Chang J, et al. mTOR-mediated immunometabolic reprogramming nanomodulators enable sensitive switching of energy deprivation-induced microglial polarization for alzheimer’s disease management. ACS Nano. (2023) 17:15724–41. doi: 10.1021/acsnano.3c03232
137. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in alzheimer’s disease. Cell. (2017) 170:649–63.e13. doi: 10.1016/j.cell.2017.07.023
138. Meyer RD and Finch RG. Community-acquired pneumonia. J Hosp Infect. (1992) 22 Suppl:A:51–9. doi: 10.1016/s0195-6701(05)80007-6
139. Wang T, Zhang J, Wang Y, Li Y, Wang L, Yu Y, et al. Influenza-trained mucosal-resident alveolar macrophages confer long-term antitumor immunity in the lungs. Nat Immunol. (2023) 24:423–38. doi: 10.1038/s41590-023-01428-x
140. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/s0140-6736(20)30183-5
141. Radhouani M, Farhat A, Hakobyan A, Zahalka S, Pimenov L, Fokina A, et al. Eosinophil innate immune memory after bacterial skin infection promotes allergic lung inflammation. Sci Immunol. (2025) 10:eadp6231. doi: 10.1126/sciimmunol.adp6231
142. Robles-Vera I, Jarit-Cabanillas A, Brandi P, Martínez-López M, Martínez-Cano S, Rodrigo-Tapias M, et al. Microbiota translocation following intestinal barrier disruption promotes Mincle-mediated training of myeloid progenitors in the bone marrow. Immunity. (2025) 58:381–96.e9. doi: 10.1016/j.immuni.2024.12.012
143. Boyle RJ and Shamji MH. Developments in the field of allergy in 2020 through the eyes of Clinical and Experimental Allergy. Clin Exp Allergy. (2021) 51:1531–7. doi: 10.1111/cea.14046
144. Zhou X, Simonin EM, Jung YS, Galli SJ, and Nadeau KC. Role of allergen immunotherapy and biologics in allergic diseases. Curr Opin Immunol. (2024) 91:102494. doi: 10.1016/j.coi.2024.102494
145. Rajput C, Han M, Ishikawa T, Lei J, Jazaeri S, Bentley JK, et al. Early-life heterologous rhinovirus infections induce an exaggerated asthma-like phenotype. J Allergy Clin Immunol. (2020) 146:571–82.e3. doi: 10.1016/j.jaci.2020.03.039
146. Lechner A, Henkel FDR, Hartung F, Bohnacker S, Alessandrini F, Gubernatorova EO, et al. Macrophages acquire a TNF-dependent inflammatory memory in allergic asthma. J Allergy Clin Immunol. (2022) 149:2078–90. doi: 10.1016/j.jaci.2021.11.026
147. Li Z, Feng Y, Liu H, Yang H, Li J, Zhou Y, et al. Single-cell transcriptomics of mouse lung reveal inflammatory memory neutrophils in allergic asthma. Allergy. (2022) 77:1911–5. doi: 10.1111/all.15286
148. Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. (2017) 18:1310–20. doi: 10.1038/ni.3857
149. Naik S and Fuchs E. Inflammatory memory and tissue adaptation in sickness and in health. Nature. (2022) 607:249–55. doi: 10.1038/s41586-022-04919-3
150. Neeland MR, Andorf S, Dang TD, McWilliam VL, Perrett KP, Koplin JJ, et al. Altered immune cell profiles and impaired CD4 T-cell activation in single and multi-food allergic adolescents. Clin Exp Allergy. (2021) 51:674–84. doi: 10.1111/cea.13857
151. Badia IMP, Wessels L, Müller-Dott S, Trimbour R, Ramirez Flores RO, Argelaguet R, et al. Gene regulatory network inference in the era of single-cell multi-omics. Nat Rev Genet. (2023) 24:739–54. doi: 10.1038/s41576-023-00618-5
152. Gialitakis M, Arampatzi P, Makatounakis T, and Papamatheakis J. Gamma interferon-dependent transcriptional memory via relocalization of a gene locus to PML nuclear bodies. Mol Cell Biol. (2010) 30:2046–56. doi: 10.1128/MCB.00906-09
153. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
154. Zhou Z, van der Jeught K, Li Y, Sharma S, Yu T, Moulana I, et al. A T cell-engaging tumor organoid platform for pancreatic cancer immunotherapy. Adv Sci (Weinh). (2023) 10:e2300548. doi: 10.1002/advs.202300548
155. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167:1354–68.e14. doi: 10.1016/j.cell.2016.09.034
156. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
157. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe. (2018) 23:89–100.e5. doi: 10.1016/j.chom.2017.12.010
158. Kehraus S, Gorzalka S, Hallmen C, Iqbal J, Müller CE, Wright AD, et al. Novel amino acid derived natural products from the ascidian Atriolum robustum: identification and pharmacological characterization of a unique adenosine derivative. J Med Chem. (2004) 47:2243–55. doi: 10.1021/jm031092g
159. Huang S. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. (2002) 2:469–76. doi: 10.1038/nrc819
160. He K, Cao X, and Deng X. Histone methylation in epigenetic regulation and temperature responses. Curr Opin Plant Biol. (2021) 61:102001. doi: 10.1016/j.pbi.2021.102001
161. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. (2013) 4:2035. doi: 10.1038/ncomms3035
162. Lee J, Kim JS, Cho HI, Jo SR, and Jang YK. JIB-04, a pan-inhibitor of histone demethylases, targets histone-lysine-demethylase-dependent AKT pathway, leading to cell cycle arrest and inhibition of cancer stem-like cell properties in hepatocellular carcinoma cells. Int J Mol Sci. (2022) 23:7657. doi: 10.3390/ijms23147657
163. Shvedunova M and Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. (2022) 23:329–49. doi: 10.1038/s41580-021-00441-y
164. Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. (2007) 13:2318–22. doi: 10.1158/1078-0432.Ccr-06-2672
165. Mann BS, Johnson JR, Cohen MH, Justice R, and Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. (2007) 12:1247–52. doi: 10.1634/theoncologist.12-10-1247
166. Rambaldi A, Finazzi G, Vannucchi AM, Martinelli V, Rodeghiero F, Nobile F, et al. A phase II study of the HDAC inhibitor givinostat in combination with hydroxyurea in patients with polycythemia vera resistant to hydroxyurea monotherapy. Blood. (2011) 118:1748–. doi: 10.1182/blood.V118.21.1748.1748
167. Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. (2012) 40:4794–803. doi: 10.1093/nar/gks066
168. Liu TF, Vachharajani VT, Yoza BK, and McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. (2012) 287:25758–69. doi: 10.1074/jbc.M112.362343
169. Rother N, Yanginlar C, Lindeboom RGH, Bekkering S, van Leent MMT, Buijsers B, et al. Hydroxychloroquine inhibits the trained innate immune response to interferons. Cell Rep Med. (2020) 1:100146. doi: 10.1016/j.xcrm.2020.100146
170. Zhao Z, Zhang Z, Li J, Dong Q, Xiong J, Li Y, et al. Sustained TNF-α stimulation leads to transcriptional memory that greatly enhances signal sensitivity and robustness. Elife. (2020) 9:e61965. doi: 10.7554/eLife.61965
171. D’Urso A and Brickner JH. Mechanisms of epigenetic memory. Trends Genet. (2014) 30:230–6. doi: 10.1016/j.tig.2014.04.004
172. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. (2012) 26:1326–38. doi: 10.1101/gad.191056.112
173. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
174. Liu X, Wang Y, Shao P, Chen Y, Yang C, Wang J, et al. Sargentodoxa cuneata and Patrinia villosa extract inhibits LPS-induced inflammation by shifting macrophages polarization through FAK/PI3K/Akt pathway regulation and glucose metabolism reprogramming. J Ethnopharmacol. (2024) 318:116855. doi: 10.1016/j.jep.2023.116855
175. Wang JZ, Zhu W, Han J, Yang X, Zhou R, Lu HC, et al. The role of the HIF-1α/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun (Lond). (2021) 41:560–75. doi: 10.1002/cac2.12158
176. Bird L. Targeting trained immunity. Nat Rev Immunol. (2019) 19:2–3. doi: 10.1038/s41577-018-0097-0
177. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2013) 71:523–30. doi: 10.1007/s00280-012-2045-1
178. Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
179. Lawrence RE and Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. (2019) 21:133–42. doi: 10.1038/s41556-018-0244-7
180. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61.e12. doi: 10.1016/j.cell.2017.11.034
181. Xu Y, Chen Y, Zhang X, Ma J, Liu Y, Cui L, et al. Glycolysis in innate immune cells contributes to autoimmunity. Front Immunol. (2022) 13:920029. doi: 10.3389/fimmu.2022.920029
182. Bettencourt IA and Powell JD. Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation, and transplantation. J Immunol. (2017) 198:999–1005. doi: 10.4049/jimmunol.1601318
183. Chen PC, Hsieh MH, Kuo WS, Wu LS, Kao HF, Liu LF, et al. Moonlighting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein of Lactobacillus gasseri attenuates allergic asthma via immunometabolic change in macrophages. J BioMed Sci. (2022) 29:75. doi: 10.1186/s12929-022-00861-8
184. Ferreira AV, Alarcon-Barrera JC, Domínguez-Andrés J, Bulut Ö, Kilic G, Debisarun PA, et al. Fatty acid desaturation and lipoxygenase pathways support trained immunity. Nat Commun. (2023) 14:7385. doi: 10.1038/s41467-023-43315-x
185. Ferreira AV, Domiguéz-Andrés J, and Netea MG. The role of cell metabolism in innate immune memory. J Innate Immun. (2022) 14:42–50. doi: 10.1159/000512280
186. Gauthier T and Chen W. Modulation of macrophage immunometabolism: A new approach to fight infections. Front Immunol. (2022) 13:780839. doi: 10.3389/fimmu.2022.780839
187. Sohrabi Y, Sonntag GVH, Braun LC, Lagache SMM, Liebmann M, Klotz L, et al. LXR activation induces a proinflammatory trained innate immunity-phenotype in human monocytes. Front Immunol. (2020) 11:353. doi: 10.3389/fimmu.2020.00353
188. van der Heijden C, Keating ST, Groh L, Joosten LAB, Netea MG, and Riksen NP. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc Res. (2020) 116:317–28. doi: 10.1093/cvr/cvz137
189. Jeon TI and Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. (2012) 23:65–72. doi: 10.1016/j.tem.2011.10.004
190. Chandrasekaran P and Weiskirchen R. The role of SCAP/SREBP as central regulators of lipid metabolism in hepatic steatosis. Int J Mol Sci. (2024) 25:1109. doi: 10.3390/ijms25021109
191. Challagundla N, Saha B, and Agrawal-Rajput R. Insights into inflammasome regulation: cellular, molecular, and pathogenic control of inflammasome activation. Immunol Res. (2022) 70:578–606. doi: 10.1007/s12026-022-09286-9
192. Khalil MI, Ali MM, Holail J, and Houssein M. Growth or death? Control of cell destiny by mTOR and autophagy pathways. Prog Biophys Mol Biol. (2023) 185:39–55. doi: 10.1016/j.pbiomolbio.2023.10.002
193. Montalbán-Hernández K, Cogollo-García A, Girón de Velasco-Sada P, Caballero R, Casanovas M, Subiza JL, et al. MV130 in the prevention of recurrent respiratory tract infections: A retrospective real-world study in children and adults. Vaccines (Basel). (2024) 12:172. doi: 10.3390/vaccines12020172
194. Cirauqui C, Benito-Villalvilla C, Sánchez-Ramón S, Sirvent S, Diez-Rivero CM, Conejero L, et al. Human dendritic cells activated with MV130 induce Th1, Th17 and IL-10 responses via RIPK2 and MyD88 signalling pathways. Eur J Immunol. (2018) 48:180–93. doi: 10.1002/eji.201747024
195. Brandi P, Conejero L, Cueto FJ, Martínez-Cano S, Dunphy G, Gómez MJ, et al. Trained immunity induction by the inactivated mucosal vaccine MV130 protects against experimental viral respiratory infections. Cell Rep. (2022) 38:110184. doi: 10.1016/j.celrep.2021.110184
196. Nieto A, Mazón A, Nieto M, Calderón R, Calaforra S, Selva B, et al. Bacterial mucosal immunotherapy with MV130 prevents recurrent wheezing in children: A randomized, double-blind, placebo-controlled clinical trial. Am J Respir Crit Care Med. (2021) 204:462–72. doi: 10.1164/rccm.202003-0520OC
197. Benito-Villalvilla C, Pérez-Diego M, Angelina A, Kisand K, Rebane A, Subiza JL, et al. Allergoid-mannan conjugates reprogram monocytes into tolerogenic dendritic cells via epigenetic and metabolic rewiring. J Allergy Clin Immunol. (2022) 149:212–22.e9. doi: 10.1016/j.jaci.2021.06.012
198. Pulendran B, S Arunachalam P, and O’Hagan DT. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov. (2021) 20:454–75. doi: 10.1038/s41573-021-00163-y
199. Bannister S, Kim B, Domínguez-Andrés J, Kilic G, Ansell BRE, Neeland MR, et al. Neonatal BCG vaccination is associated with a long-term DNA methylation signature in circulating monocytes. Sci Adv. (2022) 8:eabn4002. doi: 10.1126/sciadv.abn4002
200. Kong L, Moorlag S, Lefkovith A, Li B, Matzaraki V, van Emst L, et al. Single-cell transcriptomic profiles reveal changes associated with BCG-induced trained immunity and protective effects in circulating monocytes. Cell Rep. (2021) 37:110028. doi: 10.1016/j.celrep.2021.110028
201. Moorlag S, Rodriguez-Rosales YA, Gillard J, Fanucchi S, Theunissen K, Novakovic B, et al. BCG vaccination induces long-term functional reprogramming of human neutrophils. Cell Rep. (2020) 33:108387. doi: 10.1016/j.celrep.2020.108387
202. Giamarellos-Bourboulis EJ, Tsilika M, Moorlag S, Antonakos N, Kotsaki A, Domínguez-Andrés J, et al. Activate: randomized clinical trial of BCG vaccination against infection in the elderly. Cell. (2020) 183:315–23.e9. doi: 10.1016/j.cell.2020.08.051
203. Wimmers F, Donato M, Kuo A, Ashuach T, Gupta S, Li C, et al. The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell. (2021) 184:3915–35.e21. doi: 10.1016/j.cell.2021.05.039
204. Hilligan KL, Namasivayam S, Clancy CS, O’Mard D, Oland SD, Robertson SJ, et al. Intravenous administration of BCG protects mice against lethal SARS-CoV-2 challenge. J Exp Med. (2022) 219:e20211862. doi: 10.1084/jem.20211862
205. Hensel J, McAndrews KM, McGrail DJ, Dowlatshahi DP, LeBleu VS, and Kalluri R. Protection against SARS-CoV-2 by BCG vaccination is not supported by epidemiological analyses. Sci Rep. (2020) 10:18377. doi: 10.1038/s41598-020-75491-x
206. Wen J, Liu Q, Tang D, and He JQ. Efficacy of BCG vaccination against COVID-19: systematic review and meta-analysis of randomized controlled trials. J Clin Med. (2023) 12:154. doi: 10.3390/jcm12031154
207. Xia Z, Meng J, Wang X, Liu P, Wu Y, Xiong Y, et al. Efficacy of BCG vaccination against COVID-19 in health care workers and non-health care workers: A meta-analysis of randomized controlled trials. PloS One. (2025) 20:e0321511. doi: 10.1371/journal.pone.0321511
208. Messina NL, Pittet LF, McDonald E, Moore C, Barry S, Bonten M, et al. BCG vaccination of healthcare workers for protection against COVID-19: 12-month outcomes from an international randomised controlled trial. J Infect. (2024) 89:106245. doi: 10.1016/j.jinf.2024.106245
209. Hollm-Delgado MG, Stuart EA, and Black RE. Acute lower respiratory infection among Bacille Calmette-Guérin (BCG)-vaccinated children. Pediatrics. (2014) 133:e73–81. doi: 10.1542/peds.2013-2218
210. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun. (2014) 6:152–8. doi: 10.1159/000355628
211. Liu K, Nicoletti R, Zhao H, Chen X, Wu H, Leung CH, et al. Young age and adequate BCG are key factors for optimal BCG treatment efficacy in non-muscle-invasive bladder cancer. World J Urol. (2024) 42:547. doi: 10.1007/s00345-024-05218-4
212. Debisarun PA, Gössling KL, Bulut O, Kilic G, Zoodsma M, Liu Z, et al. Induction of trained immunity by influenza vaccination - impact on COVID-19. PloS Pathog. (2021) 17:e1009928. doi: 10.1371/journal.ppat.1009928
213. Bruxvoort KJ, Ackerson B, Sy LS, Bhavsar A, Tseng HF, Florea A, et al. Recombinant adjuvanted zoster vaccine and reduced risk of coronavirus disease 2019 diagnosis and hospitalization in older adults. J Infect Dis. (2022) 225:1915–22. doi: 10.1093/infdis/jiab633
214. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. (2018) 49:819–28.e6. doi: 10.1016/j.immuni.2018.09.008
215. Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. (2014) 5:3065. doi: 10.1038/ncomms4065
216. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
217. Hamilton JA, Cook AD, and Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. (2016) 16:53–70. doi: 10.1038/nrd.2016.231
218. Ortved KF, Austin BS, Scimeca MS, and Nixon AJ. RNA interference mediated interleukin-1β Silencing in inflamed chondrocytes decreases target and downstream catabolic responses. Arthritis. (2016) 2016:3484961. doi: 10.1155/2016/3484961
219. Stopinšek S, Ihan A, Salobir B, Terčelj M, and Simčič S. Fungal cell wall agents and bacterial lipopolysaccharide in organic dust as possible risk factors for pulmonary sarcoidosis. J Occup Med Toxicol. (2016) 11:46. doi: 10.1186/s12995-016-0135-4
220. Talreja J, Talwar H, Bauerfeld C, Grossman LI, Zhang K, Tranchida P, et al. HIF-1α regulates IL-1β and IL-17 in sarcoidosis. Elife. (2019) 8:e44519. doi: 10.7554/eLife.44519
221. Casaccia-Bonnefil P, Pandozy G, and Mastronardi F. Evaluating epigenetic landmarks in the brain of multiple sclerosis patients: a contribution to the current debate on disease pathogenesis. Prog Neurobiol. (2008) 86:368–78. doi: 10.1016/j.pneurobio.2008.09.012
222. Mastronardi FG, Noor A, Wood DD, Paton T, and Moscarello MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. (2007) 85:2006–16. doi: 10.1002/jnr.21329
223. Guan H, Nagarkatti PS, and Nagarkatti M. CD44 Reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J Immunol. (2011) 186:6955–64. doi: 10.4049/jimmunol.1004043
224. Toh ML, Bonnefoy JY, Accart N, Cochin S, Pohle S, Haegel H, et al. Bone- and cartilage-protective effects of a monoclonal antibody against colony-stimulating factor 1 receptor in experimental arthritis. Arthritis Rheumatol. (2014) 66:2989–3000. doi: 10.1002/art.38624
225. Horwood NJ, Elliott J, Martin TJ, and Gillespie MT. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol. (2001) 166:4915–21. doi: 10.4049/jimmunol.166.8.4915
226. Malemud CJ. The PI3K/Akt/PTEN/mTOR pathway: a fruitful target for inducing cell death in rheumatoid arthritis? Future Med Chem. (2015) 7:1137–47. doi: 10.4155/fmc.15.55
Keywords: trained immunity, inflammatory diseases, immune system, innate immune memory, epigenetic reprogramming, inflammation
Citation: Xu W, Guo Z, Xu T, Chen J, Chen L and Xu W (2025) Reversing inflammatory diseases via trained immunity: mechanisms, challenges, and prospects. Front. Immunol. 16:1666233. doi: 10.3389/fimmu.2025.1666233
Received: 15 July 2025; Accepted: 29 September 2025;
Published: 15 October 2025.
Edited by:
Tara Marlene Strutt, University of Central Florida, United StatesCopyright © 2025 Xu, Guo, Xu, Chen, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenan Xu, eHVfd2VuYW5Ac211LmVkdS5jbg==; Leyi Chen, cmVzdXJyZWN0aW9uQGkuc211LmVkdS5jbg==; Y2hlbmxleWkxOTk4MDhAaG90bWFpbC5jb20=