 Ziwen Qin
Ziwen Qin Wenjuan Liu2
Wenjuan Liu2 Chuanjun Huang
Chuanjun Huang Wei Zhang
Wei Zhang- 1The First Clinical Medical College, Shandong University of Traditional Chinese Medicine, Jinan, Shandong, China
- 2Department of Pulmonary and Critical Care Medicine, Yantaishan Hospital, Yantai, Shandong, China
- 3Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, China
- 4Department of Respiratory and Critical Care Medicine, Shandong University of Traditional Chinese Medicine Affiliated Hospital, Jinan, Shandong, China
Chronic obstructive pulmonary disease (COPD) is currently one of the major causes of death and hospitalization globally. Pulmonary inflammation and oxidative stress are considered important mechanisms underlying the disease. Recent studies have indicated that the metabolic processes of immune cells in COPD, notably alveolar macrophages (AMs), may undergo significant alterations, exhibiting distinct metabolic characteristics related to their functional state and polarization phenotype. This phenomenon is known as the immunometabolic reprogramming of macrophages. In this article, we review the polarization phenotype and metabolic characteristics of macrophages in COPD, as well as the mechanisms affecting macrophage metabolism, and discuss the potential significance of pathways targeting immunometabolism of AMs in the treatment of COPD.
1 Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory pulmonary disease marked by persistent airflow obstruction. Its heterogeneity is manifested in parenchymal destruction and airway remodeling, often accompanied by chronic bronchitis and emphysema, which collectively lead to a progressive and irreversible decline in lung function. COPD symptoms often encompass coughing, expectoration, dyspnea, and wheezing. Prolonged tobacco exposure, occupational exposure to particulate matter environments, or indoor air pollution may exacerbate chronic airway inflammation, intensifying clinical symptoms and hastening disease progression. Poorly controlled COPD may further progress to cor pulmonale or even respiratory failure (1). According to World Health Organization (WHO) data, COPD currently ranks as the fourth leading cause of death globally, causing 3.5 million deaths in 2021, which constitutes approximately 5% of the total global mortality (2). COPD is regarded as the consequence of dynamic gene-environment interactions; however, its specific pathophysiology remains inadequately understood. Widely recognized mechanisms include airway oxidative stress, inflammatory responses, and protease/antiprotease imbalance. Furthermore, the crucial role of macrophages in the pathogenesis of COPD has been frequently reported.
The respiratory tract is in direct contact with the external environment (3), and its immune system, particularly alveolar macrophages (AMs), plays an important role in maintaining immunological homeostasis. AMs exert crucial immune defense and surveillance functions by phagocytosing inhaled contaminants and pathogens (4, 5). In COPD, this delicate balance was disrupted, as evidenced by elevated macrophage levels in the bronchoalveolar lavage fluid (BALF) and damaged lung tissue of patients (6), which may coordinate the immune response (7) but also contributed to persistent airway inflammation and alveolar destruction. More critically, AMs in COPD patients exhibited significant phagocytic defects, which correlated with worse pulmonary function following bacterial challenges (8). These observations raise a pivotal question: why do AMs become dysfunctional despite their increased abundance? This shifts the research focus from cell numbers to the potential mechanisms regulating their functional state.
The core of this functional dysregulation might be due to macrophage immunometabolic reprogramming—an emerging and crucial research area. AMs exhibit heterogeneous phenotypes under pathological conditions (9). Various studies pointed out that the metabolic alterations in AMs enhanced the adaptability and uniqueness of their functional subsets (10, 11). Macrophages are traditionally categorized into classically activated (M1) and alternatively activated (M2) phenotypes (12), and their functional states may shaped by core metabolic pathways: M1 polarization is linked to aerobic glycolysis (the “Warburg effect”), driving pro-inflammatory mediator production, whereas M2 polarization relies on oxidative phosphorylation (OXPHOS) and the tricarboxylic acid (TCA) cycle to support anti-inflammatory and repair functions (13, 14). Consequently, alterations in macrophage metabolic patterns may dictate their inflammatory responses (13, 15). In the specific condition of COPD, the imbalance of this “metabolism-polarization-function” axis is particularly critical: M1-associated glycolysis promotes tissue destruction, while dysregulated M2-associated metabolic processes can lead to abnormal tissue repair and fibrosis. Prominent metabolic alterations in AMs may strongly correlate with disease severity in COPD patients (16), introducing the viewpoint that metabolic reprogramming may not as an epiphenomenon but as a crucial promoter of COPD pathophysiology, explaining why targeting this axis represents a promising new therapeutic strategy.
This review focuses on the phenotypes and function states of AMs associated with the pathogenesis of COPD, as well as the specific mechanisms underlying their heterogeneous immunometabolism patterns, referred to as metabolic reprogramming, hoping to be exploited to develop potential therapeutic ideas for COPD.
2 Macrophage function and metabolic characteristics
Macrophages are extensively present in various tissues throughout the body and therefore can be classified according to their anatomical location. Based on ontological origin, the macrophage populations existing in the lungs consist primarily of tissue-resident (TR-) AMs, monocyte-derived (MO-) AMs, and interstitial macrophages. Notably, TR-AMs, which originate from embryonic development, are continuously renewed within lung tissue. Circulating monocytes in the peripheral blood can be rapidly recruited to the sites of infection or injury during pulmonary inflammation and subsequently differentiate into MO-AMs (17–20). Studies of airway AMs in lung transplant patients have reinforced the key position of peripheral blood CD14+CD16- (classical) monocytes, which constitute 85% of the circulating monocyte pool, in the origin of pulmonary AMs (21–24). Furthermore, based on their activation state, human monocyte-derived macrophages (MDMs) or murine bone marrow-derived macrophages (BMDMs) cultured in vitro are categorized as M1 or M2 macrophages. These subsets exhibit distinct functional tendencies and metabolic characteristics, contributing to the inflammatory process of COPD through complex immune and metabolic mechanisms (25, 26).
2.1 Macrophage polarization and function
Macrophages eliminate pathogens through phagocytosis, initiate the innate immune response in the lung, while also orchestrating pro-inflammatory, anti-inflammatory, and tissue repair processes. Changes in local microenvironmental signals induce macrophage polarization into distinct phenotypes (27). Upon stimulation with lipopolysaccharide (LPS) and/or interferon-γ (IFN-γ), M1 macrophages highly expressed inducible nitric oxide synthase (iNOS) and pro-inflammatory cytokines to drive chronic inflammation. Besides, they also inhibited tumor growth via anti-angiogenic effects (28). However, interleukin (IL)-4-activated M2 macrophages mainly participate in inflammation suppression and tissue repair, facilitating pathological angiogenesis, organ fibrosis, tumor growth, and the progression of allergic and parasitic diseases (28).
In COPD patients who smoke, studies indicated a positive correlation between pulmonary function decline/disease severity and the dual polarization of M1 and M2 in AMs. This contrasted with healthy individuals, whose airway AMs predominantly remained in a non-polarized state (7). Similarly, an increase in both the total number of pulmonary macrophages and the M2/M1 phenotype ratio was observed in the COPD mouse model (29), highlighting the critical role of excessively polarized macrophages in COPD pathologies.
2.1.1 M1 macrophages
M1 macrophage polarization, triggered by pathogens or pro-inflammatory cytokines, is central to the immune response in COPD. It can be induced in macrophages upon stimulation with bacterial endotoxin LPS, Th1 pro-inflammatory cytokines such as IFN-γ, tumor necrosis factor α (TNF-α), or granulocyte-macrophage colony-stimulating factor (GM-CSF). This polarization is characterized by the expression of specific markers, including MHC-II molecules, CD80, and CD86 on the cell surface, alongside a cytokine profile featuring high levels of IL-12 and IL-23, but low IL-10 (30). Functionally, polarized M1 macrophages generate abundant inflammatory mediators, including reactive oxygen species (ROS), iNOS, TNF-α, IL-6, IL-1β, and the chemokines chemokine (C-X-C motif) ligand 9 (CXCL9/MIG), CXCL10/IP-10, and chemokine (C-C motif) ligand 2 (CCL2/MCP-1). Through antigen presentation, they initiated Th1-type immunity, recruiting Th1 lymphocytes in response to pathogenic microorganisms (30, 31). In COPD, this M1-driven response may become dysregulated and contribute to disease pathogenesis (1).
While M1-secreted mediators initially help clear pathogens, persistent M1 activation may lead to cytotoxic tissue injury (32). Highly expressed iNOS produced elevated nitric oxide (NO) by inducing the activation of nuclear factor kappa-B (NF-κB). Together with ROS and sustained Th1 cell recruitment, they induced substantial damage to lung tissue due to their cytotoxic effects (33). Clinically, Bazzan et al. reported an increase in both M1 and M2 macrophage proportions in the lungs of COPD patients, correlating with smoking history and disease severity. Moreover, M1 polarization was more pronounced and specifically correlated with the severity of airflow obstruction, as measured by FEV1/FVC% (7), indicating the involvement of M1 hyperpolarization in Th1 immune inflammation of COPD. Cigarette smoke (CS) further shifted macrophages toward an M1-dominated state by upregulating Wnt family member 5a (Wnt5a) and suppressing anti-inflammatory peroxisome proliferator-activated receptor γ (PPARγ), thereby reinforcing pulmonary inflammation and COPD in human and mouse models (34). Thus, the metabolic and functional profile of M1 macrophages is not merely descriptive; it underlies the sustained inflammation, tissue injury, and disease progression observed in COPD.
2.1.2 M2 macrophages
Upon induction by Th2 cytokines such as IL-4, IL-13, IL-10, and transforming growth factor-β (TGF-β), polarized M2 macrophages release anti-inflammatory factors like IL-1, IL-10, and TGF-β. This functional response, which also enhances the phagocytosis of apoptotic cells and collagen deposition, serves to protect the host from inflammatory damage and promote tissue healing (35).
During the initial phase of mycobacterium tuberculosis infection, the expression of IFN-γ and iNOS in the BALF of mice gradually increased, aligning with the observed trend of AM polarization, indicating M1-biased AM polarization. However, as the inflammation persisted, the level of M1 markers gradually reduced while the M2 markers IL-4 and arginase 1 (Arg1) secretion were substitutionally enhanced. These results indicated a dynamic polarization plasticity during inflammatory progression, as evidenced by the transition from M1-dominance in acute infection to M2-preference in chronic phases (36). In COPD, this pattern manifested as increased M2 polarization during middle and later stages, initially serving to maintain pulmonary homeostasis and mitigate the excessive inflammatory responses (37). However, chronic M2 activation becomes unadaptive. Chronic inflammatory stimulation in COPD leads to repeated processes of damage and repair in lung tissues, while emphysema, the main pathological manifestation of COPD, is closely linked to the tissue repair function of M2 AMs (25). In vivo, chronic pulmonary inflammation resulting from prolonged exposure to CS can induce the highly expressed CD206 and TGF-β in macrophages, driving the adaptive immune response of the M2 phenotype to participate in tissue repair (38). Various inflammatory mediators secreted by M2 in the lung tissues of COPD patients were reported, such as matrix metalloproteinase (MMP)-2, MMP-9, MMP-12, and cathepsin S, to cause lung parenchymal injury and eventually form emphysema, with the increase in M2 amount positively correlated with the emphysema severities (39–41). An animal study reported that PM2.5-induced M2 AM polarization upregulated the level of MMP12 via the IL-4/STAT6 pathway in mice (42), while another research indicated that IL-4-induced M2 interstitial macrophages (IMs), rather than AMs, appeared to be the major producer of MMP-12 in lungs of COPD mice (43) However, similarly, both of these studies found that M2 macrophages caused the dysfunction of the alveolar epithelial barrier and ultimately led to COPD progression, suggesting that MMP-12 secreted by M2 phenotype may play an important role in the formation of emphysema signs in COPD. In vivo and in vitro, M2-directed polarization related to the TGF-β/Smad pathway in COPD was observed (29, 44), with the M2/M1 proportions generally negatively correlated with the lung function of mice (29), demonstrating the potential significance of the polarization state biased towards M2 macrophages in COPD.
As illustrated in Figure 1, AMs play an important role in the pathogenesis of COPD by driving chronic inflammation and tissue destruction. Additionally, Table 1 summarizes the distribution of different macrophage phenotypes in COPD patients compared with non-COPD people, highlighting the characteristics in AM polarization of COPD.
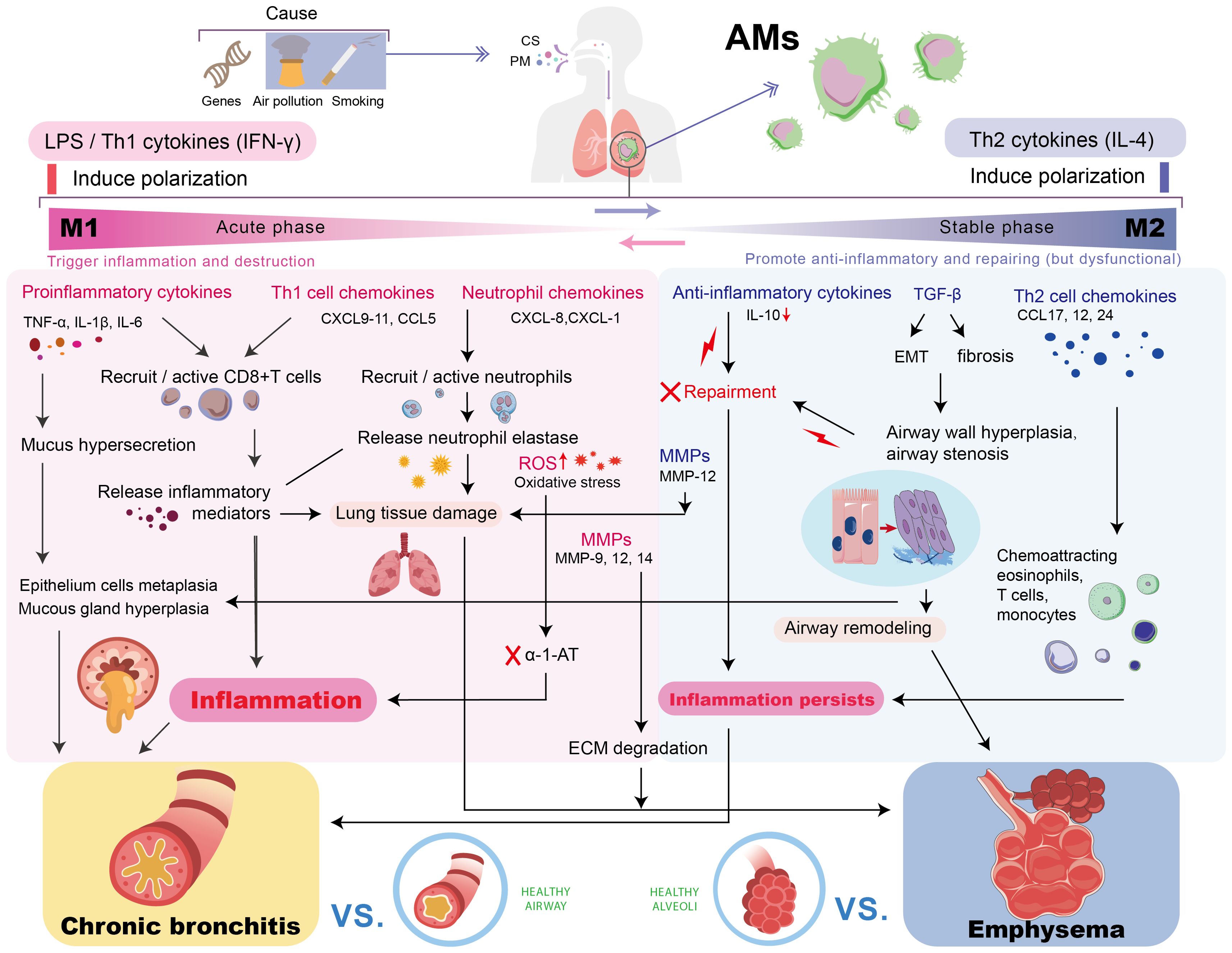
Figure 1. The Role of AMs in the pathogenesis of COPD. Chronic stimulation drives AMs to polarize into M1/M2 phenotypes. The sustained inflammation/damage mediated by M1 and the dysregulated repair/fibrosis of M2 jointly promote the key pathological processes in COPD, leading to persistent chronic inflammation, chronic bronchitis, and emphysema, ultimately resulting in progressive lung function decline. AM, alveolar macrophage; TNF-α, tumor necrosis factor-alpha; IL, interleukin; MMP, matrix metalloproteinase; ROS, reactive oxygen species; TGF-β, transforming growth factor-beta; IFN-γ, interferon-γ; CXCL, chemokine (C-X-C motif) ligand; CCL, C-C motif chemokine ligand; EMT, epithelial-mesenchymal transition; α-1-AT, α-1-antitrypsin; ECM, extracellular matrix.
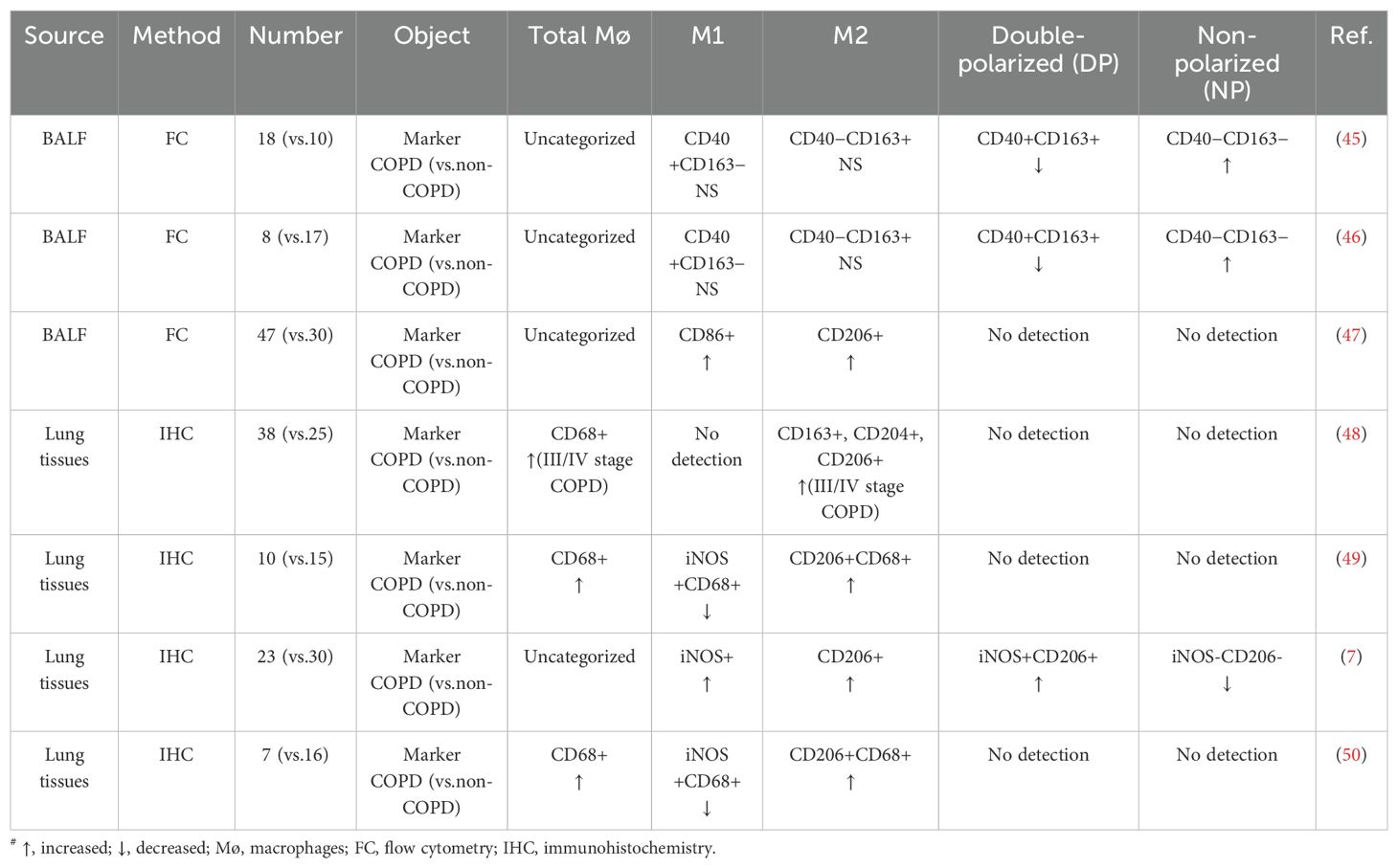
Table 1. Macrophage phenotype distribution in COPD patients#.
However, it must be pointed out that this M1/M2 classification is a simplification of the complex functional state within the body. In chronic diseases such as COPD, macrophages may exhibit mixed phenotypes or a vague state between the two (46, 51). There are apparent contradictions in evidences regarding macrophage polarization in COPD, such as the debated impact of smoking on polarization subtypes (52). These discordances may arise from disease heterogeneity, variations in clinical stages, and differences in experimental systems. Supporting this complexity, studies by Bazzan et al. and He et al. reported associations of both mixed M1/M2 polarization and phenotype skewing with COPD severity (7, 29), challenging the traditional binary classification. The dynamic alterations between M1 and M2 phenotypes underline the necessity of a balanced response across disease stages for effective immunity and repair. Consequently, a full understanding of macrophage roles in COPD requires a shift in focus from rigid categorization towards dynamic equilibrium and condition-dependent functional states.
2.2 Metabolic events that occur in macrophages
The above-mentioned functional characteristics of macrophages are closely related to intracellular metabolic reprogramming. Under the stimulation of a specific microenvironment, the metabolic pattern switches between glycolysis and OXPHOS, involving the polarization tendency of M1/M2 macrophages (14). M1 macrophage metabolism mainly shifts to glycolysis, the pentose phosphate pathway (PPP), and fatty acid (FA) synthesis to fulfill their ATP demands (10, 53, 54). Meanwhile, the functions of the TCA cycle (Krebs cycle) and OXPHOS are compromised, accompanied by the downregulated fatty acid oxidation (FAO) (10, 55, 56). Nevertheless, the M2 type mainly relies on the full TCA cycle, OXPHOS, and FAO (54).
The concept of immunometabolism was thus introduced to describe how metabolic processes within immune cells influence their functions. Specifically, these processes may not only supply energy for immune responses but also directly shape cellular activity by regulating transcriptional and post-transcriptional events. Figure 2 outlines the distinct functional states of M1 and M2 macrophages, highlighting their characteristic metabolic reprogramming.
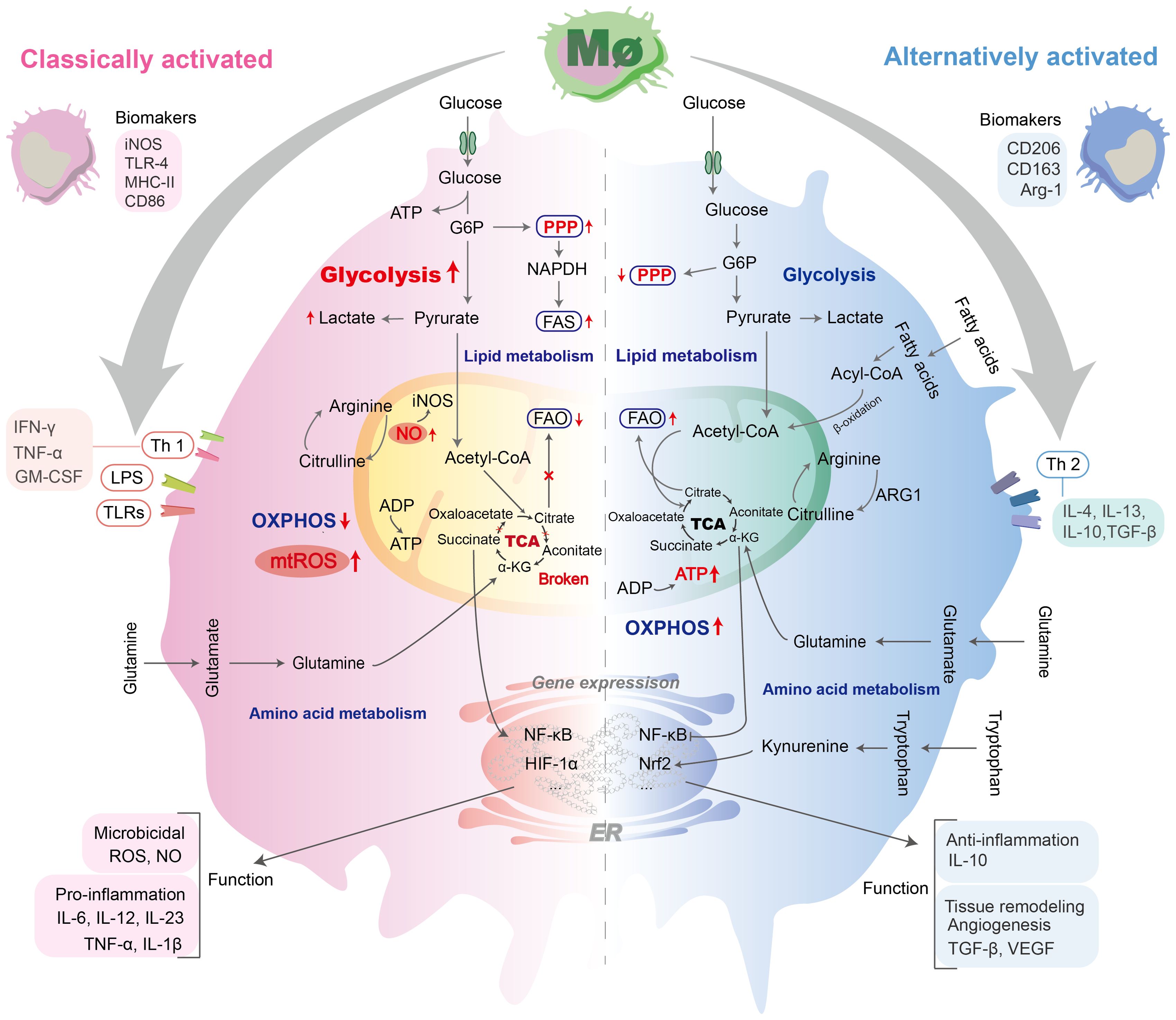
Figure 2. Polarization and metabolic alterations of M1 and M2 macrophages. M1 and M2 macrophages exhibit distinct functional phenotypes shaped by specific stimuli. M1 polarization is driven by LPS, IFN-γ, and TLRs, leading to production of pro-inflammatory mediators (e.g., IL-1β, IL-12, IL-23, ROS, TNF-α). The metabolic changes of M1 macrophages were characterized by enhanced glycolysis, disruption of the TCA cycle, and damage to OXPHOS, accompanied by downregulation of FAO. M2 polarization mainly depended on the complete TCA cycle, OXPHOS, and FAO. M2 polarization is primarily induced by IL-4, IL-13, etc., and characterized by anti-inflammatory/pro-fibrotic responses, exemplified by IL-10 and TGF-β1 release. IL, lnterleukin; MHC-II, major histocompatibility complex II; GM-CSF, granulocyte-macrophage colony-stimulating factor; Acetyl-CoA, acetyl coenzyme A; Acyl-CoA, acyl-coenzyme A; FAO, fatty acid oxidation; Glu, glucose; G6P, Glucose 6-phosphate; OXPHOS, oxidative phosphorylation; ADP, adenosine-diphosphate; ATP, adenosine-triphosphate;α-KG, α-Ketoglutaric acid; ARG1, arginase 1; IFN-γ, interferon γ; TNF-α, tumor necrosis factor-alpha; TGF-β, transforming growth factor-beta; VEGF, vascular endothelial growth factor; LPS, lipopolysaccharide; NO, nitric acid; iNOS, inducible nitric oxide synthase; PPP, pentose phosphate pathway; ROS, reactive oxygen species; mtROS, mitochondrial ROS; TCA, tricarboxylic acid; TLRs, Toll-like receptors.
2.2.1 M1 macrophages
M1 macrophage polarization may be induced by metabolic reprogramming toward aerobic glycolysis, a process with potential implications for COPD pathogenesis. This shift is characterized by several interconnected metabolic adaptations that collectively sustain pro-inflammatory responses. 6-phosphofructose-2-kinase/fructose-2, 6-bisphosphatase (PFK2) isomer (L-PFK2) was induced by M1 activation, converted into a more active form (u-PFK2), and led to the accumulation of fructose-2, 6-diphosphate, which promoted intracellular aerobic glycolysis (57). Conversely, the expression of carbohydrate kinase-like protein (CARKL) was inhibited upon the activation of the M1 phenotype, which under basal conditions supports the PPP. The over-expression of CARKL led to the decreased secretion of pro-inflammatory factors, conforming with the M2 phenotype (58). Meanwhile, the increase in the consumption of glutamine and arginine was conducive to exerting the pro-inflammatory function of M1 macrophages (59, 60). Furthermore, LPS-stimulated M1 phenotype metabolism also depends on the interrupted TCA cycle, resulting in the accumulation of several intermediate products, such as citrate, succinate, fumarate, and α-ketoglutarate, with signal transduction functions (53, 61). Among them, succinate was produced by IL-1β, regulated by hypoxia-inducible factor 1-alpha (HIF1α), and the process can be blocked by 2-deoxyglucose via inhibiting glycolysis (62). HIF1α was found by metabolomics to intervene in the expression of multiple glycolytic genes (63) and thus may become a potential target for M1 metabolism.
In brief, we might consider that metabolic events in classically activated macrophages serve a dual purpose: they rapidly provide energy and reduction equivalents to fuel bactericidal activity, and they directly participate in transcriptional regulation to shape immune responses.
2.2.2 M2 macrophages
The low glycolytic levels in IL-4-activated macrophages were reported to be compensated by elevated OXPHOS, and their metabolic activity was achieved by high OXPHOS rates, an intact TCA cycle, and FAO. Activated M2 macrophages massively induced oxidative metabolic programs, including FAO, OXPHOS, and mitochondrial respiration. In the long term, it leads to substantial ATP production via the electron transport chain (ETC) to support cellular anti-inflammatory and repair functions. Simultaneously, constituents of the ETC supported OXPHOS and introduced pyruvate into the Krebs cycle (13, 15, 53). Cellular FA levels were elevated following phagocytosis of apoptotic cells by macrophages, amplifying mitochondrial respiration and inducing NAD+-dependent signaling pathways that triggered anti-inflammatory responses for tissue healing and repair (64). This may imply that FAO is a key metabolic process underpinning M2 macrophage function (65). However, this perspective remains controversial (66), reflecting a critical need to evaluate the strength and context of the evidence. The contradiction primarily centers on whether FAO is an indispensable driver of M2 polarization or a correlative consequence of it. Therefore, while the association between FAO and M2 function is well-supported, its essentiality requires more rigorous validation. Under IL-4 stimulation, the activation of its transcription factor STAT6 induced the secretion of PPARγ-coactivator protein 1β (PGC-1β), which triggered the production of key components of mitochondrial respiration (67, 68), considered critical for the metabolic switch of M2 macrophages. The knockdown of PGC-1β impaired the metabolic profile and functions of the M2 phenotype (69). In addition, PPARs, especially PPARγ and PPARδ, are essential for phenotype maintenance as well, including the coordination of the functions of the alternative activation effect and the transcription of fatty acid β oxidation-related factors (70, 71). Moreover, TNF-α-induced protein 8-like 2 (TIPE2) was also found to induce arginine metabolism (72). Increased Arg1 catalyzed the conversion of arginine to ornithine, which is associated with the M2 polarization phenotype and repair function (13, 61).
The maintenance of M1/M2 polarization balance involves the action of multiple pathways. Inhibiting oxidative metabolism impeded M2 polarization and promoted a shift toward the M1 phenotype. Conversely, forcing oxidative metabolism in M1 macrophages reinforced the manifestation of M2 phenotype (69, 73). Thus, we consider that the state of oxidative metabolism may bidirectionally regulate macrophage polarization. On the other hand, PPP restriction in M2 macrophages led to decreased ROS and NO production, while inhibition of OXPHOS by NO suppressed the polarization from M1 to M2 phenotype under the specific microenvironment (74).
In conclusion, key metabolic diversity in macrophages under different activation states has been widely accepted. Nevertheless, the intricate molecular mechanisms that coordinate these metabolic pathways remain indistinct, and how metabolic patterns specifically regulate the polarization bias has not been fully elucidated.
3 Metabolic drivers of macrophage dysfunction in COPD
The increase in both total and polarized AMs may drive COPD pathogenesis through multiple mechanisms, including the secretion of proinflammatory cytokines, chemokines, and MMPs, along with impaired phagocytic and bactericidal functions (75). As mentioned above, the differing polarization trends and cell functions resulting from macrophage metabolic reprogramming may be closely related to the pathogenesis of COPD. In COPD, key immunometabolic features include excessive ROS/NO production, oxidative stress, and iron accumulation. These features are linked to the disease through mitochondrial phenotypic alterations and dysfunction, which appear to play a central mediating role. This connection further elucidates the critical link between macrophage metabolism and COPD pathogenesis. In addition, macrophage-associated glycolysis, amino acid metabolism, and microbial metabolism may also contribute to the pathological mechanism of COPD. Evidences for metabolic alterations of AMs in COPD are summarized in Table 2.
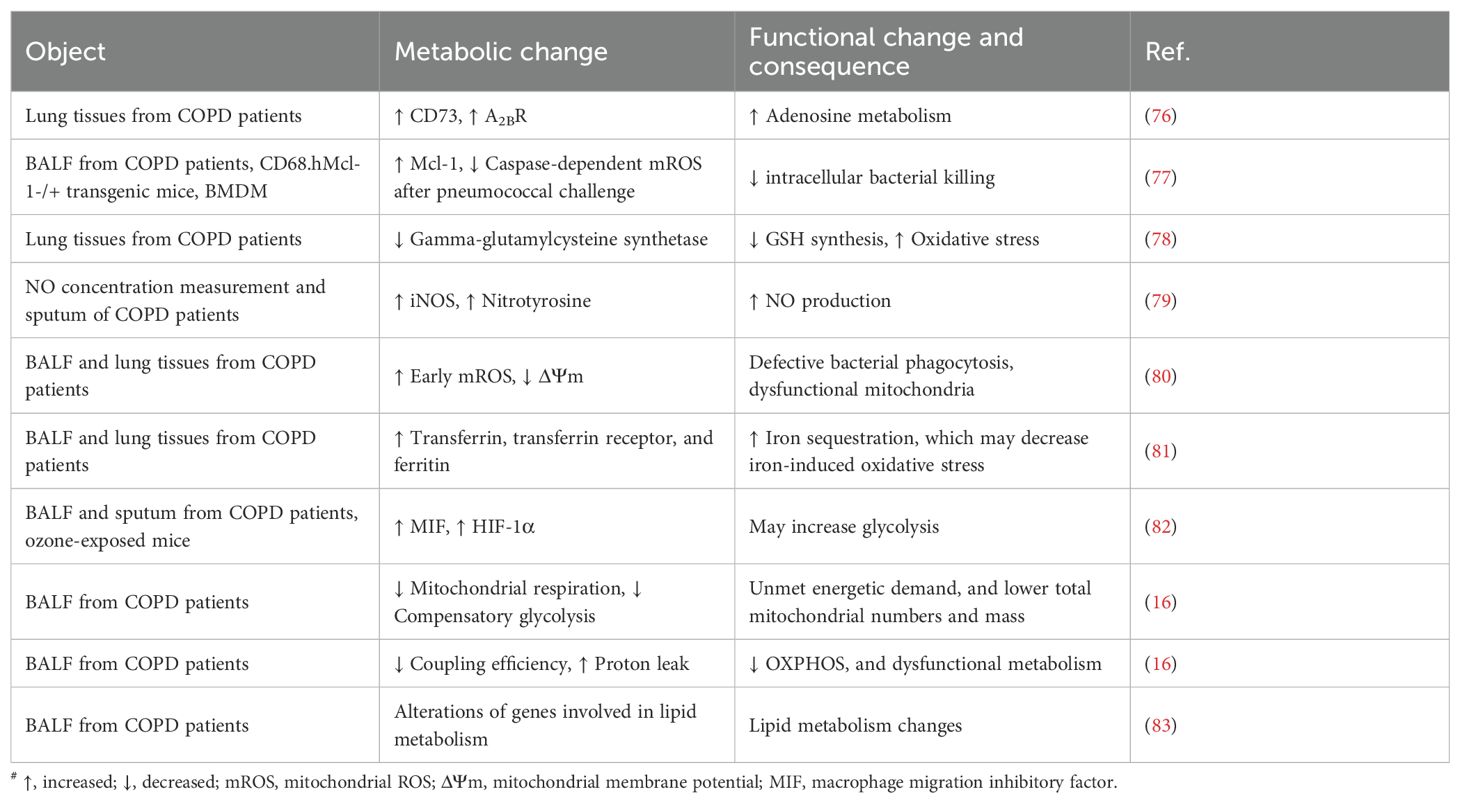
Table 2. AM metabolic alterations in COPD#.
3.1 Mitochondria-associated metabolic reprogramming of macrophages
Mitochondria serve as the central hub for macrophage metabolic plasticity, which plays an important role in energy production, immune regulation, signal transduction, and cell fate determination, intervening the inflammation regulation and macrophage repair (84). A study indicated that AMs in BALF from COPD patients exhibited lower mitochondrial respiration and compensatory glycolytic defects compared with smokers, associated with a lower predicted FEV1% (16), supporting results from several prior studies (77, 80, 85). These findings indicated that COPD patients may display abnormal macrophage responses linked to mitochondrial dysfunction. This abnormality may impair the phagocytosis and bactericidal activity of AMs against respiratory pathogens, consequently driving persistent chronic airway inflammation and lung function decline. Mitochondrial metabolism is central to the cellular metabolic network, encompassing the Krebs cycle, OXPHOS, glycolysis, FAO, amino acid metabolism, etc. (86). The intricate communication, whether direct or indirect, between mitochondrial metabolism and AM polarization may lead to changes in cellular function and help explain the shifts in the immunometabolic behavior of AMs within the context of COPD pathology.
3.1.1 ROS and oxidative stress
Chronic exposure to cigarette smoke extracts (CSE) or particulate matter resulted in elevated oxidative stress, a critical metabolic characteristic in AMs of COPD patients (87). Oxidative stress led to a significant increase in the production of mitochondrial reactive oxygen species (mtROS), superoxide anions, and hydrogen peroxide in AMs (88). Moreover, oversecreting of mtROS and a decreased ratio of mROS/superoxide dismutase 2 were linked to defective bacterial killing of AMs (77).
Subsequent research indicated that AMs from COPD patients exhibited impaired phagocytosis associated with mitochondrial dysfunction (80), possibly due to the decreased mitochondrial membrane potential, which caused cellular energy depletion, proton leakage, and overproduction of mtROS (89). Further studies revealed a strong correlation between ROS level and the polarization and metabolic characteristics of macrophages. Typically, mitochondria generate ATP via electron transport and OXPHOS. However, some electrons may escape from protein complex 1 or 3 in the ETC, resulting in the generation of superoxide anion, which is subsequently converted to ROS (such as H2O2 and hydroxyl radicals·OH). When mitochondria are metabolically active to meet the high metabolic demand, ETC is overloaded, and the likelihood of electron leakage is increased. In addition, upon activation of M1, mitochondria also markedly enhance ROS production via the reverse electron transport (RET) mechanism, thereby facilitating bactericidal and pro-inflammatory functions in cells. However, a high ROS state induces oxidative stress, leading to mitochondrial DNA (mtDNA) damage and membrane integrity disruption. Therefore, the M1 phenotype mainly relies on compensatory glycolysis for rapid energy supply, and mitochondria still maintain part of their function to support the enhanced RET, resulting in the explosively generated ROS (80). On the other hand, a low ROS state shows an OXPHOS-dominated M2 metabolic mode, featured by efficient ETC and reduced ROS production, which is conducive to the maintenance of M2 polarization and facilitates anti-inflammatory and repair functions (90). Thus, ROS plays an important role in the alteration and maintenance of M1/M2 polarization, as well as macrophages’ corresponding functions.
The key pathways of ROS generation are NADPH oxidase (NOX) and RET, which produce pathogen-killing ROS and mtROS, respectively. NOX isoforms 1, 2, 4, and 5 were all observed to remain activated in patients with end-stage COPD, whereas NOX1 and NOX4 mediated oxidative stress and inflammatory responses in mice following acute CS exposure (91). Recently, NOX2 and NOX4 were found to be involved in macrophage polarization related to ROS production. The quantity of NOX2-positive macrophages was elevated in the lungs of emphysema patients, and the elastase-induced emphysema and the high expression of ROS were prevented in NOX2-deficient mice. These findings collectively suggested a potential role of NOX2 in the pathogenesis of emphysema, probably through the sirtuin 1 (SIRT1)/MMP-9 pathway involved in macrophage-specific NOX2 (92). For macrophage polarization, NOX2-dependent ROS generation may be involved in the polarization pathway of primary macrophages to the M2 phenotype, linked to the high-mobility group box 1 (HMGB1)/Toll-like receptor 2 (TLR2)/NOX2 autophagy axis (93). Moreover, NOX4 was identified as a mediator of M1 polarization in mouse intestinal macrophages via ROS (94). Nonetheless, conclusive evidence regarding the role of NOX in AM polarization in COPD remains lacking, highlighting a key area for future research that bridges mechanistic findings to COPD pathology. The ROS degradation program exhibited a correlation with macrophage polarization as well. Peroxiredoxins (Prxs) serve as crucial antioxidant enzymes that eliminate excessive ROS in cells, thereby sustaining redox homeostasis. Deficiency of Prx5 in macrophages can induce M2 polarization and a reduction in M1-related inflammatory factor expression in lung cancer macrophages, while N-acetylcysteine (NAC), an antioxidant, was observed to suppress this tendency by inhibiting ROS production (95).
Mitophagy in macrophages, as a selective autophagy mechanism, may also influence macrophage differentiation through its impact on ROS levels. Under external environments such as ROS stress, nutrient deficiency, and cellular aging, mtDNA mutations accumulate, along with a decrease in mitochondrial membrane potential and depolarization damage (96). Damaged mitochondria are encapsulated into autophagosomes and fused with lysosomes to complete degradation. This process is called mitophagy, specifically removing dysfunctional mitochondria from the cytoplasm to maintain mitochondrial functional integrity and cellular homeostasis (97). Autophagy inhibition was observed to enhance the production of ROS-associated macrophage migration inhibitory factor (MIF), which induced and continuously promoted M1 macrophage polarization (98). Low levels of MAP kinase kinase 3 (MKK3) can enhance mitophagy and regeneration of macrophages, protecting mice from sepsis-induced lung injury (99). In contrast, another study indicated that NIX-dependent mitophagy contributes to the elimination of mitochondria during macrophage polarization to the proinflammatory and more glycolytic M1 phenotype, promoting a metabolic transition toward glycolysis in macrophage metabolism (100). However, the role of mitophagy in macrophage polarization within the context of COPD still requires further investigation.
The AM-related ROS pathway and iron accumulation in COPD have also received attention in recent years. Iron metabolism was revealed to be integral to the ROS pathway. It influenced mtROS production and lipid peroxidation, thereby activating necroptosis and ferroptosis. These forms of cell death contributed to the pathogenesis of COPD (101). With the increasing severity of COPD and emphysema, the amount of iron deposition and the percentage of iron-positive macrophages (AM as the main type) increased. It supported an iron chelation mechanism activated by AMs in COPD, potentially serving as a protective mechanism against iron-induced oxidative stress (81). Based on this, an in vitro study found that treating COPD with sulforaphane can activate the antioxidant and anti-inflammatory NRF2 pathway, restoring bacterial recognition and phagocytosis in AMs (102). Moreover, the use of a mitochondrial iron chelator or a low-iron diet can protect mice from CS-induced COPD, jointly supporting the therapeutic potential of targeting the mitochondria-iron axis in COPD (103).
3.1.2 Glucose metabolism
Macrophages can manifest with bidirectional metabolic changes between aerobic OXPHOS and anaerobic glycolysis in response to specific microenvironmental stimuli (40). Early studies in immune cell metabolism identified aerobic glycolysis in neutrophils. It is characterized by increased glucose uptake and an accelerated glycolytic rate despite sufficient oxygen, leading to excessive lactate production and suppressed OXPHOS. This metabolic shift helped to generate the bio-precursors and energy required for cell proliferation (104, 105).
A study on AM metabolic profiles indicated that AMs in COPD exhibited a diminished capacity to dynamically compensate for mitochondrial dysfunction through increasing glycolysis. This pattern manifested with significantly reduced compensatory glycolysis, non-glycolysis, and non-mitochondrial extracellular acidification rate (ECAR) in COPD smokers. The AMs of COPD patients showed impairments in mitochondrial respiration and compensatory glycolysis, correlating with the decreasing trend of lung function (16). For the direction of AM polarization, glycerol-3-phosphate dehydrogenase 2 (GPD2) was found to shift the metabolic pattern of M1 macrophages during infection and promote the activation of the M2 phenotype during tissue repair by modulating glycolysis (106). Mitochondrial dysfunction in lung epithelial cells can lead to defective ATP production and enhanced glycolysis, thereby promoting arsenic-induced massive lactate production and inducing polarization toward the M2 phenotype (107). The above results indicate that glycolysis is closely related to mitochondrial function in AMs of COPD, affecting the activation state and polarization direction of macrophages. Nonetheless, its impact on macrophage function within the COPD environment and the precise mechanisms involved in the disease process remain to be further explored.
3.1.3 Lipid metabolism
Lipids encompass both fats and lipoids, of which metabolic processes are functionally coupled with mitochondria, playing a crucial role in maintaining lung function (108). In COPD, AMs exhibited GOLD-level-dependent lipid metabolism alterations (83), further indicating the potential association between COPD lipid metabolism and the phenotype and function of AMs.
The fatty acid metabolism process involving mitochondria includes fatty acid synthesis (FAS) and FAO. Mitochondria degrade glucose in the cytoplasm via glycolysis, yielding pyruvate, which is then catalyzed by the pyruvate dehydrogenase complex (PDH) to generate acetyl coenzyme A, serving as the crucial raw material for FAS. Acetyl coenzyme A undergoes transmembrane transport through the citrate-pyruvate cycle. FAO refers to the cellular process of fatty acid degradation for energy production. At this time, acyl-coenzyme A enters the mitochondrial matrix and engages in the β-oxidation of long-chain fatty acids within the mitochondria, generating acetyl coenzyme A and reducing equivalents (NADH/FADH2), providing energy for the cell and participating in immune regulation (109). The direct effects of the FAS and FAO pathways on macrophage polarization have not been fully elucidated. Nevertheless, studies have indicated that the regulation of their states may coincide with alterations in macrophage phenotype, suggesting possible indirect or potential connections between them. For example, a study on macrophage FAS found that IL-4 can activate sterol regulatory element-binding protein 1 (SREBP1), triggering the de novo lipogenesis (DNL) program, separating it from the cell’s antioxidant defense by depleting NADPH, thus increasing ROS levels. In this process, ROS acts as a second messenger, transmitting adequate DNL signals to promote the polarization of the macrophage M2 phenotype (110). On the other hand, FAO may also be related to macrophage polarization. The Fgr tyrosine kinase may promote the M1 polarization phenotype of macrophages via the phosphorylation complex II. Additionally, the clearance of mitochondrial peroxides can inhibit Fgr activation and lead to increased FAO levels (111), indicating that mitochondrial ROS-Fgr kinase may be a key regulatory axis for pro-inflammatory macrophage activation.
The above results indicate that mitochondrial FA metabolism may play a significant role in the polarization of macrophages in COPD. Additionally, sphingolipid metabolism also plays an important role in the pathogenesis of COPD by regulating the function of AMs. Ceramide, a core product of sphingolipid metabolism, can inhibit the endocytosis of AMs by down-regulating Rac1. The excessive accumulation of ceramides may impair the cell skeleton function and consequently diminish the ability of AMs to engulf apoptotic cells, thereby amplifying the damage of emphysema (112). This process may involve its metabolite sphingosine-1-phosphate (S1P), indicating that modulating the production of sphingolipid metabolites could be a viable approach for restoring the phagocytic function of AMs to treat COPD (113). Currently, there is still inadequate evidence to illustrate its impact on the polarization phenotype of COPD macrophages.
3.1.4 Amino acid metabolism
The important role of amino acid metabolism in COPD has been confirmed (114). Studies revealed that the expression of genes related to glutathione metabolism, mitochondrial transport, pyruvate metabolism, the TCA cycle, ETC were altered in smokers and COPD patients (16). Glutathione (GSH) is a tripeptide consisting of three amino acids: glutamic acid, cysteine, and glycine. This important antioxidant can safeguard cells from oxidative damage by neutralizing free radicals. Procysteine, its precursor, has been shown to enhance the phagocytic function of AMs in COPD mice (37). In addition, the expression of iNOS in COPD AMs increased, leading to elevated levels of NO and adenosine receptor A2BR (76, 79). The rise in adenosine metabolism in COPD may be related to the increased level of HIF1α in AMs (82). Moreover, it has been observed that CS can promote the glutamine metabolism of raw cells and induce their M2 polarization phenotype (115). However, the evidence regarding amino acid metabolism’s impact on macrophage polarization and function in COPD is still insufficient and requires further research.
3.2 Microbial metabolism
The interaction between the gut microbiome, its metabolites, and the pathophysiology of pulmonary diseases is referred to as the “gut-lung axis”, which has been widely discussed in respiratory diseases (116, 117). The microbial communities colonizing the airways and alveoli, such as proteobacteria, bacteroides, firmicutes, and actinobacteria, can generate numerous metabolites, notably short-chain fatty acids (SCFAs) such as acetic acid, propionic acid, and butyric acid. These metabolites can modulate pulmonary immune-inflammatory responses, affect pulmonary barrier function, and thereby intervene in the disease progression (118). Currently, macrophages are considered the main target of SCFA produced by the microbial community (119).
According to a study by Ji et al., a probiotic mixture enhanced antiviral defense by modulating the gut-lung axis: it increased gut-derived acetate and beneficial lung bacteria (including corynebacterium and lactobacillus), which improve pulmonary microbiome dysbiosis induced by respiratory syncytial virus (RSV), and boost AM phagocytosis and IFN-β secretion. These results highlighted that the microbiome-AM axis may serve as a potential pathway for regulating AM function and its downstream factors (120). Direct evidence from multi-omics analyses of airway host-microbe interactions in COPD patients indicated that indole-3-acetic acid (IAA) derived from airway microbiota alleviated neutrophil inflammation, cell apoptosis, emphysema, and lung function decline through IL-22-mediated macrophage-epithelial cell interactions. And intranasal inoculation of two airway lactobacillus (potential producers of airway IAA) for 6 weeks can alter the lung microbiome in mice, restore IAA levels, and exert a protective effect in COPD mice (121). These studies proposed a potential link between the metabolic activities of the pulmonary microbiome and the functionality of AMs in COPD. However, the effects of SCFA on the metabolism and polarization phenotype of AMs are still in the exploration stage. Studies in experimental models further clarify the roles of specific SCFAs. Butyrate has been found to alter metabolic pathways in macrophages, leading to an increase in OXPHOS and fatty acid metabolism, and inducing polarization towards the M2 phenotype. Although the specific mechanism is still unclear, SCFAs have been reported to induce the activation of genes related to the aforementioned metabolic pathways (122, 123). Furthermore, butyrate and propionate have been reported to inhibit M2 polarization and mitigate inflammatory responses caused by allergic airway reactions in animal models. And the effects of butyrate, butyric acid, and propionate on macrophage lines may be mediated by the activation of GPR43 receptors and/or inhibition of HDAC enzymes (124). These apparent contradictory evidences regarding the pro- versus anti-M2 effects of SCFAs may be due to methodological differences. Key variables such as the specific disease context, the timing and concentration of SCFA exposure, and the source of macrophages may profoundly influence the outcome. The observed dual effects could suggest a context-dependent, homeostatic role for SCFAs in immune responses. Therefore, future research might systematically control these variables to clarify the precise conditions under which SCFAs promote or suppress M2 polarization, which is essential for evaluating their therapeutic potential.
In summary, while the precise mechanisms require further clarification, microbiome-derived metabolites, including IAA, propionate, and butyrate, may intervene in AM function and polarization in COPD. This regulation occurs via the gut-lung axis and within the local pulmonary microenvironment, representing a promising novel therapeutic target.
These evidences in Chapter 3 suggest the potential mechanisms underlying the immune-related metabolic changes in AMs. While direct longitudinal studies tracing the temporal dynamics of AM immunometabolism in human COPD are still emerging, the existing evidences allows for an assumption of this process. In the acute exacerbations or early phases driven by CS, AMs may undergo a metabolic shift towards glycolysis to meet the urgent energy demands for pro-inflammatory responses. This state is characterized by the upregulation of M1-like markers and the release of ROS and proteases, contributing to tissue damage. As COPD transitions to a more chronic phase, the sustained metabolic stress and altered microenvironment may lead to mitochondrial dysfunction and the persistence of inflammation. Notably, some populations of AMs might adopt a mixed or alternative (M2-like) activation state, potentially relying on oxidative phosphorylation to varying degrees, which could paradoxically contribute to impaired bacterial clearance and fibrotic repair processes, thus driving disease progression. Spatially, the metabolic phenotype of AMs is likely shaped by local niches. For instance, the alveolar compartment might favor certain metabolic adaptations due to its direct exposure to inhaled stimuli and unique oxygen tension, whereas the bronchial tree could present a different set of metabolic challenges and inflammatory signals. Future studies could employ single-cell technologies on longitudinally collected patient samples are crucial to validate this spatiotemporal model and uncover novel, stage-specific therapeutic targets.
4 Prospective therapies targeting macrophage immunometabolism in COPD
Based on the central role of macrophage immunometabolism in COPD, targeting this axis represents a promising therapeutic strategy. Numerous natural compounds derived from medicinal plants, animals, and fungi, such as flavonoids, terpenoids, and phenols, have shown potential in modulating the immune-metabolic state in pre-clinical models of COPD (125–135), offering the advantages of fewer toxic side effects and multi-targeted mechanisms (136). The potential mechanisms of these compounds can be categorized as follows:
First, certain flavonoids can impact macrophage polarization and abundance. Fisetin, rutin, and casticin were reported to effectively reverse the increase in neutrophil and macrophage populations in BALF of rats/mice induced by CS, mitigating oxidative stress and inflammation in lungs (127, 130, 135). Naringenin, as the main flavanone in citrus, was reported to suppress the disorder of extracellular vesicular cargoes derived from BEAS-2B induced by CSE, thereby inhibiting M1 macrophage polarization (133). Second, a key mechanism involves the enhancement of antioxidant defenses. Fisetin and oroxylin A activated the Nrf2 pathway to alleviate oxidative stress (128, 130), which was similar to the efficacy of ginsenosides (126). Third, some compounds demonstrate the ability to ameliorate mitochondrial dysfunction in macrophages. Andrographolide, a diterpene lactone compound, can alleviate mitochondrial dysfunction, inflammation, and oxidative stress in RAW 264.7 macrophages exposed to CSE by inhibiting SIRT1/ERK signaling, thereby preventing tissue damage in COPD (132). Finally, several compounds showed efficacy in specific, COPD-relevant models. Compound K (CK), a secondary ginsenoside, may act as a potential ligand for glucocorticoid receptors and a regulator of macrophage inflammatory responses, exerting potential effects on COPD (134). Triterpene acids from Eriobotrya japonica (Thunb.) Lindl. Leaf significantly inhibited NO and iNOS in AMs of chronic bronchitis rats, associated with the inhibition of p38 MAPK phosphorylation signal transduction (125). Furthermore, astaxanthin and olive kermes can reduce macrophage infiltration and tissue destruction in the BALF of COPD mice induced by CS (129, 131).
While the aforementioned findings are primarily derived from preclinical models, such as CSE-treated animals or cells, they demonstrate the possibility of using natural products to intervene in COPD pathology by regulating macrophage metabolic imbalances. However, we still lack direct evidence for the specific targets towards AMs. Future studies should consider mechanistic exploration in COPD AM models to advance clinical translation.
Micro-RNAs (miRNAs), as small, endogenous non-coding RNA molecules, are considered key regulators of metabolic homeostasis that influence macrophage function and inflammatory response in chronic lung diseases associated with mitochondrial dysfunction (137, 138). In COPD, the M1 and M2 phenotypes of AMs exhibited distinct miRNA expression profiles, implying their regulatory role in macrophage polarization (139, 140). Moreover, in Mycobacterium tuberculosis (Mtb) infection, Let-7f miRNAs can inhibit NF-κB inhibitor A20 by targeting NF-κB, promoting the polarization of M1 macrophages and NF-κB activity (141). At the same time, let-7adf can promote the expression of pro-inflammatory cytokine IL-6 and the accumulation of glycolysis and succinate through targeting the succinate metabolic pathway in LPS-activated macrophages (142). Let-7a has also been reported to target SNAP23 in colorectal cancer cells, thereby inhibiting OXPHOS (143). These results emphasize the significance of miRNAs in the metabolic reprogramming of macrophages; however, their clinical application in COPD requires a more comprehensive understanding of their mechanisms and targets, which still faces challenges.
On the other hand, targeting specific metabolic pathways may reshape the metabolism and phenotypic changes of COPD macrophages, potentially serving as a strategy to inhibit inflammation. Dimethyl fumaric acid (DMF) functions as a chemical regulator of macrophage phenotype (144). It has been found to inhibit the catalytic cysteine of glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), thereby down-regulating aerobic glycolysis in activated myeloid and lymphoid cells, altering the phenotype of macrophages (145). Given that M1 macrophages are characterized by an increased aerobic glycolysis rate and glucose demand, the inhibition of glycolysis may serve as an effective way to manipulate macrophage metabolism and suppress inflammation. 2-Deoxy-D-glucose (2DG), a glycolysis inhibitor, can competitively bind to hexokinase to inhibit glycolysis, subsequently reducing the inflammatory response triggered by M1 macrophages (61). The glycolysis regulator TEPP-46 can inhibit the pro-inflammatory effect of macrophages by obstructing the tetramerization of pyruvate kinase M2 (146) and simultaneously enhance the tolerance of macrophages to endotoxin (147). Moreover, the small molecule CPUY192018 can down-regulate glycolysis in AMs of COPD patients, thereby enhancing their phagocytic function (148).
Based on this, targeted delivery of drugs to AMs may enable the precise regulation of AM immunometabolism, allowing for the optimization of therapeutic effects and avoiding capture by mucus (149). An inhalation-based drug delivery system targeting AMs employs micro- and nanocarriers for implementation. Currently, micro- and nanospheres coated with mannose, which is based on the phagocytic activity of AMs, have been developed for the targeted delivery of antibiotics in the treatment of AM bacterial infections (150). It also provides ideas for drug delivery to AMs in other diseases, such as COPD. As mentioned in this article, chelating agents, antioxidants, miRNAs, chemical regulators, etc., may be able to regulate AM immunometabolism to treat COPD. Incorporating them into aerosolized micro- or nano-delivery systems to assist in drug administration may represent a potential idea for the development of new drugs for COPD.
5 Conclusion
In the lungs, macrophages polarize towards the M1/M2 direction under the activation of inflammatory factors, exerting pro-inflammatory/anti-inflammatory effects and participating in the COPD process of airway inflammation, lung parenchymal damage, and repair. They engulf and eliminate apoptotic and necrotic tissue cells, collaboratively shaping the microenvironment of the lungs in COPD.
However, research on the staged induction of macrophage polarization by CS remains insufficient. Further studies are required to clarify the role of AMs in the microenvironment of COPD lungs based on the polarization tendencies and functional states at different disease stages, as well as the specific molecular mechanisms involved in these mechanisms to participate in the disease, so as to provide clear directions for macrophage-targeted COPD treatment. In summary, despite its utility as a foundational framework, the M1/M2 model has limitations in COPD. Future studies may therefore apply techniques such as single-cell RNA sequencing to elucidate the full spectrum of macrophage heterogeneity across COPD stages and subtypes, to deepen the understanding of macrophage phenotypes.
On the other hand, the metabolic reprogramming of macrophages under microenvironment stimuli, involving oxidative stress, mitochondrial dysfunction, and metabolic pathways such as glycolysis, lipogenesis, amino acid metabolism, and microbial metabolism, is closely related to their polarization direction and functional expression, contributing to the pathogenesis of COPD. Interfering with metabolic patterns to reshape the phenotype of macrophages is considered a promising therapeutic strategy, but before this can be achieved, a more accurate understanding of the relationship between metabolic pathways and phenotypes is required. This review explored the mechanisms of metabolic adaptation on AM phenotypes and functions in COPD, highlighting the potential of AM metabolic plasticity for drug development and offering novel concepts for targeted treatment strategies in COPD.
Author contributions
ZQ: Conceptualization, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft. WL: Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft. CH: Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. WZ: Conceptualization, Funding acquisition, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Shandong Province (ZR2021LZY031).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
COPD: chronic obstructive pulmonary disease
WHO: World Health Organization
AMs: alveolar macrophages
BALF: bronchoalveolar lavage fluid
M1: classically activated
M2: alternatively activated
OXPHOS: oxidative phosphorylation
ROS: reactive oxygen species
TCA: tricarboxylic acid
TR-: tissue-resident
MO-: monocyte-derived
MDMs: monocyte-derived macrophages
BMDMs: bone marrow-derived macrophages
LPS: lipopolysaccharide
IFN-γ: interferon-γ
iNOS: inducible nitric oxide synthase
IL: interleukin
TNF-α: tumor necrosis factor α
GM-CSF: granulocyte-macrophage colony-stimulating factor
CXCL9/MIG: chemokine (C-X-C motif) ligand 9
CCL2/MCP-1: chemokine (C-C motif) ligand 2
NF-κB: nuclear factor kappa-B
PPARγ: peroxisome proliferator-activated receptor γ
Wnt5a: Wnt family member 5a
TGF-β: transforming growth factor-β
Arg1: arginase 1
MMP: matrix metalloproteinase
PPP: pentose phosphate pathway
FA: fatty acid
FAO: fatty acid oxidation
CARKL: carbohydrate kinase-like protein
HIF1α: hypoxia-inducible factor 1-alpha
PGC-1β: PPARγ-coactivator protein 1β
TIPE2: TNF-α-induced protein 8-like 2
CSE: cigarette smoke extracts
mtROS: mitochondrial reactive oxygen species
ETC: electron transfer chain
RET: reverse electron transport
mtDNA: mitochondrial DNA
NOX: NADPH oxidase
SIRT1: sirtuin 1
HMGB1: high-mobility group box 1
TLR2: Toll-like receptor 2
Prxs: Peroxiredoxins
NAC: N-acetylcysteine
MIF: migration inhibitory factor
MKK3: MAP kinase kinase 3
ECAR: extracellular acidification rate
GPD2: glycerol-3-phosphate dehydrogenase 2
FAS: fatty acid synthesis
PDH: pyruvate dehydrogenase complex
SREBP1: sterol regulatory element-binding protein 1
DNL: de novo lipogenesis
S1P: sphingosine-1-phosphate
GSH: Glutathione
SCFA: short-chain fatty acids
RSV: respiratory syncytial virus
IAA: indole-3-acetic acid
miRNAs: Micro-RNAs
Mtb: Mycobacterium tuberculosis
DMF: Dimethyl fumaric acid
GAPDH: glyceraldehyde-3-phosphate dehydrogenase
2DG: 2-Deoxy-D-glucose
References
1. Christenson SA, Smith BM, Bafadhel M, and Putcha N. Chronic obstructive pulmonary disease. Lancet. (2022) 399:2227–42. doi: 10.1016/s0140-6736(22)00470-6
2. Organization, W.H. Chronic obstructive pulmonary disease (COPD)(2024). Available online at: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (Accessed November 06, 2024).
3. Lloyd CM and Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity. (2017) 46:549–61. doi: 10.1016/j.immuni.2017.04.005
4. Byrne AJ, Mathie SA, Gregory LG, and Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
5. Pervizaj-Oruqaj L, Ferrero MR, Matt U, and Herold S. The guardians of pulmonary harmony: alveolar macrophages orchestrating the symphony of lung inflammation and tissue homeostasis. Eur Respir Rev. (2024) 33. doi: 10.1183/16000617.0263-2023
6. Kojima J, Araya J, Hara H, Ito S, Takasaka N, Kobayashi K, et al. Apoptosis inhibitor of macrophage (AIM) expression in alveolar macrophages in COPD. Respir Res. (2013) 14:30. doi: 10.1186/1465-9921-14-30
7. Bazzan E, Turato G, Tinè M, Radu CM, Balestro E, Rigobello C, et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res. (2017) 18:40. doi: 10.1186/s12931-017-0522-0
8. Berenson CS, Kruzel RL, Eberhardt E, and Sethi S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. (2013) 208:2036–45. doi: 10.1093/infdis/jit400
9. Ginhoux F and Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
10. Wculek SK, Heras-Murillo I, Mastrangelo A, Mañanes D, Galán M, Miguel V, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. (2023) 56:516–30.e519. doi: 10.1016/j.immuni.2023.01.011
11. Malla S, Sajeevan KA, Acharya B, Chowdhury R, and Saha R. Dissecting metabolic landscape of alveolar macrophage. Sci Rep. (2024) 14:30383. doi: 10.1038/s41598-024-81253-w
12. Svedberg FR, Brown SL, Krauss MZ, Campbell L, Sharpe C, Clausen M, et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat Immunol. (2019) 20:571–80. doi: 10.1038/s41590-019-0352-y
13. Galván-Peña S and O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
14. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
15. Van den Bossche J, O’Neill LA, and Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
16. O’Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV, et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2020) 201:735–9. doi: 10.1164/rccm.201908-1683LE
17. Shi C and Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
18. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
19. Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood. (2015) 126:1357–66. doi: 10.1182/blood-2015-01-624809
20. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
21. Thomas ED, Ramberg RE, Sale GE, Sparkes RS, and Golde DW. Direct evidence for a bone marrow origin of the alveolar macrophage in man. Science. (1976) 192:1016–8. doi: 10.1126/science.775638
22. Eguíluz-Gracia I, Schultz HH, Sikkeland LI, Danilova E, Holm AM, Pronk CJ, et al. Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax. (2016) 71:1006–11. doi: 10.1136/thoraxjnl-2016-208292
23. Nayak DK, Zhou F, Xu M, Huang J, Tsuji M, Hachem R, et al. Long-term persistence of donor alveolar macrophages in human lung transplant recipients that influences donor-specific immune responses. Am J Transplant. (2016) 16:2300–11. doi: 10.1111/ajt.13819
24. Byrne AJ, Powell JE, O’Sullivan BJ, Ogger PP, Hoffland A, Cook J, et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med. (2020) 217. doi: 10.1084/jem.20191236
25. Tetley TD. Macrophages and the pathogenesis of COPD. Chest. (2002) 121:156s–9s. doi: 10.1378/chest.121.5_suppl.156s
26. Sica A and Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/jci59643
27. Varol C, Mildner A, and Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
28. Martinez FO, Helming L, and Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
29. He S, Xie L, Lu J, and Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:3029–39. doi: 10.2147/copd.S147144
30. Lu HL, Huang XY, Luo YF, Tan WP, Chen PF, and Guo YB. Activation of M1 macrophages plays a critical role in the initiation of acute lung injury. Biosci Rep. (2018) 38. doi: 10.1042/bsr20171555
31. Arora S, Dev K, Agarwal B, Das P, and Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. (2018) 223:383–96. doi: 10.1016/j.imbio.2017.11.001
32. Mills CD, Kincaid K, Alt JM, Heilman MJ, and Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
33. Nathan C. Nonresolving inflammation redux. Immunity. (2022) 55:592–605. doi: 10.1016/j.immuni.2022.03.016
34. Feller D, Kun J, Ruzsics I, Rapp J, Sarosi V, Kvell K, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. (2018) 9:1724. doi: 10.3389/fimmu.2018.01724
35. Allard B, Panariti A, and Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol. (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
36. Redente EF, Higgins DM, Dwyer-Nield LD, Orme IM, Gonzalez-Juarrero M, and Malkinson AM. Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. J Leukoc Biol. (2010) 88:159–68. doi: 10.1189/jlb.0609378
37. Hodge S, Matthews G, Mukaro V, Ahern J, Shivam A, Hodge G, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol. (2011) 44:673–81. doi: 10.1165/rcmb.2009-0459OC
38. Oliveira da Silva C, Monte-Alto-Costa A, Renovato-Martins M, Viana Nascimento FJ, Dos Santos Valença S, Lagente V, et al. Time course of the phenotype of blood and bone marrow monocytes and macrophages in the lung after cigarette smoke exposure in vivo. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18091940
39. Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2002) 26:602–9. doi: 10.1165/ajrcmb.26.5.4685
40. Hiemstra PS. Altered macrophage function in chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2013) 10 Suppl:S180–185. doi: 10.1513/AnnalsATS.201305-123AW
41. Doherty DF, Nath S, Poon J, Foronjy RF, Ohlmeyer M, Dabo AJ, et al. Protein phosphatase 2A reduces cigarette smoke-induced cathepsin S and loss of lung function. Am J Respir Crit Care Med. (2019) 200:51–62. doi: 10.1164/rccm.201808-1518OC
42. Guo X, Yang S, Zhu H, Liu F, Li K, Li G, et al. Involvement of M2 macrophages polarization in PM2.5-induced COPD by upregulating MMP12 via IL4/STAT6 pathway. Ecotoxicol Environ Saf. (2024) 283:116793. doi: 10.1016/j.ecoenv.2024.116793
43. Shibata S, Miyake K, Tateishi T, Yoshikawa S, Yamanishi Y, Miyazaki Y, et al. Basophils trigger emphysema development in a murine model of COPD through IL-4-mediated generation of MMP-12-producing macrophages. Proc Natl Acad Sci U S A. (2018) 115:13057–62. doi: 10.1073/pnas.1813927115
44. Sun SW, Chen L, Zhou M, Wu JH, Meng ZJ, Han HL, et al. BAMBI regulates macrophages inducing the differentiation of Treg through the TGF-β pathway in chronic obstructive pulmonary disease. Respir Res. (2019) 20:26. doi: 10.1186/s12931-019-0988-z
45. Akata K, Yamasaki K, Leitao Filho FS, Yang CX, Takiguchi H, Sahin B, et al. Abundance of non-polarized lung macrophages with poor phagocytic function in chronic obstructive pulmonary disease (COPD). Biomedicines. (2020) 8. doi: 10.3390/biomedicines8100398
46. Takiguchi H, Yang CX, Yang CWT, Sahin B, Whalen BA, Milne S, et al. Macrophages with reduced expressions of classical M1 and M2 surface markers in human bronchoalveolar lavage fluid exhibit pro-inflammatory gene signatures. Sci Rep. (2021) 11:8282. doi: 10.1038/s41598-021-87720-y
47. He S, Tian R, Zhang X, Yao Q, Chen Q, Liu B, et al. PPARγ inhibits small airway remodeling through mediating the polarization homeostasis of alveolar macrophages in COPD. Clin Immunol. (2023) 250:109293. doi: 10.1016/j.clim.2023.109293
48. Kaku Y, Imaoka H, Morimatsu Y, Komohara Y, Ohnishi K, Oda H, et al. Overexpression of CD163, CD204 and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PloS One. (2014) 9:e87400. doi: 10.1371/journal.pone.0087400
49. Liu J, Zhang Z, Yang Y, Di T, Wu Y, and Bian T. NCOA4-mediated ferroptosis in bronchial epithelial cells promotes macrophage M2 polarization in COPD emphysema. Int J Chron Obstruct Pulmon Dis. (2022) 17:667–81. doi: 10.2147/copd.S354896
50. Liu T, Zhang Z, Shen W, Wu Y, and Bian T. MicroRNA let-7 induces M2 macrophage polarization in COPD emphysema through the IL-6/STAT3 pathway. Int J Chron Obstruct Pulmon Dis. (2023) 18:575–91. doi: 10.2147/copd.S404850
51. Vlahos R and Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. (2014) 5:435. doi: 10.3389/fimmu.2014.00435
52. Lugg ST, Scott A, Parekh D, Naidu B, and Thickett DR. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. (2022) 77:94–101. doi: 10.1136/thoraxjnl-2020-216296
53. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
54. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. (2021) 37:109955. doi: 10.1016/j.celrep.2021.109955
55. Funk JL, Feingold KR, Moser AH, and Grunfeld C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis. (1993) 98:67–82. doi: 10.1016/0021-9150(93)90224-i
56. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. (2012) 92:829–39. doi: 10.1189/jlb.1111537
57. Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. (2010) 185:605–14. doi: 10.4049/jimmunol.0901698
58. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
59. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
60. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
61. Kelly B and O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
62. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
63. Dang B, Gao Q, Zhang L, Zhang J, Cai H, Zhu Y, et al. The glycolysis/HIF-1α axis defines the inflammatory role of IL-4-primed macrophages. Cell Rep. (2023) 42:112471. doi: 10.1016/j.celrep.2023.112471
64. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–56.e445. doi: 10.1016/j.cmet.2018.12.004
65. Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. (2016) 17:216–7. doi: 10.1038/ni.3366
66. Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, et al. Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am J Physiol Endocrinol Metab. (2017) 312:E381–e393. doi: 10.1152/ajpendo.00408.2016
67. St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. (2003) 278:26597–603. doi: 10.1074/jbc.M301850200
68. Shao D, Liu Y, Liu X, Zhu L, Cui Y, Cui A, et al. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion. (2010) 10:516–27. doi: 10.1016/j.mito.2010.05.012
69. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
70. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
71. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
72. Lou Y, Zhang G, Geng M, Zhang W, Cui J, and Liu S. TIPE2 negatively regulates inflammation by switching arginine metabolism from nitric oxide synthase to arginase. PloS One. (2014) 9:e96508. doi: 10.1371/journal.pone.0096508
73. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
74. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
75. Brusselle GG, Joos GF, and Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. (2011) 378:1015–26. doi: 10.1016/s0140-6736(11)60988-4
76. Zhou Y, Murthy JN, Zeng D, Belardinelli L, and Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One. (2010) 5:e9224. doi: 10.1371/journal.pone.0009224
77. Bewley MA, Preston JA, Mohasin M, Marriott HM, Budd RC, Swales J, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med. (2017) 196:845–55. doi: 10.1164/rccm.201608-1714OC
78. Rahman I and MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. (1996) 21:669–81. doi: 10.1016/0891-5849(96)00155-4
79. Ichinose M, Sugiura H, Yamagata S, Koarai A, and Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med. (2000) 162:701–6. doi: 10.1164/ajrccm.162.2.9908132
80. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. (2019) 54. doi: 10.1183/13993003.02244-2018
81. Philippot Q, Deslée G, Adair-Kirk TL, Woods JC, Byers D, Conradi S, et al. Increased iron sequestration in alveolar macrophages in chronic obstructive pulmonary disease. PloS One. (2014) 9:e96285. doi: 10.1371/journal.pone.0096285
82. Russell KE, Chung KF, Clarke CJ, Durham AL, Mallia P, Footitt J, et al. The MIF antagonist ISO-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PloS One. (2016) 11:e0146102. doi: 10.1371/journal.pone.0146102
83. Fujii W, Kapellos TS, Baßler K, Händler K, Holsten L, Knoll R, et al. Alveolar macrophage transcriptomic profiling in COPD shows major lipid metabolism changes. ERJ Open Res. (2021) 7. doi: 10.1183/23120541.00915-2020
84. Nassef MZ, Hanke JE, and Hiller K. Mitochondrial metabolism in macrophages. Am J Physiol Cell Physiol. (2021) 321:C1070–c1081. doi: 10.1152/ajpcell.00126.2021
85. Gleeson LE, O’Leary SM, Ryan D, McLaughlin AM, Sheedy FJ, and Keane J. Cigarette smoking impairs the bioenergetic immune response to mycobacterium tuberculosis infection. Am J Respir Cell Mol Biol. (2018) 59:572–9. doi: 10.1165/rcmb.2018-0162OC
86. Wai T and Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. (2016) 27:105–17. doi: 10.1016/j.tem.2015.12.001
87. Liu X and Chen Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J Transl Med. (2017) 15:207. doi: 10.1186/s12967-017-1306-5
88. Harju T, Kaarteenaho-Wiik R, Soini Y, Sormunen R, and Kinnula VL. Diminished immunoreactivity of gamma-glutamylcysteine synthetase in the airways of smokers’ lung. Am J Respir Crit Care Med. (2002) 166:754–9. doi: 10.1164/rccm.2112014
89. Eapen MS, Sharma P, and Sohal SS. Mitochondrial dysfunction in macrophages: a key to defective bacterial phagocytosis in COPD. Eur Respir J. (2019) 54. doi: 10.1183/13993003.01641-2019
90. Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, and Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. (2019) 224:242–53. doi: 10.1016/j.imbio.2018.11.010
91. Wang X, Murugesan P, Zhang P, Xu S, Peng L, Wang C, et al. NADPH oxidase isoforms in COPD patients and acute cigarette smoke-exposed mice: induction of oxidative stress and lung inflammation. Antioxidants (Basel). (2022) 11. doi: 10.3390/antiox11081539
92. Trocme C, Deffert C, Cachat J, Donati Y, Tissot C, Papacatzis S, et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J Pathol. (2015) 235:65–78. doi: 10.1002/path.4423
93. Shiau DJ, Kuo WT, Davuluri GVN, Shieh CC, Tsai PJ, Chen CC, et al. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci Rep. (2020) 10:13582. doi: 10.1038/s41598-020-70137-4
94. Han C, Sheng Y, Wang J, Zhou X, Li W, Zhang C, et al. NOX4 promotes mucosal barrier injury in inflammatory bowel disease by mediating macrophages M1 polarization through ROS. Int Immunopharmacol. (2022) 104:108361. doi: 10.1016/j.intimp.2021.108361
95. Seong JB, Kim B, Kim S, Kim MH, Park YH, Lee Y, et al. Macrophage peroxiredoxin 5 deficiency promotes lung cancer progression via ROS-dependent M2-like polarization. Free Radic Biol Med. (2021) 176:322–34. doi: 10.1016/j.freeradbiomed.2021.10.010
96. Xie JH, Li YY, and Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol. (2020) 17:712–21. doi: 10.1038/s41423-020-0480-1
97. D’Arcy MS. Mitophagy in health and disease. Molecular mechanisms, regulatory pathways, and therapeutic implications. Apoptosis. (2024) 29:1415–28. doi: 10.1007/s10495-024-01977-y
98. Cotzomi-Ortega I, Nieto-Yañez O, Juárez-Avelar I, Rojas-Sanchez G, Montes-Alvarado JB, Reyes-Leyva J, et al. Autophagy inhibition in breast cancer cells induces ROS-mediated MIF expression and M1 macrophage polarization. Cell Signal. (2021) 86:110075. doi: 10.1016/j.cellsig.2021.110075
99. Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L604–619. doi: 10.1152/ajplung.00272.2013
100. Esteban-Martínez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Mariño G, Seco E, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. (2017) 36:1688–706. doi: 10.15252/embj.201695916
101. Mizumura K and Gon Y. Iron-regulated reactive oxygen species production and programmed cell death in chronic obstructive pulmonary disease. Antioxidants (Basel). (2021) 10. doi: 10.3390/antiox10101569
102. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. (2011) 3:78ra32. doi: 10.1126/scitranslmed.3002042
103. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. (2016) 22:163–74. doi: 10.1038/nm.4021
104. Warburg O, Wind F, and Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
105. Vaupel P and Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. (2021) 599:1745–57. doi: 10.1113/jp278810
106. Murphy MP. Rerouting metabolism to activate macrophages. Nat Immunol. (2019) 20:1097–9. doi: 10.1038/s41590-019-0455-5
107. Wang W, Zheng F, Lin C, and Zhang A. Changes in energy metabolism and macrophage polarization: Potential mechanisms of arsenic-induced lung injury. Ecotoxicol Environ Saf. (2020) 204:110948. doi: 10.1016/j.ecoenv.2020.110948
108. Agudelo CW, Samaha G, and Garcia-Arcos I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. (2020) 19:122. doi: 10.1186/s12944-020-01278-8
109. Qing J, Zhang Z, Novák P, Zhao G, and Yin K. Mitochondrial metabolism in regulating macrophage polarization: an emerging regulator of metabolic inflammatory diseases. Acta Biochim Biophys Sin (Shanghai). (2020) 52:917–26. doi: 10.1093/abbs/gmaa081
110. Bidault G, Virtue S, Petkevicius K, Jolin HE, Dugourd A, Guénantin AC, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. (2021) 3:1150–62. doi: 10.1038/s42255-021-00440-5
111. Acín-Pérez R, Iborra S, Martí-Mateos Y, Cook ECL, Conde-Garrosa R, Petcherski A, et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat Metab. (2020) 2:974–88. doi: 10.1038/s42255-020-00273-8
112. Petrusca DN, Gu Y, Adamowicz JJ, Rush NI, Hubbard WC, Smith PA, et al. Sphingolipid-mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. (2010) 285:40322–32. doi: 10.1074/jbc.M110.137604
113. Barnawi J, Jersmann H, Haberberger R, Hodge S, and Meech R. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: A potential link to failed efferocytosis. Respirology. (2017) 22:315–21. doi: 10.1111/resp.12949
114. Engelen MP and Schols AM. Altered amino acid metabolism in chronic obstructive pulmonary disease: new therapeutic perspective? Curr Opin Clin Nutr Metab Care. (2003) 6:73–8. doi: 10.1097/00075197-200301000-00011
115. Zhu Y, Zhang S, Sun J, Wang T, Liu Q, Wu G, et al. Cigarette smoke promotes oral leukoplakia via regulating glutamine metabolism and M2 polarization of macrophage. Int J Oral Sci. (2021) 13:25. doi: 10.1038/s41368-021-00128-2
116. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. (2017) 15:55–63. doi: 10.1038/nrmicro.2016.142
117. Wang L, Cai Y, Garssen J, Henricks PAJ, Folkerts G, and Braber S. The bidirectional gut-lung axis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2023) 207:1145–60. doi: 10.1164/rccm.202206-1066TR
118. Yao Y, Cai X, Fei W, Ye Y, Zhao M, and Zheng C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit Rev Food Sci Nutr. (2022) 62:1–12. doi: 10.1080/10408398.2020.1854675
119. Duan H, Wang L, Huangfu M, and Li H. The impact of microbiota-derived short-chain fatty acids on macrophage activities in disease: Mechanisms and therapeutic potentials. BioMed Pharmacother. (2023) 165:115276. doi: 10.1016/j.biopha.2023.115276
120. Ji JJ, Sun QM, Nie DY, Wang Q, Zhang H, Qin FF, et al. Probiotics protect against RSV infection by modulating the microbiota-alveolar-macrophage axis. Acta Pharmacol Sin. (2021) 42:1630–41. doi: 10.1038/s41401-020-00573-5
121. Yan Z, Chen B, Yang Y, Yi X, Wei M, Ecklu-Mensah G, et al. Multi-omics analyses of airway host-microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat Microbiol. (2022) 7:1361–75. doi: 10.1038/s41564-022-01196-8
122. Bailón E, Cueto-Sola M, Utrilla P, Rodríguez-Cabezas ME, Garrido-Mesa N, Zarzuelo A, et al. Butyrate in vitro immune-modulatory effects might be mediated through a proliferation-related induction of apoptosis. Immunobiology. (2010) 215:863–73. doi: 10.1016/j.imbio.2010.01.001
123. Scott NA, Andrusaite A, Andersen P, Lawson M, Alcon-Giner C, Leclaire C, et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aao4755
124. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. (2009) 461:1282–6. doi: 10.1038/nature08530
125. Huang Y, Li J, Meng XM, Jiang GL, Li H, Cao Q, et al. Effect of triterpene acids of Eriobotrya japonica (Thunb.) Lindl. leaf and MAPK signal transduction pathway on inducible nitric oxide synthase expression in alveolar macrophage of chronic bronchitis rats. Am J Chin Med. (2009) 37:1099–111. doi: 10.1142/s0192415x09007521
126. Shergis JL, Di YM, Zhang AL, Vlahos R, Helliwell R, Ye JM, et al. Therapeutic potential of Panax ginseng and ginsenosides in the treatment of chronic obstructive pulmonary disease. Complement Ther Med. (2014) 22:944–53. doi: 10.1016/j.ctim.2014.08.006
127. Lee H, Jung KH, Lee H, Park S, Choi W, and Bae H. Casticin, an active compound isolated from Vitex Fructus, ameliorates the cigarette smoke-induced acute lung inflammatory response in a murine model. Int Immunopharmacol. (2015) 28:1097–101. doi: 10.1016/j.intimp.2015.07.041
128. Li J, Tong D, Liu J, Chen F, and Shen Y. Oroxylin A attenuates cigarette smoke-induced lung inflammation by activating Nrf2. Int Immunopharmacol. (2016) 40:524–9. doi: 10.1016/j.intimp.2016.10.011
129. Kim YH, Choi YJ, Kang MK, Lee EJ, Kim DY, Oh H, et al. Oleuropein curtails pulmonary inflammation and tissue destruction in models of experimental asthma and emphysema. J Agric Food Chem. (2018) 66:7643–54. doi: 10.1021/acs.jafc.8b01808
130. Hussain T, Al-Attas OS, Alamery S, Ahmed M, Odeibat HAM, and Alrokayan S. The plant flavonoid, fisetin alleviates cigarette smoke-induced oxidative stress, and inflammation in Wistar rat lungs. J Food Biochem. (2019) 43:e12962. doi: 10.1111/jfbc.12962
131. Kubo H, Asai K, Kojima K, Sugitani A, Kyomoto Y, Okamoto A, et al. Astaxanthin suppresses cigarette smoke-induced emphysema through Nrf2 activation in mice. Mar Drugs. (2019) 17. doi: 10.3390/md17120673
132. Zhang XF, Ding MJ, Cheng C, Zhang Y, Xiang SY, Lu J, et al. Andrographolide attenuates oxidative stress injury in cigarette smoke extract exposed macrophages through inhibiting SIRT1/ERK signaling. Int Immunopharmacol. (2020) 81:106230. doi: 10.1016/j.intimp.2020.106230
133. Chen Z, Wu H, Fan W, Zhang J, Yao Y, Su W, et al. Naringenin suppresses BEAS-2B-derived extracellular vesicular cargoes disorder caused by cigarette smoke extract thereby inhibiting M1 macrophage polarization. Front Immunol. (2022) 13:930476. doi: 10.3389/fimmu.2022.930476
134. Choi S and Kim T. Compound K - An immunomodulator of macrophages in inflammation. Life Sci. (2023) 323:121700. doi: 10.1016/j.lfs.2023.121700
135. Chen J, Su J, Dong L, Bai Z, Liu Y, Bao H, et al. Rutin ameliorates cigarette smoke mediated lung inflammation in a mouse model of chronic obstructive pulmonary disease via regulating the levels of tumor necrosis factor-α, interleukin-8 and PAF. Arch Med Sci. (2025) 21:258–65. doi: 10.5114/aoms.2020.95874
136. Begnini F, Poongavanam V, Over B, Castaldo M, Geschwindner S, Johansson P, et al. Mining natural products for macrocycles to drug difficult targets. J Med Chem. (2021) 64:1054–72. doi: 10.1021/acs.jmedchem.0c01569
137. Sekar D, Johnson J, Biruntha M, Lakhmanan G, Gurunathan D, and Ross K. Biological and clinical relevance of microRNAs in mitochondrial diseases/dysfunctions. DNA Cell Biol. (2020) 39:1379–84. doi: 10.1089/dna.2019.5013
138. Purohit PK and Saini N. Mitochondrial microRNA (MitomiRs) in cancer and complex mitochondrial diseases: current status and future perspectives. Cell Mol Life Sci. (2021) 78:1405–21. doi: 10.1007/s00018-020-03670-0
139. Shen W, Wang S, Wang R, Zhang Y, Tian H, Yang X, et al. Analysis of the polarization states of the alveolar macrophages in chronic obstructive pulmonary disease samples based on miRNA-mRNA network signatures. Ann Transl Med. (2021) 9:1333. doi: 10.21037/atm-21-3815
140. Parajuli N, Subedi K, Solone XK, Jiang A, Zhou L, and Mi QS. Epigenetic control of alveolar macrophages: impact on lung health and disease. Cells. (2025) 14. doi: 10.3390/cells14090640
141. Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, et al. MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-κB pathway. Cell Host Microbe. (2015) 17:345–56. doi: 10.1016/j.chom.2015.01.007
142. Jiang S, Yan W, Wang SE, and Baltimore D. Dual mechanisms of posttranscriptional regulation of Tet2 by Let-7 microRNA in macrophages. Proc Natl Acad Sci U S A. (2019) 116:12416–21. doi: 10.1073/pnas.1811040116
143. Liu YD, Zhuang XP, Cai DL, Cao C, Gu QS, Liu XN, et al. Let-7a regulates EV secretion and mitochondrial oxidative phosphorylation by targeting SNAP23 in colorectal cancer. J Exp Clin Cancer Res. (2021) 40:31. doi: 10.1186/s13046-020-01813-6
144. Carlström KE, Ewing E, Granqvist M, Gyllenberg A, Aeinehband S, Enoksson SL, et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat Commun. (2019) 10:3081. doi: 10.1038/s41467-019-11139-3
145. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. (2018) 360:449–53. doi: 10.1126/science.aan4665
146. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
147. Yi Z, Wu Y, Zhang W, Wang T, Gong J, Cheng Y, et al. Activator-mediated pyruvate kinase M2 activation contributes to endotoxin tolerance by promoting mitochondrial biogenesis. Front Immunol. (2020) 11:595316. doi: 10.3389/fimmu.2020.595316
148. Wang L, Chen X, Li X, Liu D, Wang X, Chang X, et al. Developing a novel strategy for COPD therapy by targeting Nrf2 and metabolism reprogramming simultaneously. Free Radic Biol Med. (2021) 169:436–45. doi: 10.1016/j.freeradbiomed.2021.03.039
149. Makled S, Boraie N, and Nafee N. Nanoparticle-mediated macrophage targeting-a new inhalation therapy tackling tuberculosis. Drug Delivery Transl Res. (2021) 11:1037–55. doi: 10.1007/s13346-020-00815-3
Keywords: chronic obstructive pulmonary disease, alveolar macrophages, macrophage polarization, immunometabolic reprogramming, immunometabolism
Citation: Qin Z, Liu W, Huang C and Zhang W (2025) Metabolic reprogramming of macrophages in chronic obstructive pulmonary disease. Front. Immunol. 16:1684075. doi: 10.3389/fimmu.2025.1684075
Received: 12 August 2025; Accepted: 14 November 2025; Revised: 07 November 2025;
Published: 26 November 2025.
Edited by:
Alessio Torcinaro, National Research Council (CNR), ItalyReviewed by:
Giuliana Papoff, National Research Council (CNR), ItalyVeronica De Paolis, University of Rome Tor Vergata, Italy
Copyright © 2025 Qin, Liu, Huang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zhang, aHV4aXpoaWppYUAxMjYuY29t