 Sara Bindoli1,2*
Sara Bindoli1,2* Greta Morello-Pasin1,2Irina Guidea1,2Roberto Padoan1Luca Iorio1,2Riccardo Bixio3,4
Greta Morello-Pasin1,2Irina Guidea1,2Roberto Padoan1Luca Iorio1,2Riccardo Bixio3,4 Giovanni Orsolini4Federica Maiolini5Sara Lombardi6Sandra Lombardi7Marta Favero1,8Cristina Bernardi9Bernd Raffeiner10
Giovanni Orsolini4Federica Maiolini5Sara Lombardi6Sandra Lombardi7Marta Favero1,8Cristina Bernardi9Bernd Raffeiner10 Andrea Doria1,2
Andrea Doria1,2 Roberta Ramonda1,2
Roberta Ramonda1,2 Paolo Sfriso1,2
Paolo Sfriso1,2- 1Rheumatology Unit, Azienda Ospedale Università di Padova, Padova, Italy
- 2Department of Medicine (DIMED), Università degli Studi di Padova, Padova, Italy
- 3Unit of Rheumatology, IRCCS Sacro Cuore Don Calabria Hospital, Verona, Italy
- 4Rheumatology Unit, Department of Medicine, Università degli Studi di Verona, Verona, Italy
- 5Unit of Internal Medicine B, Department of Medicine, Università degli Studi di Verona, Verona, Italy
- 6General Medicine Unit, Girolamo Fracastoro Hospital, AULSS9 Scaligera, Verona, Italy
- 7Struttura Complessa Medicina Interna, Gorizia Hospital, Gorizia, Italy
- 8Internal Medicine 1, ULSS2 Marca Trevigiana, Ca’ Foncello Hospital, Treviso, Italy
- 9Rheumatology Unit, San Giovanni e Paolo Hospital, Venice, Italy
- 10Department of Rheumatology, Teaching Hospital of the Paracelsius Medical Hospital of Bolzano (ASAA-SABES), Bolzano, Italy
Objective: VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is a late-onset autoinflammatory disorder caused by somatic mutations in the UBA1 gene. It is characterized by systemic inflammation, a wide spectrum of rheumatologic features, including chondritis and inflammatory arthritis, dermatologic manifestations (e.g. neutrophilic dermatosis or vasculitis-like lesions), and hematologic abnormalities like macrocytic anemia and myelodysplastic syndrome. Due to its heterogeneity, diagnosis is frequently delayed. Early recognition of hallmark inflammatory symptoms, particularly by rheumatologists, is critical for timely diagnosis and management.
Methods: We conducted a retrospective analysis of 37 patients over the age of 50. Next-Generation Sequencing (Illumina HiSeq2500) was employed to assess mutations. Clinical, genetic, and demographic data were extracted from electronic medical records.
Results: Twenty patients [100% male; median age 73 years (IQR 67–77)] were confirmed to carry somatic UBA1 mutations. All patients exhibited constitutional symptoms (100%) and at least one rheumatologic manifestation, including chondritis (75%), arthralgia or arthromyalgia (50%), arthritis (30%), osteopenia or osteoporosis (15%), myalgia or myositis (10%), and tenosynovitis (5%). Dermatologic and hematologic abnormalities frequently co-occurred. Infectious complications were observed in 80% of patients and were a major contributor to overall morbidity.
Conclusion: This study underscores the need for a phenotype-driven diagnostic approach to facilitate earlier identification of VEXAS syndrome. Our findings suggest that current estimates of prevalence in rheumatology settings may significantly underestimate the true disease burden. Improved awareness and interdisciplinary collaboration, particularly among rheumatologists, hematologists, and dermatologists, are essential to enhance recognition, diagnosis, and comprehensive care for individuals affected by this complex syndrome.
Introduction
VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is an adult-onset autoinflammatory disorder caused by somatic loss-of-function mutations in the UBA1 gene within hematopoietic progenitor cells (1, 2). These mutations disrupt the function of the E1 ubiquitin-activating enzyme, leading to impaired protein degradation pathways and a broad, heterogeneous clinical phenotype. Systemic symptoms frequently include fever, night sweats, and unintentional weight loss, while organ-specific involvement most commonly affects the dermatologic and rheumatologic domains. Hematologic abnormalities, particularly those affecting the myeloid lineage, are also prevalent, with macrocytic anemia and features compatible with myelodysplastic syndrome (MDS) being the most frequently reported.
A recent systematic review (SR) by Al-Hakim et al. (3) analyzed 720 patients across 33 case reports and 21 case series. This SR confirmed that skin involvement is the most common clinical feature observed, affecting 81.8% of patients (95% CI: 78.8–84.5%), followed by constitutional symptoms in 69.4% (95% CI: 66.0–72.7%), and respiratory manifestations in 61.3% (95% CI: 57.6–64.7%). Joint involvement was instead reported in 47.3% of patients (95% CI: 43.5–51.2%), ocular inflammation in 44.3% (95% CI: 40.5–48.2%), and venous thromboembolism in 41.8% (95% CI: 38.3–45.4%). In contrast, MDS was observed in 35.8% of cases (95% CI: 32.3–39.4%), suggesting that the recurrence of inflammatory and autoimmune-like manifestations often outweighs the prominence of hematologic findings in initial clinical presentation.
Given the predominance of rheumatologic and dermatologic features, many of which mimic known autoimmune and autoinflammatory conditions, rheumatologists are likely to encounter VEXAS patients within their routine clinical practice.
The objective of this study is to describe the recurrence and distribution of key inflammatory clinical features of VEXAS syndrome, primarily as observed in rheumatological settings, to raise awareness among rheumatologists.
To this end, we collected data from patients with a high clinical suspicion of VEXAS syndrome who were referred to multiple rheumatology and internal medicine centers across the TriVeneto macro-region in northeastern Italy. This area, which has a catchment population of approximately 7,129,534 inhabitants, includes an estimated 3,386,000 individuals over the age of 50. Notably, around 1.5 million of them are males, representing 44% of this age group, and constitute the primary demographic at risk for this syndrome (4).
Patients and methods
Patients exhibiting clinical manifestations and laboratory findings consistent with a presumptive diagnosis of VEXAS syndrome were recruited from hub-and-spoke Hospitals and Outpatient Clinics across the Veneto, Trentino-Alto Adige, and Friuli Venezia Giulia Regions. The diagnosis of VEXAS syndrome was based on the identification of UBA1 mutations via Next-Generation Sequencing (NGS) (HiSeq2500 Illumina Sequencer) and demographic data were extracted from electronic medical records. Disease activity was retrospectively assessed using the recently developed VEXAS Disease Activity Index (VEXAS-DAI), based on clinical data documented at symptom’s onset (5). This score includes 12 scored domains, each with a specified number of items: inflammatory-type rash (2), chondritis (3), ophthalmologic involvement (5), periorbital involvement (1), joint (1), pulmonary (3), cardiovascular (3), genitourinary (1), neurologic (5), oral and gastrointestinal (3), renal (1), and constitutional symptoms (present or absent). Additionally, thrombosis and thromboembolism domain is included but remains unscored. Scores range from 0 to 40.
All participants provided written informed consent prior to inclusion in the study, which was conducted in accordance with the principles of the Declaration of Helsinki. The study protocol received approval by the Ethics Committee of Padova University Hospital (protocol code 5349/AO/22).
Statistical analysis
The distribution of continuous variables was assessed using the Shapiro-Wilk test. Variables that did not follow a normal distribution were reported as medians with their corresponding IQRs. Correlations between variables were evaluated using Spearman’s rank correlation coefficient. All statistical analyses were conducted using GraphPad Prism version 10 (GraphPad Software Inc., La Jolla), with a significance threshold set at p < 0.05.
Results
Clinical features
A total of 37 patients (100%) were recruited. Of these, 20 patients (54%), all male, with a median age of 73 years (IQR 67–77), were diagnosed with VEXAS syndrome based on the identification of UBA1 mutations through next-generation sequencing (NGS). Genetic variants, along with their respective variant allele fractions (VAF%), clinical manifestations, prior diagnoses, diagnostic delays, and current treatment regimens are summarized in Table 1, while the rheumatologic manifestations observed are summarized in Table 2. All patients exhibited at least one rheumatologic feature during the disease course. Constitutional symptoms, including asthenia, fatigue and weight loss, were reported in all the patients (100%), and 15 individuals (75%) experienced recurrent febrile episodes.
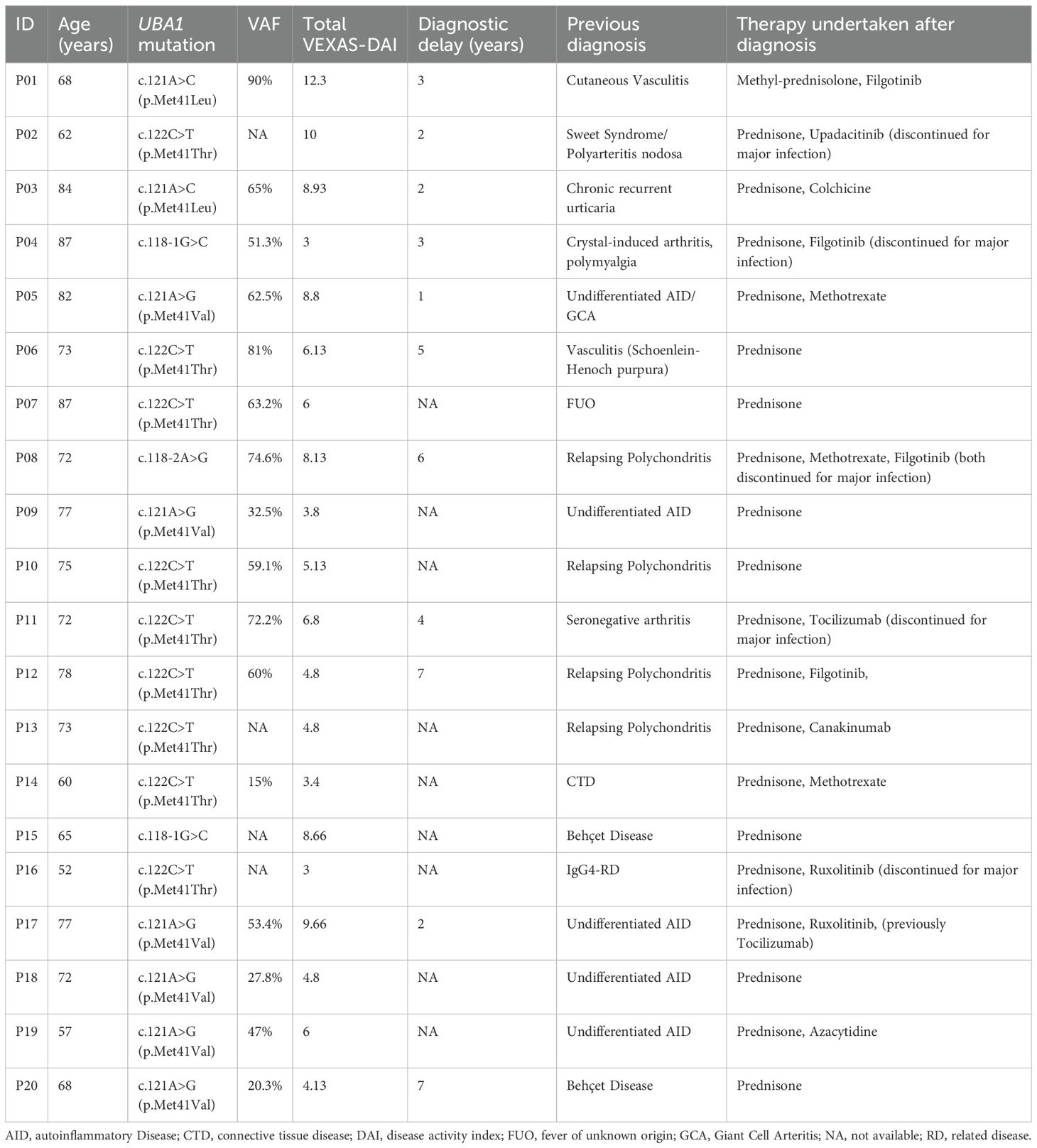
Table 1. Demographic and genetic aspects along with previous diagnosis and diagnostic delay time of patients with VEXAS syndrome.
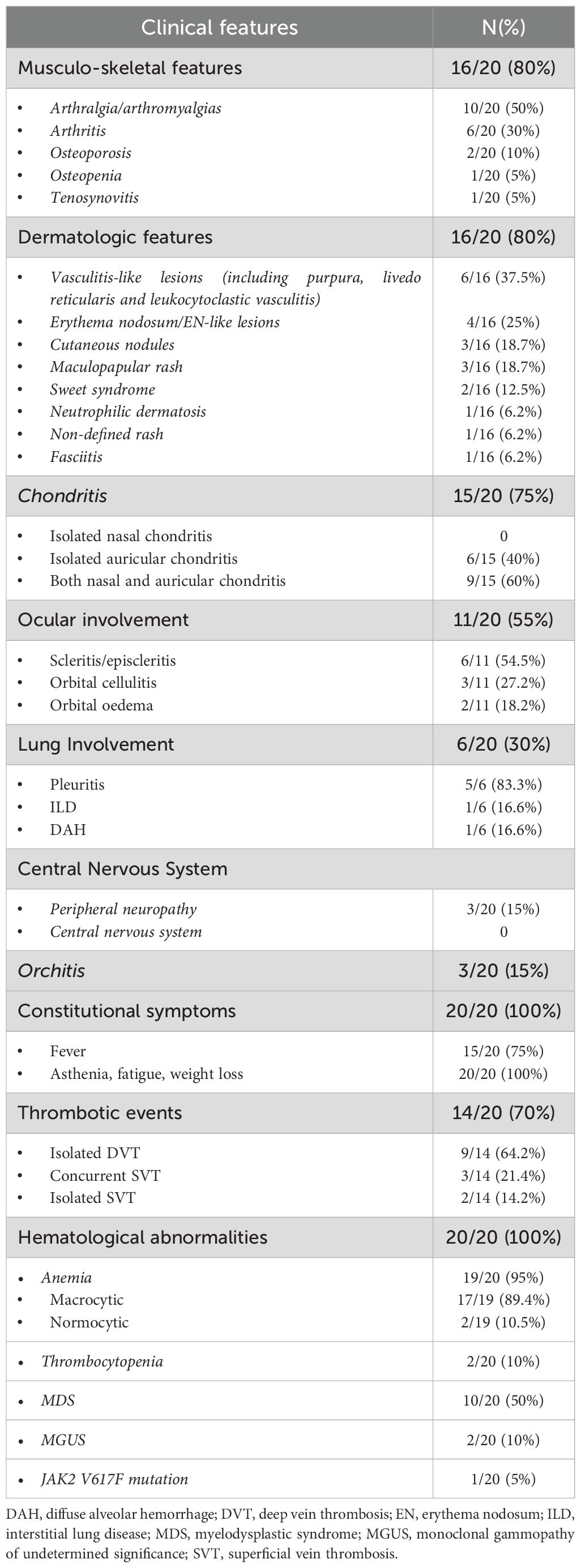
Table 2. Clinical symptoms exhibited by the patients subdivided by domain.
Arthritis was diagnosed in 6 out of 20 patients (30%), while arthralgia and/or myalgia were observed in 10 out of 20 cases (50%). Chondritis was present in 15/20 patients (75%); of these, 6/15 (40%) had isolated auricular involvement, and the remaining 9/15 (60%) presented with both auricular and nasal chondritis (Figure 1D). Notably, no cases of isolated nasal chondritis were identified. Tenosynovitis was reported in one patient (5%).
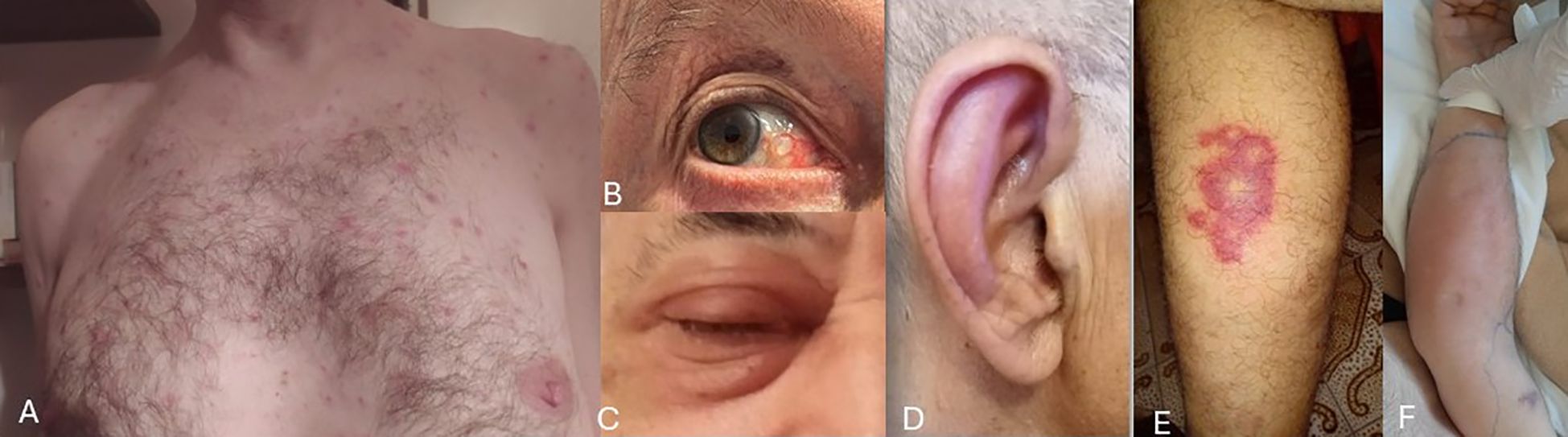
Figure 1. Images depicting selected clinical features affecting the skin and eyes in our VEXAS patients (with patients’ permission). (A) Truncal neutrophilic dermatosis; (B) Ocular scleritis; (C) Periorbital oedema; (D) Ear chondritis; (E) Vasculitic lesion on lower limbs; (F) Arm fasciitis with marked subcutaneous oedema.
Regarding bone metabolism, reduced bone mineral density was documented in a minority of patients. Overall, one individual (5%) had osteopenia, and two out of 20 patients (10%) had osteoporosis, one of whom also experienced vertebral fractures.
Cutaneous manifestations (Figures 1A, E, F) were frequent and heterogeneous, occurring overall in 16 out of 20 patients (80%). The detailed summary is provided in Table 2. Neutrophilic dermatosis, confirmed histologically by neutrophilic infiltration, was identified only in one out of 16 patients (6.2%). Sweet’s syndrome was diagnosed in 2/16 patients (12.5%), maculo-papular rash and cutaneous nodules in 3/16 patients (18.75%), respectively. Vasculitis-like lesions instead were observed in 6/16 patients (37.5%) and these included purpuric rashes, livedo reticularis and histologically confirmed leukocytoclastic vasculitis. Erythema nodosum (EN) or EN-like lesions occurred in 4/16 cases (25%). Additional dermatologic features included a non-specified rash in 1 patient out of 16 (6.2%), and bilateral arm fasciitis with significant subcutaneous oedema and diffuse redness (1 patient, 6.2%).
Although ocular involvement is not primarily managed within rheumatological practice, it is well represented in several autoinflammatory and autoimmune conditions. Indeed, also in our cohort, ocular manifestations were prominent, affecting 55% of patients (11/20). Among these eleven subjects, the majority presented with scleritis/episcleritis (54.5%), followed by orbital cellulitis (27.2%) and orbital edema (18.2%) (Figures 1B, C).
Among the non-rheumatologic features, anemia was the most consistent manifestation, observed in 19 out of 20 patients (95%), with the macrocytic form present in 89.4% of the cases, reinforcing its role as a key, although non-specific, diagnostic marker in VEXAS syndrome. Myelodysplastic syndrome (MDS) was present in 50% of cases, highlighting the pathogenic association between UBA1 somatic mutations and clonal hematopoiesis. One patient was found to carry the JAK2 Val617Phe (V617F) mutation. The presence of both macrocytic anemia and MDS, especially within a context of systemic inflammation, should be considered as strongly associated with a possible diagnosis of VEXAS syndrome. Thrombotic events were also common, occurring in 70% of the cohort (14 out of 20 subjects), a frequency higher than previously reported in the literature. Figure 2 presents a graphical summary of the clinical and biological characteristics observed in our patients.
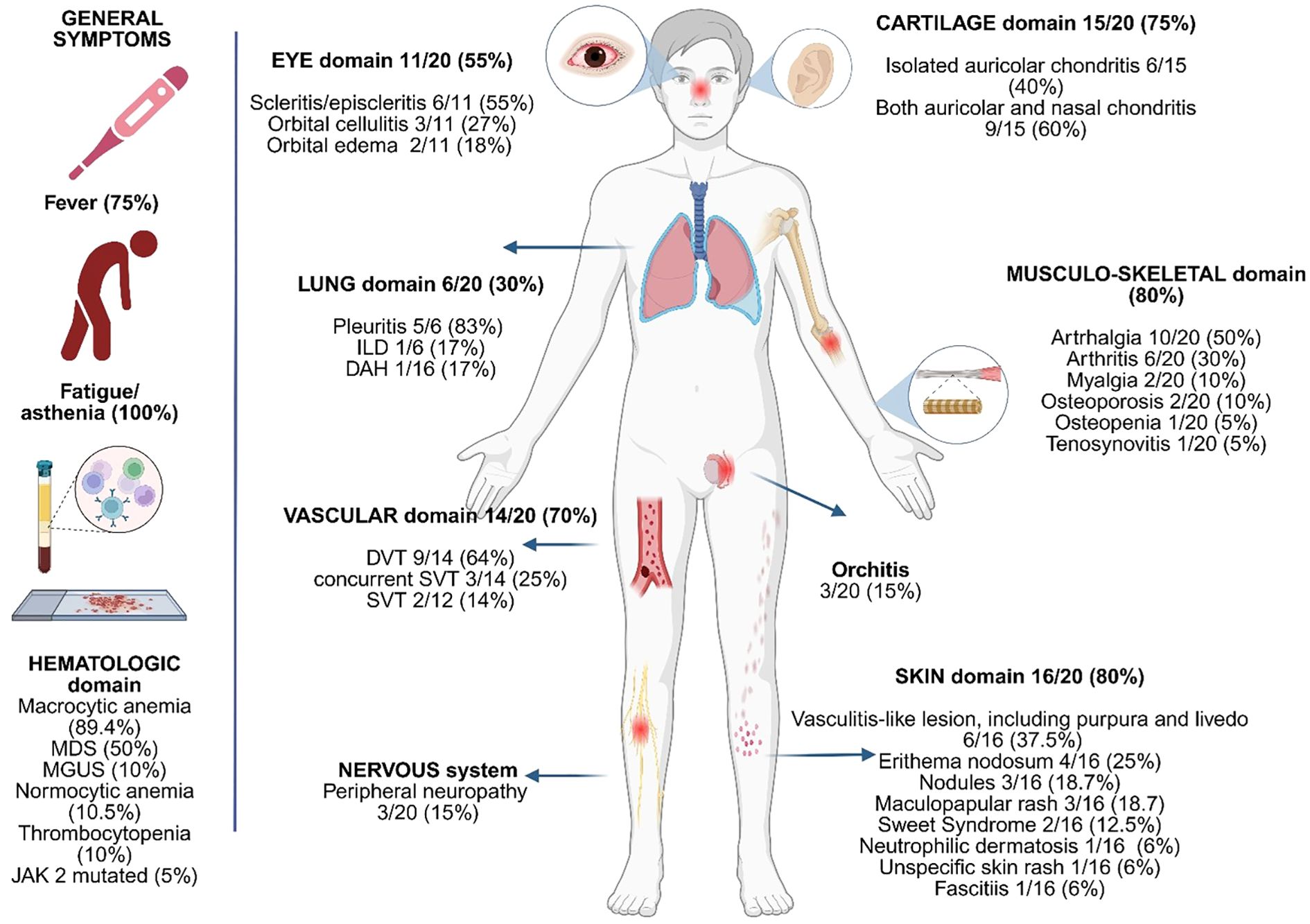
Figure 2. Graphical presentation of the clinical and biological features of the patients included.
The VEXAS-DAI score, based on clinical parameters, yielded a mean score of 6.4 and showed a statistically significant positive correlation with the variant allele fraction (r = 0.73, p = 0.0016). Both the individual patients’ VAF values and VEXAS-DAI scores are reported in Table 1.
Infections during the disease course
Of the 20 patients in our cohort, 16 (80%) experienced at least one clinically significant infection, defined as an infection requiring appropriate treatment either in the hospital or outpatient setting during follow-up. In total, 34 infections were documented (Table 3). Six patients (37.5%) had a single infectious episode, whereas 10 (62.5%) experienced multiple events throughout the disease course.
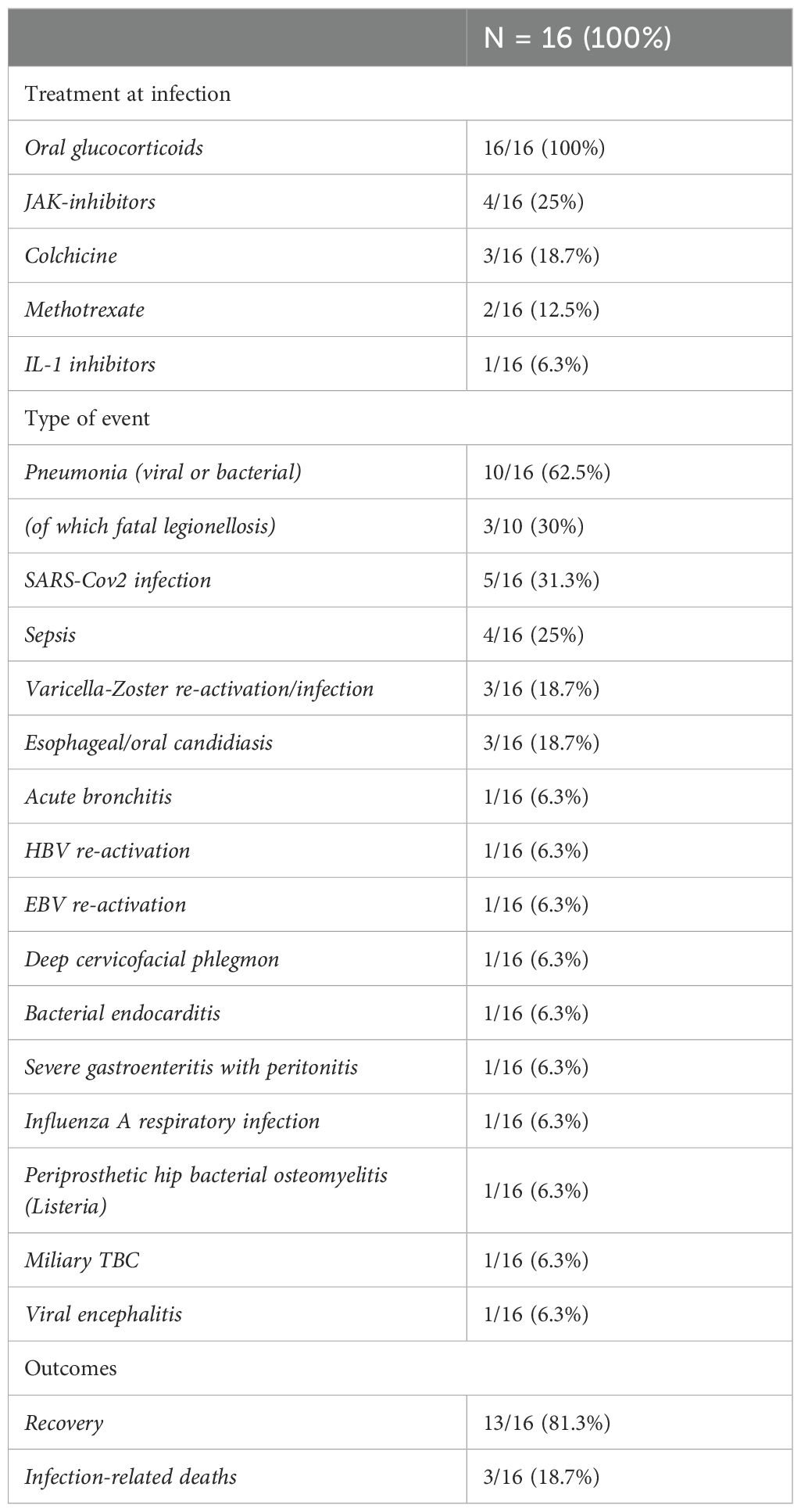
Table 3. Type of infection developed and ongoing treatment at infections.
The most frequently reported infections involved the respiratory tract, with pneumonia documented in 10 episodes. Among these, 3 cases were caused by Legionella pneumophila and resulted in fatal outcomes. Additionally, SARS-CoV-2 infection was noted in 5 episodes. Four episodes of sepsis requiring hospitalization were also observed. Other infections included herpes zoster reactivation (3 episodes) and esophageal/oral candidiasis (3 episodes). Singular events included herpes zoster virus-associated encephalitis, deep cervicofacial phlegmon, severe gastroenteritis with peritoneal signs, bacterial endocarditis, osteomyelitis, acute bronchitis, Epstein-Barr virus (EBV) infection, miliary tuberculosis, and influenza A (H1N1). Clinical outcomes were generally favorable in some cases, with 13 patients (81.3%) experiencing recovery. However, 3 patients (18.7%) died either directly or indirectly as a result of the infection. All patients (100%) were receiving oral corticosteroids at the time of the infectious episode. Additionally, some were undergoing other immunosuppressive therapies: colchicine (3 patients, 18.7%), JAK inhibitors (4 patients, 25%), methotrexate (2 patients, 12.5%), and IL-1 inhibitor canakinumab (1 patient, 6.3%).
Discussion
VEXAS syndrome is a complex, multisystem disorder characterized by a broad range of manifestations affecting rheumatologic, dermatologic, and hematologic domains. Given its diverse clinical presentation and the requirement for genetic confirmation, diagnosis is often delayed or overlooked, especially in the early disease stages.
In the initial description by Beck et al. (1) dermatologic manifestations were prominent, present in 88% of patients, with nodules being most frequent (52%). Lung involvement was detected in approximately 72%, followed by arthralgia (68%) and chondritis (64%). Similarly, the French cohort reported by Georgin-Lavialle et al. (6) confirmed cutaneous manifestations in 83% of patients, although observed lower frequencies of chondritis (36.2%) and musculoskeletal involvement (28.4%), as well as pulmonary infiltrates (40%). Ocular involvement, however, was relatively frequent, affecting around 40% of patients.
In the Swiss cohort (7), ocular and pulmonary manifestations were among the most common inflammatory features. Cutaneous involvement was again highly prevalent (86%), and musculoskeletal involvement was seen in 47%, with inflammatory arthritis being present in 35% of the patients, whereas chondritis was present in only 24% of the 17 patients included.
A recent Spanish multicenter study (8) involving 39 patients reaffirmed that cutaneous involvement was the most frequent manifestation recorded (87.2%). Notably, articular involvement in this cohort was more prevalent than in others, with polyarthritis reported in 79.4% of patients. This higher rate likely reflects the study’s rheumatologic setting, where many patients had previously received alternate diagnoses such as seronegative arthritis, relapsing polychondritis, polymyalgia rheumatica, Sweet’s syndrome, systemic lupus erythematosus, or medium-vessel vasculitis.
Across cohorts, musculoskeletal involvement showed considerable variability, ranging from 28.3% in the study by Georgin-Lavialle (6) to the nearly 80% of the Spanish cohort (8). The largest SR available (3), including 720 patients, estimated the prevalence of joint involvement at 47.3%, indicating that while it is relatively common, it is not among the most predominant features. In contrast, in the same review skin involvement (81.8%), followed by constitutional symptoms (69.4%), and respiratory manifestations (61.3%) were the most prevalent reported clinical features.
In our cohort, the predominant clinical features aligned with those typically observed in rheumatologic practice. Indeed, musculoskeletal involvement was observed in 80% of patients, with arthralgia in 50% and arthritis in 30%. Chondritis (auricular and/or nasal) and skin manifestations were particularly prevalent, affecting 75% and 80% of the patients, respectively.
Cutaneous manifestations primarily resembled vasculitis-like lesions, observed in 37.5% of patients, with palpable purpura and, more broadly, features consistent with leukocytoclastic vasculitis. EN and EN-like lesions were the next most observed. This pattern contrasts with reports from other cohorts, where maculopapular rashes, papules, and nodules were more often reported (9). Zakine et al. indeed, identified maculopapular eruptions, neutrophilic dermatosis, and arcuate plaques as key cutaneous manifestations, noting that arcuate lesions may be pathognomonic due to their high prevalence (9). In contrast, in the cohort described by Sullivan et al., 19% of the 89 patients demonstrated histopathologic evidence of small-vessel vasculitis, with most exhibiting leukocytoclastic vasculitis on skin biopsy (10). These findings suggest that, while arcuate lesions, plaques, and neutrophilic dermatosis remain distinguishing cutaneous features of VEXAS syndrome, the presence of vasculitic skin lesions should likewise prompt clinically suspicion for VEXAS syndrome. Nevertheless, such manifestations should be interpreted within the broader context of the systemic inflammatory nature of the disease.
The frequency of fever and constitutional symptoms varies considerably across cohorts, with non-infectious fever ranging from 55% (10) to 92% (1), and constitutional symptoms from 46% (10) up to 100%, as observed in our cohort. These manifestations are often self-reported, making precise estimation challenging, particularly given their nonspecific nature. Moreover, such symptoms are commonly observed across a wide spectrum of rheumatic diseases, both autoimmune and autoinflammatory. In this context, it is important to note that patients with VEXAS syndrome are typically older and often burdened with multiple comorbidities, including marked anemia, which may further contribute to the variability and nonspecific nature of these systemic symptoms.
Careful attention should also be given to pulmonary and ocular involvement, both of which are commonly observed in rheumatologic diseases. Respiratory features such as interstitial lung disease (ILD), pleural effusions, pulmonary consolidations, and nodules have been increasingly recognized in VEXAS syndrome (11). Al-Hakim et al. reported that lung involvement constitutes nearly half of all manifestations (3). In our cohort, pulmonary involvement was identified in 30%, with pleuritis being the most frequently feature reported.
Ocular involvement, well documented in VEXAS patients since the earliest reports (12), was observed in up to 59% of patients in the cohort by Wolff et al. (7). In our study, 50% of patients exhibited ocular symptoms, with scleritis/episcleritis being the most common.
Given the frequency of ocular and pulmonary inflammation in VEXAS syndrome, which are features commonly shared with other autoimmune/autoinflammatory conditions, a high index of suspicion is essential, particularly in those presenting with manifestations suggestive of vasculitis, relapsing polychondritis, Behçet’s disease, or even inflammatory arthropathies. These observations underscore the clinical heterogeneity of the disease and highlight the pivotal role of rheumatologists in its early recognition. Supporting this, a monocentric study by Muratore et al. (13) involving 147 patients with confirmed vasculitis, employed targeted UBA1 mutation screening in those presenting with overlapping rheumatologic, dermatologic, and hematologic features. This approach led to the identification of UBA1 mutations in three patients on the whole cohort, emphasizing the utility of a phenotype-driven strategy for detecting VEXAS syndrome within rheumatology practice.
Finally, although deep vein thrombosis (DVT) and superficial vein thrombosis (SVT) are not typically considered primary rheumatological manifestations, we observed a notably high incidence of thrombotic events within our cohort. While comprehensive studies on the genetic factors associated with thrombotic risk in VEXAS syndrome remain scarce, we wish to highlight the following findings: one patient tested positive for lupus anticoagulant (LAC) and anti-cardiolipin IgG at 140 GPL/mL (with negative results for IgM and anti-Beta2GP1); another patient was found to be a carrier of HLA-B51, for whom Behçet’s disease had initially been suspected; and a third patient presented with both a mutation in UBA1 and a mutation in CECR1 (or ADA2), a gene associated with DADA2 syndrome. These observations suggest the need for further investigation into the potential genetic underpinnings of thrombotic events in VEXAS syndrome, extending beyond the well-established mechanism of ‘thrombo-inflammation’ directly driven by the disease itself.
The comparison of the aforementioned cohorts, comprising more than 20 subjects with VEXAS syndrome observed within rheumatological contexts, is summarized in Table 4.
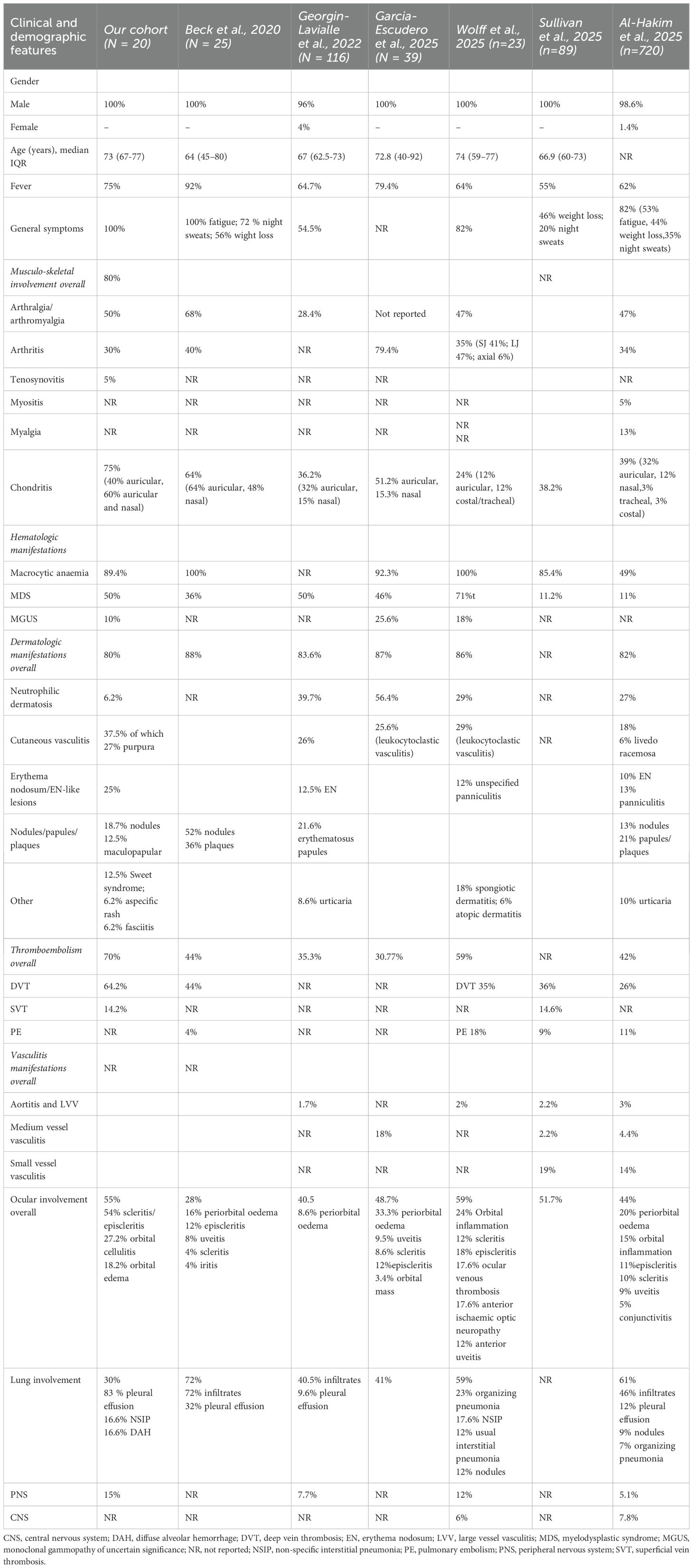
Table 4. Summary and comparison of clinical characteristics from VEXAS cohorts with over 20 patients.
Therefore, is important to avoid defining the disease solely based on the features described in the earliest reports, as this may overlook rarer or atypical manifestations. In this context, the present study takes a distinct rheumatological approach, positioning VEXAS syndrome within the broader spectrum of rheumatologic diseases. The observed differences compared to earlier cohorts may, therefore, reflect this specific focus, as seen in the variation of constitutional features.
An emerging challenge is how to quantify and assign appropriate weight to individual inflammatory manifestations. To address this and improve the classification of clinical features in patients with VEXAS syndrome, recent efforts have focused on developing a Disease Activity Index (DAI), as no validated scoring system for assessing disease activity currently exists. To date, the only proposed DAI for VEXAS syndrome is the VEXAS-CAF, which evaluates disease activity based on the presence or absence of 11 equally weighted manifestations (14). The VEXAS-DAI score was developed by an expert advisory committee using a modified Delphi methodology (5). This scoring system comprises 12 domains, with a total possible range of 0 to 40. Notably, hematologic manifestations are not included in the scoring, making the VEXAS-DAI primarily a clinical and inflammatory symptoms-based assessment tool. In our cohort, although limited to 20 subjects, the VEXAS-DAI was calculated for each patient, and a significant positive correlation was observed with the VAF% exhibited. This finding confirms and support that a higher proportion of cells carrying the UBA1 mutation is associated with increased disease severity.
From a rheumatologic perspective, it is crucial to recognize also the aspect that many therapies commonly used in autoimmune and autoinflammatory diseases carry an inherent risk of infection. This is particularly relevant in our cohort, where all patients were managed within rheumatology settings and received immunomodulatory therapies. Notably, apart from one patient who received azacytidine in combination with prednisone, none of the patients were treated with conventional hematologic therapies. These findings emphasize the need for careful selection and monitoring of treatment, as both the underlying disease and the immunosuppressive therapies may contribute to a significantly increased risk of infections (15, 16). In our study, infections occurred primarily during treatment with corticosteroids and JAK inhibitors, pointing to a heightened risk of severe infections, including common seasonal and opportunistic pathogens. This underscores the importance of preventive strategies, such as vaccination and antimicrobial prophylaxis, in the management of VEXAS syndrome. Overall, these findings reinforce the central role rheumatologists play in both recognizing and managing the disease, particularly in patients with predominant inflammatory features.
Conclusion
We collected a region-wide cohort of VEXAS patients, characterized by heterogeneous clinical manifestations, with a primary focus on the rheumatologic aspects of the disease. This study complements the longitudinal work of Gurnari et al., which provided an in-depth analysis of the hematological features of VEXAS in 41 Italian patients (17).
In this regard, a final consideration pertains to the issue of which specialist is most likely to encounter VEXAS patients initially. Although current estimates suggest that the prevalence of VEXAS syndrome in males ranges from 1 in 14,000 to as high as 1 in 4,000 in men over the age of 50 (18), our study, which identified 20 cases within rheumatological/internal medicine settings, yielded an estimated prevalence of approximately 1 in 70,000 among males over 50 in the Tri Veneto macro-region. While this likely reflects a substantial underestimation relative to current epidemiological data, it underscores a critical point: VEXAS syndrome remains markedly under-recognized, particularly outside of specialized or academic contexts. This discrepancy highlights the importance of a multidisciplinary diagnostic approach, involving rheumatologists, hematologists, dermatologists, and internists, to enhance early recognition and improve diagnostic accuracy for this complex multisystem disorder. For this reason, disease registries, such as those of the AutoInflammatory Disease Alliance (AIDA), will be indispensable for comprehensively capturing the full clinical spectrum of VEXAS syndrome (19).
Finally, a limitation of this study is the selection approach of subjects screened for genetic analysis, which, although highly specific, may have limited its sensitivity. Patients with milder or atypical phenotypes could have been excluded, possibly due to the influence of earlier literature that has shaped the disease definition. This narrow focus on initial descriptions might overlook a wider spectrum of clinical presentations, increasing the likelihood of missed diagnoses, particularly among individuals with less conventional symptoms.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of Padova University Hospital (protocol code 5349/AO/22). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SB: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing, Methodology. GM-P: Data curation, Formal analysis, Writing – original draft. IG: Data curation, Writing – review & editing. RP: Data curation, Formal analysis, Writing – review & editing. LI: Formal analysis, Writing – review & editing. RB: Data curation, Writing – review & editing. GO: Data curation, Writing – review & editing. FM: Data curation, Writing – review & editing. SRL: Data curation, Writing – review & editing. SDL: Data curation, Writing – review & editing. MF: Data curation, Writing – review & editing, Formal analysis. CB: Data curation, Writing – review & editing. BR: Data curation, Writing – review & editing. AD: Supervision, Visualization, Writing – original draft. RR: Supervision, Validation, Visualization, Writing – review & editing. PS: Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Open access funding was provided by the University of Padova within the Conference of Italian University Rectors (CRUI); Coordinamento per l'Accesso alle Risorse Elettroniche (CARE) Agreement.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
2. Ferrada MA, Savic S, Cardona DO, Collins JC, Alessi H, Gutierrez-Rodrigues F, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. (2022) 140:1496–506.
3. Al-Hakim A, Goldberg S, Gaillard S, Heiblig M, Beck DB, and Savic S. Clinical features in VEXAS syndrome: a systematic review. Rheumatology. (2025) 64:5217–229. doi: 10.1093/rheumatology/keaf293/8164134
5. Byram K, Mann H, Hammond D, Savic S, Kirino Y, Gurnari C, et al. Development of A disease activity index for the assessment of vexas syndrome (Vexas-dai). Ann Rheum Dis. (2025) 84:625–6.
6. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. (2022) 186:564–74.
7. Wolff L, Caratsch L, Lötscher F, Seitz L, Seitz P, Coattrenec Y, et al. VEXAS syndrome: a Swiss national retrospective cohort study. Swiss Med Wkly. (2024) 155:3879.
8. García-Escudero P, López-Gómez M, López BM, Dorta AG, Frade-Sosa B, Lizarzaburu MS, et al. VEXAS syndrome through a rheumatologist’s lens: insights from a Spanish national cohort. Rheumatology. (2025) 64:3747–55.
9. Zakine È, Papageorgiou L, Bourguiba R, Mekinian A, Terrier B, Kosmider O, et al. Clinical and pathological features of cutaneous manifestations in VEXAS syndrome: A multicenter retrospective study of 59 cases. J Am Acad Dermatol. (2023) 88:917–20.
10. Sullivan MM, Mead-Harvey C, Sartori-Valinotti JC, Kalantari K, Kusne YN, Patnaik MM, et al. Vasculitis associated with VEXAS syndrome. Rheumatology. (2025) 64:3889–94.
11. Borie R, Debray MP, Guedon AF, Mekinian A, Terriou L, Lacombe V, et al. Pleuropulmonary manifestations of vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome. Chest. (2023) 163:575–85.
12. Vitale A, Caggiano V, Martin-Nares E, Frassi M, Dagna L, Hissaria P, et al. Orbital/ocular inflammatory involvement in VEXAS syndrome: Data from the international AIDA network VEXAS registry. Semin Arthritis Rheum. (2024) 66:152430.
13. Muratore F, Marvisi C, Castrignanò P, Nicoli D, Farnetti E, Bonanno O, et al. VEXAS syndrome: A case series from a single-center cohort of italian patients with vasculitis. Arthritis Rheum. (2022) 74:665–70. doi: 10.1002/art.41992
14. Kirino Y, Maeda A, Asano T, Migita K, Hidaka Y, Ida H, et al. Low remission rates and high incidence of adverse events in a prospective VEXAS syndrome registry. Rheumatology. (2025) 64:3872–8. doi: 10.1093/rheumatology/keae530
15. Vitale A, Caggiano V, Leone F, Hinojosa-Azaola A, Martín-Nares E, Guaracha-Basañez GA, et al. Efficacy and safety profile of biotechnological agents and Janus kinase inhibitors in VEXAS syndrome: data from the international AIDA Network VEXAS registry. Front Pharmacol. (2025) 16:1462254. doi: 10.3389/fphar.2025.1462254
16. De Valence B, Delaune M, Nguyen Y, Jachiet V, Heiblig M, Jean A, et al. French VEXAS Group. Serious infections in patients with VEXAS syndrome: data from the French VEXAS registry. Ann Rheum Dis. (2024) 83:372–81. doi: 10.1136/ard-2023-224819
17. Gurnari C, Pascale MR, Vitale A, Diral E, Tomelleri A, Galossi E, et al. Diagnostic capabilities, clinical features, and longitudinal UBA1 clonal dynamics of a nationwide VEXAS cohort. Am J Hematol. (2024) 99:254–62. doi: 10.1002/ajh.27169
18. Beck BD, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA J Am Med Assoc. (2023) 24:318–24. doi: 10.1001/jama.2022.24836
Keywords: VEXAS syndrome, inflammation, chondritis, hematologic abnormalities, genetics
Citation: Bindoli S, Morello-Pasin G, Guidea I, Padoan R, Iorio L, Bixio R, Orsolini G, Maiolini F, Lombardi S, Lombardi S, Favero M, Bernardi C, Raffeiner B, Doria A, Ramonda R and Sfriso P (2025) VEXAS syndrome in rheumatology practice: features from a multicenter cohort in north-east Italy. Front. Immunol. 16:1700737. doi: 10.3389/fimmu.2025.1700737
Received: 07 September 2025; Accepted: 11 November 2025; Revised: 10 November 2025;
Published: 02 December 2025.
Edited by:
Piero Ruscitti, University of L’Aquila, ItalyReviewed by:
Luca Cantarini, University of Siena, ItalyPaula García-Escudero, Araba University Hospital, Spain
Copyright © 2025 Bindoli, Morello-Pasin, Guidea, Padoan, Iorio, Bixio, Orsolini, Maiolini, Lombardi, Lombardi, Favero, Bernardi, Raffeiner, Doria, Ramonda and Sfriso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Bindoli, c2FyYS5iaW5kb2xpQHVuaXBkLml0