 Wanting Wang
Wanting Wang Siyao Chang2
Siyao Chang2- 1Heilongjiang University of Chinese Medicine, Heilongjiang, Harbin, China
- 2Vascular Surgery Department, Harbin Fifth Hospital, Harbin, China
- 3Department of Peripheral Vascular Disease, First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, China
Thromboangiitis obliterans (TAO) is a non-atherosclerotic, inflammatory vasculopathy characterized by thrombotic occlusion of small- and medium-sized vessels, leading to tissue ischemia and gangrene. Emerging evidence underscores endothelial cell (EC) activation as a central driver of disease progression, mediated by immune dysregulation, oxidative stress (Nrf2/ROS imbalance), impaired nitric oxide signaling (eNOS/iNOS dysregulation), endoplasmic reticulum and mitochondrial dysfunction, and disrupted copper/iron homeostasis. These pathways collectively promote a prothrombotic, proinflammatory endothelial phenotype, perpetuating vascular injury. Current therapies primarily alleviate symptoms but fail to address underlying EC dysfunction. Recent advances, including stem cell therapy and targeted immunomodulation, offer promising avenues for restoring endothelial homeostasis. However, translating mechanistic insights into durable clinical benefits requires further research into precision medicine approaches and large-scale validation of novel therapeutics. This review summarizes the multifactorial pathogenesis of TAO, emphasizing EC activation as a therapeutic linchpin, and outlines future directions to bridge translational gaps in disease management.
1 Introduction
Thromboangiitis obliterans (TAO) is a chronic, non-atherosclerotic, segmental vasculitis characterized by inflammatory thrombi affecting small- and medium-sized arteries and veins of the extremities, often progressing to ulcers or gangrene (1). Arterial insufficiency leads to claudication, Raynaud’s phenomenon, and, in severe cases, amputation. The distal distribution and poor collateral circulation render surgical and endovascular approaches largely ineffective. Although the precise pathogenesis remains unclear, accumulating evidence suggests an autoimmune component, with various autoantibodies and immune cells targeting vascular structures (2–4). Histologically, TAO features a cellular thrombus rich in polymorphonuclear leukocytes, mononuclear cells, and giant cells, with minimal vessel wall involvement (5). Conventional inflammatory markers (ESR, CRP) and common autoantibodies may remain normal during acute episodes, yet immune dysregulation is believed to drive disease activity (6). Autoimmune features are frequently observed, including elevated anti-endothelial cell antibodies (AECAs), especially during active disease. These antibodies bind not only to surface but also intracellular endothelial antigens, implicating endothelial dysfunction in disease progression (7).
Endothelial dysfunction in TAO is often preceded by endothelial activation, marked by increased expression of adhesion molecules such as ICAM-1 and VCAM-1, which promote leukocyte adhesion and a prothrombotic vascular phenotype. Elevated circulating ICAM-1 levels further support this persistent endothelial activation (8). This process is primarily mediated by pro-inflammatory cytokines—particularly TNF and IL-6—which enhance leukocyte recruitment and adherence to the endothelium (9). Thus, endothelial activation constitutes a central mechanism in TAO pathophysiology and represents a potential therapeutic target. This review highlights recent insights into the role of endothelial cell activation in TAO and its implications for future therapeutic interventions.
2 Mechanisms of endothelial cell activation in TAO
2.1 Immune complex-mediated endothelial activation
Endothelial cell (EC) activation can be broadly classified into two types: Type I activation, a rapid response independent of new gene expression (also termed stimulation), and Type II activation, a slower but sustained response requiring de novo gene transcription. Acute inflammation involves the rapid recruitment of neutrophils within hours, driven by EC activation—a process by which resting endothelial cells acquire new functional properties (10). When acute stimuli persist, particularly under sustained adaptive immune responses, inflammation transitions toward chronicity. Type I activation is typically initiated by heterotrimeric G protein–coupled receptor ligands, whereas Type II activation is driven by proinflammatory cytokines such as tumor necrosis factor (TNF) and interleukin-1 (IL-1), involving monocytes, effector, and memory T cells. Adaptive cytokines, including interferon-γ (IFN-γ) and IL-4, further reprogram Type II–activated ECs, reinforcing T-helper cell polarization and amplifying inflammation (11). In patients with TAO, high titers of biologically active circulating mixed immune complexes have been observed, suggesting a pivotal role for immune complex–induced EC Type II activation and neutrophil-mediated endothelial injury in TAO pathogenesis (12).
2.2 Involvement of immune and inflammatory cytokines in endothelial activation
Given their anatomical interface with the circulation, endothelial cells (ECs) are persistently exposed to immune mediators and represent principal targets of pro- and anti-inflammatory cytokines. In TAO, heightened inflammation activates ECs via TNF-α, IL-6, and IL-17, thereby enhancing leukocyte adhesion and transmigration (9). Histological evidence reveals abundant CD4+ T cells in the intima and near thrombi, with macrophage infiltration observed in early disease stages (2). Differentiation of CD4+ T cells into Th1, Th2, Th17, and Treg subsets orchestrates divergent immune programs: Th1/Th17 cells amplify inflammation through IFN-γ and IL-17, whereas Th2/Treg cells secrete IL-4 and IL-10 to mediate immunoregulation. Elevated IFN-γ, IL-17, and TNF-α levels in TAO patients correlate strongly with EC activation (13). HMGB1 is markedly upregulated in TAO patient plasma, promoting inflammation and EC dysfunction by enhancing ICAM-1 expression and cytokine release (14). Moreover, immunoadsorption-based antibody removal has shown potential in attenuating disease severity. While associations with MHC class I and II alleles have been reported, genetic testing remains absent from clinical application (5).
Recent studies underscore the role of IL-33 in TAO. Endothelial cells, as IL-33 targets, respond via steroid-resistant proinflammatory signaling pathways. IL-33 binds the transmembrane receptor ST2L and recruits the adaptor MyD88, which in turn engages IRAK1/IRAK4 and TRAF6 to activate the canonical NF-κB pathway and the MAPK modules p38, JNK, and ERK (15–18), In endothelial cells, this signaling increases transcription of adhesion molecules (ICAM-1, VCAM-1, E-selectin), chemokines (CXCL1, CCL2), and prothrombotic mediators (tissue factor, PAI-1), thereby amplifying leukocyte tethering, transmigration, and thrombogenicity (19–22). Notably, IL-33–driven gene programs show relative steroid resistance, glucocorticoids incompletely suppress NF-κB/MAPK outputs downstream of MyD88/IRAK/TRAF6, providing a plausible explanation for the limited efficacy of steroid-based regimens in subsets of TAO patients with elevated circulating IL-33 (23).
2.3 Oxidative stress and endothelial activation
Excessive oxidative stress elevates intracellular reactive oxygen species (ROS) in endothelial cells, upregulating adhesion molecules including MCP-1, ICAM-1, VCAM-1, and E-selectin (24), thereby facilitating monocyte adhesion, endothelial activation, and tissue infiltration (25). Nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the Cap’n’collar basic-region leucine zipper (CNC-bZIP) transcription factor family, serves as a master regulator of the cellular antioxidant response. Under oxidative or pathological stimuli, phosphorylated Nrf2 dissociates from its cytoplasmic repressor and translocates into the nucleus, where it induces the expression of downstream genes encoding antioxidant enzymes, such as heme oxygenase-1, superoxide dismutase, and glutathione peroxidase. This cascade facilitates the detoxification of reactive oxygen species (ROS) and confers cytoprotection by mitigating oxidative stress, inflammation, and apoptosis (26). This program detoxifies ROS and mitigates oxidative stress–driven inflammation and apoptosis. Impaired Nrf2 signaling blunts these defenses, enabling ROS accumulation, mitochondrial injury, cytochrome c release, and caspase activation, thereby predisposing endothelial cells to apoptosis (27). Concurrently, sustained oxidative stress under Nrf2 deficiency augments NF-κB activity, transcriptionally upregulating ICAM-1 and VCAM-1, which amplifies leukocyte recruitment and sustains vascular inflammation (28–31). In TAO, chronic arterial occlusion and hypoxia exacerbate oxidative burden, further mismatching oxygen supply and demand (32, 33). This imbalance contributes to cellular dysfunction or death. Therefore, therapeutic strategies targeting oxidative stress and modulating Nrf2-related signaling pathways represent promising approaches for ameliorating vascular inflammation in TAO (34, 35).
2.4 Inducible nitric oxide synthase mediates endothelial cell activation
Vascular endothelial cells form a specialized barrier between blood and vessel walls, regulating vascular tone, blood pressure, coagulation balance, and vascular permeability to maintain homeostasis (10). Hemodynamic forces, particularly shear stress, play a central role in endothelial integrity. In TAO, shear stress-induced activation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production is impaired, compromising vasodilation (36). Pathological stimuli reduce eNOS expression and NO bioavailability while upregulating hypoxia-inducible factor-1 (HIF-1), endothelin-1 (ET-1), and plasminogen activator inhibitor-1 (PAI-1), collectively exacerbating endothelial dysfunction, enhancing thrombogenicity, and promoting vascular smooth muscle proliferation (37). Furthermore, the NF-κB signaling pathway is activated in TAO, inducing inflammatory mediators such as TNF-α, IL-1β, IL-6, IL-8, and matrix metalloproteinases (MMPs), which contribute to endothelial injury and atherosclerotic plaque formation (38). These factors stimulate ROS production and adhesion molecule expression, reinforcing monocyte recruitment, apoptosis, and sustained inflammation. Under normal conditions, eNOS-derived NO preserves microvascular tone; however, during inflammation, inducible nitric oxide synthase (iNOS) becomes upregulated, producing excessive NO. This reacts with ROS to form peroxynitrite and other radicals, promoting oxidative damage and thrombosis. iNOS rapidly reacts with superoxide anions to generate peroxynitrite (ONOO-), a potent oxidant capable of inducing lipid peroxidation, protein nitration, and DNA damage within endothelial cells (39, 40). Such oxidative and nitrosative stress not only exacerbates endothelial dysfunction but also promotes a prothrombotic vascular environment, thereby directly contributing to luminal narrowing and vascular occlusion (41, 42). Endothelial cells are thus likely the primary sites of early inflammatory injury in TAO. Genetic studies reveal that the protective eNOS is significantly reduced in TAO patients, implicating the T polymorphism in disease susceptibility (5, 43). In contrast, elevated iNOS expression has been detected in endothelial cells adjacent to occluded arterial segments in TAO specimens and replicated in rat TAO models (2, 44, 45). These findings underscore the pivotal role of endothelial dysfunction, driven by altered NO signaling and inflammation, in TAO pathogenesis.
2.5 Endothelial activation mediated by endoplasmic reticulum stress and mitochondrial dysfunction
Recent evidence highlights endoplasmic reticulum stress (ERS) as a pivotal driver of inflammation and apoptosis through the IRE1/ASK1/JNK cascade, which also primes NLRP3 inflammasome activation (46, 47). In endothelial cells, oxidized LDL (ox-LDL) triggers ERS and ASK1 upregulation, eliciting ROS overproduction, apoptosis, inflammatory signaling, and impaired proliferation (48). Vascular remodeling in cardiovascular disease originates from endothelial dysfunction, characterized by endothelial activation, smooth muscle cell apoptosis, and extracellular matrix degradation (49). Mitochondrial dysfunction exacerbates these processes by perturbing ROS homeostasis, calcium signaling, and bioenergetics (50, 51). Under simulated microgravity, ERS-induced calcium flux promotes mitochondrial depolarization, fragmentation, and p62/SQSTM1-mediated mitophagy, diminishing endothelial barrier integrity and NLRP3 activation (52). In TAO rat models, severe ER fragmentation and mitochondrial damage in femoral arteries suggest therapeutic potential in targeting ERS–mitochondrial interplay (53). Importantly, ERS-driven mitochondrial dysfunction serves as an upstream trigger for NLRP3 inflammasome activation, resulting in the maturation and release of IL-1β and IL-18, which amplify endothelial injury and leukocyte recruitment (54–58). This proinflammatory milieu fosters platelet activation, tissue factor expression, and fibrin deposition, thereby establishing a mechanistic link between the ERS–mitochondria–NLRP3 axis and inflammatory thrombosis in TAO (59).
2.6 Copper and iron metabolism regulates endothelial cell activation
Copper is an indispensable trace element for systemic homeostasis. Deficiency impairs growth, causes neurological dysfunction, and downregulates adhesion molecules such as ICAM-1 and VCAM-1, thereby weakening leukocyte–endothelium interactions. In contrast, copper overload promotes endothelial activation via ICAM-1 upregulation, reversible with copper chelation (60). Excess intracellular copper drives redox cycling, elevating ROS that activate NF-κB and p38/MAPK signaling, thereby inducing ICAM-1, VCAM-1, and E-selectin expression and fostering a pro-adhesive, prothrombotic phenotype (61–65). Copper activates oxidative stress pathways and the p38 mitogen-activated protein kinase (p38/MAPK) signaling cascade, leading to endothelial cell activation, DNA damage, and cell death in human umbilical vein endothelial cells (HUVECs). Copper chelation attenuates these effects, offering potential therapeutic benefit (66). Given the elevated serum copper in TAO patients, maintaining copper homeostasis is essential to limit endothelial activation and disease progression. Ferritin, composed of light and heavy chains, stores intracellular iron; its autophagic degradation increases labile iron, fueling Fenton-driven ROS production and oxidative injury. Labile iron and heme further activate endothelial and immune cells, enhancing leukocyte and erythrocyte adhesion and precipitating vasculitis and thrombosis (67). Zinc oxide nanoparticles induce ferritinophagy in endothelial cells, elevating iron, lipid peroxidation, and dysfunction in a dose- and time-dependent manner—effects reversed by iron chelators (68). Thus, targeting iron metabolism to modulate endothelial activation represents a promising strategy for mitigating vascular inflammation and thrombosis in TAO.
2.7 Glycolytic regulation of endothelial activation
Endothelial activation or injury enhances vascular permeability and leukocyte adhesion, thereby promoting thrombosis and accelerating disease progression. Owing to their low mitochondrial content, endothelial cells derive 85% of ATP from glycolysis—60% sustaining homeostasis and 40% supporting proliferation (69). Even at rest, they display high glycolytic flux, which is further elevated during migration or proliferation, generating intermediates that regulate survival and function. Dysregulated glycolysis is a central driver of endothelial dysfunction and hyperproliferation (70). Loss of the glycolytic activator PFKFB3 impairs vessel formation by limiting proliferation, filopodia/lamellipodia formation, and directional migration, partly via compartmentalization with F-actin in motile protrusions (71–73). In TAO, chronic ischemia and hypoxia in distal arteries from vascular occlusion (74, 75). The local mismatch between oxygen supply and demand caused by vascular inflammation may trigger metabolic reprogramming via enhanced glycolysis (76). Targeting glycolytic pathways to restore metabolic homeostasis offers a potential strategy to ameliorate endothelial injury and vascular dysfunction in TAO (Table 1).
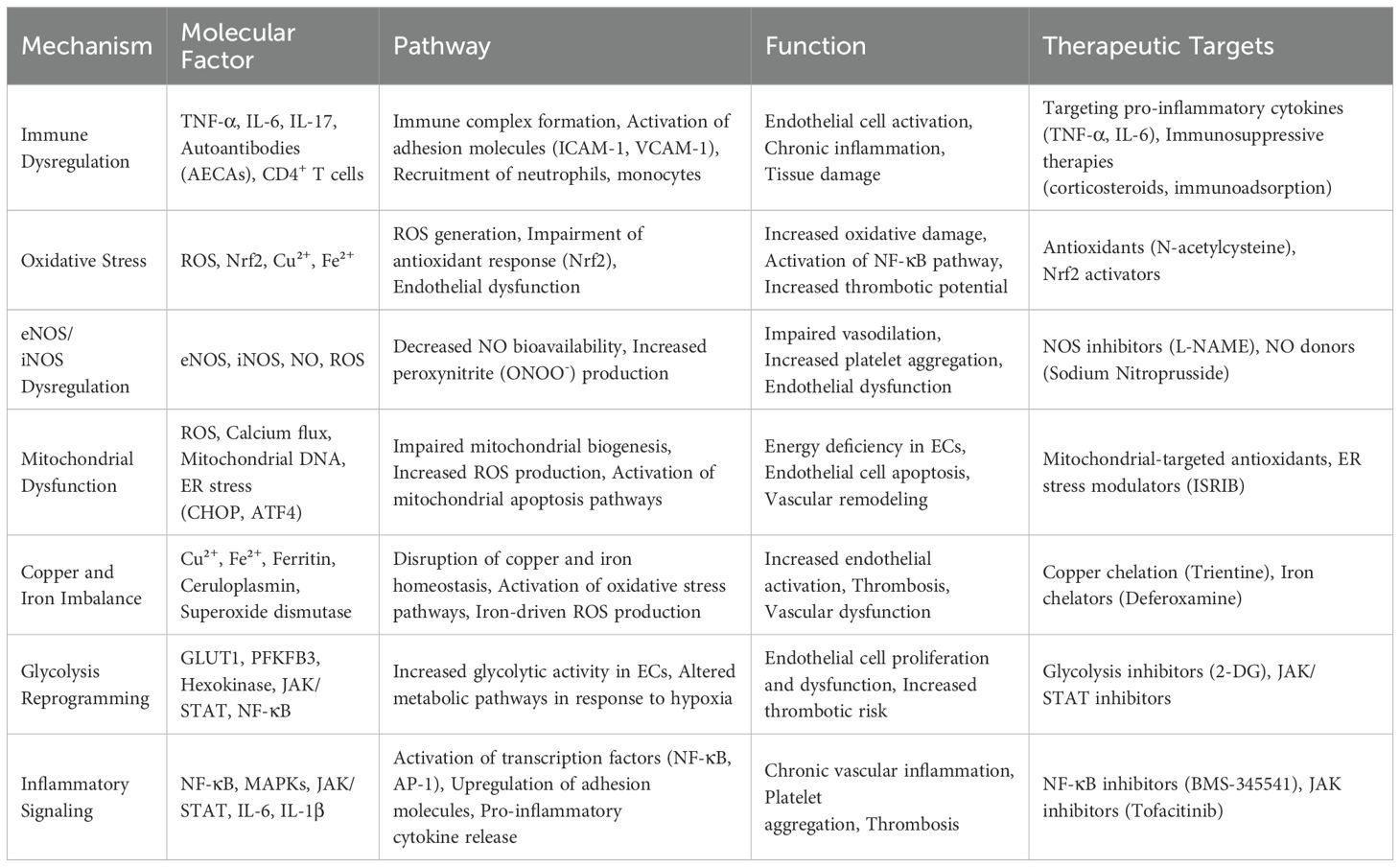
Table 1. Mechanisms of endothelial cell activation in thromboangiitis obliterans (TAO).
2.8 Inflammation-associated signaling pathways regulate endothelial cell activation
Endothelial dysfunction is a hallmark of TAO, driving inflammation, thrombosis, and vascular remodeling. In thrombohemorrhagic vasculitis, spleen tyrosine kinase (Syk) activation within vascular walls triggers neutrophil elastase release, promoting hemorrhage, fibrin deposition, and thrombosis (77). Beyond adaptive immunity, Syk orchestrates cell adhesion, innate immune activation, osteoclast differentiation, platelet aggregation, and vascular development (78), with Syk–MAPK signaling being pivotal for pro-inflammatory gene expression and NLRP3 inflammasome activation (79). Licochalcone A attenuates endothelial activation by inhibiting Syk phosphorylation, suppressing p38/JNK signaling, and reducing NLRP3-driven IL-1β and IL-18 secretion (61). Endothelial cells exhibit both antithrombotic and prothrombotic properties. Under homeostasis, they prevent thrombosis via prostacyclin, tissue plasminogen activator (t-PA), and tissue factor pathway inhibitor. However, injury shifts the balance toward thrombogenesis, enhancing platelet aggregation and thrombus formation. t-PA promotes fibrinolysis through plasmin activation, while its inhibitor, plasminogen activator inhibitor-1 (PAI-1), facilitates thrombosis by impeding fibrinolysis and enhancing platelet adhesion (80). A reduced t-PA/PAI-1 ratio critically increases thrombotic risk (81). The NF-κB pathway governs endothelial-platelet-cytokine crosstalk, disrupting coagulation-fibrinolysis homeostasis. Activation of NF-κB and AP-1 reflects upstream signaling via JNK and p38/MAPK pathways. TNF-α suppresses t-PA expression via these axes, further impairing fibrinolysis (82). Compounds such as shikonin and salvianolic acid B counteract endothelial injury and thrombosis by modulating the NF-κB/JNK/p38/MAPK pathway (83). Additionally, the JAK2/STAT3 axis regulates endothelial cytoskeletal remodeling via downstream activation of RhoA, a GTPase involved in adhesion and motility. IL-6–driven STAT3 activation enhances RhoA/ROCK signaling, altering actin dynamics and focal adhesion (84). In TAO, IL-6/STAT3 signaling disrupts endothelial homeostasis by modulating adhesion molecules and cytoskeletal structure (20). In conclusion, persistent endothelial activation in TAO is mechanistically linked to immune signaling, inflammation, and thrombosis. Disease severity correlates with the extent of endothelial injury, implicating it as a potential initiating factor. Targeting these signaling cascades may restore endothelial integrity and offer novel therapeutic strategies for TAO (Figure 1).
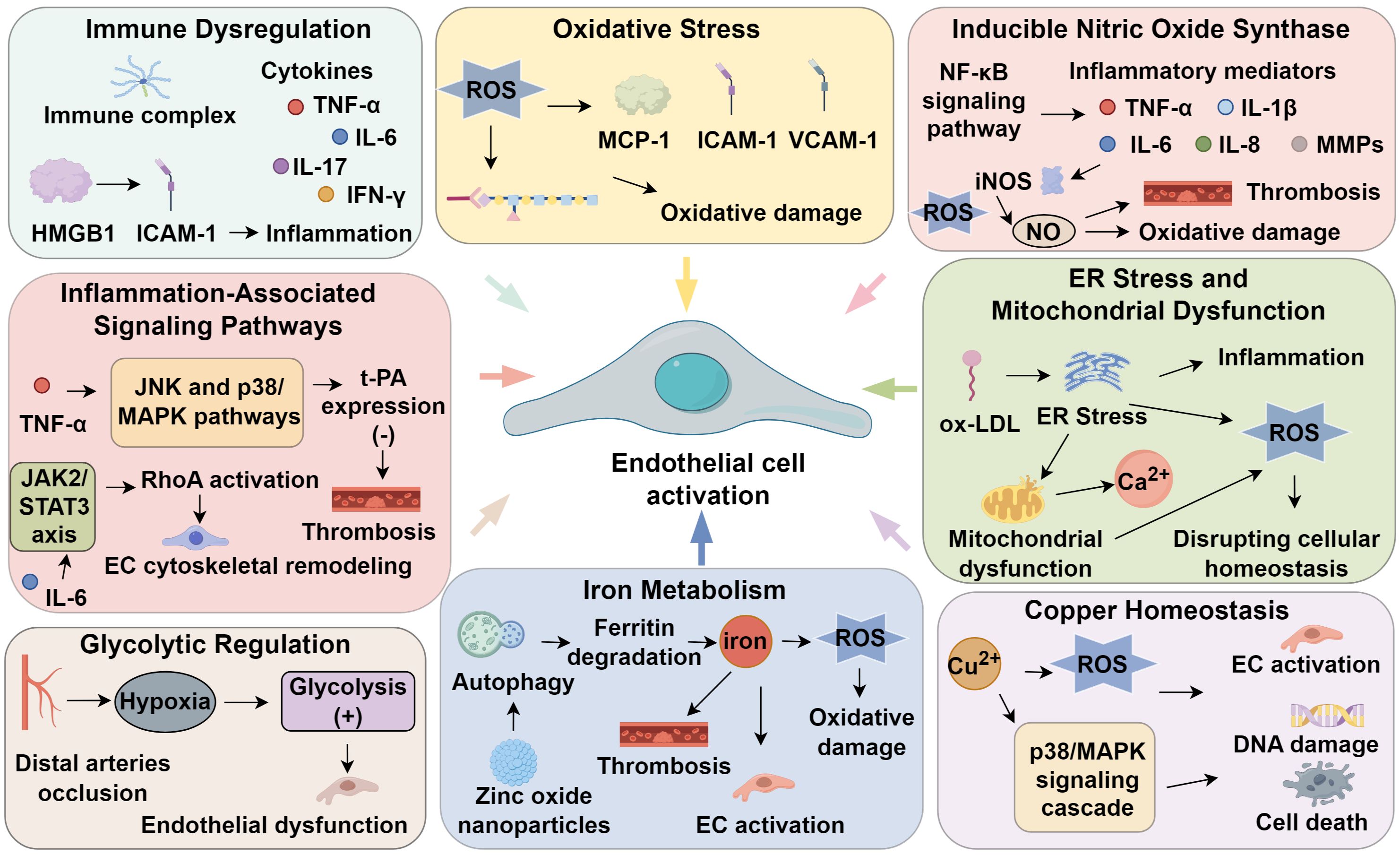
Figure 1. Endothelial activation in thromboangiitis olterans.
3 Advances in the treatment of TAO
Common oral agents for TAO include aspirin, tolazoline, batroxobin, and prostaglandin analogs (85). Argatroban, a direct thrombin inhibitor targeting its catalytic site, exhibits greater efficacy when combined with prostaglandin E1 (PGE1) than PGE1 alone (86), effectively ameliorating hypercoagulability and improving therapeutic outcomes. Prostaglandin analogs, which enhance peripheral circulation, are widely employed in ischemic limb disorders. Comparative analyses demonstrate their superiority over open lumbar sympathectomy in achieving complete ulcer healing, relieving rest pain, and reducing major amputation rates (87, 88).
Lumbar sympathectomy and endarterectomy remain cornerstone surgical interventions for TAO, targeting sympathetic denervation of the lower limbs to relieve vasospasm and enhance distal vasodilation and perfusion (89). Percutaneous transluminal angioplasty (PTA), including balloon dilation, stent placement, thrombolysis, and thrombectomy, restores vessel patency by resolving occlusive lesions (90), with balloon angioplasty being the most widely applied and demonstrating substantial efficacy (91). Clinical evidence shows that PTA significantly promotes ulcer healing, alleviates pain, and preserves limbs (92). In one series, distal perfusion improved and the limb salvage rate reached 92% (93), while another reported 100% clinical efficacy with marked gains in iliac artery flow and ankle–brachial index (64). For femoropopliteal TAO, excimer laser ablation combined with drug-coated balloon angioplasty achieved 92.0% 12-month patency (94). Although short-term outcomes are encouraging, long-term durability and complication profiles remain to be clarified. Tibial transverse bone transport (TTBT), which promotes microvascular regeneration via longitudinal tibial traction, has been shown to restore lower-limb microcirculation, reduce ischemic pain, improve hemorheology, and enhance wound healing and ankle–brachial index (95).
Stem cell transplantation offers a promising option for patients unresponsive to revascularization or conservative therapy, improving symptoms and lowering amputation rates (96, 97). Comparative studies of peripheral blood mononuclear cells (PBMC) and purified CD34+ cells (PCC) revealed satisfactory long-term outcomes, with PBMC yielding faster symptom relief and PCC causing less injection site discomfort (98, 99). Stem cell therapy attenuates endothelial activation by restoring nitric oxide synthase activity, reducing oxidative stress, and downregulating ICAM-1/VCAM-1 expression, thereby improving endothelial barrier integrity (100–103). Additionally, transplanted stem cells secrete pro-angiogenic factors such as VEGF, FGF, and angiopoietins, which stimulate neovascularization in ischemic tissues (104–106). These paracrine effects also modulate immune responses by shifting the Th1/Th17-driven pro-inflammatory milieu toward an anti-inflammatory Treg-dominant profile, potentially mitigating the autoimmune-mediated endothelial injury (107, 108). Endovascular radiofrequency ablation (ERA), which ablates sympathetic ganglia to reduce vasoconstriction, is minimally invasive and effective for ischemic pain relief (109), though current evidence suggests a higher complication rate compared with cell-based or intra-arterial approaches, warranting larger studies.
4 Conclusion
TAO is a multifactorial vasculopathy in which sustained endothelial activation represents the central pathogenic driver. This state is propagated by autoimmune processes such as anti-endothelial cell antibodies and Th17-mediated inflammation, coupled with oxidative stress from Nrf2/ROS disequilibrium, nitric oxide synthase dysregulation, and copper/iron metabolic imbalance. These insults converge to induce a pro-adhesive, pro-thrombotic endothelial phenotype that fosters leukocyte recruitment, vascular inflammation, and luminal occlusion. Endoplasmic reticulum–mitochondrial crosstalk amplifies NLRP3 inflammasome activation, while glycolytic metabolic reprogramming sustains endothelial activation under hypoxia, culminating in progressive microvascular failure and tissue ischemia.
Current medical, surgical, and endovascular interventions from prostaglandin analogues to percutaneous transluminal angioplasty offer symptomatic relief and delay amputation but do not address upstream endothelial dysfunction. Emerging strategies including stem cell transplantation, targeted immunomodulation, and metabolic correction aim to restore endothelial homeostasis by enhancing nitric oxide bioavailability, reducing oxidative stress, and promoting a Treg-skewed immune profile. Yet, their durability, safety, and patient-specific efficacy remain undefined. Future work should focus on precision medicine, integrating genetic, epigenetic, and biomarker-guided stratification to enable mechanism-based therapy, supported by large-scale longitudinal trials to transition TAO care from symptom palliation to durable disease modification.
Author contributions
WW: Writing – original draft. SC: Writing – original draft. GZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Del Conde I and Peña C. Buerger disease (thromboangiitis obliterans). Tech Vasc Interv Radiol. (2014) 17:234–40. doi: 10.1053/j.tvir.2014.11.003
2. Lee T, Seo JW, Sumpio BE, and Kim SJ. Immunobiologic analysis of arterial tissue in Buerger’s disease. Eur J Vasc Endovasc Surg. (2003) 25:451–7. doi: 10.1053/ejvs.2002.1869
3. Gulati SM, Madhra K, Thusoo TK, Nair SK, and Saha K. Autoantibodies in thromboangiitis obliterans (Buerger’s disease). Angiology. (1982) 33:642–51. doi: 10.1177/000331978203301003
4. Fazeli B, Keramat S, Assadi L, and Taheri H. Angiogenesis induction in Buerger’s disease: a disease management double-edged sword? Orphanet J Rare Dis. (2019) 14:189. doi: 10.1186/s13023-019-1166-6
5. Ketha SS and Cooper LT. The role of autoimmunity in thromboangiitis obliterans (Buerger’s disease). Ann N Y Acad Sci. (2013) 1285:15–25. doi: 10.1111/nyas.12048
6. Piazza G and Creager MA. Thromboangiitis obliterans. Circulation. (2010) 121:1858–61. doi: 10.1161/CIRCULATIONAHA.110.942383
7. Eichhorn J, Sima D, Lindschau C, Turowski A, Schmidt H, Schneider W, et al. Antiendothelial cell antibodies in thromboangiitis obliterans. Am J Med Sci. (1998) 315:17–23. doi: 10.1097/00000441-199801000-00004
8. Ostrowski SR, Haase N, Müller RB, Møller MH, Pott FC, Perner A, et al. Association between biomarkers of endothelial injury and hypocoagulability in patients with severe sepsis: a prospective study. Crit Care. (2015) 19:191. doi: 10.1186/s13054-015-0918-5
9. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. (2013) 123:540–1. doi: 10.1172/JCI66843
10. Pober JS and Cotran RS. The role of endothelial cells in inflammation. Transplantation. (1990) 50:537–44. doi: 10.1097/00007890-199010000-00001
11. Pober JS and Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
12. Somer T. Thrombo-embolic and vascular complications in vasculitis syndromes. Eur Heart J. (1993) 14:24–9.
13. Hot A, Lenief V, and Miossec P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann Rheum Dis. (2012) 71:768–76. doi: 10.1136/annrheumdis-2011-200468
14. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. (2003) 101:2652–60. doi: 10.1182/blood-2002-05-1300
15. Sun XL, Law BY, de Seabra Rodrigues Dias IR, Mok SWF, He YZ, and Wong VK. Pathogenesis of thromboangiitis obliterans: Gene polymorphism and immunoregulation of human vascular endothelial cells. Atherosclerosis. (2017) 265:258–65. doi: 10.1016/j.atherosclerosis.2017.08.009
16. Larsen KM, Minaya MK, Vaish V, and Peña MMO. The role of IL-33/ST2 pathway in tumorigenesis. Int J Mol Sci. (2018) 19:2676. doi: 10.3390/ijms19092676
17. Kang MH and Bae YS. IL-33 and IL-33-derived DC-based tumor immunotherapy. Exp Mol Med. (2024) 56:1340–7. doi: 10.1038/s12276-024-01249-4
18. Awasthi V, Vilekar P, Rao G, and Awasthi S. Anti-inflammatory mediators ST2 and SIGIRR are induced by diphenyldifluoroketone EF24 in lipopolysaccharide-stimulated dendritic cells. Immunobiology. (2020) 225:151886. doi: 10.1016/j.imbio.2019.11.021
19. Halacheva K, Gulubova MV, Manolova I, and Petkov D. Expression of ICAM-1, VCAM-1, E-selectin and TNF-alpha on the endothelium of femoral and iliac arteries in thromboangiitis obliterans. Acta Histochem. (2002) 104:177–84. doi: 10.1078/0065-1281-00621
20. Wei Z, Jiang W, Wang H, Li H, Tang B, Liu B, et al. The IL-6/STAT3 pathway regulates adhesion molecules and cytoskeleton of endothelial cells in thromboangiitis obliterans. Cell Signal. (2018) 44:118–26. doi: 10.1016/j.cellsig.2018.01.015
21. Kang HL, Várkonyi Á, Csonka Á, Szász A, Várkonyi T, Pósa A, et al. Endothelial-mesenchymal transition and possible role of cytokines in streptozotocin-induced diabetic heart. Biomedicines. (2025) 13:1148. doi: 10.3390/biomedicines13051148
22. Han G, Lee J, and Bae JS. Anti-thrombotic activity of 3-deoxysappanchalcone via inhibiting platelet aggregation and thrombin (FIIa)/activated factor X (FXa) activity. Molecules. (2025) 30:2580. doi: 10.3390/molecules30122580
23. Shoda T, Futamura K, Orihara K, Emi-Sugie M, Saito H, Matsumoto K, et al. Recent advances in understanding the roles of vascular endothelial cells in allergic inflammation. Allergol Int. (2016) 65:21–9. doi: 10.1016/j.alit.2015.08.001
24. Xu Q, Liu C, Chen S, Li X, and Xiong D. Semicarbazide conferred developmental toxicity in Oryzias melastigma embryos by oxidative stress and energy metabolism disorder. Aquat Toxicol. (2025) 287:107531. doi: 10.1016/j.aquatox.2025.107531
25. Clapp BR, Hingorani AD, Kharbanda RK, Mohamed-Ali V, Stephens JW, Vallance P, et al. Inflammation-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc Res. (2004) 64:172–8. doi: 10.1016/j.cardiores.2004.06.020
26. Lai J, Jiang J, Zhang P, Xi C, Wu L, Gao X, et al. Impaired blood-brain barrier in the microbiota-gut-brain axis: Potential role of bipolar susceptibility gene TRANK1. J Cell Mol Med. (2021) 25:6463–9. doi: 10.1111/jcmm.16611
27. Senthil KKJ, Gokila VM, and Wang SY. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget. (2017) 8:96568–87. doi: 10.18632/oncotarget.19951
28. Cai M, Zhang X, Gao X, Huo Q, Sun Y, and Dai X. Chitooligosaccharide ameliorates cognitive deficits and neuroinflammation in APP/PS1 mice associated with the regulation of Nrf2/NF-κB axis. Int J Biol Macromol. (2025) 303:140683. doi: 10.1016/j.ijbiomac.2025.140683
29. Qiao M, Zhang L, Wu Y, and Ma B. Glutathione peroxidase 3 antagonizes oxidative stress and inflammation in diabetic retinopathy via NrF2/NF-κB pathway. Int Ophthalmol. (2025) 45:231. doi: 10.1007/s10792-025-03582-7
30. Toda H, Diaz-Varela M, Segui-Barber J, Roobsoong W, Baro B, Garcia-Silva S, et al. Plasma-derived extracellular vesicles from Plasmodium vivax patients signal spleen fibroblasts via NF-kB facilitating parasite cytoadherence. Nat Commun. (2020) 11:2761. doi: 10.1038/s41467-020-16337-y
31. Wu A, Wu Y, Song M, Chen W, Xu K, Wu M, et al. Protective effects of liraglutide on hypercholesterolemia-associated atherosclerosis involve attenuation of endothelial-monocyte adhesion through down-regulating the LOX-1/NF-κB signaling pathway. Sci Rep. (2025) 15:27429. doi: 10.1038/s41598-025-13014-2
32. Galyfos G, Liakopoulos D, Chamzin A, Sigala F, and Filis K. A systematic review and meta-analysis of early and late outcomes after endovascular angioplasty among patients with thromboangiitis obliterans and chronic limb ischemia. J Vasc Surg. (2023) 77:1534–1541.e1532. doi: 10.1016/j.jvs.2022.09.017
33. Kurata A, Franke FE, MaChinami R, and Schulz A. Thromboangiitis obliterans: classic and new morphological features. Virchows Arch. (2000) 436:59–67. doi: 10.1007/PL00008199
34. Li B, Ming H, Qin S, Nice EC, Dong J, Du Z, et al. Redox regulation: mechanisms, biology and therapeutic targets in diseases. Signal Transduct Target Ther. (2025) 10:72. doi: 10.1038/s41392-024-02095-6
35. Swetha K, Indumathi MC, Kishan R, Siddappa S, Chen CH, and Marathe GK. Selenium mitigates caerulein and LPS-induced severe acute pancreatitis by inhibiting MAPK, NF-κB, and STAT3 signaling via the nrf2/HO-1 pathway. Biol Trace Elem Res. (2025) 203:4728–50. doi: 10.1007/s12011-025-04531-2
36. Joras M, Poredos P, and Fras Z. Endothelial dysfunction in Buerger’s disease and its relation to markers of inflammation. Eur J Clin Invest. (2006) 36:376–82. doi: 10.1111/j.1365-2362.2006.01646.x
37. Guber S, Ebrahimian T, Heidari M, Eliopoulos N, and Lehoux S. Endothelial nitric oxide synthase overexpressing human early outgrowth cells inhibit coronary artery smooth muscle cell migration through paracrine functions. Sci Rep. (2018) 8:877. doi: 10.1038/s41598-017-18848-z
38. Liang X, Hou X, Yang Y, Liu H, Guo R, Yang Z, et al. The feedback loop of “EMMPRIN/NF-κB” worsens atherosclerotic plaque via suppressing autophagy in macrophage. J Mol Cell Cardiol. (2018) 114:129–40. doi: 10.1016/j.yjmcc.2017.11.008
39. Hou J, Lin Y, Zhu C, Chen Y, Lin R, Lin H, et al. Zwitterion-lubricated hydrogel microspheres encapsulated with metformin ameliorate age-associated osteoarthritis. Adv Sci (Weinh). (2024) 11:e2402477. doi: 10.1002/advs.202402477
40. Liu X, Zhu J, Zhang Q, Hu H, Zhang W, Xu H, et al. Multifunctional fluorescent probe for simultaneous revealing Cys and ONOO(-) dynamic correlation in the ferroptosis. Spectrochim Acta A Mol Biomol Spectrosc. (2024) 315:124248. doi: 10.1016/j.saa.2024.124248
41. Nuchuchua O, Seephan S, Srinuanchai W, Temviriyanukul P, and Pongrakhananon V. Antioxidant and anti-inflammatory benefits of gymnema inodorum leaf extract in human umbilical vein endothelial cells under peroxynitrite stress. Antioxid (Basel). (2025) 14:427. doi: 10.3390/antiox14040427
42. Barzegar-Fallah A, Ghaffari-Bohlouli P, Nadjafi S, Razmi A, Dehpour AR, Ghaffarian-Bahraman A, et al. Tropisetron attenuates high-glucose-induced vascular endothelial dysfunction via inhibition of calcineurin/NFAT signalling. Eur J Pharmacol. (2025) 994:177389. doi: 10.1016/j.ejphar.2025.177389
43. Adigüzel Y, Yilmaz E, and Akar N. Effect of eNOS and ET-1 polymorphisms in thromboangiitis obliterans. Clin Appl Thromb Hemost. (2010) 16:103–6. doi: 10.1177/1076029609336854
44. Zhang Z, Ji J, Zhang D, Ma M, and Sun L. Protective effects and potential mechanism of salvianolic acid B on sodium laurate-induced thromboangiitis obliterans in rats. Phytomedicine. (2020) 66:153110. doi: 10.1016/j.phymed.2019.153110
45. Gu JJ, Wei YR, Ma K, Wang XQ, and Gao HL. Protective effects and potential mechanism of tongxinluo on mice with thromboangiitis obliterans induced by sodium laurate. Chin J Integr Med. (2023) 29:608–16. doi: 10.1007/s11655-023-3630-3
46. Sprenkle NT, Sims SG, Sánchez CL, and Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. (2017) 12:42. doi: 10.1186/s13024-017-0183-y
47. Place DE, Samir P, Karki R, Briard B, Vogel P, and Kanneganti TD. ASK family kinases are required for optimal NLRP3 inflammasome priming. Am J Pathol. (2018) 188:1021–30. doi: 10.1016/j.ajpath.2017.12.006
48. Hang L, Peng Y, Xiang R, Li X, and Li Z. Ox-LDL causes endothelial cell injury through ASK1/NLRP3-mediated inflammasome activation via endoplasmic reticulum stress. Drug Des Devel Ther. (2020) 14:731–44. doi: 10.2147/DDDT.S231916
49. Davidson SM and Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. (2007) 100:1128–41. doi: 10.1161/01.RES.0000261970.18328.1d
50. Quintero M, Colombo SL, Godfrey A, and Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U.S.A. (2006) 103:5379–84. doi: 10.1073/pnas.0601026103
51. Rao G, Murphy B, Dey A, Dwivedi SKD, Zhang Y, Roy RV, et al. Cystathionine beta synthase regulates mitochondrial dynamics and function in endothelial cells. FASEB J. (2020) 34:9372–92. doi: 10.1096/fj.202000173R
52. Li C, Pan Y, Tan Y, Wang Y, and Sun X. PINK1-dependent mitophagy reduced endothelial hyperpermeability and cell migration capacity under simulated microgravity. Front Cell Dev Biol. (2022) 10:896014. doi: 10.3389/fcell.2022.896014
53. Liu C, Kong X, Wu X, Wang X, Guan H, Wang H, et al. Alleviation of A disintegrin and metalloprotease 10 (ADAM10) on thromboangiitis obliterans involves the HMGB1/RAGE/NF-κB pathway. Biochem Biophys Res Commun. (2018) 505:282–9. doi: 10.1016/j.bbrc.2018.09.002
54. Ma T, Jiang D, Liu W, Li Z, Gao K, and Zhang Y. Mechanistic insights into fluoride-induced reproductive toxicity in female ovine animals: Mitochondrial dysfunction and ER stress-driven apoptosis in ovine granulosa cells. Ecotoxicol Environ Saf. (2025) 303:118830. doi: 10.1016/j.ecoenv.2025.118830
55. Zhang Q, Sun Q, Tong Y, Bi X, Chen L, Lu J, et al. Leonurine attenuates cisplatin nephrotoxicity by suppressing the NLRP3 inflammasome, mitochondrial dysfunction, and endoplasmic reticulum stress. Int Urol Nephrol. (2022) 54:2275–84. doi: 10.1007/s11255-021-03093-1
56. Samidurai M, Palanisamy BN, Bargues-Carot A, Hepker M, Kondru N, Manne S, et al. PKC delta activation promotes endoplasmic reticulum stress (ERS) and NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation subsequent to asynuclein-induced microglial activation: involvement of thioredoxin-interacting protein (TXNIP)/thioredoxin (Trx) redoxisome pathway. Front Aging Neurosci. (2021) 13:661505. doi: 10.3389/fnagi.2021.661505
57. Djebri NC, Zoudji S, Messaoud A, Messali R, Loudjedi S, and Aribi M. Metformin inhibits NF-κB p65/RelA-NLRP3 inflammasome-IL-1β axis, attenuates lipid droplet accumulation, and reprograms CD14/CD16 expression in monocytes exposed to colorectal tumor-conditioned medium. Int Immunopharmacol. (2025) 164:115299. doi: 10.1016/j.intimp.2025.115299
58. Ning C, Gao F, Wang Z, An H, Liu P, Sun Y, et al. LBH589 reduces oxidized mitochondrial DNA and suppresses NLRP3 inflammasome activation to relieve pulmonary inflammation. PloS One. (2025) 20:e0328522. doi: 10.1371/journal.pone.0328522
59. Tan J, Han X, Li S, Wang Q, Zhao L, Li Y, et al. Platelet-activating factor disrupts the nasal epithelial barrier independently of the platelet-activating factor receptor pathway. Allergy Asthma Immunol Res. (2025) 17:212–25. doi: 10.4168/aair.2025.17.2.212
60. Schuschke DA, Saari JT, and Miller FN. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol Lett. (2001) 76:139–44. doi: 10.1016/S0165-2478(01)00171-7
61. Guo H, Sun L, Ling S, and Xu JW. Levistilide A ameliorates NLRP3 expression involving the syk-p38/JNK pathway and peripheral obliterans in rats. Mediators Inflammation. (2018) 2018:7304096. doi: 10.1155/2018/7304096
62. Lv X, Zhao L, Song Y, Chen W, and Tuo Q. Deciphering the role of copper homeostasis in atherosclerosis: from molecular mechanisms to therapeutic targets. Int J Mol Sci. (2024) 25:11462. doi: 10.3390/ijms252111462
63. Zhao Q, Ma Y, and Wang S. Scutellarin alleviates cuprizone-induced demyelination by improving mitochondrial dysfunction, reducing lipid oxidation and inhibiting the p38 MAPK pathway. Antioxid (Basel). (2025) 14:723. doi: 10.3390/antiox14060723
64. Cao Y, Zhang W, Zhou B, Zhang Y, Xing M, and Wang Y. Copper exposure elicits pyroptosis and inflammation in bursa of Fabricius and omega-3 reverses this phenomenon by inhibiting NF-κB-NLRP3-GSDMD axis: In vivo, in vitro, and in silico investigations. Pestic Biochem Physiol. (2025) 212:106460. doi: 10.1016/j.pestbp.2025.106460
65. Uy GL, DeAngelo DJ, Lozier JN, Fisher DM, Jonas BA, Magnani JL, et al. Targeting hematologic Malignancies by inhibiting E-selectin: A sweet spot for AML therapy? Blood Rev. (2024) 65:101184. doi: 10.1016/j.blre.2024.101184
66. He H, Zou Z, Wang B, Xu G, Chen C, Qin X, et al. Copper oxide nanoparticles induce oxidative DNA damage and cell death via copper ion-mediated P38 MAPK activation in vascular endothelial cells. Int J Nanomed. (2020) 15:3291–302. doi: 10.2147/IJN.S241157
67. Vinchi F, Sparla R, Passos ST, Sharma R, Vance SZ, Zreid HS, et al. Vasculo-toxic and pro-inflammatory action of unbound haemoglobin, haem and iron in transfusion-dependent patients with haemolytic anaemias. Br J Haematol. (2021) 193:637–58. doi: 10.1111/bjh.17361
68. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. (2021) 17:4266–85. doi: 10.1080/15548627.2021.1911016
69. Wang R, Wang M, Ye J, Sun G, and Sun X. Mechanism overview and target mining of atherosclerosis: Endothelial cell injury in atherosclerosis is regulated by glycolysis (Review). Int J Mol Med. (2021) 47:65–76. doi: 10.3892/ijmm.2020.4798
70. Zhao X, Tan F, Cao X, Cao Z, Li B, Shen Z, et al. PKM2-dependent glycolysis promotes the proliferation and migration of vascular smooth muscle cells during atherosclerosis. Acta Biochim Biophys Sin (Shanghai). (2020) 52:9–17. doi: 10.1093/abbs/gmz135
71. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. (2013) 154:651–63. doi: 10.1016/j.cell.2013.06.037
72. Huang Y, Cong A, Li J, Zhou Z, Zhou H, Su C, et al. Glycolysis in peritubular endothelial cells and microvascular rarefaction in CKD. J Am Soc Nephrol. (2025) 36:19–33. doi: 10.1681/ASN.0000000000000488
73. Liu Z, Xu J, Ma Q, Zhang X, Yang Q, Wang L, et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci Transl Med. (2020) 12:eaay1371. doi: 10.1126/scitranslmed.aay1371
74. Fujioka A, Yanishi K, Yukawa A, Imai K, Yokota I, Fujikawa K, et al. A multicenter prospective interventional trial of therapeutic angiogenesis using bone marrow-derived mononuclear cell implantation for patients with critical limb-threatening ischemia caused by thromboangiitis obliterans. Circ J. (2023) 87:1229–37. doi: 10.1253/circj.CJ-23-0046
75. Sauvaget F, Debray M, Hervé de Sigalony JP, Fichelle JM, Farge D, Lémann M, et al. Colonic ischemia reveals thromboangiitis obliterans (Buerger’s disease). Gastroenterology. (1996) 110:900–3. doi: 10.1053/gast.1996.v110.pm8608901
76. Chen Q, Chen J, Li J, Cheng Y, Zhang R, and Liu Z. Recent advances of oxidative stress in thromboangiitis obliterans: biomolecular mechanisms, biomarkers, sources and clinical applications. Thromb Res. (2023) 230:64–73. doi: 10.1016/j.thromres.2023.08.015
77. Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, et al. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity. (2006) 25:271–83. doi: 10.1016/j.immuni.2006.05.014
78. Mócsai A, Ruland J, and Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
79. Arndt PG, Suzuki N, Avdi NJ, Malcolm KC, and Worthen GS. Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-Kinase and Syk-mediated pathways. J Biol Chem. (2004) 279:10883–91. doi: 10.1074/jbc.M309901200
80. Tsantarliotou MP, Lavrentiadou SN, Psalla DA, Margaritis IE, Kritsepi MG, Zervos IA, et al. Suppression of plasminogen activator inhibitor-1 (PAI-1) activity by crocin ameliorates lipopolysaccharide-induced thrombosis in rats. Food Chem Toxicol. (2019) 125:190–7. doi: 10.1016/j.fct.2019.01.001
81. Huebner BR, Moore EE, Moore HB, Stettler GR, Nunns GR, Lawson P, et al. Thrombin provokes degranulation of platelet α-granules leading to the release of active plasminogen activator inhibitor-1 (PAI-1). Shock. (2018) 50:671–6. doi: 10.1097/SHK.0000000000001089
82. Ulfhammer E, Larsson P, Karlsson L, Hrafnkelsdóttir T, Bokarewa M, Tarkowski A, et al. TNF-alpha mediated suppression of tissue type plasminogen activator expression in vascular endothelial cells is NF-kappaB- and p38 MAPK-dependent. J Thromb Haemost. (2006) 4:1781–9. doi: 10.1111/j.1538-7836.2006.02035.x
83. Zheng X, Liu H, Ma M, Ji J, Zhu F, and Sun L. Anti-thrombotic activity of phenolic acids obtained from Salvia miltiorrhiza f. alba in TNF-α-stimulated endothelial cells via the NF-κB/JNK/p38 MAPK signaling pathway. Arch Pharm Res. (2021) 44:427–38. doi: 10.1007/s12272-021-01325-7
84. Wei Z, Jiang X, Qiao H, Zhai B, Zhang L, Zhang Q, et al. STAT3 interacts with Skp2/p27/p21 pathway to regulate the motility and invasion of gastric cancer cells. Cell Signal. (2013) 25:931–8. doi: 10.1016/j.cellsig.2013.01.011
85. Cacione DG, Macedo CR, do Carmo Novaes F, and Baptista-Silva JC. Pharmacological treatment for Buerger’s disease. Cochrane Database Syst Rev. (2020) 5:Cd011033. doi: 10.1002/14651858.CD011033.pub4
86. Twine CP, Kakkos SK, Aboyans V, Baumgartner I, Behrendt CA, Bellmunt-Montoya S, et al. Editor’s choice - european society for vascular surgery (ESVS) 2023 clinical practice guidelines on antithrombotic therapy for vascular diseases. Eur J Vasc Endovasc Surg. (2023) 65:627–89. doi: 10.1016/j.ejvs.2023.03.042
87. Sen I, Agarwal S, Tharyan P, and Forster R. Lumbar sympathectomy versus prostanoids for critical limb ischaemia due to non-reconstructable peripheral arterial disease. Cochrane Database Syst Rev. (2018) 4:Cd009366. doi: 10.1002/14651858.CD009366.pub2
88. Cacione DG, Moreno DH, Nakano LC, and Baptista-Silva JC. Surgical sympathectomy for Buerger’s disease. JRSM Open. (2017) 8:2054270417717666. doi: 10.1177/2054270417717666
89. Chahal A, Malla S, Sharma S, Chumber S, and Madhusudhan KS. CT-guided lumbar sympathectomy as a last option for chronic limb-threatening ischemia of the lower limbs: evaluation of technical factors and long-term outcomes. AJR Am J Roentgenol. (2021) 216:1273–82. doi: 10.2214/AJR.20.23089
90. Uyanık SA, Öğüşlü U, Aminu IS, Yılmaz B, Çevik H, Atlı E, et al. Endovascular treatment of critical limb ischemia in buerger disease (Thromboangiitis obliterans) with midterm follow-up: A viable option when bypass surgery is not feasible. AJR Am J Roentgenol. (2021) 216:421–7. doi: 10.2214/AJR.20.23023
91. Serefli D and Saydam O. Endovascular treatment of Buerger’s disease in patients with critical limb ischaemia. Cardiovasc J Afr. (2022) 33:254–9. doi: 10.5830/CVJA-2022-018
92. Soliman M, Mowafy K, Elsaadany NA, Soliman R, and Elmetwally A. Thromboangiitis obliterans: Aggressive angioplasty provides a potential solution (randomized pilot study). SAGE Open Med. (2020) 8:2050312120927636. doi: 10.1177/2050312120927636
93. Modaghegh MS and Hafezi S. Endovascular treatment of thromboangiitis obliterans (Buerger’s disease). Vasc Endovascular Surg. (2018) 52:124–30. doi: 10.1177/1538574417744085
94. Jiang X, Ju S, Chen B, Jiang J, Shi Y, Ma T, et al. Safety and effectiveness of excimer laser ablation combined with drug-coated balloon for atherosclerotic obliterans in the lower extremity. J Endovasc Ther. (2023) 30:721–9. doi: 10.1177/15266028221092979
95. Hong M and Jin Q. Therapeutic effects of transverse tibial bone transport in lower limb thromboangiitis obliterans. Am J Transl Res. (2025) 17:4331–40. doi: 10.62347/HAAJ5979
96. Sun Y, Zhao J, Zhang L, Li Z, and Lei S. Effectiveness and safety of stem cell therapy for diabetic foot: a meta-analysis update. Stem Cell Res Ther. (2022) 13:416. doi: 10.1186/s13287-022-03110-9
97. Reider S, Binder L, Fürst S, Hatzl S, and Blesl A. Hematopoietic stem cell transplantation in refractory crohn’s disease: should it be considered? Cells. (2022) 11:3463. doi: 10.3390/cells11213463
98. Liu H, Pan T, Liu Y, Fang Y, Fang G, Jiang X, et al. The peripheral blood mononuclear cells versus purified CD34(+) cells transplantation in patients with angiitis-induced critical limb ischemia trial: 5-year outcomes and return to work analysis-a randomized single-blinded non-inferiority trial. Stem Cell Res Ther. (2022) 13:116. doi: 10.1186/s13287-022-02804-4
99. Shi M, Shang S, Yang Y, Li Q, and Bai XY. Establishment of PLAFMCi007-A, an induced pluripotent stem cell line, from peripheral blood mononuclear cells (PBMCs) of a healthy adult woman. Stem Cell Res. (2022) 61:102760. doi: 10.1016/j.scr.2022.102760
100. Najafi H, Abolmaali SS, Heidari R, Valizadeh H, Tamaddon AM, and Azarpira N. Integrin receptor-binding nanofibrous peptide hydrogel for combined mesenchymal stem cell therapy and nitric oxide delivery in renal ischemia/reperfusion injury. Stem Cell Res Ther. (2022) 13:344. doi: 10.1186/s13287-022-03045-1
101. Lou K, Hu J, Tong J, and Wang Z. Nanoscale therapeutics for erectile dysfunction: a meta-analysis of stem cell-derived extracellular vesicles as natural nanoparticles in diabetic rat models. Stem Cell Res Ther. (2025) 16:278. doi: 10.1186/s13287-025-04389-0
102. Zarzycka M, Korzekwa AJ, Dulińska-Litewka J, Kaingu CK, and Kotula-Balak M. Red deer (Cervus elaphus L.) antler stem cell culture medium inhibits prostate cancer cells. Histochem Cell Biol. (2025) 163:41. doi: 10.1007/s00418-025-02373-6
103. Liu H, Cui D, Huangfu S, Wang X, Yu X, Yang H, et al. VCAM-1(+) mesenchymal stem/stromal cells reveal preferable efficacy upon an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis over the VCAM-1(-) counterpart. Neurochem Res. (2024) 50:40. doi: 10.1007/s11064-024-04267-w
104. Nakamura K. Key mediators of the efficacy of mesenchymal stem cells on in vivo disease models. Cell Transplant. (2025) 34:9636897251348566. doi: 10.1177/09636897251348566
105. Lech W, Kot M, Welniak-Kaminska M, Drabik M, Buzanska L, and Zychowicz M. Dynamic changes in BDNF, VEGF, and GDNF after transplanting human protein-based scaffolds with Wharton’s Jelly MSCs in a rat brain injury model. Sci Rep. (2025) 15:22625. doi: 10.1038/s41598-025-04269-w
106. Wang Z, Shi M, Liu Z, Chen Y, Shi X, and Wang J. The stromal vascular fraction mitigates bleomycin-induced skin fibrosis in mice by modulating vascular lesions and secreting antifibrotic factors at different stages of disease. Stem Cell Res Ther. (2025) 16:328. doi: 10.1186/s13287-025-04470-8
107. Xi Y, Jingying D, Chenglong L, Hong Z, Rong Z, Xiaodong W, et al. Epigenetic therapy promotes the ratio of th1/th17 lineage to reverse immune evasion and treat leukemia relapse post-allogeneic stem cell transplantation in non-APL AML patients. Front Mol Biosci. (2020) 7:595395. doi: 10.3389/fmolb.2020.595395
108. Tian J, Zhu Q, Zhang Y, Bian Q, Hong Y, Shen Z, et al. Olfactory ecto-mesenchymal stem cell-derived exosomes ameliorate experimental colitis via modulating th1/th17 and treg cell responses. Front Immunol. (2020) 11:598322. doi: 10.3389/fimmu.2020.598322
Keywords: thromboangiitis obliterans, endothelial cell, immune dysregulation, mitochondrial dysfunction, copper/iron homeostasis, thrombogenicity, therapeutic interventions
Citation: Wang W, Chang S and Zhao G (2025) Endothelial activation in thromboangiitis obliterans: mechanisms and therapeutic horizons. Front. Immunol. 16:1668203. doi: 10.3389/fimmu.2025.1668203
Received: 17 July 2025; Accepted: 13 August 2025;
Published: 27 August 2025.
Edited by:
Jin Bin, Shandong University, ChinaReviewed by:
Zhang Hao, Zunyi Medical University, ChinaCopyright © 2025 Wang, Chang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Zhao, Z3p6eXl4MDkwOUAxNjMuY29t