 Yuan Wei1†
Yuan Wei1† Yumeng Lin2†Youjiaxi Li1†
Yumeng Lin2†Youjiaxi Li1† Jiaxuan Liu1
Jiaxuan Liu1 Yaqi Yang3
Yaqi Yang3 Haoran Chen4
Haoran Chen4 Zhongyu Han5
Zhongyu Han5 Ke Wang6Tao Qian7*Yuan Ju3*Wei Zheng3*
Ke Wang6Tao Qian7*Yuan Ju3*Wei Zheng3*- 1College of Chinese Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, China
- 2Department of Nanjing Tongren Eye Center, Nanjing Tongren Hospital, School of Medicine, Southeast University, Nanjing, China
- 3Ophthalmology Department, Affiliated Hospital of Changchun University of Traditional Chinese Medicine, Changchun,Jilin, China
- 4Chengdu Xinhua Hospital, Chengdu, China
- 5School of Medicine, Southeast University, Nanjing, China
- 6Deyang Hospital, Affiliated Hospital of Chengdu University of Traditional Chinese Medicine, Deyang, China
- 7Department of Thyroid and Breast Surgery, Affiliated Hospital of Intergrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, China
Ferroptosis, recently proposed as a novel type of cell death, is characterized by unique characteristics and recognition functions. It is involved in diverse physiological processes and in the onset and progression of various diseases and is characterized by reactions between reactive oxygen species (ROS) and iron-dependent lipid peroxidation. This process is finely regulated by a variety of metabolic pathways. Ferroptosis fundamentally differs from conventional cell death mechanisms such as apoptosis, necrosis, and autophagy. In recent years, research on ferroptosis in the field of ophthalmology has gradually emerged, and a large amount of evidence has shown that it is closely related to the occurrence and development of ophthalmic diseases such as age-related macular degeneration (AMD), diabetic retinopathy (DR), retinal ischemia–reperfusion injury (RIRI), retinitis pigmentosa, dry eye disease, cataracts, and glaucoma. This paper provides a comprehensive review of the latest advancements in ferroptosis within ophthalmological research and systematically describes the molecular mechanisms and pathophysiological significance of ferroptosis in the pathogenesis and progression of ophthalmic diseases. Exploring the mechanisms of ferroptosis holds promise for the delivery of novel molecular targets and therapeutic approaches to prevent and treat ophthalmic diseases. Additionally, its clinical translational and application are anticipated to surmount current therapeutic limitations and emerge as a significant direction for breakthroughs in the precision medicine era.
1 Introduction
Unlike most animal eyes, human eyes do more than just measure the intensity of ambient light (1). Clinical studies have shown that visual deprivation directly leads to impaired plasticity in the cerebral cortex, and this neurodevelopmental deficit is irreversible (2). The World Health Organization (WHO) ranks blinding eye disease, cardiovascular disease, and malignant tumors as the three major disabling diseases worldwide. Some surveys have indicated that eye diseases may even increase the risk of developing depression and reduce life expectancy among patients (3). When eye disease occurs, it undoubtedly affects the development of personal living standards and society, and this harm is more obvious in today’s aging society. The pathology of ocular diseases involves multiple cell death mechanisms, including apoptosis, necrosis, and autophagy (4, 5). Ferroptosis is a novel form of programmed cell death resulting from the accumulation of iron-dependent lipid peroxides (6). Its occurrence depends mainly on increases in reactive oxygen species (ROS), phospholipids containing polyunsaturated fatty acid chains (PUFA-PLs), and iron accumulation (7). Alternatively, intracellular and intercellular signaling, as well as environmental stress, can indirectly influence ferroptosis by modulating cellular metabolic processes as well as ROS levels (8).
In 1980, the membrane protein xCT5, associated with ferroptosis, was discovered by S. Bannai et al. (9). In 2003, erastin, a compound that induces cell death through non-apoptotic pathways, was first discovered and named. It is a compound with selective lethality for Rat Adenosarcoma (RAS) (10). In 2008, RSL3 and RSL5 were also shown to induce non-apoptotic cell death. Deferoxamine (DFO) and antioxidants (e.g., vitamin E) can inhibit this process (11). Ferroptosis, a term that was first used by experts such as Dixon in 2012, is characterized as a unique type of cell death that is not apoptosis and is triggered by the compound erastin, causing cells to die via iron-dependent lipid peroxidation (12). In 2014, Wan Seok Yang et al. reported that glutathione peroxidase 4 (GPX4) plays a crucial role in regulating ferroptosis (13). Since then, studies on ferroptosis have increased. Ferroptosis is not limited to mammals; it has also been detected in plants, protozoa, and mycota (14, 15).
This manuscript provides an overview of the mechanisms underlying ferroptosis, recent advancements in the field, and future directions, particularly in the context of the ocular microenvironment and ophthalmological disorders, such as age-related macular degeneration (AMD), diabetic retinopathy (DR), retinal ischemia–reperfusion injury (RIRI), retinitis pigmentosa (RP), retinoblastoma (Rb), dry eye disease (DED), corneal injury, glaucoma, and cataract. In this manuscript, we explore how ferroptosis is related to ocular pathology, as well as its mutual metabolic effects, to establish a basis for further investigation into the pathogenesis of and methods to prevent ferroptosis in ocular diseases.
2 Mechanisms governing ferroptosis
2.1 Iron metabolism
Iron homeostasis in the human body is delicately balanced; both a lack of iron and an excess of iron can be detrimental to the human body (16). Additionally, iron can readily accept and donate electrons and interconvert between iron (Fe3+) and ferrous iron (Fe2+) forms. Fe2+ is unstable and destroys tissue by promoting the transformation of hydrogen peroxide into free radicals that assault cell membranes, proteins, and DNA (17, 18). Iron absorption, iron transmembrane transport, and iron sequestration can affect ferroptosis (19) (Figure 1).
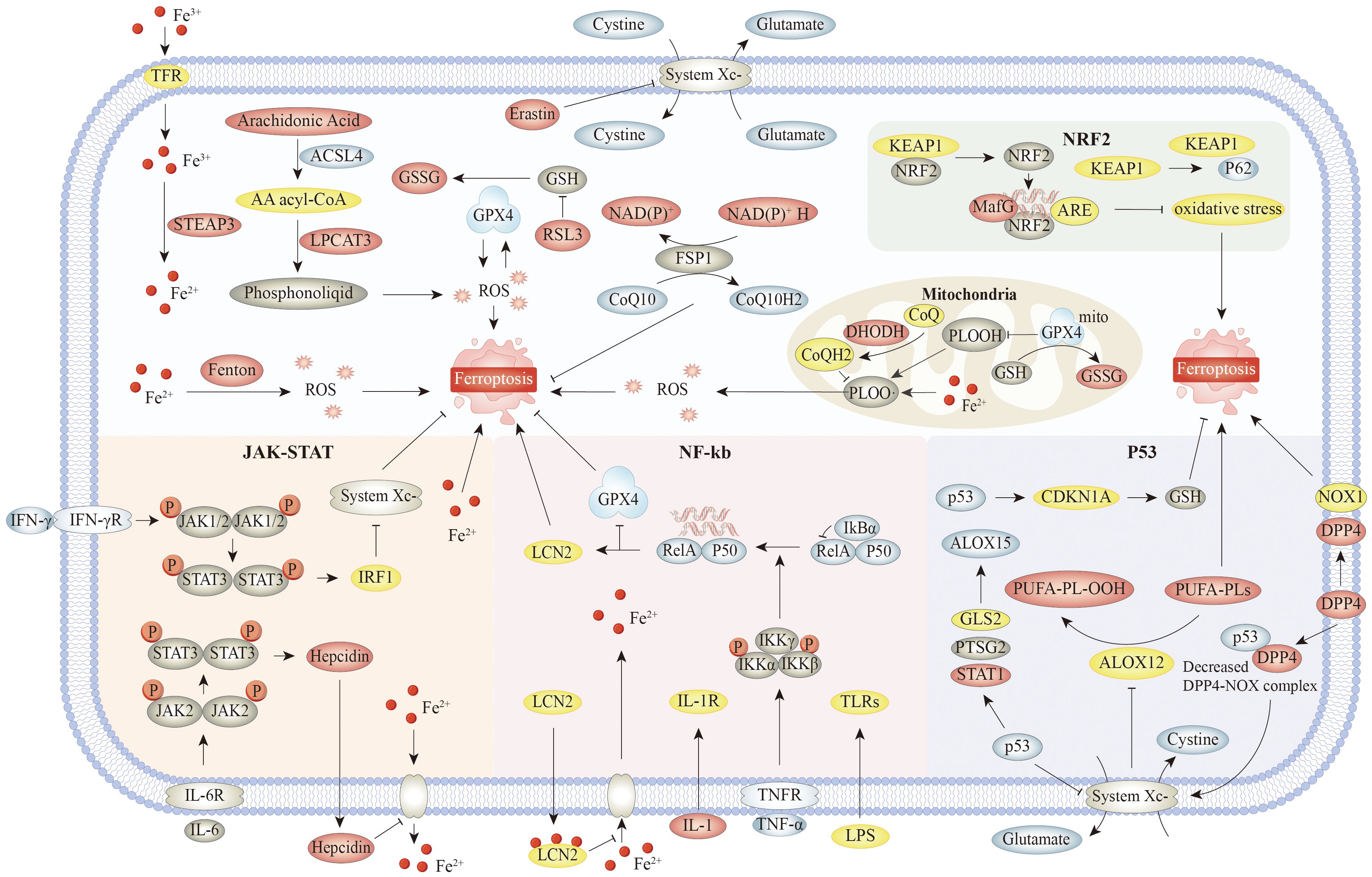
Figure 1. The main mechanisms and signaling pathways of ferroptosis. Specifically, the core mechanism involves accumulation of iron ions, and initiation and amplification of lipid peroxidation, as well as key regulatory pathways such as system Xc− and GPX4. Associated signaling pathways include Nrf2 pathway. Nrf2 inhibits ferroptosis by upregulating the expression of GPX4 and SLC7A11 and inhibiting lipid peroxidation. NF-κB is activated by Toll-like receptor ligands, TNF, IL-1, and other stimuli. IκBα binds to NF-κB dimers and inhibits NF-κB activity under resting conditions. These signaling molecules bind to the corresponding receptors and mediate phosphorylation and subsequent degradation of IκBα. Released NF-κB dimers are transported to the nucleus and regulate transcription of target genes. On the one hand, NF-κB could decrease the transcription of antioxidant molecules such as GPX4, NQO1, and HMOX1, indicating the role of NF-κB pathway in oxidative stress. On the other hand, LIFR deletion enhanced IκBα ubiquitinated degradation and positively regulated NF-κB activation, which in turn promoted LCN2 secretion and sequestrated extracellular iron. In JAK–STAT, IFN-γ promotes ferroptosis by downregulating SLC7A11 and inhibiting system Xc− through the JAK–STAT1–IRF1 axis. IL-6 upregulates hepcidin through the JAK–STAT1 axis, which can promote ferroptosis by maintaining the process outside the iron ion membrane in the cell. In addition, the JAK–STAT pathway can also suppress ferroptosis by upregulating antioxidant genes, such as GPX4, depending on the cell type and environment. On the one hand, P53 can further promote the upregulation of ALOX15 and promote ferroptosis by promoting the expression of GLS2, PTGS2, and STAT1, or can indirectly activate the function of ALOX12 by inhibiting the transcription of SLC7A11, resulting in ALOX12-dependent ferroptosis after reactive oxygen species stress. On the other hand, P53 can also inhibit the generation of lipid reactive oxygen species in cells by competitively binding DPP4 to NOX1. Meanwhile, the P53–DPP4 complex promotes the expression of SLC7A11 and CDKN1A and inhibits ferroptosis. Abbreviations: TFR, transferrin receptor; KEAP1, Kelch-like ECH-associated protein 1; NRF2, nuclear factor erythroid 2-related factor 2; GSSG, glutathione disulfide; ARE, antioxidant response element,; LPCAT3, lysophosphatidylcholine acyltransferase 3; DHODH, dihydroorotate dehydrogenase; PLOOH, phospholipid hydroperoxide; CDKN1A, cyclin-dependent kinase inhibitor 1A; NOX1, NADPH oxidase 1; DPP4, dipeptidyl peptidase-4; GLS2, glutaminase-2; IL-1R, interleukin-1 receptor; TNFR, tumor necrosis factor receptor; TNF-α, tumor necrosis factor alpha; GPX4, glutathione peroxidase 4; NQO1, NAD(P)H quinone oxidoreductase 1.
Circulating Fe3+ attaches to the transferrin receptor and is transported into the cell via transferrin receptor 1 (TFR1) (20). After entering the cell, Fe3+ is reduced and released into the labile iron pool (LIP) of the cytosol (21). Surplus iron is retained in ferritin. In the intracellular LIP, iron mostly exists as Fe2+. The instability and high reactivity of Fe2+ lead to the generation of hydroxyl radicals from excess iron by means of the Fenton reaction. These radicals can directly interact with polyunsaturated fatty acids in the cell and plasma membranes, generating substantial amounts of lipid ROS that induce cell death (22). Iron can also activate ROS-generating enzymes, such as nicotinamide adenine dinucleotide phosphate oxidase an lipoxygenase (LOX), which promote ROS generation. The overaccumulation of ROS and lipid peroxidation leads to cell membrane breakdown (23).
Iron involvement in the visual cycle was discovered by the characterization of the enzyme RPE65, an iron-dependent isomeric hydrolase critical for vision (24, 25). In the retina, iron is among the most abundant metals, and iron metabolism plays a key role in the retina (26). On the one hand, iron can contribute to the antioxidant balance by stabilizing the retina, scavenging free radicals, and shielding the retina from oxidative harm (22, 27). On the other hand, the accumulation of iron and the decrease in the cellular antioxidant protection system increase the susceptibility of the retina to oxidative stress-related cell death, which may negatively impact AMD (28). Ferroptosis induces cell death in retinal pigment epithelial (RPE) cells, retinal photoreceptor (PR) cells, and retinal ganglion cells (RGCs) and plays a role in the progression of retinal diseases such as AMD, glaucoma, and DR (29, 30). Hence, the equilibrium of iron ions is pivotal for ocular well-being.
2.2 Lipid peroxidation
Lipid proteins ensure the stability and normal function of cell membranes, which are destroyed when extensive lipid peroxidation occurs, ultimately leading to cell death (31). Lipid peroxide agglomeration is the core mechanism of ferroptosis (32). Free polyunsaturated fatty acids (PUFAs) function as substrates for lipid peroxidation. Their concentration and cellular distribution directly affect lipid peroxidation and ultimately the intensity of ferroptosis. Enzymes that can bind PUFAs to PLs play a decisive role in ferroptosis. PUFA-PLs are the most easily peroxidized lipids because the allylic carbons in PUFA-PLs are highly susceptible to attack by free radicals, LOX, and O2 (33, 34). The production of PUFA-PLs is governed by two crucial enzymes: acyl-coenzyme A synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (35). Through the esterification process mediated by ACSL4, PUFAs can bind with CoA to produce PUFA-CoA derivatives. After re-esterification, LPCAT3 integrates these PUFA-CoA intermediates into the phospholipids within the plasma membrane. This process results in the generation of PUFA-PLs, such as arachidonic acid–phosphatidylethanolamine and adrenic acid–phosphatidylethanolamine. ACSL4 phosphorylation at the Thr328 site amplifies the production of PUFA-PLs, thus promoting the buildup of lipid peroxidation byproducts (32, 36). Blocking these enzymes decreases lipid peroxidation, decreasing the risk of ferroptosis.
Ferroptosis can be significantly improved by inhibiting ACSL4 and LPCAT3. For instance, liproxstatin-1 suppresses ferroptosis by lowering ACSL4 levels in the RPE–Bruch’s membrane–choroid complex, effectively halting DR progression (37, 38). PUFAs, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), are crucial for the establishment of vision and the maintenance of retinal function. DHA is the most abundant omega-3 fatty acid in PR cells and is critical for maintaining the structural integrity and functional capacity of these cells. EPA exerts anti-inflammatory effects, which can reduce the risk of AMD and other ocular diseases. Additionally, omeg-3 PUFAs protect the retina from light-induced damage by reducing oxidative stress in ocular tissues via antioxidant actions (39, 40).
2.3 GPX4-dependent regulatory pathway
During ferroptosis, system Xc− and GPX4 can coregulate lipid oxides (41). The system Xc− cystine/glutamate exchanger is a dimeric structure with a light subunit (SLC7A11) and a heavy subunit (SLC3A2) connected via disulfide linkages (42). Cystine is transported into cells through the system Xc− transporter located on the membrane surface. Once inside, it undergoes reduction to form cysteine. This cysteine then serves as a substrate for two key enzymes, glutamate–cysteine ligase (GCLC) and glutathione synthetase (GSS), which work in sequence to produce the essential antioxidant glutathione (GSH). The glutathione peroxidase (GPX) family encompasses various isoforms, with GPX4 being a selenium-containing protein vital for mitigating intracellular lipid peroxidation damage in humans (43). Specifically, GPX4 can reduce PUFA-PL hydroperoxides (PUFA-PL-OOHs) to non-toxic PUFA-PL alcohols (PUFA-PL-OHs) using GSH, thereby playing a role in resistance to cellular ferroptosis (19).
Erastin and RSL3 are distinct types of agents that induce ferroptosis. Erastin blocks cystine absorption by suppressing system Xc−, leading to intracellular cysteine depletion, which in turn causes membrane damage and subsequent cell death (44). Notably, erastin can also activate the tumor suppressor p53, thereby inhibiting SLC7A11 and indirectly promoting the development of ferroptosis (45). RSL3 acts by inhibiting GPX4 to accumulate peroxidized phospholipids, which in turn induces the development of cellular ferroptosis (13). Cells with low GPX4 levels are more susceptible to ferroptosis than those with elevated GPX4 levels (11). GPX4 can maintain the healthy state of retinal cells by protecting cell membranes from oxidative damage (46). In GPX4-overexpressing transgenic mice, retinal integrity and function are preserved across multiple oxidative stress-induced degeneration models (47, 48). Under ocular conditions such as those of DR, a reduction in GPX4 levels can result in increased oxidative stress, consequently impacting the functionality and viability of PR cells (49). GPX4 maintains redox homeostasis and protects RPE cells (RPECs), photoreceptors, and RGCs against glutamate-induced cytotoxicity. Collectively, these findings indicate that GPX4 overexpression markedly attenuates oxidative stress-driven retinal degeneration and preserves photoreceptor outer-segment architecture. Boosting GPX4 activity, therefore, holds promise as a precise therapeutic strategy to delay or even reverse AMD progression.
2.4 GPX4-independent regulatory pathway
Although GPX4 acts as a central suppressor of ferroptosis, three other ways to inhibit ferroptosis have been found, irrespective of GPX4. Ferroptosis inhibitor protein 1 (FSP1) is an effective antiferroptosis factor and a coenzyme Q (CoQ) oxidoreductase (50). Mechanistically, FSP1 exhibits nicotinamide adenine dinucleotide + hydrogen (NADH) ubiquinone reductase activity. This mechanism facilitates the conversion of ubiquinone (CoQ10) into its reduced form, ubiquinol (CoQ10H2), which effectively curbs the production of lipid free radicals. Another pathway involves enhancing vitamin E regeneration, which in turn suppresses lipid peroxidation and prevents ferroptosis. The FSP1–CoQ10–NAD(P)H axis serves as a key cellular defense against oxidative stress and an alternative pathway for inhibiting ferroptosis (51).
In 2019, Bersuker et al. reported that tumor cells lose resistance to the ferroptosis inducer RAS-selective lethal 3 (RSL3) when they are depleted of FSP1, thereby becoming more susceptible to ferroptosis (52). FSP1 is crucial for corneal and retinal repair, enhancing tissue regeneration through cell cohesion and facilitating movement (53). Moreover, FSP1 supports RPE cells by ensuring that they remain robust and well-aligned, which are crucial for maintaining photoreceptor functionality. Furthermore, FSP1 could play a role in controlling abnormal vascular growth and scar formation in the context of vascular diseases such as DR (54). FSP1 is a vital player in fixing both the cornea and the retina. When the cornea becomes injured, FSP1 forms a temporary framework that supports the regeneration of the outer layer, which speeds up the healing process. Moreover, it is crucial in the protection of RPECs. In cases where blood vessels malfunction, like in DR, FSP1 may play a role in controlling the growth of extra blood vessels and the formation of tough scar tissue by communicating with growth factors and the cell surface receptors, which in turn affects how the extracellular matrix changes and the series of events that lead to new blood vessel formation. Furthermore, FSP1 may also be a factor in AMD by influencing the growth of abnormal blood vessels behind the retina and the development of scar tissue.
The dihydroorotate dehydrogenase (DHODH) pathway generates reduced coenzyme Q (CoQH2) within the inner mitochondrial membrane, which plays a key role in suppressing ferroptosis. By functioning as a potent radical-trapping antioxidant, CoQH2 effectively blocks lipid peroxidation—a critical mechanism that halts the progression of ferroptosis. In this process, DHODH functions concurrently with the mitochondrial GPX4, separate from the cytoplasmic GPX4 and FSP1 (55). In an experiment in which a stable DHODH knockout human corneal epithelial cell (HCEC) line was established, the balance of expression between DHODH and GPX4 closely regulated cellular ferroptosis homeostasis (56). Recently, KIO-101 ophthalmic solution, a topical DHODH inhibitor, has been shown to be safe and effective at reducing conjunctival hyperemia at low and medium doses over a 12-day period in patients with herpesvirus (HV) infection and conjunctival hyperemia (57). The enzyme DHODH acts as a safeguard against ferroptosis in eye tissues like the cornea and retina without relying on GPX4, but when this protective pathway is diminished, particularly when disrupted by oxidative stress, it could lead to damage of corneal and retinal cells by facilitating the progress of ferroptosis. In addition, researchers have deciphered the link between DHODH’s activity and several serious eye conditions, including AMD, DR, and abnormal blood vessel growth in the cornea.
Guanosine-5′-triphosphate (GTP) cyclohydrolase-1 (GCH1), a key biosynthetic enzyme, regulates tetrahydrobiopterin (BH4) production. BH4 serves as a coenzyme for critical neurotransmitter (e.g., dopamine) and nitric oxide synthesis pathways (58) and coregulates ferroptosis through two mechanisms. First, GCH1 produces the lipophilic antioxidant BH4, which functions similarly to CoQ10 to prevent lipid peroxidation. Second, GCH1 remodels the lipid membrane environment by promoting the uptake of PUFA-PL, an inducer of ferroptosis, while increasing CoQ10 (CoQ10H2) levels, thereby counteracting lipid peroxidation. In ophthalmology, the role of GCH1 could be crucial for maintaining retinal integrity and eliminating specific eye disorders, such as AMD and other retinal degenerative diseases (59).
2.5 JAK–STAT
Janus kinases (JAKs) are intracellular non-receptor tyrosine kinases that include JAK1, JAK2, JAK3, and TYK2. These kinases are widely distributed across various tissues and cells. Signal transducers and activators of transcription (STATs) act as substrates for JAKs and function as transcription factors. The STAT family includes genes such as STAT1, STAT2, and STAT3. Following phosphorylation by JAKs, STATs translocate to the nucleus to modulate gene transcription. The JAK–STAT signaling pathway is defined by these key components and is distinguished by their diverse interactions (60). When IL-6 acts on its receptor, it causes the phosphorylation of JAK2, leading to the phosphorylation of STAT3 and resulting in an increase in hepcidin expression. When excess hepcidin is transferred to the extracellular space, it inhibits the release of Fe2+ from the cell, ultimately leading to ferroptosis. IFN-γ acts on the IFN-γ receptor, resulting in the phosphorylation of JAK1/2, which triggers the phosphorylation of STAT1 and leads to an increase in interferon regulatory factor 1 (IRF1) expression, which inhibits the function of SLC7A11 with SLC3A2 (system Xc−), ultimately leading to ferroptosis (61).
In ophthalmology, corticosteroids (CSs) are a common treatment for patients with non-infectious uveitis (NIU). Potentially severe side effects may result from prolonged CS use. JAK/STAT inhibitors can provide additional therapeutic options for patients with NIU. Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disorder that mainly affects exocrine glands and contributes to DED development (62). JAK1 may influence pSS progression, offering therapeutic potential (63).
2.6 NF-κB
The classical transcription factor NF-κB was identified more than three decades ago (64). A growing body of research has indicated that ferroptosis is linked to the NF-κB signaling pathway. At rest, IκBα suppresses NF-κB by binding to its dimers. Upon external stimulation, signaling molecules bind to their receptors, leading to the activation of the IKK complex (via phosphorylation of IKKβ), which phosphorylates IκBα, causing its degradation and dissociation from NF-κB dimers. In certain pathological contexts, the nuclear translocation of NF-κB dimers can suppress the expression of antioxidant-related genes such as GPX4, thereby promoting ferroptosis (61).
Wen-Jing Liang et al. speculated that high-mobility group box 1 (HMGB1) could trigger apoptosis in retinal endothelial cells via the NF-κB pathway, leading to ischemic regions and subsequently triggering compensatory blood vessel growth and enhanced new vessel formation (65). In the mammalian retina, NF-κB signaling promotes glial reactivity and inhibits glia-mediated neuronal regeneration (66). Kaiwen Jiang et al. found that fosinopril (FOS), a TLR4 inhibitor, can inhibit NF-κB signaling in both in vivo and in vitro models. This inhibition can regulate diabetic DED, providing new therapeutic options for diabetic DED (67). Increased high-temperature requirement for A serine peptidase 1 (HTRA1) levels activate NF-κB protein synthesis, whereas HTRA1 knockdown downregulates NF-κB protein expression. Elevated NF-κB expression is a known risk factor for AMD (68).
2.7 Nrf2
With respect to nuclear transcription factors, such as Nrf2, Sun et al. reported the role of the p62–Keap1–Nrf2 antioxidant signaling cascade in safeguarding hepatoma cells against ferroptosis (69). First, the p62-mediated degradation of Keap1 activates Nrf2. Activated Nrf2 translocates to the nucleus, where it triggers the expression of antioxidants such as glutathione redox system components, enzymes that regulate iron metabolism, and other pertinent molecules, which can inhibit ferroptosis. Nrf2 cannot undergo the above manipulations when it is degraded by ubiquitination. Second, the antioxidant response element (ARE) acts as a pivotal modulator of Nrf2-driven SLC7A11 activation, with its relationship to Nrf2 occurring irrespective of p53 involvement (70). Activating the p62–Keap1–Nrf2 signaling pathway increases systemic Xc− levels, thereby mitigating lipid peroxide accumulation and inhibiting ferroptosis (71).
The eye serves as a key marker of oxidative damage (72, 73). Oxidative stress is involved in numerous eye diseases. The Keap1–Nrf2–ARE pathway serves as an antioxidant pathway. Many studies have utilized Nrf2-activating drugs to evaluate the cytoprotective effects of Nrf2 in retinal tissue, particularly in rescuing RPECs from oxidation-induced injury and death. Xu et al. enhanced bioavailability by combining quercetin with phospholipids. This approach resulted in a nearly 80% increase in the proliferation of RPECs, decreased ROS and malondialdehyde (MDA) levels, and inhibited apoptosis. The observed outcomes were facilitated through increased Nrf2 protein transport and the activation of its target genes, including heme oxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase 1 (NQO1) (74). Nrf2–Keap1 signaling is central to the complex pathology of DR (75). Empirical research has demonstrated that elevated glucose levels in diabetic mice blunt Nrf2-mediated protection; indeed, compared with wild-type mice, Nrf2-deficient diabetic rodents exhibited significantly greater retinal superoxide levels after 5 weeks of diabetic modeling (75). von Otter et al. reported that Nrf2 gene mutations may promote cataract progression but do not invariably increase the likelihood of cataract onset (76). The gene loci of Nrf2 and Keap1 were analyzed in 489 cataract patients of European ancestry. One Nrf2 haplotype, GAAAA, was associated with the progression of cataract formation. Specifically, this haplotype was significantly linked to an earlier onset of cataracts by approximately 4 years. However, the GAAGAGGC haplotype of the Nrf2 gene delayed the need for cataract surgery by 4 years (77). This undoubtedly provides new research directions for addressing the challenges posed by an aging population and the increasing demand for cataract surgery. Activating Nrf2 not only scavenges ROS but also inhibits inflammation and promotes epithelial repair, providing an ideal target for treating DED (78).
2.8 p53
The p53 gene was discovered in 1979, and studies have shown that p53 serves dual functions in ferroptosis regulation (79). On the one hand, it enhances ALOX15 expression by suppressing SLC7A11 expression, thereby deactivating ALOX12. Furthermore, it upregulates the expression of metabolic genes such as SAT1 and glutaminase-2 (GLS2); their combined activity increases lipid–ROS production and GSH turnover, thereby sensitizing cells to ferroptosis (80, 81). On the other hand, p53 suppresses ROS generation in cellular lipids by competitively binding to dipeptidyl peptidase-4 (DPP4), thereby preventing NOX1 activation. Moreover, SLC7A11 and cyclin-dependent kinase inhibitor 1A (CDKN1A) gene expression is stimulated by the p53–DPP4 interaction, thereby inhibiting ferroptosis (82). p53 activation does not noticeably affect GPX4 activity, suggesting that it does not induce ferroptosis via GPX4 (71). Recent research has indicated that wild-type p53 can promote ferroptosis in certain contexts (e.g., by repressing SLC7A11), whereas selected gain-of-function mutants may also sensitize tumor cells to ferroptosis, although many mutants confer resistance (70).
p53 can be rapidly activated to induce cell cycle arrest when retinal cells are subjected to photodamage, hypoxia, or metabolic stress, enabling DNA repair (83). When injury is extreme, p53 triggers programmed cell death to prevent the growth of damaged cells and mitigate tumor development. Wai Kit Chu presented evidence that targeting the MDM2–p53 pathway could help elucidate the pathogenesis of pterygium and develop new treatments to reduce the postoperative recurrence rate (84). Ying Chen et al. revealed a mechanism by which p53 increases FoxO3a ubiquitination levels through ubiquitin-conjugating enzyme E2 L6 (UBE2L6) and promotes aging in diabetic retinal endothelial cells, suggesting a novel therapeutic focus for the mitigation and treatment of DR (85). Moreover, p53 plays a role in modulating cellular metabolism and influences the progression of ocular disorders such as DR through its influence on blood glucose regulation and fatty acid metabolism (86).
2.9 AMPK
Energy stress is a metabolic condition characterized by ATP depletion and elevated AMP levels. Energy stress triggers the activation of AMP-activated protein kinase (AMPK). AMPK phosphorylates downstream targets to promote ATP production (87). The inhibition of ATP depletion restores energy balance. AMPK acts as a core regulator of ATP balance in cells and can have diametrically opposite effects on substrate-dependent ferroptosis. AMPK-induced BECN1 phosphorylation promotes ferroptosis by either suppressing SLC7A11 function or triggering autophagy (88, 89). In contrast, mitochondrial energy stress may suppress ferroptosis through AMPK-mediated acetyl-CoA carboxylase alpha (ACACA) phosphorylation. Experimental results have revealed a lack of significant ferroptosis when glucose concentration was insufficient, even when erastin was added, demonstrating that the energy stress-mediated AMPK pathway could inhibit ferroptosis (87).
AMPK also plays a role in controlling mitochondrial formation and inflammation. It promotes mitochondrial quality control, helping to replace dysfunctional mitochondria, which is essential for maintaining the health of eye cells. Additionally, AMPK can control the progression of ocular diseases by inhibiting the mTORC1 pathway (90, 91). Yuli Guo et al. reported that the AMPK agonist metformin could ameliorate hyperglycemia-induced meibomian gland dysfunction (MGD), demonstrating that AMPK may be a therapeutic target for diabetes-induced MGD (92). Fangli Peng et al. found that AMPK/MFF signaling plays a key role in DED progression by actively promoting mitochondrial fission and mitophagy. Suppressing excessive mitochondrial fragmentation helps mitigate oxidative stress and inflammation associated with DED, offering promising therapeutic targets for clinical intervention. These findings establish a scientific foundation for the development of novel DED treatment strategies (93).
2.10 HSPs
Heat shock proteins (HSPs) are popular representative protein families of chaperones and have traditionally been divided into nine subfamilies according to molecular weight (94). They mitigate cellular stress, provide antioxidant protection, and steer immune activity toward an anti-inflammatory state, collectively constituting key mechanisms against ferroptosis (95). The phosphorylation of heat shock protein B1 (HSPB1), also referred to as HSP25 or HSP27, mediated by protein kinase C (PKC), limits cytoskeleton-mediated iron uptake. This action restricts the death of iron-tropic cancer cells by reducing iron uptake, thereby decreasing ferroptosis (96). In addition, the phosphorylation of HSPB1 by PKC triggers the increased expression of ferritin light chain (FTL) and ferritin heavy polypeptide 1 (FTH1), both of which are important components associated with ferritin. Increased ferritin expression decreases cellular iron levels and mitigates the formation of ROS in lipids. HSPB1 also regulates TFR1 expression.
Activating the expression of heat shock protein family A (HSP70) member 5 (HSPA5) can prevent erastin-induced GPX4 degradation through the formation of HSPA5–GPX4 protein complexes, thereby inhibiting ferroptosis (97). In contrast, heat shock protein 90 can induce ferroptosis by phosphorylating receptor-interacting protein 1 (RIP1), a key regulator of necroptosis, and inhibiting GPX4 (98).
Heat shock proteins play a protective role in ocular diseases under oxidative, inflammatory, thermal, UV, or metabolic stress. T-cell reactions targeted at heat shock proteins contribute to glaucomatous neuronal degeneration (99, 100). In cases of glaucoma and neurodegenerative eye disorders, HSPs serve as a protective barrier for RGCs against mechanical stress and neurotoxicity associated with these diseases (101, 102). HSPB1 expression increases sharply when the retina is subjected to injury, such as ischemia, oxidative stress, trauma, or ocular hypertension. On the one hand, it maintains cell structural integrity; on the other hand, it inhibits apoptotic signal amplification, thereby increasing the survival rate of retinal cells under toxic stimuli (103). However, Alyce Alven et al. reported increased levels of HSPB11 and HSP60 in the tear film of DED patients after acute exposure to dry conditions, a trend that was consistently noted (104).
2.11 NADPH
NADPH plays a crucial role in maintaining cellular redox balance, and each NADPH molecule provides two electrons to ensure the integrity of antioxidant defense mechanisms (105). Glutathione reductase (GR), FSP1, NAD(P)H quinone dehydrogenase 1 (NQO1), and thioredoxin reductase (TR) maintain the reduced state of retinol, GSH, CoQ10, and vitamin K molecules through the electron supply of NADPH, effectively inhibiting the occurrence of phospholipid peroxidation (106, 107).
NADPH promotes both phospholipid synthesis and the function of heme-dependent NADPH oxidases (NOXs). These specialized enzymes transfer electrons from cytoplasmic NADPH to generate ROS that initiate lipid peroxidation. Concurrently, the same pool of NADPH fuels the GPX4, thioredoxin reductase, and lipid-remodeling pathways, both neutralizing peroxides and synthesizing protective phospholipids (PUFA-OHs) to counteract ferroptosis. Under steady-state conditions, NADPH is recruited primarily for ferroptosis defense, thereby potentially inhibiting its support of the pro-ferroptosis pathway (107). These findings indicate that NADPH plays a critical role in ferroptosis-related chemical processes (108).
NADPH functions in biosynthetic reactions, as well as maintains a reducing environment within the cell, and is crucial for the dark-phase reactions of photosynthesis in mitigating oxidative stress. In ophthalmology, ensuring NADPH levels and functionality is crucial for safeguarding ocular health and managing eye disorders (109). NADPH oxidase 4 (NOX4) contributes to DED progression by modulating IL-1β, NLRP3, and MUC5AC expression; inhibiting NOX4 using drugs and inhibitors may improve DES (110). The targeted suppression or blockage of NADPH oxidase 2 (NOX2) function significantly alleviates oxidative damage in the retina, corrects immune system imbalances, prevents damage to the inner blood–retinal barrier (iBRB), and mitigates neurovascular unit (NVU) impairment. Additionally, it reduces RGC death and optic nerve (ON) axon deterioration caused by high intraocular pressure (H-IOP). These findings suggest a promising therapeutic strategy for managing glaucoma (111).
There are many ferroptosis inhibitors. AA-861, zileuton, and PD-146176 inhibit ferroptosis by reducing the generation of LOX-induced lipid peroxidation products. ATV regulates iron content, ROS levels, and GSH to reduce lipid peroxidation. CoQ10 acts on FSP1 and GPX4 to reduce cholesterol metabolism disorders and inhibit ferroptosis. Deferoxamine and other iron chelators work by reducing iron content. Ferrostatin-1 (Fer-1) regulates ROS and the GSH/GPX4 axis to reduce lipid peroxidation. Lipstatin-1 (Lip-1) inhibits ferroptosis by suppressing the activation of lipid metabolism. Naringenin increases cellular sensitivity to ferroptosis by enhancing the SIRT1/FOXO3a signaling pathway. SAK increases the expression of antioxidant genes by activating the NRF2 signaling pathway. Selenium, sodium, and selenite increase the expression of antioxidant genes by acting on GPX4 and system Xc−. Tangeretin reduces lipid peroxidation by acting on GPX4 and NRF2. Taurine reduces iron content by regulating the OGT/GPX4 signaling pathway. Trolox and VitE reduce lipid peroxidation by scavenging hydroxyl radicals and regulating GPX4 (Table 1).
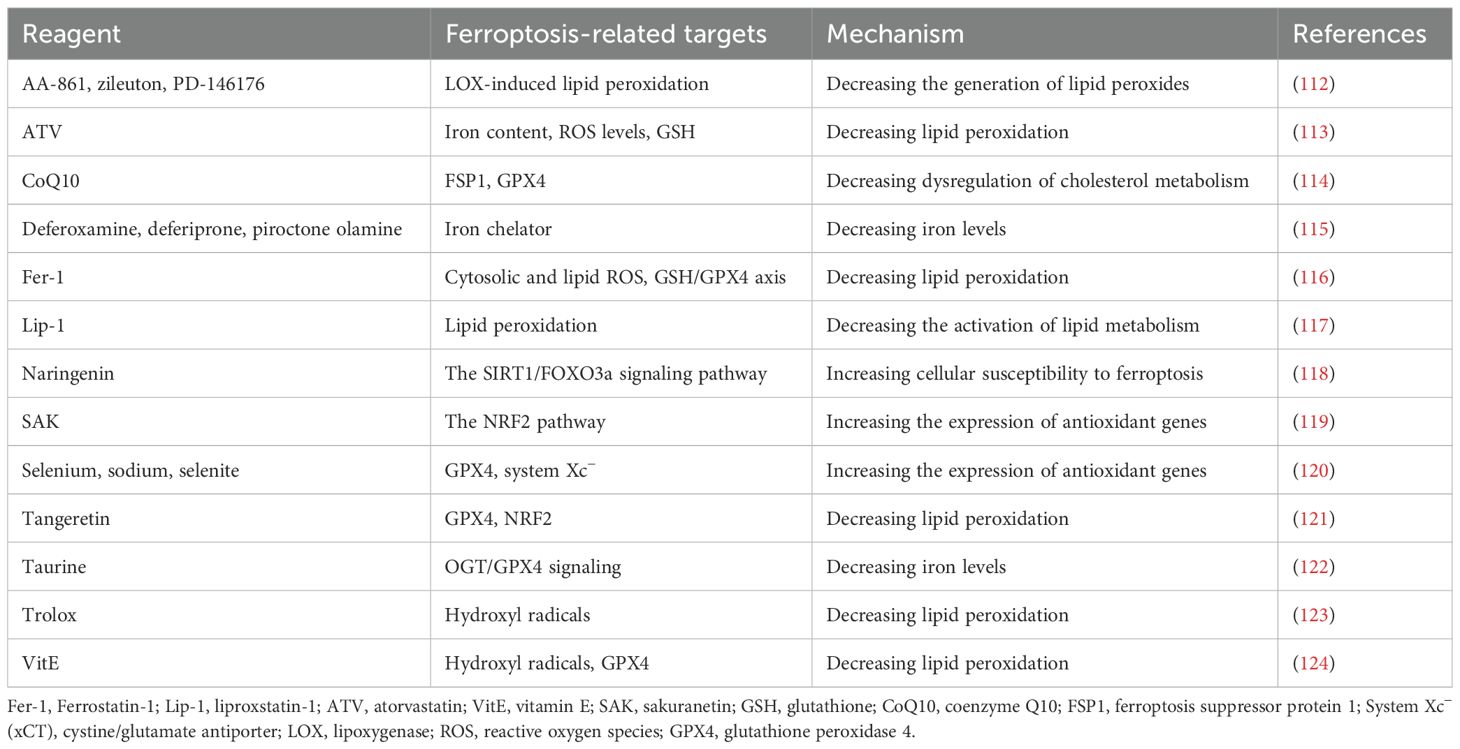
Table 1. The inhibitors of the ferroptosis pathway.
3 Ferroptosis and other forms of cell death
Ferroptosis represents an iron-driven cell elimination process that is distinct from apoptosis, marked by the excessive buildup of lipid peroxides (125). This biological mechanism plays a pivotal role in the development and progression of numerous diseases. When it comes to cellular characteristics, ferroptosis causes mitochondria to shrink, with their cristae either diminishing or completely vanishing, while also increasing membrane density. Additionally, the cell membrane becomes fragmented and develops blebbing, although the nucleus remains largely unchanged in appearance (126).
3.1 Ferroptosis and apoptosis
Apoptosis serves as a natural, programmed mechanism in the course of development, effectively eliminating unnecessary cells for an organism to function well. This biological process is key to maintaining balance within the body by selectively removing cells as needed. Additionally, it acts as a safeguard, eliminating compromised, infected, or potentially cancerous cells, and it plays a vital part in shaping and sustaining a robust immune system (127). During programmed cell death, cells systematically contract and develop blebs, bulbous protrusions resembling bubbles along their outer membrane. Concurrently, nuclear DNA disintegrates while certain internal structures, including the endoplasmic reticulum, fragment into smaller components. The cell ultimately divides into numerous membrane-encased packets known as apoptotic bodies, which are subsequently cleared by phagocytic cells like macrophages without triggering any inflammatory response (128).
Cancer treatment has recently capitalized on the combined impact of ferroptosis and apoptosis, with researchers successfully engineering a two-dimensional catalytic nanozyme known as Cu2Mo3O8 nanosheets (CMO) NSs. This innovative compound not only boosts ROS production to stimulate ferroptosis but also mimics glucose oxidase activity, essentially cutting off the tumor’s nutrient supply by breaking down glucose and generating H2O2. In addition, CMO NSs facilitate calcium release alongside ROS-induced buildup of endogenous calcium, causing calcium overload that leads to mitochondrial malfunction and apoptosis. This multi-pronged approach makes CMO NSs a promising candidate for antitumor therapy (129). Under specific conditions, ferroptosis and apoptosis can also achieve mutual transition (130). Harnessing the synergy between ferroptosis and apoptotic pathways holds immense potential for advancing cancer treatment strategies. By focusing on this interconnected cell death network, researchers are working toward a more holistic blueprint to curb tumor progression and potentially tackle other conditions linked to these mechanisms. This exciting frontier continues to be a focus of scientific exploration, with investigations delving deeper into its therapeutic possibilities (131).
In the eye field, 24 hours after retinal ischemia–reperfusion, key indicators of ferroptosis, such as lipid peroxidation and the downregulation of GPX4, rise in tandem with apoptosis markers, including cleaved caspase-3 and TUNEL-positive cells (132). Treatment with the ferroptosis inhibitor ferrostatin-1 can simultaneously reduce both types of cell death, indicating that they jointly mediate acute damage. In a chronic ocular hypertension mouse model, RGCs exhibit both ferroptosis (characterized by the upregulation of ACSL4 and increased lipid ROS) and apoptosis (marked by Bax/Bcl-2 imbalance and the release of cytochrome c). The iron chelator deferoxamine or the GPX4 activator RSL3 can concurrently reduce both types of death and decrease RGC loss by approximately 40%. In corneal epithelial cells stimulated by hyperosmosis or cigarette smoke extract (CSE), both ferroptosis and apoptosis occur simultaneously (133). The combined inhibition of Fer-1 and z-VAD more significantly restores cell viability than either agent alone. Overall, ferroptosis and apoptosis form a connection through an interplay involving ROS, mitochondrial dysfunction, and inflammation. Targeting both ferroptosis and apoptosis together has shown superior efficacy compared to single-pathway intervention, providing a new and precise therapeutic strategy for the treatment of ocular diseases.
3.2 Ferroptosis and pyroptosis
Pyroptosis is a cell death mode in which cell membrane Gasdermin family proteins form pores that lead to the release of cell contents and cell rupture (134). Pyroptosis is an important defense mechanism in the immune system that can clear pathogen-infected cells and trigger inflammatory responses. Inflammasomes and GasderminD (GSDMD) play critical roles in pyroptosis (135).
Programmed cell death (PCD) manifests in various guises, each with its own distinctive features shaped by unique molecular pathways, and these forms often cross paths in complex ways (136). Interestingly, ferroptosis and pyroptosis may be somehow connected, although the exact nature of their relationship remains to be elucidated. Recent experiments, however, have revealed their antagonistic relationship by tracking how HMGCR shifts its position during cell death and how BRCC36 controls both processes by removing ubiquitin from HMGCR. Moving forward, diving deeper into the precise mechanics of how ferroptosis and pyroptosis interact could pave the way for more targeted and effective disease treatments (137).
Inflammasomes are an important marker of pyroptosis. In recent years, the intricate relationship between ferroptosis and inflammasomes has been progressively unveiled by a growing body of research. Specifically, iron overload serves as a catalyst for lipid peroxidation, which subsequently triggers a substantial surge in the production of ROS. This surge in ROS acts as a driving force that propels the assembly and activation of inflammasomes, such as NLRP3 (138). Once activated, these inflammasomes orchestrate the release of proinflammatory cytokines IL-1β and IL-18 (139). Moreover, the formation of GSDMD pores, which are also induced by the activated inflammasomes, further exacerbates the accumulation of iron and oxidative damage within the cells. Collectively, these events culminate in a cascade amplification of cell death and inflammatory response. In the context of RPECs, which play a pivotal role in maintaining retinal homeostasis, the targeted knockdown of cGAS or STING has been shown to effectively disrupt a critical sequence of events (140). This sequence includes iron accumulation, leakage of mitochondrial DNA, and subsequent NLRP3 activation. By interrupting this pathway, the levels of ferroptosis and inflammatory factors are significantly reduced by more than 50% in photochemical injury models. This reduction underscores the potential of targeting this intersection to simultaneously block cell death and the inflammatory storm, thereby offering a novel and combined intervention strategy for retinal degenerative diseases (141).
3.3 Ferroptosis and necroptosis
Historically, apoptosis was widely regarded as the sole mechanism of programmed cell death, with necrosis dismissed as a chaotic and unregulated phenomenon. The tide began to turn in 1988 when research revealed that TNF-α could trigger both apoptotic and necrotic cell death, suggesting that necrosis may not be a mere accident but a controlled process. A pivotal study by Holler’s team later identified receptor-interacting serine/threonine-protein kinase 1 (RIPK1) as a key player in Fas-mediated cell death. The discovery of necrostatin-1 (NEC-1), a specific inhibitor of RIPK1 kinase activity, which effectively halts death receptor-induced necrosis, further cemented the idea that this type of cell demise is under strict regulation. Dubbed “necroptosis”, this programmed necrotic pathway is marked by plasma membrane disruption and shares upstream components, such as RIPK1, with caspase-8-dependent apoptosis. Its morphological hallmarks include cell swelling and ballooning, bubble-like protrusions, and rupture of the plasma membrane (142).
Necroptosis and ferroptosis differ in morphological characteristics and regulatory mechanisms. These two modes of cell death can coexist in diseases and jointly drive pathological progression (143). Studies have shown that the simultaneous inhibition of both forms of cell death can enhance therapeutic efficacy against complex necrosis-related disorders. However, targeting both types with a single compound remains challenging because they involve distinct molecular pathways.
High-level ROS not only act as a “catalyst” for ferroptosis but also directly phosphorylate RIPK3, thereby triggering necroptosis. Once the RIPK3–MLKL pores are formed, they further promote the Ca2+/Na2 influx and a mitochondrial ROS burst, which in turn exacerbates ferroptosis (144). In ARPE-19 cells, the ferroptosis inducer RSL3 suppresses GPX4 and simultaneously significantly increases RIPK3 phosphorylation. Conversely, the necroptosis inducer shikonin induces mild lipid peroxidation, indicating that the two pathways converge at RIPK3. Treatment with the RIPK1 inhibitor Nec-1 concurrently blocks both necroptosis and ferroptosis, and its protective effect is significantly superior to that of ferrostatin-1 or z-VAD alone (145). In a retinal ischemia–reperfusion model, the intravitreal administration of Nec-1 or the iron chelator deferoxamine increases RGC survival by 45% and 38%, respectively. However, their combination raises survival to 62%, suggesting synergistic protection through dual-pathway inhibition (132). In a dry-eye corneal epithelial model, hyperosmotic stress triggers both ferroptosis and RIPK3–MLKL phosphorylation, which is associated with necroptosis. Ferrostatin-1 + Nec-1 combined inhibitors restore more than 80% of epithelial integrity, significantly outperforming either agent alone. Ferroptosis and necroptosis thus form a self-amplifying, reciprocal loop mediated by ROS, RIPK3, and lipid peroxidation in ocular diseases, with RIPK3 serving as the molecular intersection (146). Combined blockade of necroptosis and ferroptosis provides synergistic protection and offers a novel multi-target intervention strategy for retinal degenerative diseases and corneal epithelial injury (132).
4 Ferroptosis in the ocular microenvironment
In the ophthalmic microenvironment, ferroptosis affects both structural and immune cells (Figure 2). Corneal epithelial cells lose the GPX4 shield under hyperosmotic conditions, and lipid ROS rapidly break through the cell membrane and induce corneal opacity. RPECs experience a rapid iron surge from ingesting outer photoreceptor segments high in PUFAs, leading to geographic atrophy. Once GSH is depleted, RGCs undergo apoptosis and signal transduction to amplify retinal degeneration in glaucoma or ischemia–reperfusion scenarios because of the dual effects of mitochondrial iron deposition and hypoxia. Moreover, infiltrating T cells upregulate Nox4 in the inflammatory microenvironment and produce a large number of extracellular ROS, such that they themselves undergo ferroptosis, thereby weakening immune surveillance. The activity of the natural killer cell-dependent and iron-dependent perforin–granzyme pathway is blocked by iron overload, thereby decreasing the killing function of these cells. After M1 polarization, macrophages release large amounts of Fe2+ via ferritin autophagy mediated by nuclear receptor coactivator 4 (NCOA4), which not only promotes ferroptosis but also causes the further release of ferroptosis-promoting factors into the cornea, RPE, and RGCs, forming a vicious cycle in the ocular microenvironment. Targeted blockade of this transcellular ferroptotic network is emerging as an innovative approach for safeguarding the visual ecosystem.
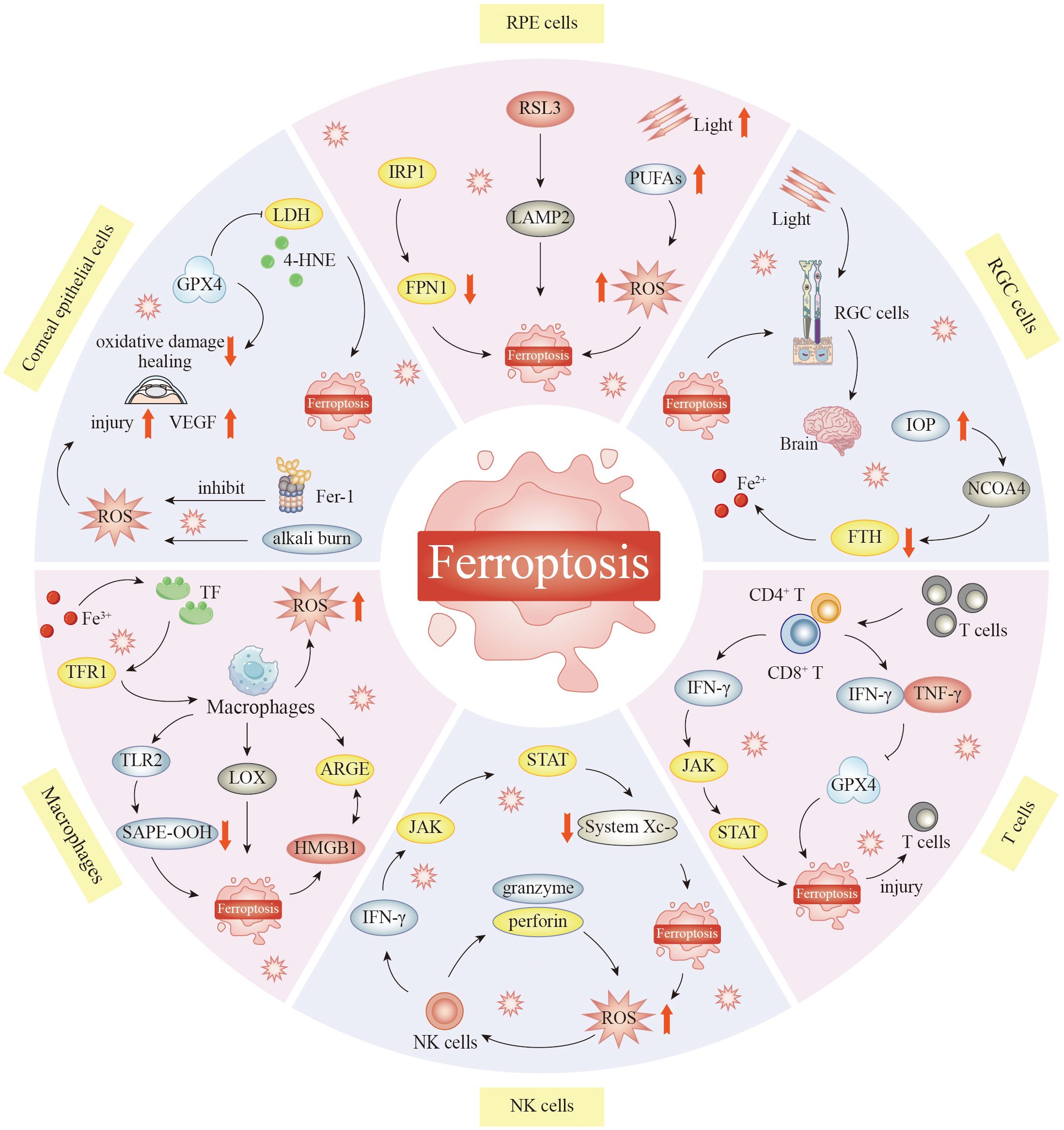
Figure 2. The role of ferroptosis in the ocular microenvironment. Ferroptosis plays an important role in keratocyte injury. Inhibition of ferroptosis can protect corneal epithelial cells from oxidative stress-induced cell death in the absence of GPX4. Corneal alkali burn activates ferroptosis. Reactive oxygen species attack mitochondria and jointly promote the occurrence of ferroptosis in corneal tissue. In RPECs, GPX4 inhibitors 1s and RSL3 can elevate LAMP2, resulting in ferroptosis. Supplementation with cysteine and glutamine restored GSH function, thereby inhibiting ROS-induced death in LAMP2 knockout RPEC. Reduction of NCOA4 leads to increased degradation of FTH1 and increased Fe2+ content in the retina. This significantly increases iron ion levels, leading to RGC damage. Macrophages can clear cells that undergo ferroptosis. HMGB1 released from ferroptosis cells can interact with AGRE on macrophages and mediate inflammatory responses in macrophages. In addition, TLR2 on macrophages recognizes and binds SAPE-OOH on the surface of ferroptosis cells to help clear ferroptosis cells. Macrophages are central to the regulation of iron homeostasis, and normally, the body can maintain the stability of iron content. When this stabilization is broken, abnormal iron metabolism may oversupply the active form of iron, ultimately leading to the development of ferroptosis. Iron accumulation and excess can lead to increased oxidative stress in natural killer cells, which triggers ferroptosis. T cells themselves may also develop ferroptosis, which may attenuate their immune response. T cells lacking GPX4 rapidly accumulate membrane lipid peroxides, leading to ferroptosis, and CD8+ cytotoxic T cells can eliminate tumor cells by inducing ferroptosis, which may enhance the effect of cancer immunotherapy. Abbreviations: RSL3, RAS-selective lethal 3; IRP1, iron-regulatory protein 1; PUFAs, polyunsaturated fatty acids; LDH, lactate dehydrogenase; LAMP2, lysosome-associated membrane protein 2; 4-HNE, 4-hydroxynonenal; VEGF, vascular endothelial growth factor; IOP, intraocular pressure; NCOA4, nuclear receptor coactivator 4; CD42 T, CD4-positive T lymphocytes; CD82 T, CD8-positive T lymphocytes; TLR2, Toll-like receptor 2; SAPE-OOH, 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine-hydroperoxide; HMGB1, high-mobility group box 1; NK cells, natural killer cells; GPX4, glutathione peroxidase 4; RPECs, retinal pigment epithelial cells; GSH, glutathione; ROS, reactive oxygen species; FTH1, ferritin heavy polypeptide 1; RGC, retinal ganglion cell.
4.1 Ferroptosis in corneal epithelial cells
The cornea is part of the optometric pathway, and corneal damage occurs when the protective function and structural integrity of the corneal epithelium are compromised (147). This disruption can result in partial or complete loss of corneal epithelial cells. Patients can present with widespread punctate lesions or surface erosions of the cornea, as well as persistent detachment and epithelial damage, and inflammatory reactions, ultimately endangering vision (148, 149). Corneal epithelial cells face greater susceptibility to oxidative damage (150).
GPX4 serves as a pivotal regulator of ferroptosis-driven processes. Nonetheless, it is a crucial antioxidant enzyme that is integral to maintaining the balance of redox reactions and fostering the repair of corneal epithelial cells. GPX4 does this by transforming harmful lipid peroxides into harmless lipid alcohols, thereby accelerating the healing process (151). Lower GPX4 levels trigger oxidative damage and cell toxicity, resulting in reduced corneal epithelial cell survival and impaired wound healing ability. Sakai introduced specific siRNAs against catalase, GPX4, superoxide dismutase 1 (SOD1), and SOD2 into HCECs (150) and found that lactate dehydrogenase (LDH) release was significantly elevated in the GPX4-, SOD1-, SOD2-, and CAT-knockdown groups, with the greatest increase observed in the GPX4-knockdown group compared with the SOD1-knockdown group. Further studies have revealed delayed wound recovery in GPX4 siRNA-treated cells 2 days after wounding. However, α-tocopherol alleviated the delay in wound healing caused by GPX4 deficiency. This study demonstrated that a decrease in GPX4 levels in HCECs resulted in elevated levels of lipid peroxidation markers such as LDH and 4-hydroxynonenal (4-HNE), which triggered ferroptosis (150). In addition, the application of the ferroptosis inhibitor Fer-1 could mitigate the decreased cell viability and elevated LDH associated with GPX4 gene knockout (152). Research has indicated that compared with their wild-type counterparts, mice with partial GPX4 deficiency exhibit delayed corneal wound healing after epithelial damage, highlighting the critical role of GPX4 in postinjury tissue repair. These findings suggest that blocking ferroptosis may shield corneal epithelial cells from oxidative stress-related cell death when GPX4 activity is compromised (153).
Corneal alkali burns have severe clinical manifestations because of their ability to dissolve proteins. Such injuries frequently result in corneal scarring, neovascular growth, and, in advanced cases, loss of vision (153). Existing therapies are limited in terms of efficacy and possible adverse reactions (154, 155). Alkali burns elevate ROS levels, upregulating the expression of genes such as Nox2, Nox4, vascular endothelial growth factor (VEGF), and matrix metalloproteinase (MMP). This upregulation exacerbates corneal injury by triggering an inflammatory response and driving the growth of new blood vessels, resulting in significant corneal damage through neovascularization (155, 156). New blood vessel growth may cause lipid leakage and buildup in the cornea (157). Elevated ROS levels induce lipid peroxidation, resulting in ferroptosis (158). Research has indicated that the ferroptosis inhibitor Fer-1 shows promise for treating conditions linked to ferroptosis. In a murine model of alkali-induced corneal injury, Fer-1 administration significantly reduced corneal opacity and abnormal neovascularization. These findings strongly implicate ferroptosis as a key mechanism in alkali burn-related corneal damage. Further studies have shown that alkali burns activate ferroptosis and that ROS attack mitochondria and jointly promote the development of ferroptosis in corneal tissue. The efficacy of Fer-1 treatment suggests that ferroptosis may be a potential target for the treatment of corneal alkali burns. Furthermore, compared with free Fer-1, Fer-1 encapsulated in liposomes was more effective at healing corneal alkali burns, and safety assessments conducted both in vitro and in vivo revealed no significant adverse effects from Fer-1 liposomes (159, 160).
Corneal injury caused by inflammation, trauma, surgery, or infection also results in the excessive production of ROS and reactive nitrogen species (RNS) (161). These compounds stimulate iron ion release and lipid oxidation, inducing ferroptosis (162). Corneal conjunctival lesions and corneal ulcers can occur in severe cases of DED, and recent studies have shown that AKR1C1 safeguards corneal epithelial cells against damage caused by oxidative stress in DED (163). Excessive oxidative stress triggers ferroptosis, which contributes to DED progression in a mouse in vivo DED model and an immortalized HCEC line. In addition, Nrf2 can trigger AKR1C1 expression to attenuate iron toxicity-induced cell injury and inflammation in HCECs.
In summary, ferroptosis is key to keratocyte damage. Thoroughly examining how central players contribute to ferroptosis and keratocyte damage and designing potent ferroptosis inhibitors could open the way for novel treatments for corneal injury.
4.2 Ferroptosis in retinal pigment epithelial cells
The RPE serves as a cornerstone of retinal structure and function, playing an indispensable part in vision. On its external side, it borders Bruch’s membrane and the choroid, whereas internally, it makes direct contact with PR outer segments. The basal surface exhibits intricate infoldings that dramatically boost cellular surface area, paving the way for streamlined material transport. Acting as the linchpin of the outer blood–retinal barrier, RPECs establish tight junctions that deliver glucose, oxygen, and vital nutrients to PRs while simultaneously clearing away their metabolic waste, keeping PRs’ visual capabilities optimal. Therefore, when ferroptosis strikes RPECs, it can wreak havoc on PRs, causing damage or even their demise, which ultimately sets the stage for the development and advancement of vision-impairing disorders. PRs detect light through specialized organelles called outer segments, where photopigments embedded in membranous discs absorb photons and initiate phototransduction (164). Blinding eye diseases such as RP and DR arise when PRs degenerate. One contributing mechanism is the ferroptosis of RPECs (151). Essential to the function of the blood–retinal barrier, RPECs play a pivotal part in sustaining PR by delivering glucose, oxygen, and a plethora of nutrients, all while removing metabolic byproducts from PRs. This dynamic exchange is paramount for preserving the visual acuity of PRs. Consequently, the onset of ferroptosis within RPECs can trigger PR damage, potentially culminating in their death. This, in turn, is a major factor in the onset and escalation of blinding eye disorders (165).
PR outer-segment membranes are highly enriched in PUFAs, particularly DHA. These PUFAs are susceptible to lipid peroxidation when excessive light exposure or mitochondrial dysfunction generates ROS, thereby amplifying oxidative stress and predisposing photoreceptors to ferroptosis. In the same manner as ROS cause PR damage, iron overload can cause lipid peroxidation and oxidative stress, leading to ferroptosis and subsequent PR damage (166). PRs are exceptionally prone to oxidative damage because of their susceptibility to lipid peroxidation, which can severely compromise their functionality. Antioxidants such as vitamin E and the enzyme GPX4 play crucial roles in shielding these cells by mitigating lipid peroxidation and bolstering their resistance to oxidative stress (167). However, the efficiency of the retinal redox system decreases significantly with age, increasing vulnerability to oxidative damage over time. Current key routes linked to RPEC ferroptosis include amino acid uptake, lipid metabolism, iron regulation, and inflammatory signaling pathways.
System Xc−, GPX4, and GSH are canonical factors involved in ferroptosis. In RPECs, GPX4 inhibitors such as 1s and 3R-RSL3 (RSL3) increase lysosome-associated membrane protein 2 (LAMP2) levels, leading to ferroptosis. Supplementation with cysteine and glutamine can restore GSH function, thereby inhibiting ROS-induced death in LAMP2-knockout RPECs (168). Elevated oxygen consumption, prolonged exposure to light, and excessive intake of PUFAs and photosensitizers may cause retinal ROS buildup and induce oxidative stress. Additionally, the excessive metabolism of iron can accelerate this process, ultimately leading to ferroptosis in RPECs (169).
HFE/Fe3+–transferrin (TF)–transferrin receptor (TFR) uptake occurs in the basolateral membrane of the RPE, and the distribution and levels of HFE and TFR are essential for iron balance in RPECs. A lack of HFE can lead to iron overload (170). Gnana-Prakasam and colleagues reported that RPECs with suppressed HFE presented traits akin to those of tumor cells, including diminished cell aging, improved motility, and increased glucose consumption (171). RPEC hypertrophy occurs in mice with iron overload, resulting in impaired RPE function.
The protein divalent metal transporter 1 (DMT1) is an endosomal membrane transporter for protons and Fe2+ (172) and is present in RPECs. Recent research has indicated that the elevated expression of the iron-regulatory protein IRP1 increases DMT1 levels and simultaneously reduces ferroportin-1 (FPN1) levels. This downregulation of FPN1 can lead to iron accumulation (173). Eventually, FPN1 downregulation promotes the accumulation of Fe2+ and induces ferroptosis in RPECs (166). In addition, DMT1 polymorphisms may serve as environmental risk markers for AMD (174).
RPE injury and inflammatory damage are associated with RPEC ferroptosis, and numerous studies have shown that ROS can cause retinal cell damage, leading to retinal disease. ROS can stimulate the generation of LOX metabolites, which then interact with ROS to trigger lipid peroxidation—a key driver of ferroptosis. Research has shown that NaIO3-triggered damage to RPECs, along with associated inflammation, occurs through ferroptotic pathways. Notably, the 5-LOX inhibitor zileuton has been shown to mitigate retinal damage and RPEC degeneration caused by NaIO3 by suppressing ferroptotic mechanisms (175).
Oxidative stress and free radical-induced damage, particularly in RPECs, drive the progression of AMD. Recent studies have shown that elevated iron levels in the RPE and Bruch’s membrane correlate with advanced AMD stages (176). The retina and RPE are rich in GSH, a vital component for safeguarding RPECs (176). GSH depletion causes cellular death. Research has shown that GSH depletion induces RPEC death via ferroptosis and autophagic mechanisms within a typical in vitro setup for AMD. In addition, the results revealed that autophagy contributes to ferroptosis triggered by GSH depletion (177). Totsuka et al. demonstrated that ferroptosis contributes to the death of RPECs (178). Through both in vivo and in vitro studies, Wei et al. highlighted the significance of ferroptosis in AMD (179). Animal studies have demonstrated that the NaIO3-induced AMD model has ferroptosis-related characteristics. This effect can be mitigated by the application of ferroptosis antagonists. Nutritional supplementation with antioxidants may help mitigate the progression of AMD. This safeguarding effect is believed to correlate with the ability of these nutrients to reduce the sensitivity of RPECs to ferroptosis and protect retinal neurons.
In high-glucose environments, the retina, a tissue with significant oxygen demand, is especially susceptible to injury. Nrf2 plays a vital role in antioxidant defense, with studies showing that its activation safeguards retinal tissue in DR models (180). Many studies in diabetic models have demonstrated that increased Nrf2 levels shield these organs from damage by inhibiting ferroptosis. Recent studies have identified initial indicators of nerve cell death and astrocyte activation in DR. Neuronal apoptosis and degeneration are associated and marked by irregular tau protein phosphorylation, a feature that is also present in Alzheimer’s disease. Research has indicated that increased tau expression and phosphorylation may induce ferroptosis in neural tissue. In addition, research has indicated that reducing GPX4 leads to decreases in the numbers of hippocampal neurons and astrocytes in adult mouse models of Alzheimer’s disease. This cell death mechanism aligns with the characteristics of ferroptosis (181, 182).
4.3 Ferroptosis in retinal ganglion cells
RGCs constitute neuronal populations situated in the inner retina that are responsible for converting light signals into neural pulses and transmitting them to the visual center of the brain (183). RGCs are neuroretinal elements that link visual sensors to the cerebral cortex to create a visual network. Several visual system disorders cause functional and structural alterations in RGCs (i.e., DR, glaucoma, demyelinating optic neuritis, and ischemic optic neuritis) (184). Ferroptosis contributes to the progression of these illnesses by compromising retinal ganglion cell functionality.
In addition, ferroptosis can induce RIRI and glaucoma by affecting RGCs. The mechanism underlying RIRI involves an initial phase of acute hypoxia (ischemia) caused by blood flow interruption, followed by a second phase of reperfusion injury when blood flow is restored, which exacerbates tissue damage. This damage leads to retinal structural alterations, RGC degeneration, and eventual functional impairment. Reperfusion leads to an overload of ROS and inflammatory cytokines, as well as RGC apoptosis. Glaucoma involves progressive optic nerve damage, leading to distinct structural alterations in the optic disc and retinal nerve fibers, accompanied by progressive RGC death, which will be specifically discussed later.
4.4 Ferroptosis in macrophages
Macrophages are white blood cells located within tissues and are derived from precursor cells in the bone marrow. Their main function is to phagocytose and digest cellular debris and pathogens in vertebrates, as well as to activate lymphocytes or other immune cells to combat pathogens. Macrophages are widely distributed in various tissues and have multiple functions, such as secreting cytokines and producing ROS. They mediate inflammatory responses; modulate iron, lipid, and amino acid metabolic processes; and significantly contribute to tissue equilibrium (185). Macrophages stimulated by different microenvironments differentiate into different subtypes, the most common of which are M1 and M2. M1 macrophages amplify inflammation, eliminate pathogens, and inflict tissue damage by secreting a repertoire of proinflammatory mediators, whereas M2 macrophages are involved mainly in tissue repair (fibrosis and neovascularization) (186). Increasing evidence suggests a strong connection between macrophages and ferroptosis.
Macrophages can clear cells that undergo ferroptosis. HMGB1 released from cells undergoing ferroptosis binds to the advanced glycation end-product receptor (AGER) on macrophages and mediates the inflammatory response in macrophages. Several molecules, such as monocyte chemoattractant protein-1 (CCL2) and macrophage inflammatory protein-1α (CCL7), can initiate macrophage recruitment and chemotaxis to increase the magnitude of immune responses. Moreover, macrophage Toll-like receptor 2 (TLR2) identifies and interacts with SAPE-OOH at the membrane of cells undergoing ferroptosis, enhancing phagocytosis and helping to clear ferroptotic cells (187). Macrophages play a key role in controlling iron balance from two main sources. First, macrophages produce iron by engulfing senescent erythrocytes and are a major source of available iron in the body. Second, extracellular iron (Fe3+) binds to TF and can enter macrophages via TFR1. Normally, the body can maintain the stability of iron content (188).
When this stabilization is broken, abnormal iron metabolism may oversupply the active form of iron, inducing ferroptosis. Moreover, cytokines secreted by macrophages modulate the activity of intracellular LOX, thereby inducing ferroptosis. Studies have shown that ferroptosis induces iron overload in macrophages, drives M1 polarization, increases inflammatory cytokine release, and impairs tissue repair and immune modulation (189). M1 and M2 macrophages differ in terms of both iron metabolism and ferroptosis susceptibility. M1 macrophages exhibit reduced susceptibility to RSL3-triggered ferroptosis because of the elevated levels of nitric oxide radicals, which inhibit lipid peroxidation.
Some investigators have developed a non-pharmacological biohybrid approach to target ferritin for synergistic ferroptotic immunotherapy by utilizing M1 macrophage microvesicles combined with HKN15-modified Prussian blue nanoparticles (190). In contrast, M2 macrophages present decreased iNOS expression and heightened susceptibility to RSL3-induced ferroptosis. Inflammatory macrophages upregulate SLC7A11 and increase cystine uptake and GSH synthesis via the NF-κB pathway, thereby maintaining intracellular redox homeostasis and resisting ferroptosis (191). According to previous studies (192), when M2 macrophages infiltrate the tumor microenvironment, they can suppress ferroptosis in neighboring cancer cells through paracrine signaling, despite being intrinsically sensitive to ferroptosis.
As people age, the number of macrophages in normal eyes gradually increases. Macrophages contribute to the production of VEGF, which can induce angiogenesis and provide routes of nutritional supply and metastasis for tumors. Because ocular melanoma is located in the eye, which lacks a lymphatic system, tumor cells must escape into the bloodstream to metastasize to other organs, such as the liver. In this process, macrophages facilitate tumor cell escape and metastasis by stimulating angiogenesis. Thus, macrophages play a role as disruptors in ocular melanoma. Experimental evidence has shown that excessive iron accumulation prompts macrophages to shift toward the M1 phenotype, triggering a cascade of inflammatory mediators. This inflammatory response affects both ocular surface structures and lacrimal gland tissue, ultimately fueling the advancement of dry eye disease pathology (193). Liposomal clodronate attenuates iron overload-induced DED by reducing the number of macrophages, particularly M1 macrophages, regulating macrophage polarization, and suppressing inflammatory responses.
4.5 Ferroptosis in natural killer cells
After T cells and B cells, natural killer (NK) cells are the third most prevalent type of lymphocyte. They are associated with antitumor activity, antiviral defense, and immune modulation, and also contribute to hypersensitivity and autoimmune disorders. NK cells originate from hematopoietic stem cells in the bone marrow and are among the core cells of the innate immune system, accounting for approximately 5%–15% of all immune cells in the blood. The majority of NK cells exhibit antitumor cytotoxicity. They produce lytic granules comprising a large number of molecules (perforin, granzyme, and human granulysin) that induce cell death in stressed cells. Macrophages can also be called by NK cells to address pathogens and damaged cells in the body, as well as mediate antibody-dependent cellular cytotoxicity (ADCC) (194). This is also an important mechanism through which common antibody drugs exert clinical effects. Owing to the broad anticancer activity of NK cells, the range of NK-cell types used for cancer treatment has become increasingly diverse in recent years (195, 196). One study showed that immune cells, mainly NK cells, eliminate senescent cells and increase animal lifespan by 20%–30% (197).
Recent research has indicated that NK cells associated with tumors exhibit characteristics of ferroptosis, including lipid peroxidation and oxidative stress. These features are associated with the inhibition of NK-cell metabolic activity in the tumor microenvironment, resulting in NK-cell impairment. In contrast, Nrf2 activation modulates the expression of multiple antioxidant molecules, thereby rescuing this dysfunction (198). In tumor-bearing mice, liproxstatin-1 enhances NK-cell viability and antitumor activity by pharmacologically inhibiting ferroptosis. These findings indicate that blocking ferroptosis improves NK-cell survival and antitumor function, suggesting that ferroptosis limits the longevity of NK cells in the tumor microenvironment (199). In ophthalmology, whether the induction of ferroptosis could be exploited as a novel therapeutic strategy against the orbital involvement of highly aggressive nasal-type extranodal NK/T-cell lymphoma remains to be investigated.
neovascular age-related macular degeneration (nvAMD) features choroidal neovascularization (CNV) development, which affects 10% of AMD patients and is the predominant factor leading to vision impairment in AMD patients (200). These irregular blood vessels penetrate the subretinal area, leading to leakage, swelling, and bleeding, which in turn cause a rapid decrease in central vision. NK cells accumulate in the diseased choroid and inhibit pathological changes in nvAMD by promoting NETosis and NET production by neutrophils (201, 202). Rituximab clears B cells through an NK-mediated lytic pathway and is indicated for B cell-driven immunoreactive thyroid eye disease (203, 204).
4.6 Ferroptosis in T cells
T lymphocytes arise from lymphoid progenitors in the bone marrow and subsequently differentiate, develop, and mature in the thymus. After receiving orderly and standardized “training”, T cells enter the blood and migrate to lymphoid tissues in the periphery. After receiving antigen stimulation, mature naive T cells develop into effector or memory cells and contribute to adaptive immune responses (205). T cells consist of two main functionally separate types: CD4+ helper T cells (Th cells) and CD8+ cytotoxic T lymphocytes (CTLs). CTLs, which are characterized by the surface expression of CD8+, eliminate infected cells and are commonly referred to as “killer T cells”. Th cells express CD4 as a surface marker and orchestrate adaptive immune responses by secreting cytokines and providing costimulatory signals (206).
In response to immunotherapy, CD82 T cells release IFN-γ, which targets system Xc−, curtails cystine uptake, and thereby sensitizes tumor cells to ferroptosis (207). Some researchers have reported that PCIF1 diminishes ferroptosis in CD8+ T cells predominantly through the upregulation of genes that regulate ferroptosis, including FTH1 and SLC3A2 (208). GPX4 is essential for protecting T cells from lipid peroxidation and ferroptosis. Notably, while T-cell growth in vitro requires system Xc−, system Xc− does not appear to be necessary for T-cell proliferation or primary and memory immune responses to tumors in vivo (209). Selenium supplementation increases GPX4 expression in follicular helper T cells (TFH cells) and decreases ferroptosis susceptibility, thus enhancing antibody responses in mice vaccinated against influenza. In accordance with these findings, mice with GPX4 deficiency specifically in T cells were unable to fight acute lymphoblastic cerebrospinal meningitis virus or Leishmania infection. This immunodeficiency could be avoided through the administration of high-dose supplements with the fat-soluble antioxidant vitamin E, which prevented ferroptosis in GPX4−/− T cells.
The eyes produce suppressed immune responses to avoid inflammation that can hinder vision. It is commonly believed that there are no T cells in the cornea; however, long-lived memory T cells reside in the cornea and can “patrol” and fight viral infections. In an experiment on corneal herpes simplex virus (HSV) infection, a team used multiphoton microscopy to obtain real-time images of living, intact biological tissue to study keratocytes in mice infected with HSV. Images revealed that long-term surviving memory T cells were generated in the eyes of the mice to fight infection. After the virus was cleared, memory T cells remained in the cornea to prevent future reinfection (210). Recent studies have provided evidence that DED is an autoimmune disorder driven by T cells (211). The retinal expression of the glycolysis-related gene LDHA markedly increased in mice with experimental autoimmune uveitis and promoted the migration of effector T cells (Teff cells). The results of these experiments revealed that LDHA inhibition could inhibit the migration of CXCR4-positive pathogenic T cells into retinal tissue to prevent the development of uveitis. Importantly, in Vogt–Koyanagi–Harada disease (VKH), the upregulation of LDHA is increased in CD4-positive T cells, and the inhibition of LDHA reduces the proliferation of CD4-positive T cells to prevent diseases (212).
5 Links between ferroptosis and ophthalmology
Ferroptosis, a lipid peroxidation-mediated, iron-dependent form of programmed cell death, has swiftly become a leading focus in ophthalmic disease research in just a few years, as it plays important roles in cataracts, glaucoma, DED, corneal injury, and thyroid-associated ophthalmopathy (TAO), among others. Ferroptosis provides not only a unified explanation for the common terminal damage associated with a variety of eye diseases but also a new window for personalized, mechanism-oriented treatment. By delving into the regulatory processes of ferroptosis—such as iron ion metabolism, ROS generation, and the equilibrium of antioxidant defenses—we can gain clearer insights into its involvement in eye diseases.
5.1 Ferroptosis in AMD
The macula, which is located at the core of the retina, is essential for focused sight (213). AMD is a multifaceted age-related eye condition marked by a decline in the architecture and functionality of the macula (214). This disease is the leading cause of blindness in individuals aged 50+ (215). The worldwide incidence of AMD is anticipated to increase from 196 million cases in 2020 to 288 million cases by 2040 (216). In addition, as the global population ages, the societal and economic impact of age-related macular degeneration is poised to increase.
Early macular degeneration is characterized by macular cysts and pigment changes. Advanced AMD is categorized into dry and neovascular (nvAMD) forms (217). Dry AMD features permanent damage to the RPE and PRs, leading to the atrophy of retinal tissue comprising supportive cells under PRs. The degeneration or dysregulation of retinal tissue is a pathogenic marker of AMD. nvAMD, also known as exudative AMD, is characterized by abnormal neovascularization, including CNV and retinal hemangiomatous hyperplasia (RAP) (218, 219), which can lead to macular leakage.
The progression of AMD is associated with oxidative stress, as well as genetic, environmental, and aging-related influences. High levels of PUFAs are located in the outer photoreceptor segment and produce substantial amounts of intracellular ROS, making the retina especially susceptible to damage from oxidative stress (166) (Figure 3). Oxidative stress can lead to RPEC loss, causing photoreceptor degeneration in AMD (220); this degeneration is also a causative factor of dry AMD. Recent research has highlighted the pivotal involvement of ferroptosis in the pathophysiology of AMD, notably through the Xc− and GPX4 pathways, and mitochondrial metabolism (221, 222).
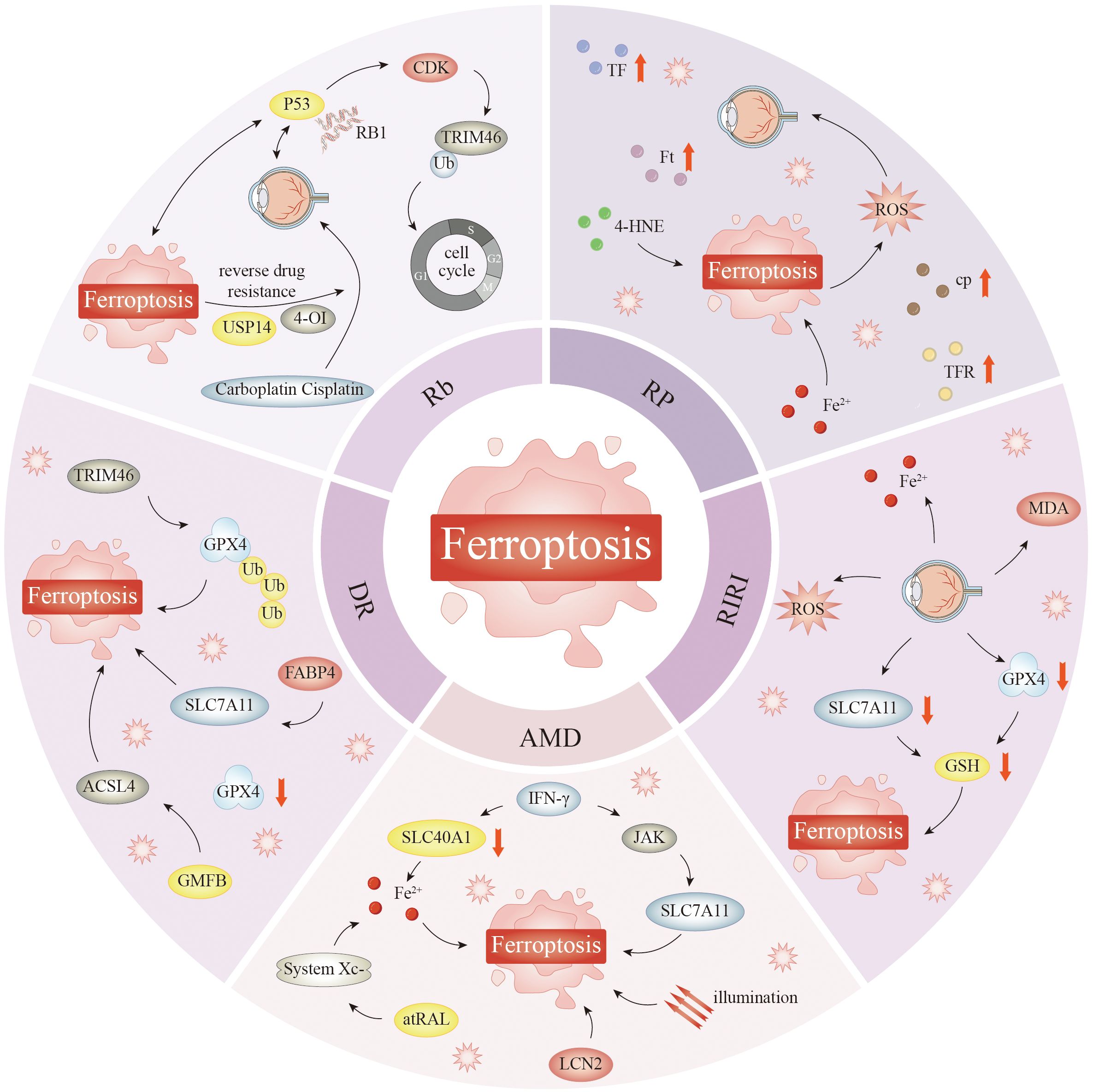
Figure 3. Ferroptosis mediates diseases such as AMD, DR, Rb, RP, and RIRI. Iron overload-induced oxidative stress triggers ferroptosis in RPECs, causing photoreceptor degeneration, a central pathway in dry AMD. Elevated LCN2 exacerbates this iron-dependent cell death by impairing iron export. In patients with AMD, excess iron accumulates in the macula, RPE, Bruch’s membrane, and drusen, while aging retinas show increased lipofuscin, 4-HNE, and MDA, lipid peroxidation markers that mirror the biochemical signature of ferroptosis. IFN-γ can also trigger ferroptosis of RPECs and promote AMD progression. Ferroptosis plays a key role in DR progression, and it promotes disease development by impairing the integrity of capillary endothelial cells. In DR, ferroptosis fuels disease progression by impairing capillary endothelial function. Under high-glucose conditions, TRIM46 is upregulated, triggering ferroptosis that destabilizes the iBRB and damages RPECs, thereby accelerating DR. Ischemia–reperfusion injury significantly downregulates SLC7A11 and GPX4 expression, leading to GSH depletion, which is the core antioxidant axis regulating ferroptosis, and dysfunction is an important mechanism of cell death in ischemia–reperfusion injury. During ischemia–reperfusion injury, ferroptosis occurs in almost all types of retinal cells. RIRI causes retinal accumulation of Fe2+, ROS, and MDA. Various studies have demonstrated that ferroptosis is a key factor leading to RP. Experimental models have established ferroptosis as a critical driver of RP. Overexpression or intraperitoneal delivery of human transferrin effectively prevents photoreceptor cell death, and iron chelators likewise rescue retinal degeneration. HO-1 is a key regulator of this process; its inhibition reduces free-iron accumulation, thereby protecting both retinal pigment epithelial cells and photoreceptors from ferroptosis-mediated degeneration. Mutations in the p53 gene and deletion of the RB1 gene have been identified to play an important role in Rb pathogenesis. Both genes have also been shown to be involved in the process of ferroptosis. 4-OI-induced ferritin degradation or USP14-targeted enhancement of ferroptosis efficiently eliminates multidrug-resistant Rb cells, offering a novel strategy to overcome chemoresistance in Rb. Abbreviations: CDK, cyclin-dependent kinase; P53, tumor protein p53; TRIM46, tripartite motif-containing protein 46; Ub, ubiquitin; USP14, ubiquitin-specific protease 14; FABP4, fatty acid-binding protein 4; AMD, age-related macular degeneration; SLC40A1, solute carrier family 40 member 1; GMFB, glia maturation factor beta; atRAL, all-trans retinol; DR, diabetic retinopathy; Rb, retinoblastoma; RP, retinitis pigmentosa; RIRI, retinal ischemia–reperfusion injury; RPECs, retinal pigment epithelial cells; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; iBRB, inner blood–retinal barrier; GPX4, glutathione peroxidase 4; ROS, reactive oxygen species.
The intraperitoneal administration of an SLC7A11 inhibitor in a laser-generated CNV model increased lipid peroxidation and triggered ferroptosis, leading to the expansion of the CNV area in ARPE-19 cells. In contrast, Fer-1 and Lip-1 rescued RPE ferroptosis and VEGF production (223). One study revealed that mice with system Xc− knockdown showed rapidly advancing retinal aging, increased retinal strain, elevated ROS levels, and compromised mitochondrial activity, providing a beneficial framework for investigating retinal aging and AMD (224). Iron, a key producer of free radicals, is closely associated with the pathogenesis of AMD (225, 226). Compared with that in young adults, the retinal iron content in older adults is significantly greater, as iron tends to accumulate in the retina as people age (225, 227). In the retinas of patients with AMD, excessive iron accumulates within the macula, RPE, Bruch’s membrane, drusen, and other structures. Aging retinas show elevated levels of lipid peroxidation-derived products (e.g., lipofuscin, 4-HNE, and MDA) that are consistent with but not exclusive to ferroptosis, which is associated with inflammation and features of AMD (228, 229). The accumulation of iron is a major cause of excess free radical generation in the RPE and is strongly linked to the development of AMD (22). Iron levels in the RPE are markedly elevated in patients with dry AMD (225). In addition, iron-binding agents such as deferiprone (DFP), DFO, and deferasirox (DFX) prevent the Fenton reaction by binding to surplus iron ions, and research has indicated that these agents serve as safeguards against retinal degeneration in various experimental setups (230, 231). GPX4 maintains redox homeostasis and protects RPECs, photoreceptors, and RGCs from glutamate-induced cytotoxicity (232). In multiple oxidative stress models of retinal degeneration, elevated GPX4 expression has been shown to protect photoreceptors and RPECs, suggesting that increasing GPX4 expression may represent a potential gene therapy approach for patients with AMD (47, 48).
Researchers have reported that mitochondrial destruction, fragmentation, and destruction occur in the RPE of patients with AMD (233). Numerous reports have shown that the degree of mtDNA damage and the number of breaks in RPECs are positively correlated with the severity of AMD (234, 235). mtDNA damage can induce ferroptosis and exacerbate both RPE degeneration and the progression of AMD (236). NAIO3, H2O2, and UV light treatment can induce a decrease in the mitochondrial membrane potential and an increase in ROS levels, leading to ferroptosis (237, 238). Ferroptosis in RPECs can be inhibited by the use of mitochondrial ROS quenchers, which can prevent the accumulation of mitochondrial ROS and lipid oxidation. Mitochondrial ROS play a crucial role in ferroptosis.
Chronic inflammation associated with aging promotes the onset and progression of AMD (239). Research has indicated a positive correlation between inflammatory responses and iron buildup. In addition, multiple inflammatory signaling cascades, including the JAK–STAT pathway, participate in ferroptosis–inflammation interactions (61). Proinflammatory factors such as IFN-γ suppress GSH production through the JAK1/2–STAT1–SLC7A11 axis, inducing ferroptosis and RPEC degeneration in vivo (179). Interferon-γ (IFN-γ) can also trigger ferroptosis in RPECs and promote the progression of AMD (179). Iron overload can lead to retinal inflammation through the downregulation of cholesterol efflux transporters, leading to increased cholesterol content in the retina (240). Notably, the activation of iron-regulated inflammasomes ultimately leads to oxidative stress and ferroptosis in RPECs (241).
Elevated iron levels increase the expression and activation of complement C3, a key inflammatory regulator (242). In vivo studies have indicated that intravenous iron administration resulted in the presence of complement C3 on the basal surface of the RPE, thereby promoting pathological changes (243). Liu and colleagues analyzed a cell model of AMD induced by sodium iodate (SI) and reported that SI can consume intracellular GSH, liberate Fe2+ from liposomes, increase cellular ROS levels, and increase lipid accumulation, leading to ferroptosis in ARPE-19 cells (244). Lee et al. reported that SI also stimulates mitochondrial ROS production and promotes ferroptosis in RPECs (175). Tang et al. found that the process of ferroptosis in RPECs triggered by SI is linked to the Nrf2/SLC7A11/HO-1 pathway. Specifically, they reported that higher levels of HO-1 expression correlate with an increase in TFR expression and a decrease in SLC40A1 expression, ultimately leading to iron accumulation (245). Liposomal accumulation of Fe2+ may increase HO-1 levels and sustain ferroptosis. Blocking HO-1 or using ZnPP can alleviate the harmful effects on the morphology and function of RPECs and PRs (246).
Ferroptosis in PRs also significantly influences the development of AMD, particularly dry AMD (247). The recycling of 11-cis-retinal (atRAL) between photoreceptors and the RPE is essential for maintaining vision (248). ATP-binding cassette transporter A4 (ABCA4) facilitates the movement of atRAL from the photoreceptor outer segment disk to the cytoplasm, whereas RDH8 enzymes are responsible for converting atRAL back into all-trans retinol (249). In rodents with genetic deletions involving the Abca4 and Rdh8 genes, photoreceptor outer-segment disc clearance is defective. After light exposure, the PR layer becomes thinner, and A2E accumulates within the retina. Additionally, GSH levels decrease, and the expression of lipid metabolism-related proteins becomes abnormal. These alterations coincide with elevated lipid peroxidation levels (221). Subsequent studies have confirmed that atRAL can trigger ferroptosis in 661W neuronal cell cultures by hindering system Xc−, increasing Fe2+ levels, and amplifying mitochondrial ROS production. Tang et al. (33) confirmed these findings, as light accelerated PR degeneration via ferroptosis in 661W cells and male Sprague–Dawley rats. Light exposure significantly diminished photoreceptor cell viability and triggered early ferroptosis markers, including iron buildup, mitochondrial shrinkage, reduced GSH levels, elevated MDA levels, and decreased SLC7A11 and GPX4 protein expression.
The transcription factor-driven reprogramming of induced RPE (iRPE) cells has been identified as a robust method to create cells that are resistant to ferroptosis. This finding not only validates the crucial role of phosphoethanolamine/phosphocholine phosphatase 1 (PHOSPHO1) as a master regulator of ferroptosis resistance but also reveals a twofold approach for combating this process. Primarily, PHOSPHO1 curtails the progression of ferroptosis by dampening PE levels within the endoplasmic reticulum (ER), effectively inhibiting lipid peroxidation. Additionally, PHOSPHO1 reduces autophagy and ferritin uptake, consequently decreasing the intracellular accumulation of free iron. In vivo rat model experiments verified that PHOSPHO1 effectively protected RPECs from ferroptosis. These findings provide a new theoretical basis and potential target for AMD treatment (250).
ZIP8 functions as a metal ion transporter crucial to the ferroptosis-driven degeneration of RPECs. ZIP8 levels are elevated in patients with AMD, as demonstrated by transcriptomic analysis. The upregulation of ZIP8 expression has also been detected in RPECs and AMD mouse models under conditions of oxidative stress. Notably, this study revealed that ZIP8 knockdown significantly inhibited the oxidative stress response to NaIO3 and the development of ferroptosis in RPECs. Specific antibody intervention effectively alleviated the ZIP8-induced obstruction of RPEC degeneration, restored retinal function, and improved visual loss in mice with NaIO3-induced AMD. The critical role of ZIP8 in RPEC ferroptosis provides a potential target for the treatment of diseases associated with retinal degeneration, such as AMD (246).
The iron content in the retinal pigment epithelium and Bruch’s membrane is significantly greater in patients with AMD than in people of the same age without AMD. However, the iron concentration in the anterior chamber fluid of patients with dry AMD was more than twice as high as the A2E concentration, and exposure to blue light revealed a role of ferroptosis in the degeneration of RPECs. Via transcriptomic sequencing, Liu et al. found that ARPE-19 RPECs showed ferroptosis-related changes after treatment with SI, and these effects could be blocked by ferrostatin-1 and DFO. Tang et al. further conducted high-throughput sequencing on ARPE-19 cells treated with SI and reported that these cells exhibited ferroptosis. They also found that zinc protoporphyrin IX, a selective heme oxygenase-1 inhibitor, could prevent ferroptosis in RPECs, thereby offering a novel therapeutic target for managing dry AMD. Elevated LCN2 in the RPE exacerbates dry AMD by suppressing autophagy, disrupting iron balance, and triggering inflammasome activity, oxidative damage, and ferroptosis in RPECs (241). Additionally, the activation of genes implicated in ferroptosis is significantly upregulated in retinal tissue in spontaneous dry age-related macular degeneration (SD-AMD), suggesting that the disruption of iron homeostasis and ferroptosis could significantly influence disease onset and development (251).
5.2 Ferroptosis in DR
DR is a unique microvascular complication of diabetes and is the leading cause of visual impairment among the working-age adult population. Approximately half of all diabetic patients will eventually develop DR (252). The incidence and speed of DR progression correlate with age in diabetic patients. One study revealed that the incidence of DR was one in five individuals in a younger-onset group and one in two individuals in an older-onset group (253). The currently employed treatment predominantly involves panretinal photocoagulation, anti-VEGF treatments, intraocular steroid administration, and vitrectomy procedures.
In the current study, increased iron content, along with higher values of Cp and TF, was observed in the retinas of DR patients (254), and increased vitreous iron levels correlated with the onset of proliferative diabetic retinopathy (PDR) (255). The reasons may be threefold: first, hyperglycemia leads to heme degradation in hemoglobin and myoglobin, resulting in the detachment of iron ions from heme. Another possibility is that retinal or vitreous hemorrhage increases iron accumulation in patients with proliferative DR. Another possibility is that elevated angiotensin II (Ang-II) levels in DR patients could upregulate iron metabolism-related genes, thereby increasing iron uptake. These studies have indicated that iron ions can contribute to the progression of DR. Iron accumulation can induce oxidative damage and neovascularization.
The aging retina becomes particularly susceptible to oxidative stress, largely because age-related changes cause a surge in the generation of ROS and RNS. In DR, capillary occlusion leads to severe hypoxia, activating hypoxia-inducible factor (HIF) and promoting neovascularization (256). Iron is involved in the HIF-mediated activation of angiogenesis-related genes. In addition, HIF plays a critical role in regulating iron metabolism by regulating the expression of iron-related proteins such as DMT1, Fpn, and TFR.
Oxidative stress is closely linked to the development and progression of DR. Recent research has shown that oxidative stress and photoreceptor impairment may occur before the initial vascular pathology in diabetes (257). PRs are rich in DHA, which is composed of long-chain PUFAs. Excess ROS can attack these lipid targets, and oxidative stress may harm the endothelial cells of retinal blood vessels, leading to vascular leakage and vitreous hemorrhage. As a tissue with high oxygen demand, the retina is especially susceptible to damage under high glucose conditions.
The researchers induced diabetes observed in iron deficiency (HFE) knockout mice, a genetic representation of iron overload. Moreover, findings from these studies have indicated that diabetes increased the degree of vascular injury and BRB leakage in HFE knockout mice. Iron overload triggers the G protein-coupled receptor 91 (GPR91) signaling cascade, increasing renin secretion. GPR91 signaling can mediate vascular growth in PDR, and renin can increase Ang-II expression (258). Ang-II activates VEGF receptors to increase vascular permeability and neovascularization (259). Excess iron-induced oxidative stress can further exacerbate diabetic macrovascular issues, such as atherosclerosis and heart disease (260). In DR, these alterations can impair retinal microvascular function.
Recent research has highlighted ferroptosis as pivotal in DR progression by damaging capillary endothelial cell function. TRIM46 gene expression is increased in diabetic patients (261). Under high-glucose conditions, TRIM46 expression increases in human retinal capillary endothelial cells (HRCECs) and promotes the ubiquitination of GPX4, which leads to ferroptosis (262). One study revealed higher levels of biomarkers associated with iron intoxication in a non-proliferative diabetic retinopathy (NPDR) group than in a PDR group. In NPDR patients, the fundus is primarily defined by capillary leakage and bleeding, and the BRB is disrupted (263). The BRB consists of inner (iBRB) and outer (oBRB) components.
Elevated glucose levels trigger ferroptosis in endothelial cells, compromising iBRB stability (264). Elevated glucose levels impair RPEC viability and function, accelerating DR progression. Investigators have shown that the expression of glial maturation factor-beta (GMFB), a neurodegenerative mediator, is elevated in DR. These researchers injected chemically synthesized GMFB into the vitreous bodies of Sprague–Dawley (SD) rats, impairing RPEC function (37). Laboratory tests have revealed that extracellular GMFB can decrease the number of RPECs and their cell–cell connections and increase ACSL4 expression (35). It promotes the generation of cytotoxic lipids in ARPE-19 cells and triggers ferroptosis. RPE ferroptosis disrupts the oBRB, resulting in macular edema, the primary factor contributing to vision loss in DR.
In addition to GMFB expression, fatty acid-binding protein 4 (FABP4) expression is positively correlated with NPDR/PDR grade and can be used as an independent prognostic biomarker (265). FABP4 drives ferroptosis by inhibiting PPARγ, and recent studies have identified early signs of neuronal apoptosis and reactive gliosis in DR (266) (267) (182).
5.3 Ferroptosis in retinal ischemia–reperfusion injury
RIRI can lead to permanent vision loss. The mechanism involves an initial phase of acute hypoxia (ischemia) due to disrupted blood circulation, followed by a second phase of reperfusion injury when blood flow is restored, which exacerbates tissue damage (268). RIRI contributes to glaucoma, retinal artery occlusion, and DR pathogenesis (269, 270). This damage leads to retinal structural changes, RGC death, and functional loss. In particular, reperfusion leads to the overproduction of ROS and inflammatory cytokines and the apoptosis of RGCs (271), causing great damage to retinal function (272, 273).
The results of previous studies confirm the involvement of ferroptosis in retinal ischemic injury in the RIRI model mice. In one study, RIRI induced the accumulation of Fe2+, ROS, MDA, and GSH; increased transferrin expression; and reduced the levels of ferritin, SLC7A11, and GPX4 in rat retinal tissue (274). Moreover, ischemia–reperfusion injury significantly inhibits SLC7A11 and GPX4 expression, causing GSH depletion. The GPX4 pathway is a central antioxidant axis that regulates ferroptosis, and its dysfunction is an important mechanism of cell death in ischemia–reperfusion injury (275). The application of the iron chelators deferoxamine and baicalin can ameliorate RIRI (276). Another study revealed that ferroptosis occurred across numerous retinal cell types during ischemia–reperfusion damage (277).
Fer-1 markedly decreased RGC death in both living organisms and laboratory cultures, thereby alleviating ischemia–reperfusion-induced retinal structural and functional damage to the retina. In summary, ferroptosis contributes to retinal ischemia–reperfusion injury, and blocking ferroptosis reduces RGC loss and inflammation. Researchers have reported that the number and diversity of retinal cells are decreased following ischemia–reperfusion-induced damage in mice. In contrast, the numbers of all types of immune cells were notably increased. A significant number of blood-dwelling immune cells invade the retinal tissue, causing blood–eye barrier damage (278). These results indicate that bone marrow cells could be key targets in RIRI.
Some researchers have developed supramolecular nanoparticles that combine the phosphodiesterase 4 (PDE4) inhibitor crisaborole and the ferroptosis inhibitor DFO. These nanoparticles can alleviate retinal IR injury by targeting the ferroptosis pathway and oxidative stress. These compounds also suppress the release of inflammatory mediators and signaling proteins while dampening glial cell activation—a hallmark pathological feature of RIRI (279).
In a mouse model of RIRI, researchers reported that Lycium barbarum polysaccharide-based nanoparticles (PLBP), a nanoparticle derived from goji polysaccharides, directly protects RGCs from ferroptosis. This protection is achieved by activating Nrf2 signaling, reducing intracellular ROS levels, and increasing the antioxidant capacity of RGCs, thereby helping to preserve visual function after ischemia–reperfusion injury. Moreover, PLBP mitigated neuroinflammation and offered indirect protection to RGCs by inhibiting microglial phagocytosis and migration and the release of proinflammatory cytokines and by activating the NF-κB pathway. These combined effects lead to notable vision restoration in mice following retinal injury. This recovery is evidenced by enhanced optomotor responses, improved dark/light preference, and stronger pupillary light reflexes (280).
5.4 Ferroptosis in retinitis pigmentosa
RP refers to a group of inherited retinal diseases whose main feature is the slow degradation of PR cells, leading to a gradual decline in the patient’s vision. In RP patients, typical characteristics of the fundus are mainly the waxy yellow color of the optic nerve, retinal vascular stenosis, and osteoid pigment dispersion. The main clinical manifestations are night blindness, reduced peripheral and central vision, disturbed color perception, and a sensation of glare. Currently, no treatment exists for this condition, and treatment is focused mainly on delaying disease progression and improving quality of life (265). RP results from the degeneration of rod and cone photoreceptors, with secondary loss of RPE cells.
In two rodent models of RP, iron overload was observed in the retina. Additionally, in rd10 mice, the levels of iron metabolism-related proteins, including TF, TFR, ferritin (Ft), and ceruloplasmin (Cp), were also elevated. Overexpression or intraperitoneal injection of human transferrin effectively inhibited PR cell death. Moreover, iron chelators have been shown to effectively reverse retinal degeneration (281, 282). Moreover, iron-chelating agents have been shown to be successful at reversing retinal atrophy (283, 284). These findings demonstrate that iron causes photoreceptor degeneration through oxidative stress, with cones being more susceptible than rods (285).
In addition, the levels of 4-HNE, which are indicative of lipid oxidation, were notably increased in the retinal tissues of RP model mice (281). Injecting Fe2+ into the eyes of mice killed photoreceptor cells and resulted in the production of superoxide free radicals, causing lipid peroxidation. It also increased 4-hydroxynonenal levels and decreased GPX4 levels, which led to ferroptosis. SOD1 is an important component of the antioxidant defense system of the retina. In genetically modified mice, if more SOD1 is present, the retina can be protected from oxidative damage; however, if not enough SOD1 is present, the oxidative damage to the retina is more severe, and the reduction in SOD1 and GPX4 levels accelerates cone cell function loss, as shown in the RP mouse model.
Liu established an SI-induced RP cell model in RPECs. After the ARPE-19 cells were treated with SI, intracellular free iron levels increased 40-fold, and lipid peroxidation increased significantly; however, GPX4 expression did not change, and the expression of GSH/cysteine was downregulated. The incorporation of ferroptosis inhibitors such as DFO mesylate and Fer-1 inhibited SI-induced cell death. Further mechanistic studies have shown that SI can deplete GSH levels, increase ROS levels, promote the release of iron chelated by iron storage proteins, and subsequently promote lipid damage, ultimately leading to ferroptosis in RPECs. The iron chelator DFO retards photoreceptor degeneration in animal models of RP, supporting a role for ferroptosis in RP progression (244). Similarly, Tang and colleagues demonstrated that ferroptosis is a pivotal cause of RP and suggested that HO-1 is a critical modulator of this process. Inhibiting HO-1 expression could prevent the degeneration of RPE and photoreceptor cells by decreasing Fe2+ accumulation.
5.5 Ferroptosis in retinoblastoma
Rb is the primary intraocular cancer in infants and children and can metastasize and be life-threatening if left untreated. Rb accounts for approximately 2% to 3% of pediatric tumors (286). Annually, there are approximately 8,000 cases worldwide, with approximately one-third of affected children having Rb that affects both eyes. A white pupil is the most common first symptom of Rb, and other complications include strabismus or more severe symptoms such as red eye pain. The progression of Rb is strongly linked to the mutation of the RB1 gene, the first identified human tumor suppressor gene (287, 288). The disease is characterized into stages A–E. The main clinical treatment is eye-protecting treatment, and the success rate of eye-protecting treatment in stages D and E is significantly reduced. At present, exploring the treatment of this disease is important.
Both p53 gene mutation and RB1 gene deletion are key factors in Rb development and are implicated in triggering ferroptosis. The RB1 gene is considered a key factor in liver cancer, and studies have shown that sorafenib, as the only first-line treatment for advanced liver cancer, can induce ferroptosis in liver cancer. The significant downregulation of the Rb protein increases the vulnerability of hepatocellular carcinoma (HCC) cells to ferroptosis and the efficacy of sorafenib, indicating that the loss of the Rb gene accelerates the progression of ferroptosis (289). These findings suggest that elevated p53 levels cause ferroptosis, whereas p21 can act as an inhibitor of ferroptosis. Furthermore, E2F overexpression inhibited ferroptosis in a p21-dependent manner, which aligns with prior findings that E2F activates p21 transcription.
Carboplatin serves as the primary first-line chemotherapy for treating human Rb; however, acquired multidrug resistance (MDR) has emerged following chronic carboplatin therapy. In these carboplatin-resistant Rb cells, the expression of proteins linked to ferroptosis was elevated, whereas the activity of necroptosis-related genes (e.g., RIPK1 and MLKL) did not significantly change. Multiple ferroptosis inducers efficiently cleared these resistant cells, suggesting that the ferroptosis mechanism could be exploited to overcome the chemoresistance of Rb (290). The investigators assessed the efficacy and safety of 4-Octyl itaconate in inducing ferroptosis in MDR Rb cells. 4-Octyl itaconate (4-OI) is a metabolite capable of inducing ferritin-dependent ferroptosis (291).
It significantly induced ferroptosis in Rb cells and increased intracellular free iron and lipid ROS levels, thereby effectively killing resistant cells. Future studies could investigate whether 4-OI induces ferroptosis in a ferritin-dependent manner in Rb cells. In addition, USP14 is one of three deubiquitinating enzymes (USP14, Uch37, and Rpn11) that bind to the proteasome. It can increase the sensitivity of Rb cells to cisplatin by mediating ferroptosis, which may represent an important target for overcoming resistance to cisplatin in Rb (292).
5.6 Ferroptosis and corneal disorders
The inhibition of ferroptosis prevents oxidative stress-induced death of corneal epithelial cells in the absence of GPX4 (153). Ferroptosis contributes to the progression of corneal alkali burns (Figure 4). Research has indicated that the specific ferroptosis inhibitor Fer-1 can ameliorate the condition of mice suffering from corneal alkali burns (158). In an alkali burn mouse corneal model, phytic acid alleviated corneal structural damage and promoted corneal function. Researchers have also reported that PA can protect limbal epithelial cells (LECs) from oxidative damage. Moreover, PA can also inhibit the induction of ferroptosis in LECs during oxidative stress in vitro (293). Keratoconus (KC) is a prevalent corneal thinning disorder and is a primary reason for transplant surgery worldwide (294, 295), but its underlying mechanisms remain unclear. ACSL4 is the most promising biomarker for keratoconus and is expressed predominantly in corneal stromal cells. Laboratory studies have additionally validated the pivotal involvement of ACSL4 in the progression of KC. ACSL4 plays an important role in the process of ferroptosis, and the results of a previous study provide ideas for the future use of ferroptosis to regulate ACSL4 to treat KC (296).
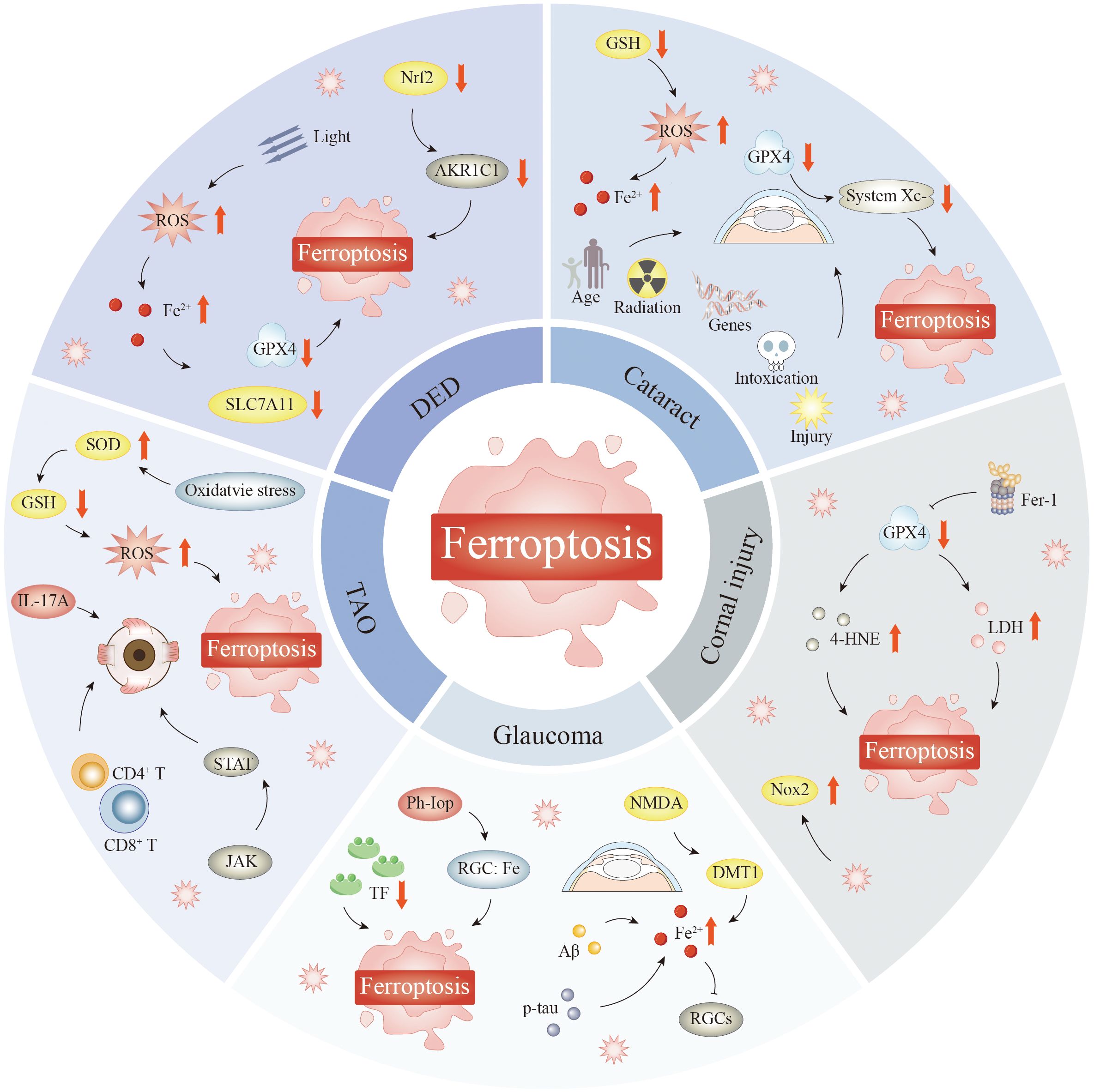
Figure 4. Ferroptosis mediates diseases such as TAO, DED, cataracts, corneal injury, and glaucoma. Inhibition of ferroptosis protects corneal epithelial cells from oxidative stress-induced cell death in the absence of GPX4, which is involved in the course of corneal alkali burns and plays a role in keratitis. Experiments in HCEC revealed that activation of the transcription factor Nrf2 upregulated AKR1C1; when AKR1C1 was knocked down, cell viability was reduced, along with increased lipid peroxidation; excessive oxidative stress-induced ferroptosis was involved in DED pathogenesis. Iron overload was found around retinal epithelial cells, RGCs, and optic nerve axons in glaucoma patients; iron was reduced in cones; TF expression levels were increased. TF could interfere with different cell death mechanisms during the pathogenesis of glaucoma, indicating that iron metabolism would affect glaucomatous neuropathy to some extent. Accumulation of reactive oxygen species and disturbance of redox homeostasis in the lens play critical roles in the development and treatment of diseases. The ability of GSH to synthesize new enzymes decreased, and patients showed impaired lens redox homeostasis, decreased GSH levels, disrupted GPX activity, increased iron redox content, and increased lipid oxidation, which suggests that age-related cataracts are closely related to ferroptosis. ROS generated during the inflammatory response can induce cell damage and play a key role in activating ferroptosis. Markers of oxidative stress are elevated in orbital tissue of TAO patients, suggesting that reactive oxygen species accumulation may trigger orbital fibroblast ferroptosis. Abbreviations: GSH, glutathione; AKR1C1, aldo-keto reductase family 1 member C1; SOD, superoxide dismutase; IL-17A, interleukin-17A; TAO, thyroid-associated orbitopathy; DMT1, divalent metal transporter 1; p-tau, phosphorylated tau protein; RGCs, retinal ganglion cells; DED, dry eye disease; GPX4, glutathione peroxidase 4; HCEC, human corneal epithelial cell; TF, transferrin receptor; ROS, reactive oxygen species.
Ferroptosis also plays an important role in keratitis. Severe eye infection and bacterial keratitis (BK) can lead to pronounced inflammation and corneal damage, resulting in decreased vision (297, 298). One study was performed using a mouse model of keratitis caused by Pseudomonas aeruginosa. Mice were infected with P. aeruginosa on Day 0 and subsequently treated from Day 1 to Day 7 with either levofloxacin (LEV) alone or LEV in combination with the ferroptosis inhibitor Fer-1. In the BK mouse model, the LEV + Fer-1 group presented lower levels of inflammatory factors, reduced corneal scarring, and lower Fe3+ levels. Notably, GPX4 and SLC7A11 levels were elevated in the LEV + Fer-1 group. In vitro experiments revealed that Fer-1 treatment effectively reversed the LPS-induced alterations in ROS, Fe2+, GPX4, and SLC7A11 levels in corneal stromal stem cells (CSSCs).
These findings indicate that ferroptosis is significantly involved in the pathogenesis of BK and that the inhibition of ferroptosis may help reduce inflammation and diminish corneal opacity, thereby improving BK patient outcomes (299). Fusarium is a primary species responsible for fungal keratitis, a prevalent cause of blindness. Oxidative stress is widely recognized as a significant factor in the development of keratitis caused by Fusarium species, and some investigators have concluded that ferroptosis can significantly affect the progression of Fusarium-induced keratitis through data collection analysis (162).
5.7 Ferroptosis and dry eye disease
DED is a multifactorial condition characterized by the loss of tear film homeostasis, which leads to a self-perpetuating cycle of ocular surface inflammation and damage (300, 301). Dysfunction of ocular structures and various systemic diseases, environmental factors, and living habits, such as long-term wearing of contact lenses, can lead to the occurrence and development of DED (302). The risks of DED, including instability, ocular surface damage, nerve paresthesia, and inflammation, have long been overlooked (303).
DED often manifests through telltale signs such as hazy vision, persistent dryness, and heightened sensitivity to light. In extreme cases, patients may develop corneal and conjunctival damage, filamentary keratitis, or even ulceration—complications that can progress to vision loss if left untreated. These debilitating effects not just compromise ocular health but also disrupt everyday activities and professional productivity, underscoring the condition’s far-reaching impact on quality of life (163). DED is also known as incurable terminal disease, for which the standard treatments include artificial tears and medications, although these may not work for all patients. Therefore, exploring the etiology of DED and developing new treatment options are very important.
Some researchers have used subcutaneous injection of scopolamine combined with a dry environment to establish DED mouse models, and immortalized human HCEC-2 cells were cultured under hypertonic conditions to establish hypertonic cell injury models and measure the levels of intracellular Fe2+, cellular ROS, and lipid peroxidation. The findings revealed significant changes in the levels of ferroptosis-related markers, intracellular iron, and lipid peroxidation in corneal epithelial cells in a hyperosmotic injury model.
Ferroptosis in hyperosmolarity-induced HCECs was suppressed when excessive oxidative stress was mitigated. In addition, the activation of the transcription factor Nrf2 in HCECs upregulated AKR1C1 under hyperosmotic conditions. When AKR1C1 was knocked down, cell viability was reduced, and lipid peroxidation was increased. Conversely, the overexpression of AKR1C1 had the opposite effect. Corneal defects enhanced inflammatory responses following the inhibition of AKR1C1 in mice. These findings indicate that ferroptosis triggered by high oxidative stress contributes to DED development. Nrf2-mediated expression of the transcription factor AKR1C1 reduced ferroptosis-induced cell injury and attenuated inflammatory responses (304).
Oxidative stress in the tear film and ocular surface increases considerably among DED patients (305). Therefore, ferroptosis can significantly affect DED development. In a previous study, researchers designed adhesive liposomes (Cyclosporine A & Fer-1 loaded adhesive liposomes decorated with chitosan and fibronectin (CF@SNPs)) that codeliver cyclosporine A (CsA) and the ferroptosis inhibitor Fer-1 to achieve synergistic anti-inflammatory and anti-ferroptotic effects on ocular tissues. First, the researchers developed an in vitro model simulating DED by exposing HCECs to hydrogen peroxide (H2O2) and a hypertonic solution (HS) to mimic a dry eye environment. The experimental findings indicated that Fer-1 markedly ameliorated cell damage in the DED model, suggesting that ferroptosis plays a pivotal role in DED and that Fer-1 may inhibit ferroptosis through the p53-SLC7A11 signaling pathway.
In addition, through RNA sequencing analysis, researchers revealed the effects of CF@SNP treatment on the expression of ferroptosis-related genes and inflammation in a mouse model of DED. They reported that CF@SNPs significantly downregulated the expression of genes related to both ferroptosis and inflammation. Immunofluorescence analysis revealed that CF@SNP treatment significantly reduced ROS levels, iron ion levels, and lipid peroxidation while decreasing inflammatory factor expression. These findings confirm the efficacy of CF@SNPs in mitigating the pathology of DED. This study systematically evaluated the potential of CF@SNPs in experiments assessing DED via both in vitro and in vivo methodologies, establishing a scientific foundation for the development of novel DED therapies (306).
Under high osmotic pressure, the small-molecule compound D609 significantly restored GPX4 expression and reduced intracellular iron ion accumulation. This activity inhibited cellular ferroptosis and protected corneal epithelial cell activity. Through combined transcriptomic and proteomic analysis, they reported that D609 inhibited hyperosmolarity-induced oxidative stress and ferroptosis by increasing melatonin (MT) protein expression and modulating the Nrf2 pathway. PAA-CD-DA increased the ocular bioavailability of D609, enabling targeted DED treatment (307).
5.8 Ferroptosis in glaucoma
Glaucoma is the number one cause of permanent vision loss worldwide and is characterized by long-term and persistent deterioration of the optic nerve. This disease causes specific changes in the optic nerve head and retinal nerve fiber layer and is accompanied by the progressive death of RGCs and visual field defects. Intraocular pressure is a key modifiable factor (308). The current primary therapeutic objective for glaucoma is to delay disease progression and maintain visual function and quality of life (309). The selective and irreversible loss of RGCs, the sole central afferent neurons of the retina, is the underlying pathological mechanism in glaucoma (310).
Compared with that in the retinas of healthy individuals, the iron distribution in the retinas of glaucoma patients varies. In glaucoma patients, iron overload is observed around RPECs, RGCs, and optic nerve fibers, whereas iron levels in cone photoreceptors are reduced (311). The levels of the iron-regulating protein TF are elevated in the retinas of monkeys and humans with glaucoma. Elevated TF levels may help protect RGCs from ocular hypertension by regulating iron homeostasis, which in turn can influence various cell death pathways, including apoptosis and ferroptosis. These findings suggest that iron metabolism can affect glaucomatous neuropathy to some extent. A recent study revealed that serum iron levels are higher in patients with acute primary angle-closure glaucoma (PACG) than in healthy people, indicating that iron metabolism may be involved in the regulation of retinal ganglion cell damage in the case of elevated intraocular pressure [pathological intraocular pressure (ph-IOP)] (312).
Ph-IOP is a key glaucoma characteristic and a major factor leading to RGC loss in patients with glaucoma (313). However, the sole management of the IOP does not entirely avert the depletion of RGCs in glaucoma. Yao et al. reported that elevated ph-IOP disrupts retinal iron balance, causing Fe2+ overload, particularly in the RGC layer (314). Elevated iron levels impair retinal redox equilibrium and induce RGC ferroptosis.
NCOA4-mediated degradation of FTH1 is important in retinal iron metabolism disorders after ph-IOP injury (314). The inhibition of NCOA4 resulted in decreased degradation of FTH1 and decreased bivalent iron content in the retina. This significantly reduces iron ion levels and alleviates RGC damage (315). When glutamate stimulates N-methyl-d-aspartate (NMDA) receptors on the surface, it activates a GTP-binding protein called Dexras1, which in turn stimulates DMT1, allowing cells to absorb more iron (316, 317). Sakamoto et al. reported increased Fe2+ levels and apoptosis in retinal ganglion cells following the intravitreal injection of NMDA (318). Iron-binding agents safeguard retinal ganglion cells against NMDA-induced excitotoxicity by decreasing intracellular iron levels and oxidative stress in rodents (318, 319).
In glaucoma, the increased levels of Aβ, p-tau, and amyloid precursor protein initiate an inflammatory response, leading to cytokine secretion and iron overload. Iron activates inflammatory factors via the Fenton reaction (320). Therefore, by further increasing the production and accumulation of Aβ, the redox state of RGCs forms a positive feedback loop with iron ions, exacerbating the loss of RGCs in glaucoma. Iron overload can damage mitochondria. The mitochondria in RGCs from glaucoma subjects and rodent glaucoma models were smaller, rounder, and more fragmented, suggesting a link between glaucoma, iron levels, and mitochondrial dysfunction (321). Mitochondrial impairment is pivotal in the degeneration of RGCs in glaucoma (322). By protecting mitochondrial function, ferroptosis inhibitors can improve the survival of RGCs and help us maintain the structure of the retina (323).
5.9 Ferroptosis in cataracts
Cataracts refer to degenerative changes in optical quality resulting from reduced transparency or color changes in the lens. The pathogenesis of cataract is complex. The lens resides in an intraocular fluid environment. Any factor that affects this intraocular environment, such as aging, genetics, metabolic abnormalities, trauma, radiation, poisoning, local nutritional disorders, and certain systemic metabolic or immune diseases, can directly or indirectly cause the lens to become opaque. Epidemiological studies have shown that ultraviolet irradiation, diabetes, hypertension, cardiovascular disease, body trauma, excessive alcohol consumption, and smoking are associated with the formation of cataracts (324). Cataracts account for 50% of global blindness and one-third of all vision impairment cases. As the global population continues to grow older, this disease continues to be a major public health burden, and further research on this disease is needed (325).
Research on age-related cataracts has revealed that the accumulation of reactive oxygen species and the destruction of the redox balance of the lens are core factors that lead to the development of diseases and affect treatment methods. In patients, the ability to synthesize GSH in the lens decreases, and patients exhibit disrupted lens redox balance, reduced GSH concentrations, impaired GPX function, elevated iron redox levels, and increased lipid peroxidation, which indicates that age-related cataracts are closely related to ferroptosis (326, 327). Research by Reddy and his collaborators revealed that mice lacking the GPX1 gene had significantly increased light scattering in the lens nuclei of their eyes (328). The mice developed age-associated cataracts over time. These extensive findings of heightened lipid peroxidation and reduced GPX activity and the accumulation of redox-active iron in aging and cataractous human lenses strongly support the occurrence of ferroptosis during lens aging and cataractogenesis. In both in vitro and ex vivo models, Wei et al. reported that the system Xc− inhibitor erastin and the GPX4 inhibitor RSL3 increased the susceptibility of lens epithelial cells to ferroptosis (329). Age-related ferroptosis sensitivity increases in both human LECs and mouse lens epithelium.
An important link in ferroptosis is lipid peroxidation, and increasing evidence suggests that lipid peroxidation contributes to cataractogenesis in the early phases. Babizhayev reported the induction of posterior subcapsular cataracts in rabbits via intravitreal phospholipid lipoperoxide application in a model study. In addition, iron overload can induce cataract formation (330). Ocular siderosis, resulting from elevated iron accumulation in eye tissues due to foreign bodies containing iron, can lead to the formation of cataracts (331). TSTA3 is a key enzyme in GDP-fucose synthesis and regulates intracellular fucosylation levels. TSTA3 overexpression or knockdown directly affects the glycosylation status of lens epithelial cells, which in turn alters their function and metabolic pathways. TSTA3-regulated fucosylation may influence ferroptosis development by regulating key nodes of these metabolic pathways, such as GPX4 expression. Researchers have reported that ferroptosis is a significant factor in high myopic cataracts (HMCs). They reported that the overexpression of discoid domain receptor tyrosine kinase 2 (DDR2) activates the Src-Hippo pathway, which in turn downregulates GPX4. This ultimately increases the ferroptosis sensitivity of the lens epithelium in highly myopic eyes, thereby promoting the formation of nuclear cataracts (332). In vivo, the injection of RSL3 into the anterior chamber of a mouse model of high myopia induced more severe lens nucleus opacification, which was partially ameliorated by ferroptosis and DDR2 inhibitors. Researchers have revealed that CD82 is strongly associated with dexamethasone (DEX)-triggered ferroptosis in lens epithelial cells. DEX induced ferroptosis via CD82-P53-induced suppression of SLC7A11 and GPX4 expression. The inhibition of iron release by Lip-1 reduces the development of glucocorticoid-induced posterior subcapsular cataracts in a DEX-treated rat model. Glucocorticoid-induced posterior subcapsular cataract (GIC) is strongly linked to ferroptosis enhancement via the CD82-P53-GPX4/SLC7A11 axis (333).
5.10 Ferroptosis in TAO
TAO is a condition characterized by an organ-selective autoimmune reaction that affects mainly the orbital muscles and surrounding structures (334). One of these antibodies reacts with thyroid cells and orbital fibroblasts, triggering inflammation in the orbital muscles, connective tissues, adipose deposits, and tear-producing glands. The disease features the infiltration of polymorphous cells with enhanced glycosaminoglycan excretion. TAO is characterized by an increase in orbital contents that can cause the orbit to expand to even eight times the normal size, with a risk of secondary intraocular pressure increase, optic nerve compression, subsequent myofiber degeneration leading to fibrosis, and restraint effects on the affected muscle leading to restrictive myopathy and diplopia (335). TAO is less prevalent in men than in women. In a previous study, most patients in a TAO cohort had mild TAO, whereas 5%–6% of patients had moderate to severe TAO (336).
Selenium is a recognized suppressor of ferroptosis, and selenium supplementation has been shown to improve the quality of life in individuals with mild TAO (337, 338). Teprotumumab, a human anti-IGF1R monoclonal antibody, has been demonstrated to be detrimental to the glycolytic phenotype in experiments with graves’ orbitopathy orbital fibroblasts (GO OFs) from patients with TAO. This leads to ferroptosis in GO OFs (339). This medication was recently approved by the Food and Drug Administration (FDA) for active, moderate-to-severe TAO, suggesting that ferroptosis may contribute to TAO progression and may become a new therapeutic target.
Oxidative stress contributes to ferroptosis and influences TAO pathogenesis. In patients with TAO, the oxidative stress response is characterized by elevated levels of SOD and lipid peroxides, whereas GSH concentrations decrease within the orbital connective tissue (340). In addition, the regulation of the Nrf2/ERK/HO-1 signaling pathway alleviates oxidative stress in orbital fibroblasts (341).
ROS produced during inflammatory reactions can incite cellular harm and significantly trigger ferroptosis. Elevated oxidative stress markers in TAO orbital tissue imply that ROS accumulation may induce ferroptosis in fibroblasts (342). At the onset of TAO, CD4+ T cells can activate and multiply orbital fibroblasts and fat cells (343). In addition, B cells, macrophages, and dendritic cells increase tissue swelling and scarring. This dysregulation of immune cells and cytokines establishes a proinflammatory environment that drives the pathogenesis of TAO. Tea-derived polyphenols have been shown to reduce inflammation triggered by lipopolysaccharides through the modulation of the NF-κB/NLR family pyrin domain 3 (NLRP3) pathway (25). In addition, the JAK–STAT axis is crucial for modulating orbital inflammation in TAO (344). Concurrently, it is pivotal in mediating ferroptosis.
Ferroptosis is also important in the fight against fibrosis. For example, triptolide reduces liver fibrosis through the HO-1 signaling pathway (345). However, excessive iron accumulation and ferroptosis in the liver were found to aggravate acetaminophen-induced fibrosis in a mouse model (346). Subsequent experiments revealed that ferroptosis may affect tissue fibrosis by modulating related signaling pathways and increasing interactions between cells. These findings provide a potential new direction for the development of antifibrotic treatments for TAO.
TAO is a complex autoimmune disease that affects structures in the orbit, such as extraocular muscles, orbital fat, and the lacrimal gland. Studies have shown that older TAO patients are more likely to experience posterior thickening of extraocular muscles. This thickening can lead to double vision and limited eye movements (347). It has been shown that cysteine deprivation and treatment with erastin induce ferroptosis and subsequently lead to orbital fibroblast proliferation (348). In TAO patients, increased ferroptosis of orbital adipose tissue is observed. Lacrimal gland tissue plays a vital role in preserving the integrity of the ocular surface. An investigation of the T-cell immunophenotype in the lacrimal glands of TAO patients revealed that one of the hallmarks of lacrimal gland inflammation is the marked infiltration of interleukin-17A (IL-17A)-producing T helper 17 (Th17) cells. IL-17A can promote the differentiation of lacrimal duct fibroblasts into myofibroblasts or adipocytes (349). In addition, a study revealed that corneal nerve damage increased ferroptosis in lacrimal tissue, which resulted in decreased tear production (350). In TAO patients, orbital fat expansion and extraocular muscle hypertrophy together lead to inflammatory infiltration and pyroptosis, and TAO-related lacrimal gland injury contributes to DED. In summary, ferroptosis clearly leads to orbital tissue injury in TAO patients and exacerbates existing orbital lesions among such individuals.
6 Targeting ferroptosis to treat eye diseases
6.1 AMD
Several inhibitors of ferroptosis exhibit protective effects on retinal neurons and hinder the progression of age-related macular degeneration. Recent research has revealed that the tumor suppressor protein p53 plays a key role in triggering ferroptosis by promoting reactive oxygen species generation while decreasing SLC7A11 expression. A groundbreaking study by Yang and colleagues demonstrated that an extract derived from Fructus Lycii and Salvia miltiorrhiza (FSE) effectively prevents photoreceptor cell death through ferroptosis. This protective mechanism occurs via the regulation of the P53/SLC7A11 pathway in 661 W cells exposed to hydrogen peroxide-induced oxidative stress (247). Research has indicated that a higher intake of omega-3 long-chain polyunsaturated fatty acids (n−3 LC-PUFAs), a component of fish, may be beneficial for patients with AMD. n−3 LC-PUFAs are essential for retinal function, notably contributing to the phototransduction process and resistance to oxidative stress, inflammation, and vascular damage—key contributors to AMD. Animal research in both laboratory and live settings has demonstrated that n−3 LC-PUFAs have the potential to maintain retinal integrity in AMD patients (351). LOXs are a family of enzymes that metabolize arachidonic acid and PUFAs, contribute to lipid peroxidation, and induce ferroptosis. Zileuton-mediated inhibition of 5-LOX reduces ROS-induced lipid peroxidation, mitochondrial damage, DNA damage, and iron-related toxicity, as verified by in vitro experiments and animal models of dry AMD.
Researchers have confirmed that ferroptosis leads to RPEC loss, disrupts mitochondrial function, and increases lipid peroxidation in ferric ammonium citrate (FAC)-treated ARPE-19 cells and mouse models. The characteristic lesions of dry AMD were highly consistent with the pathological findings of the ferroptosis experiments. These two points highlight the critical effect of ferroptosis on dry AMD, and in this study, Rhodiola rosea activated the Nrf2 pathway. The expression of the antioxidant proteins Nrf2, SLC7A11 (cystine transporter), and GPX4 (lipid peroxidase) was upregulated, and iron ion accumulation and ROS generation were inhibited. R. rosea also reduced macrophage infiltration and alleviated pathological damage in RPE cells (352). Other researchers have designed melanin-like nanoparticles called Concanavalin A-decorated melanin-like nanoparticles (ConA-MelNPs) as novel ferroptosis inhibitors. ConA-MelNPs chelated iron ions, significantly reduced mitochondrial injury triggered by oxidative stress, and prevented NaIO3-induced ferroptosis in RPECs (353).
6.2 DR
Almost all patients with diabetes develop DR. Drug therapy, such as anti-VEGF therapy, has led to drug resistance in clinical practice. The treatment of DR with drugs is still under investigation. Ferroptosis plays an important role in the pathogenesis of DR. Targeting ferroptosis to treat DR has become a new research direction.
The two key factors involved in preventing ferroptosis, namely, iron activity and lipid peroxidation, have been shown to be beneficial in DR. DFO significantly contributes to mitigating iron overload conditions. In a murine disease model, vitreous injection of DFO significantly decreased retinal iron content and reduced vascular leakage. However, the ability of DFO to penetrate the BRB is limited (354). Lactoferrin (LF) is an iron-binding glycoprotein derived from mucous cells and neutrophils. LF has a unique ability to break through the BRB (355). LF not only captures reactive iron in the retinal environment and reduces ROS production but also directly removes ROS (356). PMX500FI, an A-lipoic acid derivative studied by Moos et al., can pass through the BRB while also catching iron in the retina. It activates the Nrf2 signaling pathway and inhibits oxidative stress (357, 358). Nrf2 acts as a vital in vivo antioxidant protector, with research indicating that Nrf2 pathway activation safeguards retinal tissue in patients with diabetes (359, 360). Similarly, numerous investigations into diabetic cell targets have indicated that increasing Nrf2 levels safeguards against ferroptosis, thereby mitigating organ damage.
The inhibition of lipid peroxidation is another important strategy for preventing ferroptosis. Numerous cellular and animal studies have validated the effectiveness of ELABELA (ELA) and ferrostatin-1 in regulating the iron attenuation process in the retina through the system Xc−GPX4 pathway activation (361). The alleviation of ER stress is important for slowing DR progression (362). The alleviation of DR stress may be achieved by ameliorating oxidative stress and inflammation in cells, restoring intracellular iron homeostasis, and attenuating membrane lipid peroxidation. Wang et al. reported that blueberry anthocyanin extract inhibits ER stress, thereby providing relief for DR patients (363). In addition, the compound endoplasmic reticulum was shown to reduce ER stress and decrease inflammatory factor expression. This intervention showed promising efficacy in lowering lipid peroxidation, subsequently halting the intricate interactions among the processes that are associated with oxidative stress, endoplasmic reticulum stress, and inflammation (364).
Among natural products, quercetin inhibited TFR1, upregulated FPN, and decreased free iron levels; 50 μM quercetin reduced the percentage of ferroptotic cells from 42% to 12% in a high glucose-induced Müller cell model. Resveratrol can inhibit ferroptosis through stimulation of the Nrf2/HO-1 pathway and suppression of the p53-SLC7A11 axis. In mice with Streptozotocin (STZ)-induced diabetes, 8 weeks of oral resveratrol treatment reduced the retinal vascular leakage area by 38% and restored GPX4 expression to 1.7 times the baseline level. In addition, Tang and colleagues provided significant observations (365). Their findings revealed that astragaloside IV (AS-IV) increased Sirt1/Nrf2 activity, ultimately resulting in increased GPX4 expression, which decreased retinal lipid peroxidation.
6.3 Glaucoma
Recent studies have shown that DFO is a unique compound that can penetrate the blood–retinal barrier with ease. Following ph-IOP-induced damage, it effectively binds to excess divalent iron in retinal tissue, preventing toxic accumulation. By mitigating iron overload, DFO safeguards retinal ganglion cells from ferroptosis and helps preserve normal vision. This protective mechanism highlights the potential of DFO as a therapeutic agent for retinal neuroprotection. Currently, multifunctional nanoparticles (NPsLip-1) have been developed and applied to manage acute glaucoma. These nanoparticles can significantly reduce lipid peroxidation products and restore mitochondrial function, thereby inhibiting ferroptosis. Ultimately, they increase the survival rate of RGCs in glaucoma models by 5.1-fold. Compared with traditional iron chelators (such as DFO), NPsLip-1 address issues related to rapid metabolism, poor targeting, and systemic toxicity (366).
Glutamate-induced cell surface N-methyl-d-aspartate receptors (NMDARs) stimulate cellular iron absorption by inducing the expression of Dexras1, a GTP-binding protein that increases DMT1-mediated iron uptake (316, 317). Sakamoto et al. reported that NMDA-induced intravitreal injections caused Fe2+ buildup and cellular death within RGCs (318). Iron-chelating agents protected RGCs from NMDA excitotoxicity-mediated cell death by reducing intracellular iron levels and alleviating oxidative stress in rodents (318, 319). Suppressing NCOA4 led to reduced FTH1 breakdown and decreased bivalent iron levels in the retina. This significantly reduced iron ion levels and alleviated RGC damage (315).
Glaucoma involves cytokine release driven by inflammation triggered by abnormal processing of amyloid precursor protein and subsequent deposition of Aβ and p-Tau in retinal ganglion cells, which disrupts iron homeostasis and leads to retinal iron overload. Iron triggers inflammation via the Fenton reaction, accelerating the formation and accumulation of amyloid-beta (Aβ). Therefore, the redox balance of RGCs interacts with iron ions in a self-amplifying feedback loop, exacerbating glaucoma-induced RGC death (320). Iron overload can damage mitochondria. Mitochondria in RGCs from individuals with glaucoma and murine glaucoma models were smaller, rounder, and more fragmented, suggesting a link between glaucoma, iron levels, and mitochondrial dysfunction (321). Mitochondrial dysfunction plays a critical role in RGC loss in the context of glaucoma (322). Ferroptosis inhibitors can promote RGC survival and protect retinal structure by maintaining mitochondrial function (323).
6.4 TAO
Ferroptosis inhibitors may be effective agents for the treatment of TAO, as they have been shown to prevent cell death by alleviating oxidative stress, having anti-fibrotic effects, and inhibiting lipid peroxidation. For example, sulfasalazine, a clinically used ferroptosis inducer, is very effective against thyroid cancer cells, indicating its possible use in TAO (367, 368). Selenium serves as a vital ferroptosis regulator and an effective therapy for TAO, as it can reduce lipid hydroperoxides and inhibit lipoxygenase (369). These inhibitors have the potential to mitigate orbital inflammation and tissue remodeling, both of which are pivotal indicators of TAO. They achieve this by scavenging free radicals produced via lipid peroxidation and inhibiting the buildup of harmful lipid peroxides. The IGF1R inhibitor teprotumumab has received FDA approval for managing active, moderate-to-severe TAO.
Studies on thyroid cancer cells have shown that modulating ferroptosis may have effects on cell survival and oxidative stress. Anaplastic thyroid carcinoma cells were found to be resistant to ferroptosis (370). Targeting the ferroptosis pathway may be a viable strategy for addressing TAO, a condition in which oxidative stress is a pivotal factor. Ferroptosis inducers such as erasin, in addition to iron-binding agents, have demonstrated potential in mitigating oxidative stress-induced damage and increasing cellular viability in preclinical studies (371). Ferroptosis plays a role in TAO pathogenesis. The overexpression of SIRT6 in thyroid cancer has been shown to increase sensitivity to ferroptosis through the autophagic degradation of ferritin dependent on nuclear receptor coactivator 4, suggesting that TAO may involve a similar mechanism (372). Ferroptosis-associated lncRNAs, such as LINC01140 and ZFHX4-AS1, have been found to be differentially expressed in TAO patients (373).
6.5 Corneal disorders
Studies have demonstrated that decreased levels of GPX4 in HCECs can lead to increased LDH levels, which can trigger ferroptosis (150). The application of the ferroptosis inhibitor Fer-1 could mitigate ferroptosis mediated by this pathway (152). Research has indicated that compared with wild-type mice, GPX4-deficient mice exhibit delayed corneal wound healing following epithelial injury, highlighting the role of GPX4 in corneal wound healing. The inhibition of ferroptosis protects against oxidative stress-related mortality in corneal epithelial cells when GPX4 is absent (153).
Studies have shown that the ferroptosis antagonist Fer-1 is promising for the treatment of multiple disorders linked to ferroptosis and that Fer-1 treatment can mitigate corneal opacity and neovascularization in alkali burn mouse models. Further studies have demonstrated that alkali burns trigger ferroptosis activation and the attack of ROS on mitochondria, which together facilitate the onset of ferroptosis within corneal tissue. The effectiveness of Fer-1 indicates that ferroptosis could be a therapeutic target in corneal alkali burn management. The experiment also showed that Fer-1 liposomes were significantly more effective than free Fer-1 at managing corneal alkali injury. In addition, in vitro and in vivo safety evaluations of Fer-1 liposomes were performed, and the results revealed that they had no significant toxic side effects (159, 160). Recent studies have revealed the role of AKR1C1 in protecting corneal epithelial cells against oxidative injury in DED (163). Ferroptosis triggered by high oxidative stress contributes to DED progression in a mouse in vivo DED model and immortalized HCECs. In addition, Nrf2 triggered AKR1C1 expression to attenuate iron toxicity-induced cell injury and inflammation in HCECs.
In a BK mouse model, the LEV + Fer-1 group presented lower levels of inflammatory factors, reduced corneal scarring, and lower Fe3+ levels. Significantly, the LEV + Fer-1 group exhibited elevated levels of GPX4 and SLC7A11. In vitro experiments revealed that Fer-1 administration reversed the LPS-mediated alterations in ROS, Fe2+, GPX4, and SLC7A11 levels in CSSCs. These findings indicate that ferroptosis significantly contributes to bacterial keratitis and that the inhibition of ferroptosis may help reduce inflammation, minimize corneal damage, and improve BK patient outcomes (299).
6.6 Others
Ferroptosis is a crucial mechanism of cell death in RIRI and affects cell types such as RGCs, PRs, and RPEs. This process involves the increased expression of iron-related genes (e.g., Acsl4 and Hmox1) and decreased GPX4 and SLC7A11 levels, along with Fe2+ and ROS buildup during the pathogenesis of RIRI (374). The inhibition of ferroptosis reduces both inflammatory factors and gliosis and protects retinal structures (375). Fer-1 can reduce RGC death by 60% and significantly increase the a/b-wave amplitude according to an electroretinogram in a RIRI mouse model. In addition, MT inhibited RIR-induced ferroptosis by decreasing ROS and Fe2+ levels and increasing GSH levels through the regulation of the p53/Slc7a11/Alox12 signaling pathway (375). DFO reduces the symptoms of RIRI and has been used clinically to treat iron overload disorders, and electroretinography (ERG) and visual evoked potentials (VEP) tests have been performed to confirm that retinal visual function improves in subjects after DFO treatment (274). Supramolecular DFO–crisaborole nanoparticles, which increase GPX4 levels, downregulate the expression of ferroptosis-related genes (Acsl4 and Hmox10), reduce the expression of inflammatory markers, and reduce ROS levels (279). It has been experimentally confirmed that ferroptosis plays an indispensable role in RP, and research on RP models has indicated that iron homeostasis regulators inhibit photoreceptor ferroptosis and prevent degeneration. Drugs such as zinc deferoxamine (283); iron chelators VK28, VAR10303, and DFO (230); and hepatostatin-1, along with the ferroptosis inhibitor DFO, effectively inhibit ferroptosis. For example, DFO decreased retinal iron deposition by 50% and increased photoreceptor survival by 1.8-fold in rd10 mice, and Fer-1 induced RGC and photoreceptor death by 60% and ERG b-wave amplitude recovery in rd1 mice. Qi-Shen-Tang (QST) slows RP progression in rd10 mice. It improves retinal tissue structure and performance, increases blood flow to the tissue, and decreases iron and MDA levels while increasing SOD and GSH concentrations. Additionally, QST (Table 2) contributes to antioxidant defense and protects against ferroptosis. Also, it safeguards retinal cells by activating the Nrf2/GPX4 signaling cascade (46).
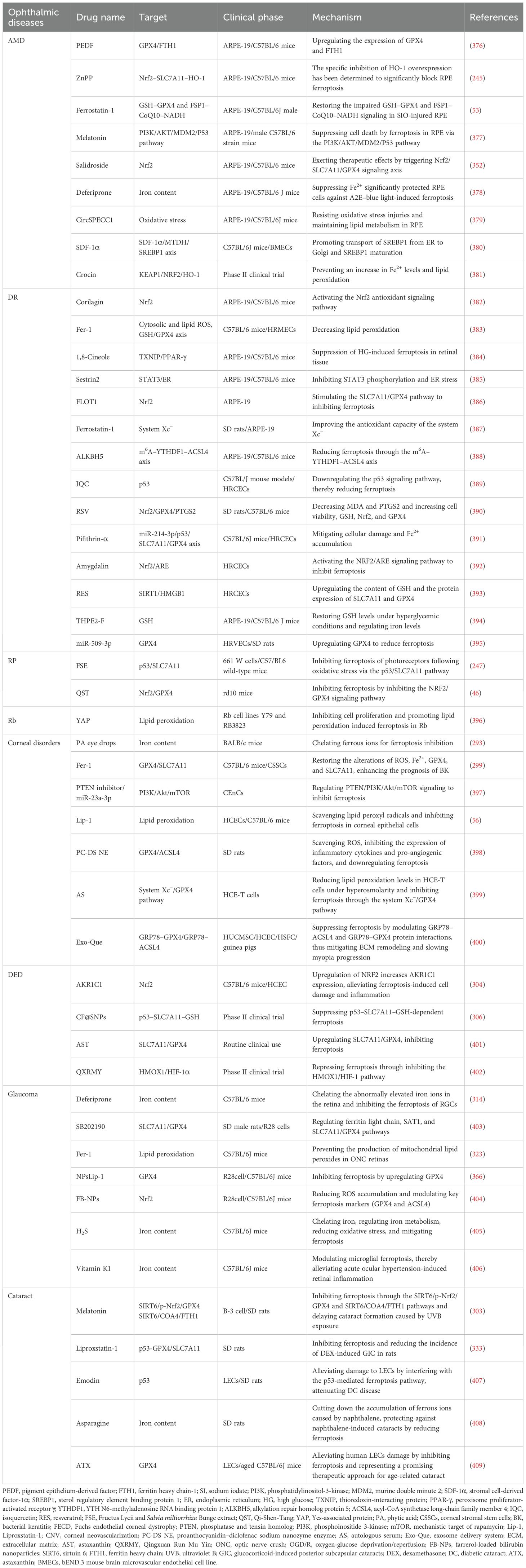
Table 2. Ferroptosis-targeting agents for ocular diseases.
Ferroptosis has been demonstrated to be an important mechanism of programmed cell death in corneal epithelial cells and lacrimal acinar cells in DED. Ferrostatin-1, when applied as an eye drop, increased corneal epithelial survival by 60%, and the levels of Fe2+ and MDA decreased in hyperosmolar models. The mucolipid CF@SNPs, which encapsulate Fer-1 and cyclosporine A, mitigated oxidative stress-induced corneal epithelial death, and treatment with the CF@SNPs resulted in superior therapeutic efficacy and improvement in clinical DED parameters in a mouse model of DED, including the alleviation of corneal injury, the restoration of lacrimal gland structure, and the proliferation of goblet cells. The ocular surfaces of DED patients have high ROS levels, increased cell death-related marker expression, and dry eye stress-induced p53 upregulation, further triggering corneal epithelial ferroptosis. The combined administration of CsA and Fer-1 substantially alleviated corneal injury and DED marker upregulation, and CF@SNPs provide a promising solution to corneal injury in DED through the disruption of the inflammatory cascade, the generation of ROS, and ferroptosis. In addition, DFO can reduce intracellular Fe2+ and ROS levels and inhibit ferroptosis (401). Astaxanthin (AST) mitigates DED via the SLC7A11/GPX4 axis in corneal epithelial cells and mouse models under hyperosmolar stress.
RB1 deletion and p53 mutation affect both Rb pathogenesis and ferroptosis. Targeting ferroptosis is a novel strategy for the treatment of Rb. The induction of ferroptosis by autophagy may lower resistance in Rb cells, whereas 4-OI triggers ferroptosis via ferritin regulation (291). After 4-OI treatment, resistant Rb cell survival was reduced by 75%, and when combined with carboplatin, the in vivo tumor burden was reduced by 90%. Erastin reduced the IC50 of carboplatin-resistant cell lines by eightfold, but did not increase normal retinal toxicity.
High levels of markers with ferroptosis were detected in patients with myopic cataracts, diabetic cataracts, and age-related cataracts. Therefore, targeting ferroptosis is a novel approach for cataract therapy. Ferrostatin-1 and dasatinib (a DDR2-Src-Hippo inhibitor) significantly alleviated RSL3-induced nuclear cataracts and restored lens transparency in HMCs (332). An Nrf2 activator (hydralazine), which reduces ROS production in LECs in mice and humans, provides cytoprotection and delays lens opacification caused by aging and oxidative stress (410). Resveratrol may protect human LECs from oxidative damage by activating antioxidant enzymes (411). Lleó et al. demonstrated that melatonin could protect cells from damage due to H2O2 and white light LED-induced death (412). Experimentally, thiol antioxidants have shown significant promise in safeguarding LECs against oxidative damage and inhibiting cataract formation (413).
7 Limitations and challenges
Most recent studies on ferroptosis in ophthalmology have focused on RPECs or RGCs, but studies on the ability of Müller cells, vascular endothelial cells, PRs, and even corneal endothelial cells to induce ferroptosis are lacking, and comprehensive single-cell maps are lacking, leaving unresolved questions like the variations in the susceptibility of various cell types to ferroptosis, which can be parallel or mutually causal with apoptosis, necrosis, and pyroptosis (414). One study revealed that erastin promotes apoptosis in cancer cells. Iron-depleting compounds trigger ER stress, resulting in alternative cell death pathways (415). Notably, ferroptosis has no specific markers identified in the eye, making it difficult to distinguish it from other types of programmed cell death. The ocular microenvironment is complex, and the immune system differs from other sites, which also poses a challenge for treating ocular diseases with ferroptosis-targeting methods.
Multiple nodes, such as TF, TFR, DMT1, FT, and FPN1, can regulate iron levels; however, these proteins exhibit distinct expression patterns across retinopathies, and it is difficult to find “universal” intervention nodes. Systemic iron chelation may increase the risk of anemia and infection, and local delivery addresses the paradox of the penetration of the blood–retinal barrier and prolonged retinal retention. Currently, only static iron deposition (MRI or tissue iron staining) has been tested and does not reflect lipid peroxidation rates or GPX4 activity fluctuations in real time. Ophthalmologic examinations, such as retinal OCT-A or autofluorescence tests, do not specifically detect ferroptosis and are difficult to use for efficacy monitoring and patient stratification. This also poses a challenge to the timing and extent of intervention with ferroptosis treatment options. If OCT is combined with targeted probes or nano-contrast agents, functional imaging is expected to be realized in the future, enabling the labeling of ferroptosis and the assessment of retinal oxidative stress distribution. Detection of apoptosing retinal cells (DARC) is a method that combines retinal imaging technology with molecular biology to identify retinal apoptosis without the need for invasive methods using advanced confocal scanning laser ophthalmoscopy (cSLO) technology. Using a similar technical approach, imaging instruments can identify ferroptosis biomarkers in living systems. This facilitates crafting customized treatment plans derived from evaluations of noted cellular events.
The currently employed validated clinical models, such as photodamage, NaIO, and STZ mouse models, cannot completely mimic the chronic, multifactorial pathogenic environment of human AMD/DR, and the extrapolation of efficacy is questionable. As of now, the study of ferroptosis in the realm of ophthalmology is primarily limited to ARPE-19 cell cultures and rodent models of acute eye diseases. Techniques like photodamage can rapidly induce retinal deterioration within a matter of hours or days, but they do not adequately replicate the prolonged, recurring mild oxidative stress and the complex interplay of metabolic, inflammatory, and genetic factors found in human AMD/DR. This disconnect between “speedy” research models and “slow-moving” diseases has left a significant translational chasm, where promising results in animals tend to fade in actual patients. Consequently, there are only a few scattered case studies on ferroptosis-focused interventions in the field of ophthalmology. What we desperately need is a two-way approach from the laboratory to the patient’s bedside, focusing on long-term, multifactorial human populations. This approach should use their extensive data to improve animal studies and, ultimately, provide patients with effective, safe, and tailored treatments for ferroptosis.
The maximum tolerated dose and retinal toxicity threshold of iron chelators (DFO and deferiprone) have not yet been established. Additionally, whether long-term inhibition of ferroptosis by inhibitors interferes with the activity of iron-dependent enzymes, such as the mitochondrial respiratory chain, still requires phase I clinical data. Ethical and safety issues still need attention. As the first ferroptosis inhibitor to enter preclinical ophthalmic development, Fer-1 eye drops were originally limited by the drug’s inherent hydrophobicity. Researchers overcame this obstacle by preparing Fer-1-loaded liposomes via thin-film hydration. The resulting formulation shows sustained-release kinetics, markedly enhanced cellular uptake, and prolonged ocular surface residence time, thereby effectively ameliorating alkali burn-induced corneal injury. Safety evaluations, both in vitro and in vivo, revealed no evident toxicity. To date, 0.1%–0.3% Fer-1 liposomal eye drops have not exhibited significant off-target effects in acute or sub-acute models. In the future, long-term safety in larger cohorts will be a key endpoint in upcoming phase I trials.
8 Opportunities
Targeting ferroptosis opens new avenues for the treatment of ocular diseases. Ferroptosis has been shown to be involved in almost all irreversible blinding eye diseases. Recent experiments confirm that most of the common terminal events in AMD, RP, glaucoma, and DR are RPE or RGC ferroptosis, and single-cell sequencing confirms that ferroptosis signals appear earlier than apoptosis. Corneal epithelial ferroptosis after alkali burn and chemical injury is a major cause of corneal opacity and neovascularization. This treatment of these diseases via ferroptosis targeting provides a new direction. At present, preclinical experiments have been completed with related drugs. Fer-1 liposome eye drops can inhibit corneal opacity and increase bioavailability fivefold in a rabbit corneal alkali burn model, which indicates that agents that target ferroptosis have the potential for clinical translation and the treatment of ocular diseases.
Advanced methods such as single-cell sequencing and spatial transcriptomics have been used to map detailed gene expression and pathway networks for each cell in the retinal microenvironment during the development of AMD (416). This method enables the detection of cell subsets potentially undergoing ferroptosis, understanding the state of cells, and treating the symptoms. Cutting-edge research has highlighted the successful use of groundbreaking sustained-release technology known as the port delivery system (PDS). This implantable device enables the steady release of ranibizumab directly into the vitreous humor, offering a novel treatment approach for individuals suffering from nvAMD (417). This innovative system holds promise for safeguarding retinal cells against ferroptosis.
Using a technical approach similar to DARC, imaging devices can identify ferroptosis biomarkers in living systems and enable personalized treatment planning (418). Progress in merging bioinformatics, computational biology, and Artificial Intelligence (AI) has streamlined the identification of ferroptosis therapeutics for ophthalmic applications. State-of-the-art approaches include molecular dynamics modeling, quantum chemical computations, machine learning techniques, and network analyses. It can be a useful tool for investigating lipid membrane properties and the function of lipid peroxides in ferroptosis-induced cell death (419). These methods are essential for pathway analysis and cellular interactions to aid target selection and drug screening. Advanced AI systems are now able to create tailored predictions for AMD progression. Within the intricate world of the eye, these computational image analyses are reshaping our understanding of retinal aging and ailments, offering the chance for swift, early screening and targeted intervention. AI is expected to integrate multi-omics data in the future, providing novel tools for predicting ferroptosis-related gene expression and optimizing drug delivery (420).
To date, natural compounds have been shown to intervene in ferroptosis to alleviate eye diseases, and Polygonatum odoratum polysaccharide can increase the GSH content and SOD activity, thus playing an antioxidant role (419). Asiaticoside suppresses IL-6, IL-1β, and TNF-α expression via the NF-κB pathway while reducing oxidative stress. Madecassoside protects ARPE-2 cells against oxidative stress caused by H2O2 through the activation of the Nrf2/HO-2 signaling cascade. Matrine has diverse pharmacological properties (421). Aloperine mitigates oxidative stress in ARPE-19 cells triggered by H2O2, and these findings highlight the therapeutic potential of aloperine in the treatment of macular degeneration.
Liu et al. reported that fucoxanthin supplements have significant potential in preventing visible light-induced retinal damage (422). The primary active ingredient in Astragalus membranaceus is astragaloside IV, also known as AS-IV. This compound has the remarkable ability to block the process of oxidative tissue damage. By doing so, AS-IV lessens oxidative stress-related damage and increases cell viability in rat RGCs. New research findings suggest that AS-IV may inhibit pathological processes associated with DR (423). Puerarin, a key monomer in kudzu vine root flavones, has numerous effects, including anti-inflammatory and antioxidative stress effects. It is beneficial for treating ischemia–reperfusion tissue damage. Zhang et al. demonstrated that puerarin suppresses Nrf2/ERK pathway activation, mitigating inflammation and oxidative damage. Ginkgo biloba is a prevalent medicinal plant in Chinese medicine and has some efficacy in scavenging oxygen free radicals (424). Li et al. reported that G. biloba extract (GBE) significantly protected against t-BHP-triggered oxidative injury in human io-m1 cells. GBE protects human RGCs against oxidative stress and is promising for inhibiting retinopathies, especially DR (425). Other studies have revealed that pretreatment with GBE significantly mitigated oxidative lipid damage and cell death (necrosis/ferroptosis) and decreased the viability of RPECs exposed to t-BHP (426).
HCE ameliorates radiation-induced ferroptosis in LO2 cells via the Nrf2-xCT/GPX4 pathway (427).
Delphinidin was shown to play a key role in iron regulation and the upregulation of elements linked to the system Xc− pathway while also curbing lipid oxidation, sustaining the integrity of cell membranes, and robustly safeguarding RPEs (661 W cells) from degeneration through the anti-ferroptosis pathway. Blueberry anthocyanins have also been shown to scavenge hydroxyl radicals, H2O2, 1,1-diphenyl-2-picrylhydrazyl (DPPH) radicals, and Fe3+ (428). Anthocyanins may be applied in ophthalmic diseases such as amblyopia, strabismus, cataracts, glaucoma, and retinopathy (429). Fucoidan shows therapeutic potential for AMD. Dörschmann et al. reported that fucoidan offers some defense against cell death triggered by erastin (430). In addition, the protective effect of fucoidan is linked to the maintenance of stable levels of the GPX4 protein, which is an important regulator of ferroptosis. These findings offer novel guidelines for managing eye disorders through ferroptosis through the uptake of natural compounds.
Author contributions
YW: Writing – original draft. YML: Writing – original draft. YJL: Writing – original draft. JL: Writing – original draft. YY: Writing – original draft. HC: Writing – original draft. ZH: Writing – original draft. KW: Writing – original draft. TQ: Writing – original draft, Writing – review & editing. YJ: Writing – original draft, Writing – review & editing. WZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nilsson DE. The diversity of eyes and vision. Annu Rev Vis Sci. (2021) 7:19–41. doi: 10.1146/annurev-vision-121820-074736
2. Baroncelli L and Lunghi C. Neuroplasticity of the visual cortex: in sickness and in health. Exp Neurol. (2021) 335:113515. doi: 10.1016/j.expneurol.2020.113515
3. Yang QH, Zhang Y, Zhang XM, and Li XR. Prevalence of diabetic retinopathy, proliferative diabetic retinopathy and non-proliferative diabetic retinopathy in asian T2dm patients: A systematic review and meta-analysis. Int J Ophthalmol. (2019) 12:302–11. doi: 10.18240/ijo.2019.02.19
4. Yuan H, Chen S, Duncan MR, De Rivero Vaccari JP, Keane RW, Dalton Dietrich W, et al. Ic100, a humanized therapeutic monoclonal anti-asc antibody alleviates oxygen-induced retinopathy in mice. Angiogenesis. (2024) 27:423–40. doi: 10.1007/s10456-024-09917-9
5. Shi X, Zhou XZ, Chen G, Luo WF, Zhou C, He TJ, et al. Targeting the postsynaptic scaffolding protein psd-95 enhances bdnf signaling to mitigate depression-like behaviors in mice. Sci Signaling. (2024) 17:eadn4556. doi: 10.1126/scisignal.adn4556
6. Tang D, Chen X, Kang R, and Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
7. Zhuo Cai J, Lan Yu Y, Biao Yang Z, Xun Xu X, Chun Lv G, Lian Xu C, et al. Synergistic improvement of humus formation in compost residue by fenton-like and effective microorganism composite agents. Biores Technol. (2024) 400:130703. doi: 10.1016/j.biortech.2024.130703
8. Jiang X, Stockwell BR, and Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
9. Bannai S and Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. (1980) 255:2372–6. doi: 10.1016/S0021-9258(19)85901-X
10. Dolma S, Lessnick SL, Hahn WC, and Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. (2003) 3:285–96. doi: 10.1016/s1535-6108(03)00050-3
11. Yang WS and Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-ras-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
12. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
13. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by gpx4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
14. Shen Q, Liang M, Yang F, Deng YZ, and Naqvi NI. Ferroptosis contributes to developmental cell death in rice blast. New Phytol. (2020) 227:1831–46. doi: 10.1111/nph.16636
15. Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging caenorhabditis elegans. eLife. (2020) 9:e56580. doi: 10.7554/eLife.56580
16. Huo G, Lin Y, Liu L, He Y, Qu Y, Liu Y, et al. Decoding ferroptosis: transforming orthopedic disease management. Front Pharmacol. (2024) 15:1509172. doi: 10.3389/fphar.2024.1509172
17. Andrews NC. Disorders of iron metabolism. N Engl J Med. (1999) 341:1986–95. doi: 10.1056/nejm199912233412607
18. Wang K, Lin Y, Zhou D, Li P, Zhao X, Han Z, et al. Unveiling ferroptosis: A new frontier in skin disease research. Front Immunol. (2024) 15:1485523. doi: 10.3389/fimmu.2024.1485523
19. Maiorino M, Conrad M, and Ursini F. Gpx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. (2018) 29:61–74. doi: 10.1089/ars.2017.7115
20. He Y, Lin Y, Song J, Song M, Nie X, Sun H, et al. From mechanisms to medicine: ferroptosis as a therapeutic target in liver disorders. Cell Communicat Signal: CCS. (2025) 23:125. doi: 10.1186/s12964-025-02121-2
21. Yang Y, Lin Y, Han Z, Wang B, Zheng W, and Wei L. Ferroptosis: A novel mechanism of cell death in ophthalmic conditions. Front Immunol. (2024) 15:1440309. doi: 10.3389/fimmu.2024.1440309
22. Zhao T, Guo X, and Sun Y. Iron accumulation and lipid peroxidation in the aging retina: implication of ferroptosis in age-related macular degeneration. Aging Dis. (2021) 12:529–51. doi: 10.14336/ad.2020.0912
23. Kajarabille N and Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci. (2019) 20:4968. doi: 10.3390/ijms20194968
24. Moiseyev G, Takahashi Y, Chen Y, Gentleman S, Redmond TM, Crouch RK, et al. Rpe65 is an iron(Ii)-dependent isomerohydrolase in the retinoid visual cycle. J Biol Chem. (2006) 281:2835–40. doi: 10.1074/jbc.M508903200
25. Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, et al. Targeting ferroptosis in bone-related diseases: facts and perspectives. J Inflammation Res. (2023) 16:4661–77. doi: 10.2147/jir.S432111
26. Picard E, Daruich A, Youale J, Courtois Y, and Behar-Cohen F. From rust to quantum biology: the role of iron in retina physiopathology. Cells. (2020) 9:705. doi: 10.3390/cells9030705
27. Han Z, Luo Y, Chen H, Zhang G, You L, Zhang M, et al. A deep insight into ferroptosis in renal disease: facts and perspectives. Kidney Dis (Basel Switzerland). (2024) 10:224–36. doi: 10.1159/000538106
28. Liu D, Liu Z, Liao H, Chen ZS, and Qin B. Ferroptosis as a potential therapeutic target for age-related macular degeneration. Drug Discov Today. (2024) 29:103920. doi: 10.1016/j.drudis.2024.103920
29. Totsuka K, Ueta T, Uchida T, Roggia MF, Nakagawa S, Vavvas DG, et al. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp Eye Res. (2019) 181:316–24. doi: 10.1016/j.exer.2018.08.019
30. Huang S, Liu K, Su Y, Wang F, and Feng T. Research progress of ferroptosis in glaucoma and optic nerve damage. Mol Cell Biochem. (2023) 478:721–7. doi: 10.1007/s11010-022-04545-7
31. Gaschler MM and Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
32. You H, Wang L, Bu F, Meng H, Huang C, Fang G, et al. Ferroptosis: shedding light on mechanisms and therapeutic opportunities in liver diseases. Cells. (2022) 11:3301. doi: 10.3390/cells11203301
33. Tang W, Guo J, Liu W, Ma J, and Xu G. Ferrostatin-1 attenuates ferroptosis and protects the retina against light-induced retinal degeneration. Biochem Biophys Res Commun. (2021) 548:27–34. doi: 10.1016/j.bbrc.2021.02.055
34. Zhao D, Li W, Han Z, Wang Z, Li D, and Li W. Exploring the role of ferroptosis in esophageal cancer: mechanisms and therapeutic implications. Cell Death Discov. (2025) 11:405. doi: 10.1038/s41420-025-02696-2
35. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. Pkcβii phosphorylates acsl4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. (2022) 24:88–98. doi: 10.1038/s41556-021-00818-3
36. Chen H, Han Z, Su J, Song X, Ma Q, Lin Y, et al. Ferroptosis and hepatocellular carcinoma: the emerging role of lncrnas. Front Immunol. (2024) 15:1424954. doi: 10.3389/fimmu.2024.1424954
37. Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J, et al. Glia maturation factor-β Induces ferroptosis by impairing chaperone-mediated autophagic degradation of acsl4 in early diabetic retinopathy. Redox Biol. (2022) 52:102292. doi: 10.1016/j.redox.2022.102292
38. Xiao R, Han Z, Jia P, Li P, Gong M, Cai Y, et al. Ferroptosis and bone health: bridging the gap between mechanisms and therapy. Front Immunol. (2025) 16:1634516. doi: 10.3389/fimmu.2025.1634516
39. Lapaquette P, Terrat S, Proukhnitzky L, Martine L, Grégoire S, Buteau B, et al. Long-term intake of lactobacillus helveticus enhances bioavailability of omega-3 fatty acids in the mouse retina. NPJ Biofilms Microbiomes. (2024) 10:4. doi: 10.1038/s41522-023-00474-5
40. Perus M, Courtaut F, Pais De Barros JP, Aires V, Hermetet F, and Delmas D. Vegf-R2/cav-1 interaction induced by resveratrol/eicosapentaenoic acid/docosahexaenoic acid-enriched formulation through functional detergent-resistant membranes is associated with decreased vegf-a release in arpe-19 cells. Mol Nutr Food Res. (2024) 68:e2300893. doi: 10.1002/mnfr.202300893
41. Shao M, Jiang Q, Shen C, Liu Z, and Qiu L. Sinapine induced ferroptosis in non-small cell lung cancer cells by upregulating transferrin/transferrin receptor and downregulating slc7a11. Gene. (2022) 827:146460. doi: 10.1016/j.gene.2022.146460
42. Xue P, Zhuang H, Bai T, Zeng X, Deng J, Shao S, et al. Iron (Ii)-based metal-organic framework nanozyme for boosting tumor ferroptosis through inhibiting DNA damage repair and system xc(). J Nanobiotechnolo. (2024) 22:228. doi: 10.1186/s12951-024-02508-2
43. Huang X, Yang X, Zhang M, Li T, Zhu K, Dong Y, et al. Selenoi functions as a key modulator of ferroptosis pathway in colitis and colorectal cancer. Adv Sci (Weinh). (2024) 11:e2404073. doi: 10.1002/advs.202404073
44. Wang Y, Huang X, Luo G, Xu Y, Deng X, Lin Y, et al. The aging lung: microenvironment, mechanisms, and diseases. Front Immunol. (2024) 15:1383503. doi: 10.3389/fimmu.2024.1383503
45. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
46. Xiong M, Ou C, Yu C, Qiu J, Lu J, Fu C, et al. Qi-shen-tang alleviates retinitis pigmentosa by inhibiting ferroptotic features via the nrf2/gpx4 signaling pathway. Heliyon. (2023) 9:e22443. doi: 10.1016/j.heliyon.2023.e22443
47. Lu L, Oveson BC, Jo YJ, Lauer TW, Usui S, Komeima K, et al. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid Redox Signal. (2009) 11:715–24. doi: 10.1089/ars.2008.2171
48. Azuma K, Koumura T, Iwamoto R, Matsuoka M, Terauchi R, Yasuda S, et al. Mitochondrial glutathione peroxidase 4 is indispensable for photoreceptor development and survival in mice. J Biol Chem. (2022) 298:101824. doi: 10.1016/j.jbc.2022.101824
49. Niu T, Shi X, Liu X, Wang H, Liu K, and Xu Y. Porous se@Sio(2) nanospheres alleviate diabetic retinopathy by inhibiting excess lipid peroxidation and inflammation. Mol Med (Cambridge Mass). (2024) 30:24. doi: 10.1186/s10020-024-00785-z
50. Yang N, Pan X, Zhou X, Liu Z, Yang J, Zhang J, et al. Biomimetic nanoarchitectonics with chitosan nanogels for collaborative induction of ferroptosis and anticancer immunity for cancer therapy. Adv Healthcare Mat. (2024) 13:e2302752. doi: 10.1002/adhm.202302752
51. Chen H, Wang C, Liu Z, He X, Tang W, He L, et al. Ferroptosis and its multifaceted role in cancer: mechanisms and therapeutic approach. Antioxid (Basel Switzerland). (2022) 11:1504. doi: 10.3390/antiox11081504
52. Nakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of fsp1 promotes ferroptosis. Nature. (2023) 619:371–7. doi: 10.1038/s41586-023-06255-6
53. Yang M, Tsui MG, Tsang JKW, Goit RK, Yao KM, So KF, et al. Involvement of fsp1-coq(10)-nadh and gsh-gpx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. (2022) 13:468. doi: 10.1038/s41419-022-04924-4
54. Liu T, Zhao J, and Lin C. Sprouty-related proteins with evh1 domain (Spred2) prevents high-glucose induced endothelial-mesenchymal transition and endothelial injury by suppressing mapk activation. Bioengineered. (2022) 13:13882–92. doi: 10.1080/21655979.2022.2086351
55. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. Dhodh-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
56. Wu MF, Peng X, Zhang MC, Guo H, and Xie HT. Ferroptosis and panoptosis under hypoxia pivoting on the crosstalk between dhodh and gpx4 in corneal epithelium. Free Radic Biol Med. (2025) 228:173–82. doi: 10.1016/j.freeradbiomed.2024.12.050
57. Schmidl D, Hommer N, Kallab M, Schlatter A, Nadvornik C, Obermayr F, et al. Safety and tolerability of kio-101 eye drops in healthy volunteers and patients with ocular surface disease-a phase I study. Pharmaceutics. (2024) 16:367. doi: 10.3390/pharmaceutics16030367
58. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. Gtp cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
59. Lohmann K, Redin C, Tönnies H, Bressman SB, Subero JIM, Wiegers K, et al. Complex and dynamic chromosomal rearrangements in a family with seemingly non-mendelian inheritance of dopa-responsive dystonia. JAMA Neurol. (2017) 74:806–12. doi: 10.1001/jamaneurol.2017.0666
60. Cai B, Cai JP, Luo YL, Chen C, and Zhang S. The specific roles of jak/stat signaling pathway in sepsis. Inflammation. (2015) 38:1599–608. doi: 10.1007/s10753-015-0135-z
61. Chen Y, Fang ZM, Yi X, Wei X, and Jiang DS. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. (2023) 14:205. doi: 10.1038/s41419-023-05716-0
62. Moutsopoulos HM. Sjögren's syndrome: autoimmune epithelitis. Clin Immunol Immunopathol. (1994) 72:162–5. doi: 10.1006/clin.1994.1123
63. Gandolfo S and Ciccia F. Jak/stat pathway targeting in primary sjögren syndrome. Rheumatol Immunol Res. (2022) 3:95–102. doi: 10.2478/rir-2022-0017
64. Zhang Q, Lenardo MJ, and Baltimore D. 30 years of nf-κb: A blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
65. Liang WJ, Yang HW, Liu HN, Qian W, and Chen XL. Hmgb1 upregulates nf-kb by inhibiting ikb-α and associates with diabetic retinopathy. Life Sci. (2020) 241:117146. doi: 10.1016/j.lfs.2019.117146
66. Palazzo I, Todd LJ, Hoang TV, Reh TA, Blackshaw S, and Fischer AJ. Nfkb-signaling promotes glial reactivity and suppresses müller glia-mediated neuron regeneration in the mammalian retina. Glia. (2022) 70:1380–401. doi: 10.1002/glia.24181
67. Jiang K, Zhang F, Chen Y, Li X, Zhao X, Jiang P, et al. Fosfenopril attenuates inflammatory response in diabetic dry eye models by inhibiting the tlr4/nf-κb/nlrp3 signaling pathway. Invest Ophthalmol Vis Sci. (2024) 65:2. doi: 10.1167/iovs.65.6.2
68. Pan S, Liu M, Xu H, Chuan J, and Yang Z. Lipopolysaccharide activating nf-kb signaling by regulates htra1 expression in human retinal pigment epithelial cells. Molecules. (2023) 28:2236. doi: 10.3390/molecules28052236
69. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-keap1-nrf2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatol (Baltimore Md). (2016) 63:173–84. doi: 10.1002/hep.28251
70. Wang D, Tang L, Zhang Y, Ge G, Jiang X, Mo Y, et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. (2022) 13:544. doi: 10.1038/s41419-022-04927-1
71. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a P53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
72. Suzuki T and Yamamoto M. Molecular basis of the keap1-nrf2 system. Free Radic Biol Med. (2015) 88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
73. Zhang H, Davies KJA, and Forman HJ. Oxidative stress response and nrf2 signaling in aging. Free Radic Biol Med. (2015) 88:314–36. doi: 10.1016/j.freeradbiomed.2015.05.036
74. Xu XR, Yu HT, Yang Y, Hang L, Yang XW, and Ding SH. Quercetin phospholipid complex significantly protects against oxidative injury in arpe-19 cells associated with activation of nrf2 pathway. Eur J Pharmacol. (2016) 770:1–8. doi: 10.1016/j.ejphar.2015.11.050
75. Xu Z, Wei Y, Gong J, Cho H, Park JK, Sung ER, et al. Nrf2 plays a protective role in diabetic retinopathy in mice. Diabetologia. (2014) 57:204–13. doi: 10.1007/s00125-013-3093-8
76. Batliwala S, Xavier C, Liu Y, Wu H, and Pang IH. Involvement of nrf2 in ocular diseases. Oxid Med Cell Longev. (2017) 2017:1703810. doi: 10.1155/2017/1703810
77. Von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergström P, et al. Nrf2-encoding nfe2l2 haplotypes influence disease progression but not risk in alzheimer's disease and age-related cataract. Mech Ageing Dev. (2010) 131:105–10. doi: 10.1016/j.mad.2009.12.007
78. Zhang YN, Ouyang WJ, Hu JY, and Liu ZG. Targeting nrf2 signaling in dry eye. Int J Ophthalmol. (2024) 17:1911–20. doi: 10.18240/ijo.2024.10.19
79. Yuan M, He Q, Xiang W, Deng Y, Lin S, and Zhang R. Natural compounds efficacy in ophthalmic diseases: A new twist impacting ferroptosis. BioMed Pharmacother. (2024) 172:116230. doi: 10.1016/j.biopha.2024.116230
80. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. Alox12 is required for P53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. (2019) 21:579–91. doi: 10.1038/s41556-019-0305-6
81. Galluzzi L, Bravo-San Pedro JM, and Kroemer G. Ferroptosis in P53-dependent oncosuppression and organismal homeostasis. Cell Death Differ. (2015) 22:1237–8. doi: 10.1038/cdd.2015.54
82. Xie T, Bai Z, Chen Z, Liang H, Liu T, Lam LK, et al. Inhibition of ferroptosis ameliorates hypertensive nephropathy through P53/nrf2/P21 pathway by taohongsiwu decoction: based on network pharmacology and experimental validation. J Ethnopharmacol. (2023) 312:116506. doi: 10.1016/j.jep.2023.116506
83. Wang S, Zhao Y, Yao F, Wei P, Ma L, and Zhang S. An anti-gd2 aptamer-based bifunctional spherical nucleic acid nanoplatform for synergistic therapy targeting mdm2 for retinoblastoma. Biomedicine Pharmacother = Biomedecine Pharmacother. (2024) 174:116437. doi: 10.1016/j.biopha.2024.116437
84. Chu WK, Choi HL, Bhat AK, and Jhanji V. Pterygium: new insights. Eye (Lond). (2020) 34:1047–50. doi: 10.1038/s41433-020-0786-3
85. Cheng Y, Zhang M, Xu R, Fu L, Xue M, Xu C, et al. P53 accelerates endothelial cell senescence in diabetic retinopathy by enhancing foxo3a ubiquitylation and degradation via ube2l6. Exp Gerontol. (2024) 188:112391. doi: 10.1016/j.exger.2024.112391
86. Zhang S, Wu J, Wang L, Mu L, Xu X, Li J, et al. Sirt1/P53 in retinal pigment epithelial cells in diabetic retinopathy: A gene co-expression analysis and he-ying-qing-re formula treatment. Front Mol Biosci. (2024) 11:1366020. doi: 10.3389/fmolb.2024.1366020
87. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated ampk activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
88. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. Ampk-mediated becn1 phosphorylation promotes ferroptosis by directly blocking system X(C)(-) activity. Curr Biol. (2018) 28:2388–99.e5. doi: 10.1016/j.cub.2018.05.094
89. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the rna-binding protein elavl1/hur to regulate ferroptosis in hepatic stellate cells. Autophagy. (2018) 14:2083–103. doi: 10.1080/15548627.2018.1503146
90. Fomo KN, Perumal N, Manicam C, Pfeiffer N, and Grus FH. Neuroretinal cell culture model as a tool for the development of new therapeutic approaches for oxidative stress-induced ocular diseases, with a focus on glaucoma. Cells. (2024) 13:775. doi: 10.3390/cells13090775
91. Dieguez HH, Romeo HE, Alaimo A, Bernal Aguirre NA, Calanni JS, Adán Aréan JS, et al. Mitochondrial quality control in non-exudative age-related macular degeneration: from molecular mechanisms to structural and functional recovery. Free Radical Biol Med. (2024) 219:17–30. doi: 10.1016/j.freeradbiomed.2024.03.024
92. Guo Y, Zhang H, Zhao Z, Luo X, Zhang M, Bu J, et al. Hyperglycemia induces meibomian gland dysfunction. Invest Ophthalmol Vis Sci. (2022) 63:30. doi: 10.1167/iovs.63.1.30
93. Peng F, Jiang D, Xu W, Sun Y, Zha Z, Tan X, et al. Ampk/mff activation: role in mitochondrial fission and mitophagy in dry eye. Invest Ophthalmol Vis Sci. (2022) 63:18. doi: 10.1167/iovs.63.12.18
94. Zhang Z, Jing J, Ye Y, Chen Z, Jing Y, Li S, et al. Characterization of the dual functional effects of heat shock proteins (Hsps) in cancer hallmarks to aid development of hsp inhibitors. Genome Med. (2020) 12:101. doi: 10.1186/s13073-020-00795-6
95. Feng Y, Chen Q, Jin C, Ruan Y, Chen Q, Lin W, et al. Microwave-activated cu-doped zirconium metal-organic framework for a highly effective combination of microwave dynamic and thermal therapy. J Controlled Release. (2023) 361:102–14. doi: 10.1016/j.jconrel.2023.07.046
96. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. Hspb1 as a novel regulator of ferroptotic cancer cell death. Oncogene. (2015) 34:5617–25. doi: 10.1038/onc.2015.32
97. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. Hspa5 regulates ferroptotic cell death in cancer cells. Cancer Res. (2017) 77:2064–77. doi: 10.1158/0008-5472.Can-16-1979
98. Zhou Y, Liao J, Mei Z, Liu X, and Ge J. Insight into crosstalk between ferroptosis and necroptosis: novel therapeutics in ischemic stroke. Oxid Med Cell Longev. (2021) 2021:9991001. doi: 10.1155/2021/9991001
99. Huang L, Hong Y, Fu X, Tan H, Chen Y, Wang Y, et al. The role of the microbiota in glaucoma. Mol Aspects Med. (2023) 94:101221. doi: 10.1016/j.mam.2023.101221
100. Saini C, Jiang S, Devlin J, Pan L, Tang Y, Tang J, et al. Association between hsp-specific T-cell counts and retinal nerve fiber layer thickness in patients with primary open-angle glaucoma. Ophthalmol Sci. (2023) 3:100310. doi: 10.1016/j.xops.2023.100310
101. Geyer O and Levo Y. Glaucoma is an autoimmune disease. Autoimmun Rev. (2020) 19:102535. doi: 10.1016/j.autrev.2020.102535
102. Tsai T, Grotegut P, Reinehr S, and Joachim SC. Role of heat shock proteins in glaucoma. Int J Mol Sci. (2019) 20:5160. doi: 10.3390/ijms20205160
103. O'Reilly AM, Currie RW, and Clarke DB. Hspb1 (Hsp 27) expression and neuroprotection in the retina. Mol Neurobiol. (2010) 42:124–32. doi: 10.1007/s12035-010-8143-3
104. Alven A, Lema C, and Redfern RL. Impact of low humidity on damage-associated molecular patterns at the ocular surface during dry eye disease. Optom Vis Sci. (2021) 98:1231–8. doi: 10.1097/opx.0000000000001802
105. Feng YQ, Liu X, Zuo N, Yu MB, Bian WM, Han BQ, et al. Nad(+) precursors promote the restoration of spermatogenesis in busulfan-treated mice through inhibiting sirt2-regulated ferroptosis. Theranostics. (2024) 14:2622–36. doi: 10.7150/thno.92416
106. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The coq oxidoreductase fsp1 acts parallel to gpx4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
107. Alves F, Lane D, Nguyen TPM, Bush AI, and Ayton S. In defence of ferroptosis. Signal Transduct Target Ther. (2025) 10:2. doi: 10.1038/s41392-024-02088-5
108. Liu N, Wu WL, Wan XR, Wang J, Huang JN, Jiang YY, et al. Regulation of fsp1 myristoylation by nadph: A novel mechanism for ferroptosis inhibition. Redox Biol. (2024) 73:103176. doi: 10.1016/j.redox.2024.103176
109. Liao J, Lai Z, Huang G, Lin J, Huang W, Qin Y, et al. Setanaxib mitigates oxidative damage following retinal ischemia-reperfusion via nox1 and nox4 inhibition in retinal ganglion cells. Biomedicine Pharmacother = Biomedecine Pharmacother. (2024) 170:116042. doi: 10.1016/j.biopha.2023.116042
110. Guo M, Liu T, Miao Y, Pan X, and Liu B. Role of nadph oxidase 4 on dry eye syndrome in mice. J Ocul Pharmacol Ther. (2024) 40:452–8. doi: 10.1089/jop.2024.0002
111. Shi X, Li P, Herb M, Liu H, Wang M, Wang X, et al. Pathological high intraocular pressure induces glial cell reactive proliferation contributing to neuroinflammation of the blood-retinal barrier via the nox2/et-1 axis-controlled erk1/2 pathway. J Neuroinflamm. (2024) 21:105. doi: 10.1186/s12974-024-03075-x
112. Liu Y, Wang W, Li Y, Xiao Y, Cheng J, and Jia J. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull. (2015) 38:1234–9. doi: 10.1248/bpb.b15-00048
113. Liu T, Shu J, Liu Y, Xie J, Li T, Li H, et al. Atorvastatin attenuates ferroptosis-dependent myocardial injury and inflammation following coronary microembolization via the hif1a/ptgs2 pathway. Front Pharmacol. (2022) 13:1057583. doi: 10.3389/fphar.2022.1057583
114. Sun Q, Liu D, Cui W, Cheng H, Huang L, Zhang R, et al. Cholesterol mediated ferroptosis suppression reveals essential roles of coenzyme Q and squalene. Commun Biol. (2023) 6:1108. doi: 10.1038/s42003-023-05477-8
115. Eberhard Y, McDermott SP, Wang X, Gronda M, Venugopal A, Wood TE, et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood. (2009) 114:3064–73. doi: 10.1182/blood-2009-03-209965
116. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3:232–43. doi: 10.1021/acscentsci.7b00028
117. Xiang X, Xu M, Liu L, Meng N, Lei Y, Feng Y, et al. Liproxstatin-1 attenuates acute hypertriglyceridemic pancreatitis through inhibiting ferroptosis in rats. Sci Rep. (2024) 14:9548. doi: 10.1038/s41598-024-60159-7
118. Zhou Y, Hu T, Zeng H, Lin L, Xie H, Lin R, et al. Naringenin inhibits ferroptosis in renal tubular epithelial cells of diabetic nephropathy through sirt1/foxo3a signaling pathway. Drug Dev Res. (2025) 86:e70044. doi: 10.1002/ddr.70044
119. Long Z, Yu X, Li S, Cheng N, Huo C, Zhang X, et al. Sakuranetin prevents acetaminophen-induced liver injury via nrf2-induced inhibition of hepatocyte ferroptosis. Drug Des Devel Ther. (2025) 19:159–71. doi: 10.2147/dddt.S497817
120. Tuo QZ, Masaldan S, Southon A, Mawal C, Ayton S, Bush AI, et al. Characterization of selenium compounds for anti-ferroptotic activity in neuronal cells and after cerebral ischemia-reperfusion injury. Neurotherapeutics. (2021) 18:2682–91. doi: 10.1007/s13311-021-01111-9
121. Zhang H, Wang Y, Wang S, Xue X, Huang K, Xu D, et al. Tangeretin alleviates sepsis-induced acute lung injury by inhibiting ferroptosis of macrophage via nrf2 signaling pathway. Chin Med. (2025) 20:11. doi: 10.1186/s13020-025-01063-8
122. Zhou X, Yang Y, Qiu X, Deng H, Cao H, Liao T, et al. Antioxidant taurine inhibits chondrocyte ferroptosis through upregulation of ogt/gpx4 signaling in osteoarthritis induced by anterior cruciate ligament transection. J Adv Res. (2025) 77:551–67. doi: 10.1016/j.jare.2025.01.010
123. Feng H and Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PloS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
124. Luo S, Zeng Y, Chen B, Yan J, Ma F, Zhuang G, et al. Vitamin E and gpx4 cooperatively protect treg cells from ferroptosis and alleviate intestinal inflammatory damage in necrotizing enterocolitis. Redox Biol. (2024) 75:103303. doi: 10.1016/j.redox.2024.103303
125. Wang J, Liu Y, Wang Y, and Sun L. The cross-link between ferroptosis and kidney diseases. Oxid Med Cell Longev. (2021) 2021:6654887. doi: 10.1155/2021/6654887
126. Li Y, Zhou Y, Liu D, Wang Z, Qiu J, Zhang J, et al. Glutathione peroxidase 3 induced mitochondria-mediated apoptosis via ampk /erk1/2 pathway and resisted autophagy-related ferroptosis via ampk/mtor pathway in hyperplastic prostate. J Trans Med. (2023) 21:575. doi: 10.1186/s12967-023-04432-9
127. Kerr JF. Shrinkage necrosis: A distinct mode of cellular death. J Pathol. (1971) 105:13–20. doi: 10.1002/path.1711050103
128. Liu S, Yao S, Yang H, Liu S, and Wang Y. Autophagy: regulator of cell death. Cell Death Dis. (2023) 14:648. doi: 10.1038/s41419-023-06154-8
129. Wang Z, Wang X, Dai X, Xu T, Qian X, Chang M, et al. 2d catalytic nanozyme enables cascade enzyodynamic effect-boosted and ca(2+) overload-induced synergistic ferroptosis/apoptosis in tumor. Adv Mater. (2024) 36:e2312316. doi: 10.1002/adma.202312316
v130. Li W, Shi J, Yu Z, Garcia-Gabilondo M, Held A, Huang L, et al. Slc22a17 as a cell death-linked regulator of tight junctions in cerebral ischemia. Stroke. (2024) 55:1650–9. doi: 10.1161/strokeaha.124.046736
131. Chen Z, Liu B, Zhou D, Lei M, Yang J, Hu Z, et al. Aqp4 regulates ferroptosis and oxidative stress of muller cells in diabetic retinopathy by regulating trpv4. Exp Cell Res. (2024) 439:114087. doi: 10.1016/j.yexcr.2024.114087
132. Qin Q, Yu N, Gu Y, Ke W, Zhang Q, Liu X, et al. Inhibiting multiple forms of cell death optimizes ganglion cells survival after retinal ischemia reperfusion injury. Cell Death Dis. (2022) 13:507. doi: 10.1038/s41419-022-04911-9
133. Otsu W, Ishida K, Chinen N, Nakamura S, Shimazawa M, Tsusaki H, et al. Cigarette smoke extract and heated tobacco products promote ferritin cleavage and iron accumulation in human corneal epithelial cells. Sci Rep. (2021) 11:18555. doi: 10.1038/s41598-021-97956-3
134. Newton K, Strasser A, Kayagaki N, and Dixit VM. Cell death. Cell. (2024) 187:235–56. doi: 10.1016/j.cell.2023.11.044
135. Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao C, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. (2022) 12:4310–29. doi: 10.7150/thno.71086
136. Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ros signaling to promote melanoma cell pyroptosis. Cell Res. (2018) 28:1171–85. doi: 10.1038/s41422-018-0090-y
137. Wang H, Shu L, Lv C, Liu N, Long Y, Peng X, et al. Brcc36 deubiquitinates hmgcr to regulate the interplay between ferroptosis and pyroptosis. Adv Sci (Weinh). (2024) 11:e2304263. doi: 10.1002/advs.202304263
138. Huang Y, Xu W, and Zhou R. Nlrp3 inflammasome activation and cell death. Cell Mol Immunol. (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
139. Meihe L, Shan G, Minchao K, Xiaoling W, Peng A, Xili W, et al. The ferroptosis-nlrp1 inflammasome: the vicious cycle of an adverse pregnancy. Front Cell Dev Biol. (2021) 9:707959. doi: 10.3389/fcell.2021.707959
140. Zou M, Ke Q, Nie Q, Qi R, Zhu X, Liu W, et al. Inhibition of cgas-sting by jq1 alleviates oxidative stress-induced retina inflammation and degeneration. Cell Death Differ. (2022) 29:1816–33. doi: 10.1038/s41418-022-00967-4
141. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the nlrp3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
142. Yan J, Wan P, Choksi S, and Liu ZG. Necroptosis and tumor progression. Trends Cancer. (2022) 8:21–7. doi: 10.1016/j.trecan.2021.09.003
143. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. (2020) 13:110. doi: 10.1186/s13045-020-00946-7
144. Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. (2022) 15:174. doi: 10.1186/s13045-022-01392-3
145. Tong Y, Wu Y, Ma J, Ikeda M, Ide T, Griffin CT, et al. Comparative mechanistic study of rpe cell death induced by different oxidative stresses. Redox Biol. (2023) 65:102840. doi: 10.1016/j.redox.2023.102840
146. Delehouzé C, Comte A, Leon-Icaza SA, Cougoule C, Hauteville M, Goekjian P, et al. Nigratine as dual inhibitor of necroptosis and ferroptosis regulated cell death. Sci Rep. (2022) 12:5118. doi: 10.1038/s41598-022-09019-w
147. Ma R, Li Y, Dong X, Zhang Y, Chen X, Zhang Y, et al. Pax6/cxcl14 regulatory axis promotes the repair of corneal injury by enhancing corneal epithelial cell proliferation. J Trans Med. (2024) 22:458. doi: 10.1186/s12967-024-05270-z
148. Sun R, Zhang J, Chen X, Deng Y, Gou J, Yin T, et al. An adaptive drug-releasing contact lens for personalized treatment of ocular infections and injuries. J Controlled Release. (2024) 369:114–27. doi: 10.1016/j.jconrel.2024.03.040
149. Liu Z, Liu K, Shi S, Chen X, Gu X, Wang W, et al. Alkali injury-induced pathological lymphangiogenesis in the iris facilitates the infiltration of T cells and ocular inflammation. JCI Insight. (2024) 9:e175479. doi: 10.1172/jci.insight.175479
150. Sakai O, Uchida T, Imai H, and Ueta T. Glutathione peroxidase 4 plays an important role in oxidative homeostasis and wound repair in corneal epithelial cells. FEBS Open Bio. (2016) 6:1238–47. doi: 10.1002/2211-5463.12141
151. Peng JJ, Song WT, Yao F, Zhang X, Peng J, Luo XJ, et al. Involvement of regulated necrosis in blinding diseases: focus on necroptosis and ferroptosis. Exp Eye Res. (2020) 191:107922. doi: 10.1016/j.exer.2020.107922
152. Liang N, Song W, and Li J. Bpa promotes lung fibrosis in mice by regulating autophagy-dependent ferroptosis in alveolar epithelial cells. Ecotoxicol Environ Saf. (2024) 278:116412. doi: 10.1016/j.ecoenv.2024.116412
153. Sharma N, Kaur M, Agarwal T, Sangwan VS, and Vajpayee RB. Treatment of acute ocular chemical burns. Surv Ophthalmol. (2018) 63:214–35. doi: 10.1016/j.survophthal.2017.09.005
154. Bizrah M, Yusuf A, and Ahmad S. An update on chemical eye burns. Eye (Lond). (2019) 33:1362–77. doi: 10.1038/s41433-019-0456-5
155. Witsberger EM and Patel SV. Alkali burn over a lasik flap. Cornea. (2021) 40:907–9. doi: 10.1097/ico.0000000000002604
156. Zhou H, Zhang W, Bi M, and Wu J. The molecular mechanisms of action of ppar-Γ Agonists in the treatment of corneal alkali burns (Review). Int J Mol Med. (2016) 38:1003–11. doi: 10.3892/ijmm.2016.2699
157. Welling JD, Pike EC, and Mauger TF. Alkali burn of the ocular surface associated with a commonly used antifog agent for eyewear: two cases and a review of previous reports. Cornea. (2016) 35:289–91. doi: 10.1097/ico.0000000000000706
158. Liu P, Wang W, Li Z, Li Y, Yu X, Tu J, et al. Ferroptosis: A new regulatory mechanism in osteoporosis. Oxid Med Cell Longev. (2022) 2022:2634431. doi: 10.1155/2022/2634431
159. Yang K, Zeng L, Yuan X, Wang S, Ge A, Xu H, et al. The mechanism of ferroptosis regulating oxidative stress in ischemic stroke and the regulation mechanism of natural pharmacological active components. BioMed Pharmacother. (2022) 154:113611. doi: 10.1016/j.biopha.2022.113611
160. Wang K, Jiang L, Zhong Y, Zhang Y, Yin Q, Li S, et al. Ferrostatin-1-loaded liposome for treatment of corneal alkali burn via targeting ferroptosis. Bioeng Transl Med. (2022) 7:e10276. doi: 10.1002/btm2.10276
161. Wang Z, Li H, Zhou W, Lee J, Liu Z, An Z, et al. Ferrous sulfate-loaded hydrogel cures staphylococcus aureus infection via facilitating a ferroptosis-like bacterial cell death in a mouse keratitis model. Biomaterials. (2022) 290:121842. doi: 10.1016/j.biomaterials.2022.121842
162. Teng X, Xiong X, Sha X, Lei Y, Diao Y, Liu J, et al. Identification of hub genes and pathways of ferroptosis in fusarium keratitis by bioinformatics methods. Front Cell infection Microbiol. (2023) 13:1103471. doi: 10.3389/fcimb.2023.1103471
163. Tavakoli A and Flanagan JL. Dry eye disease: an (in)Convenient truth. Clin Exp Optom. (2022) 105:222–9. doi: 10.1080/08164622.2021.1945410
164. Karlen SJ, Miller EB, and Burns ME. Microglia activation and inflammation during the death of mammalian photoreceptors. Annu Rev Vis Sci. (2020) 6:149–69. doi: 10.1146/annurev-vision-121219-081730
165. Yumnamcha T, Devi TS, and Singh LP. Auranofin mediates mitochondrial dysregulation and inflammatory cell death in human retinal pigment epithelial cells: implications of retinal neurodegenerative diseases. Front Neurosci. (2019) 13:1065. doi: 10.3389/fnins.2019.01065
166. Datta S, Cano M, Ebrahimi K, Wang L, and Handa JT. The impact of oxidative stress and inflammation on rpe degeneration in non-neovascular amd. Prog Retin Eye Res. (2017) 60:201–18. doi: 10.1016/j.preteyeres.2017.03.002
167. Golberg L, Martin LE, and Batchelor A. Biochemical changes in the tissues of animals injected with iron. 3. Lipid peroxidation. Biochem J. (1962) 83:291–8. doi: 10.1042/bj0830291
168. Lee JJ, Ishihara K, Notomi S, Efstathiou NE, Ueta T, Maidana D, et al. Lysosome-associated membrane protein-2 deficiency increases the risk of reactive oxygen species-induced ferroptosis in retinal pigment epithelial cells. Biochem Biophys Res Commun. (2020) 521:414–9. doi: 10.1016/j.bbrc.2019.10.138
169. Theriot CA, Westby CM, Morgan JLL, Zwart SR, and Zanello SB. High dietary iron increases oxidative stress and radiosensitivity in the rat retina and vasculature after exposure to fractionated gamma radiation. NPJ Microgravity. (2016) 2:16014. doi: 10.1038/npjmgrav.2016.14
170. Martin PM, Gnana-Prakasam JP, Roon P, Smith RG, Smith SB, and Ganapathy V. Expression and polarized localization of the hemochromatosis gene product hfe in retinal pigment epithelium. Invest Ophthalmol Vis Sci. (2006) 47:4238–44. doi: 10.1167/iovs.06-0026
171. Gnana-Prakasam JP, Veeranan-Karmegam R, Coothankandaswamy V, Reddy SK, Martin PM, Thangaraju M, et al. Loss of hfe leads to progression of tumor phenotype in primary retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2013) 54:63–71. doi: 10.1167/iovs.12-10312
172. Sterling J, Guttha S, Song Y, Song D, Hadziahmetovic M, and Dunaief JL. Iron importers zip8 and zip14 are expressed in retina and regulated by retinal iron levels. Exp Eye Res. (2017) 155:15–23. doi: 10.1016/j.exer.2016.12.008
173. Vogt AS, Arsiwala T, Mohsen M, Vogel M, Manolova V, and Bachmann MF. On iron metabolism and its regulation. Int J Mol Sci. (2021) 22:4591. doi: 10.3390/ijms22094591
174. Wysokinski D, Zaras M, Dorecka M, Waszczyk M, Szaflik J, Blasiak J, et al. An association between environmental factors and the ivs4+44c>a polymorphism of the dmt1 gene in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. (2012) 250:1057–65. doi: 10.1007/s00417-012-1966-z
175. Lee JJ, Chang-Chien GP, Lin S, Hsiao YT, Ke MC, Chen A, et al. 5-lipoxygenase inhibition protects retinal pigment epithelium from sodium iodate-induced ferroptosis and prevents retinal degeneration. Oxid Med Cell Longevity. (2022) 2022:1792894. doi: 10.1155/2022/1792894
176. Hernández-Zimbrón LF, Zamora-Alvarado R, Ochoa-De la Paz L, Velez-Montoya R, Zenteno E, Gulias-Cañizo R, et al. Age-related macular degeneration: new paradigms for treatment and management of amd. Oxid Med Cell Longev. (2018) 2018:8374647. doi: 10.1155/2018/8374647
177. Sun Y, Zheng Y, Wang C, and Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. (2018) 9:753. doi: 10.1038/s41419-018-0794-4
178. Lei G, Zhuang L, and Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
179. Wei TT, Zhang MY, Zheng XH, Xie TH, Wang W, Zou J, et al. Interferon-Γ Induces retinal pigment epithelial cell ferroptosis by a jak1-2/stat1/slc7a11 signaling pathway in age-related macular degeneration. FEBS J. (2022) 289:1968–83. doi: 10.1111/febs.16272
180. Liu H, Tang J, Du Y, Saadane A, Samuels I, Veenstra A, et al. Transducin1, phototransduction and the development of early diabetic retinopathy. Invest Ophthalmol Vis Sci. (2019) 60:1538–46. doi: 10.1167/iovs.18-26433
181. Ola MS, Alhomida AS, and LaNoue KF. Gabapentin attenuates oxidative stress and apoptosis in the diabetic rat retina. Neurotox Res. (2019) 36:81–90. doi: 10.1007/s12640-019-00018-w
182. Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, et al. α-lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301s tau transgenic mice. Redox Biol. (2018) 14:535–48. doi: 10.1016/j.redox.2017.11.001
183. Do MT and Yau KW. Intrinsically photosensitive retinal ganglion cells. Physiol Rev. (2010) 90:1547–81. doi: 10.1152/physrev.00013.2010
184. Parisi V, Oddone F, Ziccardi L, Roberti G, Coppola G, and Manni G. Citicoline and retinal ganglion cells: effects on morphology and function. Curr Neuropharmacol. (2018) 16:919–32. doi: 10.2174/1570159x15666170703111729
185. Luo X, Wang Y, Zhu X, Chen Y, Xu B, Bai X, et al. Mcl attenuates atherosclerosis by suppressing macrophage ferroptosis via targeting keap1/nrf2 interaction. Redox Biol. (2024) 69:102987. doi: 10.1016/j.redox.2023.102987
186. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
187. Yang Y, Wang Y, Guo L, Gao W, Tang TL, and Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. (2022) 13:355. doi: 10.1038/s41419-022-04775-z
188. Ma J, Zhang H, Chen Y, Liu X, Tian J, and Shen W. The role of macrophage iron overload and ferroptosis in atherosclerosis. Biomolecules. (2022) 12:1702. doi: 10.3390/biom12111702
189. Xu S, Min J, and Wang F. Ferroptosis: an emerging player in immune cells. Sci Bull (Beijing). (2021) 66:2257–60. doi: 10.1016/j.scib.2021.02.026
190. Sheng S, Zhang Y, Jin L, Sun W, Zhu D, Mei L, et al. A ferritin-targeted biohybrid triggering ferroptosis immunotherapy via activating endogenous iron and replenishing exogenous iron simultaneously. Nat Commun. (2025) 16:6045. doi: 10.1038/s41467-025-61419-4
191. Yang M, Chen X, Hu X, Li H, Huang H, Fang Y, et al. The nf-κb-slc7a11 axis regulates ferroptosis sensitivity in inflammatory macrophages. Cell Insight. (2025) 4:100257. doi: 10.1016/j.cellin.2025.100257
192. Xiao H, Du X, Tao Z, Jing N, Bao S, Gao WQ, et al. Taurine inhibits ferroptosis mediated by the crosstalk between tumor cells and tumor-associated macrophages in prostate cancer. Adv Sci (Weinh). (2024) 11:e2303894. doi: 10.1002/advs.202303894
193. Lu J, Lu F, Peng Z, Zhang Z, Jiang W, Meng X, et al. Clodronate liposome-mediated macrophage depletion ameliorates iron overload-induced dry eye disease. Exp Eye Res. (2025) 251:110204. doi: 10.1016/j.exer.2024.110204
194. Caligiuri MA. Human natural killer cells. Blood. (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
195. Valipour B, Velaei K, Abedelahi A, Karimipour M, Darabi M, and Charoudeh HN. Nk cells: an attractive candidate for cancer therapy. J Cell Physiol. (2019) 234:19352–65. doi: 10.1002/jcp.28657
196. Guillerey C. Nk cells in the tumor microenvironment. Adv Exp Med Biol. (2020) 1273:69–90. doi: 10.1007/978-3-030-49270-0_4
197. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring P16(Ink4a)-positive cells shorten healthy lifespan. Nature. (2016) 530:184–9. doi: 10.1038/nature16932
198. Lei G, Zhuang L, and Gan B. The roles of ferroptosis in cancer: tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell. (2024) 42:513–34. doi: 10.1016/j.ccell.2024.03.011
199. Kim R, Taylor D, Vonderheide RH, and Gabrilovich DI. Ferroptosis of immune cells in the tumor microenvironment. Trends Pharmacol Sci. (2023) 44:542–52. doi: 10.1016/j.tips.2023.06.005
200. Apte RS. Reducing treatment burden in amd. Cell. (2020) 180:1033. doi: 10.1016/j.cell.2020.02.028
201. Dong X, Song Y, Liu Y, Kou X, Yang T, Shi SX, et al. Natural killer cells promote neutrophil extracellular traps and restrain macular degeneration in mice. Sci Transl Med. (2024) 16:eadi6626. doi: 10.1126/scitranslmed.adi6626
202. Freud AG, Mundy-Bosse BL, Yu J, and Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
203. Lanzolla G, Marinò M, and Menconi F. Graves disease: latest understanding of pathogenesis and treatment options. Nat Rev Endocrinol. (2024) 20:647–60. doi: 10.1038/s41574-024-01016-5
204. Kulbay M, Tanya SM, Tuli N, Dahoud J, Dahoud A, Alsaleh F, et al. A comprehensive review of thyroid eye disease pathogenesis: from immune dysregulations to novel diagnostic and therapeutic approaches. Int J Mol Sci. (2024) 25:11628. doi: 10.3390/ijms252111628
205. Chapman NM, Boothby MR, and Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol. (2020) 20:55–70. doi: 10.1038/s41577-019-0203-y
206. Wik JA and Skålhegg BS. T cell metabolism in infection. Front Immunol. (2022) 13:840610. doi: 10.3389/fimmu.2022.840610
207. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. Cd8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
208. Xiang B, Zhang M, Li K, Zhang Z, Liu Y, Gao M, et al. The epitranscriptional factor pcif1 orchestrates cd8(+) T cell ferroptosis and activation to control antitumor immunity. Nat Immunol. (2025) 26:252–64. doi: 10.1038/s41590-024-02047-w
209. Bell HN, Stockwell BR, and Zou W. Ironing out the role of ferroptosis in immunity. Immunity. (2024) 57:941–56. doi: 10.1016/j.immuni.2024.03.019
210. Loi JK, Alexandre YO, Senthil K, Schienstock D, Sandford S, Devi S, et al. Corneal tissue-resident memory T cells form a unique immune compartment at the ocular surface. Cell Rep. (2022) 39:110852. doi: 10.1016/j.celrep.2022.110852
211. Bao X, Zhong Y, Yang C, Chen Y, Han Y, Lin X, et al. T-cell repertoire analysis in the conjunctiva of murine dry eye model. Invest Ophthalmol Vis Sci. (2023) 64:14. doi: 10.1167/iovs.64.3.14
212. Peng X, Li H, Zhu L, Zhao S, Li Z, Li S, et al. Single-cell sequencing of the retina shows that ldha regulates pathogenesis of autoimmune uveitis. J Autoimmun. (2024) 143:103160. doi: 10.1016/j.jaut.2023.103160
213. Woo KM, Mahrous MA, D'Amico DJ, Kiss S, and Kovacs KD. Prevalence of age-related macular degeneration in patients with chronic exposure to P2x7r inhibitors. Graefes Arch Clin Exp Ophthalmol. (2024) 262:3493–9. doi: 10.1007/s00417-024-06507-9
214. Sendecki A, Ledwoń D, Nycz J, Wąsowska A, Boguszewska-Chachulska A, Mitas AW, et al. A deep learning approach to explore the association of age-related macular degeneration polygenic risk score with retinal optical coherence tomography: A preliminary study. Acta Ophthalmol. (2024) 102:e1029–e39. doi: 10.1111/aos.16710
215. GBD 2019 Blindness and Vision Impairment Collaborators, Vision Loss Expert Group of the Global Burden of Disease Study. Trends in Prevalence of Blindness and Distance and near Vision Impairment over 30 Years: An Analysis for the Global Burden of Disease Study. Lancet Glob Health. (2021) 9:e130–e43. doi: 10.1016/s2214-109x(20)30425-3
216. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/s2214-109x(13)70145-1
217. Zarubina AV, Neely DC, Clark ME, Huisingh CE, Samuels BC, Zhang Y, et al. Prevalence of subretinal drusenoid deposits in older persons with and without age-related macular degeneration, by multimodal imaging. Ophthalmology. (2016) 123:1090–100. doi: 10.1016/j.ophtha.2015.12.034
218. Cabral De Guimaraes TA, Daich Varela M, Georgiou M, and Michaelides M. Treatments for dry age-related macular degeneration: therapeutic avenues, clinical trials and future directions. Br J Ophthalmol. (2022) 106:297–304. doi: 10.1136/bjophthalmol-2020-318452
219. Fleckenstein M, Schmitz-Valckenberg S, and Chakravarthy U. Age-related macular degeneration: A review. JAMA. (2024) 331:147–57. doi: 10.1001/jama.2023.26074
220. Adler R, Curcio C, Hicks D, Price D, and Wong F. Cell death in age-related macular degeneration. Mol Vis. (1999) 5:31.
221. Chen C, Chen J, Wang Y, Liu Z, and Wu Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J Biol Chem. (2021) 296:100187. doi: 10.1074/jbc.RA120.015779
222. Xiong L, Liu Y, Wang Y, Zhao H, Song X, Fan W, et al. The protective effect of lonicera japonica thunb. Against lipopolysaccharide-induced acute lung injury in mice: modulation of inflammation, oxidative stress, and ferroptosis. J Ethnopharmacol. (2024) 331:118333. doi: 10.1016/j.jep.2024.118333
223. Zhao X, Gao M, Liang J, Chen Y, Wang Y, Wang Y, et al. Slc7a11 reduces laser-induced choroidal neovascularization by inhibiting rpe ferroptosis and vegf production. Front Cell Dev Biol. (2021) 9:639851. doi: 10.3389/fcell.2021.639851
224. Martis RM, Knight LJ, Acosta ML, Black J, Ng R, Ji LCL, et al. Early onset of age-related changes in the retina of cystine/glutamate antiporter knockout mice. Exp Eye Res. (2023) 227:109364. doi: 10.1016/j.exer.2022.109364
225. Biesemeier A, Yoeruek E, Eibl O, and Schraermeyer U. Iron accumulation in bruch's membrane and melanosomes of donor eyes with age-related macular degeneration. Exp Eye Res. (2015) 137:39–49. doi: 10.1016/j.exer.2015.05.019
226. Ashok A, Chaudhary S, Wise AS, Rana NA, McDonald D, Kritikos AE, et al. Release of Iron-Loaded Ferritin in Sodium Iodate-Induced Model of Age Related Macular Degeneration: An in-Vitro and in-Vivo Study. Antioxid (Basel). (2021) 10:1253. doi: 10.3390/antiox10081253
227. Hahn P, Ying GS, Beard J, and Dunaief JL. Iron levels in human retina: sex difference and increase with age. Neuroreport. (2006) 17:1803–6. doi: 10.1097/WNR.0b013e3280107776
228. Nag TC, Kumar P, and Wadhwa S. Age related distribution of 4-hydroxy 2-nonenal immunoreactivity in human retina. Exp Eye Res. (2017) 165:125–35. doi: 10.1016/j.exer.2017.09.014
229. Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. (2008) 14:194–8. doi: 10.1038/nm1709
230. Song D, Song Y, Hadziahmetovic M, Zhong Y, and Dunaief JL. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic Biol Med. (2012) 53:64–71. doi: 10.1016/j.freeradbiomed.2012.04.020
231. Song D, Zhao L, Li Y, Hadziahmetovic M, Song Y, Connelly J, et al. The oral iron chelator deferiprone protects against systemic iron overload-induced retinal degeneration in hepcidin knockout mice. Invest Ophthalmol Vis Sci. (2014) 55:4525–32. doi: 10.1167/iovs.14-14568
232. Ueta T, Inoue T, Furukawa T, Tamaki Y, Nakagawa Y, Imai H, et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J Biol Chem. (2012) 287:7675–82. doi: 10.1074/jbc.M111.335174
233. Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, and Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. (2006) 27:983–93. doi: 10.1016/j.neurobiolaging.2005.05.012
234. Karunadharma PP, Nordgaard CL, Olsen TW, and Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Visual Sci. (2010) 51:5470–9. doi: 10.1167/iovs.10-5429
235. Guo J, Zhou Y, Liu D, Wang M, Wu Y, Tang D, et al. Mitochondria as multifaceted regulators of ferroptosis. Life Metab. (2022) 1:134–48. doi: 10.1093/lifemeta/loac035
236. Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, et al. Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci. (2015) 35:7304–11. doi: 10.1523/jneurosci.0190-15.2015
237. Wang S, Liu Y, Liu Y, Li C, Wan Q, Yang L, et al. Reversed senescence of retinal pigment epithelial cell by coculture with embryonic stem cell via the tgfβ and pi3k pathways. Front Cell Dev Biol. (2020) 8:588050. doi: 10.3389/fcell.2020.588050
238. Kim MH, Kwon SY, Woo SY, Seo WD, and Kim DY. Antioxidative effects of chrysoeriol via activation of the nrf2 signaling pathway and modulation of mitochondrial function. Molecules. (2021) 26:313. doi: 10.3390/molecules26020313
239. Tan W, Zou J, Yoshida S, Jiang B, and Zhou Y. The role of inflammation in age-related macular degeneration. Int J Biol Sci. (2020) 16:2989–3001. doi: 10.7150/ijbs.49890
240. Ananth S, Gnana-Prakasam JP, Bhutia YD, Veeranan-Karmegam R, Martin PM, Smith SB, et al. Regulation of the cholesterol efflux transporters abca1 and abcg1 in retina in hemochromatosis and by the endogenous siderophore 2,5-dihydroxybenzoic acid. Biochim Biophys Acta. (2014) 1842:603–12. doi: 10.1016/j.bbadis.2014.01.010
241. Gupta U, Ghosh S, Wallace CT, Shang P, Xin Y, Nair AP, et al. Increased lcn2 (Lipocalin 2) in the rpe decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry amd. Autophagy. (2023) 19:92–111. doi: 10.1080/15548627.2022.2062887
242. Li Y, Song D, Song Y, Zhao L, Wolkow N, Tobias JW, et al. Iron-induced local complement component 3 (C3) up-regulation via non-canonical transforming growth factor (Tgf)-β Signaling in the retinal pigment epithelium. J Biol Chem. (2015) 290:11918–34. doi: 10.1074/jbc.M115.645903
243. Song D, Kanu LN, Li Y, Kelly KL, Bhuyan RK, Aleman T, et al. Amd-like retinopathy associated with intravenous iron. Exp Eye Res. (2016) 151:122–33. doi: 10.1016/j.exer.2016.08.008
244. Liu B, Wang W, Shah A, Yu M, Liu Y, He L, et al. Sodium iodate induces ferroptosis in human retinal pigment epithelium arpe-19 cells. Cell Death Dis. (2021) 12:230. doi: 10.1038/s41419-021-03520-2
245. Tang Z, Ju Y, Dai X, Ni N, Liu Y, Zhang D, et al. Ho-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. (2021) 43:101971. doi: 10.1016/j.redox.2021.101971
246. Wei H, Chen C, Di F, Sun C, Wang X, Sun M, et al. Pm(2.5)-induced ferroptosis by nrf2/hmox1 signaling pathway led to inflammation in microglia. Environ pollut. (2024) 352:124130. doi: 10.1016/j.envpol.2024.124130
247. Yang Y, Wang Y, Deng Y, Lu J, Xiao L, Li J, et al. Fructus lycii and salvia miltiorrhiza bunge extract attenuate oxidative stress-induced photoreceptor ferroptosis in retinitis pigmentosa. BioMed Pharmacother. (2023) 167:115547. doi: 10.1016/j.biopha.2023.115547
248. Sohns C and Zabel M. Current role of amiodarone in antiarrhythmic therapy. Herzschrittmachertherapie Elektrophysiologie. (2010) 21:239–43. doi: 10.1007/s00399-010-0091-0
249. Chen Y, Okano K, Maeda T, Chauhan V, Golczak M, Maeda A, et al. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J Biol Chem. (2012) 287:5059–69. doi: 10.1074/jbc.M111.315432
250. Chen Z, Zhu X, Lu MM, Ou Q, Wang X, Zhao Z, et al. Phospho1 suppresses ferroptosis in retinal pigment epithelial cells by reducing the levels of phosphatidylethanolamine molecular species. Adv Sci (Weinh). (2025) 12:e2505359. doi: 10.1002/advs.202505359
251. Su W, Gao Y, Jia X, Chen X, Wu J, Wen Y, et al. Single-cell transcriptome atlas of spontaneous dry age-related macular degeneration in macaques. Fundam Res. (2025) 5:1034–46. doi: 10.1016/j.fmre.2023.02.028
252. Chaudhary K, Promsote W, Ananth S, Veeranan-Karmegam R, Tawfik A, Arjunan P, et al. Iron overload accelerates the progression of diabetic retinopathy in association with increased retinal renin expression. Sci Rep. (2018) 8:3025. doi: 10.1038/s41598-018-21276-2
253. López IM, Díez A, Velilla S, Rueda A, Alvarez A, and Pastor CJ. Prevalence of diabetic retinopathy and eye care in a rural area of Spain. Ophthalmic Epidemiol. (2002) 9:205–14. doi: 10.1076/opep.9.3.205.1516
254. Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, and Grosu P. Expression of acute-phase response proteins in retinal müller cells in diabetes. Invest Ophthalmol Vis Sci. (2005) 46:349–57. doi: 10.1167/iovs.04-0860
255. Konerirajapuram NS, Coral K, Punitham R, Sharma T, Kasinathan N, and Sivaramakrishnan R. Trace elements iron, copper and zinc in vitreous of patients with various vitreoretinal diseases. Indian J Ophthalmol. (2004) 52:145–8.
256. Zhang D, Lv FL, and Wang GH. Effects of hif-1α on diabetic retinopathy angiogenesis and vegf expression. Eur Rev Med Pharmacol Sci. (2018) 22:5071–6. doi: 10.26355/eurrev_201808_15699
257. Yang X, Huo F, Liu B, Liu J, Chen T, Li J, et al. Crocin inhibits oxidative stress and pro-inflammatory response of microglial cells associated with diabetic retinopathy through the activation of pi3k/akt signaling pathway. J Mol Neurosci. (2017) 61:581–9. doi: 10.1007/s12031-017-0899-8
258. Peters J. Cytosolic (Pro)Renin and the matter of intracellular renin actions. Front Biosci (Schol Ed). (2013) 5:198–205. doi: 10.2741/s366
259. Abcouwer SF. Angiogenic factors and cytokines in diabetic retinopathy. J Clin Cell Immunol. (2013) 1:1–12. doi: 10.4172/2155-9899
260. Saxena R, Madhu SV, Shukla R, Prabhu KM, and Gambhir JK. Postprandial hypertriglyceridemia and oxidative stress in patients of type 2 diabetes mellitus with macrovascular complications. Clinica Chimica Acta; Int J Clin Chem. (2005) 359:101–8. doi: 10.1016/j.cccn.2005.03.036
261. Bone RN, Oyebamiji O, Talware S, Selvaraj S, Krishnan P, Syed F, et al. A computational approach for defining a signature of β-cell golgi stress in diabetes. Diabetes. (2020) 69:2364–76. doi: 10.2337/db20-0636
262. Zhang J, Qiu Q, Wang H, Chen C, and Luo D. Trim46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating gpx4 ubiquitination. Exp Cell Res. (2021) 407:112800. doi: 10.1016/j.yexcr.2021.112800
263. Mu L, Wang D, Dong Z, Wu J, Wu X, Su J, et al. Abnormal levels of serum ferroptosis-related biomarkers in diabetic retinopathy. J Ophthalmol. (2022) 2022:3353740. doi: 10.1155/2022/3353740
264. Yang J and Liu Z. Mechanistic pathogenesis of endothelial dysfunction in diabetic nephropathy and retinopathy. Front Endocrinol (Lausanne). (2022) 13:816400. doi: 10.3389/fendo.2022.816400
265. Fan X, Xu M, Ren Q, Fan Y, Liu B, Chen J, et al. Downregulation of fatty acid binding protein 4 alleviates lipid peroxidation and oxidative stress in diabetic retinopathy by regulating peroxisome proliferator-activated receptor Γ-mediated ferroptosis. Bioengineered. (2022) 13:10540–51. doi: 10.1080/21655979.2022.2062533
266. Cheloni R, Gandolfi SA, Signorelli C, and Odone A. Global prevalence of diabetic retinopathy: protocol for a systematic review and meta-analysis. BMJ Open. (2019) 9:e022188. doi: 10.1136/bmjopen-2018-022188
267. Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, and Westermark GT. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and alzheimer disease. Am J Pathol. (2015) 185:834–46. doi: 10.1016/j.ajpath.2014.11.016
268. Zhang K, Wang T, Sun GF, Xiao JX, Jiang LP, Tou FF, et al. Metformin Protects against Retinal Ischemia/Reperfusion Injury through Ampk-Mediated Mitochondrial Fusion. Free Radic Biol Med. (2023) 205:47–61. doi: 10.1016/j.freeradbiomed.2023.05.019
269. Chandrasekaran PR and Madanagopalan VG. Role of curcumin in retinal diseases-a review. Graefes Arch Clin Exp Ophthalmol. (2022) 260:1457–73. doi: 10.1007/s00417-021-05542-0
270. Mathew B, Ravindran S, Liu X, Torres L, Chennakesavalu M, Huang CC, et al. Mesenchymal stem cell-derived extracellular vesicles and retinal ischemia-reperfusion. Biomaterials. (2019) 197:146–60. doi: 10.1016/j.biomaterials.2019.01.016
271. Rahimi M, Leahy S, Matei N, Burford J, Blair NP, and Shahidi M. Impairments of retinal hemodynamics and oxygen metrics in ocular hypertension-induced ischemia-reperfusion. Exp Eye Res. (2022) 225:109278. doi: 10.1016/j.exer.2022.109278
272. Lin LT, Chen JT, Tai MC, Chen YH, Chen CL, Pao SI, et al. Protective effects of hypercapnic acidosis on ischemia-reperfusion-induced retinal injury. PloS One. (2019) 14:e0211185. doi: 10.1371/journal.pone.0211185
273. Abcouwer SF, Shanmugam S, Muthusamy A, Lin CM, Kong D, Hager H, et al. Inflammatory resolution and vascular barrier restoration after retinal ischemia reperfusion injury. J Neuroinflamm. (2021) 18:186. doi: 10.1186/s12974-021-02237-5
274. Wang X, Li M, Diao K, Wang Y, Chen H, Zhao Z, et al. Deferoxamine attenuates visual impairment in retinal ischemia–Reperfusion via inhibiting ferroptosis. Sci Rep. (2023) 13:20145. doi: 10.1038/s41598-023-46104-0
275. Chen B, Caballero S, Seo S, Grant MB, and Lewin AS. Delivery of antioxidant enzyme genes to protect against ischemia/reperfusion-induced injury to retinal microvasculature. Invest Ophthalmol Vis Sci. (2009) 50:5587–95. doi: 10.1167/iovs.09-3633
276. Gehlbach P and Purple RL. Enhancement of retinal recovery by conjugated deferoxamine after ischemia-reperfusion. Invest Ophthalmol Vis Sci. (1994) 35:669–76.
277. Balla A, Tran B, Valtari A, Steven P, Scarpellini C, Augustyns K, et al. A novel ferroptosis inhibitor uamc-3203, a potential treatment for corneal epithelial wound. Pharmaceutics. (2022) 15:118. doi: 10.3390/pharmaceutics15010118
278. Li Y, Wen Y, Liu X, Li Z, Lin B, Deng C, et al. Single-cell rna sequencing reveals a landscape and targeted treatment of ferroptosis in retinal ischemia/reperfusion injury. J Neuroinflamm. (2022) 19:261. doi: 10.1186/s12974-022-02621-9
279. Lu Y, Zhu F, Zhou X, Li Y, Rong G, Liu N, et al. A supramolecular deferoxamine-crisaborole nanoparticle targets ferroptosis, inflammation, and oxidative stress in the treatment of retinal ischemia/reperfusion injury. Nano Lett. (2025) 25:1058–66. doi: 10.1021/acs.nanolett.4c05012
280. Ni Y, Hu Y, Zhu L, Jiang X, Zhang H, Liu J, et al. Lycium barbarum polysaccharide-derived nanoparticles protect visual function by inhibiting rgc ferroptosis and microglial activation in retinal ischemia–Reperfusion mice. Adv Healthc Mater. (2024) 13:e2304285. doi: 10.1002/adhm.202304285
281. Deleon E, Lederman M, Berenstein E, Meir T, Chevion M, and Chowers I. Alteration in iron metabolism during retinal degeneration in rd10 mouse. Invest Ophthalmol Vis Sci. (2009) 50:1360–5. doi: 10.1167/iovs.08-1856
282. Yefimova MG, Jeanny JC, Keller N, Sergeant C, Guillonneau X, Beaumont C, et al. Impaired retinal iron homeostasis associated with defective phagocytosis in royal college of surgeons rats. Invest Ophthalmol Vis Sci. (2002) 43:537–45.
283. Obolensky A, Berenshtein E, Lederman M, Bulvik B, Alper-Pinus R, Yaul R, et al. Zinc-desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radic Biol Med. (2011) 51:1482–91. doi: 10.1016/j.freeradbiomed.2011.07.014
284. Wang K, Peng B, Xiao J, Weinreb O, Youdim MBH, and Lin B. Iron-chelating drugs enhance cone photoreceptor survival in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. (2017) 58:5287–97. doi: 10.1167/iovs.17-22096
285. Rogers BS, Symons RC, Komeima K, Shen J, Xiao W, Swaim ME, et al. Differential sensitivity of cones to iron-mediated oxidative damage. Invest Ophthalmol Vis Sci. (2007) 48:438–45. doi: 10.1167/iovs.06-0528
286. Zeng Z, Gao ZL, Zhang ZP, Jiang HB, Yang CQ, Yang J, et al. Downregulation of cks1b restrains the proliferation, migration, invasion and angiogenesis of retinoblastoma cells through the mek/erk signaling pathway. Int J Mol Med. (2024) 54:58. doi: 10.3892/ijmm.2024.5382
287. Ancona-Lezama D, Dalvin LA, and Shields CL. Modern treatment of retinoblastoma: A 2020 review. Indian J Ophthalmol. (2020) 68:2356–65. doi: 10.4103/ijo.IJO_721_20
288. Peeler CE and Gonzalez E. Retinoblastoma. N Engl J Med. (2022) 386:2412. doi: 10.1056/NEJMicm2118356
289. Kuganesan N, Dlamini S, Tillekeratne LMV, and Taylor WR. Tumor suppressor P53 promotes ferroptosis in oxidative stress conditions independent of modulation of ferroptosis by P21, cdks, rb, and E2f. J Biol Chem. (2021) 297:101365. doi: 10.1016/j.jbc.2021.101365
290. Liu K, Huang J, Liu J, Klionsky DJ, Kang R, and Tang D. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. (2022) 13:521. doi: 10.1038/s41419-022-04974-8
291. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. (2022) 8:43. doi: 10.1038/s41420-021-00807-3
292. Liu H, Gan Q, Lai Y, Pan Z, Jin Q, Li J, et al. Usp14 increases the sensitivity of retinoblastoma to cisplatin by mediating the ferroptosis. Naunyn Schmiedebergs Arch Pharmacol. (2024) 397:8671–80. doi: 10.1007/s00210-024-03174-9
293. Gong D, Wu N, Chen H, Zhang W, Yan C, Zhang C, et al. Phytic acid-loaded polyvinyl alcohol hydrogel promotes wound healing of injured corneal epithelium through inhibiting ferroptosis. Redox Biol. (2024) 76:103354. doi: 10.1016/j.redox.2024.103354
294. Gomes JA, Tan D, Rapuano CJ, Belin MW, Ambrósio R Jr., Guell JL, et al. Global consensus on keratoconus and ectatic diseases. Cornea. (2015) 34:359–69. doi: 10.1097/ico.0000000000000408
295. Jones-Jordan LA, Walline JJ, Sinnott LT, Kymes SM, and Zadnik K. Asymmetry in keratoconus and vision-related quality of life. Cornea. (2013) 32:267–72. doi: 10.1097/ICO.0b013e31825697c4
296. Cai Y, Zhou T, Cai X, Shi W, Sun H, and Fu Y. Deciphering mitochondrial dysfunction in keratoconus: insights into acsl4 from machine learning-based bulk and single-cell transcriptome analyses and experimental validation. Comput Struct Biotechnol J. (2025) 27:1962–74. doi: 10.1016/j.csbj.2025.05.013
297. Wang H, Song F, Feng J, Qi X, Ma L, Xie L, et al. Tannin coordinated nanozyme composite-based hybrid hydrogel eye drops for prophylactic treatment of multidrug-resistant pseudomonas aeruginosa keratitis. J Nanobiotechnolo. (2022) 20:445. doi: 10.1186/s12951-022-01653-w
298. Jadi PK, Dave A, Issa R, Tabbasum K, Okurowska K, Samarth A, et al. Tetraspanin cd9-derived peptides inhibit pseudomonas aeruginosa corneal infection and aid in wound healing of corneal epithelial cells. Ocul Surf. (2024) 32:211–8. doi: 10.1016/j.jtos.2023.07.001
299. Chen Q, Wang L, Wei Y, Xu X, Guo X, and Liang Q. Ferroptosis as a potential therapeutic target for reducing inflammation and corneal scarring in bacterial keratitis. Invest Ophthalmol Vis Sci. (2024) 65:29. doi: 10.1167/iovs.65.2.29
300. Craig JP, Nichols KK, Akpek EK, Caffery B, Dua HS, Joo CK, et al. Tfos dews ii definition and classification report. Ocul Surf. (2017) 15:276–83. doi: 10.1016/j.jtos.2017.05.008
301. Jones L, Downie LE, Korb D, Benitez-Del-Castillo JM, Dana R, Deng SX, et al. Tfos dews ii management and therapy report. Ocul Surf. (2017) 15:575–628. doi: 10.1016/j.jtos.2017.05.006
302. Bron AJ, De Paiva CS, Chauhan SK, Bonini S, Gabison EE, Jain S, et al. Tfos dews ii pathophysiology report. Ocul Surf. (2017) 15:438–510. doi: 10.1016/j.jtos.2017.05.011
303. Mi Y, Wei C, Sun L, Liu H, Zhang J, Luo J, et al. Melatonin inhibits ferroptosis and delays age-related cataract by regulating sirt6/P-nrf2/gpx4 and sirt6/ncoa4/fth1 pathways. BioMed Pharmacother. (2023) 157:114048. doi: 10.1016/j.biopha.2022.114048
304. Zuo X, Zeng H, Wang B, Yang X, He D, Wang L, et al. Akr1c1 protects corneal epithelial cells against oxidative stress-mediated ferroptosis in dry eye. Invest Ophthalmol Vis Sci. (2022) 63:3. doi: 10.1167/iovs.63.10.3
305. Cheng YH, Huang HP, and Chen HH. Mucoadhesive phenylboronic acid-grafted carboxymethyl cellulose hydrogels containing glutathione for treatment of corneal epithelial cells exposed to benzalkonium chloride. Colloids Surf B Biointerfaces. (2024) 238:113884. doi: 10.1016/j.colsurfb.2024.113884
306. Zhang Y, Zhou T, Wang K, Luo C, Chen D, Lv Z, et al. Corneal mucin-targeting liposome nanoplatforms enable effective treatment of dry eye diseases by integrated regulation of ferroptosis and inflammation. Adv Sci (Weinh). (2025) 12:e2411172. doi: 10.1002/advs.202411172
307. Wang B, Zeng H, Zhao X, Zuo X, Yang X, Wang L, et al. D609-polymer-based delivery strategy targeting ferroptosis in treatment of dry eye disease. Chem Eng J. (2025) 503:158050. doi: 10.1016/j.cej.2024.158050
308. Jayaram H, Kolko M, Friedman DS, and Gazzard G. Glaucoma: now and beyond. Lancet. (2023) 402:1788–801. doi: 10.1016/s0140-6736(23)01289-8
309. Weinreb RN, Aung T, and Medeiros FA. The pathophysiology and treatment of glaucoma: A review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
310. Kang JM and Tanna AP. Glaucoma. Med Clin North Am. (2021) 105:493–510. doi: 10.1016/j.mcna.2021.01.004
311. Youale J, Bigot K, Kodati B, Jaworski T, Fan Y, Nsiah NY, et al. Neuroprotective effects of transferrin in experimental glaucoma models. Int J Mol Sci. (2022) 23:12753. doi: 10.3390/ijms232112753
312. Ramdas WD. The relation between dietary intake and glaucoma: A systematic review. Acta Ophthalmol. (2018) 96:550–6. doi: 10.1111/aos.13662
313. Stein JD, Khawaja AP, and Weizer JS. Glaucoma in adults-screening, diagnosis, and management: A review. JAMA. (2021) 325:164–74. doi: 10.1001/jama.2020.21899
314. Yao F, Peng J, Zhang E, Ji D, Gao Z, Tang Y, et al. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell Death Differ. (2023) 30:69–81. doi: 10.1038/s41418-022-01046-4
315. Hoelzgen F, Nguyen TTP, Klukin E, Boumaiza M, Srivastava AK, Kim EY, et al. Structural basis for the intracellular regulation of ferritin degradation. Nat Commun. (2024) 15:3802. doi: 10.1038/s41467-024-48151-1
316. Chen Y, Khan RS, Cwanger A, Song Y, Steenstra C, Bang S, et al. Dexras1, a small gtpase, is required for glutamate-nmda neurotoxicity. J Neurosci. (2013) 33:3582–7. doi: 10.1523/jneurosci.1497-12.2013
317. Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE 3rd, Papadopoulos V, et al. Nmda receptor-nitric oxide transmission mediates neuronal iron homeostasis via the gtpase dexras1. Neuron. (2006) 51:431–40. doi: 10.1016/j.neuron.2006.07.011
318. Sakamoto K, Suzuki T, Takahashi K, Koguchi T, Hirayama T, Mori A, et al. Iron-chelating agents attenuate nmda-induced neuronal injury via reduction of oxidative stress in the rat retina. Exp Eye Res. (2018) 171:30–6. doi: 10.1016/j.exer.2018.03.008
319. Cui QN, Bargoud AR, Ross AG, Song Y, and Dunaief JL. Oral administration of the iron chelator deferiprone protects against loss of retinal ganglion cells in a mouse model of glaucoma. Exp Eye Res. (2020) 193:107961. doi: 10.1016/j.exer.2020.107961
320. Ashok A, Singh N, Chaudhary S, Bellamkonda V, Kritikos AE, Wise AS, et al. Retinal degeneration and alzheimer's disease: an evolving link. Int J Mol Sci. (2020) 21:7290. doi: 10.3390/ijms21197290
321. Liu HT, Zhang Q, Jiang ZX, Xu YX, Wan QQ, and Tao LM. Efficacy and safety of high-dose ultrasound cyclo-plasty procedure in refractory glaucoma. Int J Ophthalmol. (2020) 13:1391–6. doi: 10.18240/ijo.2020.09.09
322. Tang J, Zhuo Y, and Li Y. Effects of iron and zinc on mitochondria: potential mechanisms of glaucomatous injury. Front Cell Dev Biol. (2021) 9:720288. doi: 10.3389/fcell.2021.720288
323. Guo M, Zhu Y, Shi Y, Meng X, Dong X, Zhang H, et al. Inhibition of ferroptosis promotes retina ganglion cell survival in experimental optic neuropathies. Redox Biol. (2022) 58:102541. doi: 10.1016/j.redox.2022.102541
324. Thompson J and Lakhani N. Cataracts. Prim Care. (2015) 42:409–23. doi: 10.1016/j.pop.2015.05.012
325. Asbell PA, Dualan I, Mindel J, Brocks D, Ahmad M, and Epstein S. Age-related cataract. Lancet (London England). (2005) 365:599–609. doi: 10.1016/s0140-6736(05)17911-2
326. Bhuyan KC, Master RW, Coles RS, and Bhuyan DK. Molecular mechanisms of cataractogenesis: iv. Evidence of phospholipid. Malondialdehyde adduct in human senile cataract. Mech Ageing Dev. (1986) 34:289–96. doi: 10.1016/0047-6374(86)90080-1
327. Huang L, Estrada R, Yappert MC, and Borchman D. Oxidation-induced changes in human lens epithelial cells. 1. Phospholipids. Free Radic Biol Med. (2006) 41:1425–32. doi: 10.1016/j.freeradbiomed.2006.07.022
328. Reddy VN, Giblin FJ, Lin LR, Dang L, Unakar NJ, Musch DC, et al. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest Ophthalmol Vis Sci. (2001) 42:3247–55.
329. Wei Z, Hao C, Huangfu J, Srinivasagan R, Zhang X, and Fan X. Aging lens epithelium is susceptible to ferroptosis. Free Radic Biol Med. (2021) 167:94–108. doi: 10.1016/j.freeradbiomed.2021.02.010
330. Babizhayev MA. Failure to withstand oxidative stress induced by phospholipid hydroperoxides as a possible cause of the lens opacities in systemic diseases and ageing. Biochim Biophys Acta. (1996) 1315:87–99. doi: 10.1016/0925-4439(95)00091-7
331. Hope-Ross M, Mahon GJ, and Johnston PB. Ocular siderosis. Eye (Lond). (1993) 7:419–25. doi: 10.1038/eye.1993.83
332. Guo D, Du Y, Liu X, Li D, Wei L, and Zhu X. Enhanced ferroptosis sensitivity promotes the formation of highly myopic cataract via the ddr2-hippo pathway. Cell Death Dis. (2025) 16:64. doi: 10.1038/s41419-025-07384-8
333. Zhang Y, Si W, Mao Y, Xu S, Li F, Liu J, et al. Upregulation of ferroptosis in glucocorticoids-induced posterior subcapsular cataracts. Commun Biol. (2025) 8:613. doi: 10.1038/s42003-025-08067-y
334. Fang S, Lu Y, Huang Y, Zhou H, and Fan X. Mechanisms that underly T cell immunity in graves' Orbitopathy. Front Endocrinol (Lausanne). (2021) 12:648732. doi: 10.3389/fendo.2021.648732
335. Mishra S, Maurya VK, Kumar S, Ankita Kaur A, and Saxena SK. Clinical management and therapeutic strategies for the thyroid-associated ophthalmopathy: current and future perspectives. Curr Eye Res. (2020) 45:1325–41. doi: 10.1080/02713683.2020.1776331
336. Bartalena L, Piantanida E, Gallo D, Lai A, and Tanda ML. Epidemiology, natural history, risk factors, and prevention of graves' Orbitopathy. Front Endocrinol (Lausanne). (2020) 11:615993. doi: 10.3389/fendo.2020.615993
337. Tian X, Li N, Su R, Dai C, and Zhang R. Selenium supplementation may decrease thyroid peroxidase antibody titer via reducing oxidative stress in euthyroid patients with autoimmune thyroiditis. Int J Endocrinol. (2020) 2020:9210572. doi: 10.1155/2020/9210572
338. Negro R, Hegedüs L, Attanasio R, and Papini E. Winther KH. A 2018 european thyroid association survey on the use of selenium supplementation in graves' Hyperthyroidism and graves' Orbitopathy. Eur Thyroid J. (2019) 8:7–15. doi: 10.1159/000494837
339. Douglas RS, Kahaly GJ, Patel A, Sile S, Thompson EHZ, Perdok R, et al. Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med. (2020) 382:341–52. doi: 10.1056/NEJMoa1910434
340. Tsai CC, Wu SB, Cheng CY, Kao SC, Kau HC, Lee SM, et al. Increased response to oxidative stress challenge in graves' Ophthalmopathy orbital fibroblasts. Mol Vis. (2011) 17:2782–8.
341. Ma C, Li H, Liu W, Lu S, Li X, Chen J, et al. Therapeutic effect of gypenosides on antioxidant stress injury in orbital fibroblasts of graves' Orbitopathy. J Immunol Res. (2022) 2022:4432584. doi: 10.1155/2022/4432584
342. Gao Y and Li W. Mechanisms of immune-related differentially expressed genes in thyroid-associated ophthalmopathy based on the geo database. Ann Transl Med. (2022) 10:926. doi: 10.21037/atm-22-3470
343. Shin HR, Cho WK, Baek IC, Lee NY, Lee YJ, Kim SK, et al. Polymorphisms of irak1 gene on X chromosome is associated with hashimoto thyroiditis in korean children. Endocrinology. (2020) 161:bqaa088. doi: 10.1210/endocr/bqaa088
344. Li K, Li H, Xu W, Liu W, Du Y, He JF, et al. Research on the potential mechanism of gypenosides on treating thyroid-associated ophthalmopathy based on network pharmacology. Med Sci Monit. (2019) 25:4923–32. doi: 10.12659/msm.917299
345. Luo P, Liu D, Zhang Q, Yang F, Wong YK, Xia F, et al. Celastrol induces ferroptosis in activated hscs to ameliorate hepatic fibrosis via targeting peroxiredoxins and ho-1. Acta Pharm Sin B. (2022) 12:2300–14. doi: 10.1016/j.apsb.2021.12.007
346. Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. (2020) 136:726–39. doi: 10.1182/blood.2019002907
347. Neag EJ and Smith TJ. 2021 Update on thyroid-Associated ophthalmopathy. J Endocrinol Invest. (2022) 45:235–59. doi: 10.1007/s40618-021-01663-9
348. Su Y, Liu X, Fang S, Huang Y, Li Y, Zhong S, et al. Age-related difference in extraocular muscles and its relation to clinical manifestations in an ethnically homogenous group of patients with graves' Orbitopathy. Graefes Arch Clin Exp Ophthalmol. (2022) 260:583–9. doi: 10.1007/s00417-021-05377-9
349. Huang Y, Wu Y, Zhang S, Lu Y, Wang Y, Liu X, et al. Immunophenotype of lacrimal glands in graves orbitopathy: implications for the pathogenesis of th1 and th17 immunity. Thyroid. (2022) 32:949–61. doi: 10.1089/thy.2021.0671
350. Liu X, Cui Z, Chen X, Li Y, Qiu J, Huang Y, et al. Ferroptosis in the lacrimal gland is involved in dry eye syndrome induced by corneal nerve severing. Invest Ophthalmol Vis Sci. (2023) 64:27. doi: 10.1167/iovs.64.7.27
351. SanGiovanni JP, Chew EY, Clemons TE, Davis MD, Ferris FL 3rd, Gensler GR, et al. The relationship of dietary lipid intake and age-related macular degeneration in a case-control study: areds report no. 20. Arch Ophthalmol. (2007) 125:671–9. doi: 10.1001/archopht.125.5.671
352. Zhu M and Yu J. Salidroside alleviates ferroptosis in fac-induced age-related macular degeneration models by activating nrf2/slc7a11/gpx4 axis. Int Immunopharmacol. (2024) 142:113041. doi: 10.1016/j.intimp.2024.113041
353. Huang K, Deng H, Wang S, Zhang F, Huang G, Wang L, et al. Melanin-like nanomedicine functions as a novel rpe ferroptosis inhibitor to ameliorate retinal degeneration and visual impairment in dry age-related macular degeneration. Adv Healthc Mater. (2024) 13:e2401613. doi: 10.1002/adhm.202401613
354. Ouyang J, Zhou L, and Wang Q. Spotlight on iron and ferroptosis: research progress in diabetic retinopathy. Front Endocrinol (Lausanne). (2023) 14:1234824. doi: 10.3389/fendo.2023.1234824
355. Singh J, Sharma M, Jain N, Aftab I, Vikram N, Singh TP, et al. Lactoferrin and its nano-formulations in rare eye diseases. Indian J Ophthalmol. (2022) 70:2328–34. doi: 10.4103/ijo.IJO_303_22
356. Chan TC, Wilkinson Berka JL, Deliyanti D, Hunter D, Fung A, Liew G, et al. The role of reactive oxygen species in the pathogenesis and treatment of retinal diseases. Exp Eye Res. (2020) 201:108255. doi: 10.1016/j.exer.2020.108255
357. Moos WH, Faller DV, Glavas IP, Harpp DN, Kamperi N, Kanara I, et al. Treatment and prevention of pathological mitochondrial dysfunction in retinal degeneration and in photoreceptor injury. Biochem Pharmacol. (2022) 203:115168. doi: 10.1016/j.bcp.2022.115168
358. Zhuang S, Ma Y, Zeng Y, Lu C, Yang F, Jiang N, et al. Mettl14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the kcnq1ot1-mir-7-5p-tfrc axis. Cell Biol Toxicol. (2023) 39:1015–35. doi: 10.1007/s10565-021-09660-7
359. Millán I, Desco MDC, Torres-Cuevas I, Pérez S, Pulido I, Mena-Mollá S, et al. Pterostilbene prevents early diabetic retinopathy alterations in a rabbit experimental model. Nutrients. (2019) 12:82. doi: 10.3390/nu12010082
360. Wang W, Zhao H, and Chen B. Dj-1 Protects Retinal Pericytes against High Glucose-Induced Oxidative Stress through the Nrf2 Signaling Pathway. Sci Rep. (2020) 10:2477. doi: 10.1038/s41598-020-59408-2
361. Wang J, Zhang Q, Chen E, Zhao P, and Xu Y. Elabela promotes the retinal angiogenesis by inhibiting ferroptosis during the vaso-obliteration phase in mouse oxygen-induced retinopathy model. FASEB J. (2022) 36:e22257. doi: 10.1096/fj.202101785RRR
362. Mazzoli V, Zhong LH, Dang VT, Shi Y, and Werstuck GH. Characterization of retinal microvascular complications and the effects of endoplasmic reticulum stress in mouse models of diabetic atherosclerosis. Invest Ophthalmol Vis Sci. (2020) 61:49. doi: 10.1167/iovs.61.10.49
363. Wang C, Wang K, and Li P. Blueberry anthocyanins extract attenuated diabetic retinopathy by inhibiting endoplasmic reticulum stress via the mir-182/ogg1 axis. J Pharmacol Sci. (2022) 150:31–40. doi: 10.1016/j.jphs.2022.06.004
364. Rani EA, Janani R, Chonche MJ, and Vallikannan B. Lactucaxanthin regulates the cascade of retinal oxidative stress, endoplasmic reticulum stress and inflammatory signaling in diabetic rats. Ocul Immunol Inflammation. (2023) 31:320–8. doi: 10.1080/09273948.2022.2027464
365. Tang X, Li X, Zhang D, and Han W. Astragaloside-iv alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of mir-138-5p/sirt1/nrf2. Bioengineered. (2022) 13:8240–54. doi: 10.1080/21655979.2022.2049471
366. Zhang Y, Zhou X, Liang G, Cui M, Qiu Z, Xu J, et al. Iron-chelating and ros-scavenging polymers with thioketal and thioether bonds delivering ferroptosis inhibitor lip-1 provide a triple therapeutic strategy for retina ganglion cells in acute glaucoma. Adv Mater. (2025) 37:e2507526. doi: 10.1002/adma.202507526
367. Wang Z, Wu S, Zhu C, and Shen J. The role of ferroptosis in esophageal cancer. Cancer Cell Int. (2022) 22:266. doi: 10.1186/s12935-022-02685-w
368. Boskovic O, Medenica S, Radojevic N, and Zarkovic M. Etanercept in the treatment of graves' Ophthalmopathy with primary hypothyroidism and rheumatoid arthritis. Cent Eur J Immunol. (2019) 44:463–5. doi: 10.5114/ceji.2019.92803
369. Conrad M and Proneth B. Selenium: tracing another essential element of ferroptotic cell death. Cell Chem Biol. (2020) 27:409–19. doi: 10.1016/j.chembiol.2020.03.012
370. D'Aprile S, Denaro S, Pavone AM, Giallongo S, Giallongo C, Distefano A, et al. Anaplastic thyroid cancer cells reduce cd71 levels to increase iron overload tolerance. J Transl Med. (2023) 21:780. doi: 10.1186/s12967-023-04664-9
371. Ma R, Gan L, Guo J, Peng Z, Wu J, Harrison AR, et al. Insights into ferroptosis: targeting glycolysis to treat graves' Orbitopathy. J Clin Endocrinol Metab. (2022) 107:1994–2003. doi: 10.1210/clinem/dgac163
372. Yang Z, Huang R, Wang Y, Guan Q, Li D, Wu Y, et al. Sirt6 drives sensitivity to ferroptosis in anaplastic thyroid cancer through ncoa4-dependent autophagy. Am J Cancer Res. (2023) 13:464–74. doi: 10.62393/ajcr2023.464
373. Chen S, Diao J, Yue Z, and Wei R. Identification and validation of ferroptosis-related genes and immune cell infiltration in thyroid associated ophthalmopathy. Front Genet. (2023) 14:1118391. doi: 10.3389/fgene.2023.1118391
374. Liu L, Lian N, Shi L, Hao Z, and Chen K. Ferroptosis: mechanism and connections with cutaneous diseases. Front Cell Dev Biol. (2022) 10:1079548. doi: 10.3389/fcell.2022.1079548
375. Zhang F, Lin B, Huang S, Wu P, Zhou M, Zhao J, et al. Melatonin alleviates retinal ischemia-reperfusion injury by inhibiting P53-mediated ferroptosis. Antioxid (Basel). (2023) 12:1173. doi: 10.3390/antiox12061173
376. Xiang W, Li L, Zhao Q, Zeng Y, Shi J, Chen Z, et al. PEDF protects retinal pigment epithelium from ferroptosis and ameliorates dry AMD-like pathology in a murine model. Geroscience. (2024) 46:2697–714. doi: 10.1007/s11357-023-01038-3
377. Wu P, Zhao L, Du Y, Lu J, He Y, Shu Q, et al. Melatonin protects retinal pigment epithelium cells against ferroptosis in AMD via the PI3K/AKT/MDM2/P53 pathway. Front Pharmacol. (2025) 16:1543575. doi: 10.3389/fphar.2025.1543575
378. Yang B, Yang K, Chen Y, Li Q, Chen J, Li S, et al. Exposure of A2E to blue light promotes ferroptosis in the retinal pigment epithelium. Cell Mol Biol Lett. (2025) 30:22. doi: 10.1186/s11658-025-00700-2
379. Chen X, Wang Y, Wang JN, Cao QC, Sun RX, Zhu HJ, et al. m(6)A modification of circSPECC1 suppresses RPE oxidative damage and maintains retinal homeostasis. Cell Rep. (2022) 41:111671. doi: 10.1016/j.celrep.2022.111671
380. Zou R, Zhang X, Dai X, Yuan Y, Dai J, and Yuan F. The SDF-1α/MTDH axis inhibits ferroptosis and promotes the formation of anti-VEGF-resistant choroidal neovascularization by facilitating the nuclear translocation of SREBP1. Cell Biol Toxicol. (2025) 41:118. doi: 10.1007/s10565-025-10066-y
381. Yang B, Yang K, Chen J, and Wu Y. Crocin protects the 661W murine photoreceptor cell line against the toxic effects of all-trans-retinal. Int J Mol Sci. (2024) 25:10124. doi: 10.3390/ijms251810124
382. Shi W, Dong Y, Liu S, Li F, and Zhu C. Corilagin alleviates ferroptosis in diabetic retinopathy by activating the Nrf2 signaling pathway. BioMed Pharmacother. (2024) 179:117409. doi: 10.1016/j.biopha.2024.117409
383. Liu Q, Liu CQ, Yi WZ, Ouyang PW, Yang BF, Liu Q, et al. Ferroptosis contributes to microvascular dysfunction in diabetic retinopathy. Am J Pathol. (2024) 194:1078–89. doi: 10.1016/j.ajpath.2024.01.019
384. Liu Z, Gan S, Fu L, Xu Y, Wang S, Zhang G, et al. 1,8-Cineole ameliorates diabetic retinopathy by inhibiting retinal pigment epithelium ferroptosis via PPAR-γ/TXNIP pathways. BioMed Pharmacother. (2023) 164:114978. doi: 10.1016/j.biopha.2023.114978
385. Xi X, Chen Q, Ma J, Wang X, Zhang J, and Li Y. Sestrin2 ameliorates diabetic retinopathy by regulating autophagy and ferroptosis. J Mol Histol. (2024) 55:169–84. doi: 10.1007/s10735-023-10180-3
386. Zhang J, Chang K, Shangguan Y, Luo R, Bi Y, Yu Z, et al. Flotillin- 1 ameliorates experimental diabetic retinopathy by inhibiting ferroptosis in blood-retinal barrier. J Mol Med (Berl). (2025) 103:671–85. doi: 10.1007/s00109-025-02544-x
387. Shao J, Bai Z, Zhang L, and Zhang F. Ferrostatin-1 alleviates tissue and cell damage in diabetic retinopathy by improving the antioxidant capacity of the Xc(-)-GPX4 system. Cell Death Discov. (2022) 8:426. doi: 10.1038/s41420-022-01141-y
388. Liao Q, Li Y, Cui M, and Liu M. m6A demethylase ALKBH5 reduces ferroptosis in diabetic retinopathy through the m6A-YTHDF1-ACSL4 axis. Diabetes Med. (2025) 42:e70033. doi: 10.1111/dme.70033
389. Cai Y, Peng S, Duan B, Shao Y, Li X, Zou H, et al. Isoquercetin alleviates diabetic retinopathy via inhibiting p53-mediated ferroptosis. Cell Biol Int. (2025) 49:852–64. doi: 10.1002/cbin.70027
390. Wang Y, Song SY, Song Y, Wang Y, Wan ZW, Sun P, et al. Resveratrol protects müller cells against ferroptosis in the early stage of diabetic retinopathy by regulating the nrf2/GPx4/PTGS2 pathway. Mol Neurobiol. (2025) 62:3412–27. doi: 10.1007/s12035-024-04496-8
391. Yuan F, Han S, Li Y, Li S, Li D, Tian Q, et al. miR-214-3p attenuates ferroptosis-induced cellular damage in a mouse model of diabetic retinopathy through the p53/SLC7A11/GPX4 axis. Exp Eye Res. (2025) 253:110299. doi: 10.1016/j.exer.2025.110299
392. Li S, Lu S, Wang L, Liu S, Zhang L, Du J, et al. Effects of amygdalin on ferroptosis and oxidative stress in diabetic retinopathy progression via the NRF2/ARE signaling pathway. Exp Eye Res. (2023) 234:109569. doi: 10.1016/j.exer.2023.109569
393. Peng Y, Hu L, Xu H, Fang J, and Zhong H. Resveratrol alleviates reactive oxygen species and inflammation in diabetic retinopathy via SIRT1/HMGB1 pathway-mediated ferroptosis. Toxicol Appl Pharmacol. (2025) 495:117214. doi: 10.1016/j.taap.2024.117214
394. Zhou J, Mei X, Zhan L, Liu X, Wang B, Xu B, et al. Structural characterization and protective effects of a glucuronogalactomannan from Tetrastigma hemsleyanum against diabetic retinopathy via targeting ferroptosis pathway. Carbohydr Polym. (2025) 366:124059. doi: 10.1016/j.carbpol.2025.124059
395. Ren M, Xu Q, Luan J, Ni Y, and Xie B. Mir-509-3p targets SLC25A13 to regulate ferroptosis and protect retinal endothelial cells in diabetic retinopathy. Acta Diabetol. (2025) 62:831–44. doi: 10.1007/s00592-024-02400-3
396. Zhong L, Meng X, Huang J, Hao W, and Zuo Y. Expression of YAP suppresses cell proliferation and elevates the sensitivity of chemotherapy in retinoblastoma cells through lipid-peroxidation induced ferroptosis. Chin Clin Oncol. (2023) 12:52. doi: 10.21037/cco-23-97
397. Chi M, Zhao Y, Yuan B, Qiu Z, Peng R, and Hong J. MiR-23a-3p targets PTEN as a novel anti-ferroptosis regulator in Fuchs endothelial corneal dystrophy. Exp Eye Res. (2025) 250:110180. doi: 10.1016/j.exer.2024.110180
398. Wang H, Yang Y, Yu H, Ma L, Qi X, Qu J, et al. Self-cascade API nanozyme for synergistic anti-inflammatory, antioxidant, and ferroptosis modulation in the treatment of corneal neovascularization. Small. (2025) 21:e2407751. doi: 10.1002/smll.202407751
399. Chu G, Wang J, Wang Z, Cai X, Shi S, Qing Q, et al. Autologous serum from sjögren's syndrome patients mitigates ferroptosis in hyperosmolarity-induced corneal epitheliopathy. J Ocul Pharmacol Ther. (2025) 41:397–407. doi: 10.1089/jop.2025.0025
400. Zhao L, Dong X, Guo B, Song J, and Bi H. Quercetin-loaded exosomes delivery system prevents myopia progression by targeting endoplasmic reticulum stress and ferroptosis in scleral fibroblasts. Mater Today Bio. (2025) 32:101896. doi: 10.1016/j.mtbio.2025.101896
401. Hou C, Xiao J, Wang Y, Pan X, Liu K, Lu K, et al. Astaxanthin activated the slc7a11/gpx4 pathway to inhibit ferroptosis and enhance autophagy, ameliorating dry eye disease. Front Pharmacol. (2024) 15:1407659. doi: 10.3389/fphar.2024.1407659
402. Wang J, Liu Y, Zong B, Zhao S, Li Y, Zhang Z, et al. Qingxuan Runmu Yin alleviates dry eye disease via inhibition of the HMOX1/HIF-1 pathway affecting ferroptosis. Front Pharmacol. (2024) 15:1391946. doi: 10.3389/fphar.2024.1391946
403. Feng L, Wang C, Zhang C, Zhang W, Zhu W, He Y, et al. p38 MAPK inhibitor SB202190 suppresses ferroptosis in the glutamate-induced retinal excitotoxicity glaucoma model. Neural Regener Res. (2024) 19:2299–309. doi: 10.4103/1673-5374.391193
404. Wang J, Liu Y, Rong R, and Xia X. Bilirubin-polymer nanocarriers enable targeted farrerol delivery for glaucoma neuroprotection via Nrf2-mediated ferroptosis/apoptosis inhibition. Mater Today Bio. (2025) 35:102304. doi: 10.1016/j.mtbio.2025.102304
405. Feng Y, Wang X, Li P, Shi X, Prokosch V, and Liu H. Exogenous hydrogen sulfide and NOX2 inhibition mitigate ferroptosis in pressure-induced retinal ganglion cell damage. Biochim Biophys Acta Mol Basis Dis. (2025) 1871:167705. doi: 10.1016/j.bbadis.2025.167705
406. Chen X, Rong Y, Jiang Y, Zhang Q, Xiang S, Chen Z, et al. Vitamin K1 alleviates retinal inflammation following acute ocular hypertension by modulating microglial ferroptosis. Invest Ophthalmol Vis Sci. (2025) 66:46. doi: 10.1167/iovs.66.4.46
407. Zuo X, Wang X, Xie J, and Jia Y. Emodin alleviates the damage to lens epithelial cells in diabetic cataract by repressing the p53-mediated ferroptosis pathway. Int Ophthalmol. (2025) 45:141. doi: 10.1007/s10792-025-03513-6
408. Cui T, Liu Y, Gao F, Wang J, Lu L, Zhang J, et al. Asparagine alleviates naphthalene-induced lens opacity by suppressing ferroptosis. Exp Eye Res. (2025) 255:110362. doi: 10.1016/j.exer.2025.110362
409. Kong D, Liu Y, Li L, Wang H, Li K, and Zheng G. Astaxanthin ameliorates oxidative stress in lens epithelial cells by regulating GPX4 and ferroptosis. Chem Biol Interact. (2023) 383:110684. doi: 10.1016/j.cbi.2023.110684
410. Chhunchha B, Kubo E, Krueger RR, and Singh DP. Hydralazine revives cellular and ocular lens health-span by ameliorating the aging and oxidative-dependent loss of the nrf2-activated cellular stress response. Antioxid (Basel). (2023) 12:140. doi: 10.3390/antiox12010140
411. Zheng Y, Liu Y, Ge J, Wang X, Liu L, Bu Z, et al. Resveratrol protects human lens epithelial cells against H2o2-induced oxidative stress by increasing catalase, sod-1, and ho-1 expression. Mol Vis. (2010) 16:1467–74.
412. Lledó VE, Alkozi HA, Sánchez-Naves J, Fernandez-Torres MA, and Guzman-Aranguez A. Melatonin counteracts oxidative damage in lens by regulation of nrf2 and nlrp3 inflammasome activity. Exp Eye Res. (2022) 215:108912. doi: 10.1016/j.exer.2021.108912
413. Babizhayev MA, Burke L, Micans P, and Richer SP. N-acetylcarnosine sustained drug delivery eye drops to control the signs of ageless vision: glare sensitivity, cataract amelioration and quality of vision currently available treatment for the challenging 50,000-patient population. Clin Interv Aging. (2009) 4:31–50.
414. Yang M, So KF, Lam WC, and Lo ACY. Novel programmed cell death as therapeutic targets in age-related macular degeneration? Int J Mol Sci. (2020) 21:7279. doi: 10.3390/ijms21197279
415. Dou J, Liu X, Yang L, Huang D, and Tan X. Ferroptosis interaction with inflammatory microenvironments: mechanism, biology, and treatment. BioMed Pharmacother. (2022) 155:113711. doi: 10.1016/j.biopha.2022.113711
416. Tan Y, Huang J, Li D, Zou C, Liu D, and Qin B. Single-cell rna sequencing in dissecting microenvironment of age-related macular degeneration: challenges and perspectives. Ageing Res Rev. (2023) 90:102030. doi: 10.1016/j.arr.2023.102030
417. Campochiaro PA, Marcus DM, Awh CC, Regillo C, Adamis AP, Bantseev V, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 ladder clinical trial. Ophthalmology. (2019) 126:1141–54. doi: 10.1016/j.ophtha.2019.03.036
418. Chen X, Comish PB, Tang D, and Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. (2021) 9:637162. doi: 10.3389/fcell.2021.637162
419. Agmon E, Solon J, Bassereau P, and Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. (2018) 8:5155. doi: 10.1038/s41598-018-23408-0
420. Liu X and Gong T. Artificial intelligence and evidence-based research will promote the development of traditional medicine. Acupuncture Herbal Med. (2024) 4:134–5. doi: 10.1097/HM9.0000000000000100
421. Zhou H, Li J, Sun F, Wang F, Li M, Dong Y, et al. A review on recent advances in aloperine research: pharmacological activities and underlying biological mechanisms. Front Pharmacol. (2020) 11:538137. doi: 10.3389/fphar.2020.538137
422. Liu M, Li W, Chen Y, Wan X, and Wang J. Fucoxanthin: A promising compound for human inflammation-related diseases. Life Sci. (2020) 255:117850. doi: 10.1016/j.lfs.2020.117850
423. Hao M, Liu Y, Chen P, Jiang H, and Kuang HY. Astragaloside iv protects rgc-5 cells against oxidative stress. Neural Regener Res. (2018) 13:1081–6. doi: 10.4103/1673-5374.233452
424. Meng F, Guo B, Ma YQ, Li KW, and Niu FJ. Puerarin: A review of its mechanisms of action and clinical studies in ophthalmology. Phytomedicine. (2022) 107:154465. doi: 10.1016/j.phymed.2022.154465
425. Boateng ID. Potentialities of ginkgo extract on toxicants, toxins, and radiation: A critical review. Food Funct. (2022) 13:7960–83. doi: 10.1039/d2fo01298g
426. Li Y, Wang K, Zhu X, Cheng Z, Zhu L, Murray M, et al. Ginkgo biloba extracts protect human retinal müller glial cells from T-bhp induced oxidative damage by activating the ampk-nrf2-nqo-1 axis. J Pharm Pharmacol. (2023) 75:385–96. doi: 10.1093/jpp/rgac095
427. Zhu Z, Wang Y, Deng Z, Lei P, Liu Q, Guo J, et al. The hemerocallis citrinaExtracts ameliorate radiation-induced ferroptosis in lo2 cells through the nrf2-xct/gpx4 pathway. Acupuncture Herbal Med. (2024) 4:513–24. doi: 10.1097/HM9.0000000000000120
428. Husain A, Chanana H, Khan SA, Dhanalekshmi UM, Ali M, Alghamdi AA, et al. Chemistry and pharmacological actions of delphinidin, a dietary purple pigment in anthocyanidin and anthocyanin forms. Front Nutr. (2022) 9:746881. doi: 10.3389/fnut.2022.746881
429. Ngamsamer C, Sirivarasai J, and Sutjarit N. The benefits of anthocyanins against obesity-induced inflammation. Biomolecules. (2022) 12:852. doi: 10.3390/biom12060852
Keywords: ferroptosis, eye, therapy, iron metabolism, TAO
Citation: Wei Y, Lin Y, Li Y, Liu J, Yang Y, Chen H, Han Z, Wang K, Qian T, Ju Y and Zheng W (2025) Redefining cell death: ferroptosis as a game-changer in ophthalmology. Front. Immunol. 16:1709354. doi: 10.3389/fimmu.2025.1709354
Received: 20 September 2025; Accepted: 10 November 2025; Revised: 05 November 2025;
Published: 28 November 2025.
Edited by:
Uzma Saqib, Devi Ahilya University, IndiaReviewed by:
Dongcheng Liu, Jinan University, ChinaYao Tong, University of California, San Francisco, United States
Copyright © 2025 Wei, Lin, Li, Liu, Yang, Chen, Han, Wang, Qian, Ju and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Qian, cXQyMTZAMTYzLmNvbQ==; Yuan Ju, anV5dWFuMDQzMUAxNjMuY29t; Wei Zheng, ZHJ6aGVuZ3dlaUAxNjMuY29t
†These authors have contributed equally to this work